
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging investigator series: surfactants, films, and coatings on atmospheric aerosol particles: a review
Kevin A.
Wokosin†
,
Emma L.
Schell
and
Jennifer A.
Faust
 *
*
Department of Chemistry, The College of Wooster, Wooster, OH, USA. E-mail: jfaust@wooster.edu
First published on 7th June 2022
Abstract
Surfactants are surface-active molecules or ions that can often be found at the surface of atmospheric aerosol particles. Some surfactants, such as biomolecules in marine aerosol and cooking oils in urban aerosol, are directly emitted into the atmosphere, whereas others form from secondary reactions in the atmosphere. Environmentally relevant surfactants have long been studied in laboratory experiments and computational models, but recent developments in mass spectrometry, spectroscopy, imaging, and colorimetry have greatly expanded measurements of surfactants in ambient aerosol. This review focuses on advances surrounding the following themes: techniques for measuring and modeling surfactants on aerosol particles, the morphology of surfactant-coated aerosol particles, the impact of surfactants on cloud droplet activation and ice nucleation, multiphase reactions and photochemical reactions on surfactant-coated particles, and finally, field observations of surfactants on ambient particulate matter. An exciting area for future work is the continued advancement of single-particle techniques to simultaneously monitor morphology and composition of individual aerosol particles. Along with an expansion of field sampling campaigns, more laboratory studies of increasingly complicated aerosol systems are needed to bridge the gap between complex observations from the field and fundamental investigations in the laboratory. Characterizing surfactants, films, and coatings on atmospheric aerosol particles can provide insights relevant to how particulate matter impacts climate, air quality, and human health.
 Kevin A. Wokosin | Kevin Wokosin is currently a graduate student in Dr Timothy Bertram's research group at the University of Wisconsin–Madison. Before beginning his graduate studies, Kevin worked with the Air Quality Research Center at University of California, Davis, and the California Air Resources Board on air quality monitoring and compliance training. He conducted undergraduate research focused on heterogeneous reactions and aerosols with Dr Jennifer Faust while earning his bachelor's in chemistry from the College of Wooster. |
 Emma L. Schell | Emma Schell is an undergraduate student at the College of Wooster, where she is pursuing a Chemistry B.A. with minors in Spanish and Latin American Studies. Her research experience includes studying the chemical kinetics of the heterogeneous oxidation of pesticides and hydrocarbons, as well as identifying the oxidation products of a novel fungicide reacting with ozone. |
 Jennifer A. Faust | Jennifer Faust is an Assistant Professor of Chemistry at the College of Wooster, where she leads an undergraduate research group in environmental chemistry. Her research interests include heterogeneous reactions at the surface of atmospheric aerosol particles and atmospheric transport of chemical contaminants through wet and dry deposition. Prior to beginning her position at Wooster, she earned a PhD in physical chemistry from the University of Wisconsin–Madison and completed a postdoctoral fellowship in atmospheric chemistry at the University of Toronto. |
Environmental significanceSurfactants, or surface-active molecules, can be found as films and coatings at the surface of aerosol particles in the atmosphere. Because surfactants change the morphology, phase state, and surface properties of aerosol particles, they alter the ability of the particles to scatter and absorb solar radiation, to serve as cloud condensation nuclei, and to act as ice nucleating particles. Each of these factors affects Earth's climate through radiative forcing. Surfactants also perturb multiphase reaction pathways across the gas-particle interface, changing sinks for reactive gases in the atmosphere and changing the evolution of particle composition with age. Characterizing surfactants, films, and coatings on atmospheric aerosol particles can provide insights for climate, air quality, and human health. |
1. Introduction
Because of its anisotropic geometry, the air–water interface presents a distinct environment from bulk gas and bulk condensed phases. Surfactants, or surface-active agents, are molecules consisting of hydrophobic and hydrophilic moieties found at the boundary between phases. They are said to be surface-active because of their favorable partitioning to the interface. Atmospheric aerosols, or particles suspended in a gas, often contain organic surfactant molecules present at the surface as monolayers, patchy islands, or thicker films and coatings.1–3 As illustrated in Fig. 1, some surfactants are directly emitted to the atmosphere from primary sources, such as biomolecules in the sea surface microlayer or oils from cooking.4–7 Other species form through secondary reactions in the atmosphere—either through heterogeneous reactions, through gas-phase reactions that produce low-volatile, poorly soluble products that adsorb to aerosol particles, or through particle-phase reactions that form surface-active species.3,8–19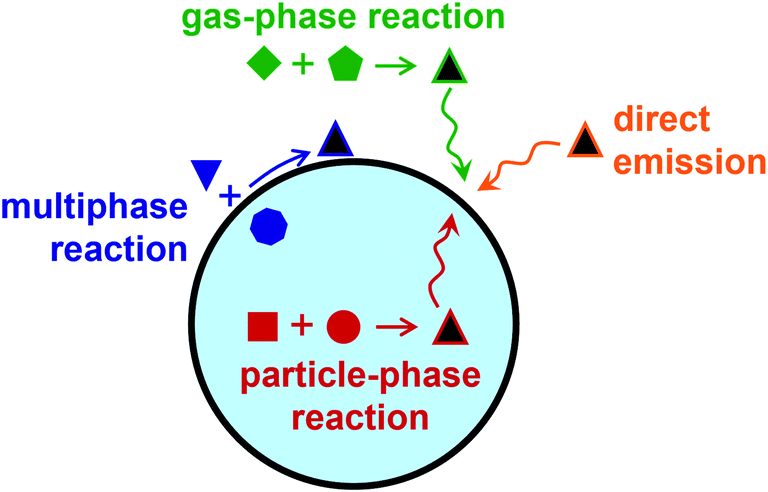 | ||
| Fig. 1 Sources of surface-active molecules (black triangles) to atmospheric aerosol particles include direct emission from primary sources, gas-phase reactions that generate products that adsorb to the particle surface, multiphase reactions across the gas-particle interface that produce surface-active species, and particle-phase reactions that form products that partition to the surface. | ||
As a class, surfactants encompass anionic, cationic, and nonionic species. Adsorption isotherms use principles of thermodynamics to model their partitioning to the air–water interface.20–23 However, aerosol particles are complex chemical environments, often with high ionic strength, low pH, and high concentrations of organic species.14,24,25 The salting out effect can markedly enhance predicted surface concentrations of organic species in the presence of inorganic salts.14,26 Furthermore, molecules ionize differently at the surface than in the bulk. This observation dates back to the 1930s for carboxylic acids27 and amines28 and to the 1950s for polypeptides and proteins,29 and it is still an active area of research today.30–36 Accordingly, surfactants in atmospheric aerosol particles present rich systems for both fundamental and applied chemical research.
Small amounts of surfactants can have dramatic implications for the physical and chemical properties of aerosol particles. Surfactants that change the optical properties of aerosol particles will affect light scattering or absorption, thereby altering radiative forcing effects on climate.2,3,37 Surfactants that change hygroscopicity or water uptake will affect the ability of the particles to grow into cloud droplets, also altering radiative forcing.2,3,38–40 Similarly, surfactants that change ice adsorption to the surface of aerosol particles will affect ice cloud formation, again altering radiative forcing.2,3,41,42 Surfactants can also affect the fate of reactive gases in the atmosphere, e.g. through loss of species like ozone (O3) and hydroxyl radicals (OH) to the surface of aerosol particles.2,3,43–46 Changes in the surface composition of aerosol particles may even affect their toxicity when inhaled into the respiratory system, although these relationships are not well understood.47–49
A seminal review of organic films on atmospheric surfaces was published by Donaldson and Vaida in 2006,2 with a comprehensive follow-up by McNeill et al. in 2013.3 Here we will focus on newer developments in the world of surfactants, films, and coatings on atmospheric aerosol particles with an emphasis on the following areas: experimental measurements and modeling techniques, particle morphology, cloud droplet activation, ice nucleation, multiphase reactions, photochemistry, and ambient observations.
2. Techniques for measuring and modeling surfactants on aerosol particles
To identify surfactants at the surface of aerosol particles, we must measure both the chemical composition and the morphology of the particles. As shown in the top portion of Fig. 2, aerosol particles can be internally or externally mixed.50 In an internal mixture, each particle is a homogeneous combination of multiple components. An external mixture, in contrast, consists of different types of particles that are each composed of a pure component. Ensemble techniques measure the average composition across all particles, whereas single-particle techniques resolve the composition on a particle-by-particle basis.50–56 Knowledge of average properties is often insufficient to predict cloud activation, gas uptake, heterogeneous reactivity, and other effects that depend on the surface composition of aerosol particles. For example, the cartoon in Fig. 2 illustrates how three populations of particles with identical bulk composition are not equally effective at forming cloud condensation nuclei (CCN).50 Directly characterizing surfactants in ambient aerosol particles is challenging because of the need to both identify the chemical species and demonstrate their surface activity.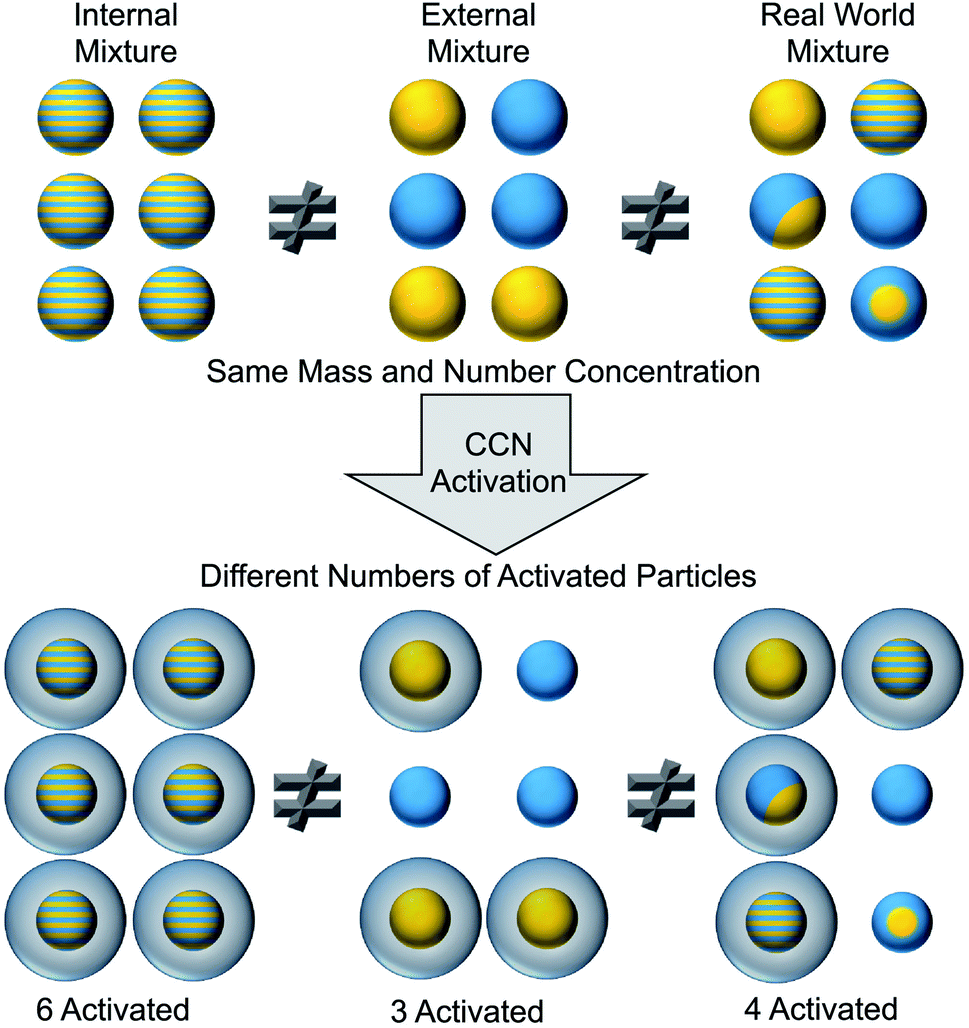 | ||
Fig. 2 Representation of three aerosol populations with the same average number and mass concentration, consisting of 50% ammonium sulfate (gold) and 50% hydrophobic organic (blue) overall. Striped particles are a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 homogeneous mixture of ammonium sulfate and hydrophobic organics. The representative real world mixture contains homogeneous particles, pure inorganic and pure organic particles, a partially engulfed (or lens) particle, and a core–shell particle. In this hypothetical scenario, ammonium sulfate must be exposed at the surface of a particle in order for it to serve as a cloud condensation nucleus. As a result, a different number of particles in each population successfully activates cloud formation. Reprinted with permission from N. Riemer, A. P. Ault, M. West, R. L. Craig and J. H. Curtis, Aerosol Mixing State: Measurements, Modeling, and Impacts, Rev. Geophys., 2019, 57, 187–249. Copyright 2019 Wiley. :50 homogeneous mixture of ammonium sulfate and hydrophobic organics. The representative real world mixture contains homogeneous particles, pure inorganic and pure organic particles, a partially engulfed (or lens) particle, and a core–shell particle. In this hypothetical scenario, ammonium sulfate must be exposed at the surface of a particle in order for it to serve as a cloud condensation nucleus. As a result, a different number of particles in each population successfully activates cloud formation. Reprinted with permission from N. Riemer, A. P. Ault, M. West, R. L. Craig and J. H. Curtis, Aerosol Mixing State: Measurements, Modeling, and Impacts, Rev. Geophys., 2019, 57, 187–249. Copyright 2019 Wiley. | ||
2.1. Laboratory techniques
Laboratory studies have a long history of providing insight into surfactant phenomena that are difficult to measure in the field. Reviews by Kolb et al.,43 Davidovits et al.,44 and Tang et al.57 provide good overviews of techniques for generating model liquid surfaces, with or without surfactants. Common setups include Langmuir troughs,58 droplet and bubble train reactors,43,44 Knudsen cell reactors,43,44,57,59,60 wetted wheels,61 liquid microjets,61,62 aerosol chambers,63,64 aerosol flow tubes,65,66 coated-wall flow reactors,60,67,68 and aerosol optical tweezers and other assorted levitation techniques.55,69,70 Of these setups, only Langmuir troughs are specifically designed to study surfactant layers, and coated-wall flow reactors are designed specifically for films. Development of methods to suspend and trap aerosol particles is a rapidly expanding area. Recent advances include the application of acoustic levitation with ultraviolet/visible (UV/vis) spectroscopy to measure changes in droplet pH,71 the use of a dual-balance electrodynamic trap to monitor phase and viscosity,72 the combination of a linear quadrupole electrodynamic balance with paper spray mass spectrometry to probe chemical composition,73 and photothermal single-particle spectroscopy to determine mass accommodation coefficients.74,75 In addition, several techniques have been developed specifically to study nascent sea spray aerosol (SSA) particles in the laboratory. These will be discussed later in Section 8.3.2.Experimental techniques to characterize the composition of aerosol particles—and by extension, the composition of surfactants, films, and coatings—fall into two main categories: mass spectrometry and spectroscopy. Depending on the instrumental setup, mass spectrometers can measure single particles or ensemble compositions.76–78 For further information about aerosol mass spectrometry, the reader is directed to reviews by Pratt and Prather,79,80 Laskin et al.,81 and Johnston and Kerecman.82
In the spectroscopy category, electronic, X-ray, and vibrational methods are all used to gain insight into aerosol properties.55 Focused reviews of X-ray photoelectron spectroscopy83–85 and X-ray scattering, including X-ray diffraction and reflectometry,86–88 are available elsewhere. Baumler and Allen reviewed vibrational spectroscopy in 2018.89 Since then, Allen and coworkers,90–92 Grassian and coworkers,93 and Du and coworkers94–100 have published many new studies on the use of infrared reflection absorption spectroscopy (IRRAS) to probe model surfactant systems relevant to sea spray aerosol. Allen and coworkers have also continued to explore the surfaces of model sea spray systems with vibrational sum frequency generation (SFG) spectroscopy.90,101 In studies relevant to the surface composition of biogenic secondary organic aerosol (SOA), Geiger and coworkers102 utilized SFG to detect β-caryophyllene ozonolysis products at the air–water interface, and Richmond and coworkers103,104 have detected methylglyoxal and pyruvic acid oligomers at the air–water interface by vibrational sum frequency spectroscopy (VSFS).
Notably, most of the work described above focuses on the air–water interface. Rao and co-workers105–109 introduced nonlinear optical spectroscopy specifically for the air-particle interface. Overcoming the challenges of low signal and small particle size, they pioneered second harmonic scattering (SHS) to determine the surface density and adsorption free energy of small organic molecules, including crystal violet and trans-4-[4-(dibutylamino)styryl]-1-methylpyridinium iodide, at the surface of ∼100 nm NaCl particles.105–107 When Rao and co-workers applied electronic sum frequency scattering (ESFS) to determine the orientation, surface population, and adsorption free energy of malachite green at the surface of aerosol particles, they observed discrepancies in adsorption energy at the particle surface compared to the air–water interface.108 The authors attributed the difference to surface curvature. In a follow-up study with aerosol particles containing the more environmentally-relevant species propionic acid, Qian et al.109 combined VSFS to probe the particle surface with hyper-Raman scattering to probe the particle bulk. The surface adsorption free energy of propionic acid was significantly more negative at the air–water interface (−15.38 ± 0.11 kJ mol−1) than at the particle surface (−12.69 ± 0.28 kJ mol−1). These in situ spectroscopic measurements challenge the long-standing practice of directly extending measurements from the air–water interface to the air-particle interface.
Finally, in new developments in Raman spectroscopy110 for analysis of surfactants, films, and coatings, Gen et al.111 used electrospray surface-enhanced Raman spectroscopy (SERS) to characterize organic brown carbon coatings formed from heterogeneous reactions, and Wang et al.112 applied micro-Raman spectroscopy to analyze ambient particulate matter from Beijing, China. Depending on the experimental setup, spectroscopy can provide insight into aerosol populations at either the single-particle or the ensemble level, as recently reviewed by Ault and Axson55 and Riemer et al.50 None of the mass spectrometry or spectroscopy techniques described here is unique to surfactant systems, but each can provide insight into the properties of surfactants, films, and coatings at particle surfaces.
A variety of imaging techniques are now in use to probe the morphology and structure of aerosol particles. Fluorescence microscopy, transmission electron microscopy (TEM), scanning electron microscopy (SEM), atomic force microscopy (AFM), scanning transmission X-ray microscopy (STXM) with near-edge X-ray absorption fine structure (NEXAFS), and Brewster angle microscopy (BAM) have all joined the suite of tools used to characterize individual aerosol particles.55,88,113,114 Applications of these techniques to map aerosol morphology and phase will be discussed in Section 3.2.
Lastly, a few more techniques for interrogating surfaces are worthy of mention: molecular beam and ion scattering,115–117 and neutron scattering and reflectometry.88,118,119 Techniques for measuring hygroscopicity (i.e., water uptake) have been reviewed by Tang et al.,120 and methods for measuring surface tension will be discussed in Section 4.2. Ultimately, the crucial goal for laboratory measurements of surfactant-coated aerosol particles remains to simultaneously determine composition and morphology at the single-particle level.
2.2. Modeling techniques
Modeling offers insights into surfactant-coated systems that cannot be gleaned from laboratory or field studies alone.121,122 Molecular dynamics (MD) simulations are a popular technique for modeling gas–liquid interfaces.123,124 MD simulations generate snapshots of molecular configurations on a femtosecond timescale based on force fields that relate positioning and potential energy. Ab initio MD and quantum mechanics/molecular mechanics (QM/MM) simulations, which are based on electronic structure, are computationally expensive and limited to systems of no more than ∼100 molecules, whereas empirical potential functions can be used to reach larger system sizes into the thousands of molecules.124 Accordingly, MD simulations have been applied to surfactant systems ranging in complexity from simple surfactant/salt mixtures125–134 to multi-component mimics for sea spray aerosol93,135–139 and SOA.140 Quantum chemical methods such as density functional theory (DFT)141 have also been applied to heterogeneous processes on atmospheric aerosol particles,142 often in combination with spectroscopic measurements.103,104,143 DFT calculates molecular properties from ground-state electron probability densities. It is the most widespread quantum chemical method for simulations, but because it requires approximation of electron–electron correlations, it cannot be systematically improved.123Mass transport models are specifically designed to model gas uptake and heterogeneous reactions, as discussed in Section 6. The traditional resistor model, named because of the mathematics of adding resistors in parallel, incorporates gas-phase diffusion, mass accommodation, Henry's law partitioning, aqueous-phase diffusion, and rate constants, but it requires steady-state conditions and homogeneous mixing.144 Shiraiwa et al.145 developed KM-GAP (Kinetic Multi-layer GAs-Particle interactions in aerosols and clouds) to also model gas-phase, interfacial, and particle-phase processes, as well as condensation, evaporation, and heat transfer. Unlike the resistor model, KM-GAP does not require steady-state, well-mixed systems. KM-GAP is easily scalable and computationally inexpensive, so it can be seen as a link between MD simulations and experimental measurements. Example applications of kinetic modeling to surfactant systems include heterogeneous reactions of long-chain acids and esters with gas-phase NO3 and O3.146,147 Reactive uptake will be treated in more detail in Section 6.3.
For systems at equilibrium, thermodynamics can be used to model a wide array of aerosol properties. Semeniuk and Dastoor148 published a comprehensive review of thermodynamic models for aerosol particles in 2020. Since then, Calderón et al. have advanced thermodynamic modeling for activity coefficients149 and density150 in aqueous solutions containing ionic surfactants and inorganic salts, and Huang et al.151 reported a unique application of AIOMFAC (Aerosol Inorganic-Organic Mixtures Functional groups Activity Coefficients, a thermodynamic model)152,153 and KM-GAP to aerosol particles with three liquid phases. The ongoing progress in thermodynamic modeling seeks to predict physical and chemical properties of aerosol particles containing increasingly complex mixtures, including those with surface coatings.
The modeling methods described so far focus on systems at the molecular level. Chemical transport models, in contrast, expand modeling to the level of the atmosphere at the regional or even global scale. Chemical transport models range in complexity from zero-dimensional box models, in which concentrations are a function only of time, to three-dimensional models, in which concentrations depend on time and x, y, z coordinates.154 It is too computationally expensive to directly incorporate thermodynamic models of mixed inorganic–organic particles into 3D air quality models like CMAQ (Community Multiscale Air Quality), but it is still possible to introduce parameterizations, e.g., for liquid–liquid phase separation.148,155,156 Such parameterizations are critical in accurately modeling gas uptake that is limited by diffusion through viscous coatings, as discussed in Section 6.3.7 for SOA formed from isoprene epoxydiols.
3. Morphology of aerosol particles
As illustrated in Fig. 1, films and coatings on atmospheric aerosol particles can originate from condensation of organic vapors onto pre-existing particles, from reactions at the gas-particle interface, or from partitioning of organic molecules from the bulk to the surface. Here we focus on the formation of organic shells on aerosol particles due to phase separation and on general measurements of particle phase and morphology.3.1. Liquid–liquid phase separation
Ambient aerosol particles consist of a combination of water, inorganic, and organic components. Depending on the composition and relative humidity (RH), particles can be homogeneously mixed or phase-separated, as illustrated for the phase transitions in Fig. 3.157 Elemental O:C ratios for chemical composition can be used as rough predictors of aerosol morphology: in general, particles with O:C < 0.56 undergo phase separation, whereas particles with O:C > 0.80 are homogeneously mixed.25,158 Additional information about chemical speciation is necessary to determine morphology for particles with intermediate O:C values,25,158 and these guidelines do have exceptions.157,159,160
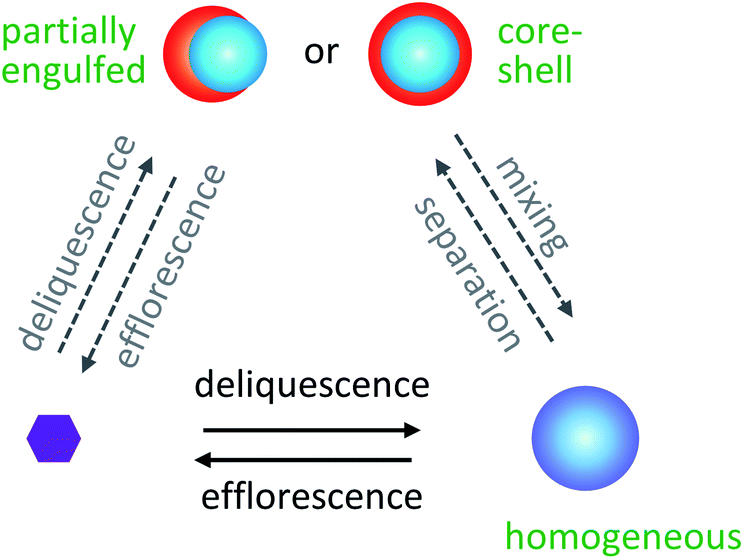 | ||
| Fig. 3 Aerosol particles undergo phase transitions between dry particles, homogeneous wet particles, and phase-separated particles. Purple represents inorganic salts, blue represents the aqueous phase, and red represents the organic phase. Deliquescence refers to the phase transition from a dry particle to a wet particle with increasing RH. Conversely, efflorescence refers to the phase transition from a wet particle to a dry particle with decreasing RH. Adapted with permission from M. A. Freedman, Liquid–Liquid Phase Separation in Supermicrometer and Submicrometer Aerosol Particles, Acc. Chem. Res., 2020, 53, 1102–1110. Copyright 2020 American Chemical Society. | ||
Phase separation is common for aqueous organic aerosol in the atmosphere.158 Lee et al.161 generated nascent SSA in a wave flume using seawater from the California coast, and they found that the core–shell morphology accounted for 31.2% of all particles detected in the 100 nm to 1.8 μm size range. Li et al.162 collected particulate matter from urban, suburban, and rural sites in China; they frequently observed an organic coating on an inorganic core for particles larger than 100 nm. More widespread measurements of ambient particles are needed to establish reproducible trends in phase separation as a function of particle composition, size, and relative humidity.
3.2. Laboratory measurements of aerosol phase
Recent laboratory results have shown that it is actually possible for aerosol particles to have three liquid phases. Kucinski et al.163 reported the first evidence that particles can be kinetically trapped in this configuration. Then, Huang et al.151 systematically investigated the formation of three liquid phases in particles containing primary organic aerosol, secondary organic aerosol, and secondary inorganic aerosol components. They found that particles with O:C < 0.8 could separate into low-polarity organic-rich, high-polarity organic-rich, and aqueous inorganic-rich phases, as shown in Fig. 4.151 Images were obtained by fluorescence microscopy with Nile red dye, which is a convenient probe for liquid–liquid phase separation.164 As reviewed by Laskin et al.,113 imaging techniques like optical microscopy,157,165 electron microscopy,157,166 AFM,167,168 and Raman microscopy169 are all commonly employed to determine the morphology of individual aerosol particles.
 | ||
| Fig. 4 Fluorescence microscopy images of single particles containing proxies for primary organic aerosol (green), secondary organic aerosol (orange), and secondary inorganic aerosol (black) as a function of relative humidity. Compositions are indicated above each row. The coloring is due to trace amounts of Nile red dye. The scale bar applies to all images. Reprinted from ref. 151 under a CC BY-NC-ND License 4.0 (https://creativecommons.org/licenses/by-nc-nd/4.0/). | ||
The particles studied by Huang et al.151 were approximately 50 μm in diameter, so it is not yet clear whether their properties can be extended down to smaller, more atmospherically relevant sizes. Freedman and coworkers157 coupled a special flash freeze flow tube with cryogenic transmission electron microscopy (cryo-TEM) to analyze particles as small as ∼10 nm. Notably, these smaller particles are less likely to undergo liquid–liquid phase separation than larger ones because of the energetic barrier to form a new phase via nucleation and growth. This conclusion is based on proof-of-concept experiments with ammonium sulfate and PEG-400,170 followed by observations in representative ambient particles consisting of ammonium sulfate and organic mixtures, such as α-pinene SOA.163 Moreover, the results are in agreement with work by Petters and Kasparoglu171 for the dependence of viscosity on particle size. More work is needed to determine when it is appropriate to extrapolate physical and chemical properties of supermicron aerosol particles down to smaller sizes.
One inherent limitation to the aerosol phase measurements discussed so far is that the particles must be deposited on a substrate, which may alter their morphology.172–174 Dutcher and co-workers175 have shown that biphasic microfluidics can be used to study morphology, and levitation methods allow for the study of particles in real time without physical contact at all. With aerosol optical tweezers, particles suspended in air are interrogated by Raman spectroscopy, and the resulting whispering gallery modes are fit with Mie theory.176–179 Following this methodology, Reid and coworkers177 demonstrated that particles composed of ammonium sulfate and C6 dicarboxylic acids (2-methylglutaric; 3-methylglutaric; 2,2-dimethylsuccinic) adopt a core–shell structure. Sullivan and coworkers70,159,180 explored how relative humidity affects the morphology of squalane and glycerol droplets coated with α-pinene SOA: particles consisting of polar α-pinene SOA and nonpolar squalane almost always adopt a core–shell structure, whereas particles consisting of α-pinene SOA and glycerol adopt a core–shell structure only at high RH.159 In another contactless technique, Haddrell et al.181 trapped single particles in an electrodynamic balance and determined morphology from forward-scattered light passed through the droplets. Their algorithm can distinguish among homogeneous, core–shell, inclusions, and non-spherical/inhomogeneous morphologies for 10–60 μm diameter droplets with a rapid time resolution of ∼10 ms.181 Finally, optical trapping and Mie scattering can be used for quantitative measurements of morphology: Jones et al.182 determined the thickness of an oleic acid coating on 2 μm diameter silica beads with a precision level of ∼1 nm. At this time, significant challenges remain in applying optical trapping to samples of ambient particles.
3.3. Modeling of phase separation
Most thermodynamic models of aerosol morphology seek to predict phase separation in particles as a function of RH and chemical and physical properties, particularly surface tension.25,158,183–185 MD simulations, in contrast, offer time-resolved predictions of phase separation. For a simple system of mixed water/butanol droplets, Hrahsheh et al.186 verified that MD and DFT gave consistent results for phase separation as a function of concentration. Karadima et al.121 applied MD to model mixtures of ammonium sulfate, water, and organics (cis-pinonic acid; 3-methyl-1,2,3-butanetricarboxylic acid; C20, C24, C30 linear alkanes; or combinations thereof). The organic species initially formed islands on the surface but then transitioned to a core–shell structure with increasing RH and/or organic mass fraction. Yu et al.187 applied machine learning and a neural network to predict phase separation in SOA generated from β-pinene, toluene, and mesitylene that was internally mixed with water and ammonium sulfate. Modeling studies, like laboratory experiments, are moving towards ever more complex systems representative of aerosols in the atmosphere.3.4. Implications of aerosol phase and morphology
The presence of organic species at the surface of aerosol particles has dramatic implications for climate and health. First, the morphology of aerosol particles can affect their optical properties, including their ability to scatter/absorb solar radiation. Fard et al.188 modeled how liquid–liquid phase separation alters the radiative forcing of brown carbon aerosols, where radiative forcing refers to a change in the energy balance of the atmosphere caused by a perturbation, and brown carbon aerosols refer to particles containing chromophores that appear brown in color. When brown carbon aerosol particles transition from well-mixed to phase-separated, their UV scattering cross sections can increase by up to 50%, and their absorption cross sections can decrease by up to 20%.188 On a regional scale, Li et al.189 showed that the CMAQ model could not accurately capture SOA formation over the United States under the simplifying assumption that all particles were homogeneous, in agreement with work by Fierce et al.190 and Wu et al.191 for black carbon. Thus, it is important for more field studies to report aerosol phase in addition to composition.A second way that the presence of organic species at the surface of aerosol particles impacts climate is through cloud formation. The ability of an aerosol particle to act as a cloud condensation nucleus depends on the propensity of water molecules to adsorb to its surface, and the ability of a particle to seed ice cloud formation depends on whether its surface acts as an appropriate template for ice nucleation. Accordingly, two particles with identical chemical composition may have different cloud activation and ice nucleation activities depending on whether each particle is well-mixed, partially engulfed, or core–shell.50,192 Finally, the surface composition of aerosol particles affects the uptake probability of reactive gases in the atmosphere. The properties of cloud droplet activation and ice nucleation will be discussed next in Sections 4 and 5, followed by chemical reactions of surfactant-coated particles in Sections 6 and 7. We will close with a review of ambient observations of aerosol particles with surfactants, films, and coatings in the atmosphere.
4. Cloud droplet activation
4.1. Köhler theory
Surfactants affect surface tension (σ), which is the amount of work required to expand a surface per unit area.193 The value of σ depends on the geometry of the surface. The Young–Laplace equation describes the vapor pressure elevation above a curved liquid surface:| Δp = p − p0 = 2σ/r | (1) |
| p = p0e2σVm/RTr | (2) |
The vapor pressure p depends not only on geometry, but also on composition. Dissolved solutes cause a decrease in vapor pressure through Raoult's law:
| p = xwp0 | (3) |
The Köhler equation incorporates both vapor pressure elevation through the Kelvin effect and vapor pressure lowering through the Raoult effect to show how the saturation vapor pressure of a dilute aqueous droplet depends on its diameter, Dp:
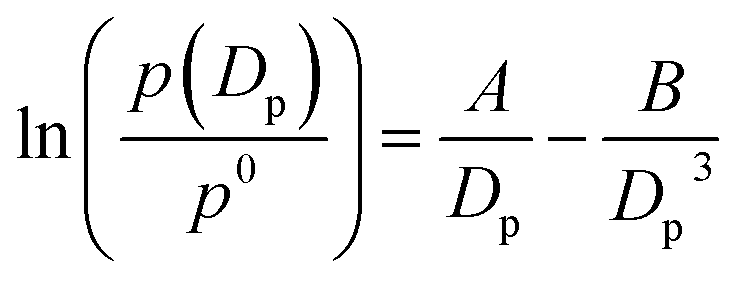 | (4) |
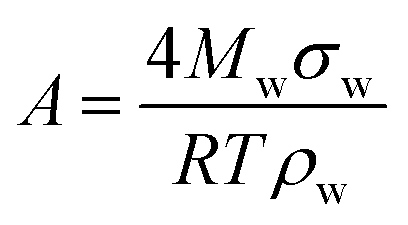 | (5) |
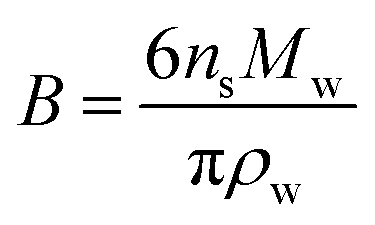 | (6) |
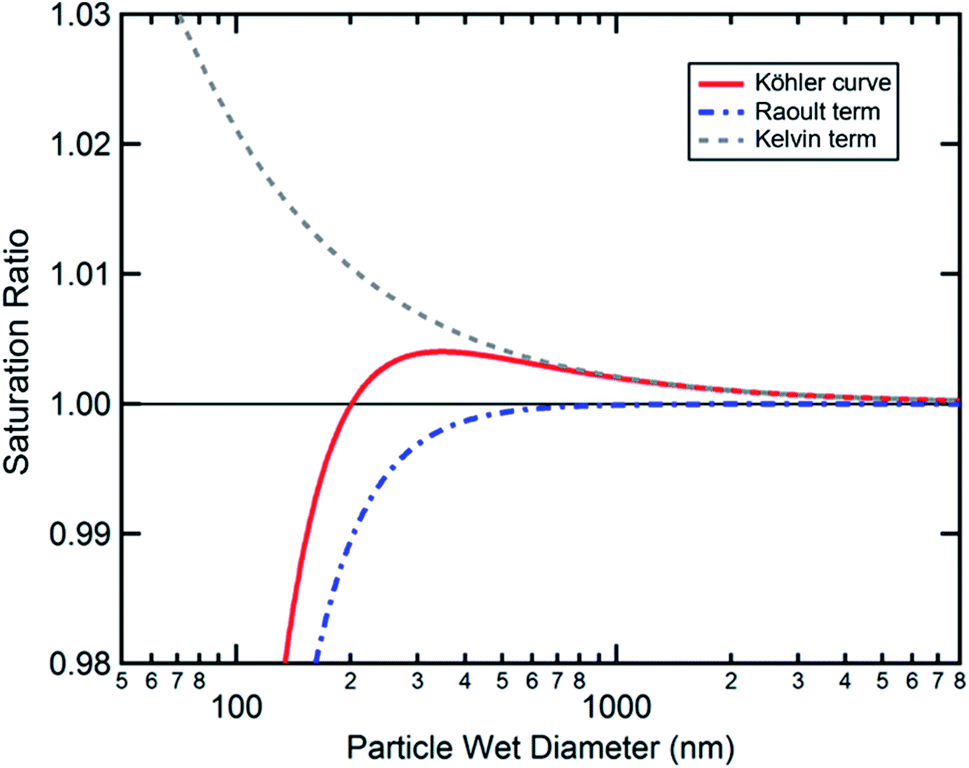 | ||
| Fig. 5 Example Köhler curve (red solid line) for an ammonium sulfate particle with a dry diameter of 50 nm, showing contributions from the Kelvin term (gray dashed line) and Raoult term (blue dash dotted line). Adapted with permission from D. K. Farmer, C. D. Cappa and S. M. Kreidenweis, Atmospheric Processes and Their Controlling Influence on Cloud Condensation Nuclei Activity, Chem. Rev., 2015, 115, 4199–4217. Copyright 2015 American Chemical Society. | ||
According to Köhler theory, surface tension influences the critical droplet diameter and critical supersaturation through the Kelvin effect term. Surfactants are thus predicted to affect cloud droplet activation because they lower the surface tension of water. Indeed, a decrease in surface tension is often used as indirect evidence that organic species are present at the surface of aqueous aerosol particles.1–3 However, because of its simplifying assumptions, the Köhler equation does not apply to most ambient aerosol particles, which consist of a complex mixture of surfactants with highly concentrated solutes. A full treatment of Köhler theory and its extensions is outside the scope of this review. Interested readers are directed to the reviews of McFiggans et al.,194 McNeill et al.,3 and Farmer et al.38 and to several subsequent publications in this area.192,195–206 Here we will focus specifically on (1) developments in measuring and modeling the surface tension of surfactant-coated solutions and (2) studies of hygroscopicity and CCN activity of coated aerosol particles, with an emphasis on work following the 2013 review by McNeill et al.3
4.2. Surface tension techniques
Experimental techniques for measuring surface tension traditionally fall into three categories: force methods (e.g., Wilhelmy plate, DuNoüy ring), shape and pressure methods (e.g., pendant drop/bubble, sessile drop, captive bubble, spinning drop/bubble), and flow methods (e.g., maximum bubble pressure, growing drop).207 Several recent techniques have been designed specifically for measuring the surface tension of atmospherically relevant droplets and aerosol particles.208 For example, Lee and Tivanski209 have reviewed applications of AFM to probe the physical and chemical properties of aerosol particles, including their surface tension.Substrate-free measurements of surface tension are possible with single-particle trapping techniques. Bzdek and co-workers have applied optical trapping to measure the surface tension of picoliter-sized droplets: when two droplets coalesce, the surface tension can be determined from the damped surface oscillations without further perturbing the gas–liquid interface by direct contact.205,210,211 Alternatively, they have shown that they can determine the surface tension of picoliter-sized droplets on a more rapid timescale (∼100 μs) using a microdroplet dispenser with stroboscopic imaging.212 The former technique is appropriate for droplets with a diameter <20 μm, whereas tests of the latter method were restricted to a diameter range of 20–50 μm.
The methods described above have been used to measure surface tension at the gas–liquid interface. In contrast, Dutcher and coworkers213,214 introduced the use of biphasic microfluidics to determine interfacial tension between immiscible aqueous solutions and organic liquids. Knowledge of liquid–liquid interfacial tension is useful in predicting phase separation in multicomponent aerosol particles. Dutcher and coworkers have also adapted the microfluidics technique to combine studies of ice nucleation activity and particle phase state.175
Developing models to accurately predict surface tension and interfacial tension of ambient aerosol particles is challenging because these particles contain many solutes at high concentrations and high ionic strength, and they often consist of multiple phases. Dutcher and coworkers214 extended an activity model to predict surface tension in systems with three or more components. The model reproduces experimental surface tension measurements for solutions containing a variety of solutes, including inorganic salts, non-dissociating organics, and diprotic acids.214 Moreover, the model performs well at realistic concentrations, even exceeding 15 molal in one test with glutaric acid.214,215 E-AIM (Extended Aerosol Inorganics Model)216–218 and AIOMFAC152,153 are two additional thermodynamic models that can be applied to predict surface tension. MD simulations have been used to model surface tension for surfactant solutions containing nonionic surfactants (fatty alcohols,219 polyethylene glycol,220 tetraethyleneglycol221), cationic surfactants (dodecyl trimethyl ammonium chloride219), and anionic surfactants (dodecylsulfate219,222,223). For more information about experimental and computational techniques for measuring and modeling surface tension, see the 2021 review by Mott et al.208
4.3. Hygroscopicity
Hygroscopicity is the ability of a particle to take up water under equilibrium conditions at RH < 100%. In κ-Köhler theory, this phenomenon is described by a single value—κ, the hygroscopicity parameter:224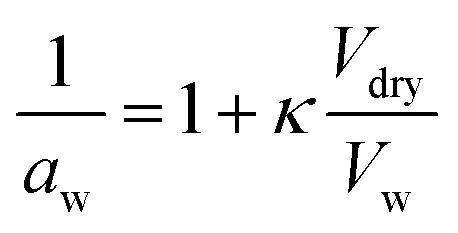 | (7) |
The effects of surfactants on hygroscopicity are not straightforward. They have been shown to both enhance and hinder water uptake.3,225–227 Indeed, water uptake by aerosol particles is highly dependent on phase and mixing state.228–232 In a relatively simple system of fatty acids on sea salt particles, Nguyen et al.233 found that palmitoleic acid (C16) and oleic acid (C18), which are liquids at room temperature, did not impede water uptake, whereas the analogous saturated fatty acids, palmitic acid (C16) and stearic acid (C18), which are solids at room temperature, did hinder water uptake. The authors attribute the opposing effects to phase. These experiments were conducted with fatty acids on artificial sea salt. For comparison, Miñambres et al.234 tested the hygroscopicity of fatty acids on single-component salt solutions: NaCl, NaBr, NaI. Water uptake depended not only on the identity of the fatty acid (hexanoic, octanoic, or lauric acid), but also on the halide anion.
For a suite of other surface-active acids, Ruehl and Wilson195 measured the hygroscopic growth of ammonium sulfate particles coated with monolayers of C3–C10 dicarboxylic acids, cis-pinonic acid, oleic acid, lauric acid, and myristic acid. The relationship between the growth factor and the organic aerosol volume fraction was not linear, as shown for two sample acids in Fig. 6.195 In fact, the configuration of the dicarboxylic acids at the air–water interface depended on parity: diacids with an even number of carbon atoms occupied ∼40 Å2 per molecule at the surface, whereas diacids with an odd number of carbon atoms occupied only ∼20 Å2 per molecule. These footprints, determined from a two-dimensional van der Waals model, are consistent with the configurations illustrated in Fig. 6, in which the even diacids are folded such that both carboxylic acid head groups are solvated in the aqueous phase, while the odd diacids are more closely packed with only one carboxylic acid group in the aqueous phase.195 Such differences in surface structure explain why even dicarboxylic acids are more hygroscopic than odd dicarboxylic acids at equivalent bulk concentrations. This work emphasizes once again the importance of applying a molecular-level view to interpret and predict trends in hygroscopicity.
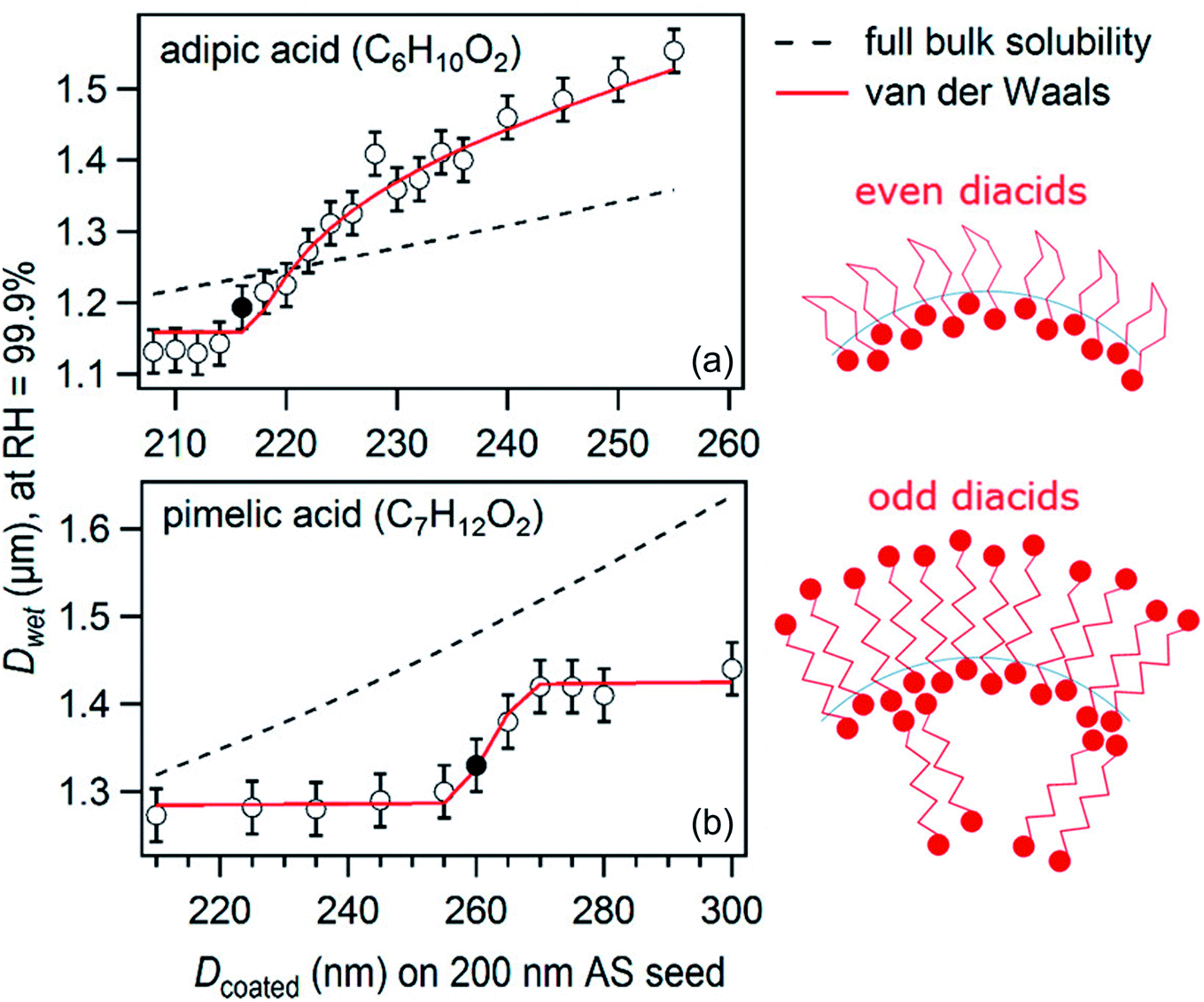 | ||
| Fig. 6 Plot of droplet diameter (Dwet) at 99.9% relative humidity for 200 nm diameter ammonium sulfate particles coated with varying amounts of (a) adipic acid or (b) pimelic acid. The filled circles mark the threshold for droplet growth, or the first coating thickness at which there is a statistically significant difference between the growth factor for the pure ammonium sulfate particle and the growth factor for the coated particle. The black dashed lines show the linear relation that would be expected if the particles were homogeneous (i.e., if the organics did not partition to the surface). The red lines show predictions from a two-dimensional van der Waals model by Ruehl and Wilson.195 The cartoons suggest how adipic (even) and pimelic (odd) dicarboxylic acid molecules are arranged at the air–water interface. Adapted with permission from C. R. Ruehl and K. R. Wilson, Surface Organic Monolayers Control the Hygroscopic Growth of Submicrometer Particles at High Relative Humidity, J. Phys. Chem. A, 2014, 118, 3952–3966. Copyright 2014 American Chemical Society. | ||
Interpreting the hygroscopicity of ambient aerosol particles from the field is challenging not only because individual particles may be phase-separated, but also because populations of particles are externally mixed. Kroflič et al.235 collected aerosol particles in the city of Ljubljana, Slovenia, but they could not predict hygroscopic growth of surfactant-coated aerosol particles without size-resolved information. Hu et al.236 measured the CCN activity of aerosol particles collected at a mountain near Beijing, China and compared the experimentally determined hygroscopicity values to thermodynamic predictions based on bulk composition. When the particulate matter came from fresh anthropogenic emissions, they needed to consider size-resolved composition to accurately predict κ. In contrast, when the particulate matter came from aged anthropogenic sources, they needed to consider surface tension lowering by surfactants to accurately predict κ. Both size and composition should be incorporated into models for hygroscopicity and CCN activity of ambient aerosols.
Work by Schill et al.237 and Cravigan et al.238 on water uptake to SSA has further highlighted the need for single particle measurements of hygroscopicity. Schill et al.237 observed a wide range in κ values for SSA generated from seawater collected in the North Atlantic Ocean, suggesting that particles varied in composition from pure salt to pure organic. Similarly, Cravigan et al.238 were unable to find a significant correlation between organic content and water uptake for SSA generated from seawater from the Southern Ocean during a phytoplankton bloom. Hendrickson et al.239 actually posit that the salts, not the organics, control κ in SSA.
Advances in single particle techniques allow quantitative measurements of hygroscopic growth factors for individual aerosol particles.240,241 Estillore et al.242 and Lee et al.243 used AFM to study water uptake on SSA particles with a core–shell morphology. They generated these particles in a wave flume with seawater collected off the coast of California, with an induced phytoplankton bloom. The organic coatings were found to inhibit water uptake, especially at low RH.243 Li et al.232 applied cryo-TEM and an individual particle hygroscopicity setup244 to measure morphology and hygroscopic growth factors for particles collected at three sites: a forest in northeastern China, urban Beijing, and urban Hangzhou. At 90% RH, growth factors ranged from 1.14–1.32. The authors attributed this wide range to variations in the thickness of the organic shell on the phase-separated particles; measurements on lab-generated aerosol particles support this conclusion.232 Therefore, because water uptake depends so strongly on surface morphology, ensemble averages for hygroscopic growth factors and κ are not meaningful for externally mixed populations of ambient aerosol particles.
4.4. CCN activation
Cloud activation, or the formation of a cloud condensation nucleus, occurs at the tipping point of the Köhler curve (Fig. 5) when the droplet diameter reaches the critical diameter or the humidity reaches the critical saturation. Because surface tension and hygroscopicity depend on surface morphology, so too does cloud droplet activation. Ovadnevaite et al.192 showed that phase separation matters for the relative contributions of the Kelvin term and the Raoult term in Köhler theory. Fig. 7 models how changes in surface morphology during droplet growth affect surface tension and therefore the shape of the Köhler curve.192 As shown in Fig. 7a, a particle at 288.15 K consisting of a 1:1 mass ratio of organic matter and sulfuric acid with an initial dry diameter of 41 nm is expected to undergo CCN activation at a critical supersaturation of 0.88% and a critical diameter of 164 nm, assuming that the surface tension is constant and equal to that of pure water (σ = 72 mN m−1). In contrast, if the surface tension is assumed to change as a function of particle size and phase, then CCN activation occurs at a much lower critical supersaturation (0.50%) and a much larger critical diameter (311 nm). The changing surface tension clearly has a dramatic effect on the shape of the Köhler curves.
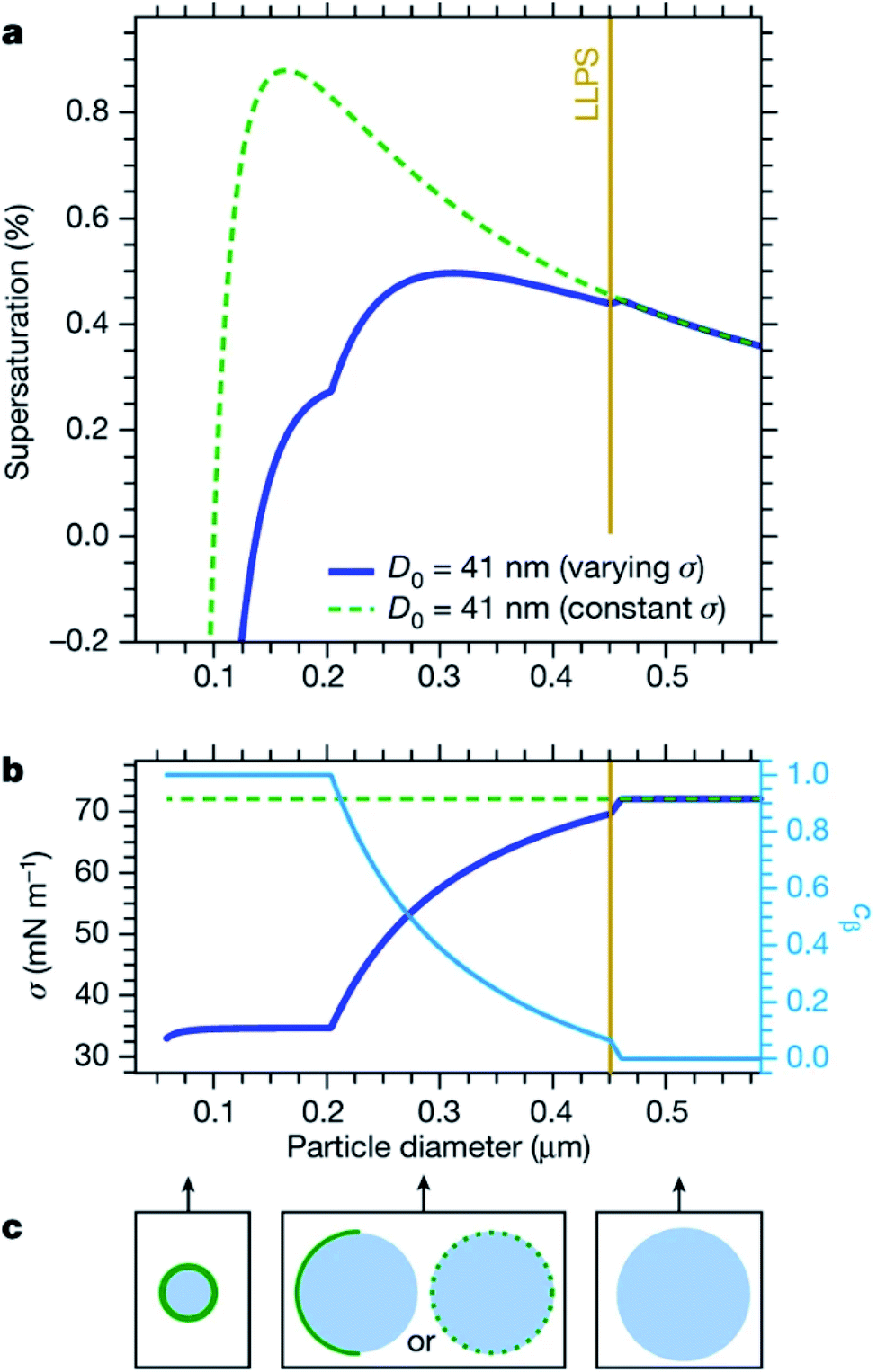 | ||
| Fig. 7 Thermodynamic equilibrium modeling of an aerosol particle consisting of a 1:1 mass ratio of organic matter to sulfuric acid at 288.15 K and relative humidity of RH = 94–101%. (a) Predicted Köhler curves of equilibrium supersaturation vs. particle diameter. The yellow vertical line marks the particle diameter at which liquid–liquid phase separation (LLPS) occurs. The dashed green curve shows the predicted Köhler curve for a droplet with a dry diameter of D0 = 41 nm and a constant surface tension equal to that of pure water (σ = 72 mN m−1). The solid blue curve shows predictions for a droplet with a dry diameter of D0 = 41 nm but a varying surface tension dependent on the size and phase of the particle. (b) The corresponding changes in surface tension as a function of particle diameter, with the predicted surface coverage of the organic layer (cβ) shown in light blue. (c) Cartoons of surface coverage as a function of particle diameter. Reprinted by permission from Springer Nature, Nature, Surface tension prevails over solute effect in organic-influenced cloud droplet activation, J. Ovadnevaite, A. Zuend, A. Laaksonen, K. J. Sanchez, G. Roberts, D. Ceburnis, S. Decesari, M. Rinaldi, N. Hodas, M. C. Facchini, J. H. Seinfeld and C. O′ Dowd, Copyright 2017. | ||
The cartoons in Fig. 7c demonstrate that as the diameter of a droplet grows, organic coverage at the surface becomes too patchy to significantly alter surface properties.192,198 Accordingly, Forestieri et al.245 observed that even though coating NaCl droplets with fatty acids significantly lowered the surface tension, the surfactants had minimal effect on the critical supersaturation for CCN activation. They conclude that fast growth of the droplets approaching critical size increased the molecular area of the surfactants enough that there was no significant change in the critical supersaturation.
Because κ-Köhler theory fails under the assumption that surface tension is constant throughout droplet growth,192,198,204,246,247 Davies et al.204 call for careful experimental measurements of surface tension for particles of varying organic fractions at constant dry diameter; this information is needed to correct for changes in the bulk/surface partitioning of organic species as a droplet takes up water. Bzdek et al.205 report just such a dependence of surface tension on droplet size for mixtures of NaCl, glutaric acid, and the detergent Triton X-100. They compared the surface tension of 14–18 μm diameter droplets (measured by holographic optical tweezers) versus the surface tension of bulk solutions of equivalent composition (measured by Wilhelmy plate). The confining effect of surface area-to-volume ratio was dramatic: the surface tension of the picoliter-sized droplets was up to 35 mN m−1 greater than that of the bulk solution. In other words, the surface tension depression was significantly less than predicted from macroscopic measurements. This work implies that surfactant concentrations in ambient particles must reach tens to hundreds of millimolar in order to appreciably affect cloud droplet activation.205 As shown by ambient measurements (Section 8), such high levels are possible in some locations.
Surface-active species in ambient particles are not limited to the classic amphiphilic species used so commonly in laboratory and modeling studies.192 In a laboratory study with particles containing NaCl and fulvic acid, Lin et al.248 observed the typical pull between the Kelvin effect and the Raoult effect. As fulvic acid partitioned to the surface, the decrease in surface tension contributed toward activation through the Kelvin effect, but the lower bulk concentration of fulvic acid limited vapor pressure lowering by the Raoult effect. There is still considerable uncertainty surrounding how best to extend κ-Köhler theory to even relatively simple mixtures of model aerosol constituents. Much more experimental and modeling research is needed in this area before we can readily predict and interpret CCN activation by ambient aerosol particles.
4.5. Implications
Aerosol-cloud interactions present the greatest uncertainty in modeling radiative forcing changes for Earth's climate.37,249 Thus, further research on CCN activation is crucial for our ability to confidently model changes in global temperature through general circulation models. Lowe et al.246 incorporated surface partitioning into a cloud parcel model to test the implications of aerosol composition, size distribution, water availability, and updraft velocity on aerosol–cloud interactions. The authors used proxies for marine and boreal aerosol based on measurements at Mace Head, Ireland and Hyytiälä, Finland, respectively. When switching from standard Köhler theory (for homogeneous particles) to a compressed surface film model (for particles with 100% surface partitioning), the authors roughly estimate that global radiative forcing could change as much as +11.5 W m−2 over sea and −0.7 W m−2 over land simply due to surfactant effects.246 These changes are dramatic given that the Sixth Assessment Report of the Intergovernmental Panel on Climate Change quotes a combined effective radiative forcing of between −1.92 W m−2 and −0.21 W m−2 for aerosol–cloud and aerosol–radiation interactions.37 Clearly more work is needed to parameterize how surface-active species affect cloud formation and therefore climate.5. Ice nucleation
Aerosol particles not only facilitate nucleation of liquid cloud droplets, they also contribute to the formation of ice clouds and mixed-phase clouds in the atmosphere. Aerosol particles that facilitate heterogeneous freezing are known as ice nucleating particles (INPs). Good INPs are larger than ∼0.1 μm in diameter and insoluble in water, but water molecules should readily adsorb to their surface. Furthermore, their surface structure should roughly match the lattice structure of ice, or they should have local active domains where ice nucleation can begin.41,250 The majority of INP studies thus far have focused on mineral dust251,252 and biological aerosol particles,253–256 such as pollen and bacteria.257,258 Organic matter, and surfactants in particular, affect heterogeneous freezing by changing the particle morphology, the glass transition temperature (Tg, the temperature at which a particle becomes so viscous that it transitions from liquid-like to solid-like), and the full deliquescence relative humidity (FDRH, the point at which the water activity of the organic particle reaches equilibrium with the relative humidity).41,259 Surfactants, films, and coatings play an outsized role in ice nucleation because surface structure is so important in determining IN activity.The field of ice nucleation has a long history of studying model surfactant systems, especially monolayers of long-chain saturated alcohols.3,260–278 Outside the laboratory, several field studies have identified ambient INPs containing surfactants. Hiranuma et al.279 collected ice residuals in Alaska that contained organic coatings on inorganic or soot cores. The organic shell consisted mainly of carboxylic acids, presumed to originate from oxidative aging of the aerosol particles. Similarly, Kupiszewski et al.280 found that ice residuals from Jungfraujoch in the Swiss Alps contained soot particles with organic coatings. In other remote field campaigns, China et al.281 observed that all INPs collected in the Azores Islands contained organic coatings, and Lata et al.282 detected organic coatings on some INPs in the free troposphere at Pico Mountain Observatory in the North Atlantic Ocean. Boose et al.283 hypothesized that the ice nucleation activity of desert dust particles collected from the Etosha salt pan in Namibia was due to biogenic organic matter, such as proteins and cellulose. Experimental measurements258 of both ambient and lab-generated INPs are challenging because of contamination concerns: trace gases that readily adsorb to aerosol particles can alter their surface properties,41 and the substrates used for measurements may also alter INP activity.278 Here we will introduce insights gained about surfactant-coated INPs through recent MD simulations, followed by a summary of measurements published after the comprehensive review by Knopf et al.41 in 2018.
5.1. A molecular-level understanding of ice nucleation
Because experimental measurements of ice nucleation are so sensitive to trace contaminants, much of our understanding about heterogeneous freezing comes from MD simulations. Qiu et al.277 present a modeling study of heterogeneous ice nucleation on organic surfaces, consisting of a series of long-chain alcohols and fatty acids. Alcohols were already known to be significantly better ice nucleators than fatty acids of equivalent chain length,260 and Qiu et al.277 were able to explain this phenomenon through molecular properties. First, they confirmed that freezing is more efficient when the organic surface has a good lattice match to ice and when surface fluctuations are minimized. Second, they correlated freezing efficiency with the free energy of binding of the organic monolayer to ice. Fig. 8 shows how alcohol headgroups can create ordered domains to initiate ice nucleation.277 In the MD snapshots, freezing begins when water molecules form hydrogen bonds to surface OH groups. Thus, MD simulations provide a molecular-level explanation for the observation that not all surfactant-coated aerosol particles act as INPs.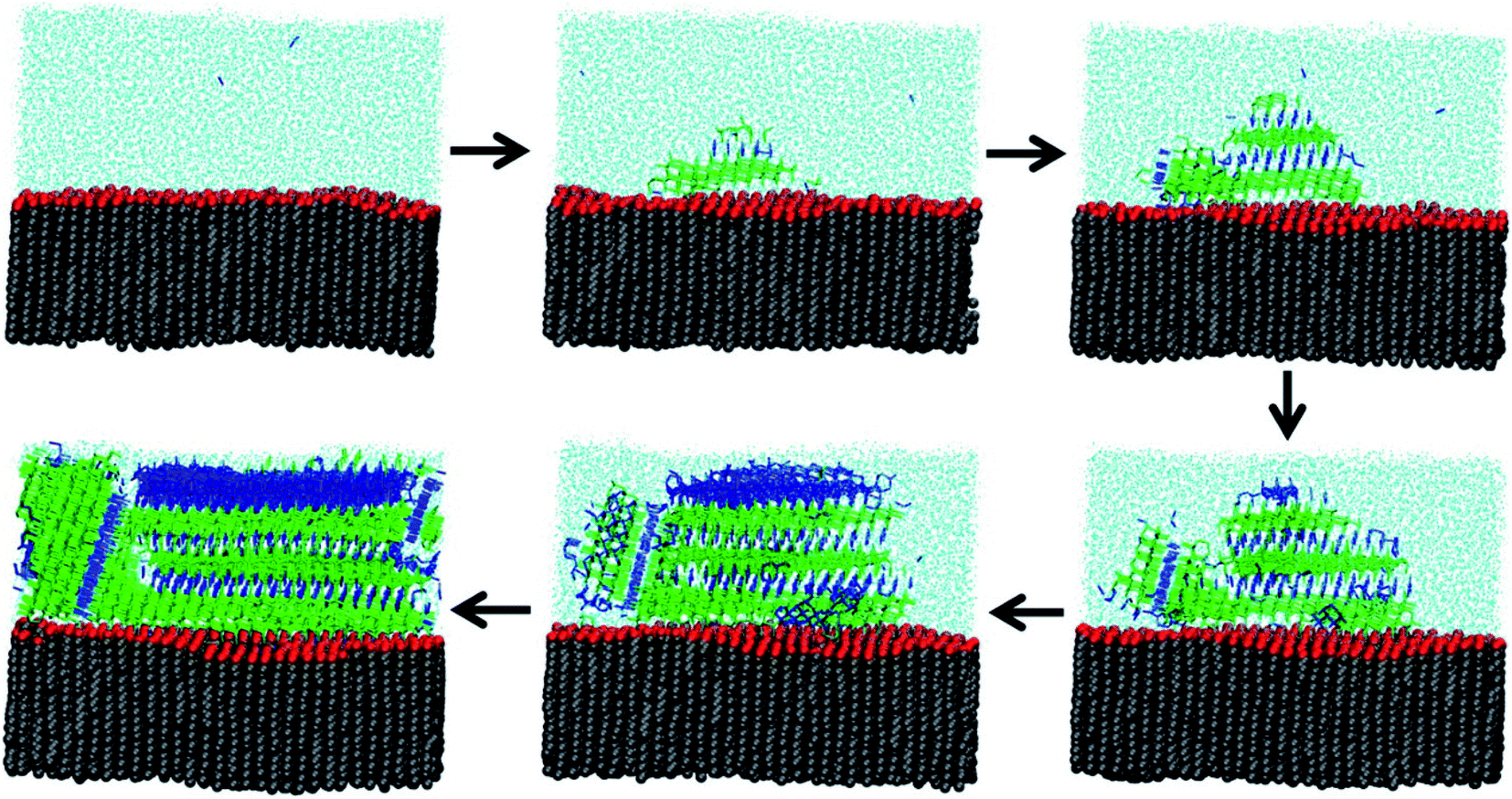 | ||
| Fig. 8 Molecular dynamics snapshots of ice nucleation at the surface of a triacontanol (C30H61OH) monolayer. The carbon backbone is shown in gray, the OH headgroup in red, liquid water molecules in light blue, hexagonal-ordered water in blue, and cubic-ordered water in green. Reprinted with permission from Y. Qiu, N. Odendahl, A. Hudait, R. Mason, A. K. Bertram, F. Paesani, P. J. DeMott and V. Molinero, Ice Nucleation Efficiency of Hydroxylated Organic Surfaces Is Controlled by Their Structural Fluctuations and Mismatch to Ice, J. Am. Chem. Soc., 2017, 139, 3052–3064. Copyright 2017 American Chemical Society. | ||
5.2. Ice nucleation by terrestrial SOA
In general, terrestrial SOA particles make for poor INPs. The discussion here will focus specifically on terrestrial aerosol particles with organic films and coatings. Recent work by Kanji et al.284 showed that coating desert dust particles with α-pinene SOA has no significant effect on ice nucleation across a range of particles sizes (0.01–3 μm) and coating thicknesses (3–60 nm). Further evidence is provided in Knopf et al.41 and references therein. However, the ability of organic aerosol particles to serve as INPs is not static over the lifetime of a particle. Jahl et al.285 generated biomass burning aerosol (BBA) from the combustion of giant cutgrass, Ponderosa pine needles, sawgrass, and birch and fatwood logs. The resulting SOA was then aged in a laboratory chamber through the following four mechanisms: (1) the evolution of time without any external perturbations, (2) oxidation by OH in the presence of NOx, (3) passage through a thermodenuder followed by OH photo-oxidation, or (4) dark ozonolysis. Jahl et al.285 observed three competing effects on ice nucleation activity. First, if aging caused the organic coating to evaporate, then ice nucleation activity increased because ice-active sites on the mineral surface were exposed. Second, if oxidative aging caused the formation of highly oxygenated species that readily dissolved in the aqueous aerosol particles, then ice nucleation activity once again increased because active sites on the mineral surface were exposed. Finally, if oxidative aging caused organic species to condense at the particle surface, then ice nucleation activity decreased because active sites on the mineral surface were blocked. Bertozzi et al.286 similarly inferred that aging enhances the ice nucleation ability of α-pinene/ammonium sulfate SOA due to changes in particle morphology and surface composition, and Boose et al.283 found that some desert dust samples become more ice nucleation-active after heating, possibly because volatile organic compounds (VOCs) evaporated from the particle surface. Piedehierro et al.287 further highlight the importance of relative humidity and water diffusivity for ice nucleation by biogenic SOA. Together, these results indicate that atmospheric conditions and particle lifetime matter when considering the ability of organic-coated aerosol particles to act as INPs.5.3. Ice nucleation by marine aerosol
Marine aerosol is likely to contain surfactants from phytoplankton and other species in the sea surface microlayer.288–290 Two experimental approaches have been employed to assess ice nucleation by SSA: in the top-down approach, genuine SSA is generated from seawater and evaluated for its ability to facilitate ice nucleation, whereas in the bottom-up approach, model systems of environmentally-relevant surfaces are prepared and tested in the laboratory. Both approaches provide valuable insights into how surfactants impact ice nucleation by marine aerosol.McCluskey et al.291 and Trueblood et al.292 followed the top-down model by generating SSA from genuine seawater samples. First, McCluskey et al.291 collected seawater from La Jolla, California, initiated a phytoplankton bloom, generated SSA with a marine aerosol reference tank, and collected the aerosol particles for ice nucleation tests. More than 30% of the resulting ice crystal residuals had organic coatings, as seen by SEM/EDS (energy dispersive spectroscopy). Trueblood et al.292 followed a similar procedure to generate SSA from Mediterranean seawater using a plunging aquarium apparatus. They classified INPs as either warm (freezing at T ≥ −22 °C) or cold (freezing at T < −22 °C). Interestingly, the cold INPs were linked to SSA that contained water-insoluble organic carbon, likely surfactants from phytoplankton in the sea surface microlayer. Both studies highlight the key contributions from surfactants to ice nucleation.
In complementary bottom-up experiments, Perkins et al.278 tested the freezing of mixed films of C16 and C18 fatty acids and alcohols, selected to represent surfactants in SSA. The authors varied the film composition, deposition amount, and salt concentration. As shown by the red and yellow data points in Fig. 9, ice nucleation occurred for all mixing ratios except the pure fatty acids. The fatty alcohols contributed to ice nucleation for T < −20 °C, in approximate agreement with measurements of ambient SSA.292 By visually examining ice nucleating domains with Brewster angle microscopy, Perkins et al.278 proposed that ice nucleation could occur in fatty acid-containing films through the refreezing effect, in which films are frozen, thawed to room temperature, and then refrozen. This cycle traps the surface in an ordered configuration that is favorable for ice nucleation. Alternatively, ice nucleation through the refreezing effect could proceed through the formation of a 3D solid phase. At this time, there is insufficient evidence to discriminate between these hypotheses explaining ice nucleation in SSA containing fatty acid surfactants.
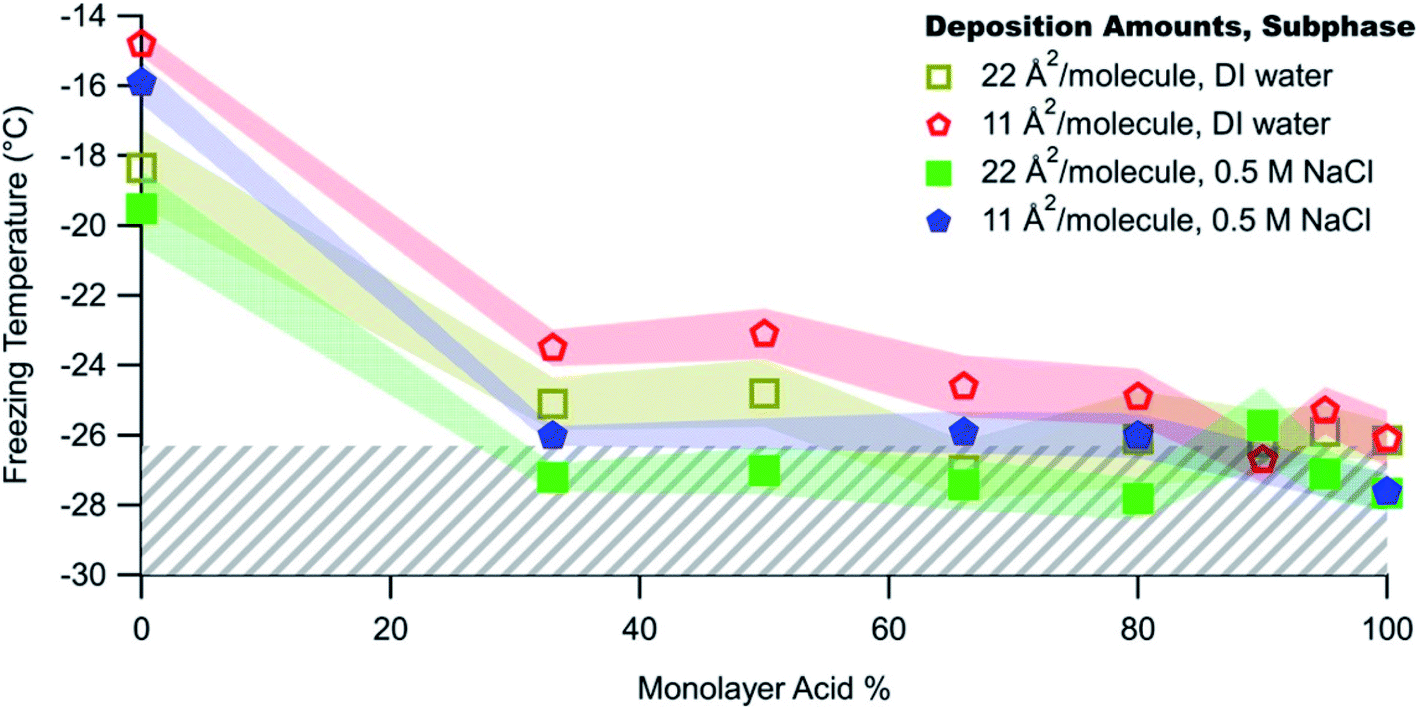 | ||
| Fig. 9 Freezing temperatures of mixed monolayers of C16 alcohols and fatty acids ranging from pure hexadecanol (C16H33OH) to pure palmitic acid (C15H31COOH) with and without salt. Greater deposition amounts correspond to smaller mean molecular areas and more tightly packed films. The colored shading marks the 95% confidence interval of 50% sample freezing. The hatched gray area for T < −26.3 °C shows the 95% confidence interval of 50% control freezing. Data have been corrected for freezing point depression in salt solutions due to colligative properties. Reprinted with permission from R. J. Perkins, M. G. Vazquez de Vasquez, E. E. Beasley, T. C. J. Hill, E. A. Stone, H. C. Allen and P. J. DeMott, Relating Structure and Ice Nucleation of Mixed Surfactant Systems Relevant to Sea Spray Aerosol, J. Phys. Chem. A, 2020, 124, 8806–8821. Copyright 2020 American Chemical Society. | ||
To better reproduce the conditions in ambient SSA, Perkins et al.278 added salt to their model surfactant films. The salt ions disrupted the surface structure of the films and thereby inhibited ice nucleation, as shown in Fig. 9. This result supports observations that marine aerosol particles are generally poor INPs.278 However, just as the ice nucleation ability of BBA evolves over its lifetime (as noted by Jahl et al.285), so too does the ice nucleation ability of SSA. As a particle takes up water, ice nucleation should become increasingly favorable because the salt concentration decreases. As the particle continues to take up water, ice nucleation will then become less favorable because the surfactant coverage on the growing droplet will be too sparse. In summary, the defining characteristics of good marine INPs are large diameter, low salt concentration, and adequate surfactant coverage.
5.4. Future needs
More work is needed to bridge observations of ice nucleation in the field with increasingly complex systems in modeling studies and laboratory experiments. In particular, measurements and models should be made for classes of surfactants beyond the traditionally studied alcohols and fatty acids. For example, surface-active per- and polyfluoroalkyl substances (PFAS)293 likely affect ice nucleation of aerosol particles.294 Interestingly, PFOA (perfluorooctanoic acid, a C8 carboxylic acid) can nucleate freezing at −16 °C, but PFOS (perfluorooctane sulfonic acid, a C8 sulfonic acid) is ineffective due to the bulky, disordered sulfonate head groups.295 Furthermore, pH matters: neutral PFOA allows for ice nucleation, but anionic PFOA does not.295 In addition, further work is needed to extrapolate room-temperature findings to the cold conditions at high altitudes in the atmosphere, where aerosol particles are glassy and viscous. And finally, more field measurements are needed to determine the composition of ice residuals and INPs in ambient aerosol.6. Multiphase reactions
The geometry of the gas–liquid interface makes for a unique reaction environment.43–45,296 Consider the approach of a molecule from the gas phase to a liquid surface, as illustrated in Fig. 10. The probability that the gaseous molecule enters the bulk liquid is characterized by the mass accommodation coefficient α, which ranges from zero to one. Mass accommodation depends on diffusion of the molecule through the gas phase, adsorption and desorption at the surface, and diffusion and solvation within the liquid phase.44,46 Shiraiwa and Pöschl297 recommend additional caution when treating the mass accommodation of gases on viscous particles because failing to account for penetration depth, volatility, and bulk-phase diffusion can have dramatic effects on the value of α. In addition to the parameters encompassed by mass accommodation, an impinging gas molecule can also undergo reaction at the surface or within the bulk. The combination of all these processes can be characterized by the uptake coefficient γ, the fraction of collisions of the incident gas molecule that lead to reaction. It may seem that γ should range from zero to one. However, it is possible for γ to be greater than one if the collision of one gas-phase molecule causes loss of more than one particle-phase molecule through secondary chemistry, especially radical–radical chain reactions.298 Surfactants create particularly rich reaction systems at the gas–liquid interface because surfactants can impede reactions through physical blocking, but they can also facilitate new reaction pathways if they participate in interfacial reactions. Here we will focus on studies of mass accommodation and uptake at surfactant-coated interfaces published after the comprehensive reviews by Donaldson and Vaida,2 Donaldson and Valsaraj,299 McNeill et al.,3 and Chapleski et al.300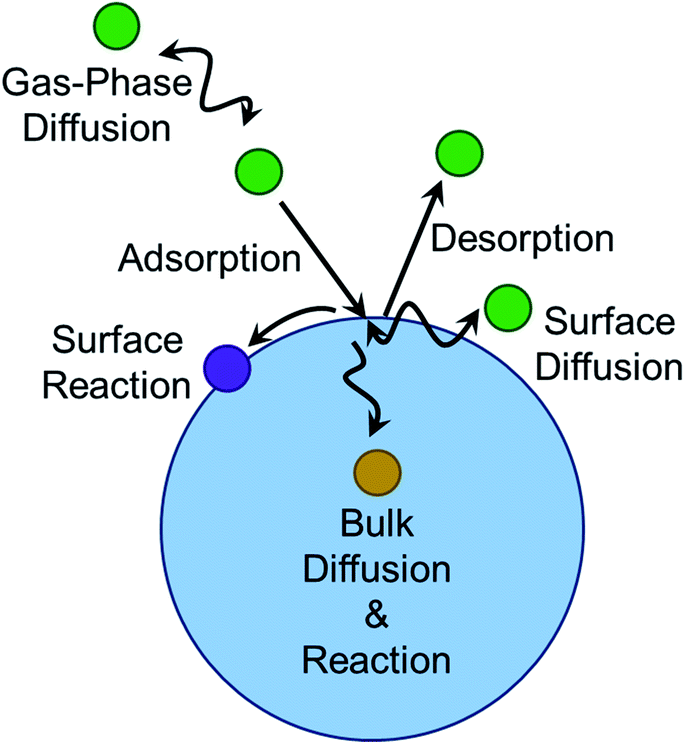 | ||
| Fig. 10 Multiphase reaction pathways. Molecules diffusing in the gas phase that encounter an aerosol particle may adsorb to the surface, diffuse within the surface region, react within the surface region, diffuse into the bulk, and react in the bulk. The impinging gas-phase molecule can also desorb without reacting, and products can remain at the surface, remain in the bulk, or desorb into the gas phase. | ||
6.1. Water uptake
The uptake of water to aerosol particles is important because condensational growth contributes to the formation of cloud condensation nuclei. In contrast, evaporation of water prevents a particle from achieving the critical supersaturation needed to seed cloud formation. By the principle of microscopic reversibility, the mass accommodation coefficient of water should be equal to its evaporation coefficient.43,301 Thus, studies of water evaporation provide insight into mass accommodation of water on surfaces, and vice versa.In the case of long-chain species like octadecanol (C18 linear fatty alcohol), surfactants can simply block the transport of water molecules across the air–water interface.97,228 Fellows et al.302 relied on modeling to present a nuanced perspective that beyond posing a physical barrier, surfactants affect water evaporation by altering surface roughness. Of course, the evaporation and adsorption of water across an interface depend on the structure of the molecules present there. Ergin and Takahama303 compared the mass accommodation coefficient of water on aqueous droplets coated with a series of alcohols: 1-decanol (C10, linear); iso-decanol (C10, one-level branching); iso-undecanol (C11, one-level branching); 3,7-dimethyl-1-octanol (C12, two-level branching); and 5,9-dimethyl-1-decanol (C12, two-level branching). MD simulations revealed a free energy barrier of 0.6 to 15 kJ mol−1 for mass accommodation of water on the alcohols with straight chains or one level of branching. Accommodation was barrierless for the alcohols with two levels of branching. The propensity of water molecules to scatter rather than stick increased with increasing carbon density in the alcohol monolayer, revealing the importance of surface structure.
Miles et al.304 measured the evaporation of water across binary fatty acid films, composed of mixtures of tridecanol + pentadecanol or tetradecanol + hexadecanol. The evaporation coefficient could initially be treated as a linear combination of the evaporation coefficients of the individual components according to their mole fractions. However, droplets shrank as evaporation progressed, and shorter-chain, more soluble alcohol molecules diffused into the bulk while longer-chain, less soluble alcohol molecules remained at the surface. Furthermore, the evaporation coefficient depended on the relative humidity because the concentration gradient in the gas phase affected heat transfer when water molecules evaporated. The cooling, which was more pronounced at low RH, caused ordering of the surface monolayer, so gas transport across the interface was inhibited.
Miles et al.304 further tested water evaporation from sucrose droplets coated with long-chain alcohols. The initial evaporation rate decreased when surfactants were present, with greater decreases for longer-chain surfactants. However, the presence of the long-chain alcohols ultimately caused more water to evaporate by the time equilibrium was reached. When Johansson et al.305 studied mass accommodation of water on butanol, they found two timescales for trapping–desorption: some water molecules rapidly desorbed within picoseconds, whereas others desorbed after a delayed period on the millisecond timescale. Unlike the room-temperature experiments of Miles et al.,304 the molecular beam experiments by Johansson et al.305 were conducted using hyperthermal water molecules (28 kJ mol−1) on a cold butanol surface (160–240 K). Nevertheless, the two cases together reveal that surfactants have both kinetic and thermodynamic effects on water evaporation, in keeping with earlier results for water on simple organic surfactants.195,306–311 Water uptake, the reverse of water evaporation, has been discussed in greater detail in Section 4.3 because of its relevance to cloud droplet activation.
6.2. Nonreactive uptake
Shaloski et al.312 performed experimental measurements of the dissociation of gas-phase hydrochloric acid in a model system: the surface of salty glycerol (C3H8O3, a triol) with and without the anionic surfactant dodecyl sulfate, CH3(CH2)11SO4−. The uptake of DCl (deuterated hydrochloric acid) decreased as the dodecyl sulfate surface coverage increased, dropping from a 70% entry probably in the absence of surfactants to just 11% when the dodecyl sulfate surface concentration was 1.8 × 1014 cm−2, or ∼34% of a monolayer. The authors attributed the decrease to physical blocking of incident DCl molecules by the surfactant chains, which prevent DCl from dissolving into the glycerol solution. In this relatively simple system, the surfactant was nothing more than a physical barrier.Li et al.313 present an elegant MD study of nonreactive uptake of water, ozone, and sulfur dioxide (SO2) onto water droplets coated with benzoic acid (C6H5COOH, aromatic acid) or lauric acid (C12 linear fatty acid). The reaction of benzoic acid with ozone and the hydrolysis of sulfur dioxide are roughly 106 to 109 times slower than the mass accommodation and solvation processes and thus were not considered here. Fig. 11 shows the free energy profiles for all three gases determined by moving the gas molecule from the gas phase to the center of the water droplet.313 Roughly monolayer coverage of the organic acids caused α for water to decrease from ∼1 in pure water to ∼0.75 with benzoic acid and ∼0.18 for lauric acid. For all cases, the minimum in the free energy profile remained within the water droplet. In contrast, the minimum in the free energy profile for ozone shifted toward the air surface, and α increased from ∼0.05 in pure water to ∼0.48 with benzoic acid and ∼0.36 with lauric acid. Finally, opposing effects were observed for SO2. The lauric acid layer caused α to decrease relative to pure water (∼0.81 vs. ∼0.90, respectively), whereas the benzoic acid layer caused α to increase slightly to ∼0.91. Accordingly, the minimum in the free energy profile was deeper within the organic layer for the lauric acid system but closer to the air surface for the benzoic acid system. The authors do not directly explain why lauric acid and benzoic acid have different and sometimes opposing effects, but they do note that lauric acid molecules have longer alkyl chains and are floppier than benzoic acid molecules. Supplementing experimental measurements (like those of Shaloski et al.312) with MD simulations (like those of Li et al.313) would provide comprehensive molecular-level insights into the complicated environment at the gas–surfactant interface.
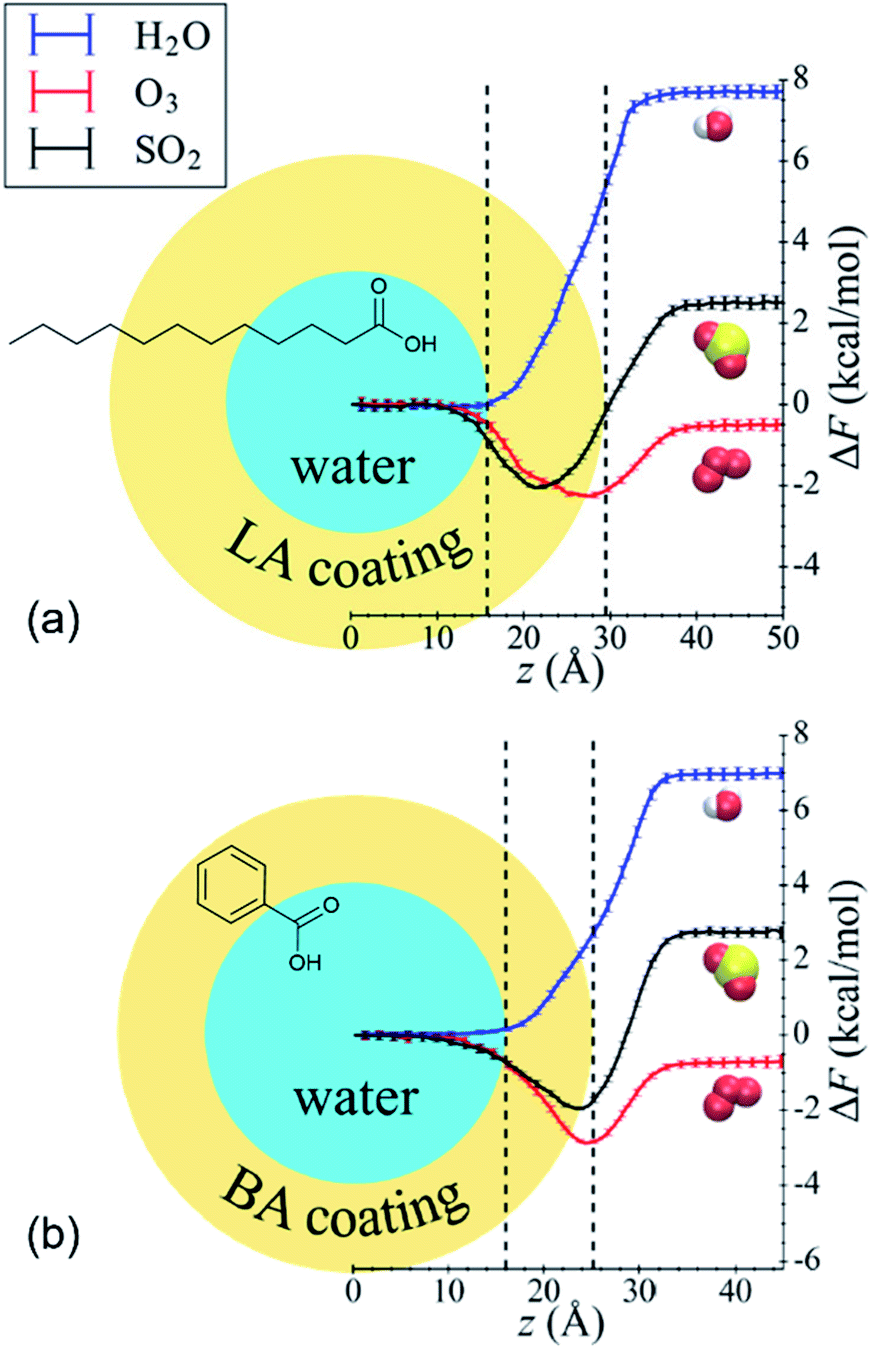 | ||
| Fig. 11 Free energy profiles for the uptake of gas-phase water (blue curves), ozone (red curves), and sulfur dioxide (black curves) by water droplets coated with (a) lauric acid (LA) or (b) benzoic acid (BA). ΔF is the change in free energy, and z is the distance between the center of mass of the gas molecule and the water droplet. The dashed vertical black lines mark the average interfacial region. Adapted with permission from W. Li, C. Y. Pak, X. Wang and Y.-L. S. Tse, Uptake of Common Atmospheric Gases by Organic-Coated Water Droplets, J. Phys. Chem. C, 2019, 123, 18924–18931. Copyright 2019 American Chemical Society. | ||
6.3. Heterogeneous reactions
Here we will present recent studies of heterogeneous reactions of O3, OH, NOy, HCl, SO2, NH3, and isoprene epoxydiols (IEPOX) at atmospherically relevant interfaces with surfactants, films, and coatings. Reactive uptake of gases to pure organic aerosol particles is outside the scope of this review.6.3.1.1 O3 + oleic acid. Unsaturated species with C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds are highly susceptible to ozonolysis. Accordingly, the oxidation of oleic acid by ozone is one of the most well-studied systems for reactive gas uptake onto surfactant films.316
C bonds are highly susceptible to ozonolysis. Accordingly, the oxidation of oleic acid by ozone is one of the most well-studied systems for reactive gas uptake onto surfactant films.316
Milsom et al.317 used time-resolved small-angle X-ray scattering (SAXS) to probe the ozonolysis of thin films of oleic acid and sodium oleate designed to mimic cooking aerosol. They prepared the films by coating the interior of a glass capillary and then passed ozone through the capillary. As shown in Fig. 12a, the reaction proceeded most rapidly on the thinnest films. Indeed, none of the reactions on films thicker than ∼5 μm reached completion, as indicated by the non-zero asymptotes in Fig. 12b. The authors propose that an inert crust of viscous oligomers forms on the surface of the oleic acid films and inhibits further reactions. Follow-up studies on acoustically levitated oleic acid/sodium oleate droplets confirmed the formation of an inert surface film.318 Zhou et al.319 observed a similar phenomenon for the multiphase ozonolysis of thin films of benzo [a]pyrene. In each case, the surface passivation effect suggests that the reactions are diffusion-limited. Indeed, ozone is not the only reactive gas to face self-limiting uptake due to the buildup of a viscous shell: reactive uptake of IEPOX can also be self-limited in this way, as discussed in Section 6.3.7.
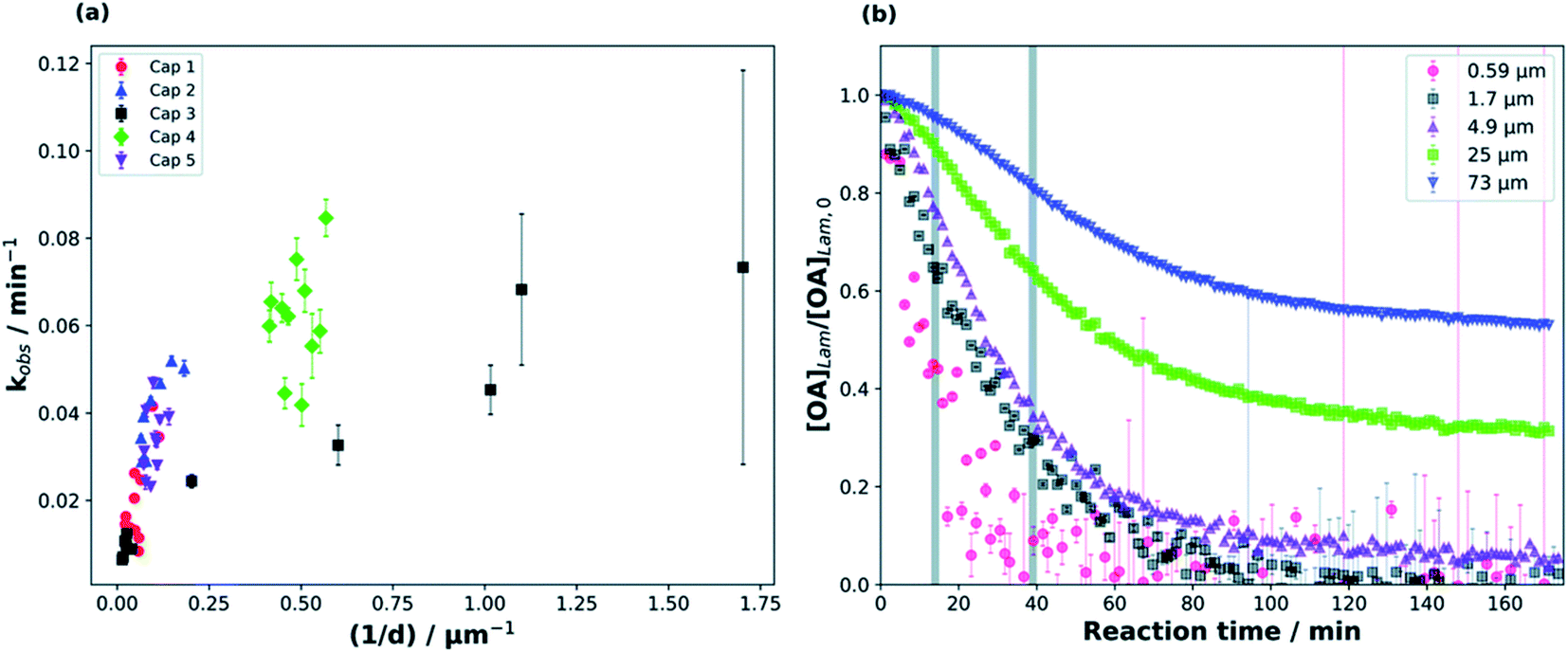 | ||
| Fig. 12 (a) Observed pseudo first-order rate constants (kobs) for the reaction of oleic acid films with 77 ± 5 ppm O3 as a function of inverse film thickness (d). The different colors and symbols represent films from different capillaries (i.e., different trials). (b) Change in lamellar oleic acid signal [OA]Lam relative to the initial signal as a function of reaction time. The gray bars mark the start and end of the reaction times used to measure kobs for films thicker than 2.5 μm. Reproduced from ref. 317 with permission from the Royal Society of Chemistry. | ||
King et al.320 further tested the influence of particle-phase diffusion on the ozonolysis of oleic acid monolayers by altering the viscosity of the subphase. These experiments were a follow-up to their 2009 work,321 which they now suspect was skewed by a surface-active contaminant in their reagent. In order to change the viscosity of the system in the 2020 experiments, King et al.320 added sodium perchlorate up to a concentration of 9.0 M, corresponding to a 7.2-fold increase in viscosity and a 9-fold increase in ionic strength. No effect on reaction rate was observed. This null result discounts the potential reaction pathway in which ozone dissolves into the bulk and then diffuses back up to the surface to react with the surfactant monolayer. As noted above, diffusion of ozone past the surface layer is a slow process even within oleic acid itself.317
Aside from serving as a model surfactant on cooking aerosol, oleic acid has also been used as a model coating for marine air–water interfaces.118,322–325 Pfrang et al.323 found that mixtures of oleic acid and sodium oleate in aqueous NaCl droplets formed 3D self-assembled nanostructures at the gas–liquid interface, and these structures slowed heterogeneous reactions with ozone. Woden et al.325 tested the influence of both temperature and salinity on the ozonolysis of oleic acid at the air–water interface. The resulting film, containing nonanoic acid, azelaic acid, and 9-oxononanoic acid, persisted at the surfaces of pure water and 36 g L−1 NaCl(aq) only if temperatures were below ∼12 °C or below freezing, respectively. Nevertheless, the rate constants seemed to be independent of temperature under their experimental conditions. On pure water, the surface rate constant was (2.2 ± 0.1) × 10−10 cm2 s−1 at 2 °C and (2.2 ± 0.4) × 10−10 cm2 s−1 at 21 °C. These results imply that there is no significant energetic barrier toward reaction. Products likely disappear because they either diffuse into the bulk or evaporate into the gas phase.
It is unlikely for ambient aerosol particles to have a coating of pure oleic acid with no other surface-active species. Skoda et al.118 previously showed that stearic acid, the linear C18 saturated fatty acid, forms islands in a 1:1 binary mixture with oleic acid. King et al.320 tested ozonolysis of oleic acid and stearic acid films across the full range of mixing ratios. The addition of stearic acid did not significantly affect the kinetics of oleic acid decay, nor was degradation of stearic acid detected, indicating that reactive intermediates from the ozonolysis of oleic acid do not accumulate at the air–water interface. Nevertheless, even a two-component mixture constitutes a relatively simple system compared to surfactant coatings on ambient particulate matter.
6.3.1.2 O3 + sesquiterpenes. Enami and coworkers have published several studies using water
:acetonitrile microjets and mass spectrometry to explore the interfacial chemistry of Criegee intermediates (CIs) generated from the ozonolysis of sesquiterpenes.326–330 Sesquiterpenes are a class of unsaturated cyclic and acyclic alkenes formed from three isoprene molecules, giving the characteristic formula C15H24. Examples used by Enami and coworkers include α-humulene and β-caryophyllene.326–330 Exposure of sesquiterpenes dissolved in the microjet to gas-phase ozone produces Criegee intermediates, which are highly reactive carbonyl oxide zwitterions or biradicals.331,332 Water readily reacts with Criegee intermediates, but Enami and coworkers have found surprising evidence that a variety of surface-active species competitively react with CIs at the surface of water:acetonitrile solutions: linear alkyl carboxylic acids329 and alcohols,326 sugars,328 and cis-pinonic acid327 all out-compete water even when their bulk concentrations are only at millimolar levels. Surface enrichment of the organic species at the gas–liquid interface makes these unexpected reactions possible.
6.3.1.3 O3 + surfactant-coated aerosol particles. Marine aerosol particles influenced by biogenic activity in the sea surface microlayer are likely to be coated with surfactants, as noted previously. Schneider et al.333 explored the impact of marine diatoms on multiphase ozonolysis by introducing gas-phase ozone into the headspace of flasks containing Thalassiosira pseudonana cultures. C7–C10 carbonyl products were detected in the gas phase by proton transfer reaction-mass spectrometry (PTR-MS), and SOA formation was observed by a scanning mobility particle sizer (SMPS) and aerosol mass spectrometer (AMS). Carbonyl products and SOA formed more rapidly with increased aging of the T. pseuodonana cultures. Cell lysis in the 21 day cultures likely released labile organic species to the air–water interface. Brüggemann et al.334 saw a similar effect of cell lysis on photochemical reactions of aged biofilms. The experiments from Schneider et al.333 were designed to mimic conditions in the sea surface microlayer, but similar considerations likely apply to reactions on SSA surfaces as well.
In contrast to the work of Schneider et al.,333 Jones et al.335 observed little reactivity of gas-phase ozone with surface seawater collected in the English Channel and aerosol particles collected in Egham, Surrey (∼30 km from London, UK). The lack of reaction is unsurprising given that the film consisted mostly of saturated species. Clearly the composition of the surface layer matters when predicting the fate of surfactant-coated aerosol particles in the presence of gas-phase ozone.
6.3.2.1 OH + biogenic SOA components. OH readily reacts with methyl jasmonate,340 which is a sparingly soluble plant VOC, and pinonic acid,341,342 which is a derivative of α-pinene, at the gas–liquid interface. Indeed, the first-order rate constant for the oxidation of methyl jasmonate is 3.5 times greater on a thin film than in bulk solution.340 This dramatic increase likely occurs because methyl jasmonate is more exposed at the gas–liquid interface, where it is only partially solvated, and because diffusion is faster in the interfacial region than in the bulk.
cis-Pinonic acid is also enriched at the air–water interface.341 Enami et al.341 were the first to identify pinonic peroxyl radicals and pinonic hydroperoxides at the air–water interface after exposing dilute solutions of pinonic acid to gas-phase OH. With knowledge of these reactive intermediates, the authors propose a new pathway to form MBTCA (3-methyl-1,2,3-butanetricarboxylic acid), which is commonly used as a tracer for biogenic SOA. The authors further estimate that 70% of the pinonic acid photo-oxidation products are volatile enough to evaporate into the gas phase, where they could potentially contribute to SOA formation. Products included alcohols and carbonyls. When Huang et al.342 exposed aqueous droplets containing cis-pinonic acid to gas-phase OH, they also detected alcohols and carbonyls, including aldehydes, ketones, and carboxylic acids. Huang et al.342 purposefully constrained the reaction to the interfacial region by adding SDS, a model anionic surfactant. SDS itself reacted with OH radicals, though slightly slower than cis-pinonic acid did (k = 2.9 × 10−8 cm2 mol−1 s−1 for SDS versus 9.4 × 10−8 cm2 mol−1 s−1 for cis-pinonic acid).342 Nevertheless, Huang et al.342 were still able to observe eight generations of oxidation products from successive oxygen additions to SDS.
Faust and Abbatt343 also used SDS as a model surfactant to study heterogeneous photo-oxidation in aqueous organic aerosol particles. Unlike pinonic acid, however, their molecule of choice was not inherently surface-active: tricarballylic acid, which is a surrogate for MBTCA, is highly soluble in water. On its own, the second-order rate constant for the reaction of aqueous-phase tricarballylic acid with gas-phase OH was found to be (1.9 ± 0.1) × 10−11 cm3 mol−1 s−1 with a reactive uptake coefficient of ∼3.0, implying that radical–radical chain reactions contribute to the loss of tricarballylic acid. For comparison, SDS on aqueous aerosol particles reacted approximately twice as rapidly as tricarballylic acid did, with a rate constant for dodecylsulfate loss of ∼4.4 × 10−11 cm3 mol−1 s−1 and a reactive uptake coefficient of ∼3.9. Fig. 13 presents a cartoon of the reaction pathways that lead to reactive uptake with and without SDS. When a gas-phase hydroxyl radical strikes the surface of an aqueous aerosol particle containing tricarballylic acid, OH can (a) react directly with tricarballylic acid at the surface, (b) diffuse and then react at the surface, or (c) diffuse and react in the bulk (see Fig. 13A). Because γ > 1, we know secondary reactions must be occurring. Fig. 13B shows how OH can also react with intermediate species at the surface (d and e) or in the bulk (f). The intermediates themselves may also undergo bulk-phase or interfacial transformations (g and h, respectively).
 | ||
| Fig. 13 Possible reaction pathways when OH collides with an aqueous aerosol particle containing tricarballylic acid (TCA, panels A and B) or tricarballylic acid and SDS (panels C–E). Steps (a) through (h) are described in the main text. Water molecules, ions, and SDS oxidation products are omitted from the cartoons for clarity. Reprinted with permission from J. A. Faust and J. P. D. Abbatt, Organic Surfactants Protect Dissolved Aerosol Components against Heterogeneous Oxidation, J. Phys. Chem. A, 2019, 123, 2114–2124. Copyright 2019 American Chemical Society. | ||
The addition of SDS to tricarballylic acid aerosol particles dramatically slowed the oxidation of tricarballylic acid. As shown in Fig. 14, the rate constant decreased by ∼60% once SDS reached monolayer coverage. In the presence of SDS, OH must penetrate through the interfacial region to encounter tricarballylic acid (Fig. 13C and D). In theory, once the amount of SDS in the aerosol particle exceeds the critical micelle concentration (CMC, i.e., the concentration at which the surfactant molecules begin to self-aggregate into micellar structures), then reactions of OH and tricarballylic acid may resume, but the experiments did not reach such high SDS levels (Fig. 13E). Across the range of surfactant concentrations under study, SDS effectively shielded bulk-phase tricarballylic acid from oxidation. The fraction of tricarballylic acid remaining at equilibrium shifted from ∼25% in the absence of SDS to ∼50% at monolayer coverage, as shown by the nonzero tricarballylic acid signals at high OH exposures in Fig. 14. Thus, the presence of surfactants on aerosol particles can extend the lifetime of molecules in the bulk.
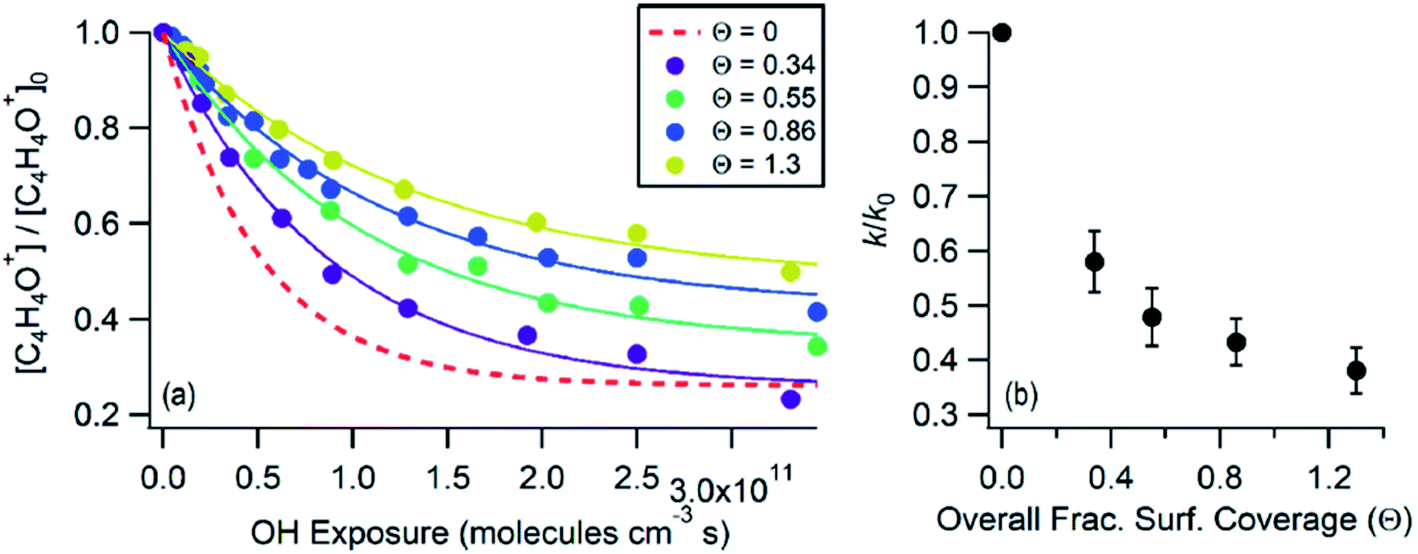 | ||
| Fig. 14 (a) Decay in normalized tricarballylic acid signal, measured as the C4H4O+ fragment by aerosol mass spectrometry, as a function of OH exposure for varying overall fractional surface coverage (Θ) by SDS. Θ is calculated as a weighted average across all particle size bins in the polydisperse aerosol population. The solid lines are fits to the equation I/I0 = I0 + (1 − I0)e−k[OH]t, where I is the C4H4O+ signal, I0 is the C4H4O+ signal at zero OH exposure, k is the effective second-order rate constant, and [OH]t is the OH exposure. The dashed red line is the fit line for tricarballylic acid particles without SDS (data points not shown). (b) Rate constants for tricarballylic acid decay in SDS-coated aerosol particles, normalized by the rate constant k0 for particles without SDS. Reprinted with permission from J. A. Faust and J. P. D. Abbatt, Organic Surfactants Protect Dissolved Aerosol Components against Heterogeneous Oxidation, J. Phys. Chem. A, 2019, 123, 2114–2124. Copyright 2019 American Chemical Society. | ||
6.3.2.2 OH + organic aerosol films and coatings. Lam et al.344 have shown that particle morphology and phase affect the kinetics of the heterogeneous reaction of OH with aqueous organic aerosol particles. They prepared a model system of aqueous droplets containing ammonium sulfate and 3-methylglutaric acid and reacted them with OH radicals in a flow tube. As they decreased the relative humidity, the particles underwent phase separation. Once the particles adopted a core–shell structure, 3-methylglutaric acid molecules in the surface shell were more readily exposed to collisions with OH, and the rate constant k increased. This effect resulted in a roughly 70% increase in k over the range of relative humidities studied, from (1.01 ± 0.02) × 10−12 cm3 mol−1 s−1 at 88% RH (when the particles were homogeneous) to (1.73 ± 0.02) × 10−12 cm3 mol−1 s−1 at 55% RH (when the particles were phase-separated).
Lim et al.345 characterized the impact of film thickness on the reaction of gas-phase OH with squalane films on ammonium sulfate particles. Squalane, a branched C30 alkane, reacted much more rapidly in the films than in pure squalane particles by up to an order of magnitude. Furthermore, the squalane-coated particles reached a higher average carbon oxidation state and experienced greater desorption of organic species in comparison to the pure squalane particles. As shown in Fig. 15, Lim et al.345 found a linear correlation between the rate constant and the surface area-to-volume ratio of the coated particle. The change in reaction rate was a purely physical phenomenon due only to the change in surface area-to-volume ratio. This observation implies that care must be taken when interpreting heterogeneous reaction rates on aerosol particles with a polydisperse size distribution.
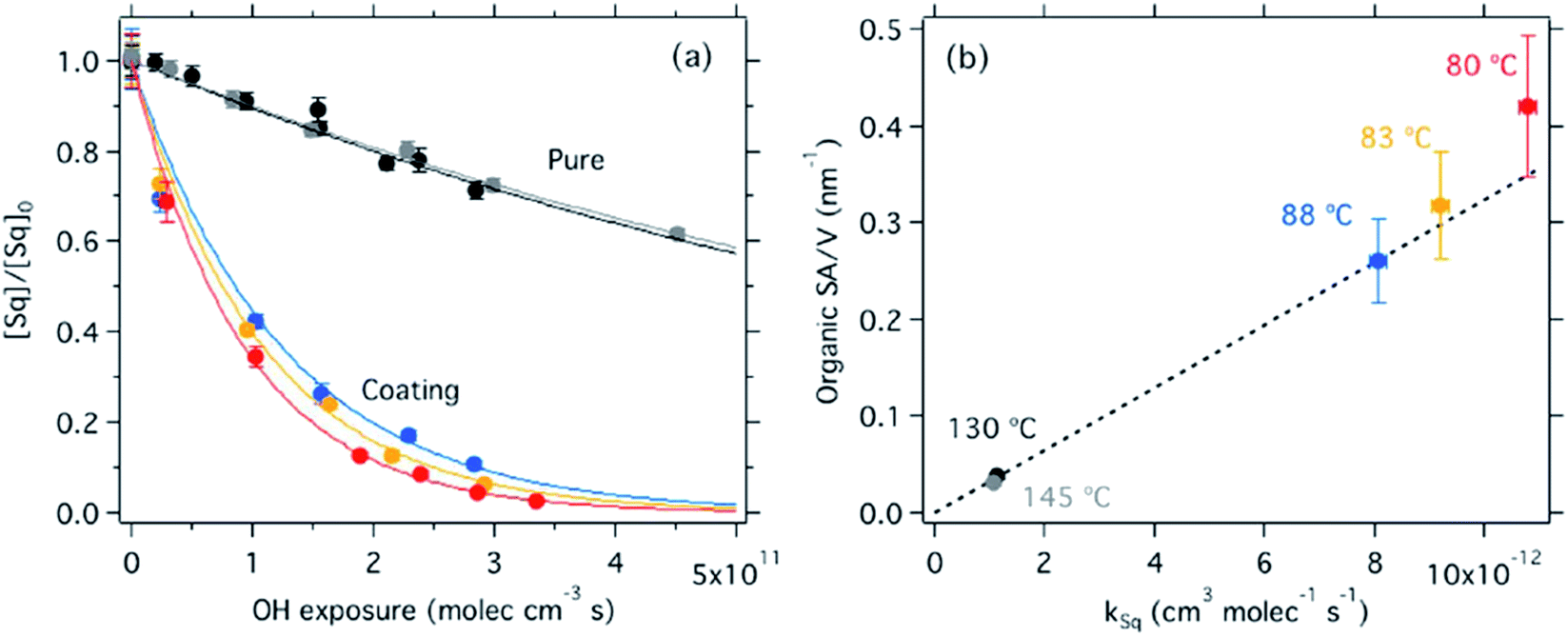 | ||
| Fig. 15 (a) Decay in normalized squalene (Sq) signal as a function of OH exposure for pure squalene particles nucleated at 145 °C (gray) and 130 °C (black) and for ∼200 nm ammonium sulfate particles coated with squalene films prepared at 88 °C (blue, 2 nm film), 83 °C (yellow, 3 nm film), and 80 °C (red, 4 nm film). The solid lines are fits to the exponential decay of squalene due to heterogeneous reaction with OH: [Sq]/[Sq]0 = e−kSq[OH]t, where kSq is the second-order rate constant. (b) Surface area-to-volume ratio (SA/V) versus rate constant kSq, corresponding to the data in panel (a). The dashed line is a weighted linear least squares fit with intercept forced through the origin. Reprinted with permission from C. Y. Lim, E. C. Browne, R. A. Sugrue and J. H. Kroll, Rapid heterogeneous oxidation of organic coatings on submicron aerosols, Geophys. Res. Lett., 2017, 44, 2949–2957. Copyright 2017 Wiley. | ||
6.3.2.3 OH + fatty acids & lipids. In the presence of sunlight, photo-oxidation of oleic acid by OH radicals is a more important loss pathway than the ozonolysis reactions discussed in Section 6.3.1. When Zhang et al.346 exposed oleic acid films at the air–water interface to gas-phase OH, they observed that the reaction of OH with the C
C bond caused mainly addition of the carbonyl group or the combination of a carbonyl group and a hydroxyl group. Fragmentation was a minor reaction route. At saturated sites in the oleic acid molecule, OH caused the addition of carbonyl and hydroxyl groups, similar to the reaction pathways in stearic acid monolayers.346 Oxygenated products were able to diffuse into the bulk and exchange with subsurface oleic acid molecules, allowing further decay of the oleic acid. This observation is in opposition to the formation of an inert crust due to dark ozonolysis of oleic acid.317,320
In a later study of lipid photo-oxidation, Zhang et al.347 found that the lipid POPG, or 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol), reacts differently with OH than oleic acid does even though both species have a cis CC bond in a C18 chain. Whereas oleic acid forms functionalization products,346 POPG forms fragmentation products.347 Furthermore, the heterogeneous reaction of OH with POPG monolayers produces Criegee intermediates, as evidenced by the formation of hydroxyhydroperoxides. The authors attribute the different reaction mechanisms to the dense packing of the POPG monolayer in comparison to the oleic acid monolayer.347 Studies with fatty acids and lipids are relevant to processes that may be occurring at the surface of primary SSA.
6.3.3.1 NO2. Recent experiments have explored uptake and reaction of NO2 to fatty acid films in the presence of salt. Sobanska et al.348 measured the uptake of gas-phase NO2 to films of stearic acid (C18 saturated fatty acid) coated on a sodium chloride crystal. The resulting surface coverage was patchy, and NO2 uptake was found to occur on exposed NaCl sites. In the presence of water vapor, sodium nitrate (NaNO3) formed on the surface. As RH increased, the authors observed a phase transition from solid NaNO3 to aqueous NaNO3 and a concomitant transition from ordered stearic acid chains to a disordered stearic acid gel at the surface.
Deng et al.349 studied uptake of gaseous NO2 to the films of the corresponding C18 monounsaturated fatty acid (oleic acid) under exposure to UVA radiation. As shown in Fig. 16, there was an inverse linear correlation between relative humidity and the reactive uptake coefficient, which ranged from γ = (1.4 ± 0.1) × 10−6 at 0% RH to γ = (7.1 ± 1.6) × 10−7 at 90% RH. The authors propose that a water layer forms on top of the oleic acid film at high RH and blocks further exposure of oleic acid molecules to NO2. Changes to the uptake coefficient upon the addition of Na2SO4 and NaNO3 are included in Fig. 16. More HONO and gas-phase nitroaromatics were produced when inorganic nitrate ions were present. Reactive uptake of NO2 to the surface of aerosol particles in the atmosphere is intrinsically linked to particle-phase nitrate chemistry.
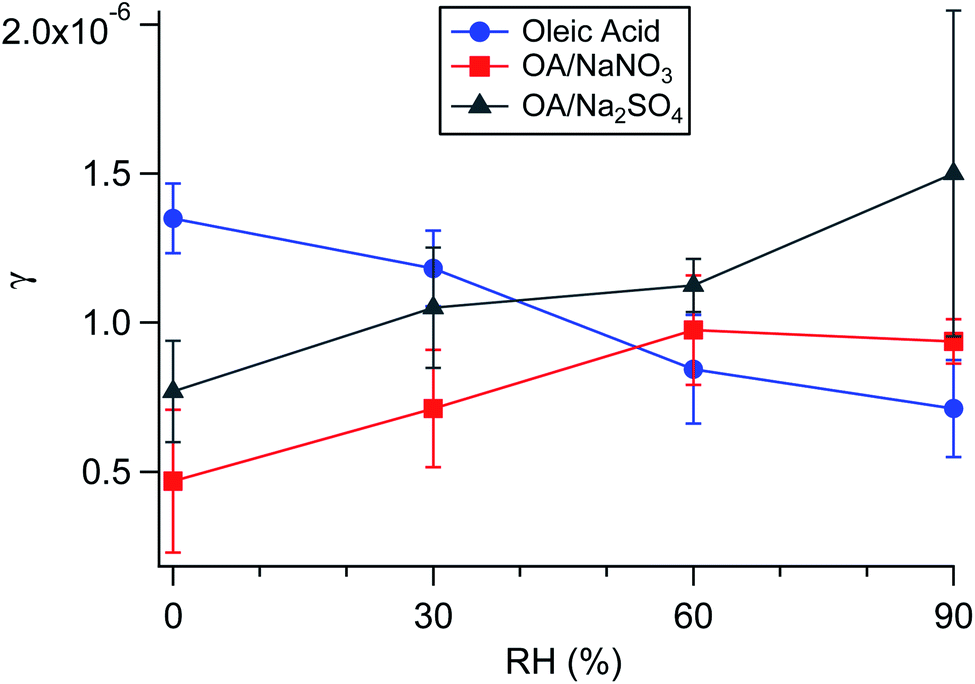 | ||
| Fig. 16 Uptake coefficient (γ) for NO2 on oleic acid (blue circles), oleic acid with sodium nitrate (red squares), and oleic acid with sodium sulfate (black triangles) as a function of relative humidity. Lines have been added to guide the eye. The NO2 mixing ratio was 40 ppb, the temperature was 296 K, and the conditions of light irradiation were 15.5 W m−2, 5.3 × 1013 photons cm−2 s−1, 300 nm < λ < 400 nm. Data from ref. 349 provided by the authors. | ||
6.3.3.2 NO3. Nitrate radicals are important drivers of nighttime chemistry in the troposphere, where they are present at concentrations up to ∼300 ppt.154,350–352 Sebastiani et al.147 quantified rate constants, characteristic lifetimes (τ), and reactive uptake coefficients for the heterogeneous reaction of NO3 with long-chain acid and ester monolayers. Their results are summarized in Table 1. Stearic acid (C18 saturated fatty acid) was four orders of magnitude less reactive than the other species, which were all unsaturated. Palmitoleic acid (C16 unsaturated fatty acid) had a longer desorption lifetime than either oleic acid (C18 unsaturated fatty acid) or methyl oleate (C18 unsaturated ester) because the shorter alkyl chains in palmitoleic acid left the reactive C
C bond more accessible to incoming NO3. Some oxidation products of the fatty acids persisted at the gas–liquid interface, but the oxidation products of methyl oleate were not surface-active. Thus, both the surfactant head group and the chain length influence reactivity. Sebastiani et al.147 considered the competition between oxidation by NO3 and O3 as nighttime sinks for fatty alcohols and esters. The reactions with NO3 were generally faster, but the effect varied significantly even for molecules of similar structure: oleic acid reacted 400 times faster with NO3 than with O3, but methyl oleate reacted only 60 times faster with NO3 than with O3. Nevertheless, concentrations of O3 generally exceed those of NO3 in the troposphere.
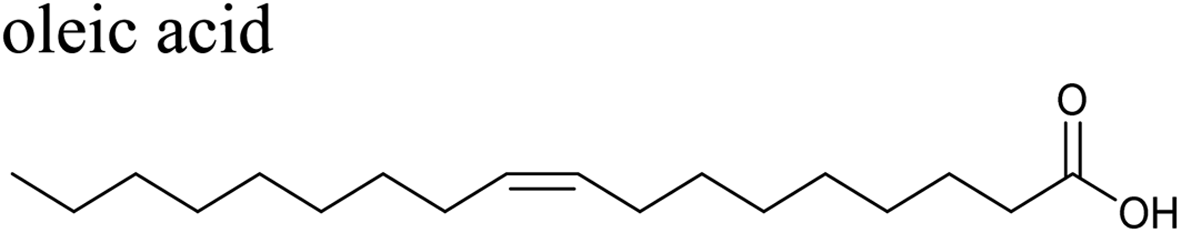
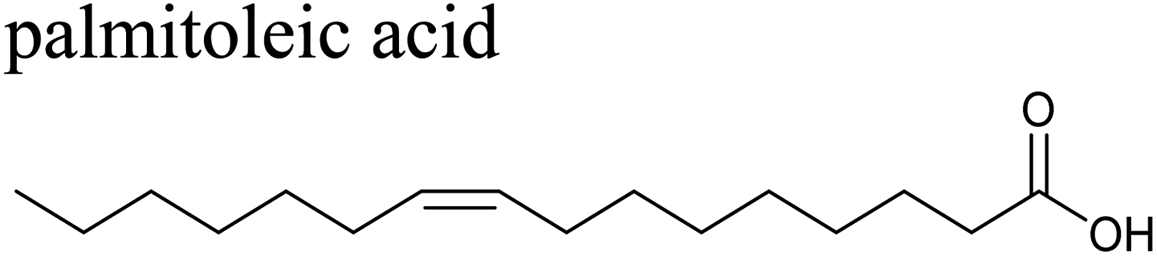

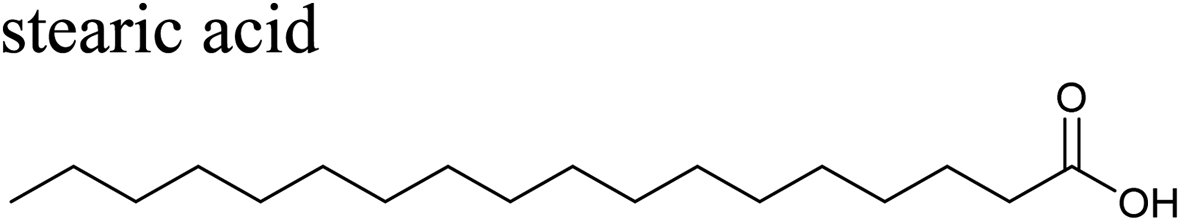
Woden et al.353 extended studies of the heterogeneous reactions of NO3 with fatty acid films to incorporate lipids. Using neutron reflectometry, IRRAS, and BAM, they monitored reactions of monolayers of galacterocerebroside (a glycolipid) on its own and in mixtures with palmitic acid or palmitoleic acid. After NO3 exposure for a period of 3–5 hours, the surface remained coated with hydrocarbon chains, and no significant changes were detected by neutron reflectometry. IRRAS measurements showed that the CC bond on the hydrocarbon tail of galacterocerebroside reacted to some extent with NO3, but adding CaCl2 to the aqueous subphase turned off the reaction because the divalent cation organized chain–chain interactions in the lipid layer. Drawing parallels to earlier work by Jones et al.335 and Zhang et al.,347 Woden et al.353 concluded that NO3 oxidation is not an important removal mechanism for lipid films at aerosol surfaces under atmospherically relevant conditions.
6.3.3.3 N2O5. Dinitrogen pentoxide (N2O5) is an important reservoir of reactive nitrogen in the atmosphere that has long served as a model system for studying heterogeneous reactions.45,351,354 Sullivan and co-workers have published a series of studies on gas-phase N2O5 uptake by BBA particles.355–358 Notably, organic coatings did not constrain reactive uptake of N2O5 at high RH by BBA particles generated from chloride-rich fuels like black needlerush.357 Furthermore, N2O5 uptake and hydrolysis at the surface of ambient aerosol particles can significantly affect particle-phase nitrate content.359 Liu et al.360 modeled nitrate formation during a heavy haze period in the Beijing-Tianjin-Hebei region of China in February 2014. Following the Riemer 2009 parameterizations for N2O5 in WRF-Chem (the Weather Research and Forecasting model coupled with Chemistry),361 Liu et al.360 were able to successfully match nitrate measurements only by incorporating the heterogeneous hydrolysis of N2O5 on hygroscopic organic coatings. Approximately 30% of total nitrate could be attributed to these reactions, which also contributed ∼12% of total PM2.5 formation.360 Accordingly, heterogeneous reactions of N2O5 on films and coatings have important implications for air quality and climate.
In a fundamental laboratory study, Sobyra et al.362 reported reactive scattering of N2O5 with surfactant-coated water microjets. They directed a near-thermal beam of N2O5 gas (10 kJ mol−1) at the surface of a supercooled aqueous jet containing bromide ions. The emission of Br2 gas indicated that an oxidation–reduction reaction had occurred as follows: N2O5(g) + 2Br−(aq) → Br2(g) + NO3−(aq) + NO2−(aq). Adding the C4 nonionic surfactant 1-butanol to the solution reduced Br2 emission somewhat. In contrast, adding even trace amounts of the C6 cationic surfactant tetrahexylammonium drastically shut down Br2 emission: adding just 9% of a tetrahexylammonium monolayer decreased the Br2 signal by 85%. The authors suggest that the tetrahexylammonium ions bring Br− toward the air–water interface and stabilize the anionic intermediate Br3− in solution. A similar stabilization was seen by Gord et al.363 for N2O5 to Br2 conversion and by Faust et al.364 for Cl2 to Br2 conversion, both on tetrahexylammonium-coated glycerol, and to a lesser extent for Cl2 to Br2 conversion on glycerol coated with the zwitterionic phospholipid dihexanoylphosphotidylcholine (DHPC).363 Chen et al.365 also saw that tetrabutylammonium ions enhanced the reactivity of gas-phase ozone with bromide ions in aqueous solutions, not only because the surfactant draws more Br− to the surface, but also because the surfactant stabilizes the bromide ozonide intermediate, increasing its lifetime by an order of magnitude. In these cases, the quaternary ammonium surfactants brought gas-phase and solution-phase reactants together in close proximity and thereby promoted the multiphase reactions. Accordingly, care must be taken when predicting the effects of ionic surfactants on interfacial reactions involving charged reactants and intermediates.
6.3.3.4 HNO3. Nitric acid (HNO3) uptake to aerosol particles is important because it facilitates acid-catalyzed reactions once dissolved in the aqueous phase. Gas-phase nitric acid has been shown to react with deprotonated carboxylate and phosphate groups in lipopolysaccharides at the surface of SSA particles.366,367 Lee et al.367 conducted flow tube experiments and MD simulations to investigate reactions of nitric acid with nascent SSA particles containing lipopolysaccharides and sodium, magnesium, and calcium ions. For the lab studies, Lee et al.367 generated SSA from real seawater under the conditions of a phytoplankton bloom. They isolated particles that were dominated by monovalent cations from those dominated by divalent cations; all particles selected for further study contained lipopolysaccharides. Lee et al.367 also prepared aerosol particles containing controlled amounts of commercial lipopolysaccharides from E. coli and NaCl, MgCl2, or CaCl2. When particles were exposed to 50 ppb HNO3 at 50% RH, the sodium-containing particles were found to be eight times more reactive than the particles containing divalent cations. Results of the MD simulations, shown in Fig. 17A, indicate that water diffuses more rapidly through the lipopolysaccharide films in the presence of monovalent cations than in the presence of divalent cations. In addition, more water molecules are present within the lipopolysaccharide layer in the presence of monovalent vs. divalent cations (Fig. 17B). The cartoons in Fig. 17C illustrate how diffusion of water through the lipopolysaccharides is critical for the ability of HNO3 to react.
 | ||
| Fig. 17 (A) Diffusion coefficients of water molecules through lipopolysaccharide (LPS) bilayers with Na+ (green), Mg2+ (blue), or Ca2+ (orange) cations. (B) Water density within lipopolysaccharide bilayers from the corresponding all-atom MD simulations averaged over 200 ns. (C) Cartoon illustrations of water diffusion (blue arrows) and HNO3 reactions within lipopolysaccharide bilayers. The yellow stars indicate that reactions occur in the presence of monovalent cations (left) but not divalent cations (right). Reprinted with permission from C. Lee, A. C. Dommer, J. M. Schiffer, R. E. Amaro, V. H. Grassian and K. A. Prather, Cation-Driven Lipopolysaccharide Morphological Changes Impact Heterogeneous Reactions of Nitric Acid with Sea Spray Aerosol Particles, J. Phys. Chem. Lett., 2021, 12, 5023–5029. Copyright 2021 American Chemical Society. | ||
In summary, reactive uptake of HNO3 to aerosol particles containing lipopolysaccharides is lower when the particles contain divalent cations like Mg2+ and Ca2+ than when they contain monovalent cations like Na+ because the divalent cations induce cross-linking of the oligosaccharide chains, altering the physical properties of the particles and slowing diffusion in the interfacial region.367
When comparing the reactivities of assorted organic films with SO2 (see Table 2), Passananti et al.368 found that acids were more reactive than alkenes, and aromatics did not react at all. Terminal CC bonds were clearly less reactive than internal CC bonds, but patterns in the reactivity of cis and trans bonds were ambiguous. The value of γ was 50 times greater for oleic acid (cis-C18) than for elaidic acid (trans-C18). However, oleic acid is a liquid at room temperature whereas elaidic acid is a solid. Based on variable temperature experiments, the authors observed that the reactive uptake coefficient dropped sharply when transitioning below the freezing point, likely due to inhibited diffusion in the condensed phase.
| Compound | Melting point [°C] | γSO2a [×10−6] |
|---|---|---|
| a Experimental conditions: 40 ppb of SO2 in dry synthetic air, flow rate = 300 mL min−1, T = 293 K. b This uptake was measured under 35% RH. c This uptake was measured at 298 K. Data from Passananti et al.368 | ||
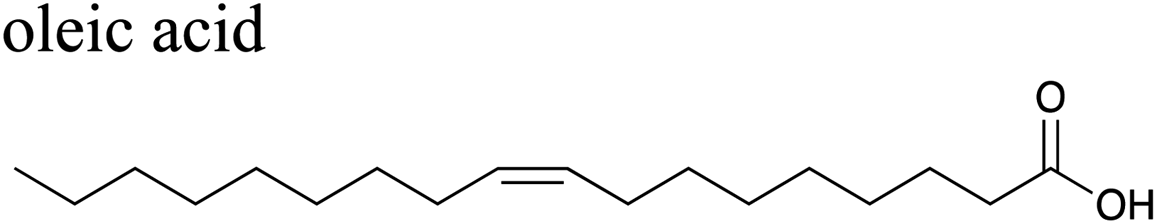
|
13–14 | 5.0 ± 0.5 |
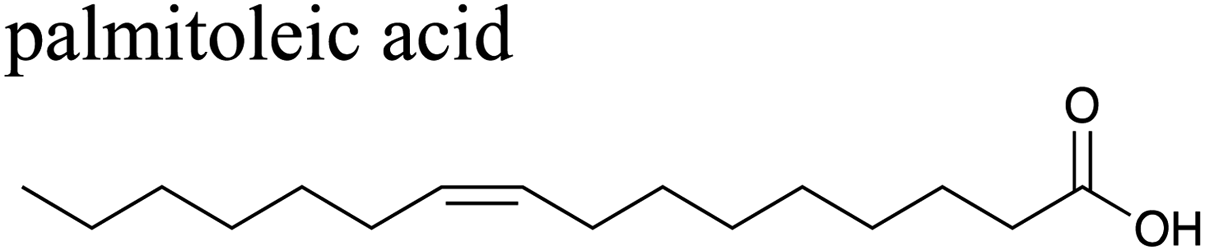
|
0.5 | 1.4 ± 0.1 |
|
−5 | 3.0 ± 0.3 |
|
−12 | 5.5 ± 0.5 |
|
42–44 | 0.10 ± 0.03b |
|
23–25 | 0.85 ± 0.05c |
|
−35 | 0.08 ± 0.01 |
|
14–16 | 0.21 ± 0.01 |
|
>300 | Not reactive |
Regardless of phase, reactions of unsaturated organic surfactants with SO2 are unlikely to play a significant role in aerosol aging under most ambient conditions, where O3 is more abundant and far more reactive.369,370 Exceptions may occur in highly polluted regions. Zhu et al.371 sampled PM2.5 in Wanqingsha, China in December 2013 and found evidence of organosulfate formation from heterogeneous reactions of SO2 gas and unsaturated fatty acids on ambient aerosol particles. These reactions contributed 7–13% of the total sulfur in all CHOS species and 0.3–0.9% of the total organic mass in all PM2.5. Thus, SO2/fatty acid reactions remain relevant under certain conditions in the atmosphere.
Atmospheric box models are unable to reproduce concentrations of IEPOX-derived SOA, however, without incorporating particle phase, especially in regions of low RH and cold temperatures.156,378–381 As IEPOX reacts at the surface of acidic sulfate particles, a viscous coating forms that inhibits further uptake. Zhang et al.382 provided experimental evidence for this phenomenon in chamber experiments of IEPOX uptake to acidified ammonium sulfate seeds, and the authors supported their conclusions with thermodynamic modeling. Drozd et al.383 previously hypothesized that uptake of α-pinene oxide to acidic sulfate aerosol was similarly limited by the formation of an organic coating, though they noted that such processes were unlikely to be relevant in the atmospheric because of the subzero pH required for reaction. IEPOX uptake, in contrast, is highly relevant to the formation and evolution of SOA in the atmosphere.376,377
Extensive laboratory studies support the importance of phase, viscosity, and diffusion for the reactive uptake of IEPOX to organic-coated aerosol particles. Gaston et al.384 observed early evidence for self-limiting IEPOX uptake when they measured reduced uptake of trans-β-IEPOX onto ammonium bisulfate particles coated with polyethylene glycol. More recent work has employed SOA coatings derived from α-pinene, toluene, and β-caryophyllene.385–388 For example, Zhang et al.386 found a 30–50% decrease in the reactive uptake coefficient of IEPOX to particles coated with just ∼15 nm of α-pinene SOA (across a relative humidity range of 15–50%).
Lei et al.388 employed a combination of AFM with photothermal infrared spectroscopy, Raman microspectroscopy, and SEM-EDX to directly show how reactive IEPOX uptake to acidified ammonium sulfate particles coated with α-pinene, β-caryophyllene, isoprene, and toluene SOA alters the physicochemical properties of the particles. IEPOX uptake significantly decreased the viscosity of the particles coated with α-pinene and β-caryophyllene SOA but did not significantly alter the viscosity of the particles coated with isoprene or toluene SOA, as shown in Fig. 18. The authors attributed the differences to molecular weight: α-pinene and β-caryophyllene have greater molecular weights than isoprene or toluene. Viscosities were inferred from the height of particles in AFM images. Taller particles with lower spreading ratios (i.e., ratio of radius to height) are assumed to be more viscous. Fig. 18e shows that IEPOX uptake altered the chemical composition of both the shell and the core of α-pinene SOA. This work by Lei et al.388 is an example of the powerful insights into heterogeneous reactions on coated particles that can be obtained by combining measurements of morphology and composition for single particles.
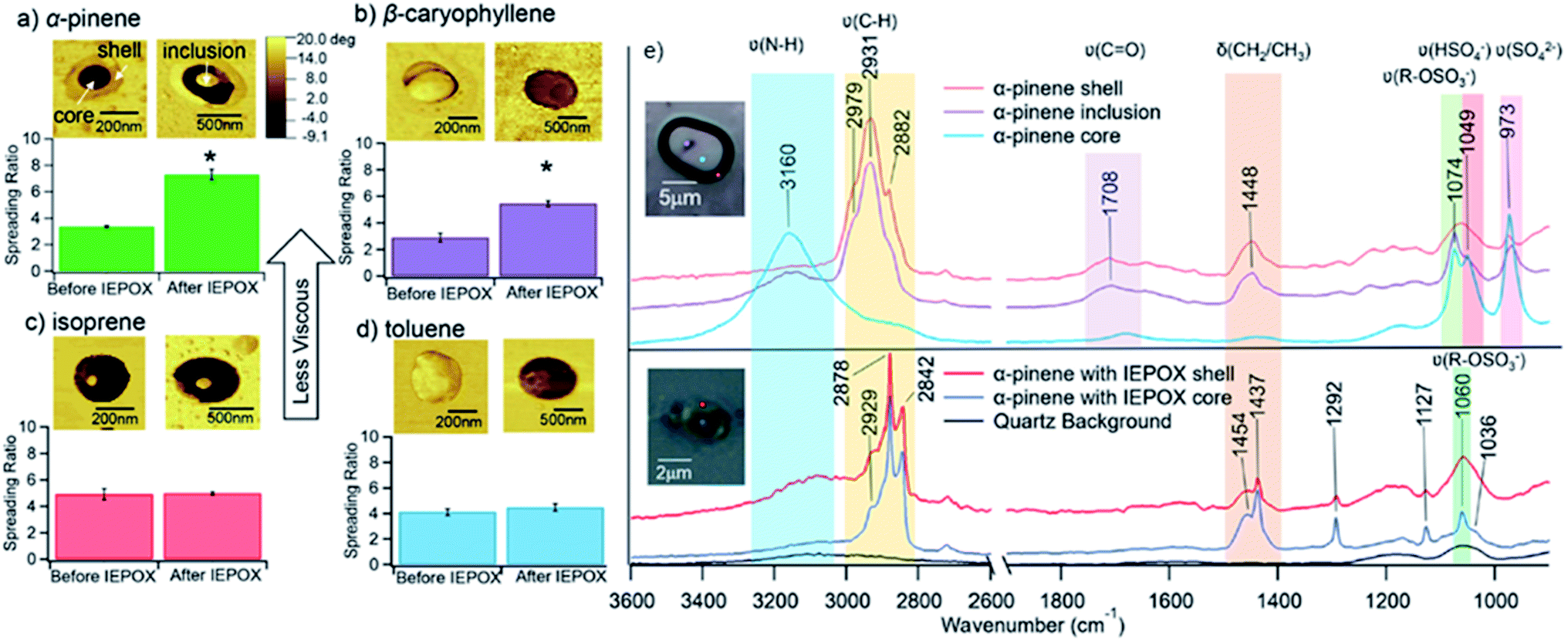 | ||
| Fig. 18 AFM phase images and spreading ratios of (a) α-pinene, (b) β-caryophyllene, (c) isoprene, and (d) toluene SOA before (left) and after (right) IEPOX uptake at 50% RH. Spreading ratios were calculated from the ratio of particle radius relative to particle height, averaged across the population of particles. Error bars show standard deviations, and asterisks denote statistically significant changes in spreading ratio after IEPOX exposure (p < 0.05). (e) Raman spectra showing the core and shell composition of α-pinene SOA before (top) and after (bottom) IEPOX exposure. The colors of the spectra correspond to the locations of the colored dots on the optical microscope images (insets). Adapted with permission from Z. Lei, N. E. Olson, Y. Zhang, Y. Chen, A. T. Lambe, J. Zhang, N. J. White, J. M. Atkin, M. M. Banaszak Holl, Z. Zhang, A. Gold, J. D. Surratt and A. P. Ault, Morphology and Viscosity Changes after Reactive Uptake of Isoprene Epoxydiols in Submicrometer Phase Separated Particles with Secondary Organic Aerosol Formed from Different Volatile Organic Compounds, ACS Earth Space Chem., 2022, 6, 871–882. Copyright 2022 American Chemical Society. | ||
6.4. Particle-phase reactions
As illustrated back in Fig. 1, some particle-phase reactions can generate surface-active species. Here we will briefly consider recent findings on particle-phase oligomerization reactions of α-keto acids, aldehydes, and terpenes to produce surface-active molecules.Pyruvic acid, a C3 α-keto acid, readily undergoes self-oligomerization at the air–water interface.103,389,390 Surface-active products include the oligomers zymonic acid and parapyruvic acid.103 These reactions occur spontaneously in the dark, but pyruvic acid also undergoes photolysis to form oligomers upon exposure to UV/visible radiation.390 Intriguingly, Rapf et al.391 showed that photolysis of C6 to C12 α-keto acids is a potential abiotic source of multi-tailed lipids at the air–water interface.
Aldehydes also undergo condensation reactions in aqueous aerosol particles.14 For example, Van Wyngarden et al.392 observed that propanal (CH3CH2CHO) forms visibly colored films on sulfuric acid solutions due to acid-catalyzed aldol condensation reactions. Major products were 2-methyl-2-pentenal (the dimer); 1,3,5-trimethylbenzene; and 2,4,6-triethyl-1,3,5-trioxane, with larger oligomers detected as well. Mixtures of multiple aldehydes form even more complicated oligomeric products because cross-reactions are often favored over self-oligomerization.392,393 Furthermore, dicarbonyl compounds such as glyoxal (CHOCHO) and methylglyoxal readily react with ammonium ions and amines to form surface-active species, such as oligomers containing imidazoles and pyrroles.15,372,394–401 These light-absorbing products belong to the class of compounds known as brown carbon.15 Brown carbon aerosols are of relevance to global climate because they absorb solar radiation, contributing to positive radiative forcing.
Terpenes, including α-pinene and isoprene, are known to form oligomers at the surface of aerosol particles during atmospheric aging.12,14,402–407 In the case of dimeric α-pinene oxidation products, the surface propensity has been shown to depend on stereochemistry.408 Ishizuka et al.409 present an interesting case study of how surfactants already present at the air–water interface affect the interfacial oligomerization of isoprene. They monitored the reaction of gas-phase isoprene with liquid microjets of aqueous solutions containing either the nonionic surfactant 1-octanol or a cationic quaternary ammonium surfactant. Both surfactants suppressed the oligomerization of isoprene, but the cationic surfactant did so to a much greater extent. The authors propose that the cation caused electrostatic exclusion of H3O+ from the interface, so proton transfer could not occur to initiate the acid-catalyzed oligomerization of isoprene. This result serves as a reminder that reactions that produce surface-active species in simple systems in laboratory experiments may behave very differently in ambient systems.
6.5. Future needs
As in all areas of atmospheric chemistry, our knowledge of multiphase reactions in and on surfactant-coated particles would benefit from continued integration of field measurements, laboratory experiments, and computational simulations, extrapolated to atmospheric chemical modeling.12,64,142,211,410–413 A continuing question surrounds the relevance of laboratory and computational studies on simple, equilibrated systems in comparison to the complex conditions in the atmosphere. Particle morphology, phase, viscosity, mixing state, ionic strength, and pH will all affect reactions both at the gas–liquid interface and in the bulk.414,415 Single-particle measurements and advances in instrumental techniques and modeling are necessary to bridge molecular-level insights and global observations. We encourage continued synergy between studies of heterogeneous reactions on aerosol particle surfaces and similar processes at the sea surface microlayer322,324,416 and on indoor films.417,4187. Photochemistry
Solar radiation that reaches the troposphere is capable of initiating reactions by exciting molecules to a higher electronic state and then triggering photodissociation, photoisomerization, energy transfer, hydrogen abstraction, free radical formation, or other chemical reactions.419,420 Photochemical processes at the air–water interface are distinct from purely gas-phase and purely condensed-phase processes because of the inherent anisotropy of the surface, where molecules are not fully solvated and can pack closely together.419,421 Here we will focus on studies of photo-driven reactions of surface-active species published from 2015 onwards, following the comprehensive review of George et al.4197.1. Fatty acids
 | ||
| Scheme 1 Direct photochemical reaction mechanism for nonanoic acid. Scheme based on ref. 422. | ||
Stirchak et al.425 compared VOC release from photochemical reactions of 2 mM nonanoic acid on simulated saltwater vs. freshwater solutions. The yield was actually slightly lower on saltwater. The authors propose three possible reasons: (1) differences in solubility of the products, (2) a decrease in the surface coverage of nonanoic acid on saltwater because of the elevated pH, and (3) interactions between nonanoic acid and the ions in saltwater.425 These results are relevant to photochemical reactions at the surface of SSA with minimal organic content.
Although George and coworkers422,424,425 have shown that it is possible for fatty acids to undergo direct photochemical reactions in highly concentrated surface films, reactions are far more likely to proceed in the presence of a photosensitizer.421 A good photosensitizer efficiently absorbs solar radiation and transfers energy to a less photochemically active species. Humic acid,426,427 4-benzoylbenzoic acid (4-BBA),427,428 and imidazole-2-carboxaldehyde (IC)428 have all been used as photosensitizers for nonanoic acid. The α-keto acids pyruvic acid and 2-oxooctanoic acid can also act as photosensitizers for nonanoic acid,429 but 2-oxooctanoic acid can undergo direct photochemical reactions on its own to form humic-like substances at the surface of acidic solutions.430
The mechanism for photodegradation of nonanoic acid in the presence of a photosensitizer is shown in Scheme 2.428 Absorption of light excites the photosensitizer to the triplet state, and it abstracts a hydrogen atom from nonanoic acid. Final products include saturated and unsaturated aldehydes, ketones, alkenes, dicarbonyls, dimers, and highly oxygenated species.426–428 Many of the same products form in the absence of photosensitizers,422 but the pathways are distinct, as seen by comparing Schemes 1 and Scheme 2. Tinel et al.428 found ∼9 times more C9H18O (identified as nonanal) in the aqueous phase and ∼14 times more C5H10O (pentanal and/or pentanone) in the gas phase in the presence of 4-BBA in comparison to when nonanoic acid was present alone. Whether the products partition to the gas phase, aqueous phase, or both depends on their volatility. Furthermore, pH affects the distribution of acidic and basic species, including nonanoic acid itself, across the air–water interface. As pH is decreased, nonanoic acid is protonated and dissolves more readily in the bulk; thus, surface photochemistry is less pronounced.426 The pH effect provides further confirmation that nonanoic acid is reacting at the gas–liquid interface rather than in the bulk.
 | ||
| Scheme 2 Indirect photochemical reaction mechanism for nonanoic acid in the presence of the photosensitizer 4-BBA (4-benzoylbenzoic acid). Scheme based on ref. 428. | ||
The reactions of nonanoic acid described so far have focused on relatively simple model systems. When Stirchak et al.425 added Suwannee River natural organic matter to nonanoic acid solutions, they found a doubling in VOC yield from photochemical reactions. Trueblood et al.427 further increased complexity by using genuine marine dissolved organic matter (DOM) as a photosensitizer for nonanoic acid. They collected water from the Pacific Ocean along the California coastline, induced a phytoplankton bloom, and then extracted the organic matter. Marine DOM was less efficient than 4-BBA and humic acid as a photosensitizer, though Trueblood et al.427 did observe some evidence that marine DOM underwent direct photolysis itself. They correlated the efficacy of the photosensitizers with aromaticity: the marine DOM was dominated by carboxylic-rich alicyclic molecules and was therefore much less efficient than 4-BBA, an aromatic molecule. Accordingly, terrestrial-influenced aerosol may be more reactive than marine DOM because of the likely presence of lignin, an aromatic species derived from plant cell walls. However, the authors caution that the marine DOM used in their work is not necessarily representative of all marine DOM.
In addition to photosensitizers, transition metal ions can also facilitate photochemical reactions of fatty acids. Huang et al.431 found that nonanoic acid reacts with Fe(III) at the air–water interface through ligand-to-metal charge transfer and subsequent decarboxylation:
| RCOOH + [FeIII(OH)x]+3−x → [RCOO – FeIII(OH)x]+2−x + H+ | (R1) |
| [RCOO – FeIII(OH)x]+2−x + hν → [RCOO˙ – FeII(OH)x]+2−x | (R2) |
| [RCOO˙ – FeII(OH)x]+2−x → R˙ + CO2 + [FeII(OH)x]+2−x | (R3) |
| R˙ + H2O/O2 → → aldehydes, alkanes | (R4) |
The major gas-phase products detected were octanal and octane. In comparison to photochemical reactions of pure nonanoic acid (Scheme 1), the reactions involving Fe(III) increase the ratio of saturated to unsaturated products from 10.2 to 31.5.431 Huang et al.431 showed that the Fe(III) reactions are prevalent at the air–water interface because nonanoic acid causes a surface enrichment of Fe(III) relative to the bulk, presumably due to electrostatic attraction between nonanoate anions and Fe3+ cations. Nonanoic acid also enriches the surface concentrations of photosensitizers like 4-BBA and IC.428 These synergistic effects highlight the unique role of surface species in the photochemistry of aerosol particles.
The interfacial photochemistry of short-to medium-chain fatty acids is of particular interest because the reactions release VOCs that can contribute to SOA formation in the atmosphere. Reactions of nonanoic acid at the air–water interface have been shown to initiate new particle formation and growth when nonanoic acid is irradiated with and without photosensitizers.432,433 Alpert et al.433 offered direct evidence for new particle formation and growth after exposing a basin of nonanoic acid on an aqueous humic acid solution to UV light and then oxidizing the resulting gas-phase products by ozone in the dark. As shown in Fig. 19a, octanal is detected in the gas phase during the period of irradiation. Once ozone is introduced into the reaction chamber, the octanal signals decay, and the number of particles in the chamber shoots up to ∼104 cm−3. The classic banana-shaped plot in Fig. 19b is characteristic of new particle formation events.434 Understanding SOA sources in the atmosphere is important because aerosol particles contribute to cloud formation, affect climate through radiative forcing, and pose health risks to humans.
 | ||
| Fig. 19 (A) Concentrations of the gas-phase species (left axis) from photochemical reactions of a nonanoic acid film on an aqueous solution containing humic acid. Signals were detected by selective reagent ion time-of-flight mass spectrometry with H3O+ ionization. The blue dots (right axis) indicate particle concentrations (Np) measured with an ultra-fine condensation particle counter (CPC). The yellow shading marks the period of illumination, and the vertical black line marks the introduction of ozone to the reaction chamber. (B) Particle size distribution (dNp/dlogDp) as a function of time after ozone injection. Dp refers to particle diameter. Reprinted from ref. 433 under a CC BY 4.0 License (https://creativecommons.org/licenses/by/4.0/). | ||
C bond by ozone is rapid enough that photodegradation is a comparatively minor sink. Nevertheless, artifacts from photochemical reactions may affect laboratory studies of unsaturated fatty acids. Parmentier et al.436 present results for direct photodegradation of oleic acid droplets in an optical trap. Reaction in the laser beam occurred via dissolved O2 within the droplets. This finding poses a cautionary warning to scientists using optical trapping of unsaturated fatty acid particles.
7.2. Alcohols
Unlike fatty acids, fatty alcohols are not photochemically active in the absence of a photosensitizer. For this reason, Lin et al.437 were surprised to observe reaction products from a film of pure nonanol…until they discovered that the film was not, in fact, pure. Contamination from nonanal allowed singlet oxygen to form and react with nonanol, releasing nonanal and 1-heptene into the gas phase. VOC production was also observed from irradiation of octanol in the presence of the photosensitizers IC and 4-BBA,438 and oligomers have been detected in the condensed phase when hexanol and nonanol are irradiated in the presence of α-keto acids.429 You et al.439 tested photosensitized reactions of octanol in the presence of brown carbon. They generated the brown carbon by mixing methylglyoxal with ammonium sulfate or glycine and then evaporating the solution, resulting in species with imidazole ring structures that acted similar to the well-known photosensitizer IC. Once the brown carbon photosensitizers were excited to the triplet state, they could abstract a hydrogen atom from octanol. The reaction proceeded through further abstraction, disproportionation, or oxidation in the presence of O2. Excited brown carbon could also generate other reactive oxygen species like HOx, superoxide, and singlet oxygen. The photosensitized reactions resulted in both unsaturated and functionalized products in the condensed phase, with more highly oxygenated products forming at longer times. The main products were octanal and octanoic acid, but C16 dimers, heptanal, hexanal, heptanoic acid, octenal, octenoic acid, pentene, heptane, and octene were also detected. If some of the more volatile products escape into the gas phase, they could contribute to SOA formation, as has been observed for photochemical reaction products from nonanoic acid.432,433 One important caveat is that the octanol/brown carbon experiments439 were carried out with UVC radiation, which is more energetic than the portion of the solar spectrum that reaches the troposphere.7.3. Phospholipids
Photochemical reactions of phospholipids are of particular interest in the marine atmosphere because they are commonly found on SSA. Li et al.440 tested the photostability of two model phospholipids, DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine) and DOPC (1,2-dioleoylsn-glycero-3-phosphocholine), at the air–water interface. No significant changes to the DSPC monolayer were observed upon irradiation in the presence of photosensitizers. In contrast, DOPC has two CC bonds and was therefore more reactive than DSPC. After irradiation with photosensitizers, the DOPC monolayer became increasingly ordered, and changes to the IR spectrum indicated the possible formation of DOPC hydroperoxide via a singlet oxygen pathway. A cartoon mechanism is presented in Fig. 20. Collins et al.441 observed the similar formation of oxygenated products when the lipid diacylglycerol phosphatidylcholine was exposed to UV radiation in the presence of docosahexaenoic acid as a photosensitizer. The work of Li et al.440 is unique because they not only used traditional model photosensitizers like IC and humic acid, they also used chamber-made SOA and ambient PM2.5 (particular matter with diameter <2.5 μm). SOA was generated from the photo-oxidation of limonene in the presence of OH, NOx, and ammonium sulfate seed particles. PM2.5 was collected in Jinan, China in January of 2016. Similar results were obtained with these more complex photosensitizers as has been observed with IC and humic acid. The Li et al.440 study marks the first time SOA and PM2.5 were tested as photosensitizers for reactions of organic Langmuir films.
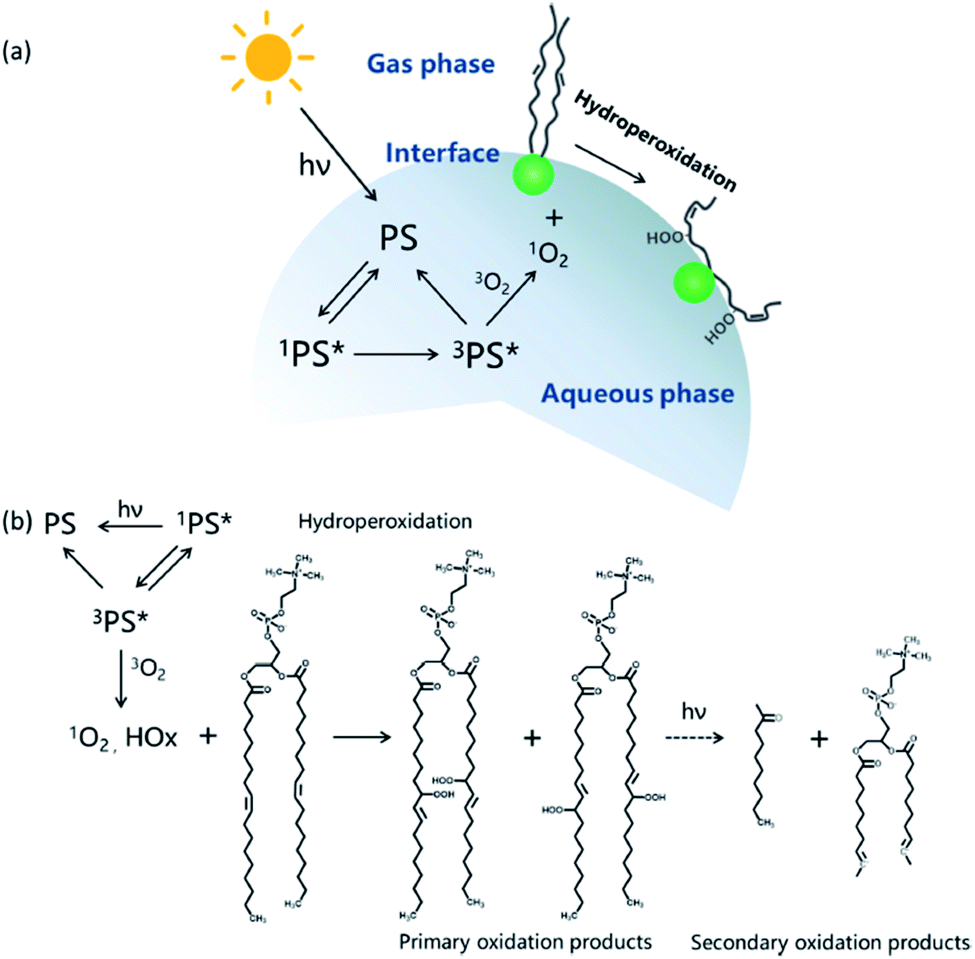 | ||
| Fig. 20 (a) Cartoon and (b) products of photochemical reactions involving the lipid DOPC and photosensitizers (PS) at the air–water interface. Reprinted from ref. 440 under a CC BY 4.0 License (https://creativecommons.org/licenses/by/4.0/). | ||
7.4. Environmental samples
The photochemical studies described so far include photosensitizers ranging in complexity from single-component surrogates to ambient samples, but all used model surfactant films. Here we will discuss two studies of direct photodegradation of ambient samples. First, Ciuraru et al.426 irradiated a water sample from the sea surface microlayer off the coast of Norway. The results were actually quite similar to their observations from the simple nonanoic acid system.426 As expected, saturated aldehydes and ketones formed in the gas phase. The authors were surprised to find unsaturated, functionalized products such as hexene, hexenal, heptadiene, and octadiene in the gas phase because these species were previously assumed to come only from biological sources in the marine environment. Brüggemann et al.334 specifically focused on biofilms as an abiotic source of VOCs. They coated groundwater with biofilms harvested from cobbles in the River Morcille and Lake Annecy, France. Oxo-fatty acids and other photosensitizers were present in the aqueous phase, allowing VOCs to form through peroxy radical pathways. More VOCs were detected in the gas phase when microbes in the samples were dead, probably because cell lysis released reactive surfactants. The authors observed new particle formation events when they oxidized the gas-phase products with ozone and OH.334 These results reinforce how photochemical reactions of surface-active species affect SOA formation and composition in the atmosphere.7.5. Future needs
Photochemical reactions of benchmark surfactants like nonanoic acid have been well characterized at atmospheric interfaces, but more work is needed on other representative species and complex mixtures. In particular, many of the experiments described here used intense UV radiation. More experimental and theoretical studies are needed with radiation relevant to the solar spectrum.8. Ambient observations
The work described so far in this review has centered on laboratory measurements and modeling studies applied to ambient aerosol particles with surfactants, films, and coatings. Now we turn at last to field measurements of coated particles in the atmosphere. The ability to confidently detect surfactants in ambient aerosol particles constitutes the most significant advance in this area in the past decade.8.1. Techniques for measuring surfactants in ambient aerosols
Direct measurements identifying surfactants in ambient aerosols were limited until Nozière and coworkers442,443 developed a double extraction method to isolate surfactants within atmospheric aerosol particles while correcting for ionic interferents. Aerosol particles are collected on filters, which are first extracted in water. Then the water extracts are extracted a second time with polydimethylsiloxane tubing, and finally they are eluted with methanol. The silicon microextraction step improves selectivity for surfactants.442 This analytical method, shared as a video protocol in the Journal of Visualized Experiments (JoVE),444 has been utilized in several notable studies of surfactants in ambient aerosol particles.442,443,445–447 For example, Gérard et al.443 coupled this targeted double extraction method with colorimetric titration using three unique dyes to determine the concentrations of anionic, cationic, and nonionic surfactants in ambient coastal PM2.5 collected from a remote island near Sweden in 2010, as summarized in Fig. 21. This study represents the first measurements of nonionic surfactants, critical micelle concentrations, and surface tension curves for ambient atmospheric surfactants.443 Their results showed significant surface tension depression via adsorption isotherms and CMC determinations as well as a potential underestimation of over 30% for total surfactant concentration in previous studies, with nonionic surfactants being especially underestimated.443 Gérard and coworkers subsequently applied their targeted extraction and colorimetric method to investigate PM1 aerosol in an expanded field scope with several urban, rural, and coastal sites in Europe focusing on amphiphilic surfactants.443,446 Various types of mass spectrometry,447–452 electrochemical methods,235,453 micro-Raman spectroscopy,454,455 AFM,456 and TEM457 have also been employed to characterize surfactants through both direct and indirect methods. These techniques provide quantitative information on ambient aerosol surfactants, which will enable improved understanding of surfactants' influence on particle activation ability.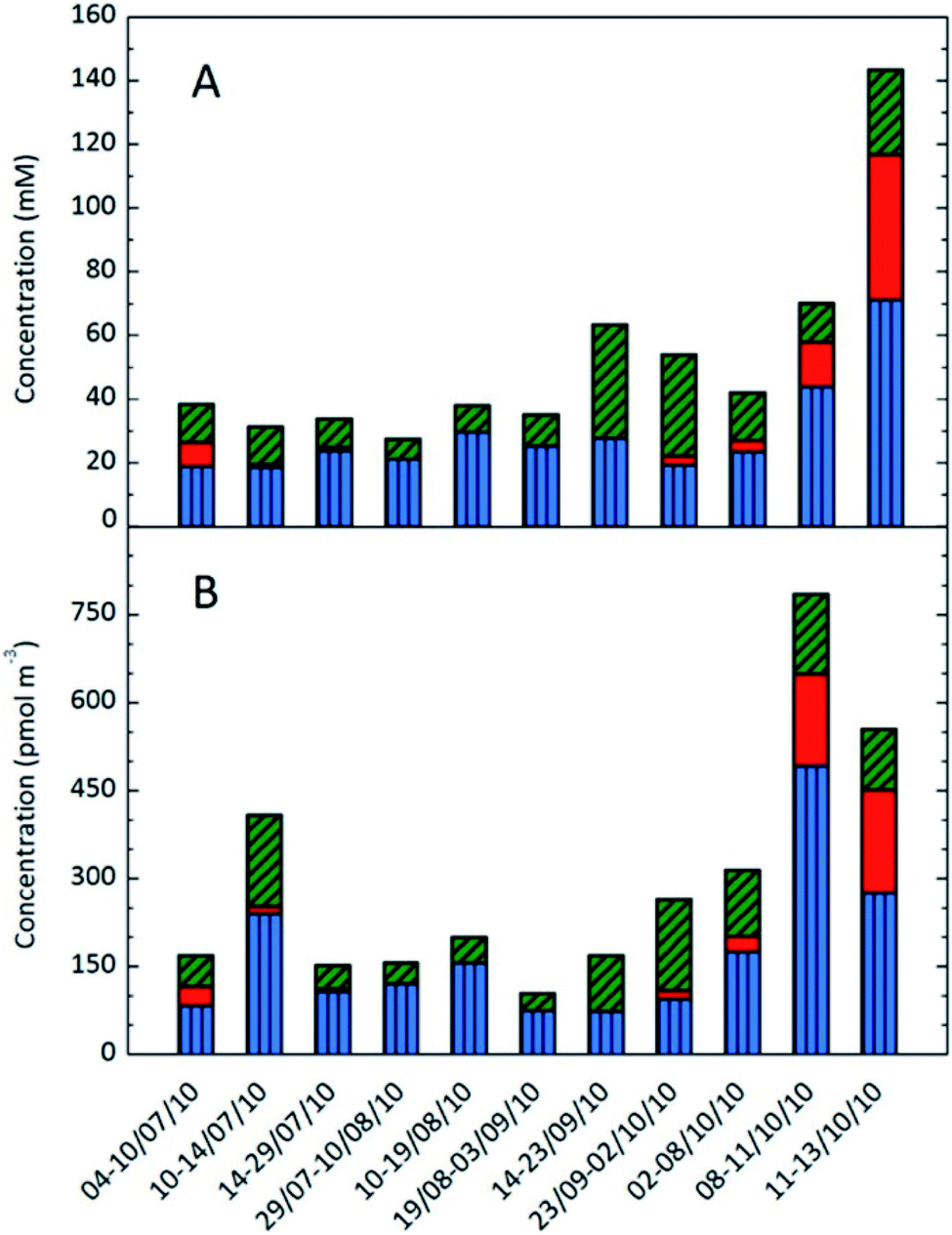 | ||
| Fig. 21 Concentrations of anionic (blue with vertical lines), cationic (solid red), and nonionic (green with diagonal lines) surfactants in (A) aerosol particles and (B) ambient air sampled at a coastal site in Sweden in summer/fall 2010, as measured by colorimetry. Reprinted with permission from V. Gérard, B. Nozière, C. Baduel, L. Fine, A. A. Frossard and R. C. Cohen, Anionic, Cationic, and Nonionic Surfactants in Atmospheric Aerosols from the Baltic Coast at Askö, Sweden: Implications for Cloud Droplet Activation, Environ. Sci. Technol., 2016, 50, 2974–2982. Copyright 2016 American Chemical Society. | ||
8.2. Coastal aerosols
Terrestrial organic matter can be transported long distances in the atmosphere over the ocean, which creates a unique mixture of terrestrial and marine aerosols in coastal environments.458 Surfactants have been identified in a variety of coastal environments using the double extraction colorimetric titration methodologies described above.444In the coastal studies from the remote island near Sweden, surfactants were attributed to biogenic origins based on correlation with chlorophyll-a.443 In contrast, Mustaffa et al.459 and Shaharom et al.460 investigated aerosol surfactants in a coastal urban environment along the Malacca Straits near Malaysia using a high volume air sampler at multiple coastal sites with varying degrees of anthropogenic activity. In both Malaysian studies, anionic and cationic surfactant concentrations were determined via ion chromatography using the established colorimetric methods, and source apportionment was investigated using principal component analysis and multiple linear regression. Mustaffa et al.459 studied both coarse mode aerosol particles (aerodynamic diameter >1.5 μm) and fine mode aerosol particles (aerodynamic diameter <1.5 μm), while Shaharom et al.460 focused exclusively on fine mode aerosols. Anthropogenically derived surfactants were most prevalent in the fine mode particles.459 Overall, anionic surfactants showed greater concentrations than cationic surfactants, and surfactants were primarily attributed to anthropogenic sources, with concentrations highest near tourism and industrial sites.459,460 Both studies also compared the northeast and southwest monsoon seasons, noting increased surfactants in fine mode aerosol particles during the southwest monsoon due to the association with biomass burning activities.459,460 Anthropogenic surfactants were identified from both terrestrial (e.g., biomass burning) and marine aerosol sources, highlighting multiple emission pathways for surfactants in coastal aerosol particles and emphasizing the complexity of source apportionment in coastal regions with concentrated anthropogenic activity. Shaharom et al.460 propose utilizing biomarkers or organic tracers to improve source apportionment for future surfactant studies.
In comparison to colorimetry, mass spectrometry offers more insight into the composition of surfactant-coated aerosol particles. Burdette et al.447 used solid-phase extraction methods coupled with mass spectrometry to determine the presence of anionic, cationic, and nonionic surfactants in atmospheric aerosol particles from Skidaway Island, GA. They observed surfactant-like material in all three ionic classes: the organic material showed surface tension depression of ∼18 mN m−1, high H:C elemental ratios (>1.5), and low O:C ratios (<0.5)—all defining characteristics of surfactants. Fig. 22 shows a plot of H:C ratio vs. O:C ratio, known as a van Krevelen diagram, comparing measurements from aerosol particle extracts with those of surfactant standards.447 Both cationic and anionic surfactant-like organics are present within the range of the surfactant standards.
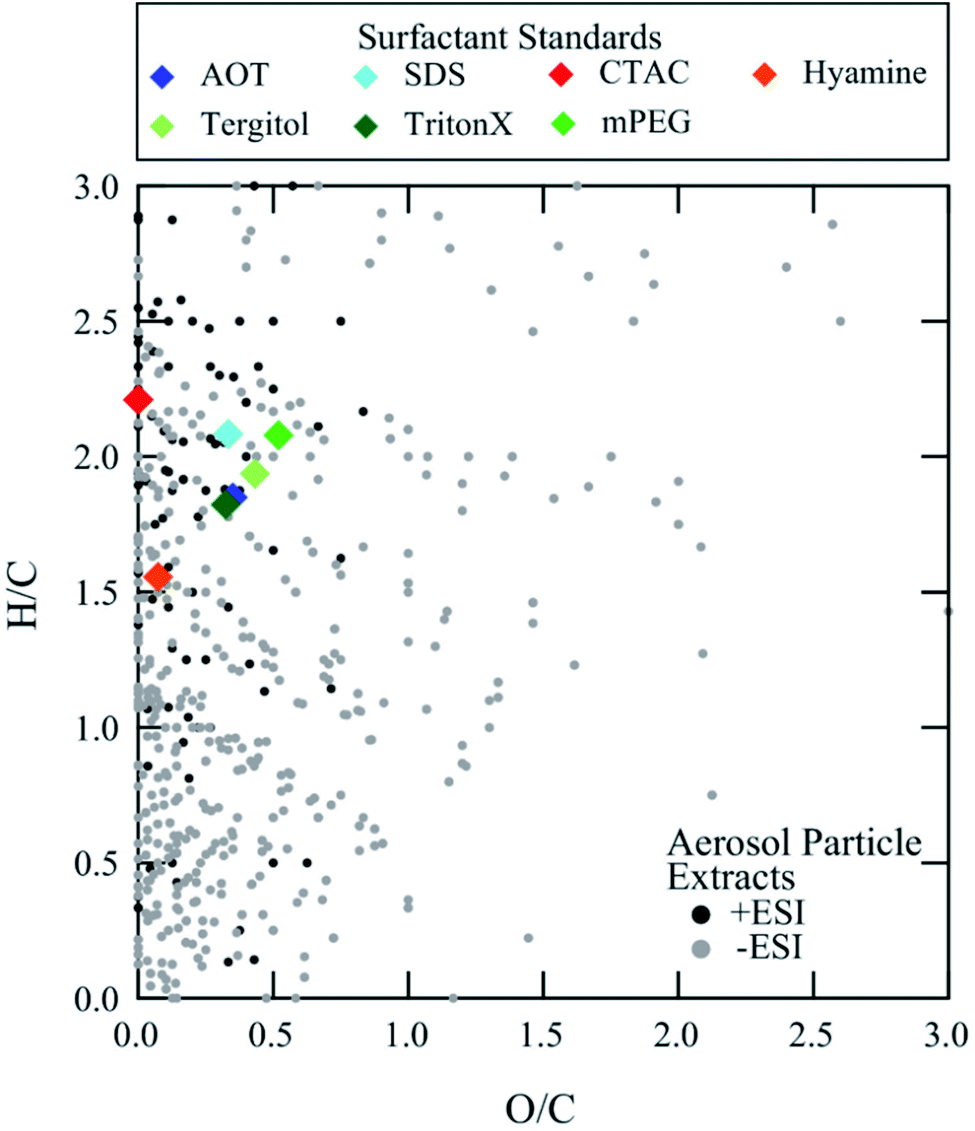 | ||
| Fig. 22 Plot of H/C ratio vs. O/C ratio for organic extracts from aerosol particles collected at Skidaway Island, Georgia in 2018. Diamond markers show the corresponding ratios from assorted surfactant standards. AOT (dioctyl sulfosuccinate sodium) and SDS (sodium dodecyl sulfate) are anionic. CTAC (cetyltrimethylammonium chloride) and Hyamine 1622 are cationic. Tergitol, TritonX, and mPEG (polyethylene glycol methyl ether) are nonionic. Reprinted from Journal of Environmental Sciences, 108, Tret C. Burdette, Amanda A. Frossard, Characterization of seawater and aerosol particle surfactants using solid phase extraction and mass spectrometry, 164–174, Copyright 2021, with permission from Elsevier. | ||
Kang et al.451 provided additional context on the complexity of source apportionment for coastal aerosols with a 2014 research cruise in the East China Sea. The authors analyzed total suspended particle samples via gas chromatograph-mass spectrometry (GC-MS) and observed a variety of surfactants, including phthalates, fatty acids, fatty alcohols, n-alkanes, and polycyclic aromatic hydrocarbons (PAHs), and noted higher daytime concentrations of surfactants, especially near coastal areas with anthropogenic activity.451 These factors, combined with tracer molecules, support anthropogenic sources, including biomass burning and fossil fuel combustion, as the main emission sources for East China Sea organic aerosols and indicate the influence of long-range transport of organic aerosol particles containing surfactants.451
8.3. Marine aerosols
Sea spray aerosol particles are a form of primary aerosol formed through wave breaking at the ocean surface that are known to contain highly enriched amounts of organic material, including surfactants, saccharides, alkanes, fatty acids, and even human lung surfactants,461 relative to the surface ocean.289,462 Several of the marine organic compound classes possess surface-active characteristics, and biosurfactants are known to significantly depress surface tension in aerosols and increase cloud nucleating efficiency.5,463 Given recent SSA reviews by Quinn et al.,5 Bertram et al.,289 and Schiffer et al.,464 we will focus primarily on content published during or after 2017.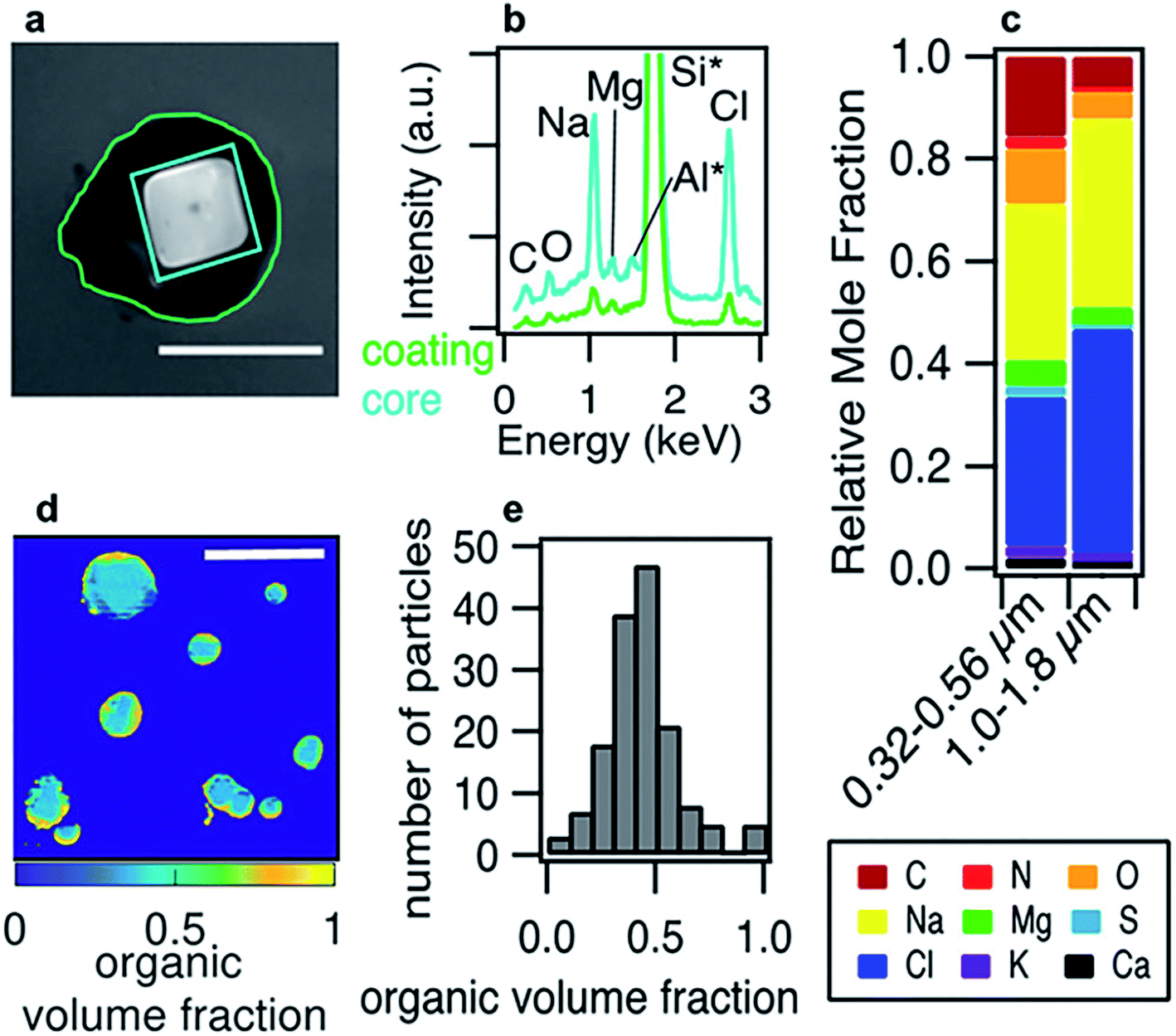 | ||
| Fig. 23 A suite of single-particle measurements of ambient Arctic sea spray aerosols. (a) SEM image of a representative particle with an organic coating on a solid crystalline core, shown with a scale bar of 3 μm. (b) Corresponding elemental composition from energy dispersive X-ray spectroscopy (EDX). The aluminum signal is due to the sample holder, and the silicon signal is due to the substrate. (c) Elemental composition averaged across 773 SSA particles in the 0.32–0.56 μm diameter size range and 918 SSA particles in the 1.0–1.8 μm diameter size range, as measured by computer-controlled SEM and EDX. (d) STXM-NEXAFS image showing the concentration of organic species at the surface of representative SSA particles. The scale bar represents 5 μm. (e) The number distribution for organic volume fraction averaged across 150 SSA particles, as measured by STXM-NEXAFS. Reprinted with permission from R. M. Kirpes, D. Bonanno, N. W. May, M. Fraund, A. J. Barget, R. C. Moffet, A. P. Ault and K. A. Pratt, Wintertime Arctic Sea Spray Aerosol Composition Controlled by Sea Ice Lead Microbiology, ACS Cent. Sci., 2019, 5, 1760–1767, https://pubs.acs.org/doi/10.1021/acscentsci.9b00541. Further permissions related to the material excerpted should be directed to the American Chemical Society. | ||
At the same time, anthropogenic influences can affect particle composition even in remote Arctic marine environments. Yu et al.466 sampled particles ranging from 100 nm to 2 μm in diameter at the Svalbard archipelago in late summer 2012. Analysis by an assortment of single-particle techniques, including TEM/EDS, SEM, secondary ion mass spectrometry (SIMS), and AFM, showed that 74% of non-sea-salt sulfate particles had organic coatings, which contributed 63% of particle volume on average.466 Approximately 20% of sulfate particles had soot inclusions within the organic coating, further highlighting anthropogenic influences on marine aerosol particles in the Arctic.
For example, Aller et al.468 collected size-fractionated aerosol particles from ambient air and from freshly generated Sea Sweep SSA in the western North Atlantic to characterize the biogenic organic components of SSA. They found that polysaccharidic and proteinaceous gels, which can behave as surface-active organics, were enriched in sub- and super-micron SSA particles. Furthermore, Aller et al.468 found minimal continental influence in the aerosol particles, based on backward trajectory modeling, and gained insights into the transport of marine biogenic material through primary particle emission, as well as size-discriminated characterization of SSA and ambient aerosol under field conditions.
MART experiments have investigated the impacts of marine bacteria on surface-active organics in laboratory SSA.291,469,470 Hasenecz et al.470 used a MART mesocosm experiment to investigate the impact of heterotrophic bacteria on saccharide enrichment in SSA and noted enrichment factor increases of 30-fold for glucose and 20-fold for arabinose in the particles relative to bulk seawater. Similarly, Rastelli et al.471 generated SSA via a bubble bursting system onboard a ship in the Northeastern Atlantic Ocean in 2006 and found that SSA is highly enriched in organic matter as compared to seawater – 140000× for lipids, 120000× for proteins, and 100000× for carbohydrates. Their bubble bursting system consisted of a vertical recirculating water jet flowing at 20 L min−1 in a 200 L tank.472 Cochran et al.473 also generated SSA via a sintered glass bubbler and quantified the selective transfer of surface-active organics from the solution to aerosol particles: they reported higher enrichment factors for more surface-active linear carboxylates (55 ± 8) vs. linear dicarboxylates (5 ± 1). Frossard et al.474 later generated laboratory SSA in a marine aerosol generator in the western North Atlantic Ocean in 2016 at two biologically productive and two oligotrophic sites. They were the first to directly pair surfactant observations in near-surface seawater, deep seawater, and SSA and found that surfactants were present in all sampled hydrographic regions and that surfactants are highly enriched in SSA compared to seawater. Comparison of surface tension isotherms revealed the preferential transfer of relatively weaker surfactants to SSA, a finding which had not previously been recorded.474
Another prominent technique used to investigate surfactants in SSA is the wave flume experiments conducted by the Center for Aerosol Impacts on Chemistry of the Environment (CAICE) at Scripps Institute of Oceanography.475,476 Several CAICE studies have investigated composition and morphology of SSA containing surface-active organics produced in real seawater with phytoplankton blooms.161,243,288,290,477 Ault et al.477 utilized Raman spectroscopy and SFG to demonstrate the presence of organic matter concentrated at the surface of SSA particles generated in a wave flume. Lee et al. investigated wave flume SSA morphology and surface tension using AFM and SEM.161,243 They observed an inorganic core–organic shell morphology and found organic enrichment during phytoplankton blooms, leading to suppressed surface tension.243 Further morphological investigations revealed size dependency and an increase in organic mass fraction with decreasing particle size, where smaller particles featured more prevalent inorganic core–organic shell morphologies.161 Additionally, Cochran et al.288 characterized classes of organic compounds in SSA, including fatty acids and polysaccharides, noting that changes in phytoplankton and heterotrophic bacteria populations coincided with changes in SSA molecular composition, and emphasized the diversity of molecular composition at the particle level. Wang et al.290 note that organic enrichment depends on a balance between phytoplankton production and bacterial degradation, providing a framework for understanding why chlorophyll-a has proven to be an inconsistent indicator for SSA organic enrichment. See section 3.2 for additional discussion of the morphology of SSA particles.
8.4. Terrestrial aerosols
Baduel et al.442 applied their double extraction method with PM10 field samples from urban Grenoble, France to investigate surfactants in aerosols. They observed pronounced seasonal variation, with summer aerosol surfactants attributed primarily to biogenic sources and giving surface tension measurements of approximately 30 mN m−1.442 Winter surfactants showed stronger surface tension depression (35–45 mN m−1) and were attributed to two groups of anthropogenic surfactants, one group of man-made surfactants similar to sodium dodecyl sulfate, and a second group of surfactants from woodburning, highlighting the importance of combustion-based surfactant emissions during the winter, in agreement with previous studies.442,479 Gérard et al. continued to apply their targeted extraction and colorimetric method to investigate PM1 aerosol in several urban, coastal, and remote regions of Europe focusing on amphiphilic surfactants.443,446 Amphiphilic surfactants were found to be present throughout the range of particle sizes and are expected to enhance the cloud droplet activation ability of submicrometer atmospheric particles.446
Frka et al.453 employed AC voltammetry to characterize and quantify surfactants in aqueous extracts from atmospheric aerosols in Europe including urban, rural, and coastal locations. An advantage of the electrochemical method is the ability to directly analyze aerosol extracts with no sample concentration required. Observed surfactant concentrations were highest for urban aerosols, and pH studies demonstrated the predominantly anionic character of surfactants.453 Zeng et al.448 investigated industrial, stationary source PM2.5 emissions via a field study to gain insights into source profiles and species-based emission factors. Palmitic acid and stearic acid, known surfactants, were the most abundant organic species in PM2.5 from industrial emissions (also commonly found in cooking emissions). Their findings showed large discrepancies with results from previous studies that did not conduct field measurements, indicating the vital importance of field research to improve emission inventories.448
8.5. Future needs
Direct measurements of surfactants in ambient aerosol particles are still relatively rare because unambiguous evidence of their presence must combine information about composition with information about morphology at the single particle level. As measurements advance, attention should be paid to temporal trends in surfactant concentrations, both on a seasonal basis and at higher-resolution time intervals, because diagnostic surfactants can be useful as tracers for source apportionment. Furthermore, as more field campaigns report surface tension, phase state, and composition of surfactant-coated particles, we can begin to bridge the gap between the behavior of complex samples from the field and the simplified model systems from laboratory experiments and modeling work.9. Outlook
The field of atmospheric chemistry is said to be built on a three-legged stool, where field studies, laboratory studies, and atmospheric modeling each represent one of the legs.411,412 However, there is growing recognition of the need to move beyond the analogy of three separate legs to more closely integrate feedback among the three traditional approaches.411–413 Such collaborative endeavors are certainly important for advancing research on surfactant-coated aerosol particles.The most important gap in our knowledge of atmospheric surfactants is simply the scarcity of field studies. We need more ambient measurements in more environments spanning more seasons. In addition to extended monitoring of long-term trends, measurements at high-temporal resolution are also needed to couple with emission inventories and source apportionment. Recent developments in instrumentation55,82,113 and sample preparation techniques444 are making novel field measurements possible.
In the laboratory, experimentalists and theoreticians are working toward ever more complex systems—essentially bringing the atmosphere into the laboratory. These lab studies and simulations are uniquely poised to offer fundamental insights into surfactant systems under controlled conditions. Accordingly, work is needed to move beyond studies of traditional surfactants to new classes of surface-active agents, such as PFAS,293 and to build layers of complexity into model systems. Valuable insights into ambient aerosol can be obtained by studying surfactant-coated particles generated under realistic conditions, e.g., SSA from a wave flume during a phytoplankton bloom,475 BBA from combustion of real fuels in an environmental chamber,483 and SOA from oxidation of VOCs like α-pinene and isoprene.13 Finally, the ways in which surfactants, films, and coatings on bioaerosols affect disease transmission (e.g., from the SARS-CoV-2 virus) remain a relatively unexplored area of research.122,484
Both field and laboratory studies feed into atmospheric models to address the question: What level of detail about particle phase and composition is needed to accurately represent climate and air quality? Because surfactants, films, and coatings affect particle morphology and phase state, they also affect the ability of aerosol particles to scatter solar radiation and their ability to act as CCN and INPs. The ideal model for climate and air quality should capture these effects while remaining as simple, elegant, and computationally inexpensive as possible.485
Across all studies of surfactant-coated aerosol particles, the greatest contributing factor toward advancing research has been the development of instrumental techniques to allow coordinated measurements of morphology and composition at the single particle level. It is our hope that continued advances in instrumentation will allow for more widespread characterization of surfactants in ambient aerosol, providing insight into broad societal concerns like climate, air quality, and health.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the College of Wooster.References
- P. S. Gill, T. E. Graedel and C. J. Weschler, Organic films on atmospheric aerosol particles, fog droplets, cloud droplets, raindrops, and snowflakes, Rev. Geophys. Space Phys., 1983, 21, 903–920 CrossRef CAS.
- D. J. Donaldson and V. Vaida, The Influence of Organic Films at the Air–Aqueous Boundary on Atmospheric Processes, Chem. Rev., 2006, 106, 1445–1461 CrossRef CAS PubMed.
- V. F. McNeill, N. Sareen and A. N. Schwier, in Atmospheric and Aerosol Chemistry, ed. V. F. McNeill and P. A. Ariya, Springer, Berlin, Heidelberg, 2013, vol. 339, pp. 201–259 Search PubMed.
- B. Gantt and N. Meskhidze, The physical and chemical characteristics of marine primary organic aerosol: a review, Atmos. Chem. Phys., 2013, 13, 3979–3996 CrossRef.
- P. K. Quinn, D. B. Collins, V. H. Grassian, K. A. Prather and T. S. Bates, Chemistry and Related Properties of Freshly Emitted Sea Spray Aerosol, Chem. Rev., 2015, 115, 4383–4399 CrossRef CAS PubMed.
- P. Renard, I. Canet, M. Sancelme, M. Matulova, I. Uhliarikova, B. Eyheraguibel, L. Nauton, J. Devemy, M. Traïkia, P. Malfreyt and A.-M. Delort, in Surfactants and Detergents, ed. A. Kumar Dutta, IntechOpen, 2019 Search PubMed.
- K. L. Abdullahi, J. M. Delgado-Saborit and R. M. Harrison, Emissions and indoor concentrations of particulate matter and its specific chemical components from cooking: a review, Atmos. Environ., 2013, 71, 260–294 CrossRef CAS.
- R. Zana, Dimeric and oligomeric surfactants. Behavior at interfaces and in aqueous solution: a review, Adv. Colloid Interface Sci., 2002, 97, 205–253 CrossRef CAS PubMed.
- N. Sareen, A. N. Schwier, T. L. Lathem, A. Nenes and V. F. McNeill, Surfactants from the gas phase may promote cloud droplet formation, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2723–2728 CrossRef CAS PubMed.
- B. Nozière, S. Ekström, T. Alsberg and S. Holmström, Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols, Geophys. Res. Lett., 2010, 37, L05806 CrossRef.
- Y.-H. Lin, S. H. Budisulistiorini, K. Chu, R. A. Siejack, H. Zhang, M. Riva, Z. Zhang, A. Gold, K. E. Kautzman and J. D. Surratt, Light-Absorbing Oligomer Formation in Secondary Organic Aerosol from Reactive Uptake of Isoprene Epoxydiols, Environ. Sci. Technol., 2014, 48, 12012–12021 CrossRef CAS PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- B. Ervens, B. J. Turpin and R. J. Weber, Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 2011, 11, 11069–11102 CrossRef CAS.
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Tropospheric aqueous-phase chemistry: Kinetics, mechanisms, and its coupling to a changing gas phase, Chem. Rev., 2015, 115, 4259–4334 CrossRef CAS PubMed.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of Atmospheric Brown Carbon, Chem. Rev., 2015, 115, 4335–4382 CrossRef CAS PubMed.
- V. F. McNeill, Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols, Environ. Sci. Technol., 2015, 49, 1237–1244 CrossRef CAS PubMed.
- G. Rao and E. P. Vejerano, Partitioning of volatile organic compounds to aerosols: a review, Chemosphere, 2018, 212, 282–296 CrossRef CAS PubMed.
- Y. S. Djikaev and E. Ruckenstein, Depletion of atmospheric organic trace gases due to their uptake by an ensemble of aqueous aerosols evolving via concurrent condensation and chemical aging, Phys. Chem. Chem. Phys., 2019, 21, 13090–13098 RSC.
- A. M. Deal, R. J. Rapf and V. Vaida, Water–Air Interfaces as Environments to Address the Water Paradox in Prebiotic Chemistry: A Physical Chemistry Perspective, J. Phys. Chem. A, 2021, 125, 4929–4942 CrossRef CAS PubMed.
- C.-H. Chang and E. I. Franses, Adsorption dynamics of surfactants at the air/water interface: a critical review of mathematical models, data, and mechanisms, Colloids Surf., A, 1995, 100, 1–45 CrossRef CAS.
- J. Eastoe and J. S. Dalton, Dynamic surface tension and adsorption mechanisms of surfactants at the air–water interface, Adv. Colloid Interface Sci., 2000, 85, 103–144 CrossRef CAS PubMed.
- A. J. Prosser and E. I. Franses, Adsorption and surface tension of ionic surfactants at the air–water interface: review and evaluation of equilibrium models, Colloids Surf., A, 2001, 178, 1–40 CrossRef CAS.
- C. M. Phan, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 79–104 Search PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- Y. You, M. L. Smith, M. Song, S. T. Martin and A. K. Bertram, Liquid–liquid phase separation in atmospherically relevant particles consisting of organic species and inorganic salts, Int. Rev. Phys. Chem., 2014, 33, 43–77 Search PubMed.
- P. K. Grover and R. L. Ryall, Critical Appraisal of Salting-Out and Its Implications for Chemical and Biological Sciences, Chem. Rev., 2005, 105, 1–10 CrossRef CAS PubMed.
- J. H. Schulman and A. H. Hughes, On the surface potentials of unimolecular films. Part IV. The effect of the underlying solution and transition phenomena in the film, Proc. R. Soc. London, Ser. A, 1932, 138, 430–450 CAS.
- E. F. Porter, Monomolecular Films of α-Aminostearic Acid, Stearic Acid, and Heptadecylamine, J. Am. Chem. Soc., 1937, 59, 1883–1888 CrossRef CAS.
- J. Glazer and M. Z. Dogan, Ionization of protein monolayers and related substances, Trans. Faraday Soc., 1953, 49, 448 RSC.
- B. A. Wellen, E. A. Lach and H. C. Allen, Surface pKa of octanoic, nonanoic, and decanoic fatty acids at the air–water interface: applications to atmospheric aerosol chemistry, Phys. Chem. Chem. Phys., 2017, 19, 26551–26558 RSC.
- T. Zhang, S. L. Brantley, D. Verreault, R. Dhankani, S. A. Corcelli and H. C. Allen, Effect of pH and Salt on Surface pKa of Phosphatidic Acid Monolayers, Langmuir, 2018, 34, 530–539 CrossRef CAS PubMed.
- M. Luo, N. A. Wauer, K. J. Angle, A. C. Dommer, M. Song, C. M. Nowak, R. E. Amaro and V. H. Grassian, Insights into the behavior of nonanoic acid and its conjugate base at the air/water interface through a combined experimental and theoretical approach, Chem. Sci., 2020, 11, 10647–10656 RSC.
- Y. B. Vysotsky, E. S. Kartashynska, D. Vollhardt and V. B. Fainerman, Surface pKa of Saturated Carboxylic Acids at the Air/Water Interface: A Quantum Chemical Approach, J. Phys. Chem. C, 2020, 124, 13809–13818 CrossRef CAS.
- R. S. Andino, J. Liu, C. M. Miller, X. Chen, S. W. Devlin, M. K. Hong, R. Rajagopal, S. Erramilli and L. D. Ziegler, Anomalous pH-Dependent Enhancement of p-Methyl Benzoic Acid Sum-Frequency Intensities: Cooperative Surface Adsorption Effects, J. Phys. Chem. A, 2020, 124, 3064–3076 CrossRef CAS PubMed.
- E. S. Kartashynska, Y. B. Vysotsky, D. Vollhardt and V. B. Fainerman, Relationship between the Bulk and Surface Basicity of Aliphatic Amines: A Quantum Chemical Approach, ACS Omega, 2020, 5, 32032–32039 CrossRef CAS PubMed.
- R. Rajagopal, M. K. Hong, L. D. Ziegler, S. Erramilli and O. Narayan, Conjugate Acid–Base Interaction Driven Phase Transition at a 2D Air–Water Interface, J. Phys. Chem. B, 2021, 125, 6330–6337 CrossRef CAS PubMed.
- P. Forster, T. Storelvmo, K. Armour, W. Collins, J. L. Dufresne, D. Frame, D. J. Lunt, T. Mauritsen, M. D. Palmer, M. Watanabe, M. Wild and H. Zhang, in Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, Cambridge University Press, 2021 Search PubMed.
- D. K. Farmer, C. D. Cappa and S. M. Kreidenweis, Atmospheric Processes and Their Controlling Influence on Cloud Condensation Nuclei Activity, Chem. Rev., 2015, 115, 4199–4217 CrossRef CAS PubMed.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 2017, 55, 509–559 CrossRef.
- L. Nandy, Y. Yao, Z. Zheng and N. Riemer, Water uptake and optical properties of mixed organic–inorganic particles, Aerosol Sci. Technol., 2021, 55, 1398–1413 CrossRef CAS.
- D. A. Knopf, P. A. Alpert and B. Wang, The Role of Organic Aerosol in Atmospheric Ice Nucleation: A Review, ACS Earth Space Chem., 2018, 2, 168–202 CrossRef CAS.
- S. M. Burrows, C. S. McCluskey, G. Cornwell, I. Steinke, K. Zhang, B. Zhao, M. Zawadowicz, A. Raman, G. Kulkarni, S. China, A. Zelenyuk and P. J. DeMott, Ice-Nucleating Particles That Impact Clouds and Climate: Observational and Modeling Research Needs, Rev. Geophys., 2022, 60, e2021RG000745 CrossRef.
- C. E. Kolb, R. A. Cox, J. P. D. Abbatt, M. Ammann, E. J. Davis, D. J. Donaldson, B. C. Garrett, C. George, P. T. Griffiths and D. R. Hanson, An overview of current issues in the uptake of atmospheric trace gases by aerosols and clouds, Atmos. Chem. Phys., 2010, 10, 10561–10605 CrossRef CAS.
- P. Davidovits, C. E. Kolb, L. R. Williams, J. T. Jayne and D. R. Worsnop, Update 1 of: Mass Accommodation and Chemical Reactions at Gas–Liquid Interfaces, Chem. Rev., 2011, 111, PR76–PR109 CrossRef CAS PubMed.
- J. P. D. Abbatt, A. K. Y. Lee and J. A. Thornton, Quantifying trace gas uptake to tropospheric aerosol: recent advances and remaining challenges, Chem. Soc. Rev., 2012, 41, 6555–6581 RSC.
- J. F. Davies and K. R. Wilson, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 403–433 Search PubMed.
- A. Rohr and J. McDonald, Health effects of carbon-containing particulate matter: focus on sources and recent research program results, Crit. Rev. Toxicol., 2016, 46, 97–137 CrossRef CAS PubMed.
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi, S. Enami, A. M. Arangio, J. Fröhlich-Nowoisky, Y. Fujitani, A. Furuyama, P. S. J. Lakey, J. Lelieveld, K. Lucas, Y. Morino, U. Pöschl, S. Takahama, A. Takami, H. Tong, B. Weber, A. Yoshino and K. Sato, Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 2017, 51, 13545–13567 CrossRef CAS PubMed.
- Z. Qi, Y. Zhang, Z.-F. Chen, C. Yang, Y. Song, X. Liao, W. Li, S. Y. Tsang, G. Liu and Z. Cai, Chemical identity and cardiovascular toxicity of hydrophobic organic components in PM2.5, Ecotoxicol. Environ. Saf., 2020, 201, 110827 CrossRef CAS PubMed.
- N. Riemer, A. P. Ault, M. West, R. L. Craig and J. H. Curtis, Aerosol Mixing State: Measurements, Modeling, and Impacts, Rev. Geophys., 2019, 57, 187–249 CrossRef.
- C. A. Noble and K. A. Prather, Real-time single particle mass spectrometry: a historical review of a quarter century of the chemical analysis of aerosols, Mass Spectrom. Rev., 2000, 19, 248–274 CrossRef CAS PubMed.
- A. Zelenyuk and D. Imre, Beyond single particle mass spectrometry: multidimensional characterisation of individual aerosol particles, Int. Rev. Phys. Chem., 2009, 28, 309–358 Search PubMed.
- B. R. Bzdek, M. R. Pennington and M. V. Johnston, Single particle chemical analysis of ambient ultrafine aerosol: a review, J. Aerosol Sci., 2012, 52, 109–120 CrossRef CAS.
- U. K. Krieger, C. Marcolli and J. P. Reid, Exploring the complexity of aerosol particle properties and processes using single particle techniques, Chem. Soc. Rev., 2012, 41, 6631–6662 RSC.
- A. P. Ault and J. L. Axson, Atmospheric Aerosol Chemistry: Spectroscopic and Microscopic Advances, Anal. Chem., 2017, 89, 430–452 CrossRef CAS PubMed.
- R. C. Sullivan, K. Gorkowski and L. Jahn, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 353–402 Search PubMed.
- M. Tang, T. Zhu, D. Zhao, C. Ye and X. Huang, in Advances in Atmospheric Chemistry, ed. J. R. Barker, A. L. Steiner and T. J. Wallington, World Scientific, 2019, vol. 2, pp. 83–167 Search PubMed.
- M. S. Aston, The study of surfactant monolayers by surface pressure–area measurement, Chem. Soc. Rev., 1993, 22, 67–71 RSC.
- D. M. Golden, G. N. Spokes and S. W. Benson, Very Low-Pressure Pyrolysis (VLPP); A Versatile Kinetic Tool, Angew. Chem., Int. Ed. Engl., 1973, 12, 534–546 CrossRef.
- R. Zellner, P. Behr, S. Seisel, H. Somnitz and L. Treuel, Chemistry and Microphysics of Atmospheric Aerosol Surfaces: Laboratory Techniques and Applications, Z. Phys. Chem., 2009, 223, 359–385 CrossRef CAS.
- J. A. Faust and G. M. Nathanson, Microjets and coated wheels: Versatile tools for exploring collisions and reactions at gas–liquid interfaces, Chem. Soc. Rev., 2016, 45, 3609–3620 RSC.
- B. Winter and M. Faubel, Photoemission from Liquid Aqueous Solutions, Chem. Rev., 2006, 106, 1176–1211 CrossRef CAS PubMed.
- R. H. Schwantes, R. C. McVay, X. Zhang, M. M. Coggon, H. Lignell, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, in Advances in Atmospheric Chemistry, ed. J. R. Barker, A. L. Steiner and T. J. Wallington, World Scientific, 2017, pp. 1–93 Search PubMed.
- S. M. Charan, Y. Huang and J. H. Seinfeld, Computational Simulation of Secondary Organic Aerosol Formation in Laboratory Chambers, Chem. Rev., 2019, 119, 11912–11944 CrossRef CAS PubMed.
- A. T. Lambe, A. T. Ahern, L. R. Williams, J. G. Slowik, J. P. S. Wong, J. P. D. Abbatt, W. H. Brune, N. L. Ng, J. P. Wright, D. R. Croasdale, D. R. Worsnop, P. Davidovits and T. B. Onasch, Characterization of aerosol photooxidation flow reactors: heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements, Atmos. Meas. Tech., 2011, 4, 445–461 CrossRef CAS.
- Z. Peng and J. L. Jimenez, Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 2020, 49, 2570–2616 RSC.
- C. J. Howard, Kinetic Measurements Using Flow Tubes, J. Phys. Chem., 1979, 83, 3–9 CrossRef CAS.
- G. Li, H. Su, U. Kuhn, H. Meusel, M. Ammann, M. Shao, U. Pöschl and Y. Cheng, Technical note: Influence of surface roughness and local turbulence on coated-wall flow tube experiments for gas uptake and kinetic studies, Atmos. Chem. Phys., 2018, 18, 2669–2686 CrossRef CAS.
- Z. Gong, Y.-L. Pan, G. Videen and C. Wang, Optical trapping and manipulation of single particles in air: Principles, technical details, and applications, J. Quant. Spectrosc. Radiat. Transfer, 2018, 214, 94–119 CrossRef CAS.
- R. C. Sullivan, H. Boyer-Chelmo, K. Gorkowski and H. Beydoun, Aerosol Optical Tweezers Elucidate the Chemistry, Acidity, Phase Separations, and Morphology of Atmospheric Microdroplets, Acc. Chem. Res., 2020, 53, 2498–2509 CrossRef CAS PubMed.
- L. Cohen, M. I. Quant and D. J. Donaldson, Real-Time Measurements of pH Changes in Single, Acoustically Levitated Droplets Due to Atmospheric Multiphase Chemistry, ACS Earth Space Chem., 2020, 4, 854–861 CrossRef CAS.
- D. S. Richards, K. L. Trobaugh, J. Hajek-Herrera and R. D. Davis, Dual-Balance Electrodynamic Trap as a Microanalytical Tool for Identifying Gel Transitions and Viscous Properties of Levitated Aerosol Particles, Anal. Chem., 2020, 92, 3086–3094 CrossRef CAS PubMed.
- R. K. Kohli and J. F. Davies, Measuring the Chemical Evolution of Levitated Particles: A Study on the Evaporation of Multicomponent Organic Aerosol, Anal. Chem., 2021, 93, 12472–12479 CrossRef CAS PubMed.
- S. Roy, M. E. Diveky and R. Signorell, Mass Accommodation Coefficients of Water on Organics from Complementary Photoacoustic and Light Scattering Measurements on Laser-Trapped Droplets, J. Phys. Chem. C, 2020, 124, 2481–2489 CrossRef.
- M. E. Diveky, M. J. Gleichweit, S. Roy and R. Signorell, Shining New Light on the Kinetics of Water Uptake by Organic Aerosol Particles, J. Phys. Chem. A, 2021, 125, 3528–3548 CrossRef CAS PubMed.
- E. Gard, J. E. Mayer, B. D. Morrical, T. Dienes, D. P. Fergenson and K. A. Prather, Real-Time Analysis of Individual Atmospheric Aerosol Particles: Design and Performance of a Portable ATOFMS, Anal. Chem., 1997, 69, 4083–4091 CrossRef CAS.
- M. R. Canagaratna, J. T. Jayne, J. L. Jimenez, J. D. Allan, M. R. Alfarra, Q. Zhang, T. B. Onasch, F. Drewnick, H. Coe, A. Middlebrook, A. Delia, L. R. Williams, A. M. Trimborn, M. J. Northway, P. F. DeCarlo, C. E. Kolb, P. Davidovits and D. R. Worsnop, Chemical and microphysical characterization of ambient aerosols with the Aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 2007, 26, 185–222 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, S. Cui, D. D. Huang and X. Ge, Aerosol Measurements by Soot Particle Aerosol Mass Spectrometer: A Review, Atmos. Meas. Tech., 2020, 6, 440–451 CAS.
- K. A. Pratt and K. A. Prather, Mass spectrometry of atmospheric aerosols-Recent developments and applications. Part I: Off-line mass spectrometry techniques, Mass Spectrom. Rev., 2012, 31, 1–16 CrossRef CAS PubMed.
- K. A. Pratt and K. A. Prather, Mass spectrometry of atmospheric aerosols-Recent developments and applications. Part II: On-line mass spectrometry techniques, Mass Spectrom. Rev., 2012, 31, 17–48 CrossRef CAS PubMed.
- J. Laskin, A. Laskin and S. A. Nizkorodov, New mass spectrometry techniques for studying physical chemistry of atmospheric heterogeneous processes, Int. Rev. Phys. Chem., 2013, 32, 128–170 Search PubMed.
- M. V. Johnston and D. E. Kerecman, Molecular Characterization of Atmospheric Organic Aerosol by Mass Spectrometry, Annu. Rev. Anal. Chem., 2019, 12, 247–274 Search PubMed.
- M. Ammann, L. Artiglia and T. Bartels-Rausch, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 135–166 Search PubMed.
- M. Ahmed and O. Kostko, From atoms to aerosols: Probing clusters and nanoparticles with synchrotron based mass spectrometry and X-ray spectroscopy, Phys. Chem. Chem. Phys., 2020, 22, 2713–2737 RSC.
- R. Dupuy, C. Richter, B. Winter, G. Meijer, R. Schlögl and H. Bluhm, Core level photoelectron spectroscopy of heterogeneous reactions at liquid–vapor interfaces: Current status, challenges, and prospects, J. Chem. Phys., 2021, 154, 060901 CrossRef CAS PubMed.
- C. Stefaniu, G. Brezesinski and H. Möhwald, Langmuir monolayers as models to study processes at membrane surfaces, Adv. Colloid Interface Sci., 2014, 208, 197–213 CrossRef CAS PubMed.
- M. K. Bera, W. Bu and A. Uysal, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 167–194 Search PubMed.
- A. Maestro and P. Gutfreund, In situ determination of the structure and composition of Langmuir monolayers at the air/water interface by neutron and X-ray reflectivity and ellipsometry, Adv. Colloid Interface Sci., 2021, 293, 102434 CrossRef CAS PubMed.
- S. M. Baumler and H. C. Allen, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 105–133 Search PubMed.
- B. A. Wellen Rudd, A. S. Vidalis and H. C. Allen, Thermodynamic versus non-equilibrium stability of palmitic acid monolayers in calcium-enriched sea spray aerosol proxy systems, Phys. Chem. Chem. Phys., 2018, 20, 16320–16332 RSC.
- M. G. Vazquez de Vasquez, K. A. Carter-Fenk, L. M. McCaslin, E. E. Beasley, J. B. Clark and H. C. Allen, Hydration and Hydrogen Bond Order of Octadecanoic Acid and Octadecanol Films on Water at 21 and 1 °C, J. Phys. Chem. A, 2021, 125, 10065–10078 CrossRef CAS PubMed.
- N. C. Auvil, M. G. Vazquez de Vasquez and H. C. Allen, Zinc–Carboxylate Binding in Mixed Octadecanoic Acid and Octadecanol Monolayers on Proxy Seawater Solution Surfaces, ACS Earth Space Chem., 2021, 5, 2947–2956 CrossRef CAS.
- M. Luo, A. C. Dommer, J. M. Schiffer, D. J. Rez, A. R. Mitchell, R. E. Amaro and V. H. Grassian, Surfactant Charge Modulates Structure and Stability of Lipase-Embedded Monolayers at Marine-Relevant Aerosol Surfaces, Langmuir, 2019, 35, 9050–9060 CrossRef CAS PubMed.
- S. Li, L. Du, N. T. Tsona and W. Wang, The interaction of trace heavy metal with lipid monolayer in the sea surface microlayer, Chemosphere, 2018, 196, 323–330 CrossRef CAS PubMed.
- S. Li, L. Du, Q. Zhang and W. Wang, Stabilizing mixed fatty acid and phthalate ester monolayer on artificial seawater, Environ. Pollut., 2018, 242, 626–633 CrossRef CAS PubMed.
- J. Li, S. Li, S. Cheng, N. T. Tsona and L. Du, Emerging investigator series: exploring the surface properties of aqueous aerosols coated with mixed surfactants, Environ. Sci.: Processes Impacts, 2018, 20, 1500–1511 RSC.
- S. Li, S. Cheng, L. Du and W. Wang, Establishing a model organic film of low volatile compound mixture on aqueous aerosol surface, Atmos. Environ., 2019, 200, 15–23 CrossRef CAS.
- S. Cheng, S. Li, N. T. Tsona, C. George and L. Du, Insights into the Headgroup and Chain Length Dependence of Surface Characteristics of Organic-Coated Sea Spray Aerosols, ACS Earth Space Chem., 2019, 3, 571–580 CrossRef CAS.
- S. Cheng, S. Li, N. T. Tsona, C. George and L. Du, Alterations in the surface properties of sea spray aerosols introduced by the presence of sterols, Sci. Total Environ., 2019, 671, 1161–1169 CrossRef CAS.
- M. Xu, N. T. Tsona, S. Cheng, J. Li and L. Du, Unraveling interfacial properties of organic-coated marine aerosol with lipase incorporation, Sci. Total Environ., 2021, 782, 146893 CrossRef CAS PubMed.
- S. K. Reddy, R. Thiraux, B. A. Wellen Rudd, L. Lin, T. Adel, T. Joutsuka, F. M. Geiger, H. C. Allen, A. Morita and F. Paesani, Bulk Contributions Modulate the Sum-Frequency Generation Spectra of Water on Model Sea-Spray Aerosols, Chem, 2018, 4, 1629–1644 CAS.
- A. G. Bé, Y. Liu, A. Tuladhar, A. D. Bellcross, Z. Wang, R. J. Thomson and F. M. Geiger, Surface-Active β-Caryophyllene Oxidation Products at the Air/Aqueous Interface, ACS Earth Space Chem., 2019, 3, 1740–1748 CrossRef.
- B. P. Gordon, F. G. Moore, L. F. Scatena and G. L. Richmond, On the Rise: Experimental and Computational Vibrational Sum Frequency Spectroscopy Studies of Pyruvic Acid and Its Surface-Active Oligomer Species at the Air–Water Interface, J. Phys. Chem. A, 2019, 123, 10609–10619 CrossRef CAS PubMed.
- B. P. Gordon, G. A. Lindquist, M. L. Crawford, S. N. Wren, F. G. Moore, L. F. Scatena and G. L. Richmond, Diol it up: the influence of NaCl on methylglyoxal surface adsorption and hydration state at the air–water interface, J. Chem. Phys., 2020, 153, 164705 CrossRef CAS PubMed.
- Y. Wu, W. Li, B. Xu, X. Li, H. Wang, V. F. McNeill, Y. Rao and H.-L. Dai, Observation of Organic Molecules at the Aerosol Surface, J. Phys. Chem. Lett., 2016, 7, 2294–2297 CrossRef CAS PubMed.
- Y. Qian, G. Deng and Y. Rao, In Situ Chemical Analysis of the Gas–Aerosol Particle Interface, Anal. Chem., 2018, 90, 10967–10973 CrossRef CAS PubMed.
- Y. Qian, G. Deng, J. Lapp and Y. Rao, Interfaces of Gas–Aerosol Particles: Relative Humidity and Salt Concentration Effects, J. Phys. Chem. A, 2019, 123, 6304–6312 CrossRef CAS PubMed.
- Y. Qian, G. Deng and Y. Rao, In Situ Spectroscopic Probing of Polarity and Molecular Configuration at Aerosol Particle Surfaces, J. Phys. Chem. Lett., 2020, 11, 6763–6771 CrossRef CAS PubMed.
- Y. Qian, J. B. Brown, Z.-C. Huang-Fu, T. Zhang, H. Wang, S. Wang, J. I. Dadap and Y. Rao, In situ analysis of the bulk and surface chemical compositions of organic aerosol particles, Commun. Chem., 2022, 5, 58 CrossRef CAS.
- Z. Liang, Y. Chu, M. Gen and C. K. Chan, Single-particle Raman spectroscopy for studying physical and chemical processes of atmospheric particles, Atmos. Chem. Phys., 2022, 22, 3017–3044 CrossRef CAS.
- M. Gen, R. Kunihisa, A. Matsuki and C. K. Chan, Electrospray surface-enhanced Raman spectroscopy (ES-SERS) for studying organic coatings of atmospheric aerosol particles, Aerosol Sci. Technol., 2019, 53, 760–770 CrossRef CAS.
- M. Wang, N. Zheng, D. Zhao, J. Shang and T. Zhu, Using Micro-Raman Spectroscopy to Investigate Chemical Composition, Mixing States, and Heterogeneous Reactions of Individual Atmospheric Particles, Environ. Sci. Technol., 2021, 55, 10243–10254 CrossRef CAS PubMed.
- A. Laskin, R. C. Moffet and M. K. Gilles, Chemical Imaging of Atmospheric Particles, Acc. Chem. Res., 2019, 52, 3419–3431 CrossRef CAS PubMed.
- D. Vollhardt, Brewster angle microscopy: a preferential method for mesoscopic characterization of monolayers at the air/water interface, Curr. Opin. Colloid Interface Sci., 2014, 19, 183–197 CrossRef CAS.
- G. Andersson and C. Ridings, Ion Scattering Studies of Molecular Structure at Liquid Surfaces with Applications in Industrial and Biological Systems, Chem. Rev., 2014, 114, 8361–8387 CrossRef CAS PubMed.
- M. A. Tesa-Serrate, E. J. Smoll, T. K. Minton and K. G. McKendrick, Atomic and Molecular Collisions at Liquid Surfaces, Annu. Rev. Phys. Chem., 2016, 67, 515–540 CrossRef CAS PubMed.
- W. A. Alexander, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 195–243 Search PubMed.
- M. W. A. Skoda, B. Thomas, M. Hagreen, F. Sebastiani and C. Pfrang, Simultaneous neutron reflectometry and infrared reflection absorption spectroscopy (IRRAS) study of mixed monolayer reactions at the air–water interface, RSC Adv., 2017, 7, 34208–34214 RSC.
- R. A. Campbell, Recent advances in resolving kinetic and dynamic processes at the air/water interface using specular neutron reflectometry, Curr. Opin. Colloid Interface Sci., 2018, 37, 49–60 CrossRef CAS.
- M. Tang, C. K. Chan, Y. J. Li, H. Su, Q. Ma, Z. Wu, G. Zhang, Z. Wang, M. Ge, M. Hu, H. He and X. Wang, A review of experimental techniques for aerosol hygroscopicity studies, Atmos. Chem. Phys., 2019, 19, 12631–12686 CrossRef CAS.
- K. S. Karadima, V. G. Mavrantzas and S. N. Pandis, Insights into the morphology of multicomponent organic and inorganic aerosols from molecular dynamics simulations, Atmos. Chem. Phys., 2019, 19, 5571–5587 CrossRef CAS.
- M. Lbadaoui-Darvas, A. Idrissi and P. Jedlovszky, Computer Simulation of the Surface of Aqueous Ionic and Surfactant Solutions, J. Phys. Chem. B, 2022, 126, 751–765 CrossRef CAS PubMed.
- F. Jensen, Introduction to computational chemistry, John Wiley & Sons, Chichester, UK, Hoboken, NJ, 3rd edn, 2017 Search PubMed.
- T.-M. Chang, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 1–40 Search PubMed.
- M. Chen, X. Lu, X. Liu, Q. Hou, Y. Zhu and H. Zhou, Temperature-Dependent Phase Transition and Desorption Free Energy of Sodium Dodecyl Sulfate at the Water/Vapor Interface: Approaches from Molecular Dynamics Simulations, Langmuir, 2014, 30, 10600–10607 CrossRef CAS PubMed.
- K. Bernardino and A. F. de Moura, Surface Electrostatic Potential and Water Orientation in the presence of Sodium Octanoate Dilute Monolayers Studied by Means of Molecular Dynamics Simulations, Langmuir, 2015, 31, 10995–11004 CrossRef CAS PubMed.
- N. Abrankó-Rideg, G. Horvai and P. Jedlovszky, Structure of the adsorption layer of various ionic and non-ionic surfactants at the free water surface, as seen from computer simulation and ITIM analysis, J. Mol. Liq., 2015, 205, 9–15 CrossRef.
- G. Ergin, M. Lbadaoui-Darvas and S. Takahama, Molecular Structure Inhibiting Synergism in Charged Surfactant Mixtures: An Atomistic Molecular Dynamics Simulation Study, Langmuir, 2017, 33, 14093–14104 CrossRef CAS PubMed.
- P. Yazhgur, S. Vierros, D. Hannoy, M. Sammalkorpi and A. Salonen, Surfactant Interactions and Organization at the Gas–Water Interface (CTAB with Added Salt), Langmuir, 2018, 34, 1855–1864 CrossRef CAS PubMed.
- M. Peng, T. T. Duignan and A. V. Nguyen, Quantifying the Counterion-Specific Effect on Surfactant Adsorption Using Modeling, Simulation, and Experiments, Langmuir, 2020, 36, 13012–13022 CrossRef CAS PubMed.
- C. Y. Pak, W. Li and Y.-L. Steve Tse, Free Energy and Dynamics of Organic-Coated Water Droplet Coalescence, J. Phys. Chem. C, 2020, 124, 8749–8757 CrossRef CAS.
- J. Werner, J. Julin, M. Dalirian, N. L. Prisle, G. Öhrwall, I. Persson, O. Björneholm and I. Riipinen, Succinic acid in aqueous solution: connecting microscopic surface composition and macroscopic surface tension, Phys. Chem. Chem. Phys., 2014, 16, 21486–21495 RSC.
- M.-M. Walz, C. Caleman, J. Werner, V. Ekholm, D. Lundberg, N. L. Prisle, G. Öhrwall and O. Björneholm, Surface behavior of amphiphiles in aqueous solution: A comparison between different pentanol isomers, Phys. Chem. Chem. Phys., 2015, 17, 14036–14044 RSC.
- V. Ekholm, C. Caleman, N. Bjärnhall Prytz, M.-M. Walz, J. Werner, G. Öhrwall, J.-E. Rubensson and O. Björneholm, Strong enrichment of atmospherically relevant organic ions at the aqueous interface: the role of ion pairing and cooperative effects, Phys. Chem. Chem. Phys., 2018, 20, 27185–27191 RSC.
- J. M. Schiffer, M. Luo, A. C. Dommer, G. Thoron, M. Pendergraft, M. V. Santander, D. Lucero, E. Pecora de Barros, K. A. Prather, V. H. Grassian and R. E. Amaro, Impacts of Lipase Enzyme on the Surface Properties of Marine Aerosols, J. Phys. Chem. Lett., 2018, 9, 3839–3849 CrossRef CAS PubMed.
- K. A. Carter-Fenk, A. Dommer, M. E. Fiamingo, J. Kim, R. Amaro and H. C. Allen, Calcium bridging drives polysaccharide co-adsorption to a proxy sea surface microlayer, Phys. Chem. Chem. Phys., 2021, 23, 16401–16416 RSC.
- W. Lin, A. J. Clark and F. Paesani, Effects of Surface Pressure on the Properties of Langmuir Monolayers and Interfacial Water at the Air–Water Interface, Langmuir, 2015, 31, 2147–2156 CrossRef CAS PubMed.
- E. M. Adams, B. A. Wellen, R. Thiraux, S. K. Reddy, A. S. Vidalis, F. Paesani and H. C. Allen, Sodium–carboxylate contact ion pair formation induces stabilization of palmitic acid monolayers at high pH, Phys. Chem. Chem. Phys., 2017, 19, 10481–10490 RSC.
- J. Lovrić, D. Duflot, M. Monnerville, C. Toubin and S. Briquez, Water-Induced Organization of Palmitic Acid at the Surface of a Model Sea Salt Particle: A Molecular Dynamics Study, J. Phys. Chem. A, 2016, 120, 10141–10149 CrossRef PubMed.
- K. S. Karadima, V. G. Mavrantzas and S. N. Pandis, Molecular dynamics simulation of the local concentration and structure in multicomponent aerosol nanoparticles under atmospheric conditions, Phys. Chem. Chem. Phys., 2017, 19, 16681–16692 RSC.
- P. Rehner, B. Bursik and J. Gross, Surfactant Modeling Using Classical Density Functional Theory and a Group Contribution PC-SAFT Approach, Ind. Eng. Chem. Res., 2021, 60, 7111–7123 CrossRef CAS.
- A. Leonardi, H. M. Ricker, A. G. Gale, B. T. Ball, T. T. Odbadrakh, G. C. Shields and J. G. Navea, Particle formation and surface processes on atmospheric aerosols: a review of applied quantum chemical calculations, Int. J. Quantum Chem., 2020, 120, e26350 CrossRef CAS.
- S. N. Wren, B. P. Gordon, N. A. Valley, L. E. McWilliams and G. L. Richmond, Hydration, Orientation, and Conformation of Methylglyoxal at the Air–Water Interface, J. Phys. Chem. A, 2015, 119, 6391–6403 CrossRef CAS PubMed.
- D. R. Hanson, Surface-Specific Reactions on Liquids, J. Phys. Chem. B, 1997, 101, 4998–5001 CrossRef CAS.
- M. Shiraiwa, C. Pfrang, T. Koop and U. Pöschl, Kinetic multi-layer model of gas-particle interactions in aerosols and clouds (KM-GAP): linking condensation, evaporation and chemical reactions of organics, oxidants and water, Atmos. Chem. Phys., 2012, 12, 2777–2794 CrossRef CAS.
- C. Pfrang, F. Sebastiani, C. O. M. Lucas, M. D. King, I. D. Hoare, D. Chang and R. A. Campbell, Ozonolysis of methyl oleate monolayers at the air–water interface: oxidation kinetics, reaction products and atmospheric implications, Phys. Chem. Chem. Phys., 2014, 16, 13220–13228 RSC.
- F. Sebastiani, R. A. Campbell, K. Rastogi and C. Pfrang, Nighttime oxidation of surfactants at the air–water interface: effects of chain length, head group and saturation, Atmos. Chem. Phys., 2018, 18, 3249–3268 CrossRef CAS.
- K. Semeniuk and A. Dastoor, Current State of Atmospheric Aerosol Thermodynamics and Mass Transfer Modeling: A Review, Atmosphere, 2020, 11, 156 CrossRef CAS.
- S. M. Calderón, J. Malila and N. L. Prisle, Model for estimating activity coefficients in binary and ternary ionic surfactant solutions: the CMC based ionic surfactant activity (CISA) model for atmospheric applications, J. Atmos. Chem., 2020, 77, 141–168 CrossRef.
- S. M. Calderón and N. L. Prisle, Composition dependent density of ternary aqueous solutions of ionic surfactants and salts: capturing the effect of surfactant micellization in atmospheric droplet model solutions, J. Atmos. Chem., 2021, 78, 99–123 CrossRef.
- Y. Huang, F. Mahrt, S. Xu, M. Shiraiwa, A. Zuend and A. K. Bertram, Coexistence of three liquid phases in individual atmospheric aerosol particles, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2102512118 CrossRef CAS PubMed.
- A. Zuend, C. Marcolli, B. P. Luo and T. Peter, A thermodynamic model of mixed organic–inorganic aerosols to predict activity coefficients, Atmos. Chem. Phys., 2008, 8, 4559–4593 CrossRef CAS.
- A. Zuend, C. Marcolli, A. M. Booth, D. M. Lienhard, V. Soonsin, U. K. Krieger, D. O. Topping, G. McFiggans, T. Peter and J. H. Seinfeld, New and extended parameterization of the thermodynamic model AIOMFAC: calculation of activity coefficients for organic–inorganic mixtures containing carboxyl, hydroxyl, carbonyl, ether, ester, alkenyl, alkyl, and aromatic functional groups, Atmos. Chem. Phys., 2011, 11, 9155–9206 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Inc, Hoboken, New Jersey, 3rd edn, 2016 Search PubMed.
- H. O. T. Pye, B. N. Murphy, L. Xu, N. L. Ng, A. G. Carlton, H. Guo, R. Weber, P. Vasilakos, K. W. Appel, S. H. Budisulistiorini, J. D. Surratt, A. Nenes, W. Hu, J. L. Jimenez, G. Isaacman-VanWertz, P. K. Misztal and A. H. Goldstein, On the implications of aerosol liquid water and phase separation for organic aerosol mass, Atmos. Chem. Phys., 2017, 17, 343–369 CrossRef CAS PubMed.
- R. Schmedding, Q. Z. Rasool, Y. Zhang, H. O. T. Pye, H. Zhang, Y. Chen, J. D. Surratt, F. D. Lopez-Hilfiker, J. A. Thornton, A. H. Goldstein and W. Vizuete, Predicting secondary organic aerosol phase state and viscosity and its effect on multiphase chemistry in a regional-scale air quality model, Atmos. Chem. Phys., 2020, 20, 8201–8225 CrossRef CAS PubMed.
- M. A. Freedman, Liquid–Liquid Phase Separation in Supermicrometer and Submicrometer Aerosol Particles, Acc. Chem. Res., 2020, 53, 1102–1110 CrossRef CAS PubMed.
- M. A. Freedman, Phase separation in organic aerosol, Chem. Soc. Rev., 2017, 46, 7694–7705 RSC.
- K. Gorkowski, N. M. Donahue and R. C. Sullivan, Aerosol Optical Tweezers Constrain the Morphology Evolution of Liquid–Liquid Phase-Separated Atmospheric Particles, Chem, 2020, 6, 204–220 CAS.
- F. Mahrt, E. Newman, Y. Huang, M. Ammann and A. K. Bertram, Phase Behavior of Hydrocarbon-like Primary Organic Aerosol and Secondary Organic Aerosol Proxies Based on Their Elemental Oxygen-to-Carbon Ratio, Environ. Sci. Technol., 2021, 55, 12202–12214 CrossRef CAS PubMed.
- H. D. Lee, S. Wigley, C. Lee, V. W. Or, E. S. Hasenecz, E. A. Stone, V. H. Grassian, K. A. Prather and A. V. Tivanski, Physicochemical Mixing State of Sea Spray Aerosols: Morphologies Exhibit Size Dependence, ACS Earth Space Chem., 2020, 4, 1604–1611 CrossRef CAS.
- W. Li, L. Liu, J. Zhang, L. Xu, Y. Wang, Y. Sun and Z. Shi, Microscopic Evidence for Phase Separation of Organic Species and Inorganic Salts in Fine Ambient Aerosol Particles, Environ. Sci. Technol., 2021, 55, 2234–2242 CrossRef CAS PubMed.
- T. M. Kucinski, J. N. Dawson and M. A. Freedman, Size-Dependent Liquid–Liquid Phase Separation in Atmospherically Relevant Complex Systems, J. Phys. Chem. Lett., 2019, 10, 6915–6920 CrossRef CAS PubMed.
- P. E. Ohno, Y. Qin, J. Ye, J. Wang, A. K. Bertram and S. T. Martin, Fluorescence Aerosol Flow Tube Spectroscopy to Detect Liquid–Liquid Phase Separation, ACS Earth Space Chem., 2021, 5, 1223–1232 CrossRef CAS.
- M. A. Dallemagne, X. Y. Huang and N. C. Eddingsaas, Variation in pH of Model Secondary Organic Aerosol during Liquid–Liquid Phase Separation, J. Phys. Chem. A, 2016, 120, 2868–2876 CrossRef CAS PubMed.
- Z. Cheng, N. Sharma, K.-P. Tseng, L. Kovarik and S. China, Direct observation and assessment of phase states of ambient and lab-generated sub-micron particles upon humidification, RSC Adv., 2021, 11, 15264–15272 RSC.
- H. D. Lee, K. K. Ray and A. V. Tivanski, Solid, Semisolid, and Liquid Phase States of Individual Submicrometer Particles Directly Probed Using Atomic Force Microscopy, Anal. Chem., 2017, 89, 12720–12726 CrossRef CAS PubMed.
- K. K. Ray, H. D. Lee, M. A. Gutierrez, F. J. Chang and A. V. Tivanski, Correlating 3D Morphology, Phase State, and Viscoelastic Properties of Individual Substrate-Deposited Particles, Anal. Chem., 2019, 91, 7621–7630 CrossRef CAS PubMed.
- R. L. Craig, A. L. Bondy and A. P. Ault, Computer-controlled Raman microspectroscopy (CC-Raman): a method for the rapid characterization of individual atmospheric aerosol particles, Aerosol Sci. Technol., 2017, 51, 1099–1112 CrossRef CAS.
- M. B. Altaf, A. Zuend and M. A. Freedman, Role of nucleation mechanism on the size dependent morphology of organic aerosol, Chem. Commun., 2016, 52, 9220–9223 RSC.
- M. Petters and S. Kasparoglu, Predicting the influence of particle size on the glass transition temperature and viscosity of secondary organic material, Sci. Rep., 2020, 10, 15170 CrossRef CAS PubMed.
- Q. Zhou, S.-F. Pang, Y. Wang, J.-B. Ma and Y.-H. Zhang, Confocal Raman Studies of the Evolution of the Physical State of Mixed Phthalic Acid/Ammonium Sulfate Aerosol Droplets and the Effect of Substrates, J. Phys. Chem. B, 2014, 118, 6198–6205 CrossRef CAS PubMed.
- O. Laskina, H. S. Morris, J. R. Grandquist, A. D. Estillore, E. A. Stone, V. H. Grassian and A. V. Tivanski, Substrate-Deposited Sea Spray Aerosol Particles: Influence of Analytical Method, Substrate, and Storage Conditions on Particle Size, Phase, and Morphology, Environ. Sci. Technol., 2015, 49, 13447–13453 CrossRef CAS PubMed.
- C. Chen, O. Enekwizu, Y. Ma, D. Zakharov and A. Khalizov, The Impact of Sampling Medium and Environment on Particle Morphology, Atmosphere, 2017, 8, 162 CrossRef.
- P. Roy, L. E. Mael, T. C. J. Hill, L. Mehndiratta, G. Peiker, M. L. House, P. J. DeMott, V. H. Grassian and C. S. Dutcher, Ice Nucleating Activity and Residual Particle Morphology of Bulk Seawater and Sea Surface Microlayer, ACS Earth Space Chem., 2021, 5, 1916–1928 CrossRef CAS.
- T. C. Preston and J. P. Reid, Accurate and efficient determination of the radius, refractive index, and dispersion of weakly absorbing spherical particle using whispering gallery modes, J. Opt. Soc. Am. B, 2013, 30, 2113–2122 CrossRef CAS.
- D. J. Stewart, C. Cai, J. Nayler, T. C. Preston, J. P. Reid, U. K. Krieger, C. Marcolli and Y. H. Zhang, Liquid–Liquid Phase Separation in Mixed Organic/Inorganic Single Aqueous Aerosol Droplets, J. Phys. Chem. A, 2015, 119, 4177–4190 CrossRef CAS PubMed.
- K. Gorkowski, H. Beydoun, M. Aboff, J. S. Walker, J. P. Reid and R. C. Sullivan, Advanced aerosol optical tweezers chamber design to facilitate phase-separation and equilibration timescale experiments on complex droplets, Aerosol Sci. Technol., 2016, 50, 1327–1341 CrossRef CAS.
- K. Gorkowski, N. M. Donahue and R. C. Sullivan, Determination of biphasic core–shell droplet properties using aerosol optical tweezers, Environ. Sci.: Processes Impacts, 2018, 20, 1512–1523 RSC.
- K. Gorkowski, N. M. Donahue and R. C. Sullivan, Emulsified and Liquid–Liquid Phase-Separated States of α-Pinene Secondary Organic Aerosol Determined Using Aerosol Optical Tweezers, Environ. Sci. Technol., 2017, 51, 12154–12163 CrossRef CAS PubMed.
- A. Haddrell, G. Rovelli, D. Lewis, T. Church and J. Reid, Identifying time-dependent changes in the morphology of an individual aerosol particle from its light scattering pattern, Aerosol Sci. Technol., 2019, 53, 1334–1351 CrossRef CAS.
- S. H. Jones, M. D. King and A. D. Ward, Atmospherically relevant core–shell aerosol studied using optical trapping and Mie scattering, Chem. Commun., 2015, 51, 4914–4917 RSC.
- S. Torza and S. G. Mason, Three-phase interactions in shear and electrical fields, J. Colloid Interface Sci., 1970, 33, 67–83 CrossRef CAS.
- Y. Qiu and V. Molinero, Morphology of Liquid–Liquid Phase Separated Aerosols, J. Am. Chem. Soc., 2015, 137, 10642–10651 CrossRef CAS PubMed.
- H. Binyaminov, F. Abdullah, L. Zargarzadeh and J. A. W. Elliott, Thermodynamic Investigation of Droplet–Droplet and Bubble–Droplet Equilibrium in an Immiscible Medium, J. Phys. Chem. B, 2021, 125, 8636–8651 CrossRef CAS PubMed.
- F. Hrahsheh, Y. Sani Wudil and G. Wilemski, Confined phase separation of aqueous–organic nanodroplets, Phys. Chem. Chem. Phys., 2017, 19, 26839–26845 RSC.
- Z. Yu, M. Jang and A. Madhu, Prediction of Phase State of Secondary Organic Aerosol Internally Mixed with Aqueous Inorganic Salts, J. Phys. Chem. A, 2021, 125, 10198–10206 CrossRef CAS PubMed.
- M. M. Fard, U. K. Krieger and T. Peter, Shortwave radiative impact of liquid–liquid phase separation in brown carbon aerosols, Atmos. Chem. Phys., 2018, 18, 13511–13530 CrossRef CAS.
- Y. Li, A. G. Carlton and M. Shiraiwa, Diurnal and Seasonal Variations in the Phase State of Secondary Organic Aerosol Material over the Contiguous US Simulated in CMAQ, ACS Earth Space Chem., 2021, 5, 1971–1982 CrossRef CAS.
- L. Fierce, T. B. Onasch, C. D. Cappa, C. Mazzoleni, S. China, J. Bhandari, P. Davidovits, D. A. Fischer, T. Helgestad, A. T. Lambe, A. J. Sedlacek, G. D. Smith and L. Wolff, Radiative absorption enhancements by black carbon controlled by particle-to-particle heterogeneity in composition, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5196–5203 CrossRef CAS PubMed.
- Y. Wu, T. Cheng and L. Zheng, Light absorption of black carbon aerosols strongly influenced by particle morphology distribution, Environ. Res. Lett., 2020, 15, 094051 CrossRef CAS.
- J. Ovadnevaite, A. Zuend, A. Laaksonen, K. J. Sanchez, G. Roberts, D. Ceburnis, S. Decesari, M. Rinaldi, N. Hodas, M. C. Facchini, J. H. Seinfeld and C. O' Dowd, Surface tension prevails over solute effect in organic-influenced cloud droplet activation, Nature, 2017, 546, 637–641 CrossRef CAS PubMed.
- K. W. Kolasinski, Surface Science: Foundations of Catalysis and Nanoscience, Wiley, Chichester, England, Hoboken, NJ, 2nd edn, 2008 Search PubMed.
- G. McFiggans, P. Artaxo, U. Baltensperger, H. Coe, M. C. Facchini, G. Feingold, S. Fuzzi, M. Gysel, A. Laaksonen, U. Lohmann, T. F. Mentel, D. M. Murphy, C. D. O'Dowd, J. R. Snider and E. Weingartner, The effect of physical and chemical aerosol properties on warm cloud droplet activation, Atmos. Chem. Phys., 2006, 6, 2593–2649 CrossRef CAS.
- C. R. Ruehl and K. R. Wilson, Surface Organic Monolayers Control the Hygroscopic Growth of Submicrometer Particles at High Relative Humidity, J. Phys. Chem. A, 2014, 118, 3952–3966 CrossRef CAS PubMed.
- T. B. Kristensen, N. L. Prisle and M. Bilde, Cloud droplet activation of mixed model HULIS and NaCl particles: experimental results and κ-Köhler theory, Atmos. Res., 2014, 137, 167–175 CrossRef CAS.
- S. S. Petters and M. D. Petters, Surfactant effect on cloud condensation nuclei for two-component internally mixed aerosols, J. Geophys. Res.: Atmos., 2016, 121, 1878–1895 CrossRef CAS.
- C. R. Ruehl, J. F. Davies and K. R. Wilson, An interfacial mechanism for cloud droplet formation on organic aerosols, Science, 2016, 351, 1447–1450 CrossRef CAS PubMed.
- N. Rastak, A. Pajunoja, J. C. Acosta Navarro, J. Ma, M. Song, D. G. Partridge, A. Kirkevåg, Y. Leong, W. W. Hu, N. F. Taylor, A. Lambe, K. Cerully, A. Bougiatioti, P. Liu, R. Krejci, T. Petäjä, C. Percival, P. Davidovits, D. R. Worsnop, A. M. L. Ekman, A. Nenes, S. Martin, J. L. Jimenez, D. R. Collins, D. O. Topping, A. K. Bertram, A. Zuend, A. Virtanen and I. Riipinen, Microphysical explanation of the RH-dependent water affinity of biogenic organic aerosol and its importance for climate, Geophys. Res. Lett., 2017, 44, 5167–5177 CrossRef CAS PubMed.
- J. J. Lin, J. Malila and N. L. Prisle, Cloud droplet activation of organic–salt mixtures predicted from two model treatments of the droplet surface, Environ. Sci.: Processes Impacts, 2018, 20, 1611–1629 RSC.
- Y. S. Djikaev and E. Ruckenstein, Effect of Heterogeneous Chemical Reactions on the Köhler Activation of Aqueous Organic Aerosols, J. Phys. Chem. A, 2018, 122, 4322–4337 CrossRef CAS PubMed.
- D. B. Collins and V. H. Grassian, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 271–313 Search PubMed.
- Y. S. Djikaev and E. Ruckenstein, Formation and evolution of aqueous organic aerosols via concurrent condensation and chemical aging, Adv. Colloid Interface Sci., 2019, 265, 45–67 CrossRef CAS PubMed.
- J. F. Davies, A. Zuend and K. R. Wilson, Technical note: The role of evolving surface tension in the formation of cloud droplets, Atmos. Chem. Phys., 2019, 19, 2933–2946 CrossRef CAS.
- B. R. Bzdek, J. P. Reid, J. Malila and N. L. Prisle, The surface tension of surfactant-containing, finite volume droplets, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8335–8343 CrossRef CAS PubMed.
- D. Roston, Molecular dynamics simulations demonstrate that non-ideal mixing dominates subsaturation organic aerosol hygroscopicity, Phys. Chem. Chem. Phys., 2021, 23, 9218–9227 RSC.
- E. I. Franses, O. A. Basaran and C.-H. Chang, Techniques to measure dynamic surface tension, Curr. Opin. Colloid Interface Sci., 1996, 1, 296–303 CrossRef CAS.
- D. M. Mott, M. Fukuyama and A. Hibara, Aerosol Droplet Surface Measurement Methods, Anal. Sci., 2021, 37, 61–68 CrossRef CAS PubMed.
- H. D. Lee and A. V. Tivanski, Atomic Force Microscopy: An Emerging Tool in Measuring the Phase State and Surface Tension of Individual Aerosol Particles, Annu. Rev. Phys. Chem., 2021, 72, 235–252 CrossRef CAS PubMed.
- B. R. Bzdek, R. M. Power, S. H. Simpson, J. P. Reid and C. P. Royall, Precise, contactless measurements of the surface tension of picolitre aerosol droplets, Chem. Sci., 2016, 7, 274–285 RSC.
- A. Marsh, G. Rovelli, Y.-C. Song, K. L. Pereira, R. E. Willoughby, B. R. Bzdek, J. F. Hamilton, A. J. Orr-Ewing, D. O. Topping and J. P. Reid, Accurate representations of the physicochemical properties of atmospheric aerosols: When are laboratory measurements of value?, Faraday Discuss., 2017, 200, 639–661 RSC.
- R. E. H. Miles, M. W. J. Glerum, H. C. Boyer, J. S. Walker, C. S. Dutcher and B. R. Bzdek, Surface Tensions of Picoliter Droplets with Sub-Millisecond Surface Age, J. Phys. Chem. A, 2019, 123, 3021–3029 CrossRef CAS PubMed.
- A. R. Metcalf, H. C. Boyer and C. S. Dutcher, Interfacial Tensions of Aged Organic Aerosol Particle Mimics Using a Biphasic Microfluidic Platform, Environ. Sci. Technol., 2016, 50, 1251–1259 CrossRef CAS PubMed.
- H. C. Boyer and C. S. Dutcher, Atmospheric Aqueous Aerosol Surface Tensions: Isotherm-Based Modeling and Biphasic Microfluidic Measurements, J. Phys. Chem. A, 2017, 121, 4733–4742 CrossRef CAS PubMed.
- H. C. Boyer, B. R. Bzdek, J. P. Reid and C. S. Dutcher, Statistical Thermodynamic Model for Surface Tension of Organic and Inorganic Aqueous Mixtures, J. Phys. Chem. A, 2017, 121, 198–205 CrossRef CAS PubMed.
- S. L. Clegg, J. H. Seinfeld and P. Brimblecombe, Thermodynamic modelling of aqueous aerosols containing electrolytes and dissolved organic compounds, J. Aerosol Sci., 2001, 32, 713–738 CrossRef CAS.
- S. L. Clegg and J. H. Seinfeld, Thermodynamic Models of Aqueous Solutions Containing Inorganic Electrolytes and Dicarboxylic Acids at 298.15 K. 1. The Acids as Nondissociating Components, J. Phys. Chem. A, 2006, 110, 5692–5717 CrossRef CAS PubMed.
- S. L. Clegg and J. H. Seinfeld, Thermodynamic Models of Aqueous Solutions Containing Inorganic Electrolytes and Dicarboxylic Acids at 298.15 K. 2. Systems Including Dissociation Equilibria, J. Phys. Chem. A, 2006, 110, 5718–5734 CrossRef CAS PubMed.
- G. Hantal, M. Sega, G. Horvai and P. Jedlovszky, Contribution of Different Molecules and Moieties to the Surface Tension in Aqueous Surfactant Solutions, J. Phys. Chem. C, 2019, 123, 16660–16670 CrossRef CAS.
- V. Sresht, E. P. Lewandowski, D. Blankschtein and A. Jusufi, Combined Molecular Dynamics Simulation–Molecular-Thermodynamic Theory Framework for Predicting Surface Tensions, Langmuir, 2017, 33, 8319–8329 CrossRef CAS PubMed.
- B. Yoo, E. Marin-Rimoldi, R. G. Mullen, A. Jusufi and E. J. Maginn, Discrete Fractional Component Monte Carlo Simulation Study of Dilute Nonionic Surfactants at the Air–Water Interface, Langmuir, 2017, 33, 9793–9802 CrossRef CAS PubMed.
- G. Hantal, M. Sega, G. Horvai and P. Jedlovszky, Role of the Counterions in the Surface Tension of Aqueous Surfactant Solutions. A Computer Simulation Study of Alkali Dodecyl Sulfate Systems, Colloids Interfaces, 2020, 4, 15 CrossRef CAS.
- G. Hantal, M. Sega, G. Horvai and P. Jedlovszky, Contribution of Different Molecules and Moieties to the Surface Tension in Aqueous Surfactant Solutions. II: Role of the Size and Charge Sign of the Counterions, J. Phys. Chem. B, 2021, 125, 9005–9018 CrossRef CAS PubMed.
- M. D. Petters and S. M. Kreidenweis, A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 2007, 7, 1961–1971 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere, Academic Press, San Diego, 2000 Search PubMed.
- A. A. Frossard, W. Li, V. Gérard, B. Nozière and R. C. Cohen, Influence of surfactants on growth of individual aqueous coarse mode aerosol particles, Aerosol Sci. Technol., 2018, 52, 459–469 CrossRef CAS.
- C. Zhang, L. Bu, F. Fan, N. Ma, Y. Wang, Y. Yang, J. Gröβ, J. Yan and A. Wiedensohler, Surfactant effect on the hygroscopicity of aerosol particles at relative humidity ranging from 80% to 99.5%: internally mixed adipic acid-ammonium sulfate particles, Atmos. Environ., 2021, 266, 118725 CrossRef CAS.
- B. R. Bzdek and J. P. Reid, Perspective: aerosol microphysics: from molecules to the chemical physics of aerosols, J. Chem. Phys., 2017, 147, 220901 CrossRef PubMed.
- S. H. Jathar, A. Mahmud, K. C. Barsanti, W. E. Asher, J. F. Pankow and M. J. Kleeman, Water uptake by organic aerosol and its influence on gas/particle partitioning of secondary organic aerosol in the United States, Atmos. Environ., 2016, 129, 142–154 CrossRef CAS.
- M. B. Altaf, D. D. Dutcher, T. M. Raymond and M. A. Freedman, Effect of Particle Morphology on Cloud Condensation Nuclei Activity, ACS Earth Space Chem., 2018, 2, 634–639 CrossRef CAS.
- S. B. Ushijima, E. Huynh, R. D. Davis and M. A. Tolbert, Seeded Crystal Growth of Internally Mixed Organic–Inorganic Aerosols: Impact of Organic Phase State, J. Phys. Chem. A, 2021, 125, 8668–8679 CrossRef CAS PubMed.
- W. Li, X. Teng, X. Chen, L. Liu, L. Xu, J. Zhang, Y. Wang, Y. Zhang and Z. Shi, Organic Coating Reduces Hygroscopic Growth of Phase-Separated Aerosol Particles, Environ. Sci. Technol., 2021, 55, 16339–16346 CrossRef CAS PubMed.
- Q. T. Nguyen, K. H. Kjær, K. I. Kling, T. Boesen and M. Bilde, Impact of fatty acid coating on the CCN activity of sea salt particles, Tellus B, 2017, 69, 1304064 CrossRef.
- L. Miñambres, E. Méndez, M. N. Sánchez, F. Castaño and F. J. Basterretxea, The effect of low solubility organic acids on the hygroscopicity of sodium halide aerosols, Atmos. Chem. Phys., 2014, 14, 11409–11425 CrossRef.
- A. Kroflič, S. Frka, M. Simmel, H. Wex and I. Grgić, Size-Resolved Surface-Active Substances of Atmospheric Aerosol: Reconsideration of the Impact on Cloud Droplet Formation, Environ. Sci. Technol., 2018, 52, 9179–9187 CrossRef PubMed.
- D. Hu, D. Liu, D. Zhao, C. Yu, Q. Liu, P. Tian, K. Bi, S. Ding, K. Hu, F. Wang, Y. Wu, Y. Wu, S. Kong, W. Zhou, H. He, M. Huang and D. Ding, Closure Investigation on Cloud Condensation Nuclei Ability of Processed Anthropogenic Aerosols, J. Geophys. Res.: Atmos., 2020, 125, e2020JD032680 CAS.
- S. R. Schill, D. B. Collins, C. Lee, H. S. Morris, G. A. Novak, K. A. Prather, P. K. Quinn, C. M. Sultana, A. V. Tivanski, K. Zimmermann, C. D. Cappa and T. H. Bertram, The Impact of Aerosol Particle Mixing State on the Hygroscopicity of Sea Spray Aerosol, ACS Cent. Sci., 2015, 1, 132–141 CrossRef CAS PubMed.
- L. T. Cravigan, M. D. Mallet, P. Vaattovaara, M. J. Harvey, C. S. Law, R. L. Modini, L. M. Russell, E. Stelcer, D. D. Cohen, G. Olsen, K. Safi, T. J. Burrell and Z. Ristovski, Sea spray aerosol organic enrichment, water uptake and surface tension effects, Atmos. Chem. Phys., 2020, 20, 7955–7977 CrossRef CAS.
- B. N. Hendrickson, S. D. Brooks, D. C. O. Thornton, R. H. Moore, E. Crosbie, L. D. Ziemba, C. A. Carlson, N. Baetge, J. A. Mirrielees and A. N. Alsante, Role of Sea Surface Microlayer Properties in Cloud Formation, Front. Mar. Sci., 2021, 7, 596225 CrossRef.
- H. S. Morris, A. D. Estillore, O. Laskina, V. H. Grassian and A. V. Tivanski, Quantifying the Hygroscopic Growth of Individual Submicrometer Particles with Atomic Force Microscopy, Anal. Chem., 2016, 88, 3647–3654 CrossRef CAS PubMed.
- L. E. Mael, H. L. Busse, G. Peiker and V. H. Grassian, Low-Temperature Water Uptake of Individual Marine and Biologically Relevant Atmospheric Particles Using Micro-Raman Spectroscopy, J. Phys. Chem. A, 2021, 125, 9691–9699 CrossRef CAS PubMed.
- A. D. Estillore, H. S. Morris, V. W. Or, H. D. Lee, M. R. Alves, M. A. Marciano, O. Laskina, Z. Qin, A. V. Tivanski and V. H. Grassian, Linking hygroscopicity and the surface microstructure of model inorganic salts, simple and complex carbohydrates, and authentic sea spray aerosol particles, Phys. Chem. Chem. Phys., 2017, 19, 21101–21111 RSC.
- H. D. Lee, H. S. Morris, O. Laskina, C. M. Sultana, C. Lee, T. Jayarathne, J. L. Cox, X. Wang, E. S. Hasenecz, P. J. DeMott, T. H. Bertram, C. D. Cappa, E. A. Stone, K. A. Prather, V. H. Grassian and A. V. Tivanski, Organic Enrichment, Physical Phase State, and Surface Tension Depression of Nascent Core–Shell Sea Spray Aerosols during Two Phytoplankton Blooms, ACS Earth Space Chem., 2020, 4, 650–660 CrossRef CAS.
- K.-H. Ahn, S.-M. Kim, H.-J. Jung, M.-J. Lee, H.-J. Eom, S. Maskey and C.-U. Ro, Combined Use of Optical and Electron Microscopic Techniques for the Measurement of Hygroscopic Property, Chemical Composition, and Morphology of Individual Aerosol Particles, Anal. Chem., 2010, 82, 7999–8009 CrossRef CAS PubMed.
- S. D. Forestieri, S. M. Staudt, T. M. Kuborn, K. Faber, C. R. Ruehl, T. H. Bertram and C. D. Cappa, Establishing the impact of model surfactants on cloud condensation nuclei activity of sea spray aerosol mimics, Atmos. Chem. Phys., 2018, 18, 10985–11005 CrossRef CAS.
- S. J. Lowe, D. G. Partridge, J. F. Davies, K. R. Wilson, D. Topping and I. Riipinen, Key drivers of cloud response to surface-active organics, Nat. Commun., 2019, 10, 5214 CrossRef CAS PubMed.
- N. L. Prisle, A predictive thermodynamic framework of cloud droplet activation for chemically unresolved aerosol mixtures, including surface tension, non-ideality, and bulk–surface partitioning, Atmos. Chem. Phys., 2021, 21, 16387–16411 CrossRef CAS.
- J. J. Lin, T. B. Kristensen, S. M. Calderón, J. Malila and N. L. Prisle, Effects of surface tension time-evolution for CCN activation of a complex organic surfactant, Environ. Sci.: Processes Impacts, 2020, 22, 271–284 RSC.
- V. Naik, S. Szopa, B. Adhikary, P. Artaxo, T. Bernsten, W. D. Collins, S. Fuzzi, L. Gallardo, A. Kiendler-Scharr, Z. Klimont, H. Liao, N. Unger and P. Zanis, in Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, Cambridge University Press, 2021 Search PubMed.
- Z. A. Kanji, L. A. Ladino, H. Wex, Y. Boose, M. Burkert-Kohn, D. J. Cziczo and M. Krämer, Overview of Ice Nucleating Particles, Meteorol. Monogr., 2017, 58, 1.1–1.33 CrossRef.
- M. A. Freedman, Potential Sites for Ice Nucleation on Aluminosilicate Clay Minerals and Related Materials, J. Phys. Chem. Lett., 2015, 6, 3850–3858 CrossRef CAS PubMed.
- M. Tang, D. J. Cziczo and V. H. Grassian, Interactions of Water with Mineral Dust Aerosol: Water Adsorption, Hygroscopicity, Cloud Condensation, and Ice Nucleation, Chem. Rev., 2016, 116, 4205–4259 CrossRef CAS PubMed.
- O. Möhler, P. J. DeMott, G. Vali and Z. Levin, Microbiology and atmospheric processes: the role of biological particles in cloud physics, Biogeosciences, 2007, 4, 1059–1071 CrossRef.
- A.-M. Delort, M. Vaïtilingom, P. Amato, M. Sancelme, M. Parazols, G. Mailhot, P. Laj and L. Deguillaume, A short overview of the microbial population in clouds: potential roles in atmospheric chemistry and nucleation processes, Atmos. Res., 2010, 98, 249–260 CrossRef CAS.
- W. Smets, S. Moretti, S. Denys and S. Lebeer, Airborne bacteria in the atmosphere: presence, purpose, and potential, Atmos. Environ., 2016, 139, 214–221 CrossRef CAS.
- S. Huang, W. Hu, J. Chen, Z. Wu, D. Zhang and P. Fu, Overview of biological ice nucleating particles in the atmosphere, Environ. Int., 2021, 146, 106197 CrossRef PubMed.
- B. J. Murray, D. O'Sullivan, J. D. Atkinson and M. E. Webb, Ice nucleation by particles immersed in supercooled cloud droplets, Chem. Soc. Rev., 2012, 41, 6519–6554 RSC.
- C. Hoose and O. Möhler, Heterogeneous ice nucleation on atmospheric aerosols: a review of results from laboratory experiments, Atmos. Chem. Phys., 2012, 12, 9817–9854 CrossRef CAS.
- T. Berkemeier, M. Shiraiwa, U. Pöschl and T. Koop, Competition between water uptake and ice nucleation by glassy organic aerosol particles, Atmos. Chem. Phys., 2014, 14, 12513–12531 CrossRef.
- M. Gavish, R. Popovitz-Biro, M. Lahav and L. Leiserowitz, Ice Nucleation by Alcohols Arranged in Monolayers at the Surface of Water Drops, Science, 1990, 250, 973–975 CrossRef CAS PubMed.
- R. Popovitz-Biro, M. Gavish, M. Lahav and L. Leiserowitz, Ice nucleation by monolayers of aliphatic alcohols, Makromol. Chem., Macromol. Symp., 1991, 46, 125–132 CrossRef CAS.
- R. J. Davey, S. J. Maginn, R. B. Steventon, J. M. Ellery, A. V. Murrell, J. Booth, A. D. Godwin and J. E. Rout, Nucleation of Crystals under Langmuir Monolayers: Kinetic and Morphological Data for the Nucleation of Ice, Langmuir, 1994, 10, 1673–1675 CrossRef CAS.
- J. Majewski, R. Popovitz-Biro, K. Kjaer, J. Als-Nielsen, M. Lahav and L. Leiserowitz, Toward a Determination of the Critical Size of Ice Nuclei. A Demonstration by Grazing Incidence X-ray Diffraction of Epitaxial Growth of Ice under the C31H63OH Alcohol Monolayer, J. Phys. Chem., 1994, 98, 4087–4093 CrossRef CAS.
- R. Popovitz-Biro, J. L. Wang, J. Majewski, E. Shavit, L. Leiserowitz and M. Lahav, Induced freezing of supercooled water into ice by self-assembled crystalline monolayers of amphiphilic alcohols at the air–water interface, J. Am. Chem. Soc., 1994, 116, 1179–1191 CrossRef CAS.
- R. Popovitz-Biro, J. Majewski, J. L. Wang, K. Kjaer, J. Als-Nielsen, M. Lahav and L. Leiserowitz, in Proceedings of the NATO Advanced Research Workshop on Hydrogen Bond Networks, ed. M.-C. Bellissent-Funel and J. C. Dore, Kiuwer Academic Publishers, Dordrecht, the Netherlands, 1994, vol. 435, pp. 395–402 Search PubMed.
- J. Majewski, R. Popovitz-Biro, W. G. Bouwman, K. Kjaer, J. Als-Nielsen, M. Lahav and L. Leiserowitz, The Structural Properties of Uncompressed Crystalline Monolayers of Alcohols CnH2n+1 OH (n = 13–31) on Water and Their Role as Ice Nucleators, Chem.–Eur. J., 1995, 1, 304–311 CrossRef CAS.
- R. Popovitz-Biro, R. Edgar, J. Majewski, S. Cohen, L. Margulis, K. Kjaer, J. Als-Nielsen, L. Leiserowitz and M. Lahav, Self-aggregation of α,ω-Alkanediols into Two and Three Dimensional Crystallites at the Air–Water Interface. Relevance to Ice Nucleation, Croat. Chem. Acta, 1996, 69, 689–708 CAS.
- J. Majewski, R. Popovitz-Biro, R. Edgar, M. Arbel-Haddad, K. Kjaer, W. Bouwman, J. Als-Nielsen, M. Lahav and L. Leiserowitz, An Insight into the Ice Nucleation Process via Design of Crystalline Ice Nucleators of Variable Size, J. Phys. Chem. B, 1997, 101, 8874–8877 CrossRef CAS.
- S. Trakhtenberg, R. Naaman, S. R. Cohen and I. Benjamin, Effect of the Substrate Morphology on the Structure of Adsorbed Ice, J. Phys. Chem. B, 1997, 101, 5172–5176 CrossRef CAS.
- M. Arbel-Haddad, M. Lahav and L. Leiserowitz, Design of Mixed Langmuir Films Exposing Segregated Hydroxyl Domains for Inducing Ice Nucleation, J. Phys. Chem. B, 1998, 102, 1543–1548 CrossRef CAS.
- Y. Dai and J. S. Evans, Molecular Dynamics Simulations of Template-Assisted Nucleation: Alcohol Monolayers at the Air–Water Interface and Ice Formation, J. Phys. Chem. B, 2001, 105, 10831–10837 CrossRef CAS.
- L. H. Seeley and G. T. Seidler, Preactivation in the nucleation of ice by Langmuir films of aliphatic alcohols, J. Chem. Phys., 2001, 114, 10464–10470 CrossRef CAS.
- E. Ochshorn and W. Cantrell, Towards understanding ice nucleation by long chain alcohols, J. Chem. Phys., 2006, 124, 054714 CrossRef PubMed.
- B. Zobrist, T. Koop, B. P. Luo, C. Marcolli and T. Peter, Heterogeneous Ice Nucleation Rate Coefficient of Water Droplets Coated by a Nonadecanol Monolayer, J. Phys. Chem. C, 2007, 111, 2149–2155 CrossRef CAS.
- X. Kong, P. U. Andersson, E. S. Thomson and J. B. C. Pettersson, Ice Formation via Deposition Mode Nucleation on Bare and Alcohol-Covered Graphite Surfaces, J. Phys. Chem. C, 2012, 116, 8964–8974 CrossRef CAS.
- L. Lupi, R. Hanscam, Y. Qiu and V. Molinero, Reaction Coordinate for Ice Crystallization on a Soft Surface, J. Phys. Chem. Lett., 2017, 8, 4201–4205 CrossRef CAS PubMed.
- Y. Qiu, N. Odendahl, A. Hudait, R. Mason, A. K. Bertram, F. Paesani, P. J. DeMott and V. Molinero, Ice Nucleation Efficiency of Hydroxylated Organic Surfaces Is Controlled by Their Structural Fluctuations and Mismatch to Ice, J. Am. Chem. Soc., 2017, 139, 3052–3064 CrossRef CAS PubMed.
- R. J. Perkins, M. G. Vazquez de Vasquez, E. E. Beasley, T. C. J. Hill, E. A. Stone, H. C. Allen and P. J. DeMott, Relating Structure and Ice Nucleation of Mixed Surfactant Systems Relevant to Sea Spray Aerosol, J. Phys. Chem. A, 2020, 124, 8806–8821 CrossRef CAS PubMed.
- N. Hiranuma, S. D. Brooks, R. C. Moffet, A. Glen, A. Laskin, M. K. Gilles, P. Liu, A. M. Macdonald, J. W. Strapp and G. M. McFarquhar, Chemical characterization of individual particles and residuals of cloud droplets and ice crystals collected on board research aircraft in the ISDAC 2008 study, J. Geophys. Res.: Atmos., 2013, 118, 6564–6579 CrossRef CAS.
- P. Kupiszewski, M. Zanatta, S. Mertes, P. Vochezer, G. Lloyd, J. Schneider, L. Schenk, M. Schnaiter, U. Baltensperger, E. Weingartner and M. Gysel, Ice residual properties in mixed-phase clouds at the high-alpine Jungfraujoch site, J. Geophys. Res.: Atmos., 2016, 121, 12343–12362 Search PubMed.
- S. China, P. A. Alpert, B. Zhang, S. Schum, K. Dzepina, K. Wright, R. C. Owen, P. Fialho, L. R. Mazzoleni, C. Mazzoleni and D. A. Knopf, Ice cloud formation potential by free tropospheric particles from long-range transport over the Northern Atlantic Ocean, J. Geophys. Res.: Atmos., 2017, 122, 3065–3079 CrossRef CAS.
- N. N. Lata, B. Zhang, S. Schum, L. Mazzoleni, R. Brimberry, M. A. Marcus, W. H. Cantrell, P. Fialho, C. Mazzoleni and S. China, Aerosol Composition, Mixing State, and Phase State of Free Tropospheric Particles and Their Role in Ice Cloud Formation, ACS Earth Space Chem., 2021, 5, 3499–3510 CrossRef CAS.
- Y. Boose, P. Baloh, M. Plötze, J. Ofner, H. Grothe, B. Sierau, U. Lohmann and Z. A. Kanji, Heterogeneous ice nucleation on dust particles sourced from nine deserts worldwide – Part 2: deposition nucleation and condensation freezing, Atmos. Chem. Phys., 2019, 19, 1059–1076 CrossRef CAS.
- Z. A. Kanji, R. C. Sullivan, M. Niemand, P. J. DeMott, A. J. Prenni, C. Chou, H. Saathoff and O. Möhler, Heterogeneous ice nucleation properties of natural desert dust particles coated with a surrogate of secondary organic aerosol, Atmos. Chem. Phys., 2019, 19, 5091–5110 CrossRef CAS.
- L. G. Jahl, T. A. Brubaker, M. J. Polen, L. G. Jahn, K. P. Cain, B. B. Bowers, W. D. Fahy, S. Graves and R. C. Sullivan, Atmospheric aging enhances the ice nucleation ability of biomass-burning aerosol, Sci. Adv., 2021, 7, eabd3440 CrossRef CAS PubMed.
- B. Bertozzi, R. Wagner, J. Song, K. Höhler, J. Pfeifer, H. Saathoff, T. Leisner and O. Möhler, Ice nucleation ability of ammonium sulfate aerosol particles internally mixed with secondary organics, Atmos. Chem. Phys., 2021, 21, 10779–10798 CrossRef CAS.
- A. A. Piedehierro, A. Welti, A. Buchholz, K. Korhonen, I. Pullinen, I. Summanen, A. Virtanen and A. Laaksonen, Ice nucleation on surrogates of boreal forest SOA particles: effect of water content and oxidative age, Atmos. Chem. Phys., 2021, 21, 11069–11078 CrossRef CAS.
- R. E. Cochran, O. Laskina, J. V. Trueblood, A. D. Estillore, H. S. Morris, T. Jayarathne, C. M. Sultana, C. Lee, P. Lin, J. Laskin, A. Laskin, J. A. Dowling, Z. Qin, C. D. Cappa, T. H. Bertram, A. V. Tivanski, E. A. Stone, K. A. Prather and V. H. Grassian, Molecular Diversity of Sea Spray Aerosol Particles: Impact of Ocean Biology on Particle Composition and Hygroscopicity, Chem, 2017, 2, 655–667 CAS.
- T. H. Bertram, R. E. Cochran, V. H. Grassian and E. A. Stone, Sea spray aerosol chemical composition: Elemental and molecular mimics for laboratory studies of heterogeneous and multiphase reactions, Chem. Soc. Rev., 2018, 47, 2374–2400 RSC.
- X. Wang, C. M. Sultana, J. Trueblood, T. C. J. Hill, F. Malfatti, C. Lee, O. Laskina, K. A. Moore, C. M. Beall, C. S. McCluskey, G. C. Cornwell, Y. Zhou, J. L. Cox, M. A. Pendergraft, M. V. Santander, T. H. Bertram, C. D. Cappa, F. Azam, P. J. DeMott, V. H. Grassian and K. A. Prather, Microbial Control of Sea Spray Aerosol Composition: A Tale of Two Blooms, ACS Cent. Sci., 2015, 1, 124–131 CrossRef CAS PubMed.
- C. S. McCluskey, T. C. J. Hill, C. M. Sultana, O. Laskina, J. Trueblood, M. V. Santander, C. M. Beall, J. M. Michaud, S. M. Kreidenweis, K. A. Prather, V. Grassian and P. J. DeMott, A Mesocosm Double Feature: Insights into the Chemical Makeup of Marine Ice Nucleating Particles, J. Atmos. Sci., 2018, 75, 2405–2423 CrossRef.
- J. V. Trueblood, A. Nicosia, A. Engel, B. Zäncker, M. Rinaldi, E. Freney, M. Thyssen, I. Obernosterer, J. Dinasquet, F. Belosi, A. Tovar-Sánchez, A. Rodriguez-Romero, G. Santachiara, C. Guieu and K. Sellegri, A two-component parameterization of marine ice-nucleating particles based on seawater biology and sea spray aerosol measurements in the Mediterranean Sea, Atmos. Chem. Phys., 2021, 21, 4659–4676 CrossRef CAS.
- J. A. Faust, PFAS on atmospheric aerosol particles: a review, Environ. Sci.: Processes Impacts, 2022 10.1039/D2EM00002D.
- M. J. Wolf, Y. Zhang, J. Zhou, J. D. Surratt, B. J. Turpin and D. J. Cziczo, Enhanced Ice Nucleation of Simulated Sea Salt Particles with the Addition of Anthropogenic Per- and Polyfluoroalkyl Substances, ACS Earth Space Chem., 2021, 5, 2074–2085 CrossRef CAS.
- R. Schwidetzky, Y. Sun, J. Fröhlich-Nowoisky, A. T. Kunert, M. Bonn and K. Meister, Ice Nucleation Activity of Perfluorinated Organic Acids, J. Phys. Chem. Lett., 2021, 12, 3431–3435 CrossRef CAS PubMed.
- M. F. Ruiz-Lopez, J. S. Francisco, M. T. C. Martins-Costa and J. M. Anglada, Molecular reactions at aqueous interfaces, Nat. Rev. Chem., 2020, 4, 459–475 CrossRef CAS.
- M. Shiraiwa and U. Pöschl, Mass accommodation and gas–particle partitioning in secondary organic aerosols: dependence on diffusivity, volatility, particle-phase reactions, and penetration depth, Atmos. Chem. Phys., 2021, 21, 1565–1580 CrossRef CAS.
- I. J. George and J. P. D. Abbatt, Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nat. Chem., 2010, 2, 713–722 CrossRef CAS PubMed.
- D. J. Donaldson and K. T. Valsaraj, Adsorption and Reaction of Trace Gas-Phase Organic Compounds on Atmospheric Water Film Surfaces: A Critical Review, Environ. Sci. Technol., 2010, 44, 865–873 CrossRef CAS PubMed.
- R. C. Chapleski, Y. Zhang, D. Troya and J. R. Morris, Heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces, Chem. Soc. Rev., 2016, 45, 3731–3746 RSC.
- P. Varilly and D. Chandler, Water Evaporation: A Transition Path Sampling Study, J. Phys. Chem. B, 2013, 117, 1419–1428 CrossRef CAS PubMed.
- C. M. Fellows, P. A. Coop, D. W. Lamb, R. C. Bradbury, H. F. Schiretz and A. J. Woolley, Understanding the role of monolayers in retarding evaporation from water storage bodies, Chem. Phys. Lett., 2015, 623, 37–41 CrossRef CAS.
- G. Ergin and S. Takahama, Carbon Density Is an Indicator of Mass Accommodation Coefficient of Water on Organic-Coated Water Surface, J. Phys. Chem. A, 2016, 120, 2885–2893 CrossRef CAS PubMed.
- R. E. H. Miles, J. F. Davies and J. P. Reid, The influence of the surface composition of mixed monolayer films on the evaporation coefficient of water, Phys. Chem. Chem. Phys., 2016, 18, 19847–19858 RSC.
- S. M. Johansson, J. Lovrić, X. Kong, E. S. Thomson, P. Papagiannakopoulos, S. Briquez, C. Toubin and J. B. C. Pettersson, Understanding water interactions with organic surfaces: environmental molecular beam and molecular dynamics studies of the water–butanol system, Phys. Chem. Chem. Phys., 2019, 21, 1141–1151 RSC.
- M. Chan and C. Chan, Mass transfer effects on the hygroscopic growth of ammonium sulfate particles with a water-insoluble coating, Atmos. Environ., 2007, 41, 4423–4433 CrossRef CAS.
- S.-C. Park, D. K. Burden and G. M. Nathanson, Surfactant Control of Gas Transport and Reactions at the Surface of Sulfuric Acid, Acc. Chem. Res., 2009, 42, 379–387 CrossRef CAS PubMed.
- S. Sakaguchi and A. Morita, Molecular Dynamics Study of Water Transfer at Supercooled Sulfuric Acid Solution Surface Covered with Butanol, J. Phys. Chem. A, 2013, 117, 4602–4610 CrossRef CAS PubMed.
- S. Takahama and L. M. Russell, A molecular dynamics study of water mass accommodation on condensed phase water coated by fatty acid monolayers, J. Geophys. Res., 2011, 116, D02203 CrossRef.
- S. Sakaguchi and A. Morita, Mass accommodation mechanism of water through monolayer films at water/vapor interface, J. Chem. Phys., 2012, 137, 064701 CrossRef PubMed.
- J. F. Davies, R. E. H. Miles, A. E. Haddrell and J. P. Reid, Influence of organic films on the evaporation and condensation of water in aerosol, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8807–8812 CrossRef CAS PubMed.
- M. A. Shaloski, T. B. Sobyra and G. M. Nathanson, DCl Transport through Dodecyl Sulfate Films on Salty Glycerol: Effects of Seawater Ions on Gas Entry, J. Phys. Chem. A, 2015, 119, 12357–12366 CrossRef CAS PubMed.
- W. Li, C. Y. Pak, X. Wang and Y.-L. S. Tse, Uptake of Common Atmospheric Gases by Organic-Coated Water Droplets, J. Phys. Chem. C, 2019, 123, 18924–18931 CrossRef CAS.
- U. Pöschl and M. Shiraiwa, Multiphase Chemistry at the Atmosphere–Biosphere Interface Influencing Climate and Public Health in the Anthropocene, Chem. Rev., 2015, 115, 4440–4475 CrossRef PubMed.
- R. Vingarzan, A review of surface ozone background levels and trends, Atmos. Environ., 2004, 38, 3431–3442 CrossRef CAS.
- T. Berkemeier, A. Mishra, C. Mattei, A. J. Huisman, U. K. Krieger and U. Pöschl, Ozonolysis of Oleic Acid Aerosol Revisited: Multiphase Chemical Kinetics and Reaction Mechanisms, ACS Earth Space Chem., 2021, 5, 3313–3323 CrossRef CAS.
- A. Milsom, A. M. Squires, B. Woden, N. J. Terrill, A. D. Ward and C. Pfrang, The persistence of a proxy for cooking emissions in megacities: a kinetic study of the ozonolysis of self-assembled films by simultaneous small and wide angle X-ray scattering (SAXS/WAXS) and Raman microscopy, Faraday Discuss., 2021, 226, 364–381 RSC.
- A. Milsom, A. M. Squires, J. A. Boswell, N. J. Terrill, A. D. Ward and C. Pfrang, An organic crystalline state in ageing atmospheric aerosol proxies: spatially resolved structural changes in levitated fatty acid particles, Atmos. Chem. Phys., 2021, 21, 15003–15021 CrossRef CAS.
- S. Zhou, B. C. H. Hwang, P. S. J. Lakey, A. Zuend, J. P. D. Abbatt and M. Shiraiwa, Multiphase reactivity of polycyclic aromatic hydrocarbons is driven by phase separation and diffusion limitations, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11658–11663 CrossRef CAS PubMed.
- M. D. King, S. H. Jones, C. O. M. Lucas, K. C. Thompson, A. R. Rennie, A. D. Ward, A. A. Marks, F. N. Fisher, C. Pfrang, A. V. Hughes and R. A. Campbell, The reaction of oleic acid monolayers with gas-phase ozone at the air water interface: The effect of sub-phase viscosity, and inert secondary components, Phys. Chem. Chem. Phys., 2020, 22, 28032–28044 RSC.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Oxidation of oleic acid at the air–water interface and its potential effects on cloud critical supersaturations, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- L. J. Carpenter and P. D. Nightingale, Chemistry and Release of Gases from the Surface Ocean, Chem. Rev., 2015, 115, 4015–4034 CrossRef CAS PubMed.
- C. Pfrang, K. Rastogi, E. R. Cabrera-Martinez, A. M. Seddon, C. Dicko, A. Labrador, T. S. Plivelic, N. Cowieson and A. M. Squires, Complex three-dimensional self-assembly in proxies for atmospheric aerosols, Nat. Commun., 2017, 8, 1724 CrossRef CAS PubMed.
- G. A. Novak and T. H. Bertram, Reactive VOC Production from Photochemical and Heterogeneous Reactions Occurring at the Air–Ocean Interface, Acc. Chem. Res., 2020, 53, 1014–1023 CrossRef CAS PubMed.
- B. Woden, M. W. A. Skoda, A. Milsom, C. Gubb, A. Maestro, J. Tellam and C. Pfrang, Ozonolysis of fatty acid monolayers at the air–water interface: organic films may persist at the surface of atmospheric aerosols, Atmos. Chem. Phys., 2021, 21, 1325–1340 CrossRef CAS.
- S. Enami and A. J. Colussi, Reactions of Criegee Intermediates with Alcohols at Air–Aqueous Interfaces, J. Phys. Chem. A, 2017, 121, 5175–5182 CrossRef CAS PubMed.
- S. Enami and A. J. Colussi, Efficient scavenging of Criegee intermediates on water by surface-active cis-pinonic acid, Phys. Chem. Chem. Phys., 2017, 19, 17044–17051 RSC.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Criegee Intermediates React with Levoglucosan on Water, J. Phys. Chem. Lett., 2017, 8, 3888–3894 CrossRef CAS PubMed.
- S. Enami and A. J. Colussi, Criegee Chemistry on Aqueous Organic Surfaces, J. Phys. Chem. Lett., 2017, 8, 1615–1623 CrossRef CAS PubMed.
- J. Qiu, S. Ishizuka, K. Tonokura, K. Sato, S. Inomata and S. Enami, Effects of pH on Interfacial Ozonolysis of α-Terpineol, J. Phys. Chem. A, 2019, 123, 7148–7155 CrossRef CAS PubMed.
- R. Criegee, Mechanism of Ozonolysis, Angew. Chem., Int. Ed., 1975, 14, 745–752 CrossRef.
- Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Bräse, Criegee Intermediates Beyond Ozonolysis: Synthetic and Mechanistic Insights, Angew. Chem., Int. Ed., 2021, 60, 15138–15152 CrossRef CAS PubMed.
- S. R. Schneider, D. B. Collins, C. Y. Lim, L. Zhu and J. P. D. Abbatt, Formation of Secondary Organic Aerosol from the Heterogeneous Oxidation by Ozone of a Phytoplankton Culture, ACS Earth Space Chem., 2019, 3, 2298–2306 CrossRef CAS.
- M. Brüggemann, N. Hayeck, C. Bonnineau, S. Pesce, P. A. Alpert, S. Perrier, C. Zuth, T. Hoffmann, J. Chen and C. George, Interfacial photochemistry of biogenic surfactants: a major source of abiotic volatile organic compounds, Faraday Discuss., 2017, 200, 59–74 RSC.
- S. H. Jones, M. D. King, A. D. Ward, A. R. Rennie, A. C. Jones and T. Arnold, Are organic films from atmospheric aerosol and sea water inert to oxidation by ozone at the air–water interface?, Atmos. Environ., 2017, 161, 274–287 CrossRef CAS.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Environmental Implications of Hydroxyl Radicals (˙OH), Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- A. D. Estillore, J. V. Trueblood and V. H. Grassian, Atmospheric chemistry of bioaerosols: heterogeneous and multiphase reactions with atmospheric oxidants and other trace gases, Chem. Sci., 2016, 7, 6604–6616 RSC.
- D. E. Heard and M. J. Pilling, Measurement of OH and HO2 in the Troposphere, Chem. Rev., 2003, 103, 5163–5198 CrossRef CAS PubMed.
- D. Stone, L. K. Whalley and D. E. Heard, Tropospheric OH and HO2 radicals: Field measurements and model comparisons, Chem. Soc. Rev., 2012, 41, 6348–6404 RSC.
- A. A. Heath and K. T. Valsaraj, An Experimental Study of the Atmospheric Oxidation of a Biogenic Organic Compound (Methyl Jasmonate) in a Thin Water Film as in Fog or Aerosols, Open J. Air Pollut., 2017, 06, 44–51 CrossRef CAS.
- S. Enami and Y. Sakamoto, OH-Radical Oxidation of Surface-Active cis-Pinonic Acid at the Air–Water Interface, J. Phys. Chem. A, 2016, 120, 3578–3587 CrossRef CAS PubMed.
- Y. Huang, K. M. Barraza, C. M. Kenseth, R. Zhao, C. Wang, J. L. Beauchamp and J. H. Seinfeld, Probing the OH Oxidation of Pinonic Acid at the Air–Water Interface Using Field-Induced Droplet Ionization Mass Spectrometry (FIDI-MS), J. Phys. Chem. A, 2018, 122, 6445–6456 CrossRef CAS PubMed.
- J. A. Faust and J. P. D. Abbatt, Organic Surfactants Protect Dissolved Aerosol Components against Heterogeneous Oxidation, J. Phys. Chem. A, 2019, 123, 2114–2124 CrossRef CAS PubMed.
- H. K. Lam, R. Xu, J. Choczynski, J. F. Davies, D. Ham, M. Song, A. Zuend, W. Li, Y.-L. S. Tse and M. N. Chan, Effects of liquid–liquid phase separation and relative humidity on the heterogeneous OH oxidation of inorganic–organic aerosols: Insights from methylglutaric acid and ammonium sulfate particles, Atmos. Chem. Phys., 2021, 21, 2053–2066 CrossRef CAS.
- C. Y. Lim, E. C. Browne, R. A. Sugrue and J. H. Kroll, Rapid heterogeneous oxidation of organic coatings on submicron aerosols: Heterogeneous Oxidation of Organic Aerosol, Geophys. Res. Lett., 2017, 44, 2949–2957 CrossRef CAS.
- X. Zhang, K. M. Barraza, K. T. Upton and J. L. Beauchamp, Time resolved study of hydroxyl radical oxidation of oleic acid at the air–water interface, Chem. Phys. Lett., 2017, 683, 76–82 CrossRef CAS.
- X. Zhang, K. M. Barraza and J. L. Beauchamp, Cholesterol provides nonsacrificial protection of membrane lipids from chemical damage at air–water interface, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3255–3260 CrossRef CAS PubMed.
- S. Sobanska, J. Barbillat, M. Moreau, N. Nuns, I. De Waele, D. Petitprez, Y. Tobon and C. Brémard, Influence of stearic acid coating of the NaCl surface on the reactivity with NO2 under humidity, Phys. Chem. Chem. Phys., 2015, 17, 10963–10977 RSC.
- H. Deng, J. Liu, Y. Wang, W. Song, X. Wang, X. Li, D. Vione and S. Gligorovski, Effect of Inorganic Salts on N-Containing Organic Compounds Formed by Heterogeneous Reaction of NO2 with Oleic Acid, Environ. Sci. Technol., 2021, 55, 7831–7840 CrossRef CAS PubMed.
- S. S. Brown and J. Stutz, Nighttime radical observations and chemistry, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- M. Ge, S. Tong, W. Wang, W. Zhang, M. Chen, C. Peng, J. Li, L. Zhou, Y. Chen and M. Liu, Important Oxidants and Their Impact on the Environmental Effects of Aerosols, J. Phys. Chem. A, 2021, 125, 3813–3825 CrossRef CAS PubMed.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS PubMed.
- B. Woden, M. Skoda, M. Hagreen and C. Pfrang, Night-Time Oxidation of a Monolayer Model for the Air–Water Interface of Marine Aerosols—A Study by Simultaneous Neutron Reflectometry and in Situ Infra-Red Reflection Absorption Spectroscopy (IRRAS), Atmosphere, 2018, 9, 471 CrossRef CAS.
- W. L. Chang, P. V. Bhave, S. S. Brown, N. Riemer, J. Stutz and D. Dabdub, Heterogeneous Atmospheric Chemistry, Ambient Measurements, and Model Calculations of N2O5: A Review, Aerosol Sci. Technol., 2011, 45, 665–695 CrossRef.
- A. T. Ahern, L. Goldberger, L. Jahl, J. Thornton and R. C. Sullivan, Production of N2O5 and ClNO2 through Nocturnal Processing of Biomass-Burning Aerosol, Environ. Sci. Technol., 2018, 52, 550–559 CrossRef CAS PubMed.
- L. A. Goldberger, L. G. Jahl, J. A. Thornton and R. C. Sullivan, N2O5 reactive uptake kinetics and chlorine activation on authentic biomass-burning aerosol, Environ. Sci.: Processes Impacts, 2019, 21, 1684–1698 RSC.
- L. G. Jahl, B. B. Bowers, L. G. Jahn, J. A. Thornton and R. C. Sullivan, Response of the Reaction Probability of N2O5 with Authentic Biomass-Burning Aerosol to High Relative Humidity, ACS Earth Space Chem., 2021, 5, 2587–2598 CrossRef CAS.
- L. G. Jahn, L. G. Jahl, B. B. Bowers and R. C. Sullivan, Morphology of Organic Carbon Coatings on Biomass-Burning Particles and Their Role in Reactive Gas Uptake, ACS Earth Space Chem., 2021, 5, 2184–2195 CrossRef CAS.
- B. Alexander, T. Sherwen, C. D. Holmes, J. A. Fisher, Q. Chen, M. J. Evans and P. Kasibhatla, Global inorganic nitrate production mechanisms: comparison of a global model with nitrate isotope observations, Atmos. Chem. Phys., 2020, 20, 3859–3877 CrossRef CAS.
- L. Liu, J. Wu, S. Liu, X. Li, J. Zhou, T. Feng, Y. Qian, J. Cao, X. Tie and G. Li, Effects of organic coating on the nitrate formation by suppressing the N2O5 heterogeneous hydrolysis: a case study during wintertime in Beijing–Tianjin–Hebei (BTH), Atmos. Chem. Phys., 2019, 19, 8189–8207 CrossRef CAS.
- N. Riemer, H. Vogel, B. Vogel, T. Anttila, A. Kiendler-Scharr and T. F. Mentel, Relative importance of organic coatings for the heterogeneous hydrolysis of N2O5 during summer in Europe, J. Geophys. Res., 2009, 114, D17307 CrossRef.
- T. B. Sobyra, H. Pliszka, T. H. Bertram and G. M. Nathanson, Production of Br2 from N2O5 and Br− in Salty and Surfactant-Coated Water Microjets, J. Phys. Chem. A, 2019, 123, 8942–8953 CrossRef CAS PubMed.
- J. R. Gord, X. Zhao, E. Liu, T. H. Bertram and G. M. Nathanson, Control of Interfacial Cl2 and N2O5 Reactivity by a Zwitterionic Phospholipid in Comparison with Ionic and Uncharged Surfactants, J. Phys. Chem. A, 2018, 122, 6593–6604 CrossRef CAS PubMed.
- J. A. Faust, L. P. Dempsey and G. M. Nathanson, Surfactant-Promoted Reactions of Cl2 and Br2 with Br− in Glycerol, J. Phys. Chem. B, 2013, 117, 12602–12612 CrossRef CAS PubMed.
- S. Chen, L. Artiglia, F. Orlando, J. Edebeli, X. Kong, H. Yang, A. Boucly, P. Corral Arroyo, N. Prisle and M. Ammann, Impact of Tetrabutylammonium on the Oxidation of Bromide by Ozone, ACS Earth Space Chem., 2021, 5, 3008–3021 CrossRef CAS PubMed.
- J. V. Trueblood, A. D. Estillore, C. Lee, J. A. Dowling, K. A. Prather and V. H. Grassian, Heterogeneous Chemistry of Lipopolysaccharides with Gas-Phase Nitric Acid: Reactive Sites and Reaction Pathways, J. Phys. Chem. A, 2016, 120, 6444–6450 CrossRef CAS PubMed.
- C. Lee, A. C. Dommer, J. M. Schiffer, R. E. Amaro, V. H. Grassian and K. A. Prather, Cation-Driven Lipopolysaccharide Morphological Changes Impact Heterogeneous Reactions of Nitric Acid with Sea Spray Aerosol Particles, J. Phys. Chem. Lett., 2021, 12, 5023–5029 CrossRef CAS PubMed.
- M. Passananti, L. Kong, J. Shang, Y. Dupart, S. Perrier, J. Chen, D. J. Donaldson and C. George, Organosulfate Formation through the Heterogeneous Reaction of Sulfur Dioxide with Unsaturated Fatty Acids and Long-Chain Alkenes, Angew. Chem., Int. Ed., 2016, 55, 10336–10339 CrossRef CAS PubMed.
- J. Shang, M. Passananti, Y. Dupart, R. Ciuraru, L. Tinel, S. Rossignol, S. Perrier, T. Zhu and C. George, SO2 Uptake on Oleic Acid: A New Formation Pathway of Organosulfur Compounds in the Atmosphere, Environ. Sci. Technol. Lett., 2016, 3, 67–72 CrossRef CAS.
- M. Brüggemann, R. Xu, A. Tilgner, K. C. Kwong, A. Mutzel, H. Y. Poon, T. Otto, T. Schaefer, L. Poulain, M. N. Chan and H. Herrmann, Organosulfates in Ambient Aerosol: State of Knowledge and Future Research Directions on Formation, Abundance, Fate, and Importance, Environ. Sci. Technol., 2020, 54, 3767–3782 CrossRef PubMed.
- M. Zhu, B. Jiang, S. Li, Q. Yu, X. Yu, Y. Zhang, X. Bi, J. Yu, C. George, Z. Yu and X. Wang, Organosulfur Compounds Formed from Heterogeneous Reaction between SO2 and Particulate-Bound Unsaturated Fatty Acids in Ambient Air, Environ. Sci. Technol. Lett., 2019, 6, 318–322 CrossRef CAS.
- J. Montoya-Aguilera, M. L. Hinks, P. K. Aiona, L. M. Wingen, J. R. Horne, S. Zhu, D. Dabdub, A. Laskin, J. Laskin, P. Lin and S. A. Nizkorodov, in ACS Symposium Series, ed. S. W. Hunt, A. Laskin and S. A. Nizkorodov, American Chemical Society, Washington, DC, 2018, vol. 1299, pp. 127–147 Search PubMed.
- R. F. Silvern, D. J. Jacob, P. S. Kim, E. A. Marais, J. R. Turner, P. Campuzano-Jost and J. L. Jimenez, Inconsistency of ammonium–sulfate aerosol ratios with thermodynamic models in the eastern US: a possible role of organic aerosol, Atmos. Chem. Phys., 2017, 17, 5107–5118 CrossRef CAS.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Reactive intermediates revealed in secondary organic aerosol formation from isoprene, roc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- Y.-H. Lin, Z. Zhang, K. S. Docherty, H. Zhang, S. H. Budisulistiorini, C. L. Rubitschun, S. L. Shaw, E. M. Knipping, E. S. Edgerton, T. E. Kleindienst, A. Gold and J. D. Surratt, Isoprene Epoxydiols as Precursors to Secondary Organic Aerosol Formation: Acid-Catalyzed Reactive Uptake Studies with Authentic Compounds, Environ. Sci. Technol., 2012, 46, 250–258 CrossRef CAS PubMed.
- R. Schmedding, M. Ma, Y. Zhang, S. Farrell, H. O. T. Pye, Y. Chen, C. Wang, Q. Z. Rasool, S. H. Budisulistiorini, A. P. Ault, J. D. Surratt and W. Vizuete, α-Pinene-Derived organic coatings on acidic sulfate aerosol impacts secondary organic aerosol formation from isoprene in a box model, Atmos. Environ., 2019, 213, 456–462 CrossRef CAS PubMed.
- D. S. Jo, A. Hodzic, L. K. Emmons, S. Tilmes, R. H. Schwantes, M. J. Mills, P. Campuzano-Jost, W. Hu, R. A. Zaveri, R. C. Easter, B. Singh, Z. Lu, C. Schulz, J. Schneider, J. E. Shilling, A. Wisthaler and J. L. Jimenez, Future changes in isoprene-epoxydiol-derived secondary organic aerosol (IEPOX SOA) under the Shared Socioeconomic Pathways: the importance of physicochemical dependency, Atmos. Chem. Phys., 2021, 21, 3395–3425 CrossRef CAS.
- M. Octaviani, M. Shrivastava, R. A. Zaveri, A. Zelenyuk, Y. Zhang, Q. Z. Rasool, D. M. Bell, M. Riva, M. Glasius and J. D. Surratt, Modeling the Size Distribution and Chemical Composition of Secondary Organic Aerosols during the Reactive Uptake of Isoprene-Derived Epoxydiols under Low-Humidity Condition, ACS Earth Space Chem., 2021, 5, 3247–3257 CrossRef CAS.
- M. Shrivastava, Q. Z. Rasool, B. Zhao, M. Octaviani, R. A. Zaveri, A. Zelenyuk, B. Gaudet, Y. Liu, J. E. Shilling, J. Schneider, C. Schulz, M. Zöger, S. T. Martin, J. Ye, A. Guenther, R. F. Souza, M. Wendisch and U. Pöschl, Tight Coupling of Surface and In-Plant Biochemistry and Convection Governs Key Fine Particulate Components over the Amazon Rainforest, ACS Earth Space Chem., 2022, 6, 380–390 CrossRef CAS.
- Y. Zhang, Y. Chen, Z. Lei, N. E. Olson, M. Riva, A. R. Koss, Z. Zhang, A. Gold, J. T. Jayne, D. R. Worsnop, T. B. Onasch, J. H. Kroll, B. J. Turpin, A. P. Ault and J. D. Surratt, Joint Impacts of Acidity and Viscosity on the Formation of Secondary Organic Aerosol from Isoprene Epoxydiols (IEPOX) in Phase Separated Particles, ACS Earth Space Chem., 2019, 3, 2646–2658 CrossRef CAS.
- G. T. Drozd, J. L. Woo and V. F. McNeill, Self-limited uptake of α-pinene oxide to acidic aerosol: the effects of liquid–liquid phase separation and implications for the formation of secondary organic aerosol and organosulfates from epoxides, Atmos. Chem. Phys., 2013, 13, 8255–8263 CrossRef.
- C. J. Gaston, T. P. Riedel, Z. Zhang, A. Gold, J. D. Surratt and J. A. Thornton, Reactive Uptake of an Isoprene-Derived Epoxydiol to Submicron Aerosol Particles, Environ. Sci. Technol., 2014, 48, 11178–11186 CrossRef CAS PubMed.
- M. Riva, D. M. Bell, A.-M. K. Hansen, G. T. Drozd, Z. Zhang, A. Gold, D. Imre, J. D. Surratt, M. Glasius and A. Zelenyuk, Effect of Organic Coatings, Humidity and Aerosol Acidity on Multiphase Chemistry of Isoprene Epoxydiols, Environ. Sci. Technol., 2016, 50, 5580–5588 CrossRef CAS PubMed.
- Y. Zhang, Y. Chen, A. T. Lambe, N. E. Olson, Z. Lei, R. L. Craig, Z. Zhang, A. Gold, T. B. Onasch, J. T. Jayne, D. R. Worsnop, C. J. Gaston, J. A. Thornton, W. Vizuete, A. P. Ault and J. D. Surratt, Effect of the Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX), Environ. Sci. Technol. Lett., 2018, 5, 167–174 CrossRef CAS.
- N. E. Olson, Z. Lei, R. L. Craig, Y. Zhang, Y. Chen, A. T. Lambe, Z. Zhang, A. Gold, J. D. Surratt and A. P. Ault, Reactive Uptake of Isoprene Epoxydiols Increases the Viscosity of the Core of Phase-Separated Aerosol Particles, ACS Earth Space Chem., 2019, 3, 1402–1414 CrossRef CAS.
- Z. Lei, N. E. Olson, Y. Zhang, Y. Chen, A. T. Lambe, J. Zhang, N. J. White, J. M. Atkin, M. M. Banaszak Holl, Z. Zhang, A. Gold, J. D. Surratt and A. P. Ault, Morphology and Viscosity Changes after Reactive Uptake of Isoprene Epoxydiols in Submicrometer Phase Separated Particles with Secondary Organic Aerosol Formed from Different Volatile Organic Compounds, ACS Earth Space Chem., 2022, 6, 871–882 CrossRef CAS.
- P. Renard, A. E. Reed Harris, R. J. Rapf, S. Ravier, C. Demelas, B. Coulomb, E. Quivet, V. Vaida and A. Monod, Aqueous Phase Oligomerization of Methyl Vinyl Ketone by Atmospheric Radical Reactions, J. Phys. Chem. C, 2014, 118, 29421–29430 CrossRef CAS.
- K. J. Kappes, A. M. Deal, M. F. Jespersen, S. L. Blair, J.-F. Doussin, M. Cazaunau, E. Pangui, B. N. Hopper, M. S. Johnson and V. Vaida, Chemistry and Photochemistry of Pyruvic Acid at the Air–Water Interface, J. Phys. Chem. A, 2021, 125, 1036–1049 CrossRef CAS PubMed.
- R. J. Rapf, R. J. Perkins, H. Yang, G. M. Miyake, B. K. Carpenter and V. Vaida, Photochemical Synthesis of Oligomeric Amphiphiles from Alkyl Oxoacids in Aqueous Environments, J. Am. Chem. Soc., 2017, 139, 6946–6959 CrossRef CAS PubMed.
- A. L. Van Wyngarden, S. Pérez-Montaño, J. V. H. Bui, E. S. W. Li, T. E. Nelson, K. T. Ha, L. Leong and L. T. Iraci, Complex chemical composition of colored surface films formed from reactions of propanal in sulfuric acid at upper troposphere/lower stratosphere aerosol acidities, Atmos. Chem. Phys., 2015, 15, 4225–4239 CrossRef CAS PubMed.
- M. Mekic, J. Liu, W. Zhou, G. Loisel, J. Cai, T. He, B. Jiang, Z. Yu, Y. G. Lazarou, X. Li, M. Brigante, D. Vione and S. Gligorovski, Formation of highly oxygenated multifunctional compounds from cross-reactions of carbonyl compounds in the atmospheric aqueous phase, Atmos. Environ., 2019, 219, 117046 CrossRef CAS.
- C. J. Kampf, A. Filippi, C. Zuth, T. Hoffmann and T. Opatz, Secondary brown carbon formation via the dicarbonyl imine pathway: nitrogen heterocycle formation and synergistic effects, Phys. Chem. Chem. Phys., 2016, 18, 18353–18364 RSC.
- P. K. Aiona, H. J. Lee, R. Leslie, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, Photochemistry of Products of the Aqueous Reaction of Methylglyoxal with Ammonium Sulfate, ACS Earth Space Chem., 2017, 1, 522–532 CrossRef CAS.
- D. O. De Haan, E. Tapavicza, M. Riva, T. Cui, J. D. Surratt, A. C. Smith, M.-C. Jordan, S. Nilakantan, M. Almodovar, T. N. Stewart, A. de Loera, A. C. De Haan, M. Cazaunau, A. Gratien, E. Pangui and J.-F. Doussin, Nitrogen-Containing, Light-Absorbing Oligomers Produced in Aerosol Particles Exposed to Methylglyoxal, Photolysis, and Cloud Cycling, Environ. Sci. Technol., 2018, 52, 4061–4071 CrossRef CAS PubMed.
- D. O. De Haan, A. Pajunoja, L. N. Hawkins, H. G. Welsh, N. G. Jimenez, A. De Loera, M. Zauscher, A. D. Andretta, B. W. Joyce, A. C. De Haan, M. Riva, T. Cui, J. D. Surratt, M. Cazaunau, P. Formenti, A. Gratien, E. Pangui and J.-F. Doussin, Methylamine's Effects on Methylglyoxal-Containing Aerosol: Chemical, Physical, and Optical Changes, ACS Earth Space Chem., 2019, 3, 1706–1716 CrossRef CAS.
- T. Beier, E. R. Cotter, M. M. Galloway and J. L. Woo, In Situ Surface Tension Measurements of Hanging Droplet Methylglyoxal/Ammonium Sulfate Aerosol Mimics under Photooxidative Conditions, ACS Earth Space Chem., 2019, 3, 1208–1215 CrossRef CAS.
- A. W. Harrison, A. M. Waterson and W. J. De Bruyn, Spectroscopic and Photochemical Properties of Secondary Brown Carbon from Aqueous Reactions of Methylglyoxal, ACS Earth Space Chem., 2020, 4, 762–773 CrossRef CAS.
- D. N. Grace, J. R. Sharp, R. E. Holappa, E. N. Lugos, M. B. Sebold, D. R. Griffith, H. P. Hendrickson and M. M. Galloway, Heterocyclic Product Formation in Aqueous Brown Carbon Systems, ACS Earth Space Chem., 2019, 3, 2472–2481 CrossRef CAS.
- Y. Li, Y. Ji, J. Zhao, Y. Wang, Q. Shi, J. Peng, Y. Wang, C. Wang, F. Zhang, Y. Wang, J. H. Seinfeld and R. Zhang, Unexpected Oligomerization of Small α-Dicarbonyls for Secondary Organic Aerosol and Brown Carbon Formation, Environ. Sci. Technol., 2021, 55, 4430–4439 CrossRef CAS PubMed.
- M. Kalberer, M. Sax and V. Samburova, Molecular Size Evolution of Oligomers in Organic Aerosols Collected in Urban Atmospheres and Generated in a Smog Chamber, Environ. Sci. Technol., 2006, 40, 5917–5922 CrossRef CAS PubMed.
- K. J. Heaton, R. L. Sleighter, P. G. Hatcher, W. A. Hall and M. V. Johnston, Composition Domains in Monoterpene Secondary Organic Aerosol, Environ. Sci. Technol., 2009, 43, 7797–7802 CrossRef CAS PubMed.
- T. B. Nguyen, J. Laskin, A. Laskin and S. A. Nizkorodov, Nitrogen-Containing Organic Compounds and Oligomers in Secondary Organic Aerosol Formed by Photooxidation of Isoprene, Environ. Sci. Technol., 2011, 45, 6908–6918 CrossRef CAS PubMed.
- S. Ishizuka, T. Fujii, A. Matsugi, Y. Sakamoto, T. Hama and S. Enami, Controlling factors of oligomerization at the water surface: Why is isoprene such a unique VOC?, Phys. Chem. Chem. Phys., 2018, 20, 15400–15410 RSC.
- S. Ishizuka, A. Matsugi, T. Hama and S. Enami, Interfacial Water Mediates Oligomerization Pathways of Monoterpene Carbocations, J. Phys. Chem. Lett., 2020, 11, 67–74 CrossRef CAS PubMed.
- Z. Yu, M. Jang, T. Zhang, A. Madhu and S. Han, Simulation of Monoterpene SOA Formation by Multiphase Reactions Using Explicit Mechanisms, ACS Earth Space Chem., 2021, 5, 1455–1467 CrossRef CAS.
- A. Bellcross, A. G. Bé, F. M. Geiger and R. J. Thomson, Molecular Chirality and Cloud Activation Potentials of Dimeric α-Pinene Oxidation Products, J. Am. Chem. Soc., 2021, 143, 16653–16662 CrossRef CAS PubMed.
- S. Ishizuka, T. Hama and S. Enami, Acid-Catalyzed Oligomerization at the Air–Water Interface Modified by Competitive Adsorption of Surfactants, J. Phys. Chem. C, 2019, 123, 21662–21669 CrossRef CAS.
- I. S. A. Isaksen, C. Granier, G. Myhre, T. K. Berntsen, S. B. Dalsøren, M. Gauss, Z. Klimont, R. Benestad, P. Bousquet, W. Collins, T. Cox, V. Eyring, D. Fowler, S. Fuzzi, P. Jöckel, P. Laj, U. Lohmann, M. Maione, P. Monks, A. S. H. Prevot, F. Raes, A. Richter, B. Rognerud, M. Schulz, D. Shindell, D. S. Stevenson, T. Storelvmo, W.-C. Wang, M. van Weele, M. Wild and D. Wuebbles, Atmospheric composition change: climate–chemistry interactions, Atmos. Environ., 2009, 43, 5138–5192 CrossRef CAS.
- J. Abbatt, C. George, M. Melamed, P. Monks, S. Pandis and Y. Rudich, New Directions: Fundamentals of atmospheric chemistry: Keeping a three-legged stool balanced, Atmos. Environ., 2014, 84, 390–391 CrossRef CAS.
- J. B. Burkholder, J. P. D. Abbatt, I. Barnes, J. M. Roberts, M. L. Melamed, M. Ammann, A. K. Bertram, C. D. Cappa, A. G. Carlton, L. J. Carpenter, J. N. Crowley, Y. Dubowski, C. George, D. E. Heard, H. Herrmann, F. N. Keutsch, J. H. Kroll, V. F. McNeill, N. L. Ng, S. A. Nizkorodov, J. J. Orlando, C. J. Percival, B. Picquet-Varrault, Y. Rudich, P. W. Seakins, J. D. Surratt, H. Tanimoto, J. A. Thornton, Z. Tong, G. S. Tyndall, A. Wahner, C. J. Weschler, K. R. Wilson and P. J. Ziemann, The Essential Role for Laboratory Studies in Atmospheric Chemistry, Environ. Sci. Technol., 2017, 51, 2519–2528 CrossRef CAS PubMed.
- P. S. Monks, A. R. Ravishankara, E. von Schneidemesser and R. Sommariva, Opinion: papers that shaped tropospheric chemistry, Atmos. Chem. Phys., 2021, 21, 12909–12948 CrossRef CAS.
- A. G. Carlton, A. E. Christiansen, M. M. Flesch, C. J. Hennigan and N. Sareen, Multiphase Atmospheric Chemistry in Liquid Water: Impacts and Controllability of Organic Aerosol, Acc. Chem. Res., 2020, 53, 1715–1723 CrossRef CAS PubMed.
- A. Tilgner, T. Schaefer, B. Alexander, M. Barth, J. L. Collett Jr, K. M. Fahey, A. Nenes, H. O. T. Pye, H. Herrmann and V. F. McNeill, Acidity and the multiphase chemistry of atmospheric aqueous particles and clouds, Atmos. Chem. Phys., 2021, 21, 13483–13536 CrossRef CAS PubMed.
- M. Cunliffe, A. Engel, S. Frka, B. Gašparović, C. Guitart, J. C. Murrell, M. Salter, C. Stolle, R. Upstill-Goddard and O. Wurl, Sea surface microlayers: a unified physicochemical and biological perspective of the air–ocean interface, Prog. Oceanogr., 2013, 109, 104–116 CrossRef.
- A. P. Ault, V. H. Grassian, N. Carslaw, D. B. Collins, H. Destaillats, D. J. Donaldson, D. K. Farmer, J. L. Jimenez, V. F. McNeill, G. C. Morrison, R. E. O'Brien, M. Shiraiwa, M. E. Vance, J. R. Wells and W. Xiong, Indoor Surface Chemistry: Developing a Molecular Picture of Reactions on Indoor Interfaces, Chem, 2020, 6, 3203–3218 CAS.
- C. Wang, D. B. Collins, C. Arata, A. H. Goldstein, J. M. Mattila, D. K. Farmer, L. Ampollini, P. F. DeCarlo, A. Novoselac, M. E. Vance, W. W. Nazaroff and J. P. D. Abbatt, Surface reservoirs dominate dynamic gas–surface partitioning of many indoor air constituents, Sci. Adv., 2020, 6, eaay8973 CrossRef CAS PubMed.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Heterogeneous Photochemistry in the Atmosphere, Chem. Rev., 2015, 115, 4218–4258 CrossRef CAS PubMed.
- C. George, M. Brüggemann, N. Hayeck, L. Tinel and J. Donaldson, in Physical Chemistry of Gas–Liquid Interfaces, ed. J. A. Faust and J. E. House, Elsevier, Amsterdam, 2018, vol. 2, pp. 435–457 Search PubMed.
- J. M. Anglada, M. T. C. Martins-Costa, J. S. Francisco and M. F. Ruiz-López, Photoinduced Oxidation Reactions at the Air–Water Interface, J. Am. Chem. Soc., 2020, 142, 16140–16155 CrossRef CAS PubMed.
- S. Rossignol, L. Tinel, A. Bianco, M. Passananti, M. Brigante, D. J. Donaldson and C. George, Atmospheric photochemistry at a fatty acid-coated air–water interface, Science, 2016, 353, 699–702 CrossRef CAS PubMed.
- H. Chen, X. Ge and Z. Ye, Aqueous-Phase Secondary Organic Aerosol Formation Via Reactions with Organic Triplet Excited States—a Short Review, Atmos. Meas. Tech., 2018, 4, 8–12 CAS.
- N. Hayeck, I. Mussa, S. Perrier and C. George, Production of Peroxy Radicals from the Photochemical Reaction of Fatty Acids at the Air–Water Interface, ACS Earth Space Chem., 2020, 4, 1247–1253 CrossRef CAS.
- L. T. Stirchak, L. Abis, C. Kalalian, C. George and D. J. Donaldson, Differences in Photosensitized Release of VOCs from Illuminated Seawater versus Freshwater Surfaces, ACS Earth Space Chem., 2021, 5, 2233–2242 CrossRef CAS.
- R. Ciuraru, L. Fine, M. van Pinxteren, B. D'Anna, H. Herrmann and C. George, Photosensitized production of functionalized and unsaturated organic compounds at the air–sea interface, Sci. Rep., 2015, 5, 12741 CrossRef CAS PubMed.
- J. V. Trueblood, M. R. Alves, D. Power, M. V. Santander, R. E. Cochran, K. A. Prather and V. H. Grassian, Shedding Light on Photosensitized Reactions within Marine-Relevant Organic Thin Films, ACS Earth Space Chem., 2019, 3, 1614–1623 CrossRef CAS.
- L. Tinel, S. Rossignol, A. Bianco, M. Passananti, S. Perrier, X. Wang, M. Brigante, D. J. Donaldson and C. George, Mechanistic Insights on the Photosensitized Chemistry of a Fatty Acid at the Air/Water Interface, Environ. Sci. Technol., 2016, 50, 11041–11048 CrossRef CAS PubMed.
- R. J. Rapf, R. J. Perkins, M. R. Dooley, J. A. Kroll, B. K. Carpenter and V. Vaida, Environmental Processing of Lipids Driven by Aqueous Photochemistry of α-Keto Acids, ACS Cent. Sci., 2018, 4, 624–630 CrossRef CAS PubMed.
- C. Gong, Y. Zhao, D. Zhang, J. Wang, C. Mu, W. Wang, S. Zhu and X. Zhang, Investigation of the Acid-Mediated Photosensitized Reactions of Amphiphilic α-Keto Acids at the Air–Water Interface Using Field-Induced Droplet Ionization Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2021, 32, 2306–2312 CrossRef CAS PubMed.
- D. Huang, J. Wang, H. Xia, Y. Zhang, F. Bao, M. Li, C. Chen and J. Zhao, Enhanced Photochemical Volatile Organic Compounds Release from Fatty Acids by Surface-Enriched Fe(III), Environ. Sci. Technol., 2020, 54, 13448–13457 CrossRef CAS PubMed.
- F. Bernard, R. Ciuraru, A. Boréave and C. George, Photosensitized Formation of Secondary Organic Aerosols above the Air/Water Interface, Environ. Sci. Technol., 2016, 50, 8678–8686 CrossRef CAS PubMed.
- P. A. Alpert, R. Ciuraru, S. Rossignol, M. Passananti, L. Tinel, S. Perrier, Y. Dupart, S. S. Steimer, M. Ammann, D. J. Donaldson and C. George, Fatty Acid Surfactant Photochemistry Results in New Particle Formation, Sci. Rep., 2017, 7, 12693 CrossRef PubMed.
- M. Kulmala and V.-M. Kerminen, On the formation and growth of atmospheric nanoparticles, Atmos. Res., 2008, 90, 132–150 CrossRef CAS.
- M. Shrestha, M. Luo, Y. Li, B. Xiang, W. Xiong and V. H. Grassian, Let there be light: stability of palmitic acid monolayers at the air/salt water interface in the presence and absence of simulated solar light and a photosensitizer, Chem. Sci., 2018, 9, 5716–5723 RSC.
- E. A. Parmentier, G. David, P. C. Arroyo, S. Bibawi, K. Esat and R. Signorell, Photochemistry of single optically trapped oleic acid droplets, J. Aerosol Sci., 2021, 151, 105660 CrossRef CAS.
- J. Lin, Q. Dai, H. Zhao, H. Cao, T. Wang, G. Wang and C. Chen, Photoinduced Release of Volatile Organic Compounds from Fatty Alcohols at the Air–Water Interface: The Role of Singlet Oxygen Photosensitized by a Carbonyl Group, Environ. Sci. Technol., 2021, 55, 8683–8690 CrossRef CAS PubMed.
- H. Fu, R. Ciuraru, Y. Dupart, M. Passananti, L. Tinel, S. Rossignol, S. Perrier, D. J. Donaldson, J. Chen and C. George, Photosensitized Production of Atmospherically Reactive Organic Compounds at the Air/Aqueous Interface, J. Am. Chem. Soc., 2015, 137, 8348–8351 CrossRef CAS PubMed.
- B. You, S. Li, N. T. Tsona, J. Li, L. Xu, Z. Yang, S. Cheng, Q. Chen, C. George, M. Ge and L. Du, Environmental Processing of Short-Chain Fatty Alcohols Induced by Photosensitized Chemistry of Brown Carbons, ACS Earth Space Chem., 2020, 4, 631–640 CrossRef CAS.
- S. Li, X. Jiang, M. Roveretto, C. George, L. Liu, W. Jiang, Q. Zhang, W. Wang, M. Ge and L. Du, Photochemical aging of atmospherically reactive organic compounds involving brown carbon at the air–aqueous interface, Atmos. Chem. Phys., 2019, 19, 9887–9902 CrossRef CAS.
- J. R. Collins, H. F. Fredricks, J. S. Bowman, C. P. Ward, C. Moreno, K. Longnecker, A. Marchetti, C. M. Hansel, H. W. Ducklow and B. A. S. Van Mooy, The molecular products and biogeochemical significance of lipid photooxidation in West Antarctic surface waters, Geochim. Cosmochim. Acta, 2018, 232, 244–264 CrossRef CAS.
- C. Baduel, B. Nozière and J.-L. Jaffrezo, Summer/winter variability of the surfactants in aerosols from Grenoble, France, Atmos. Environ., 2012, 47, 413–420 CrossRef CAS.
- V. Gérard, B. Nozière, C. Baduel, L. Fine, A. A. Frossard and R. C. Cohen, Anionic, Cationic, and Nonionic Surfactants in Atmospheric Aerosols from the Baltic Coast at Askö, Sweden: Implications for Cloud Droplet Activation, Environ. Sci. Technol., 2016, 50, 2974–2982 CrossRef PubMed.
- B. Nozière, V. Gérard, C. Baduel and C. Ferronato, Extraction and Characterization of Surfactants from Atmospheric Aerosols, J. Visualized Exp., 2017, 122, e55622 Search PubMed.
- J. Sukhapan and P. Brimblecombe, Ionic Surface Active Compounds in Atmospheric Aerosols, Sci. World J., 2002, 2, 1138–1146 CrossRef CAS PubMed.
- V. Gérard, B. Nozière, L. Fine, C. Ferronato, D. K. Singh, A. A. Frossard, R. C. Cohen, E. Asmi, H. Lihavainen, N. Kivekäs, M. Aurela, D. Brus, S. Frka and A. Cvitešić Kušan, Concentrations and Adsorption Isotherms for Amphiphilic Surfactants in PM1 Aerosols from Different Regions of Europe, Environ. Sci. Technol., 2019, 53, 12379–12388 CrossRef PubMed.
- T. C. Burdette and A. A. Frossard, Characterization of seawater and aerosol particle surfactants using solid phase extraction and mass spectrometry, J. Environ. Sci., 2021, 108, 164–174 CrossRef CAS PubMed.
- X. Zeng, S. Kong, Q. Zhang, H. Ren, J. Liu, Y. Feng, Q. Yan, S. Qin, S. Zheng, L. Yao, Z. Fan, Y. Zhang, X. Liu, Y. Yan, K. Zhu, F. Ding, W. Liu, D. Liu, S. Qi and P. Fu, Source profiles and emission factors of organic and inorganic species in fine particles emitted from the ultra-low emission power plant and typical industries, Sci. Total Environ., 2021, 789, 147966 CrossRef CAS PubMed.
- X. Zhao, Q. Hu, X. Wang, X. Ding, Q. He, Z. Zhang, R. Shen, S. Lü, T. Liu, X. Fu and L. Chen, Composition profiles of organic aerosols from Chinese residential cooking: case study in urban Guangzhou, south China, J. Atmos. Chem., 2015, 72, 1–18 CrossRef CAS.
- Y. Miyazaki, D. Gowda, E. Tachibana, Y. Takahashi and T. Hiura, Identification of secondary fatty alcohols in atmospheric aerosols in temperate forests, Biogeosciences, 2019, 16, 2181–2188 CrossRef CAS.
- M. Kang, F. Yang, H. Ren, W. Zhao, Y. Zhao, L. Li, Y. Yan, Y. Zhang, S. Lai, Y. Zhang, Y. Yang, Z. Wang, Y. Sun and P. Fu, Influence of continental organic aerosols to the marine atmosphere over the East China Sea: Insights from lipids, PAHs and phthalates, Sci. Total Environ., 2017, 607–608, 339–350 CrossRef CAS PubMed.
- C. Leck, Q. Gao, F. Mashayekhy Rad and U. Nilsson, Size-resolved atmospheric particulate polysaccharides in the high summer Arctic, Atmos. Chem. Phys., 2013, 13, 12573–12588 CrossRef.
- S. Frka, J. Dautović, Z. Kozarac, B. Ćosović, A. Hoffer and G. Kiss, Surface-active substances in atmospheric aerosol: an electrochemical approach, Tellus B, 2012, 64, 18490 CrossRef CAS.
- R. M. Kirpes, D. Bonanno, N. W. May, M. Fraund, A. J. Barget, R. C. Moffet, A. P. Ault and K. A. Pratt, Wintertime Arctic Sea Spray Aerosol Composition Controlled by Sea Ice Lead Microbiology, ACS Cent. Sci., 2019, 5, 1760–1767 CrossRef CAS PubMed.
- B. Wang, Y. Yao, Y. Wang, H. Chen and H. Sun, Per- and Polyfluoroalkyl Substances in Outdoor and Indoor Dust from Mainland China: Contributions of Unknown Precursors and Implications for Human Exposure, Environ. Sci. Technol., 2022, 56, 6036–6045 CrossRef CAS PubMed.
- H. D. Lee, A. D. Estillore, H. S. Morris, K. K. Ray, A. Alejandro, V. H. Grassian and A. V. Tivanski, Direct Surface Tension Measurements of Individual Sub-Micrometer Particles Using Atomic Force Microscopy, J. Phys. Chem. A, 2017, 121, 8296–8305 CrossRef CAS PubMed.
- K. Adachi, N. Oshima, S. Ohata, A. Yoshida, N. Moteki and M. Koike, Compositions and mixing states of aerosol particles by aircraft observations in the Arctic springtime, Atmos. Chem. Phys., 2021, 21, 3607–3626 CrossRef CAS.
- R. B. Gagosian, E. T. Peltzer and J. T. Merrill, Long-range transport of terrestrially derived lipids in aerosols from the south Pacific, Nature, 1987, 325, 800–803 CrossRef CAS.
- N. I. H. Mustaffa, M. T. Latif, M. M. Ali and M. F. Khan, Source apportionment of surfactants in marine aerosols at different locations along the Malacca Straits, Environ. Sci. Pollut. Res., 2014, 21, 6590–6602 CrossRef CAS PubMed.
- S. Shaharom, M. T. Latif, M. F. Khan, S. N. M. Yusof, N. A. Sulong, N. B. A. Wahid, R. Uning and S. Suratman, Surfactants in the sea surface microlayer, subsurface water and fine marine aerosols in different background coastal areas, Environ. Sci. Pollut. Res., 2018, 25, 27074–27089 CrossRef CAS PubMed.
- E. Van Acker, M. De Rijcke, Z. Liu, J. Asselman, K. A. C. De Schamphelaere, L. Vanhaecke and C. R. Janssen, Sea Spray Aerosols Contain the Major Component of Human Lung Surfactant, Environ. Sci. Technol., 2021, 55, 15989–16000 CrossRef CAS PubMed.
- S. Zeppenfeld, M. van Pinxteren, D. van Pinxteren, H. Wex, E. Berdalet, D. Vaqué, M. Dall'Osto and H. Herrmann, Aerosol Marine Primary Carbohydrates and Atmospheric Transformation in the Western Antarctic Peninsula, ACS Earth Space Chem., 2021, 5, 1032–1047 CrossRef CAS.
- S. Ekström, B. Nozière, M. Hultberg, T. Alsberg, J. Magnér, E. D. Nilsson and P. Artaxo, A possible role of ground-based microorganisms on cloud formation in the atmosphere, Biogeosciences, 2010, 7, 387–394 CrossRef.
- J. M. Schiffer, L. E. Mael, K. A. Prather, R. E. Amaro and V. H. Grassian, Sea Spray Aerosol: Where Marine Biology Meets Atmospheric Chemistry, ACS Cent. Sci., 2018, 4, 1617–1623 CrossRef CAS PubMed.
- M. Karl, C. Leck, F. Mashayekhy Rad, A. Bäcklund, S. Lopez-Aparicio and J. Heintzenberg, New insights in sources of the sub-micrometre aerosol at Mt. Zeppelin observatory (Spitsbergen) in the year 2015, Tellus B, 2019, 71, 1613143 CrossRef.
- H. Yu, W. Li, Y. Zhang, P. Tunved, M. Dall'Osto, X. Shen, J. Sun, X. Zhang, J. Zhang and Z. Shi, Organic coating on sulfate and soot particles during late summer in the Svalbard Archipelago, Atmos. Chem. Phys., 2019, 19, 10433–10446 CrossRef CAS.
- L. Liu, L. Du, L. Xu, J. Li and N. T. Tsona, Molecular size of surfactants affects their degree of enrichment in the sea spray aerosol formation, Environ. Res., 2022, 206, 112555 CrossRef CAS PubMed.
- J. Y. Aller, J. C. Radway, W. P. Kilthau, D. W. Bothe, T. W. Wilson, R. D. Vaillancourt, P. K. Quinn, D. J. Coffman, B. J. Murray and D. A. Knopf, Size-resolved characterization of the polysaccharidic and proteinaceous components of sea spray aerosol, Atmos. Environ., 2017, 154, 331–347 CrossRef CAS.
- M. D. Stokes, G. B. Deane, K. Prather, T. H. Bertram, M. J. Ruppel, O. S. Ryder, J. M. Brady and D. Zhao, A Marine Aerosol Reference Tank system as a breaking wave analogue for the production of foam and sea-spray aerosols, Atmos. Meas. Tech., 2013, 6, 1085–1094 CrossRef.
- E. S. Hasenecz, T. Jayarathne, M. A. Pendergraft, M. V. Santander, K. J. Mayer, J. Sauer, C. Lee, W. S. Gibson, S. M. Kruse, F. Malfatti, K. A. Prather and E. A. Stone, Marine Bacteria Affect Saccharide Enrichment in Sea Spray Aerosol during a Phytoplankton Bloom, ACS Earth Space Chem., 2020, 4, 1638–1649 CrossRef CAS.
- E. Rastelli, C. Corinaldesi, A. Dell'Anno, M. Lo Martire, S. Greco, M. Cristina Facchini, M. Rinaldi, C. O'Dowd, D. Ceburnis and R. Danovaro, Transfer of labile organic matter and microbes from the ocean surface to the marine aerosol: an experimental approach, Sci. Rep., 2017, 7, 11475 CrossRef PubMed.
- M. C. Facchini, M. Rinaldi, S. Decesari, C. Carbone, E. Finessi, M. Mircea, S. Fuzzi, D. Ceburnis, R. Flanagan, E. D. Nilsson, G. de Leeuw, M. Martino, J. Woeltjen and C. D. O'Dowd, Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates, Geophys. Res. Lett., 2008, 35, L17814 CrossRef.
- R. E. Cochran, T. Jayarathne, E. A. Stone and V. H. Grassian, Selectivity Across the Interface: A Test of Surface Activity in the Composition of Organic-Enriched Aerosols from Bubble Bursting, J. Phys. Chem. Lett., 2016, 7, 1692–1696 CrossRef CAS PubMed.
- A. A. Frossard, V. Gérard, P. Duplessis, J. D. Kinsey, X. Lu, Y. Zhu, J. Bisgrove, J. R. Maben, M. S. Long, R. Y.-W. Chang, S. R. Beaupré, D. J. Kieber, W. C. Keene, B. Nozière and R. C. Cohen, Properties of Seawater Surfactants Associated with Primary Marine Aerosol Particles Produced by Bursting Bubbles at a Model Air–Sea Interface, Environ. Sci. Technol., 2019, 53, 9407–9417 CrossRef CAS PubMed.
- K. J. Mayer, J. S. Sauer, J. Dinasquet and K. A. Prather, CAICE Studies: Insights from a Decade of Ocean–Atmosphere Experiments in the Laboratory, Acc. Chem. Res., 2020, 53, 2510–2520 CrossRef CAS PubMed.
- J. S. Sauer, K. J. Mayer, C. Lee, M. R. Alves, S. Amiri, C. J. Bahaveolos, E. B. Franklin, D. R. Crocker, D. Dang, J. Dinasquet, L. A. Garofalo, C. P. Kaluarachchi, D. B. Kilgour, L. E. Mael, B. A. Mitts, D. R. Moon, A. N. Moore, C. K. Morris, C. A. Mullenmeister, C.-M. Ni, M. A. Pendergraft, D. Petras, R. M. C. Simpson, S. Smith, P. R. Tumminello, J. L. Walker, P. J. DeMott, D. K. Farmer, A. H. Goldstein, V. H. Grassian, J. S. Jaffe, F. Malfatti, T. R. Martz, J. H. Slade, A. V. Tivanski, T. H. Bertram, C. D. Cappa and K. A. Prather, The Sea Spray Chemistry and Particle Evolution study (SeaSCAPE): overview and experimental methods, Environ. Sci.: Processes Impacts, 2022, 24, 290–315 RSC.
- A. P. Ault, D. Zhao, C. J. Ebben, M. J. Tauber, F. M. Geiger, K. A. Prather and V. H. Grassian, Raman microspectroscopy and vibrational sum frequency generation spectroscopy as probes of the bulk and surface compositions of size-resolved sea spray aerosol particles, Phys. Chem. Chem. Phys., 2013, 15, 6206–6214 RSC.
- R. Pérez Pastor, P. Salvador, S. García Alonso, A. Alastuey, S. García dos Santos, X. Querol and B. Artíñano, Characterization of organic aerosol at a rural site influenced by olive waste biomass burning, Chemosphere, 2020, 248, 125896 CrossRef PubMed.
- M. T. Latif and P. Brimblecombe, Surfactants in Atmospheric Aerosols, Environ. Sci. Technol., 2004, 38, 6501–6506 CrossRef CAS PubMed.
- M. Takhar, C. A. Stroud and A. W. H. Chan, Volatility Distribution and Evaporation Rates of Organic Aerosol from Cooking Oils and their Evolution upon Heterogeneous Oxidation, ACS Earth Space Chem., 2019, 3, 1717–1728 CrossRef CAS.
- W. F. Rogge, L. M. Hildemann, M. A. Mazurek, G. R. Cass and B. R. T. Simoneit, Sources of fine organic aerosol. 1. Charbroilers and meat cooking operations, Environ. Sci. Technol., 1991, 25, 1112–1125 CrossRef CAS.
- Q. Wang, X. He, M. Zhou, D. D. Huang, L. Qiao, S. Zhu, Y. Ma, H. Wang, L. Li, C. Huang, X. H. H. Huang, W. Xu, D. Worsnop, A. H. Goldstein, H. Guo and J. Z. Yu, Hourly Measurements of Organic Molecular Markers in Urban Shanghai, China: Primary Organic Aerosol Source Identification and Observation of Cooking Aerosol Aging, ACS Earth Space Chem., 2020, 4, 1670–1685 CrossRef CAS.
- C. D. McClure, C. Y. Lim, D. H. Hagan, J. H. Kroll and C. D. Cappa, Biomass-burning-derived particles from a wide variety of fuels – Part 1: Properties of primary particles, Atmos. Chem. Phys., 2020, 20, 1531–1547 CrossRef CAS.
- I. Ciglenečki, P. Orlović-Leko, K. Vidović and V. Tasić, The possible role of the surface active substances (SAS) in the airborne transmission of SARS-CoV-2, Environ. Res., 2021, 198, 111215 CrossRef PubMed.
- S. Fuzzi, U. Baltensperger, K. Carslaw, S. Decesari, H. Denier van der Gon, M. C. Facchini, D. Fowler, I. Koren, B. Langford, U. Lohmann, E. Nemitz, S. Pandis, I. Riipinen, Y. Rudich, M. Schaap, J. G. Slowik, D. V. Spracklen, E. Vignati, M. Wild, M. Williams and S. Gilardoni, Particulate matter, air quality and climate: Lessons learned and future needs, Atmos. Chem. Phys., 2015, 15, 8217–8299 CrossRef CAS.
Footnote |
| † Current affiliation: Dept. of Chemistry, University of Wisconsin–Madison, Madison WI, USA. |
| This journal is © The Royal Society of Chemistry 2022 |