
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDye-sensitized lanthanide containing nanoparticles for luminescence based applications
Clémence
Cheignon
 *,
Ali A.
Kassir†
,
Lohona K.
Soro†
and
Loïc J.
Charbonnière
*
*,
Ali A.
Kassir†
,
Lohona K.
Soro†
and
Loïc J.
Charbonnière
*
Equipe de Synthèse Pour l'Analyse (SynPA), Institut Pluridisciplinaire Hubert Curien (IPHC), UMR 7178 CNRS/Université de Strasbourg, ECPM, Bâtiment R1N0, 25 rue Becquerel, 67087 Strasbourg, Cedex 2, France. E-mail: ccheignon@unistra.fr; l.charbonn@unistra.fr
First published on 29th August 2022
Abstract
Due to their exceptional luminescent properties, lanthanide (Ln) complexes represent a unique palette of probes in the spectroscopic toolkit. Their extremely weak brightness due to forbidden Ln electronic transitions can be overcome by indirect dye-sensitization from the antenna effect brought by organic ligands. Despite the improvement brought by the antenna effect, (bio)analytical applications with discrete Ln complexes as luminescent markers still suffers from low sensitivity as they are limited by the complex brightness. Thus, there is a need to develop nano-objects that cumulate the spectroscopic properties of multiple Ln ions. This review firstly gives a brief introduction of the spectral properties of lanthanides both in complexes and in nanoparticles (NPs). Then, the research progress of the design of Ln-doped inorganic NPs with capping antennas, Ln-complex encapsulated NPs and Ln-complex surface functionalized NPs is presented along with a summary of the various photosensitizing ligands and of the spectroscopic properties (excited-state lifetime, brightness, quantum yield). The review also emphasizes the problems and limitations encountered over the years and the solutions provided to address them. Finally, a comparison of the advantages and drawbacks of the three types of NP is provided as well as a conclusion about the remaining challenges both in the design of brighter NPs and in the luminescence based applications.
 Clémence Cheignon | Clémence Cheignon obtained her PhD in analytical chemistry at the University Paul Sabatier (Toulouse, France) in 2016 under the supervision of Dr Fabrice Collin and Dr Christelle Hureau in Prof. Peter Faller's group, where she also carried out post-doctoral research. After one year as a Temporary Lecturer and Research assistant at the Institut des Biomolécules Max Mousseron (Montpellier, France) in the group of Prof. Christine Enjalbal, she moved to Strasbourg (France) and obtained the position of assistant professor at the Engineer School of chemistry (ECPM) of Strasbourg University (France) in 2019. She is conducting research in the SynPA group led by Dr Loïc Charbonnière at IPHC, her current research is focused on the design and characterization of luminescent lanthanide nanoparticles and their application as probes in (bio)analytical applications. |
 Ali A. Kassir | Ali Kassir obtained his BSc degree in chemistry from Lebanese University in 2017. He completed his master's in analytical sciences at Strasbourg University in 2019. Currently, he is a PhD student in the SynPA team under the direction of Dr Loic Charbonnière and the co-supervision of Dr Clémence Cheignon, and working on the development of new ELISA detection methods based on ultrabright lanthanide nanoparticles. His main interests are centered on nanomaterials and bioanalytical applications. |
 Lohona K. SORO | Lohona SORO obtained his master's degree in analytical chemistry in 2019 at the University of Strasbourg. He is now a PhD student under the supervision of Dr Loïc Charbonnière in the SynPA team. His research mainly focuses on optimizing the upconversion efficiency of lanthanide-based upconverting devices in solution. |
 Loïc J. Charbonnière | Loïc Charbonnière got a degree of engineer in chemistry from the chemistry school of Strasbourg (France) in 1991 and a master in chemistry from the University of Strasbourg in 1993. He earned the grade of Dr in 1996 for his thesis on the synthesis of dinuclear triple stranded helicates under the supervision of Prof. Alan Williams in Geneva (Switzerland). After a first post-doctoral period in Lausanne (Switzerland, 1997) with Prof. Jean-Claude Bünzli and a second one in Strasbourg (1998) with Dr Françoise Arnaud-Neu, he obtained the position of associate researcher at the French National Research Centre (CNRS) in 1998 in the team of Dr Raymond Ziessel. In 2011, he created a team dedicated to the synthesis of new chemical tools for analytical application, renamed the SynPA team in 2018. In 2011, he also earned the grade of Research Director at the CNRS. His main scientific interests lie in organic synthesis and coordination chemistry, with a strong liking for lanthanides and their exceptional properties and all their potential uses at the border of different disciplines. |
1. Introduction
Because of their exceptional electronic properties,1 lanthanide (Ln) ions are very particular ones in the spectroscopic toolbox. They display line like emission spectra which are specific signatures for each luminescent ion of the series, and their excited state lifetimes are three to six orders of magnitude longer than organic compounds or luminescent d-block coordination compounds, allowing for a very sensitive time-resolved (TR) detection. However, the same reasons providing them with these exceptional properties, i.e. the forbidden character of the electronic transitions by Laporte's and also often spin selection rules, confer on them very poor absorption properties. Molar absorption coefficients of lanthanide ions are very weak, rarely exceeding unity, apart for the exceptional case of Yb complexes.2 Fortunately, during the second world war, the discovery of Weissman3 that lanthanide ions could be indirectly photosensitized by coordinated aromatic ligands (also called antennas), changed the game. Since then, a plethora of researchers tried to improve the luminescence properties of Ln complexes, by coordinating them with adapted antenna which collect photons and transfer them to the Ln cation. It is worth noting that a proper selection of the antenna is required to ensure that it coordinates with the Ln ion, it absorbs light at the desired excitation wavelength and that its excited state is close enough to the excited state of the Ln to allow energy transfer.4 Thanks to the antenna effect, the brightness of the complexes (B), defined as the product of the molar absorption coefficient by the luminescence quantum yield,5 have begun to become acceptable in comparison with organic fluorophores.6Embedding the Ln ions into synthetic pre-organized macrocyclic structures,7 further led to kinetically and thermodynamically stable complexes which can find applications in luminescence labeling.8–10 However, even for the best of these labels and despite the advantages of TR detection, these labels are still far from the best fluorophores, such as luminescent proteins or semiconducting nanocrystals.11
An obvious further step in the development of brighter Ln based luminescent marker was then to try to gather numerous Ln ions into nanoscopic structures. The aim of this review is to highlight the three main approaches towards Ln based nanoscopic scaffolds that are illustrated in Scheme 1. These comprise the development of surface capped Ln nanoparticles (Ln-NPs) using antenna ligands (Scheme 1a), the incorporation of discrete molecular complexes into nanoscopic structures (Scheme 1b) and the preparation of nanoscopic scaffolds surface functionalized by Ln complexes (Scheme 1c). Before going into the details of each approach, we will first deal with some of the basic spectroscopic properties of luminescent Ln ion to compare them at the level of molecular complexes or at the nanoscopic scale.
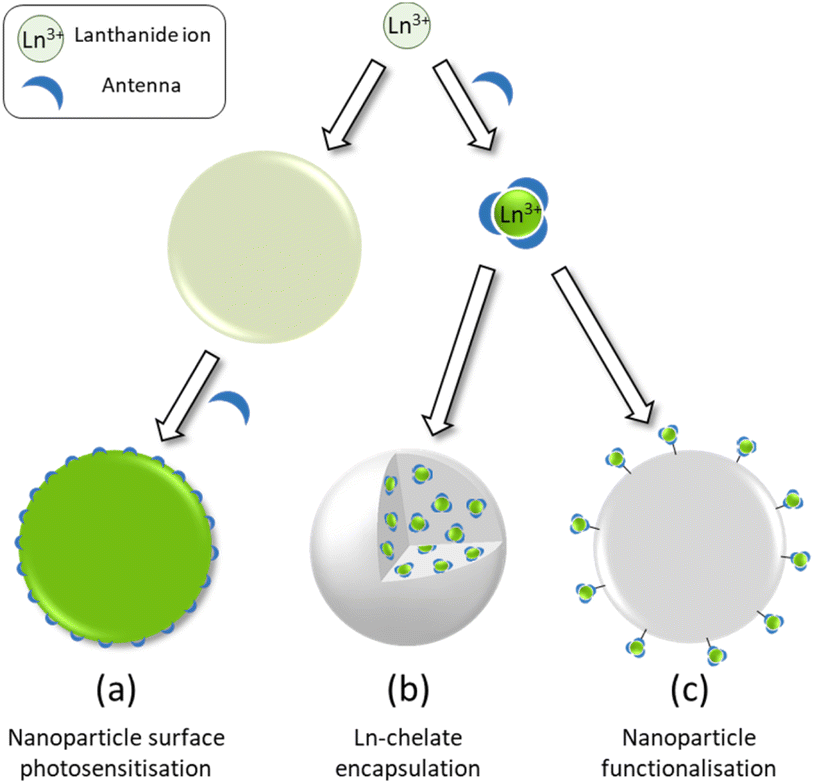 | ||
| Scheme 1 Schematic representation of the three strategies for the design of dye-sensitized lanthanide containing nanoparticles. | ||
2. Basic spectral properties of Ln ion in complexes and their comparison with Ln doped NPs
2.1. Absorption properties
The absorption A of a compound in solution is defined as its propensity to absorb photons at a certain wavelength. A is proportional to the molar absorption coefficient ελ (in mol−1 L cm−1) and is determined using the Beer–Lambert law: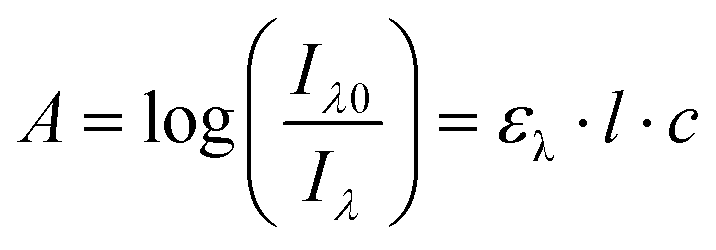 | (1) |
In which Iλ0 and Iλ are respectively the intensities of the incoming and outgoing light source at the wavelength λ, l is the length of the optical path (in cm) and c the concentration of the absorbing species in the sample (in mol L−1). Alternatively, the absorption may be related to σ the absorption cross section (in cm2) which represents the effective area that a photon needs to cross in order to be absorbed. While chemists generally prefer ε, physicists and spectroscopists prefer σ but both are related by the following relationship:
| σ = 0.382 × 10−20 × ε | (2) |
Considering the absorption properties of a Ln complex, it is important to clearly differentiate the absorption due to the ligand coordinated to the metal and the intrinsic absorption of the Ln ion itself. For the later, f–f electronic transitions are forbidden by Laporte's rule (ΔL = ±1) and for most of them by the spin multiplicity rule too (ΔS = 0). For a full detail of the selection rules in Ln compounds, the reader is invited to consider specialized literature.1 As a result, the absorption coefficients of f-f transitions in Ln complexes are very weak (<1 mol−1 L cm−1) and are rarely determined, except for some cases of Yb complexes,2,12 the corresponding 2F5/2 ← 2F7/2 electronic transition being unique and allowed by the spin selection rule. In contrast, the electronic transition centered on the ligands are generally associated to allowed 1ππ* transitions with large absorption coefficients (ε > 1000 mol−1 L cm−1). Additionally, the incorporation of multiple aromatic antenna coordinated to the Ln allows to cumulate their absorption properties.13
When Ln ions are embedded into nanoparticles, as the 4f orbitals have a small radial extension,14 and considering that these orbitals are protected from the surroundings by filled 5s and 5p orbitals, they poorly participate into the bonding with the surrounding ligands or coordinating ions and are only weakly influenced by the environment. As a result, the absorption coefficients of the f–f transitions are corresponding to the sum of the Ln atoms in the NPs and they do not benefit of any confinement effect, as it is the case for semi-conducting nanoparticles.15 Similarly, the absorption associated to the ligands coordinated to Ln-NPs are generally the result of the sum of the absorption of each ligand. In some instances, these absorptions have been determined using spectrophotometric titrations allowing to reach very large absorption coefficients for the whole NPs (ε > 107 mol−1 L cm−1),16 making such NPs of relevance for labeling and bio-analytical applications.
2.2. Luminescence properties
The luminescence quantum yield (ϕ) is defined as the ratio of emitted photons over absorbed photons. When an atom or a molecule has absorbed a photon, it reaches an excited state and can decay back to the ground state by radiative processes, resulting in luminescence, with a rate constant kr, and a radiative lifetime τr (kr = 1/τr) or by numerous other non-radiative processes, with rate constants knr. The luminescence is then related to these rate constants by: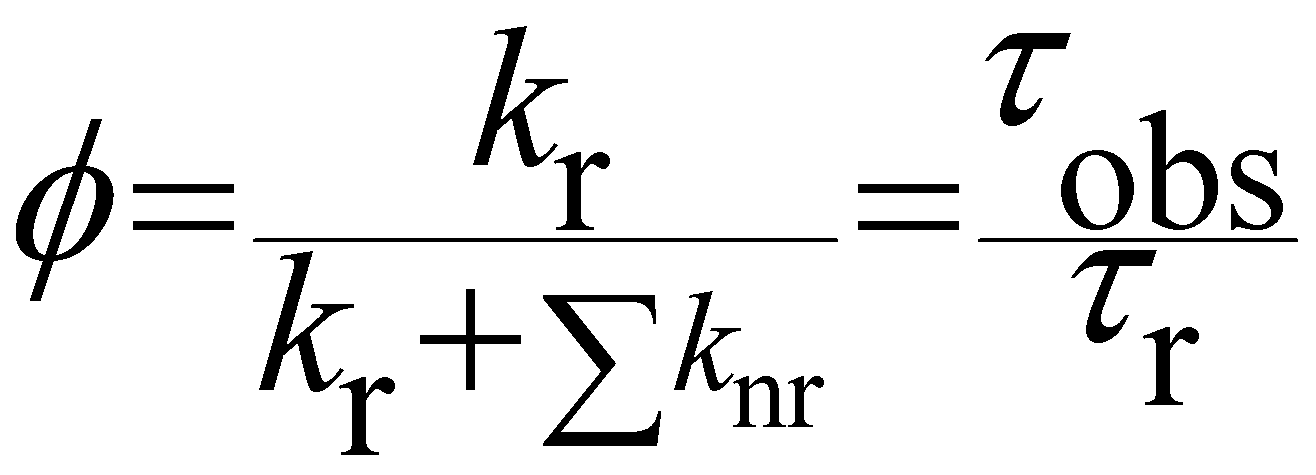 | (3) |
When one determines the excited state lifetime of the molecule by spectroscopic means, the observed decay, τobs, corresponds to the inverse of the sum of all the radiative and non-radiative phenomena leading to the right part of eqn (3). Considering eqn (3), it is obvious that the smaller the rates of non-radiative processes, the larger the luminescence quantum yield.
For discrete luminescent lanthanide complexes, the most important non-radiative phenomena are generally due to energy transfer to the overtones of high energy oscillators such as OH, NH and CH bonds of the organic ligands or of closely placed molecules of solvents.17 These energy losses have been deeply studied by different authors for visible lanthanide emitters such as Eu17,18 and Tb,17 but also for near infrared (NIR) ones such as Yb17 and Nd19 and dual NIR-visible Ln emitters Dy and Sm.20 The particular interest of these studies relies on the possibility to access to the hydration number q, i.e. the number of water molecules directly bonded to the first coordination sphere of the Ln cation. By determining the luminescence lifetime of the complexes in light water τH2O and in heavy water τD2O, q can be determined using the general formula:
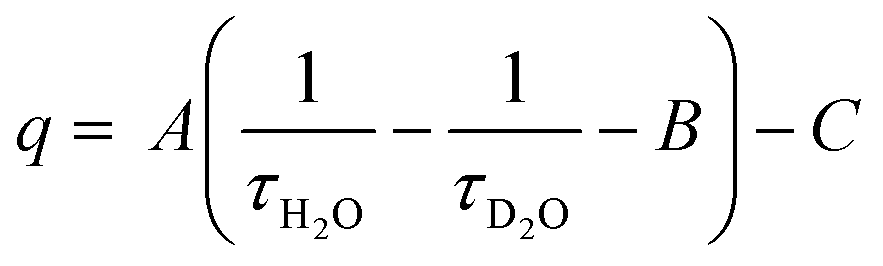 | (4) |
In which A, B and C are constants depending on the studied Ln.17–20 The impact of these non-radiative processes can be highly detrimental, especially for NIR emitters in water as the energy levels of the Ln excited states are close to the first and second overtones of the OH oscillators of water at ca. 7000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1, corresponding to ca. 1430 and 952 nm respectively.17 Although less important in general, the oscillators associated to C–H bonds present in the framework of the antenna ligands can also have a strong impact on the luminescence, justifying the importance of their deuteration to avoid the corresponding losses.21
500 cm−1, corresponding to ca. 1430 and 952 nm respectively.17 Although less important in general, the oscillators associated to C–H bonds present in the framework of the antenna ligands can also have a strong impact on the luminescence, justifying the importance of their deuteration to avoid the corresponding losses.21
In the case of Ln-NPs, two kinds of Ln environments have to be taken into account. Ln atoms in the very external layers can also be influenced by OH, NH and CH oscillators of the surrounding medium and their luminescence can be quenched by the same mechanisms as for discrete Ln complexes. In contrast, Ln atoms embedded in the core of the NPs are not influenced by the surrounding medium. However, they are still prone to non-radiative deactivation through impurities of the matrix with high-frequency vibrations, such as OH− groups22 or energy transfer processes assisted by phonons, quasiparticles having the energy of a vibration of the lattice of the crystalline core. However, phonons of the lattice are generally of much lower energy than OH, NH or CH oscillators. For example, when the OH stretching vibration of water is observed at 3500 cm−1, the highest phonon energy observed in common solid matrices do not exceed 1200 cm−1.23 The non-radiative rate constant, knr, can then be related to the energy of the phonon, ΔEp, and to the energy difference between the ground and excited states of the lanthanide ΔELn by the following relation:24
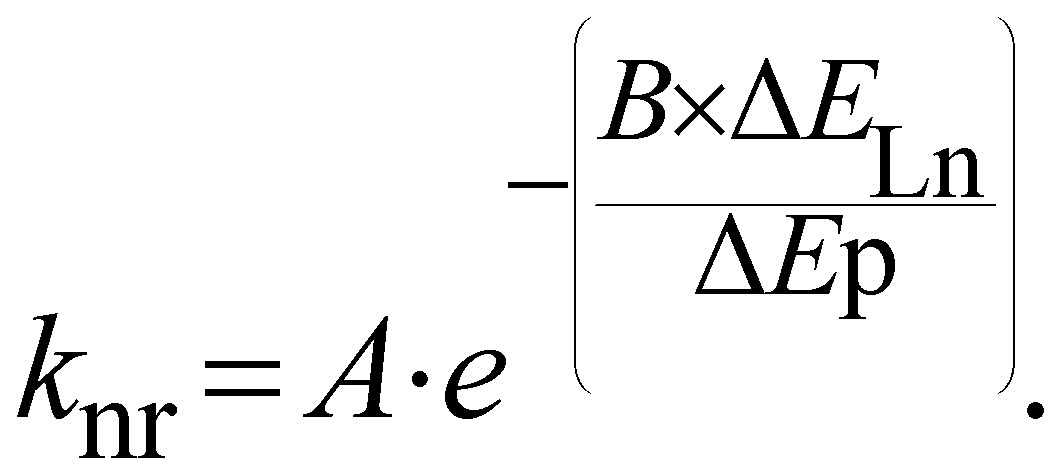 | (5) |
In which A and B are constants depending on the solid matrix. From eqn (5), one can realize that the smaller the energy of the phonon, the lower the rate constant for non-radiative decays. In some instances, the non-radiative decays can be so small that the observed luminescence lifetimes of the Ln atoms are close to the radiative lifetimes, affording an almost quantitative lanthanide centered luminescence quantum yield (eqn (3)), as it is for example observed for some Eu doped GdF3 NPs25 for which a luminescence lifetime of 10.5 ms is observed when the radiative lifetime of Eu is 9.7 ms for the aqua ion and up to 11 ms in other media.26
Considering the position of the Ln atom in the NP (outer, intermediate or inner layers), it is in some instances possible to differentiate the different environment of the Ln atoms. In the case of Eu doped LaF3 NPs,27 it was possible to extract up to three different lifetimes for the Eu emission with values of 7.04 (48%), 1.85 (38%) and 0.4 (14%) ms. These lifetimes, in agreement with the respective populations indicated in brackets, were respectively attributed to core Eu atoms, Eu atoms close to the surface and Eu atoms at the surface of the NPs, which are severely quenched by water molecules and citrate capping ligands.
The decreased luminescence properties of surface atoms might be an important issue, especially if the doping in luminescent Ln atoms becomes high. In that case, energy migration can occur within the inner Ln atoms, which might be transferred up to the surface atoms and is then partially quenched. Once again, this is particularly true for NIR Ln emitters, providing the NPs with poor luminescence efficiency. In the case of NP with upconversion (UC) properties (see below) or downconversion and downshifting in the NIR domain,28 in which NIR photons have to be accumulated, such a surface quenching effect is particularly deleterious. To overcome this problem, core/shell structures have been designed to protect the Ln emitters by an outer shell, thereby displacing the active Ln atoms from the surface of the NP. For example, NPs based on NaYF4 doped with a mixture of Nd3+, Yb3+ and Er3+ have seen their UC efficiency increased by a factor 50 when a supplementary shell of NaYF4 is added at the surface of the NPs.29 The core/shell approach can even be extended to multiple shells in order to localize finally the Ln absorbers or sensitizers and the energy acceptors with a controlled directionality of the energy transfer processes.30,31
It has to be kept in mind that, if the increase of the doping ratio of active luminescent Ln species is important to increase the NPs luminescence, increasing too much this ratio can have deleterious effects and can lead to the phenomenon of concentration quenching for which larger doping ratios lead to decreased luminescence efficiencies.32,33
The position of the emitting Ln cations into the NPs is also of large importance for energy transfer applications with organic dyes at the surface, both for ligand to Ln energy transfer (dye-sensitization) or for Ln to dye at the surface, such as for Förster resonance energy transfer (FRET) applications. In the case of dye-sensitization, although the exact mechanisms are prone to case by case debates between the Dexter and Förster mechanisms,34,35 a close contact between the sensitizing ligand and the Ln atoms at the first layers of the surface is mandatory for an efficient sensitization of the Ln atoms. In the second case of energy transfer from the Ln atoms to dyes at the vicinity of the surface, the mechanism generally assumed is that of a dipole–dipole Förster type energy transfer. Within the frame of the theoretical treatment of energy transfer with the Förster's formalism,36 the energy transfer efficiency is dependent on the donor–acceptor distance r to the inversed sixth power, thereby decreasing rapidly when the donor–acceptor distance increases. Thus Ln atoms close to the surface of the Ln NP are more efficient than those in the core of the NPs.37 Ultimately, the distance between the dye anchored at the surface of Ln-NPs can be chemically or biochemically modulated to improve the ET, for example by playing with the size of biomolecules such as full antibodies, or some of their fragments of smaller size.38
As a last point, NPs have a large advantage over Ln complexes in the possibility of mixing different Ln atoms, providing the NPs with multiplexing capabilities. One might argue that the synthesis of controlled heteropolynuclear complexes is also feasible,39,40 but generally at the expense of important synthetic efforts (see for example ref. 41 and 42). For Ln-NPs, a simple mixing of the adequate concentrations at the first step of the synthesis allows to obtain a large panel of NPs with spectroscopically unique signatures with applications in barcoding,43 multiplexing, i.e. multiple analysis with a same sample44 or multimodal analytical devices.45 On the other hand, the larger size and composition of NPs compared to Ln complexes can bring significant issues regarding toxicity,46,47 possible unwanted size dependent pharmacokinetic and bio-distribution properties,48 or simple stability troubles such as leaching in biological media.49,50
With regards to these different considerations on Ln luminescence, the next chapters aim at reviewing the main works reporting on the three principal strategies developed so far for improving Ln luminescence with the help of nanoscopic scaffolds.
3. Ln ion doped NPs with capping photosensitizers
In general, the efficiency of Ln doped NPs luminescence depends on the structure, local site symmetry, and phonon energy of the host materials.51 Low phonon energies of host materials are favorable for Ln doping to achieve intense luminescence, as it allows for low multiphonon relaxation rates and minimal nonradiative energy losses. Fluorides, owing to the low phonon energy (≈350 cm−1) and high chemical stability, are considered to be the most efficient for many applications.52 Crystal structure also significantly affects the Ln luminescence, as low-symmetry is more desirable than high-symmetry due to the higher 4f–4f transition probabilities. For example, Krämer et al. reported that the NaYF4:Yb3+/Er3+ UC efficiency of green emission is approximately 10 times stronger in hexagonal phase compared to cubic phase,53 when Quintanilla et al. revealed that the photoluminescence quantum yield of α-phase NaGdF4 Er3+,Yb3+ surpasses that of β-NaGdF4 for sizes below 20 nm, which can be related to distortion of the crystal lattice when the UC NPs become smaller.54Consequently, with typical bare NPs, it is essential to carefully select the host material with appropriate parameters of interest for achieving optimal Ln luminescence in a given application.
In the last decade, new challenges have emerged for the engineering and synthesis of ultrabright lanthanide-based NPs (Ln-NPs). One creative approach involves the coordination of fluorescent organic ligands (antennas) at the NP surface to sensitize the incorporated Ln3+, allowing light harvesting over broader wavelength ranges with greatly enhanced NPs absorption cross section. These ligands also contribute in the shielding of Ln emission from high-energy oscillators in the chemical matrix. Thus, a very efficient sensitization of Ln-NPs can be obtained through surface coverage with an adequate capping ligand and the surface-modified NPs display huge improvements of their photophysical properties and their brightness in particular.
In this case, an appropriate selection of the capping antenna becomes very important for the spectroscopic performance of the Ln-NPs as the main pathway of reaching Ln related excited state is through the energy transfer from the antenna and not from its direct excitation anymore, subsequently the nature of the host material seems to have less impact on the final luminescence performance of dye-sensitized Ln-NPs. This is illustrated by the diversity of suitable matrices doped with Ln3+ that are reported to be effective for dye-sensitization, such as oxides, fluorides, phosphates, vanadates, silicates, and hydroxides, as presented below.
This organic/inorganic dual nanomaterial received increasing attention thanks to the possibility of combining the low cost and versatility of organic molecules with the chemical and physical properties of the inorganic materials, giving a great utility for many applications, such as luminescent sensors, optical fibers, lasers, amplifiers, solar cells, electroluminescent devices, and various time-correlated luminescence applications. In this section, we will review the different spectral conversion mechanisms illustrated in Fig. 1 and investigated in dye-sensitized Ln-NPs: (1) downshifting, where one high-energy photon is transformed into one lower energy photon; (2) upconversion, where two or more low energy photons are converted into one high-energy photon; and (3) quantum cutting, also called downconversion, in which one high-energy photon is converted into several lower energy photons. A comparison of the photoluminescence properties of the various NPs (when studied) is summarized at the end of this section in Table 1, and the various dye-sensitizing ligands reported in the literature for downshifting and for both upconversion and downconversion are illustrated in Fig. 2 and Fig. 6 respectively.
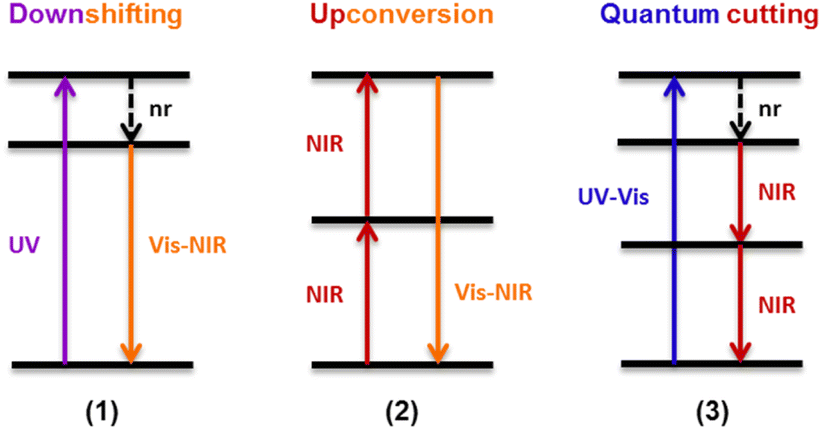 | ||
| Fig. 1 Schematic representation of different spectral conversion mechanisms possible with Ln3+-doped NPs. nr = non-radiative. | ||
 | ||
| Fig. 2 Dye-sensitizing ligands reported in the literature for surface sensitization of downshifting NPs. | ||
| Matrix | Ln | Antenna | Diameter (nm) | λ excitation (nm) | Lifetime of emitting Ln (μs) | Enhancement factora/Bb (L mol−1 cm−1) | Quantum yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Enhancement factor is the ratio of the dye-sensitized NPs luminescence to that of bare NPs. b B = Brightness. c Hydrodynamic diameter measured by DLS. | ||||||||
| LaF3 | Eu | 1 | 232 ± 65c | 305 | 125 | 135 | 1.85 ± 0.28 | 55 |
| LaF3 | Tb | 2 | 25–40 | 282 | 44 | 58 | ||
| LaF3 | Eu | 5 | 3–4 | 278 | 100 | 61 | ||
| LaF3 | Eu | 6 | 3.5 | 275 | 510 31% 1990 69% | 62 | ||
| LaF3 | Tb | 6 | 3 | 319 | 2296 55% 8305 45% | 63 | ||
| LaF3 | Tb | 7 | 3.32 | 312 | 641 37% 2604 63% | 64 | ||
| 8 | 320 | 1713 34% 3453 66% | ||||||
| LaF3 | Tb | 9 | 5.2 | 265 | 100 | 65 | ||
| LaF3 | Eu | 10 | 25 | 337 | 100 | 70 | 66 | |
| 12 | ||||||||
| LaF3 | Tb | 14 | 20–25 | 307 | 1580 48% 3760 52% | B = 2.1 × 106 | 13 | 16 |
| 15 | 337 | 1110 47% 2590 53% | B = 2.2 × 106 | 29 | ||||
| LaF3 | Eu | 16 | 10 | 350 | 180 | 61.6 | 67 and 68 | |
| LaF3 | Eu | 2 | 25–40 | 286 | 88 | 69 and 70 | ||
| 16 | 378 | |||||||
| LaF3 | Eu | 17 | 16 | 377 | 20 | 71 | ||
| 18 | ||||||||
| LaF3 | Eu | 18 | 4.4 ± 1.2 | 457 | 80 53% 320 31% 1650 16% | 13.25 | 73 | |
| LaF3 | Tb | 20 | 20–25 | 337 | 660 18% 2070 82% | 38 ± 1 | 75 | |
| 21 | ||||||||
| LaF3 | Tb | 22 | 20–25 | 335 | 726 | 8–15 | 76 | |
| CaF2 | Tb | 11 | 11 ± 4 | 311 | 2740 | 80 | ||
| NaYF4 | Tb | 24 | 60–110 | 320 | 2350 — 570— | 330 | 19 | 81 |
| Eu | 1860 — 7200— | 14 | ||||||
| LiYF4 | Eu | 25 | 90 × 40 | 365 | 2057 53% 9135 47% | 31 | 82 | |
| NaGdF4 | Eu | 26 | 317 | 10000 |
3.3 ± 0.6 | 83,84 | ||
| NaGdF4 | Eu | 2–18 | 33.9 | 277 | 27.6 | 85 | ||
| Y2O3 | Eu | 27 | 6.4 ± 1.5 | 270 | 19 | 86 | ||
| Y2O3 | Tb | 28 | 100 | 290 | 1250 | 100 | 87 | |
| YPO4 | Eu | 16 | 23.2 ± 8.8 | 350 | 4700 | 26 ± 2 | 91 | |
| YVO4 | Eu | 16 | 10–15 | 369 | 14 | 92 | ||
| LaPO4 | Eu | 16 | 7 | 350 | 850 | 33 | 22 | 94 |
| SiO2 | Eu | 29 | 207 ± 13 | 343 | 83 7% 283 31% 1085 62% | 190 | 49 | 97 |
| NaYF4 | Nd | 30 | 5.3 ± 0.6 | 340 | 4.1 20% 68 80% | 101 | ||
| Yb | 6.0 ± 0.6 | 1.1 15% 3.7 63% 12.6 22% (in DMSO) | ||||||
| NaYF4 | Yb | 31 | 5.2–6.3 | 460 | 3 6% 16.5 24% 11170% | 300 | 102 | |
| CaF2 | Yb/Nd | 33 | 4.4 ± 0.1 | 467 | 17 | 2100 | 103 | |
| NaYF4 | Yb/Er | 34 | 16 | 806 | 3300 | 0.12 | 109 | |
| NaGdF4 | Yb/Er | 34 | 12 | 806 | 33000 |
5.3 | 110 | |
| NaYF4 | Yb/Er | 35 | 20 | 783 | 80 | 111 | ||
| 36 | 820 | 70 | ||||||
| 37 | 808 | 200 | ||||||
| 38 | 845 | 100 | ||||||
| NaYF4:Yb/Er@NaYF4:Yb | Yb/Er | 34 | 35 | 806 | 1000 | 112 | ||
| NaLuF4:Yb/Er@NaLuF4:Yb,Pr | Yb/Er | 36 | 18 | 820 | 800 | 113 | ||
| NaYbF4:Tm@NaYF4:Nd | Yb/Tm | 37 | 54 | 808 | 4.8 | 114 | ||
| NaYF4:Yb/Er@NaYF4:Nd/Yb | Yb/Er | 41 | 33 | 800 | 9.2 | 116 | ||
| NaYF4:Yb/X@NaYbF4@NaYF4:Nd (X = Er, Ho, Tm or Pr) | Yb/X | 41 | 52 | 800 | 13 | 117 | ||
| NaGdF4:Yb/Er@NaGdF4:Yb@NaNdF4:Yb | Yb/Er | 37 | 56.4 | 808 | 7 | 119 | ||
| NaYF4:Yb,Er,Nd | Yb/Er | 34 | 50 | 806 | 120 | |||
| NaYF4:Yb,Er@NaYF4:Yb,Nd | Yb/Er | 35 | 16 | 783 | 34 | 1 | 121 | |
| NaYF4 | Tb/Yb | 6 | 2.71 | 272 | 1370 68% 3192 32% | 123 | ||
| NaYF4 | Pr/Yb | 42 | 22 × 55 | 397 | 21.7 | 30 | 124 | |
| NaYF4 | Tb/Yb | 43 | 30 | 405 | 2260 | 125 | ||
| NaGdF4:Nd/Yb@NaGdF4:Nd | Nd/Yb | 26 | 10 | 355 | 626 ± 48 | 126 | ||
3.1. Downshifting NPs
Most of the studies published in the field of Ln-NPs sensitized by organic antennas have been focused on the downshifting mechanism, by shifting UV-blue light to more advantageous and better exploited visible and NIR emission.Charbonnière et al. were first to report such type of spectral conversion.55 In a pioneering work of 2008, they described sensitization of 5%Eu-doped LaF3·AEP (AEP for aminoethyl-phosphate) NPs by partial AEP exchange with 6-carboxy-5′-methyl-2,2′-bipyridine (1) in water. The NPs excitation spectrum reflected the characteristic absorption bands of the ligand with a maximum at 305 nm, and the ligand to Eu3+ energy transfer produced a 135-fold enhanced emission. This general strategy was then expanded for different combinations of chromophores, Ln, and inorganic host material.
Later, Kokuoz et al. architecturally developed a new type of “core–shell nanostructure” where specific emitting Ln are constrained by an undoped layer, forming a shell protecting those in the core.56 They demonstrated that benzoic acid (2) and phthalic acid (3) could efficiently sensitize Eu3+ and Tb3+ doped LaF3 core–shell NPs, showing that an extra undoped LaF3 shell did not inhibit the sensitization of core Eu3+, proving that energy-transfer can penetrate to sites which are not directly ligand-bound, referring to a dipole–dipole mechanism.
2 was then frequently investigated as Chen et al. confirmed its ability to sensitize Eu-doped LaF3 NPs,57 while Yang et al. proved that it can also sensitize Tb-doped LaF3 NPs.58 Taking advantage of these previous works, Wang et al. used 2 in a Eu3+/Tb3+ co-doped LaF3 system in order to sensitize both Ln3+, realizing multicolor Ln-NPs by a single wavelength excitation with tunable emission spectra through controlling Eu3+/Tb3+ molar ratios.59
Then, Kokuoz et al. covered Eu-doped LaF3 NPs with 3,4-formylphenylbenzoic acid (4), and demonstrated that through a balance between ligand and lanthanide emission, these NPs exhibit color tunability from red to greenish blue as a function of selected excitation wavelengths ranging from 250 to 400 nm.60
In a similar system, Cross et al. reported a strong sensitization of 5% Eu-doped LaF3·citrate NPs upon citrate exchange by dipicolinate ligands (5), increasing the emission intensity by a factor of 100.61
In 2014, Li et al. demonstrated that 1,2,4,5-benzenetetracarboxylic acid (6), due to its high symmetry and various coordination with Ln3+, can efficiently bind to the surface of Eu-doped LaF3 NPs and strongly sensitize and protect Eu3+ luminescence,62 while Li et al. highlighted its ability to sensitize Tb as well, in Tb-doped LaF3.63 In a later study, they also introduced salicylate (7) and 5-sulfosalicylate (8) as efficient sensitizers of Tb-doped LaF3.64 Different benzoic acid derivatives were then exploited as Ghosh and Luwang described the sensitization of Tb3+ doped LaF3 NPs by p-amino-benzoic acid (9), reporting at least 100-fold enhancement in luminescence intensity.65 They also applied their model for the quantification of nitro explosive compounds in aqueous medium whereas Khudoleeva et al. aimed for bioimaging applications by performing surface modification of Eu-doped LaF3 with terephthalate (10) or 2,6-naphtalenedicarboxylate (12), increasing luminescence intensity by two orders of magnitude.66
Then, in a wide study, Goetz et al. reported the sensitization of 10% Tb-doped LaF3 using eleven different ligands derived from dipicolinic acid 5 and 2-hydroxyisophthalic acid (13) with varying coordination and photosensitizing abilities.16 The two most effective photosensitizing ligands were 5-cyano,2-hydroxyisophthalic acid (14) and the bis(2-hydroxybenzoic acid) (15) as they provided very promising brightness values up to 2.2 × 106 L mol−1 cm−1 which exceed those of QDs and semiconducting nanopolymers. The NPs were used for highly sensitive time-resolved luminescence applications, like imaging in HeLa cells by fluorescence microscopy.
Beyond carboxylic acid ligands, Janssens et al. introduced β-diketonates as efficient sensitizers for Eu3+, as they showed the possibility to sensitize 5%Eu-doped LaF3·OA (OA = oleic acid) NPs by partial surface coverage with thenoyltrifluoroacetone (16) and the resultant luminescence intensity was enhanced 180 times.67 They also succeeded to press these modified NPs at high pressure, creating transparent solid discs of high density.6816 was shown to be as efficient as 2 at photosensitizing Eu-doped LaF3 NPs, as Wang et al. observed a 40-fold luminescence intensity enhancement for both ligands.69 In a recent work, this team reported the coordination of Eu-doped LaF3 with both ligands simultaneously. This mixed hybrid possesses a broadband excitation spectrum (200∼400 nm) which perfectly covers the entire ultraviolet (UV) spectral range and can be extremely beneficial to the enhancement of the conversion efficiency of silicon solar cells.70
Advanced studies aimed to find different sensitizers for Eu3+ in such type of NPs. Safronikhin et al. proved that dibenzoylmethane (17) and 1,10-phenanthroline (18) are able to sensitize EuF3 NPs,71 while Irfanullah et al. showed the ability of 18 and 9-oxidophenalenone (19) to sensitize water dispersed 5%Eu-doped LaF3 NPs.72,73 The latest ligand ensured great protection of Eu3+ from non-radiative deactivation through high-energy vibrations of 19, as proved by the relatively long lifetime of Eu3+ emission up to 0.41 ms. In a recent study, Adusumalli et al. tested LaF3:Eu3+ and SrF2:Eu3+ NPs photosensitized by different ligands (5, 6, 10 and 13) for their haemocompatibility.74 Therefore, flow cytometry was used to analyse the possible NPs binding to the red blood cell membrane. They showed that these nanomaterials are non-cytotoxic compounds in vitro and can be further investigated for biomedical in vivo applications. Many studies claimed that these Ln-NPs are particularly appealing for bioanalytical applications, but no proof of concept was provided until Charpentier et al. recently developed novel ligands which can be simultaneously used for both sensitization of Ln-NPs and bioconjugation.75 They designed two linked hydroxy-isophthalic acid based units, where the linker was modified to introduce an activated function that react either with amine (20) or thiol (21) functions, making them linkable to many biomolecules of interest. These ligands showed important sensitization of 10%Tb-doped LaF3 leading to exceptional NP brightness up to 1.8 × 106 L mol−1 cm−1, while their bioapplicability was demonstrated in two prototypical approaches for bioimaging and biosensing. In further studies, Cheignon et al. used the same Ln-NPs for a surface coordination by p-sulfonato-sulfoxocalix[4]arene (22), which showed strong photosensitization of Tb emission upon ligand excitation.76 Such design allows to exploit the calixarene cavity to a potential inclusion of aromatic compounds. An interaction with Rhodamine 6G was proved by FRET experiments that revealed a strong association to the surface of Tb-NPs, as shown in Fig. 3.
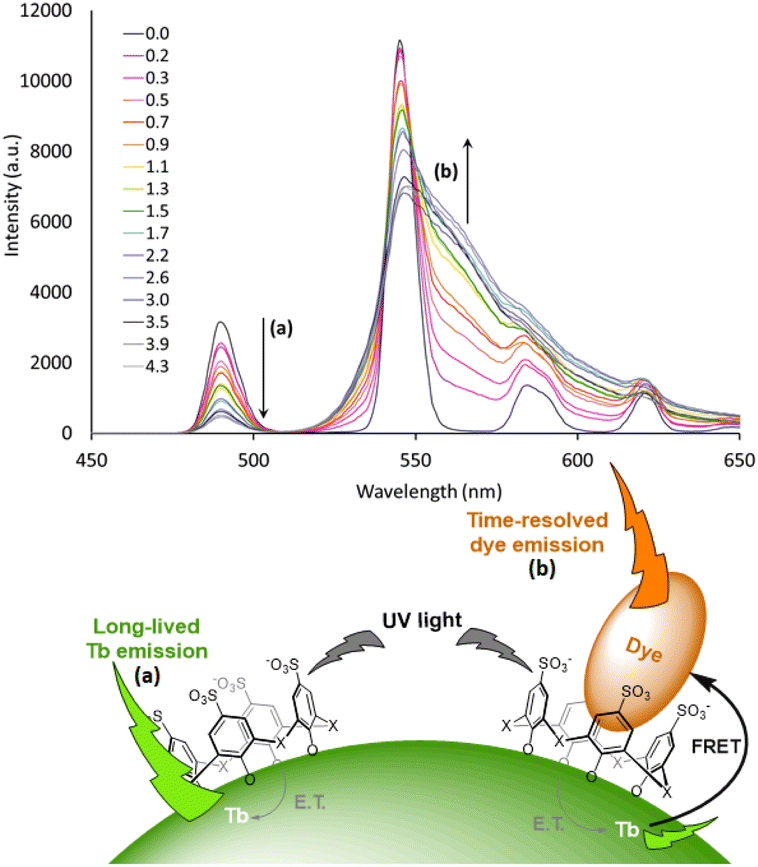 | ||
| Fig. 3 Time-resolved emission spectra of Tb NP capped with 22 upon addition of different equivalents of rhodamine 6G. The intensity decrease of the Tb emission and increase of rhodamine emission are marked with arrows a and b respectively. Inset: representation of the proposed supramolecular assembly. NP photosensitization by surface capping ligands and host–guest interaction with the aromatic charged dye. Adapted with permission from ref. 76. Copyright 2021 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Besides LaF3, alkali metal fluoride matrices have also been investigated as suitable Ln3+ host material. Wang et al. reported the sensitization properties of 2 in CaF2 doped with either Eu3+ or Tb3+, showing a drastic increase in emission intensity compared with corresponding Eu and Tb molecular complexes.77 However, those NPs could not be dispersed properly in water or organic solvents, which limited their application. Then, Song et al. synthesized Tb-doped CaF2·OA which showed good dispersibility in chloroform and toluene.78 Fluorescence measurements showed that even the surface coating OA, when excited, could slightly sensitize Tb3+,while in a later report, they performed poly(N-vinyl-2-pyrrolidone) (23) coverage of their NPs doped with Eu, Tb, or Tb/Ce, using each pyrrolidone moiety of the polymer as an individual antenna coordinated to the NP surface.7923 exhibited efficient sensitization of Tb or Eu-doped NPs, while for Tb/Ce co-doped system, simultaneous energy transfer from Ce3+ and the ligand was observed, evidencing a synergistic enhancing effect between a co-dopant and an organic antenna for the first time.
In 2019, Adusumalli et al. demonstrated that 4-mercaptobenzoic acid (11) is an efficient sensitizer for the Tb-doped CaF2 NPs.80 The prepared capped NPs were used to develop an approach for selective detection of nitroaromatic pollutants in water.
Other fluoride matrices were investigated as host material for dye-sensitized Ln-NPs. Eu or Tb-doped NaYF4 nanocrystals have been prepared by Gauthier et al. and four N-heterocyclic organic ligands were tested to promote sensitization of Eu3+ or Tb3+ luminescence.81 The terpyridine derivative 24 showed the best performance for both Ln, presenting up to a 330-fold enhancement in emission intensity. Then, Samanta et al. proposed Eu-doped LiYF4 NPs coating by 4,4,4-trifluoro-1-phenyl-1,3-butanedione (25) which provided efficient sensitization.82 They also proved the applicability of this model in Si solar cell efficiency enhancement.
On another hand, Agbo et al. detailed the photophysics of Eu-doped NaGdF4 NPs and studied their sensitization by a hydroxypyridone derivative (26), responsible for a 104-fold increase in luminescence intensity.83 They also applied their model as a tool of choice to overcome the constraints of UV solar spectrum/semiconductor bandgap mismatch.84 In a similar system, Song et al. used 2 and 1,10-phenanthroline (18) for surface modification in order to enhance the luminescence performance of Ln-doped NaGdF4 (Ln = Tb, Eu, Dy) NPs.85 Interestingly, the overlap in excitation bands for both Gd3+ ions and ligands ensured simultaneous energy transfer of Gd3+ → Ln3+ and ligands → Ln3+ under a single wavelength excitation.
Away from fluoride-based matrices, Y2O3 is one of the most studied metal–oxide matrices for Ln-NPs. First study of Eu-doped Y2O3 sensitization by acetylacetonate (27) was published by Dai et al. showing that the excitation of ligands leads to strongly enhanced white light emission arising from efficient intramolecular energy transfer to Eu3+ as well as Y2O3 oxygen vacancies.86 Those optical properties allow for applications in UV LED pumped solid-state lighting. Then, Stagi et al. proved the sensitization effectiveness of Tb-doped Y2O3 by 2,4,6-triamino-s-trazine (28) presenting a 102-fold luminescence enhancement.87
The photosensitizing ability of the well-known β-diketonate (16) has been investigated with various types of matrices. Ji et al. and Chen et al. used it to sensitize Eu-doped Y2O3 NPs, showing a greatly enhanced luminescence intensity,88,89 while Balderas et al. employed the sensitized NPs to produce transparent poly(methyl methacrylate) (PMMA) luminescent films with an extended and tunable excitation wavelength range from 200 to 550 nm.90
Chen et al. showed that 16 could also sensitize Eu-doped YPO4·(OA) NPs with a resulting ∼4700-fold brighter emission,91 while only a very low sensitization occurred for Eu-doped YVO4 NPs as demonstrated by Tang et al.92
Moreover, the sensitization of Eu-doped LaOF NPs by 16, prepared by He et al. by annealing Eu-doped LaF3 NPs, resulted in higher luminescence enhancement compared with sensitized Eu-doped LaF3 NPs.93 Thus, Vats et al. succeeded to increase the luminescence emission of Eu-doped LaPO4 NPs through 16 sensitization.94 Interestingly, photoluminescence and lifetime measurements clearly showed that sensitization takes place only at the surface of LaPO4 NPs, while the core Eu3+ remains unsensitized.
Embedding Ln3+ into a host material such as alkali halides, semiconductors, and metal oxides has been widely investigated. Gonçalves et al. introduced luminescent Eu-doped SnO2 NPs where Ln3+ were found to be essentially incorporated into the cassiterite structure, substituting Sn4+, while for high concentration they are also located at the particles surface.95 Capping with another β-diketonate 25 presented high light output under UV excitation in water, due to an efficient sensitization and protection of Eu3+ emission. In 2015, Eu-doped ZnO nanowall structures have been achieved by an electrochemical deposition method by Kang et al. where Eu3+ were uniformly distributed in the ZnO–Zn(OH)2 core/shell structure.96 Surface modification by 1,10-phenanthroline (18) generated an additional sharp Eu3+ emission while the energy transfer from ZnO to Eu3+ appears to be extremely weak without ligand capping. The results led to propose a unique cascade energy transfer model between ZnO, 18 and Eu3+.
Lately, Artizzu et al. suggested a concept model system based on purely silica-based core/shell NPs where Eu3+ ions are confined into a thin silica layer and are efficiently photosensitized through 2,2′-biquinoline-4,4′-carboxylate (29) covalently grafted on the surface of the outer shell.97 A remarkable intensity enhancement of Eu-based NPs luminescence by 190-fold was reported, showing that silica matrices are suitable and a highly performing host alternative to commonly investigated nanocrystals for the development of Ln-based luminescent materials.
The first report on the use of organic ligands to sensitize NIR luminescence of Ln-NPs was published by Zhang et al. in 2007. In this pioneering work, a strategy was established to both protect and sensitize the NIR luminescence of 20%Nd or Yb-doped NaYF4 NPs by direct coordination of tropolonate (30) ligands to the NPs surface, as proven by longer luminescence lifetimes than for the corresponding molecular complex [Ln(Trop)4]−.101 In a similar NaYF4 system, Lu et al. reported the sensitization of Yb-doped NaYF4 NPs through the excitation of capping 2-hydroxy-perfluoro-anthraquinone (31). The overall Yb3+ NIR emission intensity is increased by a factor of 300.102
In 2016, Utochnikova et al. developed a photoluminescence study on sensitization of YbF3 and EuF3, with 2,6-naphthalenedicarboxylate (12) or 9-anthracenate (32). Both ligands enhanced EuF3 luminescence intensity up to 100 times, while in the case of the NIR emitting YbF3 system, successful sensitization was achieved only with 32.104 In a later study, they reported the luminescence properties of rarely studied Dy-based NPs, DyF3 and Dy-doped LaF3, both sensitized by terephthalic acid (10) with enhanced Dy3+ NIR emission observed upon surface modification.105
Recently, Liu et al. presented a novel type of dye-sensitized CaF2:Yb3+/CaF2:Nd3+ core/shell NP, with fluorescein isothiocyanate FITC ligands (33) capping the surface of NPs and acting as efficient visible light harvester (Fig. 4a).103 The strong spectral overlap between 33 emission and Nd3+ absorption (Fig. 4b) as well as between Nd3+ emission and Yb3+ absorption (Fig. 4c), makes Nd3+ a suitable “energy bridge” to realize effective multistep sequential dye → Nd3+ → Yb3+ energy transfer. This ultraefficient cascade mechanism resulted in a remarkable enhancement of about 2100 times of the Yb3+ luminescence intensity, which is the highest figure of merit reported in literature so far for NIR-emitting analogous systems.
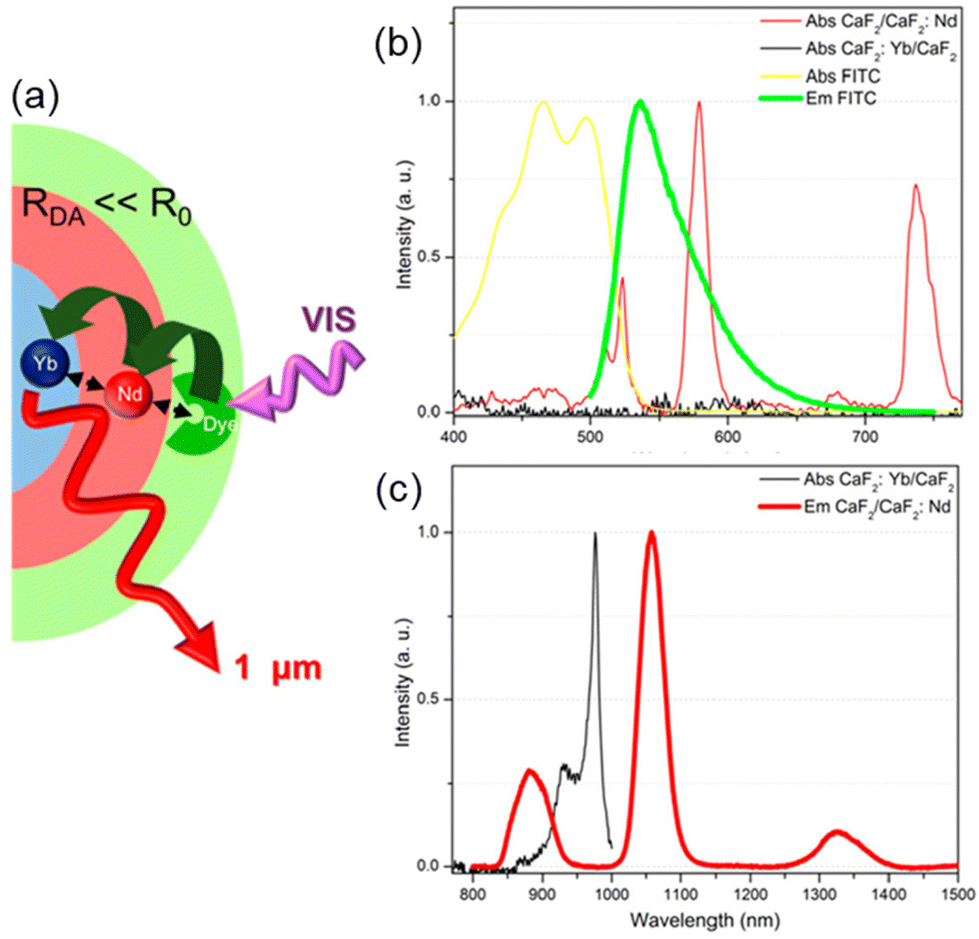 | ||
| Fig. 4 (a) Architectural design of CaF2:Yb3+/CaF2:Nd3+ core/shell NP dye-sensitized by 33 and (b–c) spectral overlaps between donor–acceptor emission–absorption bands for multistep sequential energy transfer. Adapted with permission from ref. 103. Copyright 2019, American Chemical Society. | ||
3.2. Dye-sensitized upconversion NPs
In the last decade, lanthanide-doped upconversion NPs (UCNPs) have attracted worldwide attention due to their capability of generating shorter-wavelength photons under infrared light excitation, with promising applications in biomedical imaging, photodynamic therapy, solar cells, and display technologies.106Generally, upconversion NPs consist of a couple of lanthanide dopants (typically Yb3+/Ho3+, Yb3+/Er3+, or Yb3+/Tm3+) in an organic host material. Yb3+ (or alternatively Nd3+) ions are usually doped to function as sensitizer ions, absorbing the 980 nm (or 808 nm for Nd) laser irradiation and then successively transfer their excitation energy to nearby co-doping activators (Ho3+, Er3+, Tm3+…), finally leading to the emission of upconverted light (UCL).108 However, the weak and narrow absorption bands of lanthanide ions pose a fundamental limit of UCNPs to withhold their brightness, creating a long-standing hurdle for the field. Dye-sensitization emerged once again to address this performance-limiting problem.
In 2012, Zou et al. reported the first attempt to enhance the brightness of NaYF4:Yb3+/Er3+ UCNPs through organic carboxylated cyanine dye IR-806 (34) sensitization.109 FRET occurs from the excited 34 to the Yb3+ absorption centers on the surface of NPs, which further sensitized Er3+ to produce UCL. The overall upconversion is dramatically enhanced by a factor of 3300. In a similar system, Garfield et al. tested the same typical Yb3+/Er3+ couple and 34 in a NaGdF4 inorganic host lattice, and reported a 33000 times increase in brightness and a 100-fold increase in efficiency over uncapped UCNPs.110 Later, this approach was extended and applied to a set of NaYF4:Yb3+/X3+ (X = Er, Tm or Ho) NPs and many NIR dyes with distinctive absorption ranges for more efficient absorption and a tunable excitation band in a wide spectral range. In their study, Wu et al. used a series of NIR dyes and showed their ability to sensitize NaYF4:20%Yb3+, 2%Er3+ UCNPs.111 These included commercially available dyes IR-783 (35) and IR-820 (36) and their respective carboxylate derivatives IR-808 (37) and IR-845 (38) which were synthesized.
Thanks to the modular nature of NIR dyes, Lee et al. suggested a simultaneous use of multiple types of dyes on the surface which can dramatically widen the photon absorption window of NaYF4:20%Yb3+,0.5%Tm3+ UCNPs, to the entire visible and NIR range.107 Three types of dye sensitizers, 34, BODIPY-FL (39) and Cy3.5 (40), were chosen to perform sensitization. The sufficient spectral overlap between the emission spectrum of one sensitizer and the absorption spectrum of another enabled a cascade FRET sensitization of the UCNPs, while empowering collective absorption spectra of the three ligands as shown in Fig. 5. In 2016, Wu et al. introduced core/shell nanostructured NaYF4:Yb3+/Er3+@NaYF4:Yb3+ UCNPs where Yb3+ sensitizer was directly doped in the shell.112 Through a sensitization by 34, and Ln3+ extra protection provided by the core/shell design, they succeeded to amplify upconversion efficiency, and aimed for their application in controlling neuronal activity and bioimaging.
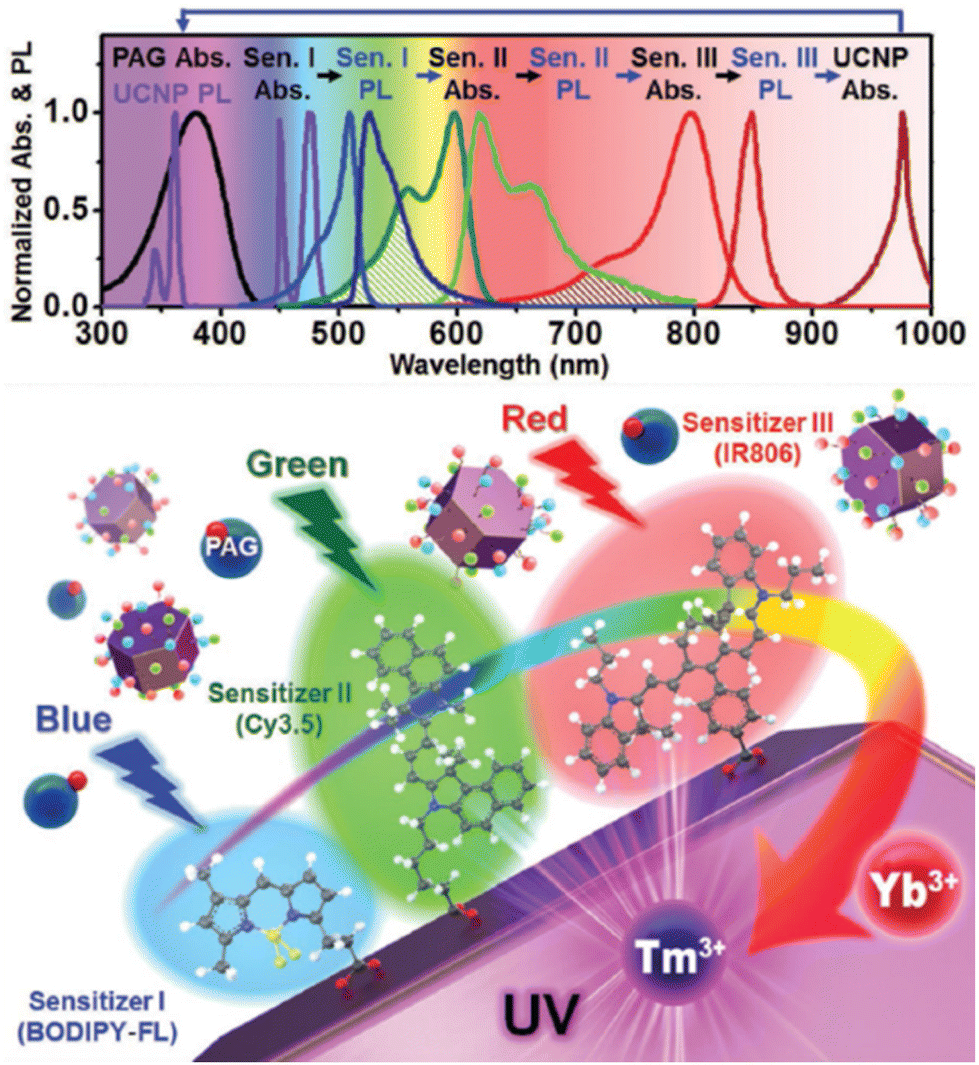 | ||
| Fig. 5 Design of multi-dye-sensitized UCNPs for wide-range photo-absorption and upconversion. Reprinted with permission from ref. 107. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Similar results were also observed by Yin et al. using 36 sensitized NaLuF4:Yb3+/Er3+@NaLuF4:Yb3+,Pr3+ core/shell UCNPs, with a 800 times UCL enhancement.113
Despite recent progress, several drawbacks persist as the limitation of spectral overlap between the emission of NIR dyes and the absorption of the typically used sensitizer Yb3+ ions, restraining energy transfer efficiency from the dye to the UCNPs. Then, Chen et al. proposed further advances involving the concept of multistep cascade energy transfer in which 37 is used to sensitize Nd3+ doped in the shell layer, whereas Yb3+ and Tm3+ are a second sensitizer and activator respectively, in the core of an epitaxially designed core/shell (NaYbF4:Tm3+0.5%) @NaYF4:Nd3+ UCNPs.114 They demonstrated that a multistep cascade energy transfer dye → Nd3+ → Yb3+ is about 1.5 times more efficient than the direct energy transfer from 37 to Yb3+ yielding an upconversion efficiency nearly ∼100 times higher than typically reported for Ln-UCNPs.
Later, Wei et al. showed that optimal doping concentration of Nd3+ in such UCNPs system could be increased from 2 to 20 mol% resulting in additional 10 times higher upconversion brightness.115
In additional studies, Chen et al. proved that the achieved UCL intensities from the sensitization of NaYF4:Yb3+/Er3+@NaYF4:Nd3+/Yb3+ core/shell NPs by indocyanine green organic dye (41) were 2–3 times higher than that from sensitized NPs incorporating only Yb3+ or Nd3+ in the shell layer.116 This was attributed to the strong overlap of the emission spectrum of this ligand with the absorption peaks of Nd3+ and Yb3+ exhibiting a synergistic effect from the multidimensional energy transfer involving multiple pathways: 41 → shell Yb3+ → core Yb3+; 41 → shell Nd3+ → coreYb3+; 41 → shell Nd3+ → shell Yb3+ → core Yb3+. Shao et al. reported a higher luminescence enhancement (×28) with NaYF4:Yb3+/Er3+@NaYF4:Yb3+/Nd3+ core/shell NPs sensitized by 34117 and then extended the core/shell concept by presenting NaYF4:Yb3+/X3+@NaYbF4@NaYF4:Nd3+ core/shell/shell nano-structure (X = Er, Ho, Tm or Pr) sensitized by 41.118 Thanks to a cascade energy transfer pathway: 41 → Nd3+ (outer shell) → Yb3+(inner shell) → Yb3+/X3+(core), the brightness was 4-times increased with a broad excitable spectral range which facilitates their use in bioapplications.
Finally, by combining cascade and multidimensional energy transfer pathways, Xu et al. demonstrated that the UCL intensity of NaGdF4:Yb3+/Er3+@NaGdF4:Yb3+@NaNdF4:Yb3+ NPs could be enhanced over 7-fold by sensitization with 37.119 The advantage of this core/shell/shell design with Nd3+ and Yb3+ co-doped in the outer shell is that nearly 100% Nd3+ allow efficient extraction of the excited-dye energy, while the Yb-containing shell layer minimizes possible back energy transfer from the core to the surrounding environment, thus maximizing the UCL efficiency.
Such dye-sensitized UCNPs have attracted worldwide attention due to their promising applications. Among recent works, Wei et al. found that the temperature increases significantly through energy conversion of 34 dye sensitized NaYF4:Yb3+,Er3+,Nd3+ UCNPs. When applied to a tumor, NPs are found successfully distributed to tumor cells, which can be killed efficiently based on the photothermal effect of the NPs.120 Therefore, they are promising to be used for effective thermal therapy of tumors with real-time temperature monitoring.
Dye-sensitized UCNPs found interest in biophotonic applications like biological imaging, multimodal imaging and photodynamic therapy. In addition, the use of these UCNPs in solar cells have achieved rapid progresses. The highest power conversion efficiency was reported by Bi et al. at 21.1% using 35 dye sensitized core/shell NaYF4:Yb3+,Er3+@NaYF4:Yb3+, Nd3+ UCNPs and coupled with plasmonic Au nanorods film.121 This work indicates that insulating dye-sensitized UCNPs are of great significance for the future of solar cells devices.
3.3. Dye-sensitized quantum cutting NPs
Another conversion mechanism known as quantum cutting or downconversion (DC, not to be confused with the conventional downshifting), allows to transform the energy of one absorbed photon into two or more emitted low-energy photons, with quantum efficiency potentially larger than 100% (Fig. 1). This phenomenon has been demonstrated in various Ln3+–Yb3+ co-doped couples (Ln = Tb, Tm, Pr, Ho or Dy), where Yb3+ functions as an ideal acceptor with its unique NIR emission band at around 1000 nm, specifically important for photovoltaic applications.122While the low absorption cross section of Ln hampers practical application, organic–inorganic hybrids are an alternative option to sensitize NIR quantum cutting in Ln-NPs. This approach has been described for the first time in 2013 by Li et al. with a combination of a UV-absorbing sensitizer 6 and Tb3+/Yb3+ co-doped NaYF4 NPs.123 Upon absorption of 6, the energy is non-radiatively transferred to coordinated Tb3+, and by cooperative energy transition through downconversion, Tb3+ transfers the energy to Yb3+, which in turn undergoes a multiphoton relaxation and subsequent emission in the near infrared region. As a downconversion luminescent converter, this kind of material meant to be useful in Si-based solar cells to reduce thermalization loss and enhance conversion efficiency of solar cells via spectral modification. But this type of spectral conversion mechanism remains rarely studied. In 2018, Wang et Meijerink reported another Ln combination for an efficient dye-sensitized downconversion using a strong UV/blue absorbing Coumarin dye (42) to sensitize quantum cutting of Pr3+/Yb3+ couple in NaYF4 NPs.124 Photoluminescence and lifetime measurements, demonstrated a FRET from Coumarin to Pr3+ followed by downconversion, resulting in Yb3+ NIR emission with ∼30 times enhancement with respect to the bare NPs.
The efficiency of downconversion emission in these NPs is limited by the concentration quenching effect due to nonradiative recombination paths set by intramolecular motions mechanisms, common to most aromatic hydrocarbons and their derivatives.115 Shao et al. introduced the concept of an ultimate photosensitization by aggregation-induced enhanced emission luminophores (AIEE LP) dyes to overcome this limitation.125 This concept illustrated in Fig. 7 was demonstrated by completely covering the surface of Yb3+/Tb3+ co-doped NaYF4 NPs with a dicyanostilbene derivative AIEE dye (43), which is designed for efficient attachment to the NPs at high density to maximize absorbance while passivating the surface. The energy transfer resulted in a 2260-fold enhancement of multiphoton downconversion by quantum cutting relative to the dye-free NPs. In a prototypical application, this quantum cutting of UV photons to NIR photons matching with Si solar cells bandgap produced a 4% increase in efficiency under concentrated solar illumination.
 | ||
| Fig. 6 Reported photosensitizing ligands for upconversion and downconversion related NPs. | ||
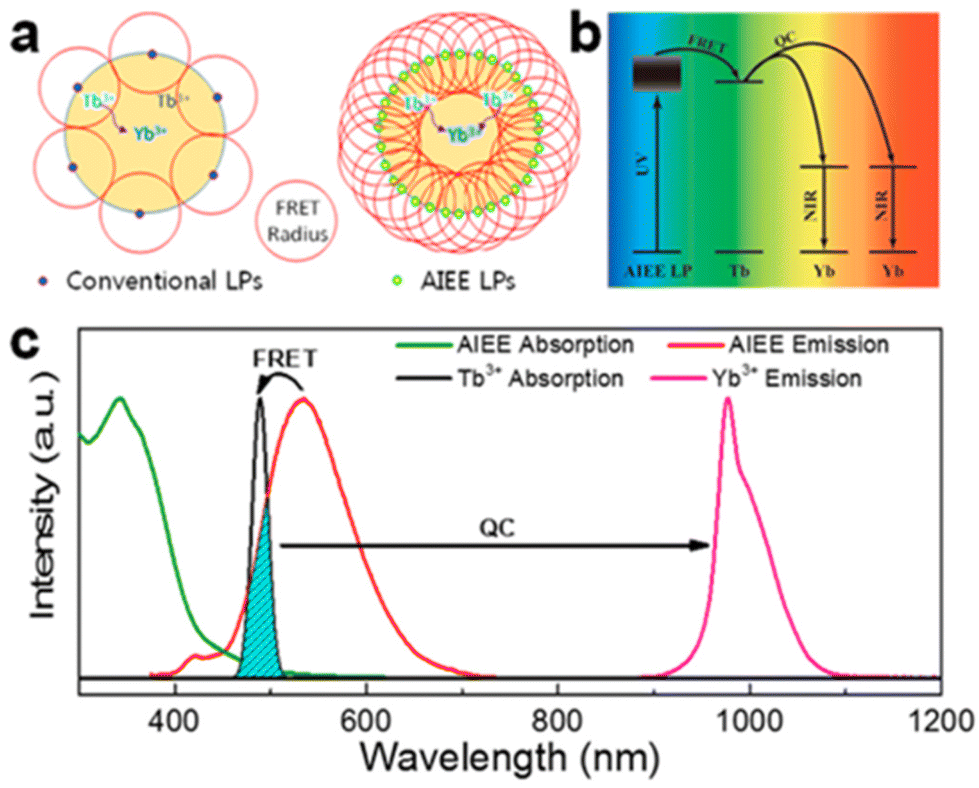 | ||
| Fig. 7 (a) Schematic illustration of the difference between conventional emission and AIEE in the sensitization of quantum cutting. (b) Energy-transfer pathway from the AIEE LPs to NPs. (c) Normalized absorption and photoluminescence spectra of the AIEE dye and NPs. Reprinted with permission from ref. 125. Copyright (2018) American Chemical Society. | ||
Recently, Agbo et al. demonstrated the role that core/shell structures can play in this type of spectral conversion. They used 26 as a UV photosensitizer of NaGd1−x−yNdxYbyF4/NaGd1−xNdxF4 core/shell NPs,126 enabling an efficient, stepwise ligand-donor–acceptor (26-Nd3+–Yb3+) energy transfer that mitigates the possibility of efficiency losses arising from the direct sensitization of Yb3+ by 26. Using a detailed power dependence study, they described a spectral transformation through partial utilization of decay channels that permit the production of two NIR photons per UV photon absorbed. These results are important to challenge the energetic mismatches between solar illumination and the spectral response of commercial photovoltaic cells. However, dye-sensitized quantum cutting, with external quantum efficiency larger than 100%, is yet to be reported.
To summarize, Ln incorporation into inorganic matrix covered with capping antennas offers a very high protection from environment related deactivation, as highlighted by long excited-state lifetimes exhibited by numerous NPs (see Table 1). Various types of organic matrices can host Ln and the doping level can be tuned as desired. However, despite these important and promising merits, this type of nano-objects still has some drawbacks, such as the stabilization of the organic dyes on the surface of NPs, as the coordination of the ligands to metallic ions on the surface suggested an electrostatic, non-stable bonding nature. Moreover, the NP (bio)functionalization for analytical applications is a tricky point as significant organic synthesis work is needed in order to obtain appropriate antennas with an active function. The photostability of the organic dyes over time on the surface of NPs could also be a point to be considered as they are not protected. To overcome these points, organic/inorganic systems could be isolated from the environment (by encapsulation inside polymeric or silica coatings) to prevent dissociations or photochemical reactions and facilitate the functionalization.
4. Ln-Complexes encapsulated into NPs
A second strategy for cumulating the properties of Ln complexes is their encapsulation inside NPs. Incorporation of several Ln complexes into one NP has two advantages: (i) it allows to drastically increase the brightness of the entity compared to the single complex, by cumulating the absorption properties of chelates, and (ii) the complexes are protected from their environment and thus suffer less from deactivation.In order to optimize the synthesis and obtain NPs with the best luminescence properties, it is important to have an estimation of the number of Ln-complexes (or in some cases the weight percentage (wt%) relative to the polymer) in a NP. While the complex encapsulation is often assumed to be quantitative (the initial and final wt% are considered to be equal), an estimation may be done by assuming that no quenching occurs in the NPs and thus by directly comparing the photoluminescence intensities of NPs with complexes in the solution at the same entity concentration.127 Proper quantifications of the number of complexes into a NP can be realized by comparing the luminescence intensity of released complexes after NP dissociation with Ln-complex standards,128 or by Ln quantification by Inductively Coupled Plasma Atomic Emission Spectroscopy of the remaining supernatant or after NP digestion.129
In this part of the review, we will introduce the different examples of Ln-complex doped NPs described in the literature over the years. The photosensitizing ligands encountered in the literature are reported in Fig. 8. The main reports are related to encapsulation in either polymer or silica NPs. The photoluminescence properties of the various NPs (when studied) are summarized at the end of this section in Table 2.
 | ||
| Fig. 8 Photosensitizing ligands present in the Ln complexes encapsulated in the NPs. | ||
| Matrix | Ln | Antenna | Complexes per NP | Diameter (nm) | ε/B (L mol−1 cm−1) | QY (%) | Lifetime (μs) | λ excitation (nm) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Hydrodynamic diameter measured by DLS. 2PE = two-photon excitation. | |||||||||
| Polymer | Eu | 48 | 107 | 720 | 128 | ||||
| Eu | 49; 53 | 1400 | 45 | 618 | 340 | 144 | |||
| Tb | 1000 | 695 | 320 | ||||||
| Sm | 250 | 89 | 341 | ||||||
| Eu | 16; 54 | 3500 | 52 | ε = 6.5 × 104B = 7.0 × 107 | 31 | 586 | 412 (2PE @ 832) | 146 | |
| Eu | 16; 18 | 2 wt% | 52 to 94 | 720 | 343 | 129 | |||
| Eu | 18; 50 | 60 wt% | 15 | 31.5 | 509 | 342 | 149 | ||
| Eu | 59 | 2 wt% | 376a | 60 | 2920 | 300 | 154 | ||
| Eu | 16; 18 | 40 wt% (5000) | 34 | B = 4 × 107 | 26 | 1300 | 350 | 155 | |
| Eu | 16; 55 | 156 | 15 | 14 | 756 | 410 | 157 | ||
| Silica | Eu | 62 | 37 ± 3 | 384 | 336 | 170 | |||
| Eu | 63 | 50 ± 5 | 1.1 | 770 | 336 | 172 | |||
| Tb | 66 | ∼1500 | 42 ± 3 | 1520 | 320 | 127 | |||
| Tb | 66 | 45 ± 3 | 10 | 2000 | 324 | 173 | |||
| Tb | 67 | 50 ± 3 | 1500 | 328 | 175 | ||||
| Eu | 68 | 55 ± 3 | 600 | 328 | 176 | ||||
| Eu | 53; 68 | 10 | 21 | 397 | 335 | 177 | |||
| Eu | 53; 61 | 36 ± 3 | 66 | 390 | 406 | 178 | |||
| Eu2 | 54; 69 | 42 ± 3 | 31 | 346 | 345–390 | 179 | |||
| Tb | 71 | 40 ± 5 | 1950 | 280 | 180 | ||||
| Eu | 1200 | ||||||||
| Eu | 61 | 45 ± 5 | 450 | 380 | 181 | ||||
| Tb | 72 | 50 | 2060 | 279 | 182 | ||||
| Eu | 1230 | ||||||||
| Yb | 74 | 0.11 | 4.17 | 375 | |||||
| Nd–Yb | 72; 74 | 0.095 (Yb) 0.018 (Nd) | 0.058 (Nd) | 272 | |||||
| 0.13 (Yb) | 4.01 (Yb) | 375 | |||||||
| Tb | 64 | 56 ± 4 | 1200 | 335 | 183 | ||||
| Eu | 1280 | ||||||||
| Eu2 | 75 | 2380 | 90 ± 10 | 28 | 2400 | 330 | 185 | ||
| Eu | 56 | 1000 | 10 | ε = 5 × 107 B = 3 × 106 | 6 | 243 | 350 | 196 | |
| Eu | 56 | 2 wt% | 20–30 | 250 (25 °C) | 400 | 186 | |||
| Eu | 76 | 5 wt% | 70 ± 20a | 750 ± 20 | 370 (2PE@740) | 187 | |||
| Tb | 77 | 22 ± 3 | 3 | 330 | 191 | ||||
| Tb-Yb | 77 | 59 ± 4 | 1116 | 330 | 192 | ||||
| Tb-Gd | 1247 | ||||||||
| Zirconia | Tb | 66 | 33 ± 4 | 8.9 | 2000 | 324 | 174 | ||
4.1. Polymer NPs
Polymeric NPs are a well-suited matrix for the encapsulation of Ln complexes. Extensively studied as biomaterials for biomedical applications130,131 in particular for drug delivery132 because of their remarkable stability in biological environment and their biocompatibility, they are also used as matrix for dye-encapsulated NPs.133 The techniques of preparation of the dye-loaded polymer NPs are beyond the scope of this review but are well addressed in other publications.133,134 Very briefly, they can be obtained by either polymerization of monomer units (such as emulsion, micro-emulsion…) or by preparation from preformed polymers (e.g. self-assembly, nano-precipitation), in the presence of the dye to be encapsulated.The first attempt of Tb-chelate and Eu-chelate doped polymer NPs synthesis in the literature was reported by Tamaki et al. in 2002.135 Sub-micron PEG-coated polystyrene (PSt) particles (diameter of circa 200 nm) were encapsulating β-diketonate (44 or 45) ternary Tb3+ or Eu3+ complexes coordinated to an acryl derivative of phenanthroline (46 or 47). The acryl function enabled to covalently bind the complexes to the host matrix thanks to its involvement in the copolymerization process and to avoid a possible leakage from the particles over time. At the same period, [Eu(48)3] doped PSt NPs were also commercialized and have been extensively used by Härmä and colleagues over the years for improving bioanalytical methods (immunoassays,136–139 cell imaging,140 protein quantification141 and protein aggregation142,143). The 107 nm NPs in particular were found to contain 31000 chelates and to exhibit an Eu-based luminescence emission lifetime of circa 720 μs.128 Härmä's group was also the first to describe the synthesis and characterization of Sm(III) and Dy(III) chelates doped polymer based NPs with respectively β-diketonate derivative 49 and dipicolinic acid derivatives (51, 52 or 53) as photosensitizing ligands, as well as their Eu(III) and Tb(III) counterparts.144 NPs of around 45 nm were obtained, exhibiting photoluminescence emission in the visible (from green to red depending on the Ln ion) with lifetimes of 618, 695 and 89 μs for Eu, Tb and Sm doped polymeric NPs respectively, Dy-related lifetime being too short to be detected here. Their potential application as fluorescent label was highlighted with a sandwich immunoassay after surface functionalization by streptavidin.
Tamaki et al. studied the impact of the use of PSt and PMMA as polymer matrix for encapsulation of [Eu(16)3(18)] and [Ln(25)3(18)] (Ln = Eu, Tb, Sm) complexes by precipitation.145 Only [Ln(25)3(18)] with PSt matrix resulted in relatively uniform and luminescent NPs, probably thanks to the interaction between the phenyl moieties of 25 and styrene, but even there, very large particles in the 1–2 μm range were obtained. This highlights the complexity of the NP preparation and complex encapsulation steps for preparing luminescent NPs with size uniformity in the nm range.
Yuan Wang and colleagues designed poly(methylmethacrylate-co-methacrylic acid) NPs embedding circa 3500 [Eu(16)3(54)] complexes per NP, for application as cell imaging probe.146 52 nm water-dispersible nanospheres were obtained by co-precipitation, exhibiting two-photon-sensitized luminescence properties: Eu ion emits red light through two-photon excitation of 54 at 832 nm via a singlet energy-transfer pathway predominantly. With high two-photon excitation action cross sections (δ × Φ estimated at 1.2 × 105 GM), they are promising probes for two-photon excitation microscopy, as demonstrated by the NP bioconjugation and use as bionanoprobe for imaging of live cancer cells.
In 2012, Desbiens et al. demonstrated that the doping level of Ln-complex in the PSt NPs has a strong influence on the monomer conversion degree (conversion of monomeric carbon–carbon double bonds into polymeric carbon–carbon single bonds) and the Ln content in the NPs.129 In this study, PSt NPs were synthesized by mini-emulsion polymerization in the presence of various concentrations of [Eu(16)3(18)] complex (from 2 to 7 wt% relatively to styrene) and the results indicated that only a maximal doping level of 2% by weight in final particles could be achieved. At higher doping levels, neither the monomer conversion degree nor the final Eu content in the NPs are reproducible anymore, probably due to the limited solubility of the complex in styrene. This assumption has been supported by the study of Aikawa et al.,147 where two complexes [Eu(16)3(TOPO)2] (TOPO = Tri-Octyl Phosphine Oxyde, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(n-octyl)3) and [Eu(16)3(H2O)2], were incorporated into PSt NPs. The styrene conversion was remarkably reduced with the latter complex compared with the former one, as it was not dissolved in styrene. Thanks to the high solubility of [Eu(16)3(TOPO)2] in styrene, a maximal loading ratio of 15 wt% Eu-chelate was achieved, but the NPs started to aggregate at upper Eu content.
P(n-octyl)3) and [Eu(16)3(H2O)2], were incorporated into PSt NPs. The styrene conversion was remarkably reduced with the latter complex compared with the former one, as it was not dissolved in styrene. Thanks to the high solubility of [Eu(16)3(TOPO)2] in styrene, a maximal loading ratio of 15 wt% Eu-chelate was achieved, but the NPs started to aggregate at upper Eu content.
At the same period, new types of lanthanide doped polymer based NPs have emerged. Tan et al. described the use of lanthanide coordination polymer nanoparticle composed of adenine and dipicolinic acid (5) coordinated to Tb(III), as probe for Hg2+ detection.148 Photoinduced energy transfer between adenine and 5 caused by hydrogen bonds between the two prevents Tb photosensitization by 5, but the presence of mercuric ion leads to an enhancement of Tb luminescence, originating from the coordination of the mercuric cation to adenine. The detection has been shown to be specific to Hg (no luminescence enhancement for other metals ions) with a detection limit of 0.2 nM.
Chiu and colleagues designed Eu-doped polymer dots with a matrix able to photosensitize the Eu-chelate via FRET.149 High amounts (up to 80 wt%) of two different Europium-(β-diketonate) complexes ([Eu(50)3(18)] and [Eu(56)3(TOPO)2]) were embedded in a fluorescent poly(9-vinylcarbazole) (PVK) matrix, giving rise to Eu-related red emission luminescence through excitation of PVK at 342 nm. A maximal QY of 33.5% was obtained with 60 wt% [Eu(50)3(18)], the latter decreasing for higher doping ratios, possibly due to concentration quenching. The polymer dots were employed as probe for living cell imaging. They also proposed the preparation of polymer dots containing [Eu(16)3(18)] complex covalently linked to the matrix, with the aim of avoiding complex leakage.150
Complex coacervate core micelles (nanometric colloidal complexes produced by the co-assembly of ionic-neutral block copolymers with oppositely charged species151) containing Eu and Gd ions were proposed by Wang et al. for potential applications both in luminescence spectroscopy and magnetic relaxation measurements.152 Both Ln ions are chelated by a dipicolinate derivative (57), an anionic coordination polymer that electrostatically interacts with a cationic neutral diblock polymer to form micelles in aqueous solution. Micelle-like NPs were also proposed by Thévenaz et al. by coordinating Eu(III) or Tb(III) with a dipicolinate moiety functionalized with diblock copolymers based on poly(ethylene glycol) (PEG) and poly(ε-caprolactone) (PCL) segments (58).153 Solvent displacement allowed to form green or red emitting NPs composed of a PCL core (solid sphere < 47 nm in diameter) containing Ln-complexes and a PEG corona (vesicle >47 nm).
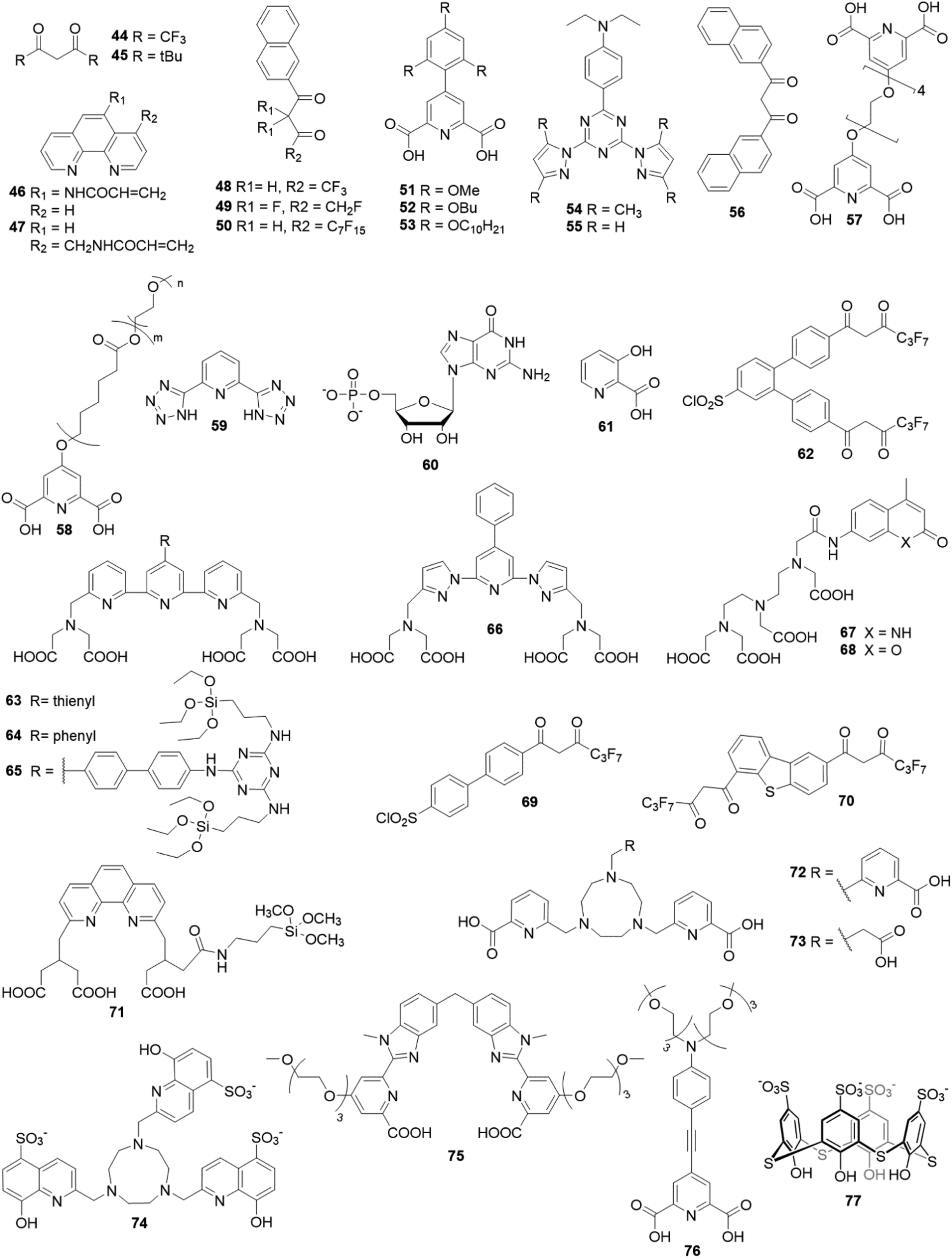
In 2013, Wartenberg et al. reported the use of pyridine-bistetrazolate ligand (59) as sole photosensitizer of Tb, Eu, Dy and Sm in Vis-NIR luminescent polymeric NPs containing the four [Ln(59)3]3− complexes.154 The authors highlighted the importance of using a cationic surfactant to incorporate efficiently the anionic complexes into PMMA matrix and obtain luminescent NPs. Once again, the doping level is a limiting parameter, as a maximum of 2 wt% was reached without destabilizing the NPs, similar to the doping level of [Eu(16)3(18)] in Pst NPs seen previously.129 6 years later, Cardoso Dos Santos et al. have overcome this issue as they succeeded to encapsulate up to 40 wt% of [Eu(16)3(18)] complex into NPs (corresponding to 5000 complexes per NP) composed of PMMA copolymers bearing sulfonate (PMMA-SO3H; 1 or 3 mol%) or carboxylate (PMMA-COOH; 10 mol%) groups (Fig. 9).155 Two emission lifetimes were measured, a short one (around 0.5 ms, similar to the free complex in acetonitrile) attributed to the complex at the surface of the NP and a long one (up to 1.3 ms) attributed to the complexes protected in the core of the NP, the lifetime exceeding that of Eu complex in solid state (see Fig. 9a for decay profiles). With a quantum yield up to 26% (Fig. 9b) and a remarkable brightness exceeding 107 L mol−1 cm−1, the authors have reported a very promising candidate for single-particle and live-cell imaging.
 | ||
| Fig. 9 Top: schematic representation of the preparation of Eu-complex doped polymer NPs for single particle and live-cell imaging. Bottom: (a) time-resolved emission decay profiles of Eu (λexc = 350 nm, λem = 620 nm) measured for solutions of [Eu(16)3(18)] in acetonitrile and of PMMA-COOH 10% NPs loaded with different doping rates of [Eu(16)3(18)] and (b) photoluminescence quantum yield as a function of Eu-chelate doping level. Adapted with permission from ref. 155. Copyright (2019) American Chemical Society. | ||
Various analytical and bioanalytical applications of Ln-complex doped polymeric NPs have been made in the last years. A ratiometric nanothermometer for intracellular temperature measurement in real time was reported by Takei et al.156 Thermosensitive [Eu(16)3] complex and thermoinsensitive rhodamine 101 were incorporated into PMMA NPs, and the ratio of both emission intensities is related to the temperature of the NP environment. Wang et al. developed another two-photon excitation polymeric nanoprobe encapsulating [Eu(16)3(55)] complex157 very similar to the previous one ([Eu(16)3(54)])146 with a lower two-photon excitation cross section (estimated at 3.7 × 103 GM for excitation at 800 nm). Functionnalized with a tumor-targeting agent, the NPs have been used as nanocarrier and luminescent probe for imaging. Gao et al. designed a coordination polymer NP composed of Tb and Eu ions with 60 as Tb photosensitizer, for the ratiometric detection of the anthrax biomarker dipicolinic acid (DPA, 5).158 The presence of DPA induces both an enhancement of Tb and a quenching of Eu emissions by coordinating and photosensitizing Tb ions in addition to preventing Tb to Eu energy transfer. Finally, immunochromatographic assays have also been developed with [Eu(17)3(18)] doped PSt NPs for detection of procalcitonin,159 an indicator of bloodstream infections and sepsis, and even more recently for the trendy detection of anti-SARS-CoV-2 IgG.160
4.2. Silica NPs
Silica NPs are commonly employed as biomaterial because of their water solubility, high stability, limited toxicity and good biodegradability. They are also easily produced with a tunable size and their surface functionalization is simple and versatile.161 Besides their application as nanocarriers for drug delivery,161,162 silica NPs have also emerged as a matrix of choice for dye encapsulation as it is transparent to visible light and not involved in energy/electron transfer processes.163 The preparation of dye-doped silica NPs is well described in the literature164–166 and will not be addressed here. Dye-Doped silica NPs have been reported as luminescent probe for numerous applications (chemosensor, bioprobe in imaging or immunoassay, (bio)marker quantification…),163,165 but one major limitation of these short-lived emitting nanoprobes is the media autofluorescence that generates a high background signal and thus decreases the method sensitivity. To overcome this issue, long-lived emission of Ln-complex is a very promising alternative.The first attempt of Eu-chelate doped silica NPs synthesis have been proposed by Trindade and colleagues in 2003.167 SiO2 NPs doped with complexes of Eu3+ ion coordinated by three 3-hydroxypicolinic acids (61) were prepared by a sol–gel method adapted from the Stöber process,168 leading to quite large NPs of 130 nm in diameter with an Eu emission lifetime of 0.5 ms. However, the experimental results strongly suggest that the Eu3+ complexes would be located at the surface of the NP, Eu3+ being also coordinated by silanol groups. Using a similar method based on Stöber process, Zhao et al. succeeded to encapsulate [Eu(17)3(18)] complexes into Silica particles, but here again, with a very large size (300 nm).169 These studies highlight the difficulty of efficiently and homogeneously encapsulating complexes into NPs.
Jingli Yuan's group designed 20 to 50 nm silica NPs doped with various chelates (Eu3+ with 62170,171 or 63,172 Tb with 66127,173) by water-in-oil microemulsion. Encapsulation was realized either with free Ln-complex127,171–173 or by copolymerization of Ln-complex bound to 3-aminopropyl(triethoxy)silane (APTES).170 With the latter, Ln complexes are covalently bound to silicon atom, thus more protected from dye leaking and less sensitive to the different handling steps (washing, surface modification, bioconjugation…). The encapsulation of Ln-complex is beneficial as less photobleaching is observed, however emission lifetimes and quantum yields are much lower than those of the free complexes once encapsulated in the NPs. These results could originate from the exciting light absorbance by the matrix and/or from a concentration quenching due to a high concentration of Ln-complexes in the NPs. These NPs, functionalized with amino groups, were then conjugated with a biomarker (streptavidin or antibody) prior to be used as luminescent probes in fluoroimmunoassays.
Encapsulation of Tb chelated by 66 in zirconia NPs as an alternative to silica NPs has also been proposed in parallel.174 Doped zirconia NPs have been shown to be more stable than their silica counterpart127 in high pH aqueous media and to have a slightly longer Tb emission lifetime (2.0 vs. 1.5 ms), however the limit of detection (LOD) of the immunoassay realized with ZrO2 NPs is much higher. As the NP conjugation is non-covalent (Lewis acid–base conjugation), dissociation probably occurs at low concentrations or during washing steps, explaining the high LOD compared with covalently-conjugated silica NPs.
Chen et al. proposed the synthesis of silica NPs embedding Tb3+ coordinated to a polyaminocarboxylate derivative with a Carbostyril 124 as photosensitizer [Tb(67)]175 and its Eu counterpart [Eu(68)] with a coumarin120.176 These 50 nm large NPs exhibit emission lifetime of 1.5 ms and 0.6 ms for Tb and Eu respectively, upon excitation of the antenna (λexc = 328 nm), similar to the ones in free complexes. Each NP is reported to be as bright as about 340 free Tb complexes and 1300 free Eu complexes respectively, highlighting a potential 100 to 1000-fold increase of sensitivity for biomarker detection and quantification. DNA sandwich hybridization assays were successfully realized with the detection-oligonucleotide labelled luminescent NPs and capture-oligonucleotide linked to a magnetic bead to detect and quantify the targeted DNA, with a detection 100-fold (for Tb) and 50-fold (for Eu) more sensitive than the Fluorescein isothiocyanate (FITC) technique.
From 2008, Jingli Yuan's group focused on the design of red-emitting Eu-doped silica NPs with excitation in UV-Vis range (from 200 to 450 nm) with [Eu(54)–(69)3] complex177 and excitation in visible light (406 nm) with [Eu(54)–(62)] complex.178 Both 62 and 69 were linked to APTES to be covalently bound to the matrix. The best properties were obtained with [Eu(54)–(62)] complex, the related NPs exhibiting a Eu emission lifetime of 0.39 ms and a QY of 66%. Other UV-Vis excited NPs were proposed with a dinuclear [Eu2(70)3(55)2] complex (excitation up to 475 nm) with a similar lifetime but a QY of 31%.179 These three different NPs were utilized as luminescent probe for the detection of environmental pathogens by time-gated luminescence imaging.
In the late 2000s, several studies also emerged with NPs containing two different Ln ions. The first report was made by Zhang et al. with the preparation of NPs encapsulating different ratios of Tb/Eu ions both chelated and photosensitized by (71) and exhibiting longer emission lifetime than the related free complexes (1.2 ms vs. 0.42 ms for Eu, 1.95 ms vs. 0.64 ms for Tb).180 Six years after their first attempt,167 Trindade and colleagues proposed a different strategy of [Ln(61)3] complexes (Ln = Tb, Eu or Tb-Eu) encapsulation based on the formation of a core of complexes and a shell of amorphous silica as a protective layer, by reverse microemulsion.181 This new method is more adapted, as the photoluminescence results confirmed the presence of Ln-complexes in the NPs and the size of NPs is more suitable for bioassays (45 nm vs. 127 nm previously167). In 2010, Samuel et al. proposed two types of dual-mode lanthanide doped luminescent silica NPs with a tunable size between 10 and 100 nm.182 Visible and NIR emitting nanospheres containing Tb(72)–Eu(72) and Nd(72)–Yb(74) respectively exhibited emission lifetimes similar to their corresponding free complexes and the emission of the NIR emitting NPs could be tuned by varying the excitation wavelength, suggesting a potential application as multiluminescent bar codes. Jiang et al. designed NPs with emission colors from green to red by co-binding different molar ratios of Eu(64) and Tb(64).183 Wartenberg et al. designed their own Tb–Eu co-doped silica NPs with 72 or 73 as antennas,184 and proposed a new method to study the efficiency of the incorporation process by using 152Eu chelates for radioactivity measurements.
The effect of Ln-complex encapsulation on the ligand triplet-state energy level was questioned by Eliseeva et al., and [Eu2(75)3] binuclear helicates incorporation into silica NPs was shown to have no impact on it.185
The beginning of the 2010s’ was also marked by the breakthrough of new kind of probes in this field. To the best of our knowledge, the first thermometer composed of Ln-complex encapsulated in silica matrix was presented by Wolfbeis and colleagues in 2010. They first tried to encapsulate a high content of [Eu(56)3(TOPO)2] complex into 2-bis(trimethoxysilyl)decane but the resulting luminescence intensity was reduced by concentration quenching.186 To overcome this issue, an hybrid NP was prepared by using PMMA polymeric material as co-matrix to compensate the decrease of Eu complex concentration. This hybrid nanoprobe was employed for sensing and imaging of physiological temperatures (from 25 to 45 °C with a precision of ±0.3 °C) thanks to the dependence of both photoluminescence intensity and emission lifetime to temperature upon visible–light excitation.186 One year later, Philippot et al. presented the first Ln-complex doped silica NPs with two-photon excitation.187 The encapsulation of [Eu(76)3]3− complex was beneficial as the NPs are luminescent even in aqueous solution whereas the free complex is not, because of ligand substitution by water molecules. The 5 wt% doped NPs exhibited typical Eu red emission upon excitation into 76 at 370 nm, and they are also promising probes for two-photon microscopy imaging thanks to the two-photon absorption ability of 76 in the NIR (740 nm).
The last five years have been marked by a decrease of the number of publications compared with the beginning of the century. The published studies are more focused on the analytical/bioanalytical applications, such as the detection of Cardiac Troponin I by [Eu(65)] doped NPs189 or anthrax biomarker (dipicolinic acid (5), named DPA) detection by a time-resolved ratiometric fluorescent probe.188 In this study, the surface of silica NPs encapsulating Tb(III) ions coordinated to DPA (Tb/DPA@SiO2 in Fig. 10a) was functionalized with a guanosine monophosphate bound to Eu(III) (Eu/GMP). In the presence of DPA in solution, Eu ions are chelated by the latter through the replacement of a water molecule in the Eu/GMP complex. Upon DPA excitation, both Tb(III) and Eu(III) ions are dye-sensitized, thus emitting green and red light respectively. The green emitting light related to Tb emission serves as a stable reference, and the DPA in solution is detected and quantified by measuring the ratio of Eu over Tb emission intensities as shown by the emission spectra for various DPA concentrations (Fig. 10b).
 | ||
| Fig. 10 Anthrax biomarker (DPA) detection via a dual-lanthanide based silica NPs. (a) Representation of the luminescent probe. (b) Emission spectra of Tb/DPA@SiO2-Eu/GMP probe to different concentrations of DPA upon DPA excitation. (c) Fluorescence color image of the designed paper-based visual sensor over various concentrations of DPA solution under 254 nm UV light excitation and standard curve obtained with a smartphone. Reprinted from ref. 188, Copyright (2017), with permission from Elsevier. | ||
This time-resolved ratiometric fluorescent assay has revealed a good selectivity and sensitivity (LOD of 7.3 nM) towards DPA detection in biological samples. Moreover, a paper-based visual sensor (Fig. 10c) was developed for fast detection by naked eyes or with a smartphone under a UV lamp, but with a low sensitivity that has to be improved (LOD of 1 μM). A similar method was also developed for mercury detection.190
Finally, in 2019, p-sulfonatothiacalix[4]arene (77) was used as (i) coordinating photosensitizer of Tb ion in green emitting doped silica NPs for immunoassay application191 and as (ii) photosensitizer of Tb ion and chelator of Yb ion in dual visible-NIR luminescent silica NPs.192 In the case of Tb–Yb doped NPs, besides the Tb-related visible emission, a Tb(III) → Yb(III) energy transfer occurs and leads to NIR emission through UV excitation of 77 (320 nm). The internalization into cells of these dual visible–NIR emitting NPs highlighted their possible use as probes in cellular imaging.
To sum up, encapsulation of Ln-complexes into NPs is a good strategy to cumulate the spectroscopic performances of the luminescent complexes and protect them from quenching. However, to control the size of NPs and to obtain an appropriate loaded amount of complex in the NPs without destabilizing the polymerization reaction and complex leakage is a real challenge, as highlighted by the literature. The question of the quantity of Ln-complex to be encapsulated to have the brightest NPs has been studied for years, and a subtle equilibrium has to be respected to encapsulate the highest number of complexes to cumulate their properties without suffering from concentration quenching. The use of bulky groups or bulky counterions is also a good strategy to control the organization of complexes in the NPs and decrease the risk of concentration quenching.133 Complex leakage over time can also be avoided by using complexes linked to a monomeric unit to be covalently linked to the matrix.
Finally, these NPs are well suited for (bio)analytical applications as (i) the host matrix (polymer or silica) is biocompatible and (ii) reactive groups (such as amino or carboxylate groups) can be introduced at the surface of NPs during their synthesis or with an extra polymerization step, allowing for (bio)functionalization.
5. NPs surface functionalized by Ln-complexes
The functionalization of NPs with lanthanide complexes gained interest over the years. The combination of their properties led to new systems very promising for applications such as medical imaging or luminescent probes. In this part of the review, we will mainly overview the different systems presented in the literature. The photosensitizing ligands used to elaborate those systems are illustrated in Fig. 11 and 15. | ||
| Fig. 11 Photosensitizing ligands used to form the Ln complexes and functionalize the NPs, part 1. | ||
5.1. Functionalized quantum dots
Quantum dots (QDs) are used for almost three decades for their size dependent optical properties with high absorption cross sections193 and narrow emission bands.194,195QDs offer an improvement in term of stability toward photobleaching in comparison to organic fluorophores197–199 and can be used as an energy donor with dyes but also interact with luminescent donors as fluorescent proteins,200 luminescent polymers,201 or lanthanide complexes in the case of the FRET process. Despite those positive points, the FRET process is particularly limited by donor–acceptor distance as its efficiency depends on it.202 This part of the review will be mainly focused on FRET with QDs as acceptors and lanthanides as donors. The photoluminescence properties of those systems are summarized in Table 3.
| Ln | Antenna | Number of coated complexes | Lifetimea (μs) | FRET Distance (nm) | FRET efficiency (%) | λ exc (nm) | Ref. |
|---|---|---|---|---|---|---|---|
| a Ln3+ excited-state lifetimes after functionalization of the QDs. | |||||||
| Tb | 78 | 5 to 7 | 170 and 560 | 7.2–7.6 | 63 | 308 | 203 |
| Tb | 78 | 6 | 8.4 | 315 | 204 | ||
| Tb | 78 | 5.6 | 10 | up to 70 | 308 | 205 | |
| Eu | 78; 79 | 9.6 | |||||
| Tb | 80 | 6 | 5 to 11 | 340 | 209 | ||
| Tb | 80 | 6 to 10 | 339 | 208 | |||
| Tb | 80 | 10.1 and 7.5 | 340 and 400 | 213 | |||
| Tb | 80 | 6.3 to 8.1 | up to 94 | 339 and 400 | 214 | ||
| Tb | 80 | 70 to 170 | 6 to 7.5 | up to 97 | 308 and 340 | 211 | |
| Tb | 80 | 5.5 to 11.5 | 212 | ||||
| Tb | 80 | 5.4 to 11.1 | 340 | 217 | |||
| Tb | 80 | 10 | 337 | 218 | |||
| Tb | 80 | 14 | 340 | 216 | |||
| Tb | 80 | 740 and 1820 | 10.3 and 12.2 | 330–340 | 43 | ||
| Eu | 81 | 610 and 1090 | 9.2 and 11.1 | ||||
| Tb | 80 | 10 | 337 | 219 | |||
| Tb | 80 | 1 to 10 | 215 | ||||
| Tb | 86 | 75–80 | 275–320 | 223 | |||
| Eu | |||||||
| Eu | 89; 90; 91; 92 | Up to 1430 | 267–305 | 225 | |||
| Tb | Up to 1470 | ||||||
| Eu, Tb, Yb | 93 | 0.005 to 0.009 | 220 to 440 | 226 | |||
Löhmannsröben and Hildebrandt firstly worked on this subject by introducing a system composed of CdSe/ZnS QDs as acceptor functionalized with [Tb(78)] chelates as donor into a FRET process based on a Time-Resolved Fluoroimmunoassay format (TR-FIA).203 This assay relies on biotin–streptavidin recognition process where Tb-chelate labelled streptavidin interacts with biotins coated at the surface of the QDs. The use of a donor with a long-lived excited state such as Tb (1.5 ms) is a great advantage for energy transfer to the QD, leading to a FRET efficiency of 63 ± 4%, and allowed the use of time-resolved acquisition thanks to the increased decay time of the QDs (few hundreds of μs) enhancing the sensitivity of the detection procedure. These results were confirmed in a more detailed analysis of the decay time of the FRET process.204 Eu complexes were also successfully employed as energy donor in FRET experiments with QDs, however the use of [Tb(78)] chelates showed better results than [Eu(78)] and [Eu(79)] complexes, with a LOD under 2 pM and generally a better FRET efficiency (up to 70%).205
Those pioneer reports illustrating the Ln-complex functionalization of QDs and the related FRET process have paved the way for many researchers, in particular Hildebrandt and his collaborators whose work has largely contributed to the scientific progress of the field. Various studies were realized with a commercial [Tb(80)] complex (named Lumi4-Tb) labelled with streptavidin and biotin tagged QDs for the development of new ultrasensitive assays with multiplexed detection. This method is based on the FRET interaction between different QDs emitting at different wavelengths and [Tb(80)] present in the same sample. Their system is expected to contribute to applications in biology and medicine for example, as it provides a LOD in the sub-picomolar level.206
A new assay for multiplexed ultrasensitive detection allowing the measurement of manifold clinical parameters simultaneously within one sample (Fig. 12, left panel) was reported,207 with a LOD lower than 1 pM, offering a better sensitivity than a well-established homogeneous immunoassay FRET pair (40 to 240-fold higher). A spectroscopic ruler208 was also designed, which permitted to correlate the efficiency of the energy transfer with the donor–acceptor distance, and consequently to provide information about the size and shape of the QD (as illustrated in Fig. 12, right panel).202 Such tool was realized by optimizing the energy transfer between QDs emitting at different wavelengths and [Tb(80)] complexes without the help of a FRET enhancer.209,210 A good energy transfer with distance over 10 nm and 11 nm where obtained without and with FRET enhancer respectively, making possible applications in life sciences and nanotechnology.208 The spectroscopic ruler measurement can also be set by others methods,211,212 such as QDs coated with [Tb(80)] functionalized peptides or maltose-binding proteins, allowing to increase the precision in shape and size determination compared to other analytical methods like TEM, DLS and HPLC.212
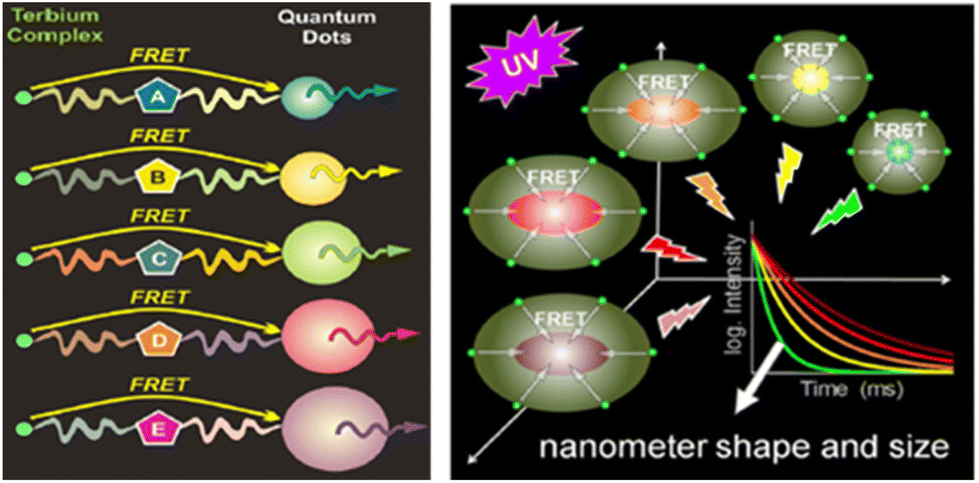 | ||
| Fig. 12 Principle of multiplexed Tb-complex to QDs FRET applied for ultra-sensitive diagnostics (left) and spectroscopic ruler measurements (right). Reprinted with permission from ref. 207 and 208. Copyright (2010), Wiley-VCH Verlag GmbH & Co. KGaA. | ||
In 2012, they demonstrated the ability of QDs to be, in the same system, both an acceptor and a donor for FRET relays (Fig. 13).213 QDs were functionalized with [Tb(80)] and a dye with the help of peptides. In such a system, excited Tb-complex transfers the energy to the QD which then transfers it to the dye. This process has been used for protease activity and DNA hybridization by using trypsin and chymotrypsin labelled with the Tb-complex and the dye, respectively. Moreover, despite the presence of only one QD, this system was also shown to be employable for multiplexed protease sensing with time-gated FRET relay by conjugating the QDs with dye/Tb-complex labelled peptides (Fig. 13).214 The system was studied with the use of trypsin which gave a very high FRET efficiency (around 94%).
 | ||
| Fig. 13 Principle of the time-gated Tb to QD to Dye FRET relay. Reprinted from ref. 214. Copyright (2012) American Chemical Society. | ||
Recently, Xu, Qiu and Hildebrandt combined nucleic acid amplification and NPs for microRNA (miRNA) biosensing.215 [Tb(80)]-to-QD related FRET was combined with rolling circle amplification (RCA) for a specific miR-21 assay. A subpicomolar LOD, a multiplexing capability and FRET with donor–acceptor distances of about 1 to 10 nm were obtained. This work also highlighted that the development of such systems requires many modification and optimization steps and is more complex than what can be found in the literature.
In addition to the biotin–streptavidin recognition system, other methods for the QDs functionalization by Ln-complexes were investigated by Hildebrandt and co-workers.216–218 The team proposed a system where [Tb(80)] and a dye were linked to the QDs using DNA components, as shown in Fig. 14.216 The dye was used as a bridge for energy transfer from the Tb-complex to the QDs, allowing to overcome the limitations of energy transfer due to the donor–acceptor distance and the long lifetime of the QDs, by reaching a FRET distance of ca. 14 nm but with a lower FRET efficiency (between 10 and 20%) compared to the previously mentioned systems.
 | ||
| Fig. 14 Representation of the four [Tb-complex](green)–Dye(yellow)–QD-DNA systems with the different distances in between fluorophores. Reprinted with permission from ref. 216. Copyright (2017) American Chemical Society. | ||
A system using peptides functionalized with [Tb(80)] complex on one side and a hexahistidine tag on the other side was also proposed. They were assembled to a zinc-rich surface of dihydrolipoic acid capped QDs. Both this system and another based on biotin–streptavidin recognition were used to evaluate the influence of plasma on photophysical properties by comparing them in buffer and plasma.217 Biotin–streptavidin binding system was shown to be the most suitable for clinical assays as it reached a low picomolar range of LOD when zinc–histidine interaction related LOD was 100 times higher. A few years later, they proposed another system made of QDs with a SiO2 shell of different thickness (6 and 12 nm) on which two maleimide functionalized [Tb(80)] and [Eu(81)] complexes were attached forming one single nanoparticle,43 allowing for the detection of different cells in a single view thanks to four specific photoluminescence decays given by Tb and Eu complexes. Thanks to a Red/Green/Blue (RGB) coding by the NPs, the living cells can be labelled and distinguished individually in mixtures, and the coding range can be extended with the different QDs colors, increasing the possibilities of fluorescence encoding. More recently, they succeeded to accomplish sensitive quantification of biological targets and long-distance conformational analysis with a sub-nanometer resolution for DNA and peptides.219 Meanwhile, the surface bio-functionalisation of the NPs was extended to antibodies and their fragments, to offer applications into the field of biological diagnosis applied to the detection of prostate specific antigens.220,221
In parallel to the work realised by Hildebrandt and collaborators, different research groups reported the use of Ln-complex functionalized QDs. In 2007, Weller and co-workers worked on competitive binding assays with CdTe QDs coated with streptavidin and [Eu(82)], [Eu(83)], [Eu(84)] and [Tb(85)] complexes coupled to biotin or estradiol.222 A few years later, Mazzanti and co-workers covalently attached [Tb(86)] and [Eu(86)] complexes to QDs.223 Their main interest was the binding of a Gd-chelate for MRI probe but their study on luminescent Tb/Eu-complexes attached to QDs opened the possibility to produce a bimodal probe made with Tb/Eu complexes and the QDs for their luminescent aspect and Gd-chelates for their magnetic aspect. They also proposed a new series of bi-luminescent hybrid materials with 87 and 88 complexing Ln(III) (Ln = Eu, Tb, Gd and Yb).224 The attachment of the complexes was allowed by the functionalization of the ligands with thiol functions. In this case, the FRET process was made with the QDs emitting in a range of 525–540 nm as a donor and the Ln complexes as an acceptor. Bettencourt-Dias and co-workers aimed to synthesize surface-modified QDs by capping them with different ligands (89, 90, 91 and 92) complexing both Tb(III) and Eu(III).225 QDs were capped with the ligands and then mixed with the desired amount of Ln(III) in a one-pot synthesis method. Overall emission efficiencies, up to 79% and 36% for [Tb(90)] and [Eu(90)] respectively, are obtained making those frameworks suitable for imaging and sensing applications. Zhu and co-workers worked on the functionalization of Carbon Quantum Dots (CDots) with Ln complexes.226 CDots were chosen because they are free of heavy metals and display better biocompatibility and lower toxicity than QDs.227 The complexes were made with Ln(III) ions (Eu, Tb, Yb) and ligand 93 and were covalently linked to the CDots thanks to branched polyethylenimine forming an amine bond. This system has been characterized and could be used for multicolour imaging as they exhibited high fluorescence quantum yield (10 to 15%).
5.2. Functionalized silica NPs
As presented in section 4.2, the use of silica NPs is widely used for the encapsulation of Ln-complexes. However, it is also possible to directly coat the surface of the silica NPs with the Ln-complexes to combine the properties of the lanthanide and the nanoscale silica chemistry,228,229 as demonstrated by the unexpected silica NP functionalization by [Ln(61)3] obtained by Trindade and colleagues in 2003 (instead of complex encapsulation, see Section 4.2 for more details).167 To perform such systems, two methods are mainly used: (i) the organic modification of silica NPs surface using either amino, dipyridyl groups228 or β-diketonate230 ligands prior to Ln ion or chelate complexation and (ii) the grafting of organosilylated Ln(III) complexes on the surface of the silica NPs. The photoluminescence properties of the functionalized silica NPs are summarized in Table 4.| Ln | Antenna | QY (%) | Lifetimea (ms) | Enhancement factorb | λ exc (nm) | Ref. |
|---|---|---|---|---|---|---|
| a Ln3+ lifetimes after functionalization of the NPs. b Luminescence enhancement factor: comparison between the free Ln3+-complex intensity and the functionalized NPs intensity. | ||||||
| Tb | 94 | 2.2 | 306 | 231 | ||
| Eu | 95 | 0.398 | 232 | |||
| Tb | 96 | 2.224 | ||||
| Eu | 17 | 0.9 to 1.07 | 245 and 395 | 233 | ||
| Tb | 26 | 0.58 to 1.03 | 295 and 345 | |||
| Eu | 97 | 0.31 | 324 | 234 | ||
| Tb | 0.76 | 290 | ||||
| Eu | 98; 99 | 43 | 1.05 | 385 | 235 | |
| Eu | 18; 100 | 30 | 0.786 | 440 | 236 | |
| Eu | 101 | 0.37 and 0.89 | 285 | 237 | ||
| Tb | 102 | 0.86 and 1.45 | 309 | |||
| Dy | 18 | 0.017 | 3.4 | 313 | 238 | |
| 104 | 0.019 | 2.7 | 306 | |||
| Eu | 103 | 0.57 and 0.7 | 2.02 and 2.14 | 293 and 294 | 239 | |
| Eu, Tb | 16, 17 | 0.45 to 0.52 | 240 | |||
| Eu | 114 | 0.26 | 365 | 241 | ||
| 16; 108 | 0.65 | 242 | ||||
Using the former method, Wang and co-workers were the first to propose a facile strategy to design Ln-complex coated silica NPs,231 one year after Trindade's report. The coating was firstly made with the ligand 94 and Tb(III) was added to form the complexes at the silica NPs surface. As compared to the free complex, the use of silica NPs increased the lifetime of the Tb(III), (1.3 to 2.2 ms) considering that the structure of the resulting nanocomposite is relatively rigid and limits the non-radiative transitions due to the vibrations of the ligands. This successful approach opened the gate to new systems combining silica NPs and Ln-complex.
In 2007, Ye Xu and Qingge Li covalently coated the surface of the silica NPs with [Eu(95)] and [Tb(96)] complexes in order to design a luminescent probe for a time-resolved immunofluorometric assay (TR-FIA) for detection of hepatitis B with a LOD 20- to 30-fold lower than ELISA.232 Moreover, their Eu3+/Sm3+ dual-labelling related TR-FIA was shown to be less sensitive than the individual Eu(III) TR-FIA, due to the low fluorescence yield and short emission lifetime of Sm. The next year, Bing Yan and Bing Zhou designed another system where [Eu(17)] and [Tb(26)] complexes were covalently attached to mesoporous MCM-41 silica NPs.233 The ligands were first functionalized with tri-alkoxysilyl group allowing the bonding to the silica NPs before complexing the lanthanide. This was the first report presenting modified β-diketones derivatives bonded mesoporous host. A similar system has also been proposed with 97 as ligand complexing either Eu3+ or Tb3+.234 In 2009, the team of Reddy designed a hybrid material with the MCM-41 silica NPs.235 They covalently linked a [Eu(98)3(99)] to the silica NPs by grafting 98 to a coupling agent by a co-condensation route. This system exhibited an overall quantum yield of 43% and an intrinsic quantum yield reaching 81% for an energy transfer efficiency of 53%. The ligand replacement by 18 and 100 resulted into lower overall and intrinsic quantum yields (30% and 53%) with a quite similar energy transfer efficiency (57%).236 Huo and co-workers immobilized [Eu(101)3(H2O)2] and [Tb(102)3(H2O)2] complexes onto mesoporous silica NPs with the use of a silane compound to obtain new luminescent hybrid nanomaterials.237 The nanomaterial containing Tb exhibited higher lifetimes (864 and 1450 μs) than the one containing Eu (372 and 893 μs). The absence of emission from the ligand in the emission spectrum of [Tb(102)3(H2O)2] indicated a more efficient energy transfer from the ligand to the Tb(III). Li and co-workers proposed silica NPs functionalization using [Dy(103)(18)2(ClO4)3(H2O)2] and [Dy(103)(104)2(ClO4)3(H2O)2].238103 was used as a silane coupling agent to link Dy to the silica NPs, enhancing the fluorescence emission of the lanthanide by 2.71 and 3.41 times for [Dy(103)(18)2(ClO4)3(H2O)2] and [Dy(103)(104)2(ClO4)3(H2O)2], respectively. The grafting of Eu complex instead of Dy on the same NPs, using a saline coupling linked technique to graft the complex to the silica NPs surface, led to a less important increase (approximately 2 times improvement).239 In 2018, Qian and co-workers presented the self-assembly of nanocomposites made with Ln3+ complexes and silica NPs.240 The complexes were designed with β-diketone ligands as 16 and 17 and were coordinated to the NPs by the intermediary of 107 which was used to functionalize the silica NPs and to sensitize the Ln ions. Their first method presented good results of forming Ln(III) complexes coated silica NPs but it is limited by the heterogeneous surface and a low grafting ratio. To overcome this issue, a second method consisting in grafting of organosilylated Ln(III) complexes on the surface of the silica NPs was developed. Menu and co-workers were the first to propose a system with this method, by using dipyridine derivatives with an alkoxysilane group to form [Eu(108)3(109)] and [Eu(108)3(110)] complexes prior to attach them on the surface of the silica NPs.228 By comparison with free complexes, the grafted complexes on the silica NPs were found to be more efficient when excited in the UV (260 nm) or in the near UV (340 nm). Two new probes for E. coli bacteria bioimaging were also proposed, based on the functionalization of silica NPs with [Eu(114)3] and [Eu(16)3(108)],236,237 before proposing a new tool for biological analysis with a Vis-NIR-emitting probe.239 A [Yb(16)3(115)] complex was linked to [Ru(104)(109)], resulting in a [Yb(16)3(115)Ru(104)(109)]Cl2 complex that was covalently bonded to the silica NPs by the intermediary of 109. This system confers the possibility of having emission in the visible and in the NIR by excitation into the singlet Metal–Ligand Charge Transfer transition of the Ru(II). An energy transfer of 73% is obtained from Ru(II) to Yb(III). Both good luminescence and biological properties were demonstrated, essential qualities for a potential biomarker.
Taylor and Lin presented a method for the detection of dipicolinic acid (DPA, 5) using different EDTA based ligands for Tb(III) coordination.243 [Tb(111)(H2O)3], [Tb(112)(H2O)3] and [Tb(113)(H2O)3] complexes were individually covalently attached to silica NPs and in the presence of DPA, Tb(III) emission was observed thanks to the replacement of water molecules by DPA. The selectivity of the method has also been tested and presented high selectivity for DPA.
5.3. Functionalized metal NPs
This section will be dedicated to the various types of metallic NPs functionalized with Ln complexes, namely iron, silver and gold NPs. A comparison of the photoluminescence properties of the systems (when applicable) will be presented in Table 5.| NP composition | Ln | Antenna | Functionalized NP diameter (nm) | QY (%) | Lifetimea (ms) | Enhancement factorb | λ exc (nm) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Ln3+ lifetime after functionalization of the NPs. Percentages are the relative distribution of the lifetimes obtained. b Luminescence enhancement factor between the free complex and the functionalized NPs. | ||||||||
| Fe-NPs | Tb | 116 | 120 to 160 | 5.6 | 0.9 | 307 | 252 | |
| Eu | 117 | 0.35 | 334 | 254 | ||||
| Tb | 118 | 0.096 | 1.15 | 325 | 256 | |||
| 118; 119 | ||||||||
| Eu | 16 | 26 | 14 | 0.23 | 372 | 258 | ||
| Eu | 16; 98 | 0.35 | 365 | 259 | ||||
| Eu | 25 | 0.37 | 0.35 (solid) and 0.64 (suspension) | 270 and 310 | 260 | |||
| Eu | 120 | 1.4128% and 0.6672% | 332 | 261 | ||||
| Tb | 0.4363% and 1.6637% | 338 | ||||||
| Eu | 16 | 29 | 0.28 | 340 and 370 | 262 | |||
| Tb | 27 | 13 | 0.2 | 330 | ||||
| Au-NPs | Eu | 106 | 0.96 | 0.36 | 320 | 265 | ||
| Tb | 0.38 | 0.73 | ||||||
| Eu | 121 | 8 × 10−4 | 266 | 266 | ||||
| Eu | 122 | 0.29 | 340 | 268 | ||||
| Eu | 48 | 230 | 0.19 and 0.67 | 395 | 269 | |||
| Eu | 48 | 120 | 0.1614%0.6790% | 330 | 270 | |||
| Eu | 124 | 0.54 | 279 | 271 | ||||
| 48 | 12.2 | 0.63 | ||||||
| Tb | 80 | 4 to 6.3 | 371 | 272 | ||||
| Ag-NPs | Eu | 16 | 22 | 21 | 0.37 | 343 | 274 | |
| Eu | 16; 104 | 0.62 | 21 | 343 | 275 | |||
| Sm | 2; 18; 104 | 3.8 | 427, 441, 463 | 276 | ||||
| Dy | 5.9 | |||||||
| Eu | 2; 129 | 2 to 6 | 277 | |||||
| Tb | 6.5 and 23 | 3 to 26 | ||||||
Jingli Yuan's team designed NPs with magnetic and optical properties by combining Fe3O4 NPs and [Eu(117)] complex.254 The NPs were firstly coated with poly(vinylpyrrolidone) (PVP) to stabilize the Fe-NPs before [Eu(117)] copolymerization with free 3-aminopropyltriethoxysilane (APS), tetraethyl orthosilicate and Fe-NPs which led to a covalent bond between Eu-complexes and the NPs thanks to the presence of APS. With a S/N ratio 4 times higher when using time-resolved fluorescence imaging than steady-state fluorescence imaging and a SM of 2.5 emu g−1 which is enough for bio-separation,255 it has been proposed as a probe for biological labelling, detection and cell separation. A few years after, Yang and his team coated two [Tb(118)(119)] complexes on the surface of Fe3O4 NPs.256 Functionalized NPs were used as luminescent probe for targeted HeLa cells imaging thanks to the affinity between 119 and the folate receptor of the cells, and would be able to provide contrast in MRI. Another luminomagnetic system was proposed by Ni et al., composed of Fe3O4 NPs functionalized with smaller NPs containing [Eu(17)3(18)]Cl3 complexes (Fig. 16), both NPs owning a protective silica shell.257 These functionalized Fe-NPs with an average size of 570 nm can be used in optical imaging as well as for effective magnetic separation. In 2014, Malta and co-workers chose a different path to functionalize the Fe-NPs by using calixarene-Ln(III) complexes.258 Thanks to the one-pot method, [Eu(16)] and [Tb(27)] complexes were successfully attached to the Fe-NPs with a calixarene as intermediate (Fig. 17) for the stabilization of the NPs. The efficiency obtained with this system was lower than the complex i.e. 14% and 29% for the Fe3O4@calix-Eu(16) and the free [Eu(16)3(H2O)2] complex, respectively. Two years later, Ribeiro and his team chose to work with other SPIO NPs, Fe2O3, for designing their bifunctional magnetic luminescent particles composed of [Eu(16)(98)] complexes grafted on the surface of the silica shell of the NPs.259
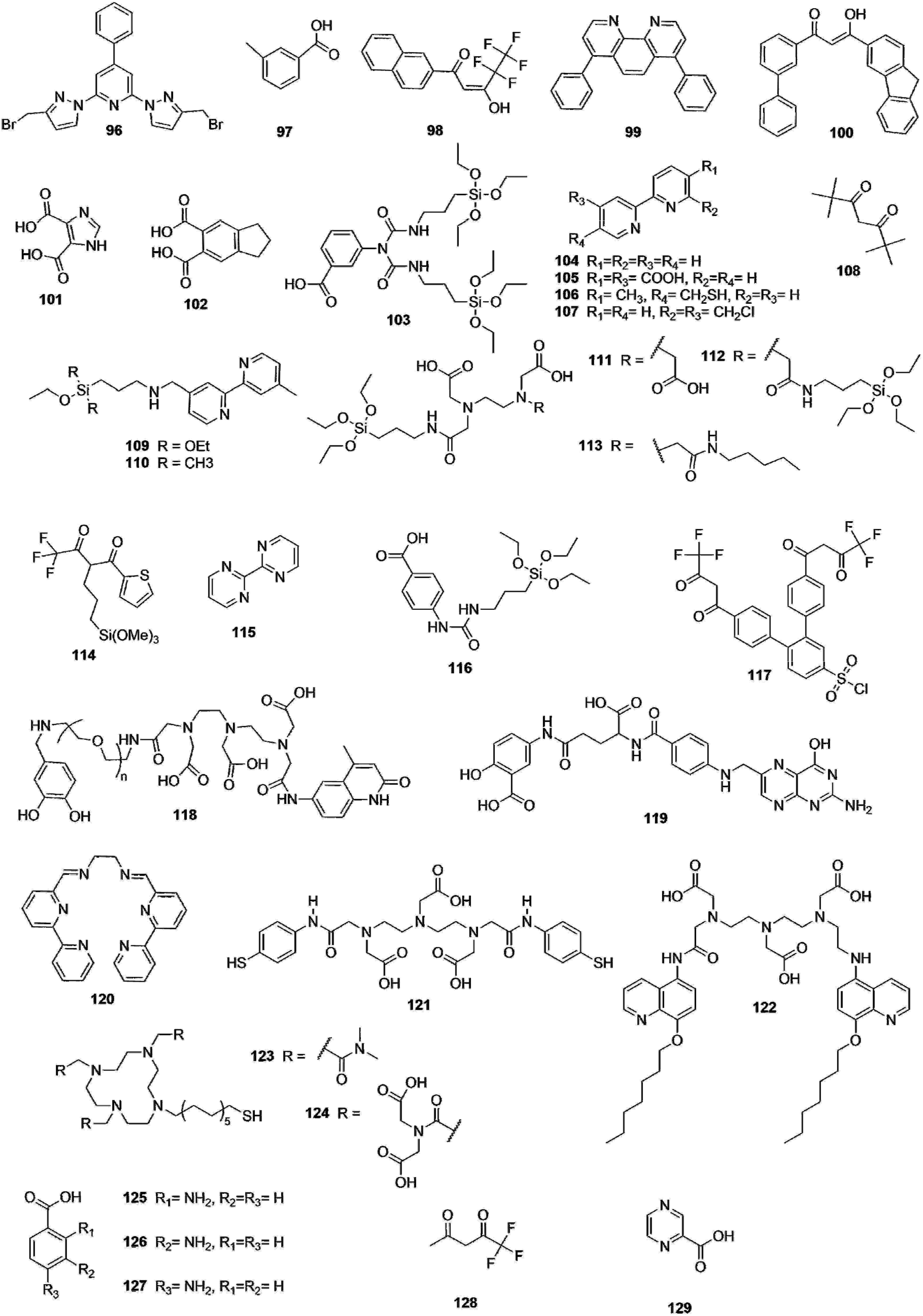 | ||
| Fig. 15 Ligands used to form the Ln complexes and functionalize the NPs, part 2. | ||
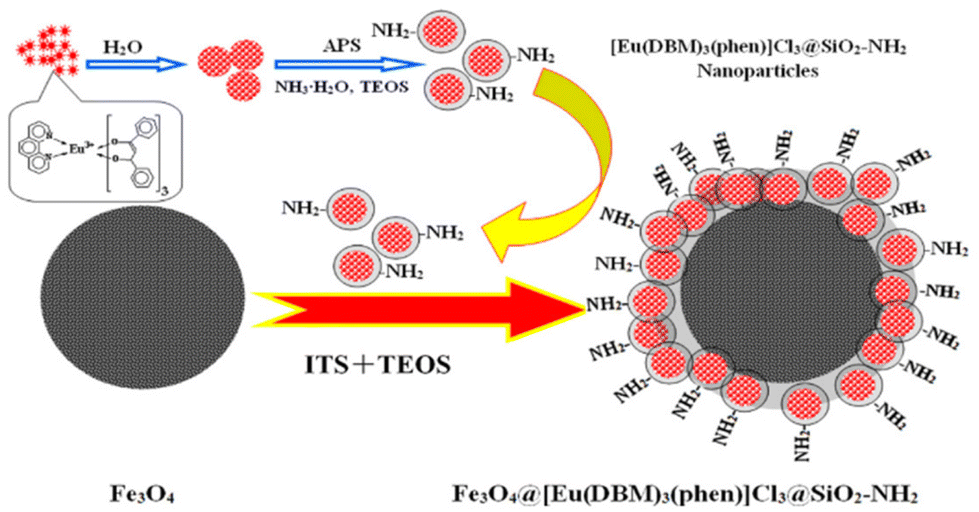 | ||
| Fig. 16 Preparation of a functionalized Fe-NPs with Ln-complexes containing NPs Reprinted from ref. 257, Copyright (2013), with permission from Elsevier. | ||
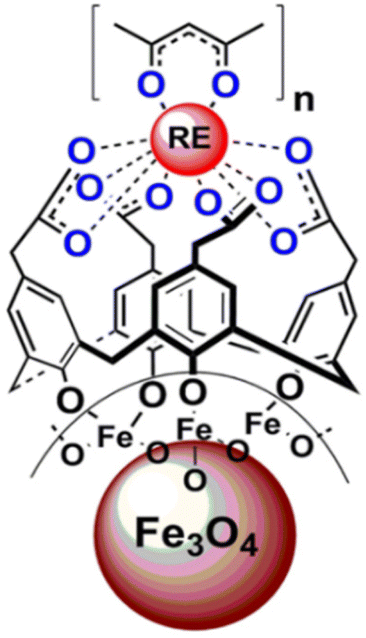 | ||
| Fig. 17 Structure of Fe3O4@calix-Ln(β-diketonate). Reprinted with permission from ref. 258. Copyright (2014) American Chemical Society. | ||
By comparison with the original complex, the luminescence lifetime decreased from 0.60 ms to 0.35 ms which might be due to some defects on the SPIO NPs, and the SM decreased from 60 emu g−1 to 0.12 emu g−1. In 2019, Carlos Geraldes and his team proposed a system with maghemite covered by a silica shell and impregnated with [Eu(25)3(H2O)2], acting as a luminescent probe,260 exhibiting enhancement of the quantum efficiency from 0.19 (free complex) to 0.37 for the NP. [Gd(25)3((H2O)2] was also added, affording a bimodal system able to act as a MRI contrast agent in addition to its luminescent role. The following year, Stefan Lis and his team proposed a new system with the expectation of improving the bifunctionality (magnetism and luminescence), that Fe-NPs and Ln-complexes can provide.261 They prepared Fe3O4@silica core–shell NPs in which [Ln(120)] (Ln = Tb or Eu) are linked to the NPs by the intermediary of terephtalic acid. Unfortunately, a decrease of both luminescence intensity and lifetime of the system is observed in comparison to the respective Ln-complexes at R.T and at 77 K, explained by the low content of chain complexes and by the high absorption of magnetic cores. Recently, new luminescent-magnetic architectures were developed by covering magnetite NPs with biocompatible chitosan (CS) and glutathione (GSH) functionalized with either [Eu(16)3(H2O)2] or [Tb(27)3(H2O)3], leading to Fe3O4-CS-Eu/Tb and Fe3O4-GSH-Eu/Tb, with promising bioapplications.262 Quantum emission efficiencies of 29% and 13% and lifetimes of 0.28 ms and 0.2 ms were obtained for the Fe3O4–CS–Eu and Fe3O4–GSH–Eu NPs, respectively. The values of the former are close to the free Eu-complex (29% and 0.26 ms) and a decrease is observed with those of the latter. These results can be related to the number of water molecules found in the first coordination sphere of the Eu atoms which are of 2.5 for Fe3O4–CS–Eu and 5 for Fe3O4–GSH–Eu.
The first examples were reported in 2006, when Thomas and his team coated the surface of the Au-NPs with [Ln(106)3] (Ln = Eu(III) or Tb(III)).265 The phosphorescent nanomaterial was proposed as sensor for biologically important cations such as Ni(II), Ca(II), Mg(II) and others. Different metal ions were added to solutions of functionalized NPs, which may cause a replacement of Ln(III) by the metal ion depending on its affinity for the ligand, leading to a decrease of Ln photoluminescence intensity. The presence of transition metal ions has been shown to induce a higher decrease of the luminescence intensity compared to alkaline earth metals. In parallel, Pikramenou's team designed a nanomaterial composed of Au-NPs coated with [Eu(121)] complexes.266 The ligand conferred a strong affinity with the surface of Au-NPs thanks to the presence of thiol functions, known to be poorly coordinating towards Ln atoms.267 A luminescent pH probe for human platelets was then designed, composed of Au-NPs coated by [Eu(122)] complexes and by peptides.268 The Eu related luminescent properties of the NPs make them suitable optical probes for imaging in cells whereas the peptides allow the NPs to cross the cell membrane. By the intermediary of the peptide, the nano-objects associate with the cell membrane at a pH of 7.4, and upon reduction of the pH (≤6.5), the NPs enter the cell membrane. This pH-mediated internalization is triggered by the pH-dependent peptide translocation across the membrane. [Eu(122)] complex was chosen for its neutrality, its good stability, and its inertness towards the peptide. The NPs luminescence was monitored in platelets and displayed luminescence lifetimes of 288 μs and 277 μs in spread and fixed platelets respectively, which are close to the lifetimes of the NPs in solution (290 μs) and confirmed the presence of the NPs in the cells. In 2008, Gunnlaugsson and co-workers presented their own Ln-complex functionalized Au-NPs as a probe for luminescent sensing of biological substrates.269 [Eu(123)] functionalized Au-NPs were prepared from DMAP-Au-NPs by exchange of DMAP (4-(dimethylamino)-pyridine) with Eu-chelate. The NPs exhibited no luminescence when the ligand was excited but after the addition of ligand 48, this antenna sensitized the Eu excited state, leading to the observation of red emission. The interaction of this system with Bovine Serum Albumin (BSA) was also studied, resulting in the quenching of the Eu(III) luminescence, explained by the strong interaction between BSA and ligand 48.270 With the same sensitizer, [Eu(124)] coated Au-NPs were also proposed for analysis of the structure of microdamaged bovine bones.271 In 2018, Werts and co-workers used the system of [Tb(80)] labelled streptavidin (mentioned in QDs part) to coat the surface of biotinylated Au-NPs for nanosurface energy transfer,272 an energy-transfer mechanism measuring (bio)molecular interactions at longer distances than FRET.273
Ag colloidal NPs exhibited a luminescence as intense as the free Eu-complex with similar lifetimes and quantum yield (0.386 ms and 23.1% for the complex and 0.368 ms and 21% for the Ag colloidal NPs) due to the low energy transfer between the Eu-complex and the metal, leading to a weak quenching effect, caused by the aggregation of the complexes at the surface of the Ag-NPs which induced a red-shift of the absorption band (J-aggregation).278,279 The ligand 104 was then employed in addition to 16 as chelator in Ln complexes to substitute the water molecules.275 In addition of [Eu(16)3104], [Tb(16)3104] was used to stabilize the nanocomposite as it helps reducing the quenching effect. An enhancement of 21% for the luminescence emission intensity of the [Eu(16)3104] complex was then obtained compared to the one of the complex itself. Following their steps, Chu and co-workers studied the effect of the presence of the silica shell between Ln complexes and Ag-NP surface on Ln luminescence.276 Three different core–shell Ag@SiO2 NPs were prepared with varying silica shell thicknesses (10, 25 and 80 nm) and with Ln complexes attached at the surface of the NPs. Sm(III) and Dy(III) complexes were chosen with 2, 18 and 104 as photosensitizing ligands. All the compounds exhibited improved photoluminescence properties than the free Ln complexes with a 5.9-fold and 3.8-fold enhancement of emission intensity for the Dy(III) and Sm(III) complexes coated Ag-NPs, respectively. The impact of the position of substituents in aromatic ligands on the luminescence of Sm(III) and Dy(III) complexes functionalized Ag@SiO2 NPs was also studied with 125 (–NH2 in ortho position), 126 (–NH2 in meta position), and 127 (–NH2 in para position) as chelators.280 A stronger emission was observed with amino group in meta position (ligand 126) as the positive conjugation effect of amino group is weaker.
In 2014, Yan and colleagues proposed a new approach to obtain a white emitting soft material using [Eu(16)4], [Sm(17)4] or [Eu(128)4] complexes linked to Ag-NPs thanks to a thiol-functionalized ionic liquid (SHIL).281 By changing the number of complexes bound to Ag-NPs, the luminescence colour of the soft hybrid material was adjusted thanks to the band gap differences between the Ln(III) complexes and the NPs. Among the different systems, only Ag-SHIL-[Eu(128)4]3, Ag-SHIL-[Sm(17)4]3 and Ag-SHIL-[Eu(16)4]3 systems displayed white luminescence.
To the best of our knowledge, the last report was made by Zhao's team in 2020,277 where the different factors impacting the metal-enhanced luminescence of NPs were investigated. For this purpose, Ag-NPs were synthesized with different thickness of the silica shell upon which [Eu(2)3], [Eu(129)3], [Tb(2)3] and [Tb(129)3] complexes were separately adsorbed. Excitation wavelength has been considered as a key factor with a luminescent enhancement from 1.55 to 14.97 times when changing the excitation wavelength from 255 to 380 nm between the nanocomposite and the free complex. The silica shell thickness was also considered as a key factor as the distance between the complex and the core of the NPs influences the luminescence of Ln. The best quantum yield (23%) was obtained with a thickness of 24 nm compared to the free complex (3.1%) and a shell with nearly the double of the thickness (6.5%).
In this section, the functionalization of different NPs by Ln(III) complexes have been overviewed. Those new nanomaterials were useful for multiple applications such as imaging and sensing, and led to increased luminescence properties (lifetimes, emission intensity) of the complexes compared to the free ones, particularly in the case of the silica NPs, Au-NPs and Ag-NPs. Limitations related to the FRET distance were overcome with these types of NPs by reaching a distance up to 14 nm with the use of a dye as a bridge.216 However, this improvement comes with a limited FRET efficiency (less than 20%). Bifunctional nano-objects were also designed with Fe-NPs, making possible to combine magnetic and luminescence properties. However, to the best of our knowledge, no Gd based NPs with Ln-complex functionalized surface that could combine photoluminescence and MRI properties has been described up to date. Such a luminescence/MRI bimodal probe would be of great interest,282 especially with NIR-light absorbing and emitting Ln complexes.
6. Outlook and conclusion
Bringing the antenna effect of photosensitizing ligands to the level of Ln-NPs has created a new playground for chemists and physicists with extremely bright nano-objects. The complementary advantages of long-lived excited states and spectral signatures of each Ln ions further afford highly sensitive luminescence detection and the capacity of multiplexed detection with numerous (bio)analytical applications.A direct comparison of the performances between the different types of NPs and even in the same category is quite difficult as few studies present a full spectroscopic characterization (excited-state lifetime, quantum yield and brightness). However, it can be highlighted that each type of NPs has its advantages and limitations that make them the most suitable depending on the application. For example, by comparing the higher Tb lifetimes of each category of NPs, Ln-doped NPs with surface capping antennas exhibit the highest Tb lifetimes (up to 8.3 ms – quite close to the radiative lifetime283 – vs. 2.06 ms for Ln-complex encapsulated NPs and 1.66 ms for Ln-complex surface functionalized NPs). Embedding the atoms in the core of the NPs clearly affords a better Ln protection from deactivation caused by the environment, the latter being still important for the encapsulated or grafted complexes. However, (bio)functionalization remains a limiting factor for analytical applications as numerous/complex steps of organic synthesis are required to form functionalized antennas, when surface with active function can be more easily obtained for polymer or silica NPs. Finally, surface decoration of NPs with Ln complexes has a unique advantage of offering bifunctional detections by the combination of luminescent properties of Ln complexes with the specific properties of the NPs (such as surface plasmon resonance for Ag-NPs, magnetism for Fe-NPs) or to present multiplexed optical detection and FRET possibilities with QDs. However, in this case, Ln complexes are not protected from the environment and consequently are more impacted by solvent related deactivations like free complexes, potentially leading to lower spectroscopic performances compared with the first two types of NPs.
As seen in the last three sections, most of the light harvesting range of dye-sensitized NPs is in the UV-Vis zone, which might provide biocompatibility issues. The development of NIR-excited Ln containing NPs would be of particular importance to comply with the biological window and allow non-invasive study of biological specimens. Several strategies, already illustrated in few studies but demanding deeper development, can be considered: the use of NIR-light absorbing antennas in combination with NIR absorbing Ln (such as Yb or Er) either for upconversion or downshifting, or two-photon excitation antennas excited in the NIR range.
Considering the relative youth of the field, it is surmised that new systems will be developed to better tune the up and down energy transfer processes, with surface ligands playing the rule of energy bridges between the core NPs and the external surroundings,284 or with new spectroscopic properties arising from the interaction of the NPs with chemical entities at the surface as observed for the triplet excitons enhancement occurring for some organic dye interacting with Ln-NPs.285
The maturation stage of these new tools will probably soon open them the doors of commercial applications. In particular, the bio-analytical fields are already tackling the use of Ln-NPs for in vitro applications such as lateral flow immunoassays,286 ELISA like detection,287 or luminescence microscopy.288 However, the high sensitivity obtained with Ln-NPs is based on the development of dedicated instruments, working with a time-delayed detection mode, that will require dedicated time-resolved readers to unravel the best of the sensitivity of these nano-objects. Additionally, if few reports studied their long term chemical or colloidal stabilities,46,47 and, if applications have already been found in in vitro and in cellulo diagnostics, numerous questions remain to be answered concerning the biocompatibility and toxicity of these nano-objects before any eventual long term in vivo applications could be safely envisaged.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the French National Research Agency (ANR) through the contract number ANR-11-LABX-0058_NIE within the investissement d'Avenir program ANR-10-IDEX-0002-02 and through the project ANR-21-CE19-0016-01. The French Ministère de l'enseignement Supérieur, de la Recherche et des Technologies is gratefully acknowledged for financial support (LKS).References
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- C. Doffek and M. Seitz, Angew. Chem., Int. Ed., 2015, 54, 9719–9721 CrossRef CAS PubMed.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, D. Parker, J. W. Walton and J. M. Zwier, Dalton Trans., 2015, 44, 4791–4803 RSC.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- N. Sabbatini, S. Perathoner, V. Balzani, B. Alpha and J.-M. Lehn, in Supramolecular Photochemistry, ed. V. Balzani, Springer Netherlands, Dordrecht, 1987, pp. 187–206 Search PubMed.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750–1756 RSC.
- N. Weibel, L. J. Charbonnière, M. Guardigli, A. Roda and R. Ziessel, J. Am. Chem. Soc., 2004, 126, 4888–4896 CrossRef CAS PubMed.
- J. Xu, T. M. Corneillie, E. G. Moore, G.-L. Law, N. G. Butlin and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 19900–19910 CrossRef CAS PubMed.
- P. Reiss, M. Protière and L. Li, Small, 2009, 5, 154–168 CrossRef CAS PubMed.
- N. Souri, P. Tian, C. Platas-Iglesias, K.-L. Wong, A. Nonat and L. J. Charbonnière, J. Am. Chem. Soc., 2017, 139, 1456–1459 CrossRef CAS.
- L. Charbonnière, S. Mameri, P. Kadjane, C. Platas-Iglesias and R. Ziessel, Inorg. Chem., 2008, 47, 3748–3762 CrossRef.
- A. J. Freeman and R. E. Watson, Phys. Rev., 1962, 127, 2058–2075 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- J. Goetz, A. Nonat, A. Diallo, M. Sy, I. Sera, A. Lecointre, C. Lefevre, C. F. Chan, K.-L. Wong and L. J. Charbonnière, ChemPlusChem, 2016, 81, 526–534 CrossRef CAS PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- R. M. Supkowski and W. DeW. Horrocks, Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- T. Kimura and Y. Kato, J. Alloys Compd., 1998, 271–273, 867–871 CrossRef CAS.
- T. Kimura and Y. Kato, J. Alloys Compd., 1995, 225, 284–287 CrossRef CAS.
- C. Doffek, N. Alzakhem, C. Bischof, J. Wahsner, T. Güden-Silber, J. Lügger, C. Platas-Iglesias and M. Seitz, J. Am. Chem. Soc., 2012, 134, 16413–16423 CrossRef CAS PubMed.
- C. Homann, L. Krukewitt, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and M. Haase, Angew. Chem., Int. Ed., 2018, 57, 8765–8769 CrossRef CAS PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- Q. Ju, Y. Liu, D. Tu, H. Zhu, R. Li and X. Chen, Chem. – Eur. J., 2011, 17, 8549–8554 CrossRef CAS PubMed.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- M. Irfanullah, N. Bhardwaj and A. Chowdhury, Dalton Trans., 2016, 45, 12483–12495 RSC.
- Y. Zhong and H. Dai, Nano Res., 2020, 13, 1281–1294 CrossRef CAS PubMed.
- A. Lay, C. Siefe, S. Fischer, R. D. Mehlenbacher, F. Ke, W. L. Mao, A. P. Alivisatos, M. B. Goodman and J. A. Dionne, Nano Lett., 2018, 18, 4454–4459 CrossRef CAS PubMed.
- Z. Zhuo, Y. Liu, D. Liu, P. Huang, F. Jiang, X. Chen and M. Hong, Chem. Sci., 2017, 8, 5050–5056 RSC.
- X. Cheng, D. Tu, W. Zheng and X. Chen, Chem. Commun., 2020, 56, 15118–15132 RSC.
- F. Auzel, G. Baldacchini, L. Laversenne and G. Boulon, Opt. Mater., 2003, 24, 103–109 CrossRef CAS.
- Z. Liu, Q. Meng, H. Liu, C. Yao, Q. Meng, W. Liu and W. Wang, Opt. Mater., 2013, 36, 384–389 CrossRef CAS.
- P. A. Tanner, L. Zhou, C. Duan and K.-L. Wong, Chem. Soc. Rev., 2018, 47, 5234–5265 RSC.
- M. W. Mara, D. S. Tatum, A.-M. March, G. Doumy, E. G. Moore and K. N. Raymond, J. Am. Chem. Soc., 2019, 141, 11071–11081 CrossRef CAS PubMed.
- N. Hildebrandt and I. L. Medintz, FRET - Förster Resonance Energy Transfer: From Theory to Applications, Wiley-VCH Verlag GmbH & Co. KGaA, First edition., 2014 Search PubMed.
- S. Bhuckory, E. Hemmer, Y.-T. Wu, A. Yahia-Ammar, F. Vetrone and N. Hildebrandt, Eur. J. Inorg. Chem., 2017, 2017, 5186–5195 CrossRef CAS.
- K. D. Wegner, Z. Jin, S. Lindén, T. L. Jennings and N. Hildebrandt, ACS Nano, 2013, 7, 7411–7419 CrossRef CAS PubMed.
- E. Debroye and T. N. Parac-Vogt, Chem. Soc. Rev., 2014, 43, 8178–8192 RSC.
- A. M. Nonat and L. J. Charbonnière, Coord. Chem. Rev., 2020, 409, 213192 CrossRef CAS.
- S. Faulkner and S. J. A. Pope, J. Am. Chem. Soc., 2003, 125, 10526–10527 CrossRef CAS PubMed.
- M. Sy, D. Esteban-Gómez, C. Platas-Iglesias, A. Rodríguez-Rodríguez, R. Tripier and L. J. Charbonnière, Eur. J. Inorg. Chem., 2017, 2017, 2122–2129 CrossRef CAS.
- C. Chen, L. Ao, Y. T. Wu, V. Cifliku, M. Cardoso Dos Santos, E. Bourrier, M. Delbianco, D. Parker, J. M. Zwier, L. Huang and N. Hildebrandt, Angew. Chem., Int. Ed., 2018, 57, 13686–13690 CrossRef CAS.
- X. Liu, W. Wu, D. Cui, X. Chen and W. Li, Adv. Mater., 2021, 33, 2004734 CrossRef CAS PubMed.
- H. Xing, W. Bu, S. Zhang, X. Zheng, M. Li, F. Chen, Q. He, L. Zhou, W. Peng, Y. Hua and J. Shi, Biomaterials, 2012, 33, 1079–1089 CrossRef CAS PubMed.
- E. Fröhlich, J. Nanobiotechnol., 2017, 15, 84 CrossRef.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561–1584 RSC.
- V. Mittelheisser, P. Coliat, E. Moeglin, L. Goepp, J. G. Goetz, L. J. Charbonnière, X. Pivot and A. Detappe, Adv. Mater., 2022, e2110305 CrossRef.
- O. Dukhno, F. Przybilla, V. Muhr, M. Buchner, T. Hirsch and Y. Mély, Nanoscale, 2018, 10, 15904–15910 RSC.
- S. Lahtinen, A. Lyytikäinen, H. Päkkilä, E. Hömppi, N. Perälä, M. Lastusaari and T. Soukka, J. Phys. Chem. C, 2017, 121, 656–665 CrossRef CAS.
- W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhu and X. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- S. Heer, K. Kömpe, H.-U. Güdel and M. Haase, Adv. Mater., 2004, 16, 2102–2105 CrossRef CAS.
- K. W. Krämer, D. Biner, G. Frei, H. U. Güdel, M. P. Hehlen and S. R. Lüthi, Chem. Mater., 2004, 16, 1244–1251 CrossRef.
- M. Quintanilla, E. Hemmer, J. Marques-Hueso, S. Rohani, G. Lucchini, M. Wang, R. R. Zamani, V. Roddatis, A. Speghini, B. S. Richards and F. Vetrone, Nanoscale, 2022, 14, 1492–1504 RSC.
- L. J. Charbonnière, J.-L. Rehspringer, R. Ziessel and Y. Zimmermann, New J. Chem., 2008, 32, 1055–1059 RSC.
- B. Kokuoz, C. Kucera, J. R. DiMaio and J. Ballato, Opt. Mater., 2009, 31, 1327–1330 CrossRef CAS.
- N. Chen, Y. He and G. Du, Mater. Sci. Semicond. Process., 2014, 24, 62–67 CrossRef CAS.
- Y. Yang, B. Liu, L. Tang and N. Chen, Mater. Sci. Semicond. Process., 2015, 30, 513–517 CrossRef CAS.
- J. Wang, W. Wang, L. Zhang, H. Yao and Z. Li, Mater. Lett., 2013, 106, 281–283 CrossRef CAS.
- B. Kokuoz, J. Dimaio, C. Kucera, J. Evanoff David and J. Ballato, J. Am. Chem. Soc., 2008, 130, 12222–12223 CrossRef CAS.
- A. M. Cross, P. S. May, F. C. J. M. van Veggel and M. T. Berry, J. Phys. Chem. C, 2010, 114, 14740–14747 CrossRef CAS.
- S. W. Li, H. J. Ren and S. G. Ju, J. Nanosci. Nanotechnol., 2014, 14, 3677–3682 CrossRef CAS PubMed.
- S. Li, X. Zhang, Z. Hou, Z. Cheng, P. Ma and J. Lin, Nanoscale, 2012, 4, 5619–5626 RSC.
- S. Li, X. Li, Y. Jiang, Z. Hou, Z. Cheng, P. Ma, C. Li and J. Lin, RSC Adv., 2014, 4, 55100–55107 RSC.
- D. Ghosh and M. N. Luwang, RSC Adv., 2015, 5, 10468–10478 RSC.
- V. Yu. Khudoleeva, V. V. Utochnikova, A. S. Kalyakina, I. M. Deygen, A. A. Shiryaev, Ł. Marciniak, V. A. Lebedev, I. V. Roslyakov, A. V. Garshev, L. S. Lepnev, U. Schepers, S. Bräse and N. P. Kuzmina, Dyes Pigm., 2017, 143, 348–355 CrossRef CAS.
- S. Janssens, G. V. M. Williams and D. Clarke, J. Appl. Phys., 2011, 109, 023506 CrossRef.
- S. Janssens, G. Williams, D. Clarke and M. Sharp, Basic Solid State Phys., 2011, 248, 439–443 CrossRef CAS.
- X. Wang, Y. Yang, N. Chen, B. Liu and G. Liu, J. Nanosci. Nanotechnol., 2016, 16, 3729–3734 CrossRef CAS PubMed.
- X. Wang, Y. Yang, G. Du, N. Chen and A. Zhang, J. Nanosci. Nanotechnol., 2018, 18, 1876–1881 CrossRef CAS PubMed.
- A. Safronikhin, H. Ehrlich, N. Kuzmina and G. Lisichkin, Appl. Surf. Sci., 2014, 307, 482–488 CrossRef CAS.
- M. Irfanullah, N. Bhardwaj and A. Chowdhury, Dalton Trans., 2016, 45, 12483–12495 RSC.
- M. Irfanullah, D. K. Sharma, R. Chulliyil and A. Chowdhury, Dalton Trans., 2015, 44, 3082–3091 RSC.
- V. N. K. B. Adusumalli, L. Mrówczyńska, D. Kwiatek, Ł. Piosik, A. Lesicki and S. Lis, ChemMedChem, 2021, 16, 1640–1650 CrossRef CAS PubMed.
- C. Charpentier, V. Cifliku, J. Goetz, A. Nonat, C. Cheignon, M. Cardoso Dos Santos, L. Francés-Soriano, K.-L. Wong, L. J. Charbonnière and N. Hildebrandt, Chem. – Eur. J., 2020, 26, 14602–14611 CrossRef CAS PubMed.
- C. Cheignon, M. Heurté, R. C. Knighton, A. A. Kassir, A. Lecointre, A. Nonat, A. Boos, C. Christine, Z. Asfari and L. J. Charbonnière, Eur. J. Inorg. Chem., 2021, 2021, 3761–3770 CrossRef CAS.
- J. Wang, Z. Wang, X. Li, S. Wang, H. Mao and Z. Li, Appl. Surf. Sci., 2011, 257, 7145–7149 CrossRef CAS.
- L. Song and L. Xue, Appl. Surf. Sci., 2012, 258, 3497–3501 CrossRef CAS.
- L. Song, J. Gao and J. Li, J. Lumin., 2014, 151, 18–21 CrossRef CAS.
- V. N. K. B. Adusumalli, H. V. S. R. M. Koppisetti, N. Madhukar and V. Mahalingam, Microchim. Acta, 2019, 186, 389 CrossRef PubMed.
- N. Gauthier, O. Raccurt, D. Imbert and M. Mazzanti, J. Nanopart. Res., 2013, 15, 1723 CrossRef.
- T. Samanta, S. K. Jana, A. E. Praveen and V. Mahalingam, Dalton Trans., 2017, 46, 9646–9653 RSC.
- P. Agbo, T. Xu, M. Sturzbecher-Hoehne and R. J. Abergel, ACS Photonics, 2016, 3, 547–552 CrossRef CAS.
- P. Agbo and R. J. Abergel, Inorg. Chem., 2016, 55, 9973–9980 CrossRef CAS PubMed.
- Y. Song, B. Shao, Y. Feng, W. Lü, G. Liu and H. You, Dalton Trans., 2016, 45, 9468–9476 RSC.
- Q. Dai, M. E. Foley, C. J. Breshike, A. Lita and G. F. Strouse, J. Am. Chem. Soc., 2011, 133, 15475–15486 CrossRef CAS PubMed.
- L. Stagi, D. Chiriu, A. Ardu, C. Cannas, C. M. Carbonaro and P. C. Ricci, J. Appl. Phys., 2015, 118, 125502 CrossRef.
- L. Ji, N. Chen, G. Du, M. Yan and W. Shi, Ceram. Int., 2014, 40, 3117–3122 CrossRef CAS.
- N. Chen, L. Ji and G. Du, J. Lumin., 2014, 153, 259–263 CrossRef CAS.
- U. Balderas, S. Carmona, L. Mariscal, I. Martínez and C. Falcony, Chem. Phys., 2018, 511, 1–6 CrossRef CAS.
- J. Chen, Q. Meng, P. S. May, M. T. Berry and C. Lin, J. Phys. Chem. C, 2013, 117, 5953–5962 CrossRef CAS.
- L. Tang, W. Gui, K. Ding, N. Chen and G. Du, J. Alloys Compd., 2014, 590, 277–282 CrossRef CAS.
- Y. He, N. Chen and G. Du, J. Am. Ceram. Soc., 2014, 97, 1931–1936 CrossRef CAS.
- B. G. Vats, M. Shafeeq, P. Singhal and S. Neogy, ChemistrySelect, 2018, 3, 4930–4938 CrossRef CAS.
- R. R. Gonçalves, Y. Messaddeq, M. A. Aegerter and S. J. L. Ribeiro, J. Nanosci. Nanotechnol., 2011, 11, 2433–2439 CrossRef PubMed.
- J.-S. Kang, Y.-K. Jeong, J.-G. Kang, L. Zhao, Y. Sohn, D. Pradhan and K. T. Leung, J. Phys. Chem. C, 2015, 119, 2142–2147 CrossRef CAS.
- F. Artizzu, D. Loche, D. Mara, L. Malfatti, A. Serpe, R. V. Deun and M. F. Casula, J. Mater. Chem. C, 2018, 6, 7479–7486 RSC.
- J. Huang and K. Pu, Chem. Sci., 2021, 12, 3379–3392 RSC.
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
- T. Fix, A. Nonat, D. Imbert, S. Di Pietro, M. Mazzanti, A. Slaoui and L. J. Charbonnière, Prog. Photovolt. Res. Appl., 2016, 24, 1251–1260 CrossRef CAS.
- J. Zhang, C. M. Shade, D. A. Chengelis and S. Petoud, J. Am. Chem. Soc., 2007, 129, 14834–14835 CrossRef CAS PubMed.
- H. Lu, Y. Peng, H. Ye, X. Cui, J. Hu, H. Gu, A. N. Khlobystov, M. A. Green, P. J. Blower, P. B. Wyatt, W. P. Gillin and I. Hernández, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- J. Liu, A. M. Kaczmarek, F. Artizzu and R. Van Deun, ACS Photonics, 2019, 6, 659–666 CrossRef CAS.
- V. V. Utochnikova, A. S. Kalyakina, L. S. Lepnev and N. P. Kuzmina, J. Lumin., 2016, 170, 633–640 CrossRef CAS.
- V. Y. Khudoleeva, V. V. Utochnikova, A. S. Goloveshkin, Ł. Marciniak, A. V. Knotko, L. S. Lepnev and N. P. Kuzmina, Dyes Pigm., 2019, 160, 890–897 CrossRef CAS.
- S. Wilhelm, ACS Nano, 2017, 11, 10644–10653 CrossRef CAS.
- J. Lee, B. Yoo, H. Lee, G. D. Cha, H.-S. Lee, Y. Cho, S. Y. Kim, H. Seo, W. Lee, D. Son, M. Kang, H. M. Kim, Y. I. Park, T. Hyeon and D.-H. Kim, Adv. Mater., 2017, 29, 1603169 CrossRef.
- K. L. Reddy, R. Balaji, A. Kumar and V. Krishnan, Small, 2018, 14, 1801304 CrossRef PubMed.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- D. J. Garfield, N. J. Borys, S. M. Hamed, N. A. Torquato, C. A. Tajon, B. Tian, B. Shevitski, E. S. Barnard, Y. D. Suh, S. Aloni, J. B. Neaton, E. M. Chan, B. E. Cohen and P. J. Schuck, Nat. Photonics, 2018, 12, 402–407 CrossRef CAS.
- X. Wu, H. Lee, O. Bilsel, Y. Zhang, Z. Li, T. Chen, Y. Liu, C. Duan, J. Shen, A. Punjabi and G. Han, Nanoscale, 2015, 7, 18424–18428 RSC.
- X. Wu, Y. Zhang, K. Takle, O. Bilsel, Z. Li, H. Lee, Z. Zhang, D. Li, W. Fan, C. Duan, E. M. Chan, C. Lois, Y. Xiang and G. Han, ACS Nano, 2016, 10, 1060–1066 CrossRef CAS PubMed.
- D. Yin, Y. Liu, J. Tang, F. Zhao, Z. Chen, T. Zhang, X. Zhang, N. Chang, C. Wu, D. Chen and M. Wu, Dalton Trans., 2016, 45, 13392–13398 RSC.
- G. Chen, J. Damasco, H. Qiu, W. Shao, T. Y. Ohulchanskyy, R. R. Valiev, X. Wu, G. Han, Y. Wang, C. Yang, H. Ågren and P. N. Prasad, Nano Lett., 2015, 15, 7400–7407 CrossRef CAS PubMed.
- W. Wei, G. Chen, A. Baev, G. S. He, W. Shao, J. Damasco and P. N. Prasad, J. Am. Chem. Soc., 2016, 138, 15130–15133 CrossRef CAS PubMed.
- G. Chen, W. Shao, R. R. Valiev, T. Y. Ohulchanskyy, G. S. He, H. Ågren and P. N. Prasad, Adv. Opt. Mater., 2016, 4, 1760–1766 CrossRef CAS.
- Q. Shao, X. Li, P. Hua, G. Zhang, Y. Dong and J. Jiang, J. Colloid Interface Sci., 2017, 486, 121–127 CrossRef CAS PubMed.
- W. Shao, G. Chen, A. Kuzmin, H. L. Kutscher, A. Pliss, T. Y. Ohulchanskyy and P. N. Prasad, J. Am. Chem. Soc., 2016, 138, 16192–16195 CrossRef CAS PubMed.
- J. Xu, M. Sun, Y. Kuang, H. Bi, B. Liu, D. Yang, R. Lv, S. Gai, F. He and P. Yang, Dalton Trans., 2017, 46, 1495–1501 RSC.
- Y. Wei, S. Liu, C. Pan, Z. Yang, Y. Liu, J. Yong and L. Quan, Int. J. Nanomed., 2020, 15, 1409–1420 CrossRef CAS PubMed.
- W. Bi, Y. Wu, C. Chen, D. Zhou, Z. Song, D. Li, G. Chen, Q. Dai, Y. Zhu and H. Song, ACS Appl. Mater. Interfaces, 2020, 12, 24737–24746 CrossRef CAS PubMed.
- X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2012, 42, 173–201 RSC.
- S. Li, Z. Hou, Z. Cheng, H. Lian, P. Ma, C. Li and J. Lin, RSC Adv., 2013, 3, 5491–5497 RSC.
- Z. Wang and A. Meijerink, J. Phys. Chem. Lett., 2018, 9, 1522–1526 CrossRef CAS PubMed.
- W. Shao, C.-K. Lim, Q. Li, M. T. Swihart and P. N. Prasad, Nano Lett., 2018, 18, 4922–4926 CrossRef CAS.
- P. Agbo, J. S. Kanady and R. J. Abergel, Front. Chem., 2020, 8, 579942 CrossRef CAS.
- Z. Ye, M. Tan, G. Wang and J. Yuan, Anal. Chem., 2004, 76, 513–518 CrossRef CAS PubMed.
- H. Härmä, T. Soukka and T. Lövgren, Clin. Chem., 2001, 47, 561–568 CrossRef.
- J. Desbiens, B. Bergeron, M. Patry and A. M. Ritcey, J. Colloid Interface Sci., 2012, 376, 12–19 CrossRef CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS.
- A.-I. Moreno-Vega, T. Gómez-Quintero, R.-E. Nuñez-Anita, L.-S. Acosta-Torres and V. Castaño, J. Nanotechnol., 2012, 2012, 936041 Search PubMed.
- K. M. El-Say and H. S. El-Sawy, Int. J. Pharm., 2017, 528, 675–691 CrossRef CAS PubMed.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC.
- K. Tamaki and M. Shimomura, Int. J. Nanosci., 2002, 01, 533–537 CrossRef CAS.
- T. Soukka, H. Härmä, J. Paukkunen and T. Lövgren, Anal. Chem., 2001, 73, 2254–2260 CrossRef CAS PubMed.
- T. Soukka, J. Paukkunen, H. Härmä, S. Lönnberg, H. Lindroos and T. Lövgren, Clin. Chem., 2001, 47, 1269–1278 CrossRef CAS.
- T. Soukka, K. Antonen, H. Härmä, A.-M. Pelkkikangas, P. Huhtinen and T. Lövgren, Clin. Chim. Acta, 2003, 328, 45–58 CrossRef CAS.
- L. Kokko, K. Sandberg, T. Lövgren and T. Soukka, Anal. Chim. Acta, 2004, 503, 155–162 CrossRef CAS.
- V. Väisänen, H. Härmä, H. Lilja and A. Bjartell, Luminescence, 2000, 15, 389–397 CrossRef.
- A. Valanne, J. Suojanen, J. Peltonen, T. Soukka, P. Hänninen and H. Härmä, Analyst, 2009, 134, 980–986 RSC.
- S. Pihlasalo, T. Deguchi, M. Virtamo, J. Jacobino, K. Chary, F. R. López-Picón, G. Brunhofer-Bolzer, R. Huttunen, A. Fallarero, P. Vuorela and H. Härmä, Anal. Chem., 2017, 89, 2398–2404 CrossRef CAS PubMed.
- S. Pihlasalo, J. Kirjavainen, P. Hänninen and H. Härmä, Anal. Chem., 2011, 83, 1163–1166 CrossRef CAS PubMed.
- P. Huhtinen, M. Kivelä, O. Kuronen, V. Hagren, H. Takalo, H. Tenhu, T. Lövgren and H. Härmä, Anal. Chem., 2005, 77, 2643–2648 CrossRef CAS PubMed.
- K. Tamaki, H. Yabu, T. Isoshima, M. Hara and M. Shimomura, Colloids Surf., A, 2006, 284–285, 355–358 CrossRef.
- G. Shao, R. Han, Y. Ma, M. Tang, F. Xue, Y. Sha and Y. Wang, Chem. – Eur. J., 2010, 16, 8647–8651 CrossRef CAS.
- T. Aikawa, A. Mizuno, M. Kohri, T. Taniguchi, K. Kishikawa and T. Nakahira, Colloids Surf., B, 2016, 145, 152–159 CrossRef CAS PubMed.
- H. Tan, B. Liu and Y. Chen, ACS Nano, 2012, 6, 10505–10511 CrossRef CAS PubMed.
- W. Sun, J. Yu, R. Deng, Y. Rong, B. Fujimoto, C. Wu, H. Zhang and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 11294–11297 CrossRef CAS PubMed.
- Q. Li, J. Zhang, W. Sun, J. Yu, C. Wu, W. Qin and D. T. Chiu, Langmuir, 2014, 30, 8607–8614 CrossRef CAS PubMed.
- I. K. Voets, A. de Keizer and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2009, 147–148, 300–318 CrossRef CAS PubMed.
- J. Wang, A. H. Velders, E. Gianolio, S. Aime, F. J. Vergeldt, H. V. As, Y. Yan, M. Drechsler, A. de Keizer, M. A. C. Stuart and J. van der Gucht, Chem. Commun., 2013, 49, 3736–3738 RSC.
- D. C. Thévenaz, C. A. Monnier, S. Balog and G. L. Fiore, Biomacromolecules, 2014, 15, 3994–4001 CrossRef PubMed.
- N. Wartenberg, O. Raccurt, D. Imbert, M. Mazzanti and E. Bourgeat-Lami, J. Mater. Chem. C, 2013, 1, 2061–2068 RSC.
- M. Cardoso Dos Santos, A. Runser, H. Bartenlian, A. M. Nonat, L. J. Charbonnière, A. S. Klymchenko, N. Hildebrandt and A. Reisch, Chem. Mater., 2019, 31, 4034–4041 CrossRef CAS.
- Y. Takei, S. Arai, A. Murata, M. Takabayashi, K. Oyama, S. Ishiwata, S. Takeoka and M. Suzuki, ACS Nano, 2014, 8, 198–206 CrossRef CAS.
- W. Yang, L.-M. Fu, X. Wen, Y. Liu, Y. Tian, Y.-C. Liu, R.-C. Han, Z.-Y. Gao, T.-E. Wang, Y.-L. Sha, Y.-Q. Jiang, Y. Wang and J.-P. Zhang, Biomaterials, 2016, 100, 152–161 CrossRef CAS.
- N. Gao, Y. Zhang, P. Huang, Z. Xiang, F.-Y. Wu and L. Mao, Anal. Chem., 2018, 90, 7004–7011 CrossRef CAS.
- T. Liao, F. Yuan, C. Shi, C.-X. He and Z. Li, RSC Adv., 2016, 6, 103463–103470 RSC.
- Z. Chen, Z. Zhang, X. Zhai, Y. Li, L. Lin, H. Zhao, L. Bian, P. Li, L. Yu, Y. Wu and G. Lin, Anal. Chem., 2020, 92, 7226–7231 CrossRef CAS PubMed.
- L. B. Capeletti, L. M. D. Loiola, A. S. Picco, M. da Silva Liberato and M. B. Cardoso, in Smart Nanoparticles for Biomedicine, ed. G. Ciofani, Elsevier, 2018, pp. 115–129 Search PubMed.
- A. Bitar, N. M. Ahmad, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 1147–1154 CrossRef CAS PubMed.
- S. Bonacchi, D. Genovese, R. Juris, M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Angew. Chem., Int. Ed., 2011, 50, 4056–4066 CrossRef CAS.
- G. Yao, L. Wang, Y. Wu, J. Smith, J. Xu, W. Zhao, E. Lee and W. Tan, Anal. Bioanal. Chem., 2006, 385, 518–524 CrossRef CAS PubMed.
- V. Gubala, G. Giovannini, F. Kunc, M. P. Monopoli and C. J. Moore, Cancer Nanotechnol., 2020, 11, 1 CrossRef CAS.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- P. C. R. Soares-Santos, H. I. S. Nogueira, V. Félix, M. G. B. Drew, R. A. Sá Ferreira, L. D. Carlos and T. Trindade, Chem. Mater., 2003, 15, 100–108 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- D. Zhao, W. Qin, C. Wu, G. Qin, J. Zhang and S. Lü, Chem. Phys. Lett., 2004, 388, 400–405 CrossRef CAS.
- X. Hai, M. Tan, G. Wang, Z. Ye, J. Yuan and K. Matsumoto, Anal. Sci., 2004, 20, 245–246 CrossRef CAS PubMed.
- Z. Ye, M. Tan, G. Wang and J. Yuan, J. Mater. Chem., 2004, 14, 851 RSC.
- M. Tan, Z. Ye, G. Wang and J. Yuan, Chem. Mater., 2004, 16, 2494–2498 CrossRef CAS.
- Z. Ye, M. Tan, G. Wang and J. Yuan, Talanta, 2005, 65, 206–210 CAS.
- Z. Ye, M. Tan, G. Wang and J. Yuan, J. Fluoresc., 2005, 15, 499–505 CrossRef CAS PubMed.
- Y. Chen, Y. Chi, H. Wen and Z. Lu, Anal. Chem., 2007, 79, 960–965 CrossRef CAS PubMed.
- Y. Chen and Z. Lu, Anal. Chim. Acta, 2007, 587, 180–186 CrossRef CAS PubMed.
- J. Wu, G. Wang, D. Jin, J. Yuan, Y. Guan and J. Piper, Chem. Commun., 2008, 365–367 RSC.
- J. Wu, Z. Ye, G. Wang, D. Jin, J. Yuan, Y. Guan and J. Piper, J. Mater. Chem., 2009, 19, 1258–1264 RSC.
- L. Tian, Z. Dai, L. Zhang, R. Zhang, Z. Ye, J. Wu, D. Jin and J. Yuan, Nanoscale, 2012, 4, 3551–3557 RSC.
- H. Zhang, Y. Xu, W. Yang and Q. Li, Chem. Mater., 2007, 19, 5875–5881 CrossRef CAS.
- K. O. Iwu, P. C. R. Soares-Santos, H. I. S. Nogueira, L. D. Carlos and T. Trindade, J. Phys. Chem. C, 2009, 113, 7567–7573 CrossRef CAS.
- J. Samuel, G. Tallec, P. Cherns, W. L. Ling, O. Raccurt, O. Poncelet, D. Imbert and M. Mazzanti, Chem. Commun., 2010, 46, 2647–2649 RSC.
- H. Jiang, G. Wang, W. Zhang, X. Liu, Z. Ye, D. Jin, J. Yuan and Z. Liu, J. Fluoresc., 2010, 20, 321–328 CrossRef CAS.
- N. Wartenberg, O. Raccurt, E. Bourgeat-Lami, D. Imbert and M. Mazzanti, Eur. J. Inorg. Chem., 2013, 2013, 1493–1498 CrossRef CAS.
- S. V. Eliseeva, B. Song, C. D. B. Vandevyver, A.-S. Chauvin, J. B. Wacker and J.-C. G. Bünzli, New J. Chem., 2010, 34, 2915–2921 RSC.
- H. Peng, M. I. J. Stich, J. Yu, L. Sun, L. H. Fischer and O. S. Wolfbeis, Adv. Mater., 2010, 22, 716–719 CrossRef CAS PubMed.
- C. Philippot, A. Bourdolle, O. Maury, F. Dubois, B. Boury, S. Brustlein, S. Brasselet, C. Andraud and A. Ibanez, J. Mater. Chem., 2011, 21, 18613–18622 RSC.
- Q.-X. Wang, S.-F. Xue, Z.-H. Chen, S.-H. Ma, S. Zhang, G. Shi and M. Zhang, Biosens. Bioelectron., 2017, 94, 388–393 CrossRef CAS.
- K. R. Kim, Y. D. Han, H. J. Chun, K. W. Lee, D.-K. Hong, K.-N. Lee and H. C. Yoon, Biosensors, 2017, 7, 48 CrossRef.
- H. Tan, Q. Li, C. Ma, Y. Song, F. Xu, S. Chen and L. Wang, Biosens. Bioelectron., 2015, 63, 566–571 CrossRef CAS PubMed.
- E. Fanizza, N. Depalo, S. Fedorenko, R. M. Iacobazzi, A. Mukhametshina, R. Zairov, A. Salatino, F. Vischio, A. Panniello, V. Laquintana, M. L. Curri, A. Mustafina, N. Denora and M. Striccoli, Int. J. Mol. Sci., 2019, 20, 3139 CrossRef CAS PubMed.
- S. Fedorenko, D. Gilmanova, A. Mukhametshina, I. Nizameev, K. Kholin, B. Akhmadeev, A. Voloshina, A. Sapunova, S. Kuznetsova, A. Daminova, S. Katsyuba, R. Zairov and A. Mustafina, J. Mater. Sci., 2019, 54, 9140–9154 CrossRef CAS.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019–7029 CrossRef CAS.
- P. Reiss, J. Bleuse and A. Pron, Nano Lett., 2002, 2, 781–784 CrossRef CAS.
- H. Peng, C. Wu, Y. Jiang, S. Huang and J. McNeill, Langmuir, 2007, 23, 1591–1595 CrossRef CAS PubMed.
- A. R. Kortan, R. Hull, R. L. Opila, M. G. Bawendi, M. L. Steigerwald, P. J. Carroll and L. E. Brus, J. Am. Chem. Soc., 1990, 112, 1327–1332 CrossRef CAS.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Nat. Biotechnol., 2003, 21, 47–51 CrossRef CAS PubMed.
- N. N. Mamedova, N. A. Kotov, A. L. Rogach and J. Studer, Nano Lett., 2001, 1, 281–286 CrossRef CAS.
- I. Potapova, R. Mruk, C. Hübner, R. Zentel, T. Basché and A. Mews, Angew. Chem., Int. Ed., 2005, 44, 2437–2440 CrossRef CAS PubMed.
- L. Stryer and R. P. Haugland, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 719–726 CrossRef CAS PubMed.
- N. Hildebrandt, L. J. Charbonnière, M. Beck, R. F. Ziessel and H. G. Löhmannsröben, Angew. Chem., Int. Ed., 2005, 44, 7612–7615 CrossRef CAS PubMed.
- N. Hildebrandt, L. J. Charbonnière and H.-G. Löhmannsröben, J. Biomed. Biotechnol., 2007, 2007, 1–6 CrossRef PubMed.
- L. J. Charbonnière, N. Hildebrandt, R. F. Ziessel and H. G. Löhmannsröben, J. Am. Chem. Soc., 2006, 128, 12800–12809 CrossRef PubMed.
- N. Hildebrandt, K. D. Wegner and W. R. Algar, Coord. Chem. Rev., 2014, 273274, 125–138 CrossRef.
- D. Geissler, L. J. Charbonnière, R. F. Ziessel, N. G. Butlin, H. G. Löhmannsröben and N. Hildebrandt, Angew. Chem., Int. Ed., 2010, 49, 1396–1401 CrossRef CAS.
- F. Morgner, D. Geißler, S. Stufler, N. G. Butlin, H. G. Löhmannsröben and N. Hildebrandt, Angew. Chem., Int. Ed., 2010, 49, 7570–7574 CrossRef CAS PubMed.
- D. Geissler, N. G. Butlin, D. Hill, H.-G. Löhmannsröben and N. Hildebrandt, in Optics InfoBase Conference Papers, ed. I. Georgakoudi, J. Popp and K. Svanberg, 2009, vol. 7368, p. 73680P Search PubMed.
- I. Gryczynski, J. Malicka, Z. Gryczynski, J. R. Lakowicz and C. D. Geddes, J. Fluoresc., 2002, 12, 131–133 CrossRef CAS.
- D. Geissler, H.-G. Löhmannsröben, L. J. Charbonnière, R. F. Ziessel, N. G. Butlin, I. L. Medintz, H. Mattoussi and N. Hildebrandt, Proc. SPIE 7575, Colloidal Quantum Dots for Biomedical Applications V, 2010, 75750E Search PubMed.
- K. D. Wegner, F. Morgner, E. Oh, R. Goswami, K. Susumu, M. H. Stewart, I. L. Medintz and N. Hildebrandt, Chem. Mater., 2014, 26, 4299–4312 CrossRef CAS.
- W. R. Algar, A. P. Malanoski, K. Susumu, M. H. Stewart, N. Hildebrandt and I. L. Medintz, Anal. Chem., 2012, 84, 10136–10146 CrossRef CAS PubMed.
- W. R. Algar, D. Wegner, A. L. Huston, J. B. Blanco-Canosa, M. H. Stewart, A. Armstrong, P. E. Dawson, N. Hildebrandt and I. L. Medintz, J. Am. Chem. Soc., 2012, 134, 1876–1891 CrossRef CAS.
- J. Xu, X. Qiu and N. Hildebrandt, Nano Lett., 2021, 21, 4802–4808 CrossRef CAS PubMed.
- S. A. Díaz, G. Lasarte Aragonés, S. Buckhout-White, X. Qiu, E. Oh, K. Susumu, J. S. Melinger, A. L. Huston, N. Hildebrandt and I. L. Medintz, J. Phys. Chem. Lett., 2017, 8, 2182–2188 CrossRef.
- F. Morgner, S. Stufler, D. Geißler, I. L. Medintz, W. R. Algar, K. Susumu, M. H. Stewart, J. B. Blanco-Canosa, P. E. Dawson and N. Hildebrandt, Sensors, 2011, 11, 9667–9684 CrossRef CAS PubMed.
- T. Hallaj, M. Amjadi, X. Qiu, K. Susumu, I. L. Medintz and N. Hildebrandt, Nanoscale, 2020, 12, 13719–13730 RSC.
- C. Léger, A. Yahia-Ammar, K. Susumu, I. L. Medintz, A. Urvoas, M. Valerio-Lepiniec, P. Minard and N. Hildebrandt, ACS Nano, 2020, 14, 5956–5967 CrossRef PubMed.
- L. Mattera, S. Bhuckory, K. D. Wegner, X. Qiu, F. Agnese, C. Lincheneau, T. Senden, D. Djurado, L. J. Charbonnière, N. Hildebrandt and P. Reiss, Nanoscale, 2016, 8, 11275–11283 RSC.
- S. Bhuckory, L. Mattera, K. D. Wegner, X. Qiu, Y.-T. Wu, L. J. Charbonnière, P. Reiss and N. Hildebrandt, Chem. Commun., 2016, 52, 14423–14425 RSC.
- H. Härmä, T. Soukka, A. Shavel, N. Gaponik and H. Weller, Anal. Chim. Acta, 2007, 604, 177–183 CrossRef.
- G. J. Stasiuk, S. Tamang, D. Imbert, C. Poillot, M. Giardiello, C. Tisseyre, E. L. Barbier, P. H. Fries, M. De Waard, P. Reiss and M. Mazzanti, ACS Nano, 2011, 5, 8193–8201 CrossRef CAS PubMed.
- J. K. Molloy, C. Lincheneau, M. M. Karimdjy, F. Agnese, L. Mattera, C. Gateau, P. Reiss, D. Imbert and M. Mazzanti, Chem. Commun., 2016, 52, 4577–4580 RSC.
- R. A. Tigaa, G. J. Lucas and A. De Bettencourt-Dias, Inorg. Chem., 2017, 56, 3260–3268 CrossRef CAS PubMed.
- F. Wu, L. Yue, L. Yang, K. Wang, G. Liu, X. Luo and X. Zhu, Colloids Surf., B, 2019, 175, 272–280 CrossRef CAS PubMed.
- H. Su, Y. Liao, F. Wu, X. Sun, H. Liu, K. Wang and X. Zhu, Colloids Surf., B, 2018, 170, 194–200 CrossRef CAS PubMed.
- S. Cousinié, M. Gressier, C. Reber, J. Dexpert-Ghys and M. J. Menu, Langmuir, 2008, 24, 6208–6214 CrossRef PubMed.
- L. J. Charbonnière, N. Weibel, C. Estournes, C. Leuvrey and R. Ziessel, New J. Chem., 2004, 28, 777–781 RSC.
- E. DeOliveira, C. R. Neri, O. A. Serra and A. G. S. Prado, Chem. Mater., 2007, 19, 5437–5442 CrossRef CAS.
- L. Lu, F. Liu, G. Sun, H. Zhang, S. Xi and H. Wang, J. Mater. Chem., 2004, 14, 2760–2762 RSC.
- Y. Xu and Q. Li, Clin. Chem., 2007, 53, 1503–1510 CrossRef CAS PubMed.
- B. Yan and B. Zhou, J. Photochem. Photobiol., A, 2008, 195, 314–322 CrossRef CAS.
- Y. Li and B. Yan, J. Solid State Chem., 2008, 181, 1032–1039 CrossRef CAS.
- D. B. Ambili Raj, S. Biju and M. L. P. Reddy, J. Mater. Chem., 2009, 19, 7976–7983 RSC.
- V. Divya, S. Biju, R. L. Varma and M. L. P. Reddy, J. Mater. Chem., 2010, 20, 5220–5227 RSC.
- D. Zhang, X. Wang, Z. A. Qiao, D. Tang, Y. Liu and Q. Huo, J. Phys. Chem. C, 2010, 114, 12505–12510 CrossRef CAS.
- W. X. Li, Y. S. Zheng, X. F. Cao, J. Bai, Z. F. Fu, J. R. Bao and Y. L. Li, J. Lumin., 2016, 178, 470–478 CrossRef CAS.
- Z. F. Fu, W. X. Li, J. Bai, J. R. Bao, X. F. Cao and Y. S. Zheng, Luminescence, 2017, 32, 327–333 CrossRef CAS PubMed.
- Y. Y. Ma, H. X. Huang, M. Chen and D. J. Qian, J. Lumin., 2018, 203, 277–285 CrossRef CAS.
- A. P. Duarte, M. Gressier, M. J. Menu, J. Dexpert-Ghys, J. M. A. Caiut and S. J. L. Ribeiro, J. Phys. Chem. C, 2012, 116, 505–515 CrossRef CAS.
- A. P. Duarte, L. Mauline, M. Gressier, J. Dexpert-Ghys, C. Roques, J. M. A. Caiut, E. Deffune, D. C. G. Maia, I. Z. Carlos, A. A. P. Ferreira, S. J. L. Ribeiro and M. J. Menu, Langmuir, 2013, 29, 5878–5888 CrossRef CAS PubMed.
- K. M. L. Taylor and W. Lin, J. Mater. Chem., 2009, 19, 6418–6422 RSC.
- S. Mornet, S. Vasseur, F. Grasset, P. Veverka, G. Goglio, A. Demourgues, J. Portier, E. Pollert and E. Duguet, Prog. Solid State Chem., 2006, 34, 237–247 CrossRef CAS.
- Y. Jun, J. Choi and J. Cheon, Chem. Commun., 2007, 1203–1214 RSC.
- D. Patel, J. Y. Moon, Y. Chang, T. J. Kim and G. H. Lee, Colloids Surf., A, 2008, 313–314, 91–94 CrossRef.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397–415 CrossRef CAS PubMed.
- M. Ma, Y. Zhang, W. Yu, H. Y. Shen, H. Q. Zhang and N. Gu, Colloids Surf., A, 2003, 212, 219–226 CrossRef CAS.
- X. Shen, X. Fang, Y. Zhou and H. Liang, Chem. Lett., 2004, 33, 1468–1469 CrossRef CAS.
- J. Choi, J. C. Kim, Y. B. Lee, I. S. Kim, Y. K. Park and N. H. Hur, Chem. Commun., 2007, 1644 RSC.
- S. Y. Yu, H. J. Zhang, J. B. Yu, C. Wang, L. N. Sun and W. D. Shi, Langmuir, 2007, 23, 7836–7840 CrossRef CAS.
- H. M. Lu, W. T. Zheng and Q. Jiang, J. Phys. D: Appl. Phys., 2007, 40, 320–325 CrossRef CAS.
- J. Wu, Z. Ye, G. Wang and J. Yuan, Talanta, 2007, 72, 1693–1697 CrossRef CAS PubMed.
- H. Lu, G. Yi, S. Zhao, D. Chen, L. Guo and J. Cheng, J. Mater. Chem., 2004, 14, 1336 RSC.
- B. Wang, J. Hai, Q. Wang, T. Li and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 3063–3066 CrossRef CAS PubMed.
- Y. Liu, G. Cheng, Z. Wang, J. Zhang, D. Sun, G. Hong and J. Ni, Mater. Lett., 2013, 97, 187–190 CrossRef CAS.
- L. U. Khan, H. F. Brito, J. Hölsä, K. R. Pirota, D. Muraca, M. C. F. C. Felinto, E. E. S. Teotonio and O. L. Malta, Inorg. Chem., 2014, 53, 12902–12910 CrossRef CAS.
- R. R. Silva, A. P. Duarte, R. M. Sábio, J. M. A. Caiut, M. Gressier, M. J. Menu, A. Franco and S. J. L. Ribeiro, ChemistrySelect, 2016, 1, 5923–5928 CrossRef CAS.
- S. L. C. Pinho, J. Sereno, A. J. Abrunhosa, M.-H. Delville, J. Rocha, L. D. Carlos and C. F. G. C. Geraldes, Inorg. Chem., 2019, 58, 16618–16628 CrossRef CAS PubMed.
- S. Goderski, S. Kanno, K. Yoshihara, H. Komiya, K. Goto, T. Tanaka, S. Kawaguchi, A. Ishii, J. Shimoyama, M. Hasegawa and S. Lis, ACS Omega, 2020, 5, 32930–32938 CrossRef CAS PubMed.
- A. de Espindola, C. S. Chagas, E. Barbosa, C. E. Castro, F. L. A. Fonseca, P. S. Haddad and C. Molina, J. Magn. Magn. Mater., 2022, 545, 168751 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- Y. Zhao, Y. Tian, Y. Cui, W. Liu, W. Ma and X. Jiang, J. Am. Chem. Soc., 2010, 132, 12349–12356 CrossRef CAS PubMed.
- B. I. Ipe, K. Yoosaf and K. G. Thomas, J. Am. Chem. Soc., 2006, 128, 1907–1913 CrossRef CAS PubMed.
- D. J. Lewis, T. M. Day, J. V. MacPherson and Z. Pikramenou, Chem. Commun., 2006, 1433–1435 RSC.
- L. Prodi, M. Montalti, N. Zaccheroni, G. Pickaert, L. Charbonnière and R. Ziessel, New J. Chem., 2003, 27, 134–139 RSC.
- A. Davies, D. J. Lewis, S. P. Watson, S. G. Thomas and Z. Pikramenou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1862–1867 CrossRef CAS.
- J. Massue, S. J. Quinn and T. Gunnlaugsson, J. Am. Chem. Soc., 2008, 130, 6900–6901 CrossRef CAS PubMed.
- S. Comby and T. Gunnlaugsson, ACS Nano, 2011, 5, 7184–7197 CrossRef CAS PubMed.
- E. M. Surender, S. Comby, B. L. Cavanagh, O. Brennan, T. C. Lee and T. Gunnlaugsson, Chem, 2016, 1, 438–455 CAS.
- C. Chen, C. Midelet, S. Bhuckory, N. Hildebrandt and M. H. V. Werts, J. Phys. Chem. C, 2018, 122, 17566–17574 CrossRef CAS.
- A. Samanta, Y. Zhou, S. Zou, H. Yan and Y. Liu, Nano Lett., 2014, 14, 5052–5057 CrossRef CAS PubMed.
- Y. Sun, H. Jiu, D. Zhang, J. Gao, B. Guo and Q. Zhang, Chem. Phys. Lett., 2005, 410, 204–208 CrossRef CAS.
- S. Wu, Y. Sun, X. Wang, W. Wu, X. Tian, Q. Yan, Y. Luo and Q. Zhang, J. Photochem. Photobiol., A, 2007, 191, 97–103 CrossRef CAS.
- J. Kang, Y. Li, Y. Chen, A. Wang, B. Yue, Y. Qu, Y. Zhao and H. Chu, Mater. Res. Bull., 2015, 71, 116–121 CrossRef CAS.
- Y. Zhao, A. Wang, J. Kang, H. Chu, H. Zhang and Y. Zhao, J. Photochem. Photobiol., A, 2020, 400, 112678 CrossRef CAS.
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS.
- H. Fukumoto and Y. Yonezawa, Thin Solid Films, 1998, 327–329, 748–751 CrossRef CAS.
- J. Kang, Y. Zhao, H. Chu and Y. Zhao, J. Photochem. Photobiol., A, 2018, 365, 119–124 CrossRef CAS.
- Y. Mei and B. Yan, Inorg. Chem. Commun., 2014, 40, 39–42 CrossRef CAS.
- W. Mnasri, M. Parvizian and S. Ammar-Merah, Nanomaterials, 2021, 11, 354 CrossRef CAS PubMed.
- W. T. Carnall, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1979, vol. 3, pp. 171–208 Search PubMed.
- C. Lin, Z. Xia, L. Zhang, X. Chen, Q. Sun, M. Lu, Z. Yuan, X. Xie and L. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 31783–31792 CrossRef CAS PubMed.
- S. Han, R. Deng, Q. Gu, L. Ni, U. Huynh, J. Zhang, Z. Yi, B. Zhao, H. Tamura, A. Pershin, H. Xu, Z. Huang, S. Ahmad, M. Abdi-Jalebi, A. Sadhanala, M. L. Tang, A. Bakulin, D. Beljonne, X. Liu and A. Rao, Nature, 2020, 587, 594–599 CrossRef CAS PubMed.
- F. Mousseau, C. F. Tarisse, S. Simon, T. Gacoin, A. Alexandrou and C. I. Bouzigues, Nanoscale, 2021, 13, 14814–14824 RSC.
- S. Zhou, W. Zheng, Z. Chen, D. Tu, Y. Liu, E. Ma, R. Li, H. Zhu, M. Huang and X. Chen, Angew. Chem., 2014, 126, 12706–12710 CrossRef.
- L. Francés-Soriano, N. Hildebrandt and L. J. Charbonnière, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2022. https://doi.org/10.1016/B978-0-12-823144-9.00095-9 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |