
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atom-efficient synthesis of a benchmark electrolyte for magnesium battery applications†
Scott A.
Brown
ac,
Serena A.
Cussen
*b,
Rhiannon
Kennard
b,
Sofia
Marchesini
c,
Jethro J.
Pryke
b,
Annabel
Rae
a,
Stuart D.
Robertson
 *a,
Rudra N.
Samajdar
ac and
Andrew J.
Wain
*c
*a,
Rudra N.
Samajdar
ac and
Andrew J.
Wain
*c
aWestCHEM, Department of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: stuart.d.robertson@strath.ac.uk
bDepartment of Materials Science & Engineering, The University of Sheffield, Sheffield, S1 3JD, UK. E-mail: s.cussen@sheffield.ac.uk
cDepartment of Electromagnetic & Electrochemical Technologies, National Physical Laboratory, Hampton Road, Teddington, TW11 0LW, UK. E-mail: andy.wain@npl.co.uk
First published on 3rd October 2022
Abstract
The benchmark magnesium electrolyte, [Mg2Cl3]+ [AlPh4]−, can be prepared in a 100% atom-economic fashion by a ligand exchange reaction between AlCl3 and two molar equivalents of MgPh2. NMR and vibrational spectroscopy indicate that the reported approach results in a simpler ionic composition than the more widely adopted synthesis route of combining PhMgCl with AlCl3. Electrochemical performance has been validated by polarisation tests and cyclic voltammetry, which demonstrate excellent stability of electrolytes produced via this atom-efficient approach.
In the pursuit of long-term sustainable energy storage, magnesium has the potential to contribute towards alleviating pressure on incumbent lithium, the dominant player in the rechargeable battery arena.1–5 This can, in part, be attributed to the substantial abundance advantage magnesium holds (it is approximately a thousand times more abundant than Li in the Earth's crust), coupled with a superior theoretical capacity and a competitive voltage window. That said, many challenges remain to be surmounted before magnesium can be considered a viable alternative. One such challenge is the development of suitable electrolytes.6 The viability of many different types of Mg electrolyte has been tested over the last thirty years, including Grignard reagents,7 wholly inorganic salts8–11 and alkyl/amido trichloroaluminates12–14 amongst others. Among the currently leading candidates is a magnesium aluminate solution in tetrahydrofuran (THF) known as the ‘all-phenyl complex’ (APC). The primary active species associated with APC solutions has been identified as [Mg2Cl3]+ [AlPh4]− (1).15 However, the APC electrolyte is typically generated and used in situ by combining the Grignard reagent16 PhMgCl with AlCl3 in THF (eqn (1)). Such a combination can never deliver species 1 with 100% atom economy since the ratio of Ph to Mg in the starting materials is unity, yet this is not the case in the final product, meaning that other species are also present in solution that can potentially interfere with the electrochemistry. This is supported by the identification of other Mg/Al complexes by X-ray diffraction from such combinations, for example [MgCl]+ [AlCl4]− (eqn (2)) and [Mg2Cl3]+ [Ph2AlCl2]−,17 and by 27Al NMR spectroscopic studies which display a variety of resonances indicative of multiple species being present in solution.18 Consequently, we pursued a more atom-efficient pathway to generate complex 1. For this, we utilised Ph2Mg and AlCl3 in a 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, thus matching exactly the empirical formula of the target APC product (eqn (3)). This approach did indeed yield [Mg2Cl3·6THF]+ [AlPh4]− in a high yield (31% crystalline yield, 99% overall yield) as determined by comparison of unit cell parameters with published data (entry AVIXAF in the CCDC; unit cell 16.524 Å, 16.358 Å, 39.263 Å, 90°, 111.13°, 90°),15 with NMR spectroscopic studies on the mother liquor (both 1H and 27Al) confirming that this was the only organometallic species generated by the reaction (see Fig. S1–S8, ESI†). Specifically, the only resonance in the 27Al NMR spectrum was a sharp singlet at 132.9 ppm, which was one of the many resonances observed previously in the Grignard/AlCl3 synthesis (eqn (1)).15 The narrow half-height line width is indicative of a symmetric tetrahedral aluminium centre. The 1H NMR spectrum exhibited only one set of phenyl resonances (7.67, 6.96, 6.90 ppm) consistent with the presence of a single product. Integration of the resonances for the solid samples suggested the presence of 5 molecules of THF, consistent with loss of one when drying under high vacuum, such formulation also being confirmed by C and H microanalyses.
:1 ratio, thus matching exactly the empirical formula of the target APC product (eqn (3)). This approach did indeed yield [Mg2Cl3·6THF]+ [AlPh4]− in a high yield (31% crystalline yield, 99% overall yield) as determined by comparison of unit cell parameters with published data (entry AVIXAF in the CCDC; unit cell 16.524 Å, 16.358 Å, 39.263 Å, 90°, 111.13°, 90°),15 with NMR spectroscopic studies on the mother liquor (both 1H and 27Al) confirming that this was the only organometallic species generated by the reaction (see Fig. S1–S8, ESI†). Specifically, the only resonance in the 27Al NMR spectrum was a sharp singlet at 132.9 ppm, which was one of the many resonances observed previously in the Grignard/AlCl3 synthesis (eqn (1)).15 The narrow half-height line width is indicative of a symmetric tetrahedral aluminium centre. The 1H NMR spectrum exhibited only one set of phenyl resonances (7.67, 6.96, 6.90 ppm) consistent with the presence of a single product. Integration of the resonances for the solid samples suggested the presence of 5 molecules of THF, consistent with loss of one when drying under high vacuum, such formulation also being confirmed by C and H microanalyses.| 4PhMgCl + 2AlCl3 → [Mg2Cl3]+ [AlPh4]− + 2MgCl2 + AlCl3 | (1) |
| PhMgCl + 2AlCl3 → [MgCl]+ [AlCl4]− + PhAlCl2 | (2) |
| 2MgPh2 + AlCl3 →[Mg2Cl3]+ [AlPh4]− | (3) |
Having identified an atom-economic approach, for clarity we will now refer to APC solutions generated according to eqn (1) as 1A, those prepared in situ according to eqn (3) as 1B and those generated by dissolving solid APC crystallised from eqn (3) in THF as 1C.
With an efficient synthetic protocol for the active APC electrolyte, we proceeded to perform chemical characterisation of the electrolyte solution using FTIR and Raman spectroscopy (which have been demonstrated to be powerful techniques for the speciation of Mg electrolytes10,15,19), in order to compare samples 1A and 1B. For 1A, we mirrored the concentration adopted by Aurbach,15 combining equal volumes of a 0.5 M solution of PhMgCl and a 0.25 M solution of AlCl3 for a product concentration of 0.0625 M on account that the product stoichiometry is ¼ that of the original Grignard (eqn (1)) and the complex is further diluted since the combination of both solutions doubles the volume. For our atom-efficiently prepared sample 1B we targeted a 0.14 M solution of the desired product in THF (marginally lower than the maximum concentration we could generate by dissolving 1C in THF, 0.15 M) by mixing equal volumes of 0.56 M MgPh2 and 0.28 M AlCl3, both in THF, according to eqn (3).
Fig. 1 shows the resulting FTIR and Raman spectra for electrolyte solutions 1B (Fig. 1a and c) compared with 1A (Fig. 1b and d). FTIR spectra (Fig. 1a and b) are dominated by multicomponent modes arising from the solvent, THF, here classified according to the dominant vibration: νC–O–C (asymmetric) at 1066 cm−1 and νCα–Cβ (asymmetric) at 909 cm−1, with satellite peaks at 875 cm−1 (βC–H vibrations and δ ring modes) and 1028 cm−1 (βC–H; νC–Cα/B and δ ring modes).20 These modes arise from the C2 symmetry of molecular THF in bulk liquid. The satellite modes are inherently weak in the pure solvent (Fig. S9, ESI†) and intensify in the presence of the electrolyte due to distortion from bulk liquid equilibrium symmetry in the presence of coordination centres. Fig. 1a shows an additional weak peak at 889 cm−1 which can be attributed to the νC–O–C (asymmetric) mode of coordinated THF. Comparison with the FTIR spectrum of the electrolyte solution 1A (Fig. 1b) indicates an increase in absorbance associated with the satellite peaks, and the appearance of new bands at 1040 cm−1 (distorted νC–C) and 845 cm−1 (distorted βC–H; see discussion in ESI† and Fig. S10 for the deconvolution), consistent with the presence of multiple coordinated THF species and the formation of local clusters which are commonly associated with coordination centres.19,21
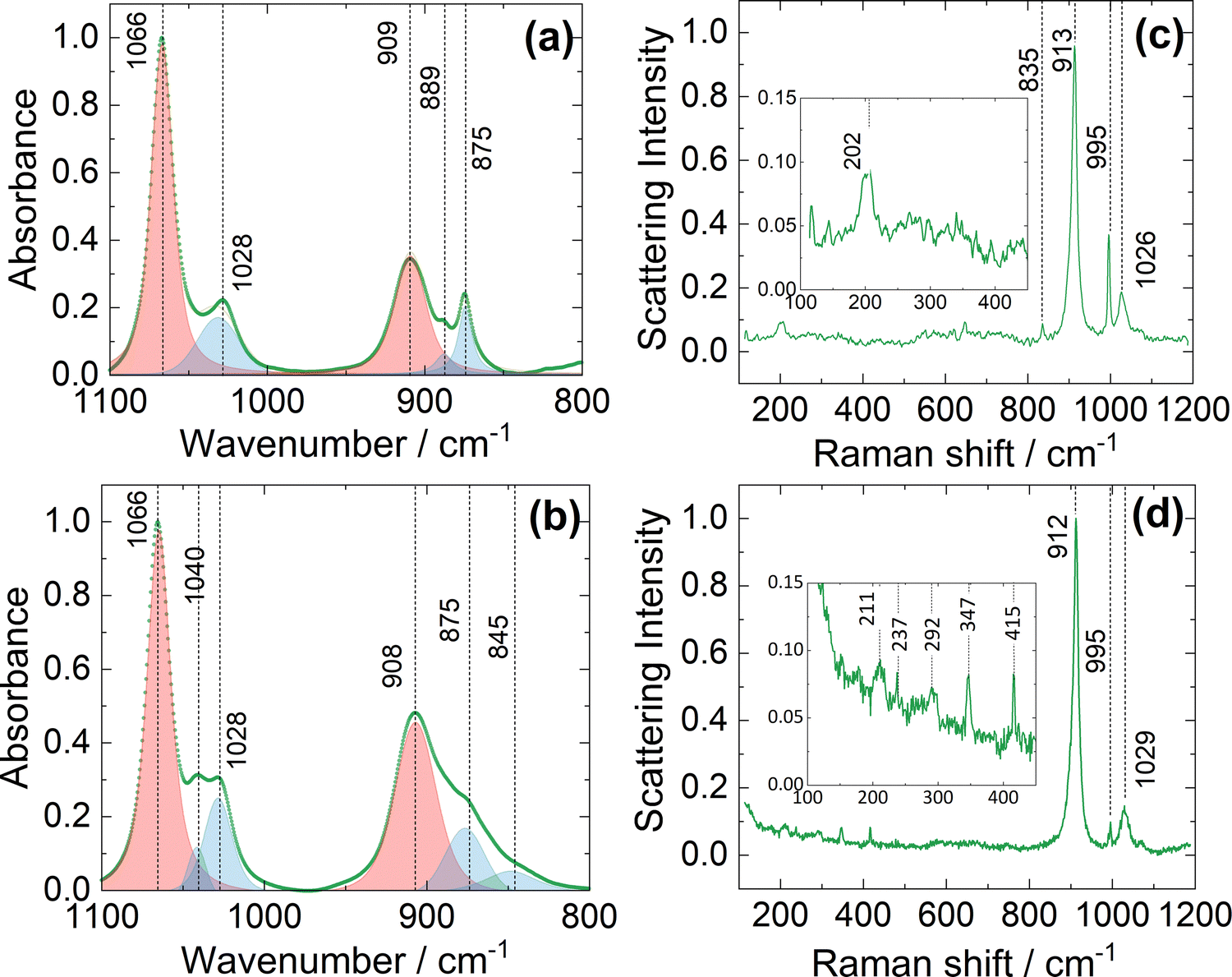 | ||
| Fig. 1 Vibrational spectra of electrolytes 1B (a and c) and 1A (b and d). Panels (a) and (b) show FTIR spectra in which the absorbance scale is normalised to the most intense peak and the dominant peaks are deconvoluted using a Voigt profile fitting: vibrational modes dominant in pure THF are shaded in red and those associated with coordinated THF are shaded in blue. Panels (c) and (d) show Raman spectra in which the scattering intensity scale is normalised to the most intense peak. The lower Raman shift regions are magnified in the insets of (c) and (d). | ||
The Raman spectrum of the pure solvent (Fig. S9, ESI†) is dominated by a strong peak at 913 cm−1 (Cα–Cβ asymmetric stretching), and weaker modes at 1030 cm−1 (Cα–Cβ symmetric stretching) and 1069 cm−1 (C–O–C asymmetric stretching).20 Electrolyte 1B exhibits the same major Raman peaks, albeit with a slight downshift of the 1030 cm−1 band to 1026 cm−1 (Fig. 1c), likely reflecting the different coordination environment of the THF. In addition, a single Raman peak is observed at 202 cm−1 (arising from solvated Mg cationic species), and features at 835 cm−1 and 995 cm−1 which arise from the metal-phenyl ring vibrations present in the anion [AlPh4]−.15,22 For electrolyte 1A, Raman spectra (Fig. 1d) show multiple low wavenumber scattering modes (shown as inset). The peak at 211 cm−1 indicates presence of Mg2+ in solution, while peaks at 237 cm−1 and 292 cm−1 are thought to arise from multiple organohaloaluminate anions.15 The 347 cm−1 mode is attributed to solvated chloroaluminate species.15 We are not able to unambiguously assign a vibrational mode to the peak at 415 cm−1, but this has been reported at low temperatures in 1:1 stoichiometric solutions of PhMgCl:AlCl3 in THF.15 Together, these observations indicate the presence of fewer coordination environments and a cleaner chemical equilibrium in solution for the electrolyte 1B synthesised by the atom-efficient method reported here. The presence of multiple species in equilibrium may result in poor electrochemical performance, since organochloroaluminates are particularly known for reducing anodic stability and corroding current collectors. Moreover, the complex speciation associated with the uncontrolled stoichiometry makes mechanistic interpretation more challenging.
The ionic conductivity of electrolytes prepared according to eqn (3) by in situ generation (1B) and from crystallized samples dissolved in THF (1C), were then determined at room temperature and compared with that of the sample generated according to eqn (1) (1A; Fig. S11, ESI†). The atom-efficient samples exhibited conductivities of 1.046 mS cm−1 (1B) and 1.045 mS cm−1 (1C) (at 0.1 M with respect to the formula [Mg2Cl3]+ [AlPh4]−), which is comparable to that for 1A (1.060 mS cm−1, using 0.5 M PhMgCl/0.25 M AlCl3, i.e. 0.0625 M [Mg2Cl3]+ [AlPh4]−). However, the ionic conductivities are complicated by the presence of additional, potentially more mobile, charge-carrying by-products in 1A compared to 1B and 1C, so a quantitative comparison is not recommended. Nevertheless, to observe acceptable ionic conductivity whilst simplifying the ionic profile and reducing the potentially corrosive chloride byproduct in the electrolyte solution is a clear benefit of our approach.
To evaluate the plating and stripping behaviour of the APC electrolyte synthesised according to our atom-efficient method, polarisation tests were performed in symmetric cells using Mg-metal electrodes separated by fibreglass separators at a current density of 200 μA cm−2. Initial electrochemical tests demonstrated that the electrolytes are stable up to 3.5 V (vs. Mg), in accord with previously studied APC solutions.23 Voltage profiles of the APC electrolyte prepared according to eqn (3), both from an in situ generated sample (1B) and by dissolving a crystallised sample in THF (1C), are shown in Fig. 2a and b, respectively. For the electrolyte sample 1C, the average cell polarisation is largely constant, indicating excellent stability. This observed profile is similar to previous constant current cycle reports for solutions of electrolyte 1A.24 For the in situ generated APC sample 1B, there is an observed increase in cell voltage with cycling which suggests a slight increase in resistance towards plating and stripping of Mg. Electrochemical performance for each solution was further analysed by CV against a Mg-metal anode and Mo6S8 cathode, acquired at a scan rate of 0.1 mV s−1 (Fig. 2c and d). The cathode intercalation process in the Chevrel phase follows a two-step mechanism but for both solutions this is manifested as a single broad cathodic peak. Both CV profiles demonstrate chemical reversibility over the 30 cycles collected, albeit with a slight shift in peak position and decrease in peak current with cycling for the in situ generated solution 1B. The reasons for this are unclear but it is possible that small differences in electrolyte concentration affects the interfacial properties of the Chevrel phase cathode. Gratifyingly, there is no discernible decline in electrochemical performance when comparing our atom-efficient electrolytes with respect to APC solutions reported previously.18
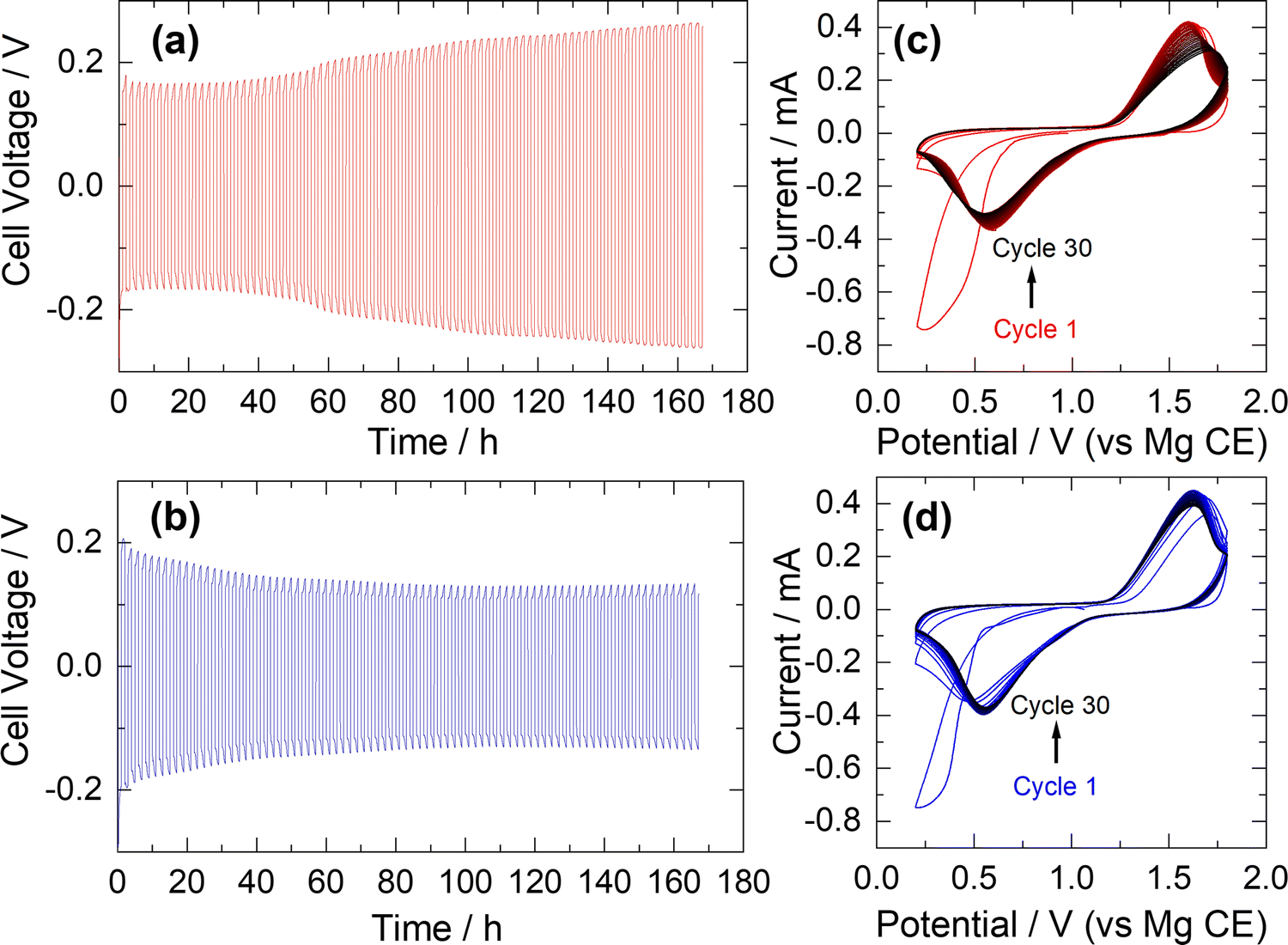 | ||
| Fig. 2 Polarisation measurements of symmetrical cells with Mg-metal electrodes and (a) in situ generated APC, 1B, and (b) crystallised APC dissolved in THF, 1C, at current densities of 200 μA cm−2. Cyclic voltammograms of (c) in situ generated APC, 1B, and (d) crystallized APC dissolved in THF, 1C, at scan rates of 0.1 mV s−1 in cells comprising the electrolyte, a Mg-metal anode and the Mo6S8 Chevrel phase cathode material (75% cathode active material, 15% carbon black, 10% PTFE binder). | ||
In summary, an atom-economic synthetic protocol has been reported which provides high-yielding access to a pure sample of the all-phenyl complex, [Mg2Cl3]+ [AlPh4]−, paving the way to facile modification of this benchmark electrolyte for rechargeable magnesium batteries. Electrochemical performance measurements indicate good stability, with both the recrystallised and in situ generated electrolytes facilitating reversible Mg plating and stripping, consistent with the previously reported behaviour of APC. Vibrational spectroscopy demonstrates that the new synthetic protocol leads to tighter control over the range of solution phase species compared to the more established method of producing APC. Our new synthetic approach therefore offers a potential route to improved reproducibility in performance, supporting enhanced mechanistic understanding of the behaviour of this electrolyte during electrochemical cycling.
Strathclyde and NPL groups thank the EPSRC Impact Accelerator Account (Grant EP/R51178X/1) for generous funding. SAC and JJP gratefully acknowledge the EPSRC Centre for Doctoral Training in Energy Storage (EP/L016818/1); SAC and RK acknowledge support from the Faraday Institution (grant FIRG018). We also acknowledge the National Measurement System of the UK Department of Business, Energy and Industrial Strategy for support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Muldoon, C. B. Bucur, A. G. Oliver, J. Zajicek, G. D. Allred and W. C. Boggess, Energy Environ. Sci., 2013, 6, 482–487 RSC.
- R. Mohtadi and F. Mizuno, Beilstein J. Nanotechnol., 2014, 5, 1291–1311 CrossRef PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- C. You, X. Wu, X. Yuan, Y. Chen, L. Liu, Y. Zhu, L. Fu, Y. Wu, Y.-G. Guo and T. van Ree, J. Mater. Chem. A, 2020, 8, 25601–25625 RSC.
- Z. Liang and C. Ban, Angew. Chem., Int. Ed., 2021, 60, 11036–11047 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Angew. Chem., Int. Ed., 2017, 56, 12064–12084 CrossRef CAS PubMed.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775–780 CrossRef CAS.
- P. Canepa, S. Jayaraman, L. Cheng, N. N. Rajput, W. D. Richards, G. S. Gautam, L. A. Curtiss, K. A. Persson and G. Ceder, Energy Environ. Sci., 2015, 8, 3718–3730 RSC.
- E. N. Keyzer, H. F. J. Glass, Z. Liu, P. M. Bayley, S. E. Dutton, C. P. Grey and D. S. Wright, J. Am. Chem. Soc., 2016, 138, 8682–8685 CrossRef CAS PubMed.
- K. A. See, Y.-M. Liu, Y. Ha, C. J. Barile and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2017, 9, 35729–35739 CrossRef CAS PubMed.
- Y. Li, S. Guan, H. Huo, Y. Ma, Y. Gao, P. Zuo and G. Yin, Adv. Funct. Mater., 2021, 31, 2100650 CrossRef CAS.
- Z. Zhao-Karger, X. Zhao, O. Fuhr and M. Fichtner, RSC Adv., 2013, 3, 16330–16335 RSC.
- E. V. Brouillet, A. R. Kennedy, K. Koszinowski, R. McLellan, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2016, 45, 5590–5597 RSC.
- E. V. Brouillet, M. Amores, S. A. Corr and S. D. Robertson, Inorg. Chem. Front., 2020, 7, 2305–2312 RSC.
- N. Pour, Y. Gofer, D. T. Major and D. Aurbach, J. Am. Chem. Soc., 2011, 133, 6270–6278 CrossRef CAS PubMed.
- D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef CAS.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- O. Mizrahi, N. Amir, E. Pollak, O. Chusid, V. Marks, H. Gottlieb, L. Larush, E. Zinigrad and D. Aurbach, J. Electrochem. Soc., 2008, 155, A103 CrossRef CAS.
- I.-T. Kim, K. Yamabuki, M. Sumimoto, H. Tsutsumi, M. Morita and N. Yoshimoto, J. Power Sources, 2016, 323, 51–56 CrossRef CAS.
- B. Cadioli, E. Gallinella, E. Coulombeau, H. Jobic and G. Berthier, J. Phys. Chem., 1993, 97, 7844–7856 CrossRef CAS.
- D. T. Bowron, J. L. Finney and A. K. Soper, J. Am. Chem. Soc., 2006, 128, 5119–5126 CrossRef CAS PubMed.
- T. Liu, J. T. Cox, D. Hu, X. Deng, J. Hu, M. Y. Hu, J. Xiao, Y. Shao, K. Tang and J. Liu, Chem. Commun., 2015, 51, 2312–2315 RSC.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef CAS.
- X. Chen, S. Wei, F. Tong, M. P. Taylor and P. Cao, Electrochim. Acta, 2021, 398, 139336 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, FTIR and Raman spectra. See DOI: https://doi.org/10.1039/d2cc03290b |
| This journal is © The Royal Society of Chemistry 2022 |