
Non-precious transition metal single-atom catalysts for the oxygen reduction reaction: progress and prospects
Penggang
Jiao
a,
Donghao
Ye
b,
Chunyou
Zhu
c,
Shuai
Wu
a,
Chunling
Qin
a,
Cuihua
An
 *a,
Ning
Hu
d and
Qibo
Deng
*a
*a,
Ning
Hu
d and
Qibo
Deng
*a
aKey Laboratory of Hebei Province on Scale-span Intelligent Equipment Technology, and School of Mechanical Engineering, School of Materials Science and Engineering, Hebei University of Technology, Tianjin 300401, China. E-mail: ancuihua@hebut.edu.cn; qibodeng@hebut.edu.cn
bWuhan Marine Electric Propulsion Research Institute, Wuhan 430064, China
cHunan Aerospace Kaitian Water Services Co., Ltd., Changsha 410100, China
dState Key Laboratory of Reliability and Intelligence Electrical Equipment, Hebei University of Technology, Tianjin, 300130, China
First published on 7th September 2022
Abstract
The massive exploitation and use of fossil resources have created many negative issues, such as energy shortage and environmental pollution. It prompts us to turn our attention to the development of new energy technologies. This review summarizes the recent research progress of non-precious transition metal single-atom catalysts (NPT-SACs) for the oxygen reduction reaction (ORR) in Zn–air batteries and fuel cells. Some commonly used preparation methods and their advantages/disadvantages have been summarized. The factors affecting the ORR performances of NPT-SACs have been focused upon, such as the substrate type, coordination environment and nanocluster effects. The loading mass of a metal atom has a direct effect on the ORR performances. Some general strategies for stabilizing metal atoms are included. This review points out some existing challenges of NPT-SACs, and also provides ideas for designing and synthesizing NPT-SACs with excellent ORR performances. The large-scale preparation and commercialization of NPT-SACs with excellent ORR properties are prospected.
1. Introduction
Environmental pollution and increasing energy demand have forced mankind to turn to renewable and clean energy to gradually replace fossil fuels.1–10 A variety of devices have been developed for energy storage and conversion.11–16 Metal–air batteries and fuel cells have broad application prospects due to their excellent performances.17–20 As an important part of the electrode reaction, the reaction thermodynamics and kinetics of the ORR have determined the actual working efficiency of metal–air batteries and fuel cells.21–27 According to the number of transferred electrons, the ORR can be divided into two pathways, namely a four-electron reaction and a two-electron reaction.25 The four-electron reaction has a higher oxygen utilization rate than the two-electron reaction, allowing the maximum efficiency to be obtained in metal–air batteries and fuel cells. The overall equation of the four-electron reaction in acidic and alkaline electrolytes can be expressed in the following forms, respectively.28| O2 + 4H+ + 4e− → 2H2O (Acidic) |
| O2 + 2H2O + 4e− → 4OH− (Alkaline) |
The four-electron process in an alkaline electrolyte can be represented by the following form.
| O2 (g) + * → *O2 |
| *O2 + H2O(l) + e− → *OOH + OH− |
| *OOH + e− → *O + OH− |
| *O + H2O(l) + e− → *OH + OH− |
| *OH + e− → *OH− + * |
* indicates an active site. The ORR occurs in the electrode–electrolyte interface, which involves mass transport and electron transfer processes, reactant/intermediate/product adsorption and desorption processes, etc. Each step of the aforementioned processes may limit the reaction rate of the ORR. Therefore, it is very necessary to select efficient and stable catalysts to accelerate the reaction process. Recently, the emergence and development of in situ techniques and density functional theory (DFT) have been of great significance for the study of the reaction process of the ORR.29 For the ORR, platinum-based catalysts are the most efficient catalysts.30 A wide range of platinum-based ORR catalysts have been successfully prepared, which exhibit excellent catalytic performances.31 For example, a nanoporous gold substrate coated Pt layer has been prepared and its ORR catalytic activity has been improved by compressing the lattice parameters of Pt through strain engineering.32 This core–shell structured PtAuCu@Pt catalyst has been obtained by preparing a Pt shell on the PtAuCu core.33 Yang et al. have successfully prepared a Pt–Fe alloy with a hollow structure for an ORR catalyst via alloying Pt and transition metal iron.34 For platinum single crystals, the ORR activity of different crystal faces is also different in acidic or alkaline solutions.35 High-entropy alloys (HEAs) of noble metals such as platinum with other transition metals have been reported to exhibit high ORR activity.36 The mass current density of PdCuPtNiCo HEAs formed by annealing of PdCu@PtNiCo is nearly 1 time that of the former.37 In 2011, Zhang et al. creatively proposed the concept of “single atom” and found that the electron transfer between Pt single atoms and the substrate improved the activity and stability of the catalyst.38 Liu et al. used a catalyst formed by anchoring Pt single atoms with defects on carbon substrates for the ORR.39
However, precious metals such as the platinum group have the disadvantages of scarcity of reserves and high prices. In order to reduce the cost of catalysts, the amount of precious metals should be reduced and their intrinsic activities should be improved.40–43 Another idea is to use non-precious metals directly instead of Pt. Non-precious transition metal elements (NPTs) are abundant and inexpensive, and NPT catalysts have also been found to exhibit excellent ORR catalytic performances.44–47 Nanoscale catalysts are typically heterogeneous catalysts. Compared with homogeneous catalysts, only the surface part of nanoparticles plays a catalytic role.48 The composition, structure, morphology and size of electrochemical catalysts have been extensively explored.49–52 Traditional homogeneous catalysts have good catalytic efficiency but poor stability, and are difficult to separate. Heterogeneous catalysts don't require separation and have good stability but low catalyst utilization. Single-atom catalysts (SACs) have become a bridge between homogeneous and heterogeneous catalysts due to their special properties.53–59 Fe-Based single-atom catalysts (Fe-SACs) are found to exhibit excellent ORR performance, so researchers have conducted extensive explorations to optimize the performance of Fe-SACs.60 Other single-atom catalysts such as Mn-SACs, Co-SACs, etc. have similar properties. For these reasons, researchers’ enthusiasm for research on NPT-SACs has not diminished in recent years. The catalytic performances of ORR catalysts are mainly affected by some factors, such as the intrinsic activity of the active site, the density of the active site, and the structure of the substrate material. Therefore, the density of active sites can be greatly increased by manipulating the size of metal particles. When reduced to the size of a single atom, if these exposed single-atom active sites are all accessible, the metal utilization can approach 100%.61 From blocks to nanoparticles to single atoms, the surface energy increases with the decrease of the volume, resulting in easy agglomeration of metal atoms.62 For this reason, increasing the single-atom loading while maintaining atomic dispersion is difficult to achieve. By introducing nitrogen-rich molecules into ZIF-8 in situ to trap Fe single atoms, the loading mass of Fe single atoms increases to about 3.5 wt% content.63 The Fe atomic loading and ORR performance of SA-Fe-NHPC are investigated. The results show that in the range of 1.07–1.25 wt%, the half-wave potential of SA-Fe-NHPC increases with the increase of Fe atom loading.64 FeSA catalysts with an Fe loading of 3.5 wt% have onset potentials as high as 0.95 V (vs. RHE).65 In addition, when the metal is reduced to atomic size, its electronic structure changes. By adjusting the coordination environment of single atoms and the interaction between single atoms and substrates, the intrinsic activity and stability of catalysts can be changed, and the final ORR performances can be enhanced.66 N is doped on a carbon substrate and then coordinated with Co atoms to form Co–N4 sites. The coordinated N changed the electronic structure of the central atom Co, and then the catalyst shows excellent ORR catalytic activity and stability.58,67 Compared with Fe(Fc)-N-C without S doping, Fe(Fc)-N/S-C has a higher onset potential and half-wave potential, indicating that S doping can enhance the ORR activity of Fe(Fc)-N-C.68 Cu-SA/NC(meso)-x can adjust the ratio of Cu1+/Cu2+ by changing the ratio of Cu salt to urea in the precursor. DFT calculation results show that the Cu-N2C2 sites formed by Cu1+ have higher ORR activity than the Cu–N4 sites formed by Cu2+.69 The mass transfer process greatly limits the rate of the ORR, which requires the substrate material to have a microstructure that is conducive to mass transfer. Co-SAs@NHOPC catalysts with a hierarchical pore structure were prepared by further pyrolysis of ZnCo-ZIFs grown in polyethylene microsphere templates.70 Co-SAs@NHOPC exhibits excellent ORR performances due to this ordered hierarchical pore structure favoring mass transfer. The petal-like structure of FeNC-D0.5 possesses high mesoporosity, large external surface area and appropriate hydrophobicity. The assembled ZAB (Zn–air battery) and MFC (methanol fuel cell) using FeNC-D0.5 exhibit extremely high power densities.71 The ordered mesoporous structure of Fe–N–C/N-OMC substrates facilitates the formation of densely exposed active sites and facilitates mass and electron transfer processes. Fe–N–C/N-OMC exhibits excellent ORR performance in both acidic and alkaline electrolytes.72
In this review, we first emphasize the significance of developing NPT-SACs for ORR catalysts and some major factors that affect the ORR performance of NPT-SACs. Some common methods for synthesizing NPT-SACs are also summarized in this review. The morphology and properties of the prepared samples as well as the advantages and existing problems of these preparation methods are described in section II. The effects of coordination between metal atoms and the substrate, metal atoms, and metal atoms and nanoclusters are discussed in detail in section III. Aiming at the problems of easy migration and agglomeration of metal atoms and low loading, several strategies for stabilizing metal atoms (steric hindrance strategy, defect stabilization strategy and coordination strategy) are summarized. At the end, some problems existing in the field of NPT-SACs catalyzing the ORR are pointed out and prospects are proposed.
2. Preparation method
It has been more than ten years since the concept of SACs is proposed and developed, during which a large number of methods have been tried to prepare SAC catalysts. In this section, we summarize several of the most commonly used preparation methods, and describe the advantages and disadvantages of each method. Understanding these will help readers to evaluate the preparation of SACs from multiple perspectives, such as optimization of the ORR performance of SACs, the economics of preparing SACs, and the feasibility of mass production (Table 1).| Electrocatalyst | Electrolyte | E 1/2 V vs. RHE | Tafel slope (mV dec−1) | Kinetic current density (JK) mA cm−2 | Ref. |
|---|---|---|---|---|---|
| Z8@DA-FIP-950-C | 0.1 M HClO4 | 0.828 | 64.1 | 21.342 (0.8 V) | 74 |
| FeNP@Fe–N–C-950 | 0.1 M KOH | 0.84 | 67 | 4.9 (0.85) | 75 |
| Mn–N–C | 0.1 M KOH | 0.86 | 89.2 | — | 76 |
| Mn–N–C–OAc-10-second | 0.1 M HClO4 | 0.80 | 69.38 | 0.41 (0.85) | 79 |
| 0.1 M KOH | 0.94 | 64.24 | 96.86 (0.85) | ||
| M15-FeNC-NH3 | 0.1 M HClO4 | 0.782 | 58.3 | — | 80 |
| CAN-Pc(Fe/Co) | 0.1 M KOH | 0.84 | 54 | — | 82 |
| FeN4CB | 0.1 M HClO4 | 0.82 | 85.41 | — | 83 |
| 0.1 M KOH | 0.84 | 79.66 | — | ||
| Co-CMS | 0.1 M KOH | 0.83 | 53.97 | — | 84 |
| FeNC-900-8 | 0.1 M KOH | 0.88 | 63 | — | 85 |
| Co@NCB | 0.1 M KOH | 0.851 | — | — | 86 |
| SA-Fe-NC | 0.1 M KOH | 0.88 | 68 | — | 93 |
| 0.1 M HClO4 | 0.79 | 87 | — | ||
| Fe–N/P–C-700 | 0.1 M KOH | 0.867 | — | — | 95 |
| Co1–N3PS/HC | 0.1 M KOH | 0.920 | 31 | 25.8 (0.775 V) | 96 |
| 0.5 M H2SO4 | 0.790 | 43 | 10.0 (0.775 V) | ||
| Co@DMOF-900 | 0.1 M KOH | 0.866 | — | 97 | |
| Fe–N/S–C | 0.1 M KOH | 0.882 | 49.5 | 5.87 (0.88 V) | 99 |
| 0.1 M HClO4 | 0.751 | — | — | ||
| Fe–N/C-SAC | 0.1 M KOH | 0.91 | 52 | 55 (0.85 V) | 100 |
| Fe–N4-PN | 0.1 M KOH | 0.91 | 58 | 24.04 (0.85 V) | 113 |
| Co-PTS-COPs@MWCNTs | 0.1 M KOH | 0.835 | 71 | — | 114 |
| CoSAs/N-CNS | 0.1 M KOH | 0.91 | 73.2 | 33.17 (0.85 V) | 115 |
| Fe1/d-CN | 0.1 M KOH | 0.950 | 54 | 22.7 (0.9 V) | 116 |
| 0.5 M H2SO4 | 0.781 | 64 | 25.6 (0.75 V) | ||
| 0.1 M PBS | 0.605 | 98 | 17.1 (0.55 V) | ||
| Fe/Ni–NX/OC | 0.1 M KOH | 0.938 | 59.9 | 28.1 (0.90 V) | 102 |
| 0.1 M HClO4 | 0.840 | 60.1 | — | ||
Co-NCA@F127–1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.56 :0.56 |
0.1 M KOH | 0.805 | 73.2 | — | 117 |
| Fe–N–C-900 | 0.1 M KOH | 0.90 | 83.6 | 19.69 (0.85 V) | 106 |
| Fe-ACSA@NCs | 0.1 M KOH | 0.90 | 78 | — | 123 |
| Co-SAs/SNPs@NC | 0.1 M KOH | 0.898 | 65 | 60.68 (0.85 V) | 124 |
| Fe3O4@FeNCs | 0.1 M KOH | 0.890 | 58.8 | — | 125 |
| Mn–Fe–N–C | 0.1 M HClO4 | 0.799 | 98.6 | — | 126 |
2.1 High temperature pyrolysis method
High-temperature pyrolysis is a common step for preparing single-atom catalysts, generally including template-assisted pyrolysis, metal–organic framework pyrolysis, special metal precursor complex/mixture pyrolysis, etc.73 High-temperature pyrolysis is often accompanied by nitrogen doping, pyrolysis and carbonization of the substrate framework, formation of active sites, and subsequent acid treatment. The pyrolysis temperature has a significant effect on the performances of the prepared catalysts. Issues such as the migration and agglomeration of metal atoms during pyrolysis are also worthy of our attention. Therefore, the choice of an appropriate method is very important to ensure that pyrolysis forms a high density of atomically dispersed active sites.There's no need for acid treatment after pyrolysis, in line with the concept of green chemistry. The precursor Z8@DA-FIP is obtained by polymerizing and coating the outer layer of ZIF-8 with dopamine (DA) and Fe-phenanthroline complexes.74 Z8@DA-FIP-950-C has been obtained by pyrolysis of the Z8@DA-FIP precursor (Fig. 1a). Z8@DA-FIP-950-C has been observed to have a dodecahedral structure similar to ZIF-8, as shown in Fig. 1b. A large number of pores and defects are formed during the pyrolysis process. The macroporous and mesoporous structures are beneficial for the exposure of active sites and mass transfer. The resulting Z8@DA-FIP-950-C forms a high density of Fe–N4 sites, as shown in Fig. 1c, due to the restricted Fe aggregation by PDA. DFT calculations suggest that the presence of defects leads to better activity at Fe–N4 sites. The excellent ORR performance of Z8@DA-FIP-950-C is fully demonstrated in Fig. 1d and e. The half-wave potential (E1/2) of Z8@DA-FIP-950-C in 0.1 M HClO4 electrolyte reaches 0.828 V (vs. RHE), which is close to that of commercial Pt/C. The kinetic current density (JK) of Z8@DA-FIP-950-C is 21.342 mA cm−2 (at 0.8 V). Z8@DA-FIP-950-C has a high peak power density in a H2/air PEMFC (proton exchange membrane fuel cell). A bifunctional catalyst FeNP@Fe–N–C-950 for the ORR/OER has been prepared by pyrolysis of the ZIF-8 precursor containing melamine and ferrous ions (Fig. 1f).75 Interestingly, the pyrolysis product (FeNP@Fe–N–C-950) forms an intertwined CNT structure and doesn't maintain the morphology of ZIF-8 (Fig. 1g). The Fe single atoms are attached to the outer wall of the carbon nanotubes, and the Fe nanoclusters (acid treatment can remove) are confined in the carbon nanotubes. Melamine promotes the formation of carbon nanotubes during the pyrolysis process. The formation of dense Fe–N4 active sites benefits from the large specific surface area of the substrate (Fig. 1h). The massive mesoporous structure of carbon nanotubes is more favorable for mass transfer than the single microporous structure of traditional MOFs. As depicted in Fig. 1i, the pyrolysis temperature also leads to different ORR performances of the catalysts. FeNP@Fe–N–C-950 with a pyrolysis temperature of 950 °C has the highest E1/2 (0.84 V vs. RHE). The low pyrolysis temperature will lead to the residual Zn species, and the high temperature is not beneficial for the formation of single-atom active sites, which are not conducive to the ORR performance of the catalyst.75 Precursors have been obtained from manganese nitrate, phenanthroline, etc. Mn–N–C catalysts with mesoporous amorphous carbon supports have been prepared from the pyrolysis of precursors.76 The atomic dispersion of Mn can be promoted by adjusting the ratio of manganese salt to phenanthroline to form specific coordination complexes. The resulting product of pyrolysis shows no nanoparticle formation and therefore doesn't require acid treatment, which would otherwise destroy the substrate structure. The ORR test of Mn–N–C in 0.1 M KOH solution shows that the E1/2 of Mn–N–C is 0.86 V (vs. RHE). The comprehensive performance of Mn–N–C is similar to that of Pt/C. This method is environmentally friendly and can be mass-produced, in line with the concept of green chemistry.
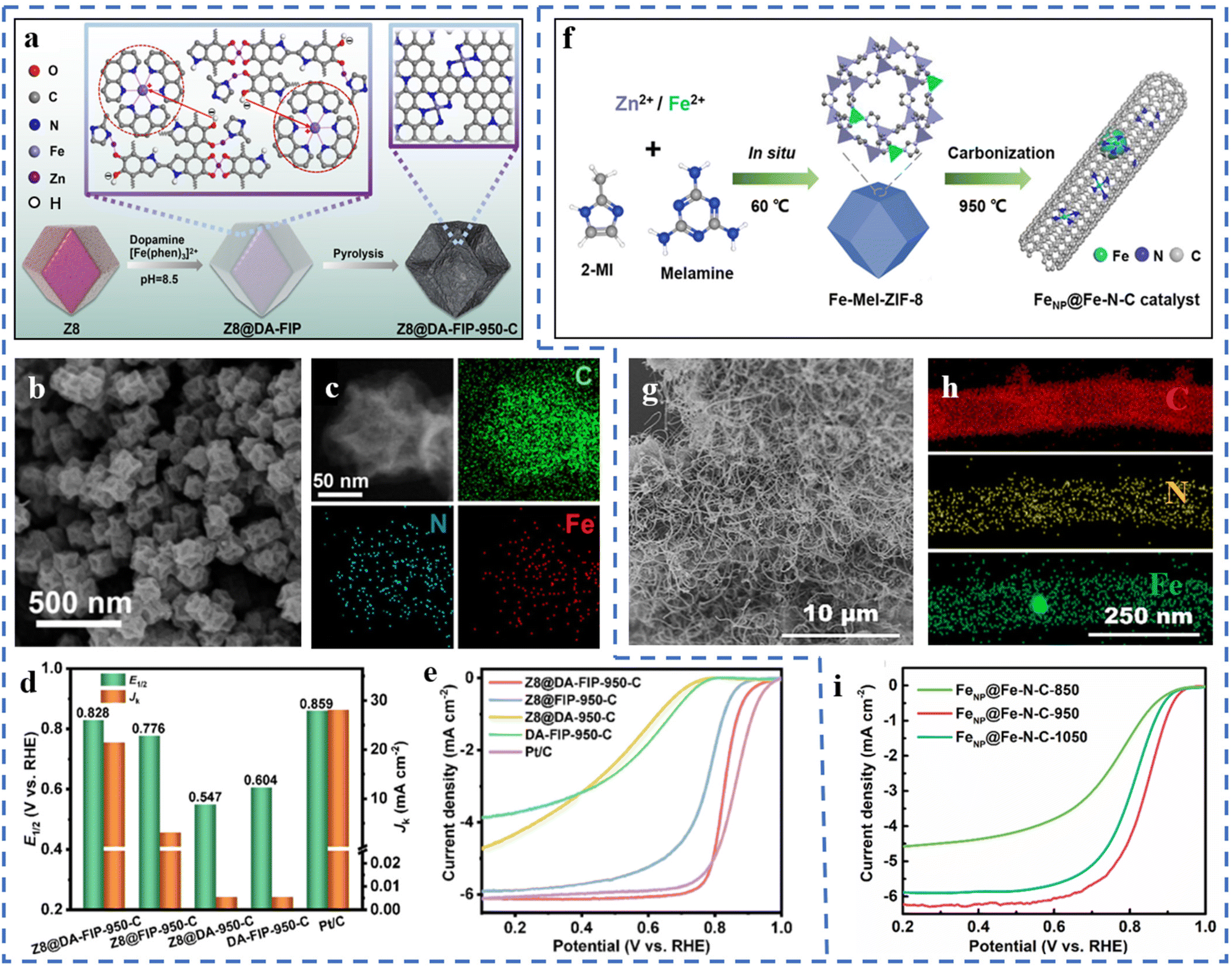 | ||
| Fig. 1 (a) Schematic of the production of Z8@DA-FIP-950-C. (b) SEM images of Z8@DA-FIP-950-C. (c) HAADF-STEM images and the corresponding EDS mapping images of Z8@DA-FIP-950-C. (d) E1/2 and JK of Z8@DA-FIP-950-C and reference catalysts in 0.1 M HClO4 with a rotation rate of 1600 rpm at 0.8 V. (e) LSV curves of Z8@DA-FIP-950-C and reference catalysts in 0.1 M HClO4 with a rotation rate of 1600 rpm. Reproduced with permission from ref. 74. Copyright © 2022, ELSEVIER. (f) Schematic diagram of the synthesis of FeNP@Fe–N–C catalysts. (g) SEM images of the FeNP@Fe–N–C catalyst. (h) EDX elemental mapping of the FeNP@Fe–N–C catalysts. (i) LSV curves of the catalysts obtained at different pyrolysis temperatures in 0.1 M KOH. Reproduced with permission from ref. 75. Copyright © 2022, ELSEVIER. | ||
2.2 Ball milling method
The mechanical energy generated by high-energy ball milling enables the molecules to acquire enough energy to cause the breaking of chemical bonds and the formation of new bonds.77 The advantages of high-energy ball milling to synthesize SACs are simplicity, low cost, large-scale preparation, and easy adjustment of technical parameters. The disadvantages of high-energy ball milling are that the energy consumption is high and impurities are easily introduced, resulting in unsatisfactory product performance.78ZIF-8 is often used as a precursor for the synthesis of single-atom catalysts. Mn is uniformly dispersed on ZIF-8 by ball milling, which effectively avoids the formation of inert compounds during subsequent pyrolysis. A Mn–N–C–OAc-10-second catalyst has been successfully prepared for the ORR (Fig. 2a).79 The selection of manganese salts for the preparation of precursors also plays an important role in whether the Mn species can easily form inert compounds during the pyrolysis process. The X-ray diffraction results are given in Fig. 2b. Mn(OAc)2·4H2O was used to synthesize precursors by ball milling, which can effectively avoid the formation of inert compounds. The E1/2 of Mn–N–C–OAc-10-second is found to be 0.80 V (vs. RHE) and 0.944 V (vs. RHE) in 0.1 M KClO4 and KOH solution, respectively (Fig. 2c and d). A M15-FeNC-NH3 catalyst has been prepared for the ORR via ball milling, microwave-assisted pyrolysis, and pyrolysis.80 The ORR performances in 0.1 M HClO4 solution are excellent: E1/2 = 0.782 V (vs. RHE), restricted diffusion-limited current density JL = 6.14 mA cm−2, and the Tafel slope is 58.3 mV dec−1. The advantage of this method is that ball milling and microwave assistance make the achievement of large-scale production easy. The performance of the as-produced catalyst has a slight gap compared to that of commercial Pt/C. The catalytic performance of M15-FeNC-NH3 is expected to improve in large-scale production by optimizing the equipment to achieve sufficient contact activation of M15-FeNC and NH3. Different from the physical mixing of traditional ball milling, a top-down abrasion method to prepare single atoms has been developed. Ball milling in this experiment is mainly used to generate mechanochemical reactions. This method can be used to synthesize Fe, Co, Ni, Cu and other metals that can form stable chemical bonds with the matrix. The shape and loading of metal atoms can be regulated by changing the parameters of ball milling (such as the rotation speed and time) and adjusting the amount of graphite added. This preparation method is also suitable for some oxide-based SACs. No acid is required throughout the process, which is environmentally friendly and easy to mass-produce.81 Metal phthalocyanine material contains natural M–N4 sites. According to the process of Fig. 2e, the two-dimensional metal phthalocyanine CAN-Pc(Fe/Co) can be obtained by ball milling and peeling off metal polyphthalocyanines with different Fe/Co atomic ratios.82 In the two-dimensional metal phthalocyanine, atomically dispersed Co atoms and Fe atoms exist in the same conjugate plane. The exfoliation effect of ball milling increases the specific surface area of 2D metal phthalocyanine compared with polyphthalocyanine, and has a high density of active sites. It can be seen in Fig. 2f and g that the two-dimensional CAN-Pc(Fe/Co) exhibits superior ORR performances compared to commercial Pt/C.
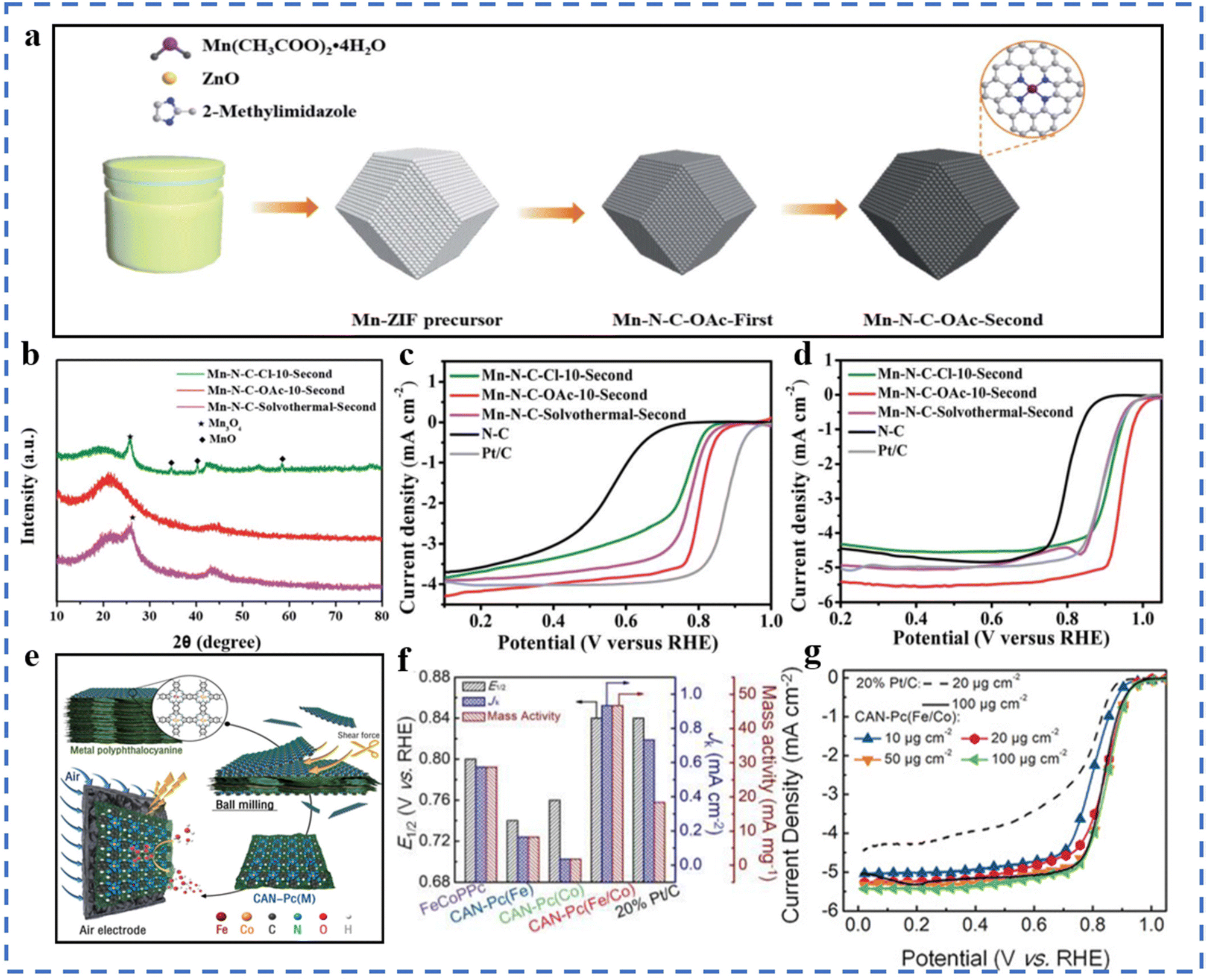 | ||
| Fig. 2 (a) Illustration of the mechanochemical synthesis of Mn–N–C catalysts. (b) XRD patterns of the Mn–N–C–OAc-10-second and the reference catalysts. (c) LSV curves of the Mn–N–C–OAc-10-second and reference catalysts in 0.1 M HClO4 solution and (d) in 0.1 M KOH solution. Reproduced with permission from ref. 79. Copyright © 2022, the Royal Society of Chemistry. (e) Schematic of the production of the CAN-Pc(M) electrocatalysts. (f) The E1/2 and JK at 0.9 V (vs. RHE), and mass activity at 0.9 V (vs. RHE) of CAN-Pc(Fe/Co) and reference catalysts. (g) LSV curves of CAN-Pc(Fe/Co) and reference catalysts in 0.1 M KOH with a sweep rate of 5 mV s−1 at 1600 rpm. Reproduced with permission from ref. 82. Copyright © 2019, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
2.3 Wet chemical method
Wet chemical methods include hydrothermal method, impregnation method, co-precipitation, sol–gel method, etc. The method is often used to prepare precursors of SACs, and the obtained precursors often require subsequent drying and calcination. The wet chemical process is simple and easy to achieve mass production, but there are also some problems: (1) the metal atoms in the as-prepared SACs are easily entrapped by the matrix, resulting in a reduced number of exposed active sites.73 (2) The wet chemical method needs to be carried out in a solution environment. It is inevitable to use organic/inorganic solvents, which is not conducive to environmental protection.Using ZIF-8 as the supporting framework, the boron source Cs2[B12H12] has been introduced by the impregnation method to obtain ZIF-8@B12. After impregnation–pyrolysis treatment, the product FeN4CB maintains the basic structure of ZIF-8. FeN4CB exhibits excellent ORR performance not only in alkaline electrolytes but also in acidic electrolytes.83 A molecular sieve CMS has been synthesized by a hydrothermal method using MCM-48 as a template. During the impregnation process, the hydroxyl groups and micropores in the molecular sieve CMS can trap Co precursors. Co-CMS is obtained after the pyrolysis of the precursor. Its ORR performance has been tested in 0.1 M KOH solution: the E1/2 is 0.83 V (vs. RHE), and the Tafel slope is 53.97 mV dec−1.84 An Fe3+-containing hydrogel precursor has been prepared by a sol–gel method. The aerogel containing Fe3+ is obtained by freeze-drying the hydrogel precursor, followed by pyrolysis and acid treatment to obtain the final product FeNC-900-8. During the formation of the hydrogel, Fe3+ are dispersed and stabilized by the coordination effect of sodium alginate (SA). During the formation of aerogels, the removal of water molecules leads to a large number of pore structures. In 0.1 M KOH solution, its E1/2 is 0.88 V (vs. RHE), and the Tafel slope is 63 mV dec−1, both better than those of commercial Pt/C.85 SiO2@RF microspheres have also been synthesized using the sol–gel method. A template with a specific bowl-like structure was obtained by etching SiO2. The Co2+ salt is combined with the template by the impregnation method. The impregnated template has been finally pyrolyzed under an NH3 (Ar/H2) atmosphere to obtain Co@NCB (Co@CB) (Fig. 3a). Compared with Co@CB, Co@NCB has Co-N4 in addition to Co nanoparticles (a coated carbon layer on the outside), which explains the effect of the final pyrolysis atmosphere on the formation of atomically dispersed Co atoms (Fig. 3b). The hollow bowl-like structure has good electrical conductivity, which is also conducive to the ORR performance. The ORR performance test has been carried out in 0.1 M KOH solution. As shown in Fig. 3c and d, the E1/2 of Co@NCB is 0.851 V (vs. RHE).86
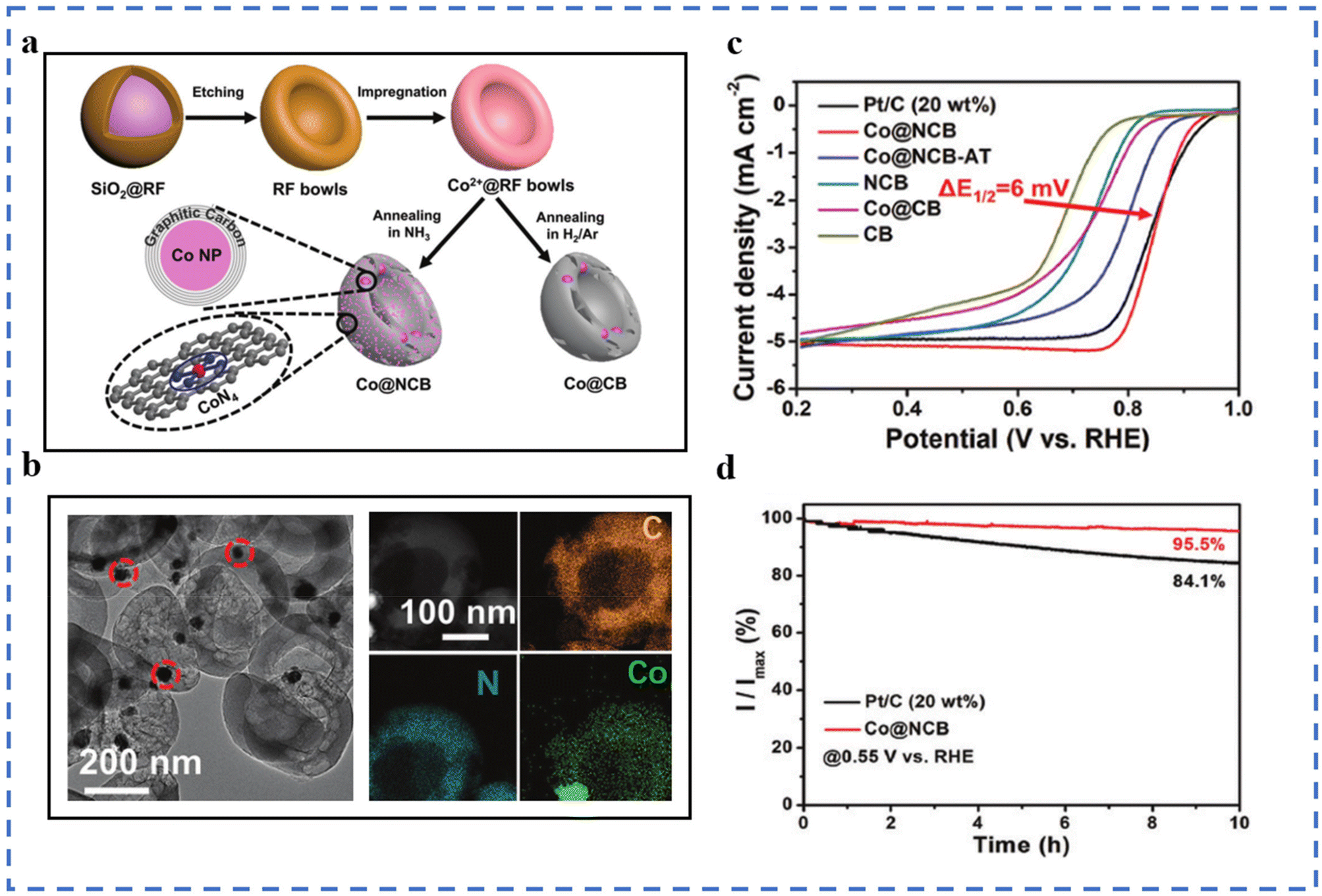 | ||
| Fig. 3 (a) Schematic of the production of Co@NCB. (b) TEM image of Co@NCB and the corresponding EDX mappings. (c) LSV curves of Co@NCB and the reference catalysts in 0.1 M KOH at 5 mV s−1 and 1600 rpm. (d) Current–time chronoamperometric responses of Co@NCB and Pt/C in O2-saturated 0.1 M KOH at 1600 rpm. Reproduced with permission from ref. 86. Copyright © 2020, the Royal Society of Chemistry. | ||
2.4 Atomic layer deposition method
Atomic layer deposition (ALD) is a top-down approach to fabricate SACs. The method usually involves modification of the substrate, loading of metal atoms on the substrate by ALD to form single-atom sites, and sometimes removal of the organic ligand portion of the metal precursor.63 ALD can control the deposition amount on the substrate material by adjusting the number of cycles. The deposition can be well and uniformly dispersed. ALD also has some problems: (1) high cost; (2) slow deposition rate; (3) vacuum and high temperature environment; (4) need to precisely control the anchoring metal atoms to form active sites.77,78Atomic layer deposition can be used to prepare non-precious metal SACs, but the high dissociation barrier of Co metal precursors often leads to the aggregation of directly deposited Co atoms or low Co loadings (Fig. 4a). Considering that Pt can easily achieve atomically dispersed loading, Pt single atoms have been first deposited on N-doped carbon nanosheets. Then Fe, Co, and Ni atoms are loaded on the Pt-loaded N-doped carbon nanosheets using the same method (Fig. 4b). The experimental results show that atomically dispersed Pt single atoms can promote the atomic dispersion of Fe, Co, and Ni, thereby increasing their loadings. Co(Cp)2 is easy to dissociate on the metal surface, so atomically dispersed Pt may also reduce the dissociation energy of Co(Cp)2, which has been confirmed by DFT calculations.87,88 Although the ORR catalytic performances of Fe1Pt1/NCNS, Ni1Pt1/NCNS, and Co1Pt1/NCNS have not been tested, this provides a way for us to prepare high-load atomically dispersed non-precious metal ORR catalysts in the future. This research also gives us some insights: (1) whether it is possible to replace Pt with other non-precious metals to reduce the cost; (2) whether it is possible to change the substrate type or perform special treatment on the substrate to promote the dispersion of metal atoms.
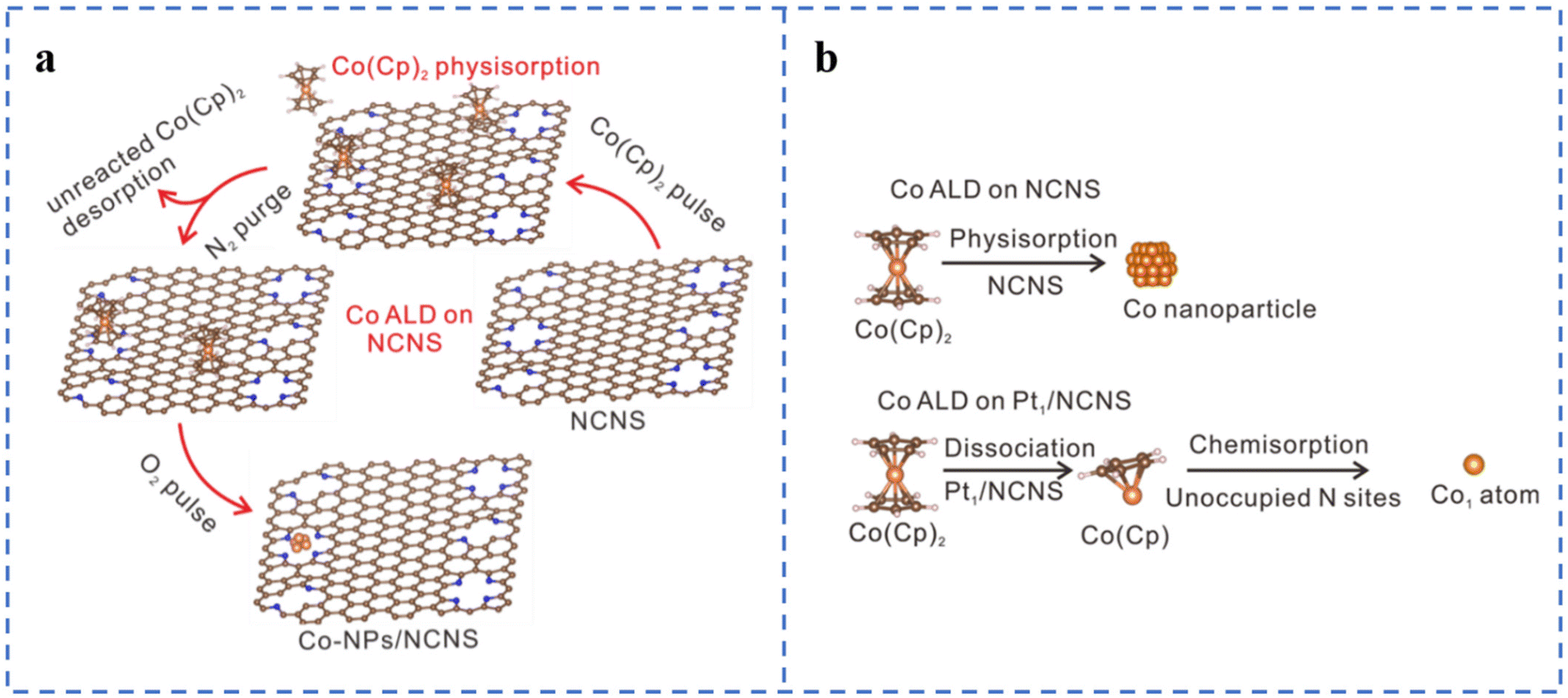 | ||
| Fig. 4 (a) Schematic of the Co ALD process on NCNS. White represents H, blue represents N, brown represents C, and orange spheres represent Co. (b) Assumption of the Co ALD process on pristine NCNS and Pt1/NCNS. Reproduced with permission from ref. 88. Copyright © 2021, Nature Publishing Group. | ||
3. Factors affecting the ORR performances of NPT-SACs
The metal atoms in NPT-SACs need to be anchored on the specific substrates, so the interaction between the substrate and metal atoms is very essential for the ORR catalytic performances of NPT-SACs.89 The mass transport and electron transfer capabilities of the substrate structure will also affect the final catalytic performances of NPT-SACs. On the other hand, the coordination environment of the metal atom is closely related to the intrinsic activity and stability of the active site. Therefore, it is of great significance to understand the effect of the coordination environment and substrate on the ORR performances of NPT-SACs. The synergy between the metal atoms and the adjacent metal nanoclusters can also be used to modify the ORR performance of NPT-SACs. Understanding these will help researchers design NPT-SACs with excellent ORR performances.3.1 Effects of the coordination environment
The coordination configuration formed by the central metal atom and its coordination atoms is closely related to the electronic and geometric structures of the active site. It will change the ORR catalytic activity of NPT-SACs. From an energetic point of view, most of these factors affect the ORR catalytic process of NPT-SACs by changing the adsorption free energy of the active site for O2 or oxygen-containing intermediates.Taking Fe–NX as an example, different numbers of N coordinated to the central metal atom will lead to different coordination structures. It will lead to differences in the ORR performances of the active sites. C–Fe–N2 is found to have a good two-electron process, which is favorable for the generation of H2O2.90 However, Fe–N4 sites mostly have a good four-electron process in the ORR, and are often used in the cathode reaction of fuel cells. Fe–CXN1−X with different coordination configurations have been synthesized and theoretical calculations have been performed. The results showed that their ORR activities are different. Among them, an Fe–N4 type structure has the highest ORR activity, and an Fe–N5 type structure has the lowest activity.91 However, in some studies, the quadrangular pyramid Fe–N5 is considered to have better activity and stability than the planar Fe–N4.92 SA-Fe-NCs supported on N-doped defect carbon nanosheets were prepared by the pyrolysis method. The extended X-ray absorption fine structure fitting (EXAFS) results show that the coordination number of N coordinated with Fe was 4.4. Due to the thermodynamic instability of other coordination configurations other than Fe–N4 and Fe–N5, it is inferred that Fe atoms are most likely to exhibit these two coordination types. The fitted coordination structures are shown in Fig. 5a. The SA-Fe-NC has excellent ORR catalytic activity and stability in both alkaline and acidic electrolytes. In 0.1 M KOH solution, its onset potential (Eonset) is 1.04 V (vs. RHE), the E1/2 is 0.88 V, and the Tafel slope is 68 mV dec−1 (Fig. 5b). In 0.1 M HClO4 solution, its Eonset is 0.92 V (vs. RHE), the E1/2 is 0.79 V, and the Tafel slope is 87 mV dec−1.93
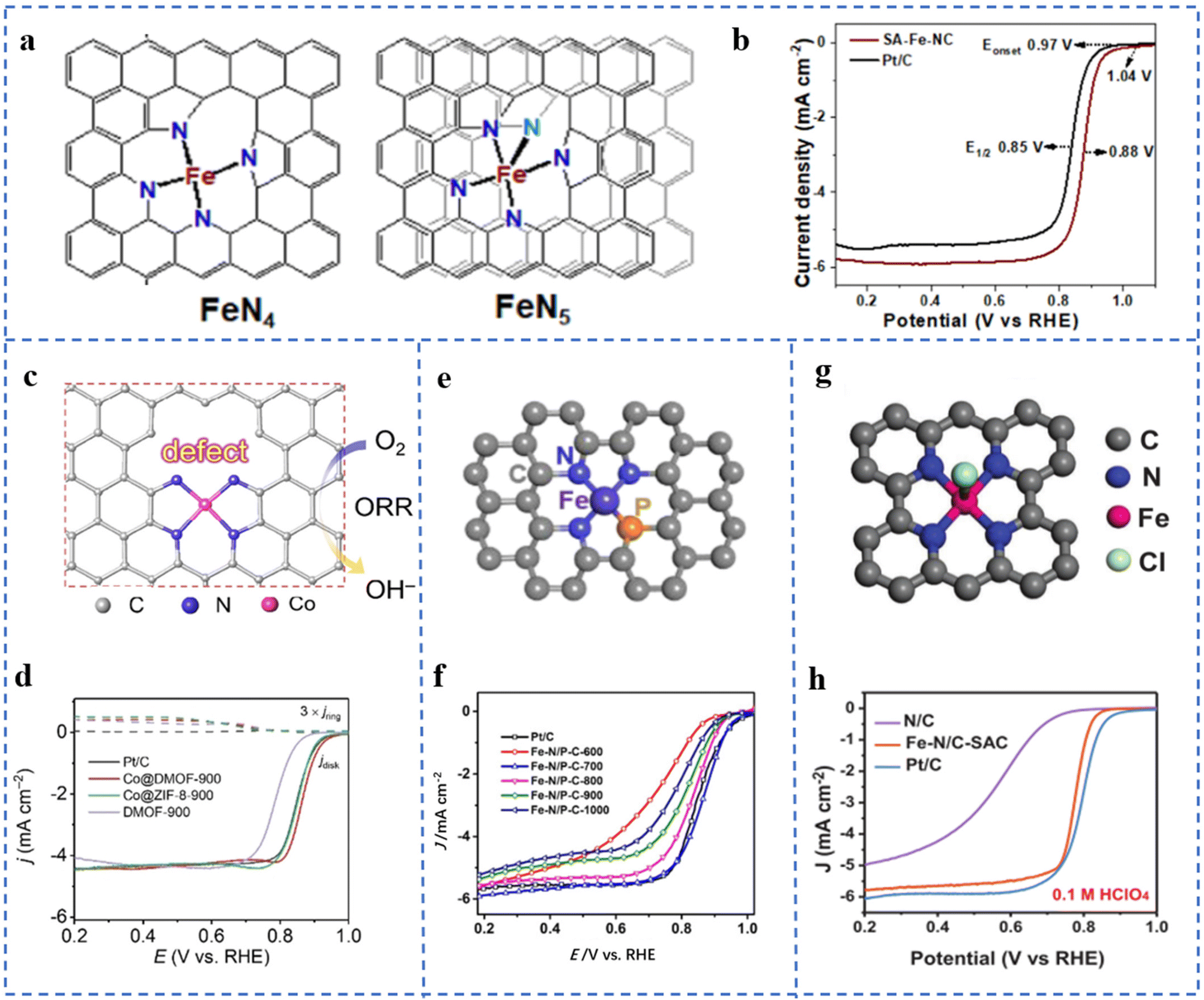 | ||
| Fig. 5 (a) The models of the Fe–N4 and Fe–N5 structures of SA-Fe-NCs. (b) LSV curves of SA-Fe-NCs and Pt/C at 1600 rpm. Reproduced with permission from ref. 93. Copyright © 2021, American Chemical Society. (c) The model of active sites near defects in Co@DMOF. (d) LSV plots of Pt/C, Co@DMOF-900, Co@ZIF-8-900, and DMOF-900 in 0.1 M KOH. Reproduced with permission from ref. 97. Copyright © 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (e) Fe–N/P–C structural model fitted by EXAFS. (f) ORR polarization curves of Fe–N/P–C catalysts and reference catalysts at 1600 rpm in 0.1 M KOH. Reproduced with permission from ref. 95. Copyright © 2020, American Chemical Society. (g) EXAFS spectral curve fitting analysis model of the Fe–N/C-SAC. (h) LSV plots of the Fe–N/C-SAC in 0.1 M KClO4. Reproduced with permission from ref. 100. Copyright © 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The electronegativity of P and S is different from that of N. The coordination of atoms such as P and S in M–NX will change the local electron distribution of the active site, thereby enhancing the ORR performances of M–NX.94 Fe–N/P–C-700 with N and P co-coordination has been prepared by pyrolysis of an Fe3+-containing polypyrrole hydrogel. It can be seen in Fig. 5e that EXAFS fitting results confirm that the coordination situation is Fe–N3P. Fig. 5f shows that Fe–N/P–C-700 exhibits excellent ORR performances. Density functional theory (DFT) calculation results show that the doping of P changes the electronic structure of Fe which improves its ORR intrinsic activity and reaction kinetics.95 An optimal model CoN3PS@PS has been obtained through DFT calculations. The calculation results show that CoN3PS@PS has the lowest overpotential for the reaction rate-determining step. The coordinated coordination of P and S reduces the d-band center of Co, changes its electronic density of states, and enhances its ORR activity. CoN3PS@HC with a CoN3PS coordination structure has been obtained by pyrolysis of a ZIF-8-derived core–shell structure (containing P and S). In 0.1 M KOH solution, the Eonset and E1/2 of CoN3PS@HC are 1.00 V (vs. RHE) and 0.92 V, respectively. In 0.5 M H2SO4 solution, the E1/2 is 0.790 V (vs. RHE).96
Active sites (M–NX) near defects, including C vacancies, substrate edges, N vacancies, etc. often exhibit excellent ORR performances. A carboxylate-containing MOF-derived precursor has been pyrolyzed. Carboxylate groups in the precursor are converted to COX species detached from the substrate during pyrolysis and form defects. The obtained catalyst is named Co@DMOF-T (Fig. 5c). XPS and XAFS results show that the coordination of Co atoms is Co–N4. The comparative sample Co@ZIF-8-900 undergoes no decarboxylation-induced defect generation process, so the defect contents are less. As depicted in Fig. 5d, the E1/2 of Co@DMOF-900 is 0.866 V (vs. RHE) in 0.1 M KOH solution. The Co and N contents of Co@DMOF-900 are much lower than those of Co@ZIF-8-900. However, both have similar active specific surface areas Cdl and Tafel slopes. The deduced mass activity (jm) and turnover frequency (TOF) of Co@DMOF-900 are about 8–9 times higher than those of Co@ZIF-8-900. In the DFT calculation, several Co–N4 models with different degrees of defects have been established. The results show that the defects can weaken the adsorption free energy of the intermediate at the active site (Co–N4) which improves its ORR catalytic activity.97
Atoms or groups that form axial coordination with metal atoms have a similar effect. Penta-coordinated O–Fe–N2C2 has been prepared using mesoporous SiO2 as a template. O coordination changes the electronic density of Fe, which optimizes the free energy of Fe adsorption for oxygen-containing intermediates.98 Fe–N/S–C supported on N,S-doped porous carbon nanostructures has been obtained using ZnS nanospheres as self-sacrificial templates. Characterization results show that the active site is Fe–N3S1OH-1, and Fe is coordinated with 3 N, 1 S, and 1 axial hydroxyl group. In 0.1 M KOH solution, the E1/2 is 0.882 V (vs. RHE), the Tafel slope is 49.5 mV dec−1, and the limiting exchange current density JK = 5.87 mA cm−2. The ORR performance is better than that of commercial Pt/C. DFT calculations show that the hydroxyl group on the fifth coordination axis changes the electron filling degree of the partially occupied 3d orbital of Fe, which is beneficial for the adsorption and activation of O2.99 A five-fold coordination Fe–N/C-SAC was prepared by the molten salt method. The coordination situation of the Fe–N/C-SAC obtained by EXAFS curve fitting is depicted in Fig. 5g, which shows that Fe is coordinated with four N atoms and one axial Cl atom in the plane. As shown in Fig. 5h, the Fe–N/C-SAC exhibits excellent ORR performance. The decrease of E1/2 and the increase of the Tafel slope after dechlorination of the Fe–N/C-SAC indicate the positive effect of axially coordinated Cl in promoting ORR performance. DFT calculations show that the introduction of the axial Cl atom changes the electronic structure of the active site, thereby improving the oxygen adsorption energy of the active site and lowering the intermediate reduction reaction barrier.100
3.2 Effects of the substrate
The mass transfer ability, electron transfer ability of the substrate and the interaction between the substrate and the active site significantly affect the ORR performances of NPT-SACs.101–105 In addition, the substrate also plays an important role in forming high-density active sites and regulating the selectivity and stability of NPT-SACs. Most carbon substrates have excellent electrical conductivity, easy modification, structural diversity and economy. Carbon substrates are currently the most widely studied substrate materials in the field of NPT-SACs. Carbon nanotubes,106,107 graphene and graphene-like66 carbon nanosheets,108 MOF derived materials,109–111 triazine skeleton,112etc. are commonly used to assemble NPT-SACs.It can be seen in Fig. 6a and b that Fe–N4-PN was obtained by introducing pyrrolic N(PN) near the active site Fe–N4 on the substrate, which changed the original symmetric electric field of Fe–N4. In 0.1 M KOH solution, the E1/2 of Fe–N4-PN is 0.91 V, with excellent ORR catalytic performances (Fig. 6c). DFT calculations indicate that pyrrolic N on carbon substrates can capture the electrons of Fe–N4 and redistribute them around the active site, resulting in a higher ORR activity of Fe–N4-PN.113 A core–shell structure single-atom Co catalyst has been designed and synthesized without pyrolysis. Tetraamine porphyrin (TPP) and thieno/thiophene-2,5-dicarboxaldehyde (bTDA) assemble on multi-walled carbon nanotubes. The macrocyclic porphyrin structure in the shell layer anchors Co atoms to form Co-PTS-COPs@MWCNTs. The shell layer with appropriate thickness facilitates mass transfer and exposure of active sites, and carbon nanotubes improve the overall conductivity of the catalyst. The Eonset and E1/2 of optimal Co-PTS-COPs@MWCNTs in 0.1 M KOH solution is 0.930 V (vs. RHE) and 0.835 V, respectively.114 A Co single-atom supported on graphene-like ultrathin carbon nanosheets (CoSAs/N-CNS) with a hierarchical porous structure has been synthesized using g-CN as an in situ sacrificial template. Both Raman spectroscopy and XPS results show that the presence of Co atoms makes the substrate have a higher degree of graphitization. In 0.1 M KOH electrolyte, CoSAs/N-CNS has an E1/2 of 0.91 V (vs. RHE), a Tafel slope of 73.2 mV dec−1, an ECSA of 47.9 m2 g−1, a charge transfer impedance of 65.70 Ω, and excellent stability. This substrate has a high degree of graphitization, a loose porous structure, and abundant edges, which are conducive to the formation of a large number of active sites and mass transport/electron transfer.115 Fe1/d-CN has been obtained by growing another layer of Fe-containing ZIF-8 outside the ZIF-8, followed by pyrolysis. A higher ID/IG value indicates more defects in Fe1/d-CN. In 0.1M KOH solution, the E1/2 of Fe1/d-CN is 0.950 V (vs. RHE), and the Tafel slope is 54 mV dec−1, which is better than that of commercial Pt/C. In 0.5 M H2SO4 solution and 0.1 M PBS electrolyte, the E1/2 are 0.781 V and 0.605 V (vs. RHE), respectively, and the Tafel slopes are 64.0 mV dec−1 and 98.0 mV dec−1, respectively. The hierarchical pore structure and a large number of defects formed by the pyrolysis of this bilayer MOF help to enhance the ORR performance of Fe1/d-CN in a wide pH range.116 A bimetallic single-atom catalyst Fe/Ni–NX supported on highly ordered hierarchical porous N-doped carbon has been prepared by the template-impregnation–pyrolysis method using polystyrene microspheres as sacrificial templates and ZIF-8 as a matrix/OC. The highly ordered hierarchical pore structure and the high density of metal atom loadings can be observed in Fig. 6d and g. The substrate with a special structure is favorable for the formation of exposed active sites, which also greatly facilitates the mass transfer process. Benefiting from the synergistic effect of bimetallic sites and the unique substrate structure, Fe/Ni–NX/OC exhibits excellent ORR performances in both acidic and alkaline electrolytes (Fig. 6e, f, h and i).102
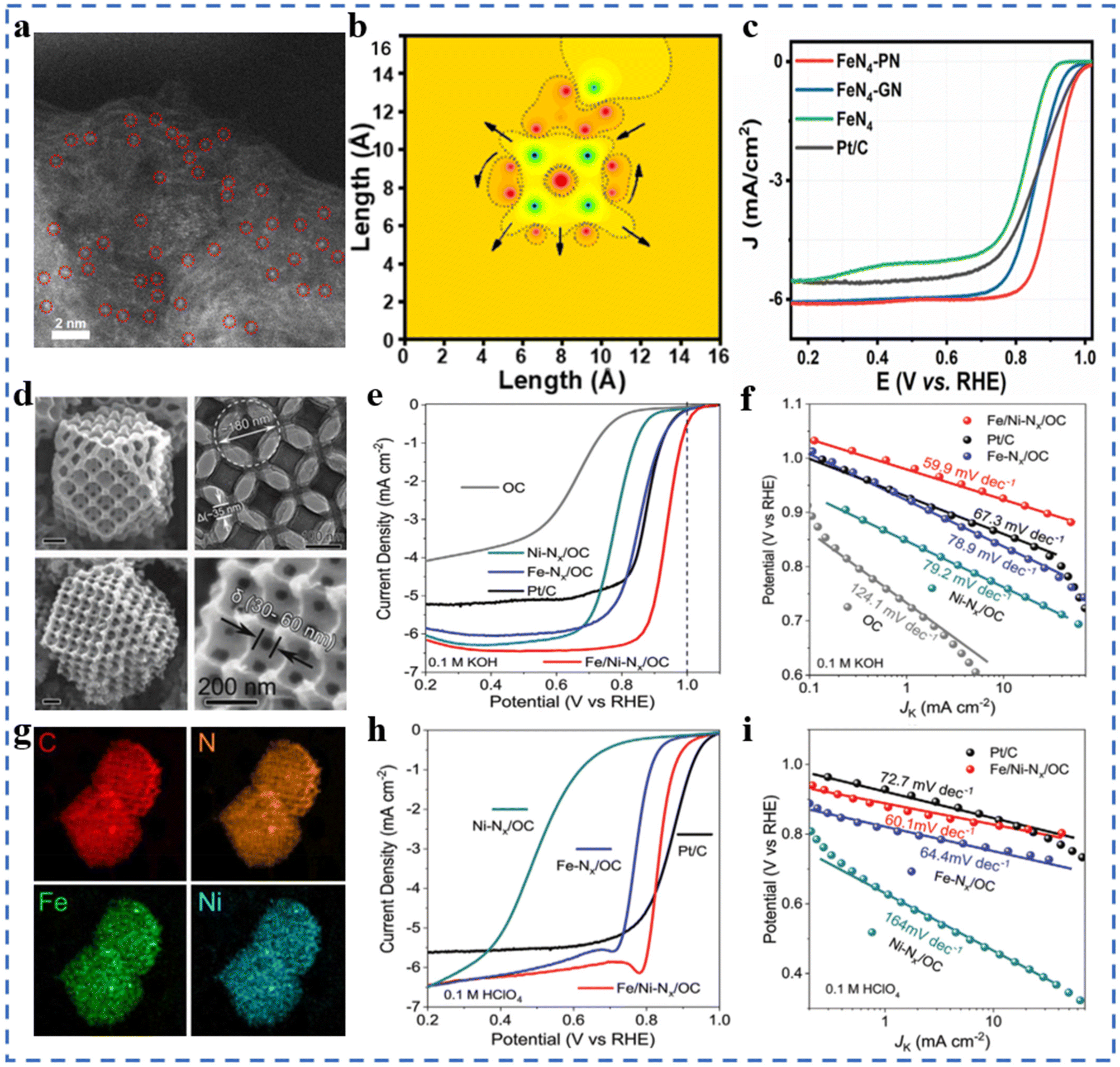 | ||
| Fig. 6 (a) SAC-HAADF-STEM image of FeN4-PN (brilliant dots represent Fe SACs, circled in red). (b) MATLAB simulations of a local electric field on FeN4-PN in two dimensions. (c) ORR polarization curves of FeN4-PN and reference catalysts. Reproduced with permission from ref. 113. Copyright © 2021, American Chemical Society. (d) SEM and TEM images of Fe/Ni–NX/OC. (e) ORR polarization curves of Fe/Ni–NX/OC and reference catalysts in 0.1 M KOH at 1600 rpm, and (f) the corresponding Tafel plots. (g) EDS mapping images of C, N, Fe, and Ni (in HAADF-STEM) (h) ORR polarization curves of Fe/Ni–NX/OC and reference catalysts in 0.1 M HClO4 at 1600 rpm, and (i) the corresponding Tafel plots. Reproduced with permission from ref. 102. Copyright © 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In addition to these materials, researchers have also conducted extensive explorations on other possible substrate materials. Carbon aerogels have large specific surface areas, hierarchical porous structures and good structural stability. Co-NCA@F127-1:0.56 has been prepared with a carbon aerogel as a substrate with an E1/2 of 0.935 V (vs. RHE).117 A series of transition metal atoms are introduced in TM@Mo2B2 through first-principles calculations. The transition metal atoms are embedded in specific vacancies and they bond with the surrounding B atom. By calculating the ORR overpotential of TM@Mo2B2, it is found that Cu@Mo2B2 has the smallest overpotential (0.34 V), which is comparable to that of Pt noble metals.118 Transition metal sulfides also have large specific surface areas and electrical conductivity. MoS2 exhibits excellent HER performance.119 Metal atoms supported on monolayer MoS2 (1T′-MoS2) have been analysed using DFT. The results show that MoS2 loaded with specific atoms such as Co and Cu exhibits excellent ORR and HER catalytic activities. It is a good idea for the construction of bifunctional and even multifunctional SACs.120 Two-dimensional black phosphorus nanomaterials have excellent electrical conductivity, easily tunable electronic structures, and active lone pair electrons. These properties make them have great potential in the field of electrocatalysis. Single-atom catalysts supported on two-dimensional black phosphorus substrates have been analysed by DFT calculations. NiN3-BP SACs exhibit the best ORR activity with an overpotential of 0.44 V. By adjusting the number of N with the central metal atom, the catalytic activity of the active site can be effectively improved.121
3.3 Effects of nanoclusters/particles
Due to the high surface energies, metal atoms usually tend to agglomerate easily during pyrolysis.73,122 In the previous sections, we have introduced some preparation methods to avoid the agglomeration of metal atoms. It is generally believed that the agglomeration to form nanoclusters/particles will lead to low atom utilization and is not conducive to the ORR catalytic process. However, some in-depth studies have shown that the synergy between single-atom metal sites and other single-/multi-core metal sites may alter the electron distribution around the active site (metal substrate). Changes of the electronic structure can optimize the adsorption behaviour of active sites and intermediates.Fe–N–C–T has been obtained by pyrolysis of Fe-ZIF-8-C in argon at different temperatures. Atomically dispersed atoms and nanoclusters of Fe in Fe–N–C–T coexist on hollow carbon nanotubes. In 0.1 M KOH solution, the E1/2 of Fe–N–C–T is 0.90 V (vs. RHE). After the removal of Fe nanoclusters by acid treatment, the E1/2 shows a significant negative shift, indicating the synergistic effect between Fe single atoms and Fe nanoclusters.106 Fe-ACSA@NCs with dispersed metal single atoms coexisting with metal clusters have been prepared by coating–pyrolysis–etching. After acid etching, it can be seen in Fig. 7a that the Fe nanoparticles in Fe-NPSA@NCs have been removed, leaving only Fe single atoms and Fe nanoclusters, named Fe-ACSA@NCs. The ORR performance of Fe-ACSA@NCs has been tested in 0.1 M KOH solution, and the Eonset is found to be 1.03 V (vs. RHE), the E1/2 to be 0.90 V, the limiting diffusion current density (JL) to be 5.00 mA cm−2, and the Tafel slope to be 78 mV dec−1. The ORR performance of Fe-ACSA@NCs is better than that of Fe-SA@NCs without nanoclusters, Fe-NPSA@NCs without acid etching, and commercial Pt/C (Fig. 7b). DFT theoretical calculation results show that the presence of nano-clusters of Fe induces charge redistribution between single-atom Fe and the substrate. It enhances the desorption ability of the Fe–NX active site for intermediate *OH, resulting in excellent ORR catalytic performance (Fig. 7c).123 A Co-SAs/SNPs@NC with the coexistence of Co single atoms (Co-SAs) and small nanoparticles of Co (Co-SNPs) on N-doped porous carbon nanocages has been synthesized (Fig. 7d). As shown in Fig. 7e, in 0.1 M KOH solution, the E1/2 is 0.898 V (vs. RHE), the JK is 60.68 mA cm−2, and the Tafel slope is 65 mV dec−1. DFT calculations show that, thanks to the charge transfer between Co-SAs and Co-SNPs, Co-SNPs can reduce the free energy of oxygen adsorption at the Co–N4 site and reduce the energy barrier of the ORR process. When the nanoparticles are larger, the adsorption strength of the Co–NX active sites for oxygen-containing intermediates will be further weakened. However, too small or too large adsorption strength is not conducive to the ORR (Fig. 7f).124
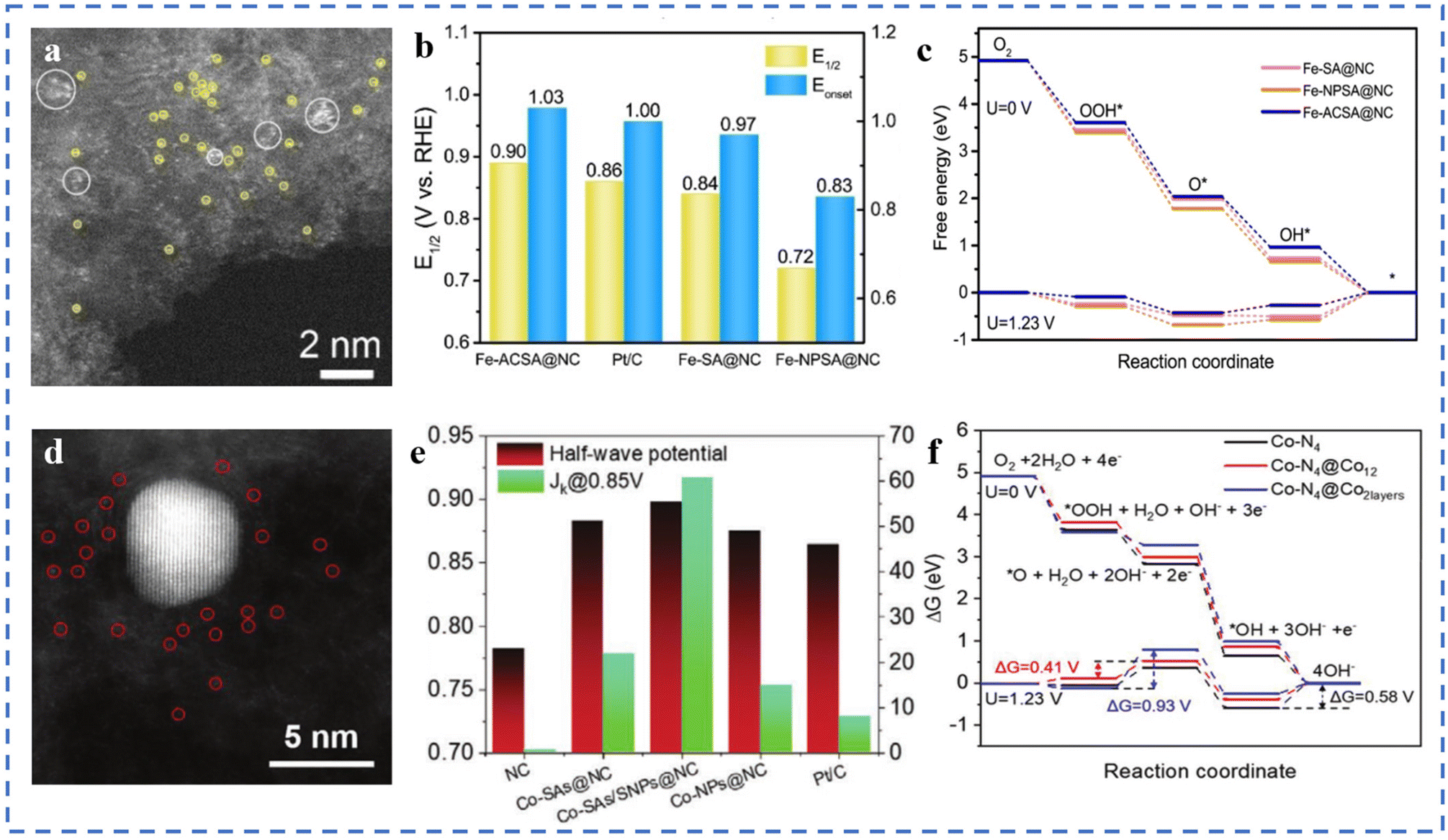 | ||
| Fig. 7 (a) The aberration-corrected STEM image of Fe-ACSA@NCs. (b) Comparison of E1/2 and Eonset. (c) Free energy diagram of the ORR for Fe-SA@NCs and reference catalysts at 1.23 V and 0 V vs. RHE (theoretical analysis). Reproduced with permission from ref. 123. Copyright © 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (d) HAADF-STEM image of the Co-SAs/SNPs@NC. (e) ORR E1/2 (V vs. RHE) potential and JK for the Co-SAs/SNPs@NC and the references. (f) ORR free energy diagrams for Co–N4 (black line), Co–N4@Co12 (red line), and Co–N4@Co2layers (blue line) at U = 0 V, and 1.23 V along the 4-electron pathway. Reproduced with permission from ref. 124. Copyright © 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Using SiO2 and ZnCl2 as pore formers, Fe3O4@FeNCs with a hierarchical pore structure coexisting with Fe atoms and Fe3O4 nanoparticles have been prepared. In 0.1M KOH electrolyte, the Eonset is 1.007 V, the E1/2 is 0.890 V (vs. RHE), the Tafel slope is 58.8 mV dec−1, and the JL is close to 6 mA cm−2. The microporous structure of the substrate is conducive to the nucleation of Fe3O4. Macropores and mesopores are conducive to the exposure of active sites and mass transfer. These factors synergistically improve the ORR performances of the catalyst.125 The two-electron pathway of ORR leads to the generation of H2O2, which greatly affects the stability of Fe–N–C on carbon nanotubes containing MnOX species. The results show that the introduction of Mn species clusters significantly increases the half-wave potential of Mn–Fe–N–C compared with Fe–N–C without MnOX. The yield of H2O2 (≤0.80%) on Mn–Fe–N–C is extremely low in the voltage range of 0.2–0.8 V, due to the excellent four-electron process of Mn–Fe–N–C and the disproportionation of MnOX species that can decompose off the generated H2O2. These lead to the reduction of the Fenton reaction, resulting in the excellent stability of Mn–Fe–N–C. The E1/2 of Mn–Fe–N–C has only a negative shift of 6 mV after 10000 cycles.126
4. General strategies for stabilizing metal atoms
Increasing the number of active sites can significantly improve the ORR performances of NPT-SACs.74,75 The active sites in NPT-SACs are composed of metal single atoms and their surrounding coordination structures. It is crucial to improve the catalytic performances of NPT-SACs by increasing the metal loading and ensuring its atomic dispersion. Two commonly used strategies for stabilizing metal atoms in NPT-SACs are summarized in this section, which are named “space confinement” and “defect stabilization” (Table 2). In addition, the “coordination strategy” is also a powerful strategy for stabilizing metal atoms. One is that specific molecules and metals in precursors coordinate with in situ generated active sites.51 Another is non-metallic elements (N, B, S, etc.) on the substrate coordinate with metal atoms during pyrolysis to anchor metal atoms. The coordination strategy is widely used. In the process of preparing single-atom catalysts using N-doped carbon materials as substrates, increasing the amount of N doping can promote the formation of M–NX active sites (M: metal).124,127 However, the “coordination strategy” is often used in conjunction with other strategies, which are not listed separately in this section.| Electrocatalyst | Electrolyte | Metal content | E 1/2 V vs. RHE | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|
| Fe–N–Si-CNFs | 0.1 M KOH | 0.53 at% | 0.86 | 65 | 128 |
| NR-CNT@FeN-PC800 | 0.1 M KOH | 0.8 at% | 0.88 | 51 | 129 |
| Mo SACs/N–C | 0.1 M KOH | 2.51 wt% | 0.83 | — | 130 |
| Fe/pNC@HCF | 0.1 M KOH | 1.34 wt% | — | 68 | 131 |
| PA@Z8-Fe–N–C | 0.1 M KOH | 5.49 wt% | 0.88 | 75 | 132 |
| Cu-SAs@N-CNS | 0.1 M KOH | 1.1 wt% | 0.90 | 66 | 136 |
| Fe, Co SAs-PNCF | 0.1 M KOH | 9.80 wt% (Co and Fe) | 0.93 | 77 | 137 |
| 0.1 M HClO4 | 0.78 | 126 | |||
| GO-Fe–N | 0.1 M KOH | 2.2 wt% | 0.87 | 66.2 | 138 |
| Fe–N–C-KCl | 0.1 M KOH | 0.92 at% | 0.877 | 78 | 139 |
4.1 Space restriction
The space restriction effect of some specific structures of the substrates (such as micropores, etc.) can effectively reduce the migration of metal single atoms, thereby reducing the agglomeration of metal atoms.77 During the synthesis of SACs, the introduction of other sterically hindered moieties into the matrix can also effectively limit the formation of metal particles/clusters. These effects are beneficial for obtaining NPT-SACs with high-density atomically dispersed active sites.Nanofibers loaded with uniformly distributed Fe atoms have been prepared by electrospinning technology. Then a layer of SiO2 is coated on the outer layer of the fibers. Fe–N–Si-CNFs were prepared by removing SiO2 with HF acid after pyrolysis treatment (Fig. 8a). Many Fe–NX sites are formed after pyrolysis due to the SiO2 layer hindering the migration of Fe atoms. The XPS results show that the Fe–NX site content in Fe–N–Si-CNFs is 1.24%, while that in the Fe–N-CNFs prepared by an uncoated SiO2 layer is only 0.71%. In alkaline solution, the E1/2 of Fe–N–Si-CNFs is 0.86 V (vs. RHE), which is more stable than commercial Pt/C.128 As shown in Fig. 8b, polyaniline (PANI) polymerizes on the surface of carbon nanotubes to form a shell. The PANI shell is used to anchor Fe atoms. The SiO2 in the outermost layer of the shell can hinder the migration and aggregation of Fe atoms and reduce the loss of N during the pyrolysis process. The formation of a large number of Fe–NX sites requires sufficient Fe–N coordinative anchoring to facilitate their atomic dispersion. Dicyandiamide (DCD) and PANI constitute a double nitrogen source, which can solve the problem of insufficient nitrogen content of polyaniline. The catalyst prepared by this method is named NR-CNT@FeN-PC800. NR-CNT@FeN-PC800 has an E1/2 (0.88 V vs. RHE) superior to commercial Pt/C in 0.1 M KOH solution. However, XPS elemental analysis indicates that the Fe content coordinated to N in NR-CNT@FeN-PC800 was only 0.8 at%.129
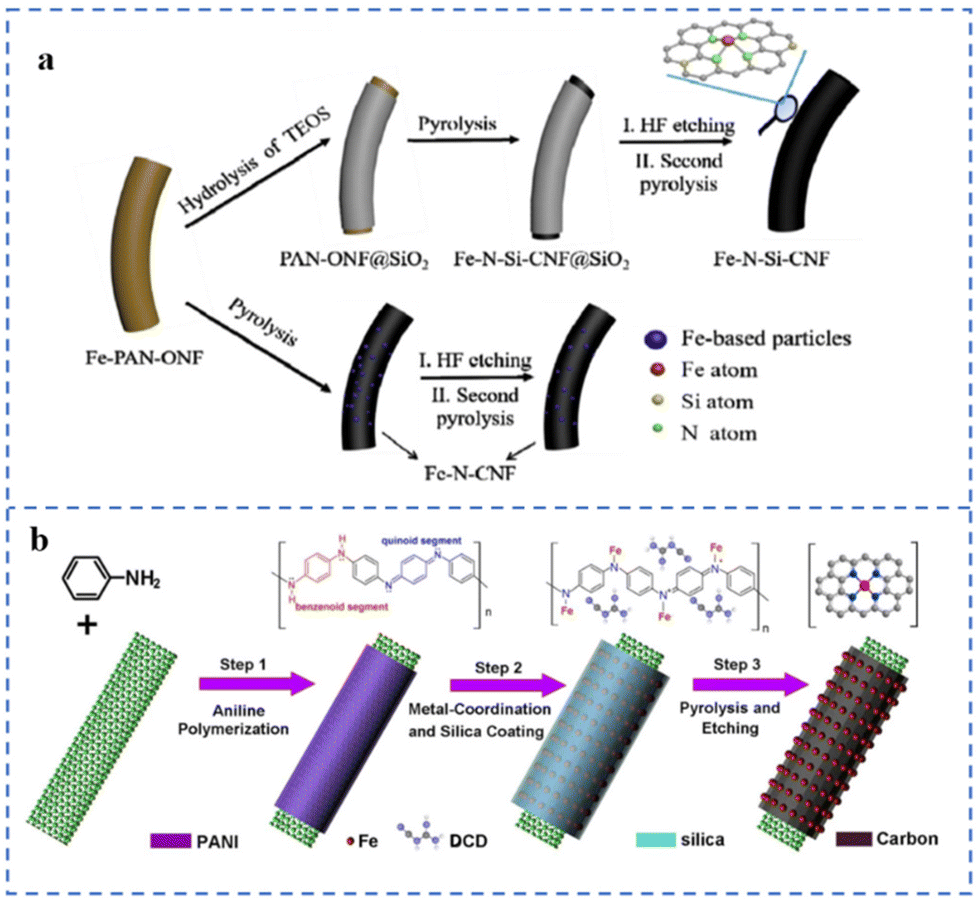 | ||
| Fig. 8 (a) Space restriction strategy of the Fe–N–Si-CNFs and references. Reproduced with permission from ref. 128. Copyright © 2020, ELSEVIER. (b) Space restriction strategy of NR-CNT@FeN-PC. Reproduced with permission from ref. 129. Copyright © 2021, IOP Publishing Ltd. | ||
ZIF-8 has many cavity structures that can serve as a place to hold metal species. A caged pyrolysis strategy has been proposed by Kou et al.130 Mo species (POM) matching the cavity size of ZIF-8 are added during the preparation of ZIF-8 to obtain POM@ZIF-8 in one step. POM/ZIF-8 has been prepared by impregnating and adsorbing POM with ZIF-8. Compared with POM/ZIF-8, the Mo species in POM@ZIF-8 are more likely to form small nanoclusters or single-atom morphologies in the subsequent pyrolysis. Pyrrole nitrogen-rich carbon (pNC) is coated on ZIF-8 for anchoring Fe atoms, and then Fe/pNC@HCF is prepared by pyrolysis (Fig. 9a). Since the pyrrole N in pNC is negatively charged and the Fe ion is positively charged, the electrostatic interaction between the two is conducive to the capture of Fe ions by pNC. The N and Fe species in the pNC layer form a large number of Fe–NX sites during the subsequent pyrolysis process. Inductively coupled plasma-atomic absorption spectroscopy (ICP-AAS) and XPS results show that the Fe atomic content and the number of Fe–NX sites increased by about 20% due to the addition of pNC. Fe/pNC@HCF has been used for an anion exchange membrane fuel cell (AEMFC) with a comparable power density (343 mW cm−2) at 0.6 V to an AEMFC using 40% Pt/C.131 A highly loaded Fe single-atom catalyst for the ORR has been prepared on the ZIF-8 framework according to the scheme in Fig. 9b. ZIF-8 as the basic framework supporting single atoms is the first barrier. During the synthesis of ZIF-8, phenylboronic acid is added to form PA@ZIF-8. Phenylboronic acid is used as the second barrier to prevent the migration of Fe atoms. Due to the steric hindrance effect of these two barriers, the Fe single-atom loading in the as-prepared PA@Z8-Fe–N–C is as high as 5.49 wt%. The E1/2 of PA@Z8-Fe–N–C in 0.1 M KOH solution is 0.88 V (vs. RHE) and the Tafel slope is 75 mV dec−1.132
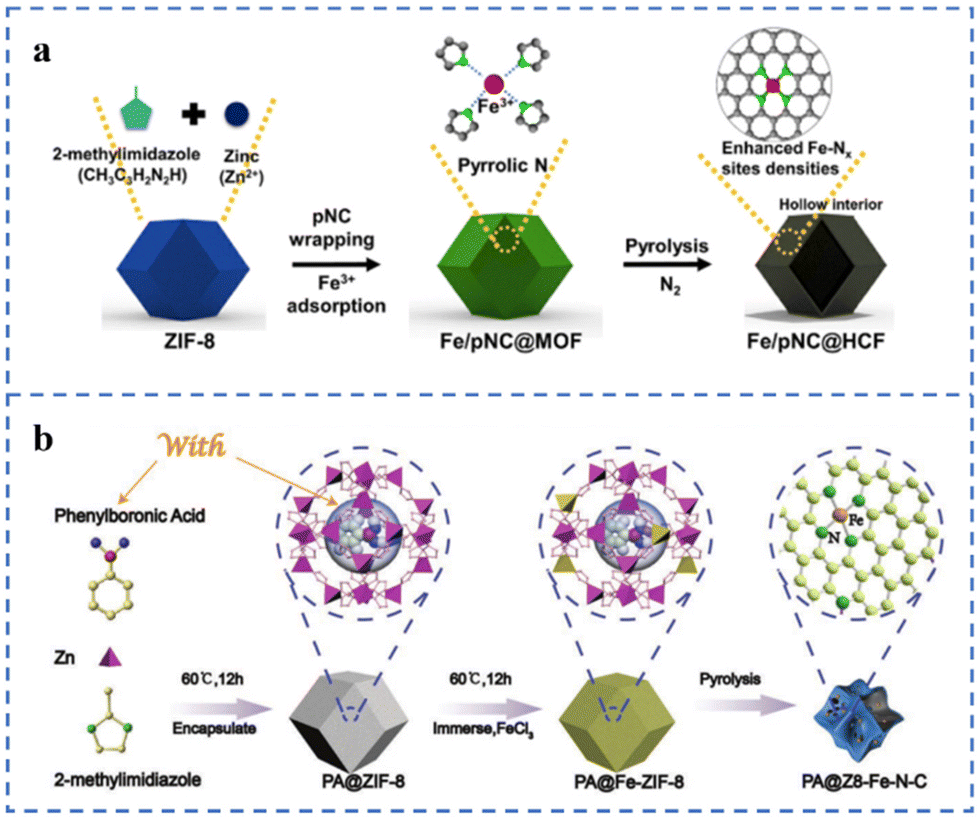 | ||
| Fig. 9 (a) Space restriction strategy of Fe/pNC@HCF-1000. Reproduced with permission from ref. 131. Copyright © 2022, ELSEVIER. (b) Space restriction strategy of PA@Z8-Fe–N–C catalysts. Reproduced with permission from ref. 132. Copyright © 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
4.2 Defect stabilization
Defect engineering can be used to prepare SACs with good stability and activity. Defect centers can serve as sites for trapping and anchoring metal atoms. In addition, electron transfer between defects and single atoms may enhance the activity or stability of the active sites. Therefore, the study of the relationship between substrate defects and single-atom loading and stability has become a hot field.39,133,134 Steps, vacancies, defects, etc. are expected to enhance the stability of single metal atoms on hydroxides,55 metal oxides,135 metal sulfides,134 carbon supports and other substrates.According to the process of Fig. 10a, several NPT-SACs (Cu, Fe, Co, Ni) have been prepared by artificially creating defects on carbon nanospheres, named TM-SAs@N-CNS (TM: transition metal). The hydrothermally prepared carbon nanospheres (CSs) have been transformed into porous carbon nanospheres (CNSs) after KOH activation. Compared with CSs, CNSs have an increased pore structure. The ID/IG value of CNSs is greater than 1, indicating a large number of defects. These defect sites can selectively adsorb transition metal ions, which is conducive to the formation of M–NX structures to anchor metal atoms. Among them, Cu-SAs@N-CNS exhibits the best ORR performance, with an E1/2 of 0.90 V (vs. RHE) and a limiting diffusion current density of −5.50 mA cm−2 in 0.1 M KOH solution.136 The as-prepared nanofibers are treated with N2 plasma to increase the number of defects and the N doping amount in the carbon substrate (Fig. 10b). A large number of carbon defects will act as anchor sites for Fe and Co atoms. A sufficient amount of N doping will further promote the formation of M–NX sites, resulting in a large number of atomically dispersed metal active sites. Due to the synergistic effect of defects and doped N and bimetallic sites, the Fe/Co loading has increased to 9.80 wt%. The prepared catalyst is named Fe, Co SAs-PNCF. The E1/2 is 0.93 V (vs. RHE) in 0.1 M KOH solution and the Eonset is 0.94 V (vs. RHE) in 0.1 M HClO4 solution.137 It can be seen in Fig. 10c that graphene oxide (GO) and reduced graphene oxide (rGO) are used as substrates to support single-atom Fe, respectively. Compared with GO, the number of surface defects and oxygen-containing groups of rGO decreased to a certain extent after thermal reduction. N atoms have been doped into carbon defects in GO and rGO during pyrolysis to form GO-N and rGO-N. Fe atoms are anchored by N at these defect sites, and the corresponding catalysts are named GO-Fe–N and rGO-Fe–N, respectively. The element content of GO-Fe–N is determined by EDS. It was found that the N and Fe contents in GO-Fe–N are 5.83 wt% and 2.2 wt%, respectively. While the N and Fe contents in rGO-Fe–N are 4.18 wt% and 1.52 wt%, respectively. This result illustrates the positive effect of carbon defects on anchoring Fe atoms to form Fe–NX sites. The E1/2 of GO-Fe–N and rGO-Fe–N in 0.1 M KOH solution are 0.87 V (vs. RHE) and 0.81 V, respectively.138 Muhammad Arif Khan et al. used KCl as an auxiliary template to prepare M–N–C (M: Fe, Co) for ORR catalysts. During the pyrolysis process, KCl acts as a template to hinder the migration of metal atoms. Moreover, K+ would be reduced to K atoms during pyrolysis. The insertion and removal of K atoms in the carbon support would lead to the increase of defects in the carbon support. These defects can easily anchor metal atoms to form M–NX sites. The E1/2 of Fe–N–C-KCl-4 is 0.877 V (vs. RHE).139
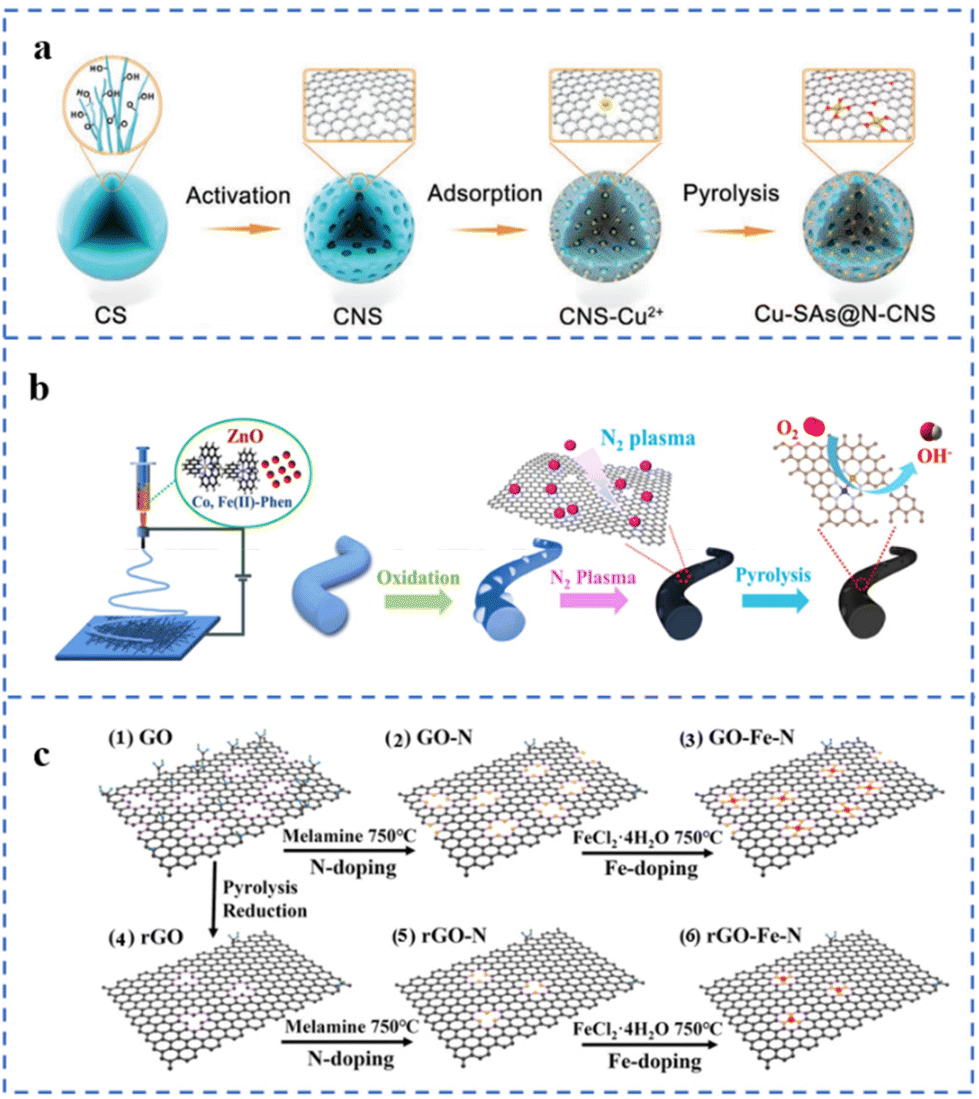 | ||
| Fig. 10 (a) Defect stabilization strategy of Cu-SAs@N-CNS. Reproduced with permission from ref. 136. Copyright © 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) Defect fabrication and defect stabilization strategies in substrates for the preparation of Fe, Co SAs-PNCF. Reproduced with permission from ref. 137. Copyright © 2022, ELSEVIER. (c) Illustration of the preparation of graphene-based catalysts with different defect degrees. (1–3) The GO doping process and (4–6) the rGO doping process. Reproduced with permission from ref. 138. Copyright © 2022, American Chemical Society. | ||
5. Conclusions
The increasingly severe environmental and energy problems have prompted mankind to turn attention to the development of new energy technologies. Zn–air batteries and fuel cells have attracted widespread attention due to their excellent performances in energy conversion. However, the slow kinetics of the ORR greatly limits the development of these devices. The Pt catalyst, which is considered to exhibit the best ORR catalytic performances, has limited application due to its low reserves and high cost. In recent years, non-precious transition metal single-atom catalysts (NPT-SACs) have shown excellent performances and great potential in the ORR field. There are still some problems related to NPT-SACs to be solved: (1) it is difficult to increase the metal loading in NPT-SACs. When the metal reaches a certain content, it will agglomerate into nanoclusters/particles instead of just dispersing atomically. Metal atomic agglomeration would lead to a decrease in the number of active sites, which might degrade the ORR performances of NPT-SACs. Therefore, the preparation of atomically uniformly dispersed NPT-SACs with high metal loadings on substrates for the ORR remains a major challenge. (2) The ORR performances of NPT-SACs are different in acidic and alkaline electrolytes. The performances of many NPT-SACs in acidic electrolytes are unsatisfactory. Therefore, it is still a big challenge to develop SAC catalysts with high activity and stability in electrolytes in a wide pH range; (3) the final catalytic performances of NPT-SACs are related to many factors, such as the number of active sites, the coordination environment of metal atoms, and the substrate structure. However, there is no clear structure–activity relationship between the catalytic performances of SACs and these factors. The heterogeneity of active sites and the existence of substrate defects further increase the difficulty of analysis. (4) There is a lack of uniform standards for the electrochemical tests of the as-prepared NPT-SACs. The lack of unification of the specifications of commercial Pt/C for reference, the test conditions of RDE, and the film production process and quality make comparison difficult.In this review, we summarize some recent advances in NPT-SACs, and classify and summarize these NPT-SACs according to their preparation methods. This review focuses on some factors affecting the ORR performances of NPT-SACs, and on strategies for stabilizing metal atoms. We discuss in detail the coordination environment of metal atoms, the influence of substrates, and the synergy between metal atoms and nanoclusters. Ultimately, the following conclusions come into being: (1) the commonly used preparation methods of NPT-SACs include wet chemical method, high temperature pyrolysis method, high energy ball milling method, atomic layer deposition method, etc. Among them, the wet chemical method has low equipment requirement and it is easy to achieve large-scale production through this method. However, using this method, it is difficult to ensure atomic dispersion or the formation of exposed active sites; high-temperature pyrolysis of metal-containing precursors can obtain NPT-SACs supported on specific substrates (such as MOF, ZIF, graphene, etc.). The pyrolysis process is expected to enhance the activity of active sites. But the pyrolysis process needs to be carried out at a suitable temperature. If the temperature is too low, the carbonization of the substrate may be insufficient. If the temperature is too high, the metal atoms will tend to agglomerate and may even cause damage to the substrate structure. Neither too high nor too low temperature are conducive to the ORR performances of NPT-SACs. High-energy ball milling helps to break bonds and form new bonds, but generally takes a long time and may introduce impurities. The atomic layer deposition method can achieve uniform loading of metal atoms, but it requires complex equipment and higher cost. Therefore, it is necessary to explore a general preparation method to ensure the precise synthesis of atomically active sites. It is very meaningful to streamline the process and reduce the cost, and finally realize the large-scale commercial synthesis of NPT-SACs. (2) The ORR performances of NPT-SACs can be improved by increasing the number of metal active sites. However, the high surface energy of a single metal atom makes it prone to agglomerate during the preparation process, which makes it difficult to synthesize NPT-SACs with high metal loading. Both space restriction strategies and defect stabilization strategies can hinder the migration of metal atoms, making it easier to prepare NPT-SACs with higher metal loadings. However, we still have a long way to go to increase the loading and keep the atoms evenly dispersed. (3) The influence of the substrate on the catalytic activity of NPT-SACs is reflected in loading capacity, charge transfer capacity, mass transfer capacity, and the interaction between the substrate and the metal atoms. Therefore, the selection of suitable substrate materials is very critical to improve the ORR catalytic performances of NPT-SACs. Carbon materials, sulfides, oxides, MXenes, etc. are often used as substrates for SACs, among which carbon materials are the most widely studied. By adjusting the coordination environment around the metal atom, the d-band center of the central metal atom is shifted relative to the Fermi level to improve the selective catalytic activity and stability of NPT-SACs. It is key to obtain non-precious transition metal SACs with excellent ORR performances by selecting the appropriate matrix material, selecting the precursor according to the matrix material, and adjusting the coordination environment of the central atom. (4) Although researchers have tried numerous methods to improve the metal loading of single-metal single-atom catalysts, the loading of metal atoms is still low. The metal atom loading in bimetallic single-atom catalysts can often be about two times that of single-metal single-atom catalysts. Hence, the development of bimetallic/polymetallic single-atom catalysts is a new trend.
In conclusion, NPT-SACs have great potential in the ORR. It requires us to reveal a clear structure–activity relationship between the structure of NPT-SACs and ORR performances. Therefore, researchers should consider preparing catalysts with well-defined active sites, whose characterization results could support the establishment of corresponding models for theoretical calculations. This research approach will provide guidance for designing non-precious transition metal SACs with excellent ORR performances. It is believed that in the near future, non-precious transition metal SACs will strongly promote the development of fuel cells.
Author contributions
Conceptualization, P. J. and C. A.; investigation, Q. D.; resources, C. Z.; writing—original draft preparation, P. J.; writing—review and editing, C. A.; visualization, D. Y.; supervision, C. Q.; project administration, S. W.; funding acquisition, N. H. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Program of Local Science and Technology Development under the Guidance of Central (216Z4402G), the National Natural Science Fund of China (grant No.: 11632004, 52005151, and U1864208), the National Science and Technology Major Project (2017-VII-0011-0106), the Science and Technology Planning Project of Tianjin (20ZYJDJC00030), the Key Program of Research and Development of Hebei Province (202030507040009), the Fund for Innovative Research Groups of Natural Science Foundation of Hebei Province (A2020202002), the Key Project of Natural Science Foundation of Tianjin (S20ZDF077) and the Opening Foundation of State Key Laboratory of Tribology in Tsinghua University (SKLTKF20B03). Q. D. and C. A. acknowledge support from the “Yuanguang” Scholar Program of Hebei University of Technology.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Zhou, H. L. Wang and S. J. Guo, Chem. Soc. Rev., 2016, 45, 1273–1307 RSC.
- Y. Q. Yin, T. T. Liu, D. Liu, Z. Wang, Q. B. Deng, D. Y. Qu, Z. Z. Xie, H. L. Tang and J. S. Li, J. Colloid Interface Sci., 2018, 530, 595–602 CrossRef CAS PubMed.
- Z. Cao, T. T. Zhou, X. R. Ma, Y. L. Shen, Q. B. Deng, W. Zhang and Y. F. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 11007–11015 CAS.
- K. Wang and Q. B. Deng, Front. Chem., 2020, 8, 6 CrossRef CAS PubMed.
- Q. N. Zhu, Z. Y. Wang, J. W. Wang, X. Y. Liu, D. Yang, L. W. Cheng, M. Y. Tang, Y. Qin and H. Wang, Rare Met., 2021, 40, 309–328 CrossRef CAS.
- C. H. An, M. Y. Wang, W. Q. Li, Q. B. Deng, Y. J. Wang, L. F. Jiao and H. T. Yuan, Electrochim. Acta, 2017, 250, 135–142 CrossRef CAS.
- Y. F. Zhao, X. R. Ma, P. L. Li, Y. Lv, J. F. Huang, H. X. Zhang, Y. L. Shen, Q. B. Deng, X. Z. Liu, Y. Ding and Y. Han, J. Mater. Chem. A, 2020, 8, 3491–3498 RSC.
- W. J. Qiu, C. H. An, Y. W. Yan, J. Xu, Z. J. Zhang, W. Guo, Z. Wang, Z. J. Zheng, Z. B. Wang, Q. B. Deng and J. S. Li, J. Power Sources, 2019, 423, 98–105 CrossRef CAS.
- C. H. An, W. Q. Li, M. Y. Wang, Q. B. Deng and Y. J. Wang, Electrochim. Acta, 2018, 283, 603–610 CrossRef CAS.
- F. Zhao, D. H. Zheng, Y. Liu, F. D. Pan, Q. B. Deng, C. L. Qin, Y. Y. Li and Z. F. Wang, Chem. Eng. J., 2021, 415, 12 Search PubMed.
- W. Q. Wei, B. Q. Liu, Y. Q. Gan, H. J. Ma and D. W. Cui, Rare Met., 2021, 40, 409–416 CrossRef CAS.
- X. Q. Li, Y. F. Xing, J. Xu, Q. B. Deng and L. H. Shao, Chem. Commun., 2020, 56, 364–367 RSC.
- Y. W. Liu, C. Gao, L. Dai, Q. B. Deng, L. Wang, J. Y. Luo, S. Liu and N. Hu, Small, 2020, 16, 13 Search PubMed.
- Z. J. Xie, L. M. Zhang, L. Li, Q. B. Deng, G. X. Jiang, J. Q. Wang, B. Q. Cao and Y. J. Wang, Prog. Nat. Sci., 2021, 31, 207–214 CrossRef CAS.
- A. X. Wang, Q. B. Deng, L. J. Deng, X. Z. Guan and J. Y. Luo, Adv. Funct. Mater., 2019, 29, 7 Search PubMed.
- H. Q. Ji, M. F. Wang, S. S. Liu, H. Sun, J. Liu, T. Qian and C. L. Yan, Electrochim. Acta, 2020, 334, 10 Search PubMed.
- Z. H. Meng, S. C. Cai, R. Wang, H. L. Tang, S. Q. Song and P. Tsiakaras, Appl. Catal., B, 2019, 244, 120–127 CrossRef CAS.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 17832–17852 CrossRef CAS PubMed.
- S. S. Ren, X. D. Duan, S. Liang, M. D. Zhang and H. G. Zheng, J. Mater. Chem. A, 2020, 8, 6144–6182 RSC.
- C. C. Wang, Y. X. Chen, M. X. Zhong, T. L. Feng, Y. Liu, S. Y. Feng, N. Zhang, L. Shen, K. Zhang and B. Yang, J. Mater. Chem. A, 2021, 9, 22095–22101 RSC.
- Y. Q. Wang, K. Liu, J. Li, X. T. Yang, J. H. Hu, T. S. Chan, X. Q. Qiu, W. Z. Li and M. Liu, Chem. Eng. J., 2022, 429, 9 Search PubMed.
- L. P. Yang, X. Zhang, L. X. Yu, J. H. Hou, Z. Zhou and R. T. Lv, Adv. Mater., 2022, 34, 10 Search PubMed.
- E. Yeager, J. Mol. Catal., 1986, 38, 5–25 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. T. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- X. L. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- Q. Zhang and J. Guan, Energy Environ. Mater., 2020, 4, 307–335 CrossRef.
- Z. W. Chen, L. X. Chen, C. C. Yang and Q. Jiang, J. Mater. Chem. A, 2019, 7, 3492–3515 RSC.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
- C. Wang, C. H. An, C. L. Qin, H. Gomaa, Q. B. Deng, S. Wu and N. Hu, Nanomaterials, 2022, 12, 23 Search PubMed.
- J. Zhang, S. Yin and H. M. Yin, ACS Appl. Energy Mater., 2020, 3, 11956–11963 CrossRef CAS.
- Y. Sohn, N. Jung, M. J. Lee, S. Lee, K. S. Nahm, P. Kim and S. J. Yoo, J. Ind. Eng. Chem., 2019, 79, 210–216 CrossRef CAS.
- Z. J. Yang, L. Shang, X. Y. Xiong, R. Shi, G. I. N. Waterhouse and T. R. Zhang, Chem. – Eur. J., 2020, 26, 4090–4096 CrossRef CAS PubMed.
- A. M. Gomez-Marin, R. Rizo and J. M. Feliu, Catal. Sci. Technol., 2014, 4, 1685–1698 RSC.
- X. F. Huang, G. X. Yang, S. Li, H. J. Wang, Y. H. Cao, F. Peng and H. Yu, J. Energy Chem., 2022, 68, 721–751 CrossRef.
- Y. F. Chen, X. Zhan, S. L. A. Bueno, I. H. Shafei, H. M. Ashberry, K. Chatterjee, L. Xu, Y. W. Tang and S. E. Skrabalak, Nanoscale Horiz., 2021, 6, 231–237 RSC.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. Liu, M. G. Jiao, B. B. Mei, Y. X. Tong, Y. P. Li, M. B. Ruan, P. Song, G. Q. Sun, L. H. Jiang, Y. Wang, Z. Jiang, L. Gu, Z. Zhou and W. L. Xu, Angew. Chem., Int. Ed., 2019, 58, 1163–1167 CrossRef CAS PubMed.
- J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu and S. Sun, Joule, 2019, 3, 124–135 CrossRef CAS.
- Q. B. Deng, M. Smetanin and J. Weissmuller, J. Catal., 2014, 309, 351–361 CrossRef CAS.
- Q. B. Deng, V. Gopal and J. Weissmuller, Angew. Chem., Int. Ed., 2015, 54, 12981–12985 CrossRef CAS PubMed.
- J. He, Y. L. Shen, M. Z. Yang, H. X. Zhang, Q. B. Deng and Y. Ding, J. Catal., 2017, 350, 212–217 CrossRef CAS.
- J. M. Luo, X. C. Qiao, J. T. Jin, X. L. Tian, H. B. Fan, D. M. Yu, W. L. Wang, S. J. Liao, N. Yu and Y. J. Deng, J. Mater. Chem. A, 2020, 8, 8575–8585 RSC.
- Q. Dong, H. Wang, S. Ji, X. Y. Wang, Q. B. Liu, D. J. L. Brett, V. Linkov and R. F. Wang, Adv. Sustainable Syst., 2019, 10, 1900085 CrossRef.
- Y. H. Lei, N. Tan, Y. Z. Zhu, D. Huo, S. B. Sun, Y. L. Zhang and G. H. Gao, J. Electrochem. Soc., 2020, 167, 9 Search PubMed.
- K. Maiti, N. H. Kim and J. H. Lee, Chem. Eng. J., 2021, 410, 13 CrossRef.
- D. Bhalothia, L. Krishnia, S. S. Yang, C. Yan, W. H. Hsiung, K. W. Wang and T. Y. Chen, Appl. Sci., 2020, 10, 19 Search PubMed.
- L. Zhang, W. X. Niu and G. B. Xu, Nano Today, 2012, 7, 586–605 CrossRef CAS.
- C. Z. Zhu, Q. R. Shi, S. F. Fu, J. H. Song, H. B. Xia, D. Du and Y. H. Lin, Adv. Mater., 2016, 28, 8779–8783 CrossRef CAS PubMed.
- C. Z. Zhu, D. Du, A. Eychmuller and Y. H. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, S. Kuhl, P. Strasser and B. Roldan Cuenya, Nat. Rev. Mater., 2016, 1, 14 Search PubMed.
- Y. J. Chen, S. F. Ji, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- X. Wang, W. X. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. X. Ju, J. C. Dong, L. R. Zheng, W. S. Yan, X. S. Zheng, Z. J. Li, X. Q. Wang, J. Yang, D. S. He, Y. Wang, Z. X. Deng, Y. E. Wu and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS.
- J. Zhang, X. Wu, W. C. Cheong, W. X. Chen, R. Lin, J. Li, L. R. Zheng, W. S. Yan, L. Gu, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Nat. Commun., 2018, 9, 8 CrossRef PubMed.
- C. Gao, S. M. Chen, Y. Wang, J. W. Wang, X. S. Zheng, J. F. Zhu, L. Song, W. K. Zhang and Y. J. Xiong, Adv. Mater., 2018, 30, 9 Search PubMed.
- R. H. Lin, D. Albani, E. Fako, S. K. Kaiser, O. V. Safonova, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2019, 58, 504–509 CrossRef CAS PubMed.
- Z. S. Shi, W. Q. Yang, Y. T. Gu, T. Liao and Z. Q. Sun, Adv. Sci., 2020, 7, 26 Search PubMed.
- K. Ye, Z. Zhou, J. Shao, L. Lin, D. Gao, N. Ta, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 4814–4821 CrossRef CAS PubMed.
- Y. Yan, H. Cheng, Z. Qu, R. Yu, F. Liu, Q. Ma, S. Zhao, H. Hu, Y. Cheng, C. Yang, Z. Li, X. Wang, S. Hao, Y. Chen and M. Liu, J. Mater. Chem. A, 2021, 9, 19489–19507 RSC.
- J. Lin, A. Q. Wang, B. T. Qiao, X. Y. Liu, X. F. Yang, X. D. Wang, J. X. Liang, J. X. Li, J. Y. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- R. X. Qin, P. X. Liu, G. Fu and N. F. Zheng, Small Methods, 2018, 2, 21 Search PubMed.
- Y. Z. Zhou, G. B. Chen, Q. Wang, D. Wang, X. F. Tao, T. R. Zhang, X. L. Feng and K. Mullen, Adv. Funct. Mater., 2021, 31, 9 Search PubMed.
- G. B. Chen, P. Liu, Z. Q. Liao, F. F. Sun, Y. H. He, H. X. Zhong, T. Zhang, E. Zschech, M. W. Chen, G. Wu, J. Zhang and X. L. Feng, Adv. Mater., 2020, 32, 7 Search PubMed.
- Y. Cheng, J. Zhang, X. Wu, C. Tang, S. Z. Yang, P. Su, L. Thomsen, F. Zhao, S. Lu, J. Liu and S. P. Jiang, Nano Energy, 2021, 80, 105534 CrossRef CAS.
- H. L. Fei, J. C. Dong, D. L. Chen, T. D. Hu, X. D. Duan, I. R. Shakir, Y. Huang and X. F. Duan, Chem. Soc. Rev., 2019, 48, 5207–5241 RSC.
- X. R. Zhang, X. M. Xu, S. X. Yao, C. Hao, C. Pan, X. Xiang, Z. Q. Tian, P. K. Shen, Z. P. Shao and S. P. Jiang, Small, 2022, 18, 16 Search PubMed.
- X. Li, X. Yang, L. Liu, H. Zhao, Y. Li, H. Zhu, Y. Chen, S. Guo, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2021, 11, 7450–7459 CrossRef CAS.
- T. Sun, Y. Li, T. Cui, L. Xu, Y. G. Wang, W. Chen, P. Zhang, T. Zheng, X. Fu, S. Zhang, Z. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 6206–6214 CrossRef CAS PubMed.
- Y. C. Guo, F. Liu, L. Feng, X. M. Wang, X. Zhang and J. S. Liang, Chem. Eng. J., 2022, 429, 12 Search PubMed.
- C. Shao, S. Zhuang, H. Zhang, Q. Jiang, X. Xu, J. Ye, B. Li and X. Wang, Small, 2021, 17, e2006178 CrossRef PubMed.
- J. X. Han, H. L. Bao, J. Q. Wang, L. R. Zheng, S. R. Sun, Z. L. Wang and C. W. Sun, Appl. Catal., B, 2021, 280, 10 CrossRef.
- M. M. Liu, L. L. Wang, K. N. Zhao, S. S. Shi, Q. S. Shao, L. Zhang, X. L. Sun, Y. F. Zhao and J. J. Zhang, Energy Environ. Sci., 2019, 12, 2890–2923 RSC.
- F. Liu, L. Shi, X. N. Lin, D. L. Yu, C. Zhang, R. Xu, D. Liu, J. S. Qiu and L. M. Dai, Appl. Catal., B, 2022, 302, 11 Search PubMed.
- C. Yang, S. S. Shang, Q. F. Gu, J. Shang and X. Y. Li, J. Energy Chem., 2022, 66, 306–313 CrossRef.
- G. L. Li, Z. F. Lu, X. Wang, S. Cao and C. Hao, ACS Sustainable Chem. Eng., 2022, 10, 224–233 CrossRef CAS.
- B. W. Zhang, Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Small Methods, 2019, 3, 25 Search PubMed.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- Z. Y. Kong, T. Liu, K. Hou and L. H. Guan, J. Mater. Chem. A, 2022, 10, 2826–2834 RSC.
- X. L. Xu, X. M. Zhang, Z. X. Xia, R. L. Sun, H. Q. Li, J. H. Wang, S. S. Yu, S. L. Wang and G. Q. Sun, J. Energy Chem., 2021, 54, 579–586 CrossRef.
- G. F. Han, F. Li, A. I. Rykov, Y. K. Im, S. Y. Yu, J. P. Jeon, S. J. Kim, W. H. Zhou, R. Ge, Z. M. Ao, T. J. Shin, J. H. Wang, H. Y. Jeong and J. B. Baek, Nat. Nanotechnol., 2022, 17, 403–407 CrossRef CAS PubMed.
- S. X. Yang, Y. H. Yu, M. L. Dou, Z. P. Zhang, L. M. Dai and F. Wang, Angew. Chem., Int. Ed., 2019, 58, 14724–14730 CrossRef CAS PubMed.
- X. Zhao, X. Li, Z. H. Bi, Y. W. Wang, H. B. Zhang, X. H. Zhou, Q. Wang, Y. T. Zhou, H. S. Wang and G. Z. Hu, J. Energy Chem., 2022, 66, 514–524 CrossRef.
- S. Liang, L. C. Zou, L. J. Zheng, F. Li, X. X. Wang, L. N. Song and J. J. Xu, Adv. Energy Mater., 2022, 12, 2103097 CrossRef CAS.
- R. Hao, S. Gu, J. Chen, Z. Wang, Q. Gan, Z. Wang, Y. Huang, P. Liu, K. Zhang, K. Liu, C. Liu and Z. Lu, Mater. Today Energy, 2021, 21, 100826 CrossRef CAS.
- H. B. Zhong, C. W. Shi, J. T. Li, R. H. Yu, Q. Yu, H. Y. Liu, Y. Yao, J. S. Wu, L. Zhou and L. Q. Mai, Chem. Commun., 2020, 56, 4488–4491 RSC.
- J. Choi and P. A. Dowben, Surf. Sci., 2006, 600, 2997–3002 CrossRef CAS.
- J. J. Li, Y. F. Jiang, Q. Wang, C. Q. Xu, D. J. Wu, M. N. Banis, K. R. Adair, K. Doyle-Davis, D. M. Meira, Y. Z. Finfrock, W. H. Li, L. Zhang, T. K. Sham, R. Y. Li, N. Chen, M. Gu, J. Li and X. L. Sun, Nat. Commun., 2021, 12, 11 CrossRef PubMed.
- B. Z. Lu, Q. M. Liu and S. W. Chen, ACS Catal., 2020, 10, 7584–7618 CrossRef CAS.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CrossRef CAS.
- Y. C. Li, X. F. Liu, L. R. Zheng, J. X. Shang, X. Wan, R. M. Hu, X. Guo, S. Hong and J. L. Shui, J. Mater. Chem. A, 2019, 7, 26147–26153 RSC.
- Y. M. Zhao, P. C. Zhang, C. Xu, X. Y. Zhou, L. M. Liao, P. J. Wei, E. Liu, H. Q. Chen, Q. G. He and J. G. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 17334–17342 CrossRef CAS PubMed.
- X. Liang, Z. Li, H. Xiao, T. Zhang, P. Xu, H. Zhang, Q. Gao and L. Zheng, Chem. Mater., 2021, 33, 5542–5554 CrossRef CAS.
- Y. J. Chen, R. Gao, S. F. Ji, H. J. Li, K. Tang, P. Jiang, H. B. Hu, Z. D. Zhang, H. G. Hao, Q. Y. Qu, X. Liang, W. X. Chen, J. C. Dong, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- K. Yuan, D. Lutzenkirchen-Hecht, L. B. Li, L. Shuai, Y. Z. Li, R. Cao, M. Qiu, X. D. Zhuang, M. K. H. Leung, Y. W. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- S. Yuan, J. Zhang, L. Hu, J. Li, S. Li, Y. Gao, Q. Zhang, L. Gu, W. Yang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 21685–21690 CrossRef CAS PubMed.
- H. Shen, H. Peng, R. Cao, L. Yang, Y. Gao, A. Turak, T. Thomas, X. Guo, Y. Zhu, J. Wang and M. Yang, J. Phys. Chem. Lett., 2020, 12, 517–524 CrossRef PubMed.
- L. B. Li, S. H. Huang, R. Cao, K. Yuan, C. B. Lu, B. Y. Huang, X. N. Tang, T. Hu, X. D. Zhuang and Y. W. Chen, Small, 2022, 18, 12 Search PubMed.
- C. Xin, W. Shang, J. Hu, C. Zhu, J. Guo, J. Zhang, H. Dong, W. Liu and Y. Shi, Adv. Funct. Mater., 2022, 32, 2108345 CrossRef CAS.
- Y. S. Xu, L. P. Zhu, X. X. Cui, M. Y. Zhao, Y. L. Li, L. L. Chen, W. C. Jiang, T. Jiang, S. G. Yang and Y. Wang, Nano Res., 2020, 13, 752–758 CrossRef CAS.
- Z. J. Zhu, H. J. Yin, Y. Wang, C. H. Chuang, L. Xing, M. Y. Dong, Y. R. Lu, G. Casillas-Garcia, Y. L. Zheng, S. Chen, Y. H. Dou, P. Liu, Q. L. Cheng and H. J. Zhao, Adv. Mater., 2020, 32, 8 Search PubMed.
- Z. P. Zhang, J. T. Sun, F. Wang and L. M. Dai, Angew. Chem., Int. Ed., 2018, 57, 9038–9043 CrossRef CAS PubMed.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2019, 58, 12469–12475 CrossRef CAS PubMed.
- Z. Zhu, H. Yin, Y. Wang, C.-H. Chuang, L. Xing, M. Dong, Y.-R. Lu, G. Casillas-Garcia, Y. Zheng, S. Chen, Y. Dou, P. Liu, Q. Cheng and H. Zhao, Adv. Mater., 2020, 32, 2004670 CrossRef CAS PubMed.
- L. G. Ma, J. L. Li, Z. W. Zhang, H. Yang, X. Q. Mu, X. Y. Gu, H. H. Jin, D. Chen, S. L. Yan, S. L. Liu and S. C. Mu, Nano Res., 2022, 15, 1966–1972 CrossRef CAS.
- Y. L. Wu, G. F. Liang, D. Chen, Z. L. Li, J. C. Xu, G. J. Huang, M. Z. Yang, H. Zhang, J. Chen, F. Y. Xie, Y. S. Jin, N. Wang, S. H. Sun and H. Meng, ACS Appl. Mater. Interfaces, 2021, 13, 48923–48933 CrossRef CAS PubMed.
- K. Qin, Z. Y. Zhu, M. J. Yi, S. Y. Hu, F. X. Ma and J. H. Zhang, ChemElectroChem, 2021, 8, 4804–4809 CrossRef CAS.
- H. B. Xu, H. X. Jia, H. Z. Li, J. Liu, X. W. Gao, J. C. Zhang, M. Liu, D. L. Sun, S. L. Chou, F. Fang and R. B. Wu, Appl. Catal., B, 2021, 297, 9 CrossRef.
- X. Y. Xie, L. Shang, X. Y. Xiong, R. Shi and T. R. Zhang, Adv. Energy Mater., 2022, 12, 8 Search PubMed.
- H. B. Li, M. D. Zhang, W. Zhou, J. G. Duan and W. Q. Jin, Chem. Eng. J., 2021, 421, 9 Search PubMed.
- S. Zhou, Z. Xiao, Q. Yang, X. Huang, Y. Niu, Y. Ma and L. Zhi, Sci. China Mater., 2021, 64, 2221–2229 CrossRef CAS.
- Y. Lin, K. Liu, K. Chen, Y. Xu, H. Li, J. Hu, Y.-R. Lu, T.-S. Chan, X. Qiu, J. Fu and M. Liu, ACS Catal., 2021, 11, 6304–6315 CrossRef CAS.
- R. Ma, X. Cui, Y. L. Wang, Z. Y. Xiao, R. Luo, L. K. Gao, Z. N. Wei and Y. K. Yang, J. Mater. Chem. A, 2022, 10, 8 Search PubMed.
- D. Wang, M. W. Yuan, J. S. Xu, Y. Y. Li, K. F. Shi, H. Yang, H. F. Li and G. B. Sun, ACS Sustainable Chem. Eng., 2021, 9, 16956–16964 CrossRef CAS.
- M. Zhao, H. Liu, H. Zhang, W. Chen, H. Sun, Z. Wang, B. Zhang, L. Song, Y. Yang, C. Ma, Y. Han and W. Huang, Energy Environ. Sci., 2021, 14, 6455–6463 RSC.
- Z. Q. Mai, Z. Liu, S. C. Liu, X. F. Zhang, Z. M. Cui and Z. H. Tang, Int. J. Hydrogen Energy, 2021, 46, 36836–36847 CrossRef CAS.
- T. Zhang, B. Zhang, Q. Peng, J. Zhou and Z. Sun, J. Mater. Chem. A, 2021, 9, 433–441 RSC.
- C. H. An, W. Kang, Q. B. Deng and N. Hu, Rare Met., 2022, 41, 378–384 CrossRef CAS.
- Y. Wang, M. Wang, Z. Lu, D. Ma and Y. Jia, Nanoscale, 2021, 13, 13390–13400 RSC.
- X. M. Zhao and Y. Pei, J. Phys. Chem. C, 2021, 125, 12541–12550 CrossRef CAS.
- H. He, H. H. Wang, J. J. Liu, X. J. Liu, W. Z. Li and Y. N. Wang, Molecules, 2021, 26, 26 Search PubMed.
- H. Huang, D. Yu, F. Hu, S. C. Huang, J. Song, H. Y. Chen, L. L. Li and S. Peng, Angew. Chem., 2022, 61, e202116068 CAS.
- Z. Wang, C. Zhu, H. Tan, J. Liu, L. Xu, Y. Zhang, Y. Liu, X. Zou, Z. Liu and X. Lu, Adv. Funct. Mater., 2021, 31, 2104735 CrossRef CAS.
- S. Q. Hu, W. P. Ni, D. H. Yang, C. Ma, J. H. Zhang, J. F. Duan, Y. Gao and S. G. Zhang, Carbon, 2020, 162, 245–255 CrossRef CAS.
- S. Ding, Z. Lyu, E. Sarnello, M. Xu, L. Fang, H. Tian, S. E. Karcher, T. Li, X. Pan, J. McCloy, G. Ding, Q. Zhang, Q. Shi, D. Du, J. C. Li, X. Zhang and Y. Lin, J. Mater. Chem. A, 2022, 10, 5981–5989 RSC.
- Y. X. Wang, H. Y. Su, Y. H. He, L. G. Li, S. Q. Zhu, H. Shen, P. F. Xie, X. B. Fu, G. Y. Zhou, C. Feng, D. K. Zhao, F. Xiao, X. J. Zhu, Y. C. Zeng, M. H. Shao, S. W. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- J. Su, J. Zang, P. Tian, S. Zhou, X. Liu, R. Li, Y. Wang and L. Dong, Int. J. Hydrogen Energy, 2020, 45, 28792–28799 CrossRef CAS.
- Y. Q. Cui, J. X. Xu, Y. Zhao and L. H. Guan, Nanotechnology, 2021, 32, 305402 CrossRef CAS PubMed.
- Z. Kou, W. Zang, Y. Ma, Z. Pan, S. Mu, X. Gao, B. Tang, M. Xiong, X. Zhao, A. K. Cheetham, L. Zheng and J. Wang, Nano Energy, 2020, 67, 104288 CrossRef CAS.
- G. S. Kang, J. H. Jang, S. Y. Son, Y. K. Lee, D. C. Lee, S. J. Yoo, S. Lee and H.-I. Joh, J. Power Sources, 2022, 520, 230904 CrossRef CAS.
- L. S. Gao, X. Gao, P. Jiang, C. Y. Zhang, H. Guo and Y. H. Cheng, Small, 2022, 18, 10 Search PubMed.
- J. W. Wan, W. X. Chen, C. Y. Jia, L. R. Zheng, J. C. Dong, X. S. Zheng, Y. Wang, W. S. Yan, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Adv. Mater., 2018, 30, 8 Search PubMed.
- G. L. Liu, A. W. Robertson, M. M. J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y. C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS PubMed.
- J. Jones, H. F. Xiong, A. T. Delariva, E. J. Peterson, H. Pham, S. R. Challa, G. S. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernandez, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- L. Zong, K. Fan, W. Wu, L. Cui, L. Zhang, B. Johannessen, D. Qi, H. Yin, Y. Wang, P. Liu, L. Wang and H. Zhao, Adv. Funct. Mater., 2021, 31, 2104864 CrossRef CAS.
- M. Jiang, F. Wang, F. Yang, H. He, J. Yang, W. Zhang, J. Y. Luo, J. Zhang and C. P. Fu, Nano Energy, 2022, 93, 10 Search PubMed.
- J. He, T. L. Zheng, D. J. Wu, S. M. Zhang, M. Gu and Q. G. He, ACS Catal., 2022, 12, 1601–1613 CrossRef CAS.
- M. Khan, C. Sun, J. Cai, D. Ye, K. Zhao, G. Zhang, S. Shi, L. Shah, J. Fang, C. Yang, H. Zhao, S. Mu and J. Zhang, ChemElectroChem, 2021, 8, 1298–1306 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |