
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomoleptic phenoxy-imine pyridine zinc complexes: efficient catalysts for solvent free synthesis and chemical degradation of polyesters†
Sara
D'Aniello
a,
Sidonie
Laviéville
b,
Federica
Santulli
a,
Malaury
Simon
b,
Michele
Sellitto
a,
Consiglia
Tedesco
 a,
Christophe M.
Thomas
*b and
Mina
Mazzeo
*a
a,
Christophe M.
Thomas
*b and
Mina
Mazzeo
*a
aDipartimento di Chimica e Biologia “Adolfo Zambelli”, Università di Salerno, via Giovanni Paolo II, 132, 84084 Fisciano, SA, Italy. E-mail: mmazzeo@unisa.it
bChimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France. E-mail: christophe.thomas@chimieparistech.psl.eu
First published on 29th August 2022
Abstract
The synthesis and structural characterization of six homoleptic zinc complexes supported by monoanionic phenoxy-imine pyridine ligands bearing different substituents on the phenoxy moiety are described. All complexes have been tested as catalysts for the ring-opening polymerization of L-lactide under mild or industrially relevant conditions using technical grade L-lactide and excess alcohol. Interestingly, the Zn(II) catalysts were stable under severe conditions, showing activities comparable to those of industrial catalysts. The same complexes were also revealed to be active in the depolymerization by alcoholysis of polylactide and poly(ethylene terephthalate). For both processes, a structure–reactivity relationship has been found, depending on the substituents introduced on the ligand backbone. Tests for the catalyst recycling in solvent-free alcoholysis were performed, offering new opportunities for the implementation of a circular (bio)plastics economy through procedures of low environmental impact.
Introduction
Synthetic plastics derived from fossil resources are highly versatile materials that find numerous uses in all industrial productions, from everyday packaging and textiles to more specialized applications in biomedicine and electronics.1The extreme longevity of traditional oil-derived polymers and the lack of efficient recycling policies have led to an excessive accumulation of plastics in the environment.2,3 Currently, about 70% of all plastics produced follow a linear economy. If current production and waste management trends continue, it is estimated that by 2050, approximately 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mt of plastic waste will be massively landfilled or released into the environment,1 with dramatic consequences in terms of environmental pollution. Therefore, the transition from a linear to a circular economy for plastics is mandatory.4,5
000 Mt of plastic waste will be massively landfilled or released into the environment,1 with dramatic consequences in terms of environmental pollution. Therefore, the transition from a linear to a circular economy for plastics is mandatory.4,5
At the same time, the progressive depletion of fossil resources and the current energy crisis, with the resulting variability of their costs, are important issues that must be addressed.
For global problems of such complexity, there is no unique solution, but various strategies may be adopted that consider both the massive market introduction of biodegradable plastics, preferably from renewable resources,6–9 as well as the implementation of efficient procedures for the recycling of traditional plastics10,11 and new materials.4,10,12–16
A polymer that fits well with the demands of a circular economic model is polylactide (PLA).17 It is a biodegradable and compostable polymer produced from annually renewable resources. However, despite its potential sustainability, there are still some criticisms related to its life cycle.
Regarding PLA production, it is obtained industrially by ring-opening polymerization (ROP) of lactide, promoted by tin catalysts that are highly toxic.18 Thus, the development of efficient, easily synthesized, low-cost and highly robust catalytic systems is of crucial importance.19–22 In this context, zinc23 and magnesium24–26 complexes represent potent catalysts to promote the ring-opening polymerization of lactide with high efficiency and excellent control.27,28
In terms of end-of-life options, PLA waste is currently being collected for composting, although high temperature and moisture content are required. Since the recovery of lactic acid from PLA waste is more energetically favorable than the production by fermentation of glucose from biomass,29 chemical depolymerization is emerging as a powerful strategy for recycling PLA.30–32 The degradation by hydrolysis or alcoholysis produces lactic acid or alkyl lactates as final products, the former being a chemical platform or starting monomer for PLA production and the latters having uses as green solvents in agriculture, polymers, and pharmaceuticals.33
Among traditional polyesters, poly(ethylene terephthalate) (PET) is a high consumption material and its recycling is therefore crucial from an economic and environmental point of view.34,35 The chemical depolymerization of PET has been studied with a variety of metal salts and organic catalysts.36 Depending on the method used, PET can be converted into different products that can be used for the synthesis of virgin PET or other value-added chemicals. However, the drastic conditions used do not offer the opportunity of a selective degradation of polymeric mixtures. In this context, discrete homogeneous metal complexes with defined structures may offer great opportunities but they are still poorly investigated as catalysts.37–41
Recently, several homoleptic zinc complexes stabilized by monoanionic (NNO) ligands have been reported by Jones as efficient catalysts for the synthesis and alcoholysis of PLA.42–44 For these systems the neutral amine of the ligands seems to have a crucial role in assisting the depolymerization via H bond activation of the alcohol.
In this context, we developed a phenoxy-imine ligand bearing an additional pyridine moiety, a weaker base than an amine, which can therefore behave as a hemilabile donor to the metal. We recently described both homoleptic and heteroleptic complexes of zinc and magnesium stabilized by this ligand.45,46 These studies put in evidence the potentiality of this class of ligands for the synthesis of homoleptic zinc complexes as catalysts for the PLA synthesis and alcoholysis.
In this work, we describe homoleptic zinc(II) complexes bearing phenoxy-imine pyridine ligands with different substituents that were tested as catalysts for the synthesis of PLA, under mild or industrial conditions, and in the degradation of PLA and PET by alcoholysis.
Their efficiency for LA polymerization and degradation processes was studied under different reaction conditions and related to steric and electronic characteristics of the ancillary ligands.
Results and discussion
Synthesis of complexes
A series of phenoxy-imine pyridine proligands bearing different substituents on the phenolate rings were obtained via simple condensation reactions (Scheme 1) in yields always above 77%. Diagnostic resonances in 1H NMR spectra were the singlet at δ = 8 ppm for the imine proton, the singlet at 14 ppm for the phenolic proton and the doublet at ca. 8.5 ppm for the proton adjacent to the pyridine nitrogen.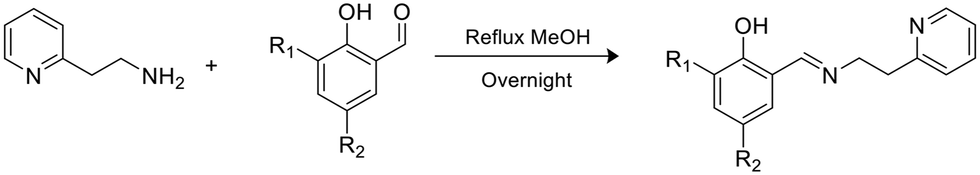 | ||
| Scheme 1 Synthesis of the proligands. | ||
Homoleptic zinc complexes were obtained by direct reaction between two equivalents of the proligands with ZnEt2, in anhydrous toluene or benzene, at room temperature (Scheme 2).
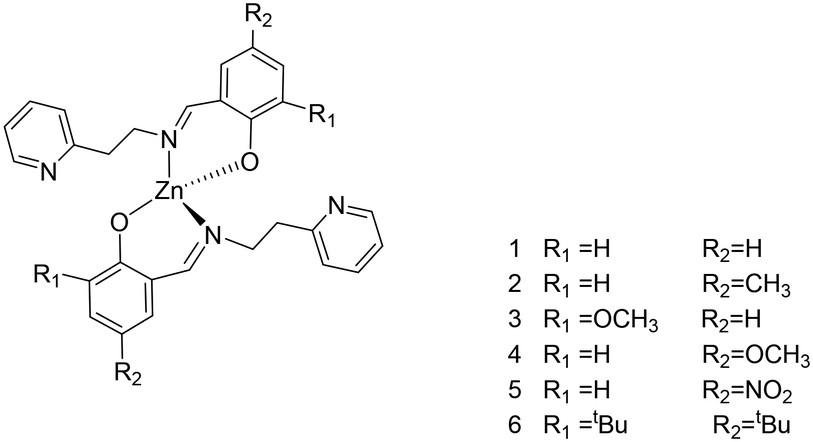 | ||
| Scheme 2 Structures of the homoleptic Zn(II) complexes 1–6. | ||
The products were purified via crystallization from a benzene/pentane solution or by washing with hexane. All complexes were characterized by elemental analysis and 1H and 13C NMR spectroscopy.
Complexes 1, 2 and 4 were characterized by single crystal X-ray diffraction. Suitable single crystals were obtained by vapor diffusion of pentane in benzene solutions of the compounds at room temperature. The X-ray molecular structures are shown in Fig. 1, selected bond distances and angles are listed in Table 1, the crystal data and refinement details are reported in Table S1.† In the solid state, the complexes are monomeric and feature a four-coordinated zinc center in a distorted tetrahedral geometry.
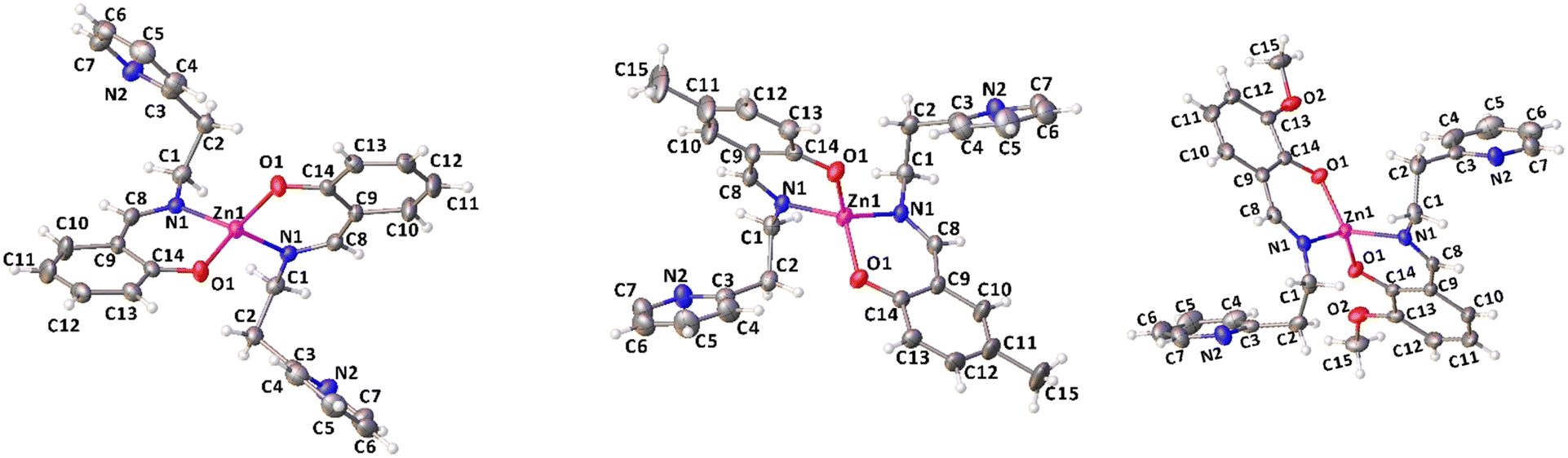 | ||
| Fig. 1 X-ray molecular structures of complexes 1, 2 and 4 (ORTEP). Ellipsoids were drawn at 20% probability level. | ||
| 1 | 2 | 4 | |
|---|---|---|---|
| Zn1–O1 | 1.9121(15) | 1.914(2) | 1.9073(15) |
| Zn1–N1 | 1.9914(16) | 1.992(3) | 1.9992(18) |
| C8–C9 | 1.440(3) | 1.444(5) | 1.435(4) |
| C9–C14 | 1.413(3) | 1.412(5) | 1.419(3) |
| O1–C14 | 1.309(2) | 1.303(4) | 1.302(2) |
| N1–C1 | 1.470(3) | 1.464(4) | 1.468(3) |
| N1–C8 | 1.282(3) | 1.273(4) | 1.277(3) |
| O1–Zn1–N1 | 97.11(6) | 97.55(10) | 96.76(7) |
| O1–Zn1–O1* | 106.99(10) | 107.84(16) | 112.75(9) |
| N1–Zn1–N1* | 110.01(9) | 111.53(15) | 102.84(10) |
| O1–Zn1–N1* | 123.96(7) | 122.09(11) | 124.96(8) |
| O1*–Zn1–N1* | 97.11(6) | 97.55(10) | 96.75(7) |
| O1*–Zn1–N1 | 123.96(7) | 122.09(11) | 124.96(8) |
The Zn atom lies on a crystallographic two-fold axis, with two bidentate ligands almost perpendicular to each other and one pendant pyridine group uncoordinated to the metal center. The τ4 values, calculated using Houser's method, are close to 1 for all the complexes investigated, regardless of the substituents on the phenolate groups.47 The dihedral angle between the mean planes of the ligands is 72.4° in 1, 74.6° in 2, and 73.7° in 4. The mean plane is defined for each ligand by considering N1, C8, C9, C14 and O1 atoms.
As shown in Fig. 2 and S19–S22 in the ESI,† all compounds assemble in the solid state in a similar way through CH⋯O and CH⋯N hydrogen bonds (in 1 the distances are C5H5⋯N2 2.432 Å and C1H1b⋯O1 2.634 Å, C5–H5⋯N2 166.2° and C1–H1b⋯O1 148.5°) forming an infinite chain of molecules along the shortest axis (i.e., the b axis). CH–π and π–π interactions account for the assembly along the other directions. The addition of a protruding methyl group in complex 2 determines a less compact assembly and allows the formation of a cavity where one (disordered) benzene molecule is located (Fig. S19–S21†).
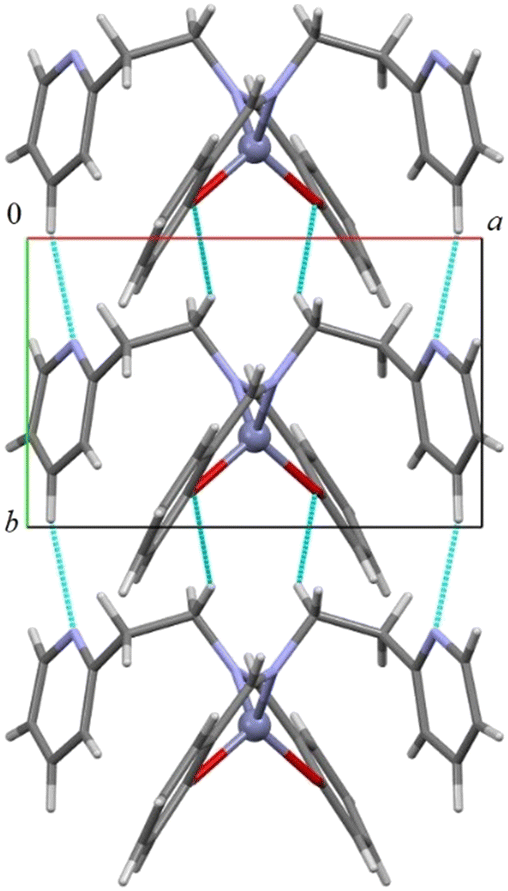 | ||
| Fig. 2 Columnar assembly along the b-axis in compound 1. CH⋯O and CH⋯N short distances are depicted in light blue. | ||
The 1H NMR spectra, measured at 298 K, revealed symmetric structures in solution for all complexes, as observed in the solid state. Resonances related to protons adjacent to the nitrogen of the pyridyl fragment showed chemical shifts comparable to those of the free ligands (8.40–8.45 ppm) suggesting structures in which the pyridyl groups were not coordinated.48,49
All complexes, in the solid form, were found to be very stable in air, with no degradation observed after several days at room temperature.50 To further verify the feasibility of using these complexes for catalytic processes at high temperatures, we studied their thermal stability at 180 °C by an isothermal thermal gravimetric analysis (TGA) for 18 h in the presence of air (Table S2 and Fig. S23†). At this temperature, the weight loss of the complexes was negligible (less than 5 wt%) while decomposition was observed after 40 min at 250 °C. Subsequently, new tests of stability were performed by heating the solid complexes at 150 °C in air for 15 hours. Then, the solids were analyzed by 1H NMR spectroscopy. The spectra resulted equivalent to those of the initial complexes (see Fig. S24† for complex 2). Weak resonances were observed as degradation products (quantified as 7%) corresponding to the ancillary ligand.
The excellent thermal stability of complexes 1–6 suggests their potential application as industrial catalysts for polymerization of lactide.
Polymerization studies
All zinc complexes were tested as catalysts for the polymerization of lactide (L-LA) both in solution and under industrial conditions, namely in the absence of solvent and at elevated temperatures (Tables 2 and 3, respectively).| Entrya | Cat | Conv (%) | TOFb (h−1) | M nGPC (103) | Đ | M thn (103) |
|---|---|---|---|---|---|---|
| a All reactions were carried out by using 10 μmol of Zn catalyst and BnOH, and 1 mmol of L-LA at 80 °C in toluene solution (1 mL); reaction time 20 min. b TOF values were evaluated by NMR after 2 min (Table S3†). c Experimental Mn and Đ values were determined by GPC analysis in THF using polystyrene standards and were corrected by the factor 0.58. d M thn = [LA]/[Zn] × conv × 144. e Bimodal distribution, only major peak is reported. | ||||||
| 1 | 1 | 94 | 1470 | 4.4 | 1.6 | 13.5 |
| 2 | 2 | >99 | 1530 | 4.6 | 1.8 | 14.4 |
| 3 | 3 | 83 | 815 | 3.9 | 1.6e | 12.0 |
| 4 | 4 | >99 | 1950 | 4.1 | 1.7 | 14.4 |
| 5 | 5 | 84 | 1380 | 3.9 | 1.7e | 12.1 |
| 6 | 6 | 85 | 1255 | 4.2 | 1.6 | 12.2 |
| Cat | T (°C) | k app (10−2 min−1) | R 2 |
|---|---|---|---|
|
a Reaction conditions: [Zn] = 5 μmol; [L-LA]:[Zn]:[BnOH] = 100:1:1; 0.5 mL of CDCl3.
|
|||
| 1 | 25 | 0.38 | 0.973 |
| 1 | 50 | 1.09 | 0.998 |
| 2 | 50 | 1.71 | 0.996 |
| 3 | 50 | 0.34 | 0.995 |
| 4 | 50 | 29.95 | 0.997 |
| 5 | 50 | 4.33 | 0.995 |
| 6 | 50 | 0.65 | 0.994 |
Initially, the polymerization experiments were performed at 80 °C in toluene solution, using lactide purified only by crystallization from toluene, and one equivalent of benzyl alcohol as a co-initiator (Table 2).
A clear reactivity scale was observed for which the most active catalysts were complexes 1, 2 and 4 in which electron donating groups were present on the para positions of the phenoxy rings, while a slight decrease in reactivity was observed when an electron withdrawing group was introduced (complex 5).
Steric effects seem to be more significant than electronic ones: indeed, complexes 3 and 4, bearing the same methoxy substituent in different positions of the phenolate ring (ortho and para, respectively), showed very different activities, highlighting the importance of structure–activity relationships for this class of catalysts (entries 3 and 4, Table 2).
Kinetic studies for complexes 1–6 in CDCl3 at 50 °C confirmed this activity trend (Table 3 and Fig. 3 and S25†). The little differences observed can be attributed to the diverse polarity of solvent. Plots of ln([LA]0/[LA]t) versus time showed linear relationships in agreement with a pseudo-first order reaction with respect to monomer consumption.
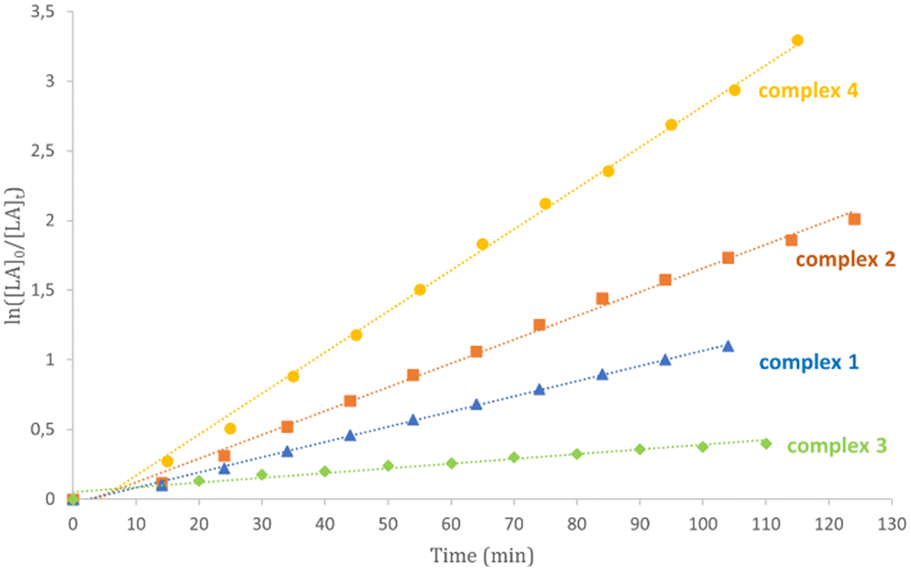 | ||
| Fig. 3 Kinetic plots for ROP of L-LA promoted by Zn catalysts depicting reaction orders of unity with respect to the monomer concentration. Reaction conditions: Zn = 5 μmol; [L-LA]:[Zn]:[BnOH] = 100:1:1; T = 50 °C; 0.5 mL of CDCl3. | ||
As expected, the catalytic activity reduced with decreasing temperature. An activated-monomer mechanism was demonstrated for the ROP of LA promoted by this class of complexes in which the pendant pyridine groups of the ancillary ligands could have an active role during the polymerization reactions activating the hydroxyl functionality of the growing chain (Scheme 3) for the nucleophilic attack to the monomer activated by coordination at the zinc centre.46
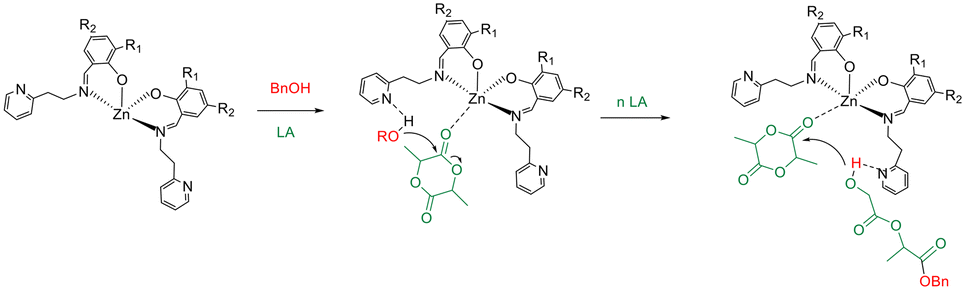 | ||
| Scheme 3 Possible mechanism of polymerization of lactide with complexes 1–6. | ||
After the polymerization runs, the catalysts preserve their structure as evidenced by 1H NMR analysis of the polymerization medium (Fig. S26†).
Subsequently, complexes 1–6 were tested in the ROP of L-LA under industrially relevant melt conditions at 180 °C using technical grade purity of the monomer and a high concentration of alcohol as a chain transfer agent to minimize the amount of catalyst used (Table 4). Under these conditions, all zinc complexes were active and, for most of them, quantitative monomer conversion was achieved. In contrast to what was observed at 50 °C, the most active catalysts were complexes 5 and 6. It is not straightforward to rationalize the reason for this inversion, but it could be a consequence of a different stability of these complexes.
| Entry | Cat | Time (h) | Conv (%) | TOFb (h−1) | M nGPC (103) | M thn (103) | Đ |
|---|---|---|---|---|---|---|---|
|
a All reactions were carried out by using 10 μmol of Zn and technical grade monomer [L-LA]:[cat]:[BnOH] 5000:1:50 and at 180 °C, reaction times were not optimized.
b TOF were evaluated after 20 min see Table S4.†
c Experimental Mn and Đ values were determined by GPC analysis in THF using polystyrene standards standards and were corrected by the factor 0.58.
d
M
thn = [LA]/[BnOH] × conv × 144.
|
|||||||
| 1 | 1 | 3 | >99 | 6.00 × 103 | 1.2 | 14.4 | 1.6 |
| 2 | 2 | 3 | >99 | 5.40 × 103 | 0.8 | 14.4 | 1.4 |
| 3 | 3 | 18 | >99 | 7.51 × 102 | 1.3 | 14.4 | 1.7 |
| 4 | 4 | 8 | >99 | 2.25 × 103 | 0.9 | 14.4 | 2.2 |
| 5 | 5 | 2 | >99 | 9.00 × 103 | 6.2 | 14.4 | 1.4 |
| 6 | 6 | 0.3 | 67 | 1.00 × 104 | 2.2 | 9.6 | 1.5 |
The molecular masses of the PLA samples were much lower than the theoretical values showing rather broad distributions, a consequence of the presence of protic impurities in the monomer that act as chain transfer agents and/or of extensive transesterification phenomena. On the other hand, when the polymerization experiment was performed by using lactide purified by standard procedures (crystallization from toluene and sublimation), and for not quantitative conversions, an efficient control of molecular masses and of nature of chain end groups was achieved as reported in the a previous study.46
In this case the MALDI-ToF spectra of low molecular weight polymer samples obtained with all catalysts showed a single sequence of –OBn and H end-capped chains with a peak separation of 144 g mol−1 corresponding to the monomer unit (Fig. 4).
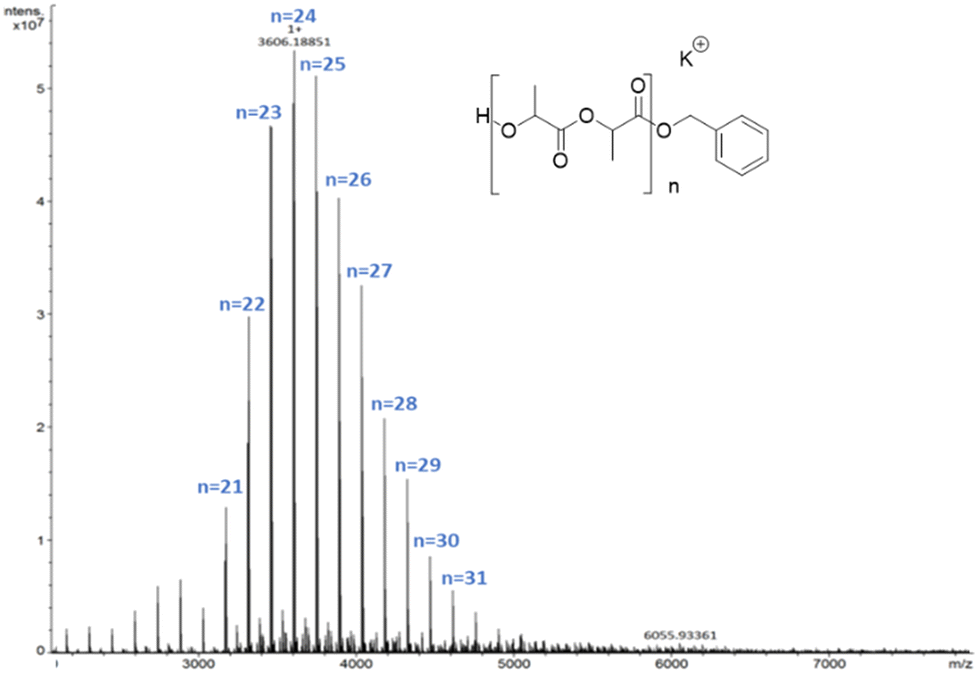 | ||
| Fig. 4 MALDI-ToF-MS spectrum of PLA synthesized by 1 ([LA]/[1] = 20, T = 80 °C). | ||
However, transesterification phenomena were observed by extending the reaction time beyond the complete conversion of the monomer (Fig. S27†). Thus, the catalysts were able to selectively promote the polymerization reaction in the presence of the monomer but, after the complete consumption of LA, the zinc species were still active and promoted rapid transesterification reactions of the polymer chains. This observation highlighted the potential of these complexes as catalysts for the depolymerization of PLA. Thus degradation studies of PLA samples were performed.
Chemical degradation of PLA
Chemical recycling of polyesters by alcoholysis is a method that has attracted great interest in recent years.43,51–53 Alcoholysis is a transesterification reaction that leads, under mild conditions, to the formation of molecules of high commercial interest.Given the high stability of the synthetized complexes, we decided to explore their behavior in the alcoholysis of commercial PLA plastic cups (MM = 58 KDa).
Initially, all zinc complexes were investigated in the metal-mediated degradation of PLA to Me–LA in the presence of MeOH (Scheme 4). The degradation experiments were performed in the air in THF at room temperature (Table 5).
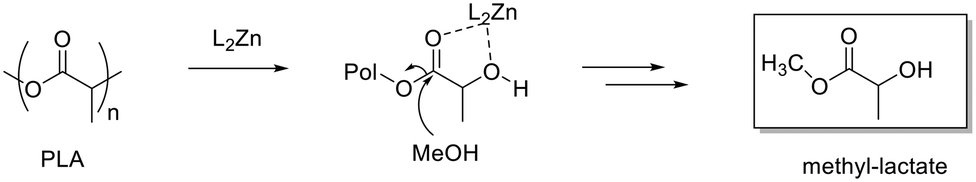 | ||
| Scheme 4 Mechanism of degradation of PLA. | ||
| Entry | Cat | Time (h) | X int (%) | S Me–La (%) | Y Me–La (%) |
|---|---|---|---|---|---|
| a All reactions were carried out by using 10 μmol of Zn catalyst (0.6 mol% relative to ester linkages) in 1.8 mL of THF, with 0.2 mL of MeOH, T = 25 °C. b Determined by 1H NMR. | |||||
| 1 | 1 | 3 | 54 | 10 | 5 |
| 2 | 2 | 3 | 64 | 16 | 10 |
| 3 | 3 | 3 | 39 | 5 | 2 |
| 4 | 4 | 3 | 75 | 17 | 13 |
| 5 | 5 | 3 | 19 | 0 | 0 |
| 6 | 6 | 3 | 36 | 15 | 10 |
The degradation parameters, PLA conversion of internal methine units (Xint), selectivity for Me–La (SMe–La) and yield of Me–La (YMe–La), were calculated by integration of the diagnostic signals from the 1H NMR spectra (Fig. S28†).
As previously reported, for alcoholysis reactions performed in THF solution, the degradation process proceeds via a random scission of polymeric chains that are initially converted to oligomers and gradually to methyl lactate (Fig. 5).45
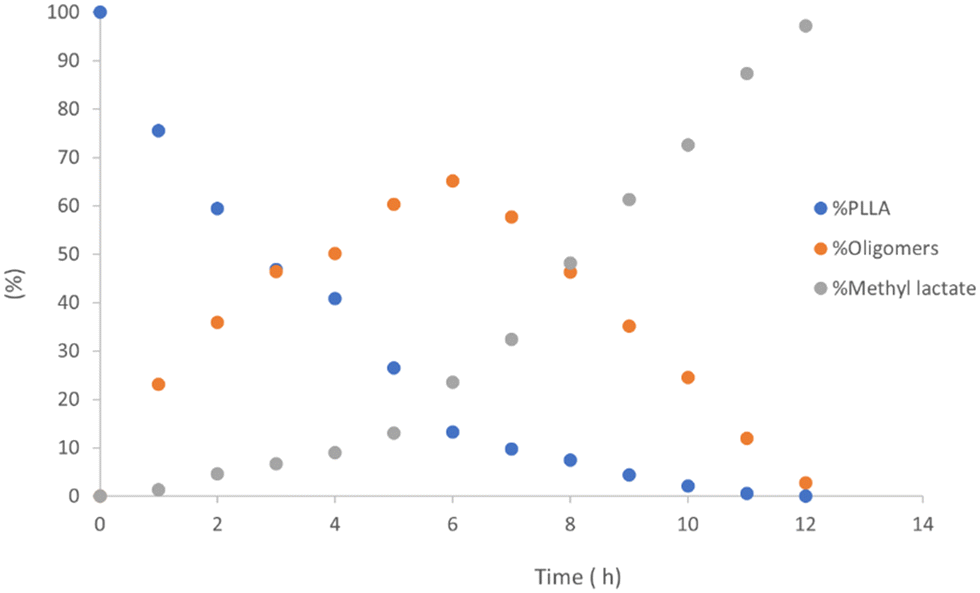 | ||
| Fig. 5 Methanolysis of PLLA. Reaction conditions: 30 μmol of 4, (0.6 mol% relative to ester linkages) in 10 mL of THF, with 1.2 mL of MeOH, T = 25 °C. | ||
As observed for the polymerization experiments, the conversions were related to substituent effects in the catalyst structures: the efficiency of the catalyst is reduced in the presence of ortho substituents on the phenoxy moiety. The best performances were observed for complexes with electron-donating substituents in the para position, while the presence of electron-withdrawing substituents produced detrimental effects. The activities displayed by these complexes were higher than those achieved with homoleptic phenoxy-imino-amine complexes which were able to convert 86% of PLA only after six hours at 40 °C.42
Additional degradation experiments were performed in the absence of solvent, under reaction conditions that reduced the environmental impact of the procedure (Table 6). Due to the insolubility of PLA and the catalysts in methanol, the reactions were performed at 65 °C. Also under these reaction conditions, complexes 1–6 revealed better performances in comparison to the zinc complex bearing a (imidazole[1,5-a]pyrid-3-yl)phenolate ligand.54
| Entry | Cat | ROH | T (°C) | X int (%) | S R-La (%) | Y R-La (%) |
|---|---|---|---|---|---|---|
| a All reactions were carried out by using 10 μmol of Zn catalyst (0.6 mol% relative to ester linkages), PLLA (115 mg) with 2 mL of ROH, time = 1 h. b Determinated by 1H NMR. c Double amounts of all reagents were used. | ||||||
| 1 | 1 | MeOH | 65 | 100 | 96 | 96 |
| 2 | 2 | MeOH | 65 | 100 | 95 | 95 |
| 3 | 3 | MeOH | 65 | 100 | 89 | 89 |
| 4 | 4 | MeOH | 65 | 100 | 92 | 62 |
| 5 | 5 | MeOH | 65 | 78 | 81 | 63 |
| 6 | 6 | MeOH | 65 | 100 | 100 | 100 |
| 7 | 4 | EtOH | 78 | 85 | 28 | 24 |
| 8c | ZnO | MeOH | 65 | 5 | — | — |
Degradation in neat methanol proceeded very rapidly; the complete degradation of the PLA samples was observed after one hour and the full conversion to methylactate was achieved after 1.5 hours for almost all catalysts.
The reaction was obviously slower when performed in neat ethanol (entry 7, Table 6), probably due to the steric hindrance of ethanol compared to methanol on approaching to the carbonyl centre.55,56
A control experiment of methanolysis was performed with ZnO (entry 8, Table 6). After 1 hour, only 5% of PLA was degraded, as a result of the low solubility of the zinc salt under these reaction conditions.57
Finally, we verified the possibility of recycling the catalyst. A solvent free alcoholysis was performed according to the general procedure using (576 mg of PLA and 25.6 mg of 1) 50 μmol of 1 with 0.6 mol% relative to the ester linkages with 10 mL of MeOH.
After two hours, a complete degradation to methyl lactate was achieved. The residual methanol and methyl lactate were removed under vacuum to recover the undecomposed catalyst as a solid residue (51% of the catalyst was recovered). This was verified by 1H NMR analysis. Equal amounts of methanol (10 ml) and polylactide (576 mg) were added and refluxed for an additional 2 hours. After this time, the PLA degradation, as determined by 1H NMR analysis, was approximately 30% (see Fig. S29–S31†).
A second test of catalyst reuse was performed, with the first degradation step performed as previously described. After two hours and complete degradation of PLA to methyl lactate, equal amounts of PLA and alcohol were added to the solution. After another two hours, 94% conversion was observed with 74% of selectivity; thus, no decrease in process efficiency was detected despite the higher dilution. This suggests the possibility of using this catalyst for a continuous degradation process.
Glycolysis of PET
Zinc complexes 1–6 were also tested in the degradation of a commercial sample of PET (Mn = 42.000 g mol−1, Đ = 1.7).Alcoholysis performed in the presence of ethylene glycol (EG) is an efficient procedure to convert PET waste into well-established commercial products such as bis(2-hydroxyethyl)terephthalate (BHET) and EG, which constitute the monomeric units for PET synthesis (Scheme 5).58,59
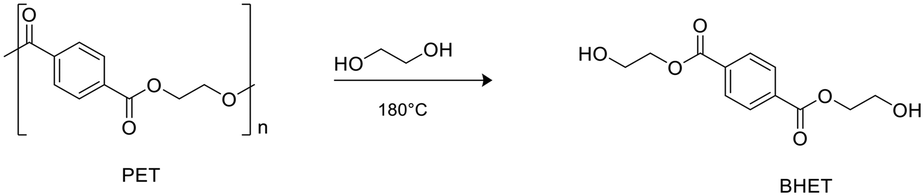 | ||
| Scheme 5 Glycolysis of PET with production of BHET. | ||
As reference tests, depolymerization experiments were initially performed with the commercial catalyst Zn(OAc)2·2H2O (entry 1, Table 7), or with ZnO (ref. 60 and 61) (entry 10, Table 7), and EG at 180 °C (0.07 equivalents of zinc and 27.5 equivalents of EG with respect to ester functions). The conversion of PET was evaluated as the ratio of the difference between the initial weight of PET and the weight of residual PET. The degradation products were added to distilled water, mixed and then filtered. The selectivity of the degradation process was calculated as the percentage of BHET obtained by crystallization from distilled water. The degradation products were isolated as solids and characterized by 1H NMR spectroscopy.
| Entry | Cat | Cat eq. (%) | WPET (mg) | t (h) | Conv (%) | Sel BHET (%) | Yield BHET (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: PET bottle (Mn ≈ 42.000 g mol−1), 1.6 mL of EG (27.8 equivalents relative to the ester bonds) and zinc catalyst (0.013 equivalents relative to the ester bonds). b 2 mL of EG (27.5 equivalents related to ester bonds), Zn(OAc)2 2H2O = 8% by weight (0.02 g, 7% in mol with respect to ester bonds). c 1 = 0.02 g, 2.5% moles with respect to the ester bonds. | |||||||
| 1b | Zn(OAc)2·2H2O | 7 | 250 | 4 | >99 | 43 | 44 |
| 2c | 1 | 2.5 | 250 | 1 | >99 | 58 | 58 |
| 3 | 1 | 1.3 | 200 | 1 | 45 | 58 | 26 |
| 4 | 2 | 1.3 | 200 | 1 | 27 | 39 | 29 |
| 5 | 3 | 1.3 | 200 | 1 | 29 | 60 | 25 |
| 6 | 4 | 1.3 | 200 | 1 | 27 | 21 | 30 |
| 7 | 5 | 1.3 | 200 | 1 | 95 | 57 | 60 |
| 8 | 6 | 1.3 | 200 | 1 | 93 | 64 | 60 |
| 9 | — | — | 200 | 1 | 8 | — | — |
| 10 | ZnO | 1.3 | 100 | 4 | 72 | 28 | 20 |
Complex 1, under the same reaction conditions but with only one-third the amount of catalyst, was found to be more active than the industrially used catalyst; in fact, complete degradation of the polymer sample was achieved after only one hour (entry 2, Table 7).
To investigate the influence of the catalyst structure on the catalytic activity, zinc complexes 1–6 were tested under the same conditions and by using a very low catalyst loading (0.013 equivalents relative to ester bonds). In this process, complexes 5 and 6 were the most active, showing high conversions and excellent selectivity (up to 60%).
The ZnO catalyst60 was definitively less active than Zn(OAc)2 and complexes 1–6. Indeed, the degradation of 72% of PET was reached only after four hours (entry 10, Table 7).
The effect of reaction temperature and molar amount of EG on PET degradation was also studied (Table 8).
| Entry | Cat | EG eq. (%) | Conv (%) | Sel BHET (%) | Yield BHET (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.100 g of PET bottle (Mn ≈ 42.000 g mol−1), zinc catalyst = 0.013 equivalents relative to the ester bonds. Reaction time 24 h. | |||||
| 1 | 1 | 27.8 | >99 | 43 | 27 |
| 2 | 2 | 27.8 | >99 | 52 | 25 |
| 3 | 3 | 27.8 | 45 | 54 | 30 |
| 4 | 4 | 27.8 | 27 | 29 | 55 |
| 5 | 5 | 27.8 | 29 | 68 | 23 |
| 6 | 6 | 27.8 | 27 | 58 | 29 |
| 7 | 6 | 13.8 | 95 | 59 | 27 |
| 8 | 6 | 41.3 | 93 | 49 | 32 |
As expected, the reaction temperature clearly influences the degradation of PET. For reactions performed at 130 °C, good conversions were obtained only after 24 hours (entries 1–6, Table 8).
By decreasing the amount of EG, the effect on the conversion was minimal, which is particularly interesting from an industrial point of view because it allows reducing the amount of reagent/solvent used in the reaction, a fundamental aspect to manage volumes in industrial processes.
To verify the stability of the zinc complexes during the glycolysis of PET, a test was performed with complex 2, which was treated with EG at 150 °C for one hour. The 1H HMR spectrum still showed the resonances of the starting complex 2, although additional resonances of lower intensity relating to the ligand were identified (Fig. S32†).
Experimental
General
Toluene and methanol were refluxed over Na and distilled under nitrogen. Benzene, hexane and tetrahydrofuran were distilled under nitrogen over sodium benzophenone (THF). Monomers (Sigma-Aldrich) were purified before use.
Deuterated solvents, CDCl3, C7D8 and C6D6 were purchased from Eurisotop were dried over molecular sieves. All other reagents and solvents were purchased from Aldrich and used without further purification.
The zinc precursor ZnEt2 was purchased from Aldrich and was used as received. L-Lactide were purchased from Aldrich and crystallized by dry toluene and afterward stored at −20 °C in a glovebox. All other chemicals were commercially available and used as received unless otherwise stated.
Instruments and measurements
The resonances are reported in ppm (δ) and the coupling constants in Hz (J) and are referenced to the residual solvent peak at δ = 7.16 ppm for C6D6 and δ = 7.27 for CDCl3. 13C NMR spectra are referenced using the residual solvent peak at δ = 128.06 for C6D6 and δ = 77.23 for CDCl3. Spectra recording was performed using Bruker-TopSpin v2.1 software. Data processing was performed using TopSpin v2.1 or MestReNova v6.0.2 software.
GPC. Size exclusion chromatography (SEC) of polymers was performed in THF at 35 °C using an Agilent 1260 Infinity Series GPC (ResiPore 3 μm, 300 × 7.5 mm, 1.0 mL min−1, RI (PL-GPC 220) detectors) at Chimie ParisTech. The number average molecular masses (Mn) and dispersity (Đ) of the polymers were calculated with reference to a universal calibration vs. polystyrene standards (limits Mw = 200 to 400
000 g mol−1).
A MALDI-ToF-MS. A MALDI-ToF-MS analysis was performed on a Waters Maldi Micro MX equipped with a 337 nm nitrogen laser. An acceleration voltage of 25 kV was applied. The polymer sample was dissolved in THF with Milli-Q water containing 0.1% formic acid at a concentration of 0.8 mg mL−1. The matrix used was 2,5-dihydroxybenzoic acid (DHBA) (Pierce) and was dissolved in THF at a concentration of 30 mg mL−1.
X-ray crystallography
Suitable crystals of compounds 1–2, 4 were selected and mounted on a cryoloop with paratone oil and measured at 293 K with a Bruker D8 QUEST diffractometer equipped with a PHOTON100 detector using CuKα radiation (λ = 1.54178 Å). Indexing was performed using APEX3.62 Data integration and reduction were performed using SAINT.62 Absorption correction was performed by multi-scan method in SADABS.62For all compounds the structures were solved by direct methods using SIR2014 (ref. 63) and refined by means of full matrix least-squares based on F2 using the program SHELXL.64
For all compounds non-hydrogen atoms were refined anisotropically, hydrogen atoms were positioned geometrically and included in structure factors calculations, but not refined.
Compound 2 features one benzene molecules per molecule of Zn complex. The benzene molecule lies on a crystallographic inversion centre and is disordered, two possible orientations were considered and refined as rigid group with isotropic displacement parameters.
Crystal structures were drawn using Mercury.65
Synthesis of the ligands
1H NMR (CDCl3, 400 MHz, 298 K): δ 8.50 (1H, d, –N–CH = pyridine), 8.20 (1H, s, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 7.52 (1H, td, Ar), 7.15 (4H, m, Ar), 6.86 (2H, m, Ar), 3.94 (2H, t, N–CH2–CH2–C), 3.09 (2H, t, N–CH2–CH2–C).
N), 7.52 (1H, td, Ar), 7.15 (4H, m, Ar), 6.86 (2H, m, Ar), 3.94 (2H, t, N–CH2–CH2–C), 3.09 (2H, t, N–CH2–CH2–C).
1H NMR (C6D6, 600 MHz, 298 K): δ 8.50 (1H, d, –N–CH = pyridine), 7.70 (1H, s, HCN), 7.10 (1H, td, Ar), 6.90 (2H, m Ar), 6.82(1H, m, Ar), 6.70 (1H, m, Ar), 6.64 (1H, m, Ar), 6.58 (1H, m, Ar), 3.70 (2H, t, N–CH2–CH2–C), 2.87 (2H, t, N–CH2–CH2–C).
13C NMR (C6D6, 150 MHz, 298 K): δ 165.5, 162.0, 149.7, 135.8, 132.3, 131.5, 128.2, 127,9, 123.5, 121.3, 119.3, 118.4, 117.4, 59.1 (N–CH2CH2), 39.14 (N–CH2CH2–).
1H NMR (CDCl3, 300 MHz, 298 K): 8.57 (1H, d, –N–CHortho pyridine), 8.24 (1H, s, HCN), 7.60 (1H, td, Ar), 7.15 (3H, m Ar), 6.98 (1H, s, Ar), 6.84 (1H, m, Ar), 4.02 (2H, t, N–CH2–CH2–C), 3.18 (2H, t, N–CH2–CH2–C), 2.27 (3H, s, C–CH3).
1H NMR (C6D6, 600 MHz, 298 K): 8.48 (1H, d, –N–CHortho pyridine), 7.74 (1H, s, HCN), 7.16 (s, C6D6), 7.04 (1H, td, Ar), 6.97 (1H, m, Ar), 6.84 (1H, m, Ar), 6.71 (1H, m, Ar), 6.57 (2H, m, Ar), 3.74 (2H, t, N–CH2–CH2–C), 2.89 (2H, t, N–CH2–CH2–C), 2.03 (3H, s, C–CH3).
13C NMR (C6D6, 150 MHz, 298 K): δ 165.5, 162.0, 149.7, 135.8, 132.3, 131.5, 128.2, 127.8 (aromatic), 123.5, 121.3, 119.3, 118.4, 117.4, 59.14 (N–CH2CH2), 39.1 (N–CH2CH2–), 20.7 (CH3).
N), 7.49 (1H, td, Ar), 7.27 (s, CDCl3), 7.06 (2H, m Ar), 6.72 (3H, m, Ar), 3.93 (2H, t, N–CH2–CH2–C), 3.78 (3H, s, O–CH3), 3.07 (2H, t, N–CH2–CH2–C).
1H NMR (C6D6, 600 MHz, 298 K): 8.45 (1H, d, –N–CH = ortho pyridine), 7.74 (1H, s, HCN), 6.96 (1H, td, Ar) 6.70 (1H, m, Ar), 6.65 (1H, m, Ar), 6.62 (2H, m, Ar), 6.55 (2H, m, Ar), 3.68 (2H, t, N–CH2–CH2–C), 3.47 (3H, s, O–CH3), 2.84 (2H, t, N–CH2–CH2–C).
13C NMR (C6D6, 150 MHz, 298 K): 166.1, 160.0, 153.3, 150.1, 149.7, 136.2, 124.0, 123.8, 121.6, 119.8, 118.2, 116.0, 59.1, 56.5, 39.9.
N), 7.50 (1H, td, Ar), 7.06 (2H, m Ar), 6.78 (2H, m, Ar), 6.61 (1H, s, Ar), 3.93 (2H, t, N–CH2–CH2–), 3.66 (3H, s, O–CH3), 3.08 (2H, t, N–CH2–CH2–C).
1H NMR (C6D6, 600 MHz, 298 K): 8.47 (1H, d, –N–CH ortho pyridine), 7.65 (1H, s, HCN), 7.50 (1H, td, Ar), 7.01 (2H, m Ar), 6.98 (1H, td, Ar), 6.73 (2H, m Ar), 6.57 (1H, m, Ar), 6.45 (1H, s, Ar), 3.72 (2H, t, N–CH2–), 3.31 (3H, s, O–CH3), 2.89 (2H, t, N–CH2–CH2–).
13C NMR (C6D6, 150 MHz, 298 K): δ 165.3, 159.6, 152.3, 149.7, 135.8, 123.6, 121.3, 119.2, 118.1, 115.4, 59.1 (N–CH2CH2–pyridine), 55.3 (CH3–O–) 39.1 (N–CH2CH2).
N), 8.07 (2H, m, Ar), 7.52 (1H, td, Ar), 7.07 (1H, m, Ar), 6.82 (1H, m, Ar), 4.04 (2H, t, N–CH2), 3.13 (2H, t, N–CH2–CH2–C).
1H NMR (C6D6, 600 MHz, 298 K): 8.44 (1H, d, CH = ortho pyridine), 7.79 (1H, d,), 7.64 (1H, s, HCN), 7.00 (1H, m, Ar), 6.98 (1H, td, Ar), 6.58 (3H, m, Ar), 3.72 (2H, t, N–CH2–), 3.31 (3H, s, O–CH3), 2.89 (2H, t, N–CH2–CH2–C).
13C NMR (C6D6, 150 MHz, 298 K): δ 168.1, 164.4, 158.8, 149.9, 158.8, 135.8, 121.5, 118.2, 59.1, 57.5 (CH3–O), 38.1 (–CH2CH2–pyridine).
1H NMR (400 MHz, C6D6, 298 K): δ 14.2 (s, 1H, –OH), 8.45 (dd, J1 = 4.8 Hz, J2 = 1.7 Hz, 1H, Py–H), 7.75 (s, 1H, CHN), 7.56 (d, J = 2.4 Hz, 1H, Ph–H), 6.95 (dt, J1 = 7.5 Hz, J2 = 2.1 Hz, 1H, Py–H), 6.87 (d, J = 2.4 Hz, 1H, Ph–H), 6.70 (d, J = 7.7 Hz, 1H, Py–H), 6.55 (dt, J1 = 7.2 Hz, J2 = 2.1 Hz, 1H, Py–H), 3.75 (dt, J1 = 7.2 Hz, J2 = 1.0 Hz, 2H, CH2), 2.92 (t, J = 7.1 Hz, 2H, CH2), 1.66 (s, 9H, tBu), 1.30 (s, 9H, tBu).
13C NMR (100 MHz, C6D6, 298 K): δ 166.75, 159.76, 158.88, 149.83, 139.98, 137.01, 135.82, 126.82, 126.40, 123.50, 121.29, 118.63, 59.12, 39.75, 35.43, 34.26, 31.74, 31.37, 29.79, 29.48 ppm.
Synthesis of zinc complexes
1H NMR (400 MHz, C6D6, 298 K): 8.43 (2H, d, –N–CH = pyridine), 7.59 (2H, s, HCN), 7.24 (2H, td, Ar), 6.93 (4H, m Ar), 6.76 (4H, m, Ar), 6.56 (4H, m, Ar), 3.72 (4H, t, N–CH2–CH2–C), 3.08 (4H, t, N–CH2–CH2–C).
13C NMR (75 MHz, C6D6, 298 K): δ 171.5, 158.5, 135.9, 134.8, 127.8, 124.0, 123.6, 121.2, 118.3, 113.9, 60.5, 38.3.
Elemental analysis calculated for: C28H26N4O2Zn (%): C, 65.19; H, 5.08; N, 10.86. Found: C, 64.32; H, 5.01; N, 10.93.
1H NMR (300 MHz, C6D6, 298 K): 8.40 (2H, d, –N–CH = pyridine), 7.55 (2H, s, HCN), 7.21 (2H, td, Ar), 7.00 (4H, m Ar), 6.86 (2H, m, Ar), 6.48 (4H, m, Ar), 3.73 (4H, t, N–CH2–CH2–C), 3.05 (4H, t, N–CH2–CH2–C), 2.08 (6H, s, C–CH3).
13C NMR (75 MHz, C6D6, 298 K): δ 171.4, 158.3, 149.2, 136.1, 135.8, 135.0, 123.4, 122.0, 121.0, 117.5, 99.46, 60.2, 38.2, 19.8.
Elemental analysis calculated for: C30H30N4O2Zn (%): C, 66.24; H, 5.56; N, 10.30; found: C, 66.11; H, 5.79; N, 10.48.
1H NMR (300 MHz, C6D6, 298 K): 8.35 (2H, d, –N–CH = ortho-pyridine), 7.55 (2H, s, HCN), 7.09 (2H, m, Ar), 6.84 (2H, td, Ar), 6.74 (2H, m Ar), 6.44 (4H, m, Ar), 3.66 (4H, t, N–CH2–CH2–C), 3.54 (6H, s, –O–CH3), 3.02 (4H, t, N–CH2–CH2–C).
13C NMR (100 MHz, C6D6, 298 K): δ 171.3, 159.8, 159.2, 149.5, 136.8, 136.2, 135.4, 123.8, 121.4, 117.2, 113.1, 60.2, 55.5, 39.1 ppm. Elemental analysis calculated for: C30H30N4O4Zn (%): C, 62.56; H, 5.25; N, 9.73; found: C, 62.47; H, 5.39; N, 9.61.
1H NMR (400 MHz, C6D6, 298 K): 8.45 (2H, d, –CH ortho-pyridine), 7.57 (2H, s, HCN), 7.09 (2H, td, Ar), 6.99 (2H, m, Ar), 6.93 (2H, m, Ar), 6.55 (2H, m, Ar), 6.33 (2H, m, Ar), 3.79 (4H, t, N–CH2–CH2–C), 3.44 (6H, s, –O–CH3), 3.13 (4H, t, N–CH2–CH2–C).
13C NMR (75 MHz, C6D6, 298 K): δ 171.4, 163.4, 158.6, 153.2, 149.0, 135.8, 124.3, 120.9, 118.9, 116.3, 112.7, 60.1, 55.9, 38.0.
Elemental analysis calculated for: C30H30N4O4Zn (%): C, 62.56; H, 5.25; N, 9.73; found: C, 62.63; H, 5.18; N, 9.80.
N imide), 7.81 (2H, m, Ar), 7.52 (2H, td, Ar), 7.08 (2H, d, Ar), 6.98 (2H, t, Ar), 6.47 (2H, d, Ar), 4.04 (4H, t, N–CH2–CH2–C), 3.33 (4H, t, N–CH2–CH2–C). Yield 75%.
13C NMR (75 MHz, CDCl3, 298 K): δ 172.1, 163.4, 158.6, 153.2, 149.0, 135.8, 124.3, 120.9, 118.9, 116.3, 112.7, 55.9, 38.0.
1H NMR (400 MHz, C6D6, 298 K): δ 8.37 (d, J = 4.8 Hz, 1H, Ha), 7.67 (m, 2H, Hi–g), 6.84 (dt, J1 = 7.6 Hz, J2 = 1.8 Hz, 1H, Hc), 6.76 (d, J = 2.6 Hz, 1H, Hh), 6.59 (d, J = 7.8 Hz, 1H, Hd), 6.48 (t, J = 6.1 Hz, 1H, Hb), 3.77 (t, J = 7.4 Hz, 2H, Hf), 3.01 (m, 2H, He), 1.73 (s, 9H, tBum), 1.37 (s, 9H, tBul).
13C NMR (100 MHz, C6D6, 298 K): δ 172.6, 169.4, 158.9, 149.6, 141.7, 136.0, 135.2, 130.2, 129.8, 123.7, 121.4, 117.9, 60.3, 39.2, 36.0, 34.1, 31.9, 30.0.
Elemental analysis calculated for: C44H58N4O2Zn (%): C, 71.38; H, 7.90; N, 7.57; found: C, 71.16; H, 7.88; N, 7.65.
Test of stability
In the glovebox 0.0070 g (11 μmol) of complex 2 were weighted and then transferred to a vial. The stability test was conducted in air at 180 °C for 15 hours. Then, the solid was dissolved in 0.5 mL of C6D6 and analyzed by 1H NMR spectroscopy.Polymerization of L-LA performed in toluene solution
In glove box, a 10 mL Schlenk tube was charged with 0.144 g (1 mmol, 100 eq.) of L-LA, (10 μmol,) of zinc complex and 1.25 mL of toluene. Then 0.10 mL of BnOH 0.1 M solution in toluene was added (10 μmol, 1 eq.). The reaction mixture was stirred in a at the opportune temperature. For each test, an aliquot of the reaction mixture was analyzed by 1H NMR analysis.Polymerization reaction: industrial conditions
In glove box, a 50 mL Schlenk tube was charged with technical grade L-LA (7.207 g, 50 mmol, 5000), 0.05 mL of benzylic alcohol (500 μmol) and 10 μmol of catalyst. The Schlenk tube was closed and thermostated in an oil bath at 180 °C. The polymerization mixture was cooled at room temperature, CHCl3 was added to solubilize the polymer and the resulting solution was analyzed by 1H NMR to establish the conversion.Kinetic studies
The L-LA amount (0.0721 g, 500 μmol, 100 eq.) was weighted in a 10 mL vial and 0.3 mL of CDCl3 were added than 0.1 mL of BnOH 0.05 M solution in CDCl3 (5 μmol, 1 eq.) was added. In another 10 mL vial, the catalyst amount (5 μmol) was weighted and 0.1 mL of CDCl3 were added. The two solutions were transferred in a J-Young NMR tube. NMR spectra were after the opportune times.General procedure of PLA alcoholysis in solution
For each experiment, 0.1152 g (1.6 mmol, 160 eq.) of PLA plastic cups, 1.8 mL of THF, 0.2 mL of methanol and the predicted amount of catalyst (10 μmol, 1 eq.) were added to a 10 mL vial with a magnetic stirrer. Methanolysis reactions were carried out at 25 °C under air atmosphere. After three hours, the reaction mixture was analyzed by 1H NMR spectroscopy.General procedure of PLA solvent free alcoholysis
For each experiment, 0.1152 g (1.6 mmol, 160 eq.) of PLA plastic cups, 2 mL of alcohol and the predicted amount of catalyst (10 μmol, 1 eq.) were added to a 10 mL Schlenk tube. Alcoholysis reactions were carried out at 65 °C. After one hour, the reaction mixture was analyzed by 1H NMR spectroscopy.The conversion of PLA, methyl lactate and oligomers were calculated from 1H NMR, by the following equations:
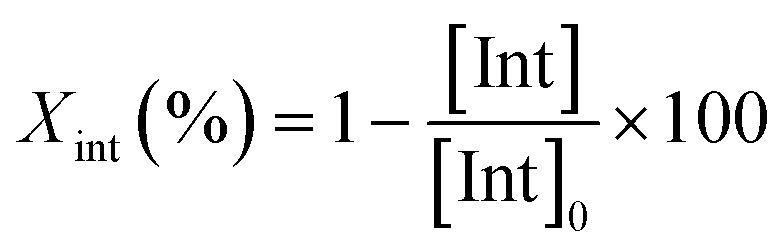
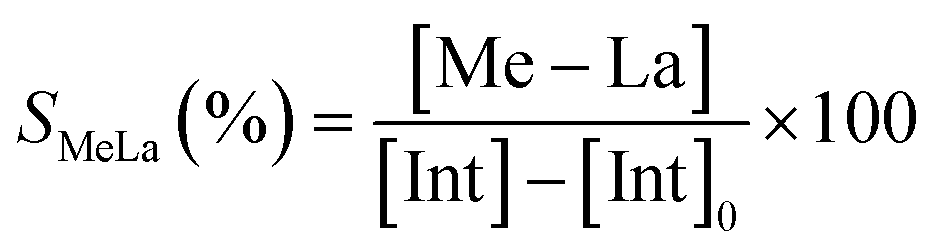
| YMeLa(%) = XintSMeLa |
General procedure of PET glycolysis
For each experiment, 0.250 g or of 0.200 g PET particles, the opportune volume of ethylene glycol, and the predicted amount of catalyst were added to a 25 mL reaction tube with a magnetic stirrer. Glycolysis reactions were carried out at the opportune temperature (180 °C or 130 °C) for the predicted time. The reaction mixture was cooled to room temperature and about 8 mL of distilled water were added, the resulting mixture was stirred and then filtered to eliminate the residual PET that were dried at 60 °C in vacuo to constant weight.The conversion of PET is calculated by the following equation:
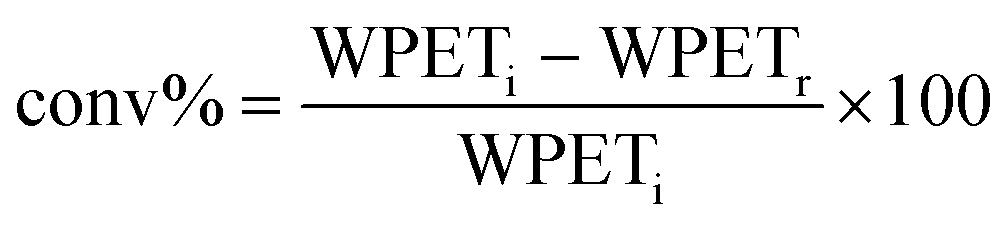
The solution was concentrated by using a vacuum rotary evaporator at 70 °C and then refrigerated at 0 °C for 12 h to obtain white crystals of pure BHET. Yield of BHET refers to the isolated yield of crystals followed by drying at 90 °C for 3 h in vacuo.
Sel. and yield of BHET were calculated according to the following equations:
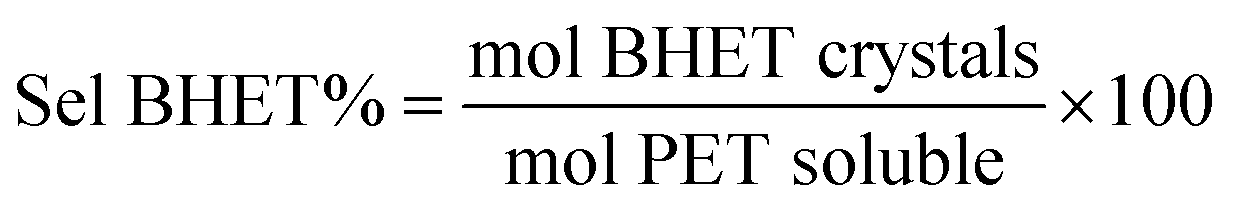
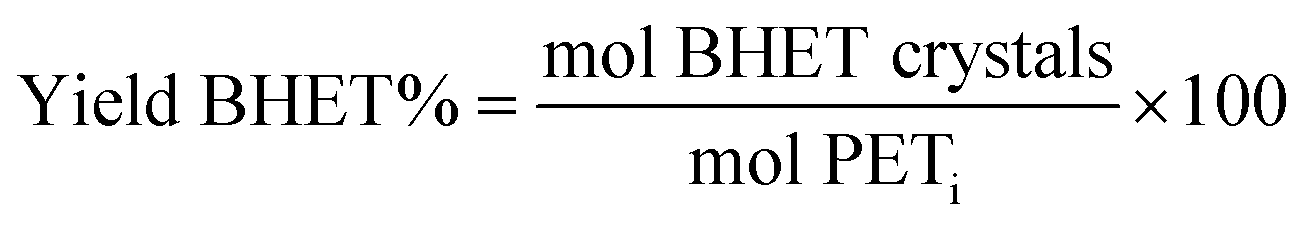
Conclusions
Despite the economic and environmental benefits of promoting the chemical recycling of polymers that are continuously produced, this type of procedure still requires improvement. In this paper, we describe the synthesis and characterization of homoleptic Zn(II) complexes based on tridentate phenoxy-imine pyridine ligands that have been shown to be efficient in the ROP of L-LA and in the degradation of commercial polyesters such as PLA and PET. All complexes were tested in the ROP of L-lactide at room temperature, demonstrating a clear structure–reactivity relationship, dependent on the structure of the substituents introduced on the phenolate fragment of the ancillary ligands. While electron donating groups increased the polymerization rate, the steric hindrance of the substituents generally had a greater impact reducing the activity. All complexes exhibited high thermal stability and thus were successfully used to produce isotactic PLLA under industrially relevant melt conditions.In the depolymerization reactions of PLLA via alcoholysis, the same structure–reactivity trend was observed. In preliminary PET glycolysis tests, all complexes were found to be more efficient than Zn(OAc)2, even when a lower catalyst loading, and shorter reaction times are used, thus demonstrating the high potential of these systems in depolymerization processes.
Author contributions
Prof. Mina Mazzeo and Prof. Christophe M. Thomas: conceptualization and writing. Miss Sara D'Aniello, Miss Sidonie Laviéville, Miss Malaury Simon, Mr Michele Sellitto, Miss Federica Santulli: investigation. Prof. Consiglia Tedesco: investigation and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Patrizia Iannece for Maldi ToF spectra and Dr Patrizia Oliva for NMR assistance.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- L. Lebreton and A. Andrady, Palgrave Commun., 2019, 5, 6 CrossRef.
- E. MacArthur, Science, 2017, 358, 843–843 CrossRef PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501 CrossRef CAS.
- J.-P. Lange, M. Dusselier and S. De Wildeman, in Biodegradable Polymers in the Circular Plastics Economy, 2022, pp. 1–16, DOI:10.1002/9783527827589.ch1.
- M. J. L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- M. A. Hillmyer, Science, 2017, 358, 868–870 CrossRef CAS PubMed.
- K. Schroder, K. Matyjaszewski, K. J. T. Noonan and R. T. Mathers, Green Chem., 2014, 16, 1673–1686 RSC.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084 RSC.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- C. Jehanno, M. M. Perez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- P. B. V. Scholten, J. Cai and R. T. Mathers, Macromol. Rapid Commun., 2021, 42, 2000745 CrossRef CAS PubMed.
- A. Lancien, R. Wojcieszak, E. Cuvelier, M. Duban, P. Dhulster, S. Paul, F. Dumeignil, R. Froidevaux and E. Heuson, ChemCatChem, 2021, 13, 247–259 CrossRef CAS.
- C. Bruneau and C. Fischmeister, in Organometallics for Green Catalysis, ed. P. H. Dixneuf and J. F. Soule, 2019, vol. 63, pp. 77–102 Search PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- A. S. Narmon, L. M. Jenisch, L. M. Pitet and M. Dusselier, in Biodegradable Polymers in the Circular Plastics Economy, 2022, pp. 205–271, DOI:10.1002/9783527827589.ch7.
- M. Helou, N. Ajellal, M. Bouyahyi, C. M. Thomas, A. Trifonov, S. M. Guillaume and J.-F. Carpentier, PMSE Prepr., 2009, 100, 387–388 CAS.
- R. Platel, L. Hodgson and C. Williams, Polym. Rev., 2008, 48, 11 CrossRef CAS.
- M. Strianese, D. Pappalardo, M. Mazzeo, M. Lamberti and C. Pellecchia, Dalton Trans., 2020, 49, 16533–16550 RSC.
- C. Fliedel, S. Dagorne and E. Le Roux, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 410–464, DOI:10.1016/B978-0-08-102688-5.00089-1.
- I. D'Auria, M. Lamberti, M. Mazzeo, S. Milione, G. Roviello and C. Pellecchia, Chem. – Eur. J., 2012, 18, 2349–2360 CrossRef.
- D. Wannipurage, T. S. Hollingsworth, F. Santulli, M. Cozzolino, M. Lamberti, S. Groysman and M. Mazzeo, Dalton Trans., 2020, 49, 2715–2723 RSC.
- I. D'Auria, C. Tedesco, M. Mazzeo and C. Pellecchia, Dalton Trans., 2017, 46, 12217–12225 RSC.
- A. Pilone, M. Lamberti, M. Mazzeo, S. Milione and C. Pellecchia, Dalton Trans., 2013, 42, 13036–13047 RSC.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2020, 59, 21778–21784 CrossRef CAS.
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- P. McKeown and M. D. Jones, Sustainable Chem., 2020, 1, 1 CrossRef.
- C. T. Bowmer, R. N. Hooftman, A. O. Hanstveit, P. W. M. Venderbosch and N. Van Der Hoeven, Chemosphere, 1998, 37, 1317–1333 CrossRef CAS.
- D. Paszun and T. Spychaj, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185 CrossRef CAS.
- J. Payne, P. McKeown, O. Driscoll, G. Kociok-Kohn, E. A. C. Emanuelsson and M. D. Jones, Polym. Chem., 2021, 12, 1086–1096 RSC.
- K. Troev, G. Grancharov, R. Tsevi and I. Gitsov, J. Appl. Polym. Sci., 2003, 90, 1148 CAS.
- J. M. Payne, G. Kociok-Köhn, E. A. C. Emanuelsson and M. D. Jones, Macromolecules, 2021, 54, 8453–8469 CrossRef CAS.
- J. M. Payne, M. Kamran, M. G. Davidson and M. D. Jones, ChemSusChem, 2022, 15, e202200255 CAS.
- J. Stewart, M. Fuchs, J. Payne, O. Driscoll, G. Kociok-Kohn, B. D. Ward, S. Herres-Pawlis and M. D. Jones, RSC Adv., 2022, 12, 1416–1424 RSC.
- L. A. Roman-Ramirez, P. McKeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef CAS.
- P. McKeown, L. A. Roman-Ramirez, S. Bates, J. Wood and M. D. Jones, ChemSusChem, 2019, 12, 5233–5238 CrossRef CAS PubMed.
- H. Shere, P. McKeown, M. F. Mahon and M. D. Jones, Eur. Polym. J., 2019, 114, 319–325 CrossRef CAS.
- F. Santulli, M. Lamberti and M. Mazzeo, ChemSusChem, 2021, 14, 5470–5475 CrossRef CAS PubMed.
- F. Santulli, G. Gravina, M. Lamberti, C. Tedesco and M. Mazzeo, Mol. Catal., 2022, 528, 112480 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- S. Friedrich, M. Schubart, L. H. Gade, I. J. Scowen, A. J. Edwards and M. McPartlin, Chem. Ber., 1997, 130, 1751–1759 CrossRef CAS.
- M. A. Bahili, E. C. Stokes, R. C. Amesbury, D. M. C. Ould, B. Christo, R. J. Horne, B. M. Kariuki, J. A. Stewart, R. L. Taylor, P. A. Williams, M. D. Jones, K. D. M. Harris and B. D. Ward, Chem. Commun., 2019, 55, 7679–7682 RSC.
- The RX analysis was repeated on the same crops of crystals stored in air for a week demonstrating their stability.
- M. Noda and H. Okuyama, Chem. Pharm. Bull., 1999, 47, 467–471 CrossRef CAS.
- L. D. Brake, U.S. Pat., US52646171993, 1993 Search PubMed.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- M. C. D'Alterio, I. D'Auria, L. Gaeta, C. Tedesco, S. Brenna and C. Pellecchia, Macromolecules, 2022, 55, 5115–5122 CrossRef.
- L. A. Román-Ramírez, M. Powders, P. McKeown, M. D. Jones and J. Wood, J. Polym. Environ., 2020, 28, 2956–2964 CrossRef.
- D. Bykowski, A. Grala and P. Sobota, Tetrahedron Lett., 2014, 55, 5286–5289 CrossRef CAS.
- C. Alberti and S. Enthaler, ChemistrySelect, 2020, 5, 14759–14763 CrossRef CAS.
- J. W. Chen and L.-W. Chen, J. Appl. Polym. Sci., 1999, 73, 35–40 CrossRef CAS.
- M. Imran, D. H. Kim, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and S. M. Ramay, Polym. Degrad. Stab., 2013, 98, 904–915 CrossRef CAS.
- Y. Hu, W. A. Daoud, B. Fei, L. Chen, T. H. Kwan and C. S. Ki Lin, J. Cleaner Prod., 2017, 165, 157–167 CrossRef CAS.
- J.-T. Du, Q. Sun, X.-F. Zeng, D. Wang, J.-X. Wang and J.-F. Chen, Chem. Eng. Sci., 2020, 220, 115642 CrossRef CAS.
- J. Pflugrath, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1718–1725 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357–361 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2171735–2171737. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cy01092e |
| This journal is © The Royal Society of Chemistry 2022 |