
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to high value sp3-rich frameworks using photocatalyzed [2 + 2]-cycloadditions of γ-alkylidene–γ-lactams†‡
Dimitris
Kalaitzakis§
 ,
Ioannis
Kampouropoulos§
,
Manolis
Sofiadis§
,
Tamsyn
Montagnon
and
Georgios
Vassilikogiannakis
*
,
Ioannis
Kampouropoulos§
,
Manolis
Sofiadis§
,
Tamsyn
Montagnon
and
Georgios
Vassilikogiannakis
*
Department of Chemistry, University of Crete, Vasilika Vouton, 71003, Crete, Greece. E-mail: vasil@uoc.gr
First published on 22nd June 2022
Abstract
By harnessing an energy transfer process, new photocatalyzed [2 + 2]-cycloadditions occurring between γ-alkylidene–γ-lactams and unsaturated substrates have been developed. The reaction mode is particularly powerful because it leads to the formation of different high value sp3-rich frameworks and further diversity can be introduced through cascade sequences wherein strain releasing opening of the cyclobutane intermediates gives access to complex polycyclic alkaloid frameworks.
Achieving greater structural novelty1 and a higher sp3 carbon count2 have been identified as key ways to improve both the quality of pharmaceutical hits and the frequency with which they can be found. Thus, these two features have become essential prerequisites when developing new screening sets. As a result, three-dimensionally rich spirocyclic compounds3 have attracted particular attention. Interest that has been bolstered by the fact that the rigidity of these highly saturated frameworks endows the compounds with further benefits; such as, the lowering of the entropy penalty incurred when docking at a receptor site3a and the potential to reduce receptor promiscuity.1,2b
During our work on functionalizing γ-lactams,4 we noticed that lactams of type 1 (1a or 1b, Scheme 1A) had an inclination to isomerize substantially (from E to Z) when irradiated with blue light in the presence of [Ru(bpy)3]Cl2 (1a/b → 2a/b). This behavior was also seen with Ir(ppy)3, albeit with lower final Z/E ratios; whereas, the direct irradiation of 1 without a catalyst led to only 8.5% isomerization. Lactam 1a′ proved to be inert to the isomerization conditions. Interestingly, this observation is in keeping with the known isomerization of the structurally similar heam-metabolite, bilirubine, which forms the basis of light therapy for neonatal jaundice.5 Thus, we began to develop a strategy predicated on the hypothesis that this isomerization involved an energy transfer reaction which might be harnessed to give a series of novel photocatalytic [2 + 2]-cycloadditions.
 | ||
| Scheme 1 (A) Observed E–Z isomerization of unsaturated lactams of type 1. (B) Photocatalytic [2 + 2]-cycloaddition reaction modes. | ||
The initial target groups would be architecturally complex sp3-rich cyclobutanes formed by intra- or intermolecular reactions. Cyclobutanes not only feature in many biologically active compounds,6 but are also useful intermediates due to their potential to undergo ring strain releasing reactions.7 There are other methods to make cyclobutanes,8 but these are mostly eclipsed by the widespread use of [2 + 2]-photocycloadditions.9 Within this class the historic variant involves the olefin absorbing UV light directly.9 Milder and newer methods using visible light and a photocatalyst operate either via an energy transfer (EnT)10 or single electron transfer (SET) mechanisms.11 The photocatalytic variants require some form of extended conjugation because it affords the substrates with lower triplet state energies and lower redox potentials to facilitate the generation of stabilized open shell intermediates (resonance stabilization, Scheme 1B). This feature has, therefore, often limited reaction scope to substrates bearing aromatic substituents. Photocatalyzed isomerization of conjugated alkenes from E to Z is also an energy transfer process sometimes used to access cis alkenes from their more readily synthesized trans analogs.12 It was for this reason that we believed the previously observed isomerization might indicate that γ-alkylidene–γ-lactams could be uniquely suitable non-aromatic substrates for photocatalyzed [2 + 2]-cycloadditions to form new cyclobutanes via an energy transfer mechanism. Notably, a number of the N-substituted cyclobutane products would share key structural features with natural alkaloids and pharmaceuticals.3a,13
We began our investigation with the facile synthesis of the γ-alkylidene–γ-lactam 4a from furan 3a and allylamine using our previously developed photocatalytic protocol (Scheme 2).4f Substrate 4a has a double bond on the amide side chain that could partner with the exocyclic double bond in a cross [2 + 2]-cyclization to yield the tightly packed sp3-rich polycycle 5a. Indeed, when 4a was treated with PC1 in CH3CN (0.1 M) and irradiated with blue LEDs, the desired reaction occurred; however, the reactions did not reach completion even after 24 h (entries 1 and 2). In surveying photocatalysts, it was found that organic dyes PC2, PC3 and methylene blue (MB), all of which have only moderate triplet energies,14 were not competent catalysts for the reaction (entries 3–5). In contrast, PCs 4 and 5, which have high triplet energies,14 proved to be highly efficient converting 4a into 5a within 8 h in high yield (92 and 94% yield, respectively). The reaction did not proceed in the absence of the photocatalyst or light; although with the former, a small degree of double bond isomerisation did take place (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z, 2.4:1, entry 8). Extended conjugation in the substrates is a requirement as shown by the inert nature of 4a′ to the reaction conditions (entry 10). We expanded the set of substrates 4 and showed that they all reacted efficiently (85–95% yield, 1.6:1–2:1 dr, Scheme 3). Substrates that include groups sometimes sensitive to radicals and/or other photocatalyzed reactions they also successfully provided the desired products (5d and 5e). Interestingly, framework 5 contains the skeleton of the natural amino acid 2,4-methanoproline,15 which, along with its analogs, has been shown to exhibit a range of biological activities.16 It was also possible to relocate the partner double bond in these intramolecular [2 + 2]-photocycloadditions; for example, substrates of type 6 afforded polycyclic products 7 with excellent diastereoselectivity (7a–7c, 92–96% yield, Scheme 3).
:Z, 2.4:1, entry 8). Extended conjugation in the substrates is a requirement as shown by the inert nature of 4a′ to the reaction conditions (entry 10). We expanded the set of substrates 4 and showed that they all reacted efficiently (85–95% yield, 1.6:1–2:1 dr, Scheme 3). Substrates that include groups sometimes sensitive to radicals and/or other photocatalyzed reactions they also successfully provided the desired products (5d and 5e). Interestingly, framework 5 contains the skeleton of the natural amino acid 2,4-methanoproline,15 which, along with its analogs, has been shown to exhibit a range of biological activities.16 It was also possible to relocate the partner double bond in these intramolecular [2 + 2]-photocycloadditions; for example, substrates of type 6 afforded polycyclic products 7 with excellent diastereoselectivity (7a–7c, 92–96% yield, Scheme 3).
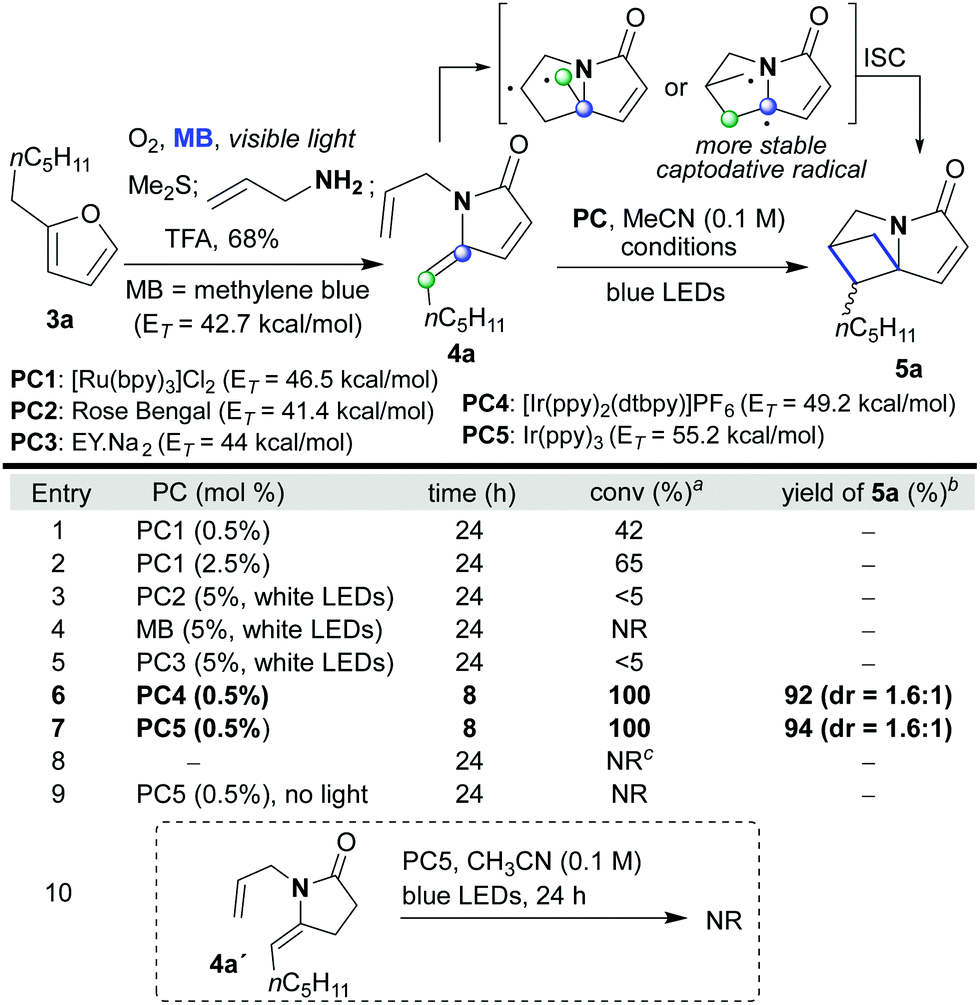 | ||
| Scheme 2 Optimization of the intramolecular [2 + 2]-cycloaddition of 4a. aDetermined by 1H-NMR of the crude reaction mixture. bIsolated yield. cE/Z isomerization was observed (E/Z = 2.4:1). | ||
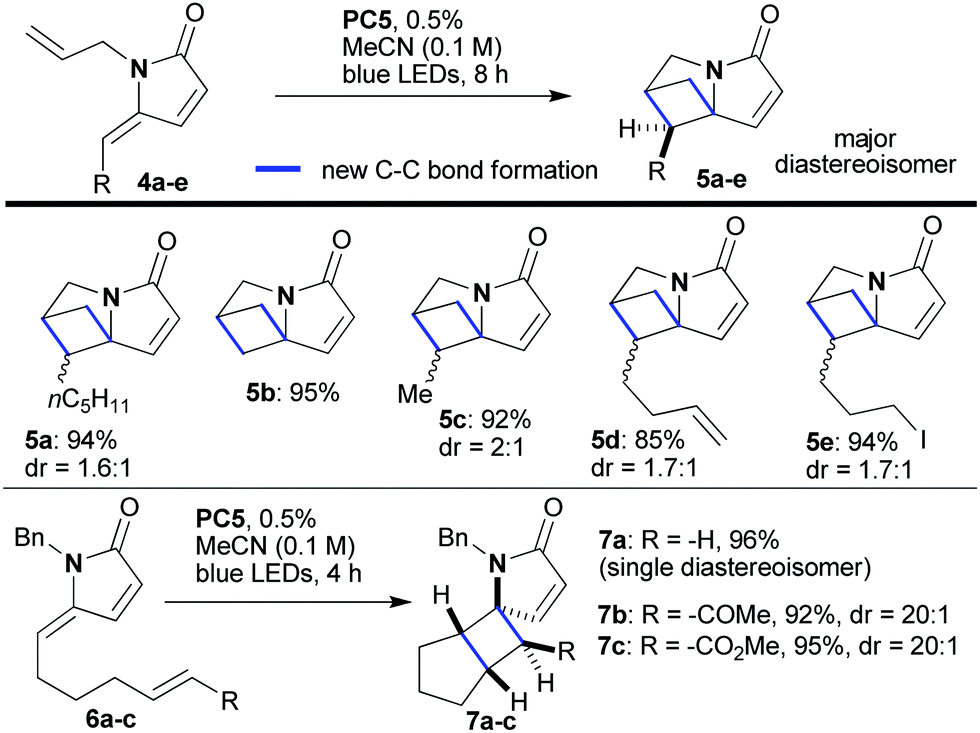 | ||
| Scheme 3 Photocatalytic intramolecular [2 + 2]-cycloaddition reactions of unsaturated lactams of type 4 and 6. | ||
Next, we sought to move on to the more challenging intermolecular variant of the reaction in which the γ-alkylidene–γ-lactam would react with a second molecule containing an electron deficient double bond. The reaction would give us access to relatively rare 5,4-spirocycles of type 9 (Scheme 4). A variety of combinations were tested in which lactams 8 were combined with an excess (10 equiv.) of the unsaturated carbonyl compound under the previously optimized conditions (0.5% of PC5 in CH3CN). All the reactions worked well, affording the products 9 in good to high yield (9a–9g, 60–82%) and with excellent regioselectivity. Despite the fact that every reaction furnished a mixture of diastereomers (1.6:1, by 1H NMR), the products with a keto or aldehyde group were epimerized to a single stereoisomer upon chromatographic purification (9a–9c, 9e–9g). Only in the case of 9d (bearing an ester group less able to drive epimerization) did the reaction afford a 1:1 ratio of diastereomers which remained unchanged during purification. The intermolecular [2 + 2]-cycloaddition with the more electron-rich alkene styrene also works, but it is quite messy.
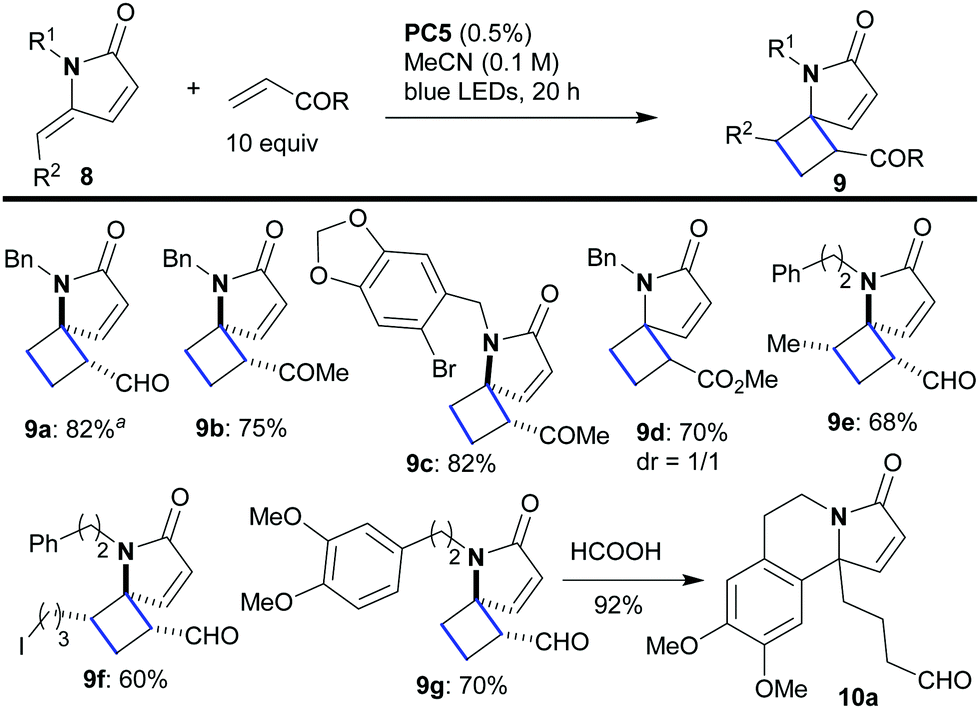 | ||
| Scheme 4 Photocatalytic synthesis of spirocyclic compounds of type 9via intermolecular [2 + 2]-cycloaddition. aThe reaction occurred similarly using 0.5% of PC4. | ||
An interesting result emerged when product 9g was treated with formic acid. The acidic conditions catalyzed a retro-Mannich reaction, presumably, driven by opening of the strained cyclobutane ring, which was followed by nucleophilic attack of the aromatic group on the resulting N-acyliminium cation, to furnish lactam 10a as a sole product (92% yield). Such N-containing aromatic polycycles of type 10 constitute the basic skeleton of many natural alkaloids (10a has been used as a precursor for the synthesis of erysotramidine, Scheme 5).17a,b The retro-Mannich ring opening of 9g7 was intriguing because it opened up the possibility for developing cascade sequences that could diversify the type of products accessible to us through this methodology (8 → 10–12, Scheme 5). In practice, lactams 8, with internal nucleophilic groups at either R1 or R2, first underwent the [2 + 2]-photocycloaddition using an excess (10 equiv.) of acrolein or methyl vinyl ketone. The intermediates of type 9 were not isolated, but, instead, acid was added to attain a range of alternative products. More specifically, when R1 bore an electron rich aromatic group, treatment with formic acid catalyzed further transformation into products of type 10 (overall 8 → 10a–10c in one pot with yields ranging from 60 to 69%). Similarly, when an appropriate nucleophile was appended to R2, various products could be attained depending on the nature of that nucleophile. For example, spirocycles 11a and 11b were formed diastereoselectively in good yields in a single operation starting from lactam 8 (for 11a formic acid was used while for 11b PTSA was employed, Scheme 5). These compounds constitute the skeleton of marine Clavelina17c and marineosins17d alkaloids. Furthermore, chain homologation (a form of C–H activation) could be achieved for substrates with no internal nucleophile through a similar acid catalyzed cyclobutane ring opening reaction sequence. Thus, lactams of type 12 could be synthesized in one pot (8 → 12a–12c, yields 61–72%, Scheme 5). The retro-Mannich ring opening does not work with ester 9d, because the ester group is less electron withdrawing compared to aldehydes and ketones.
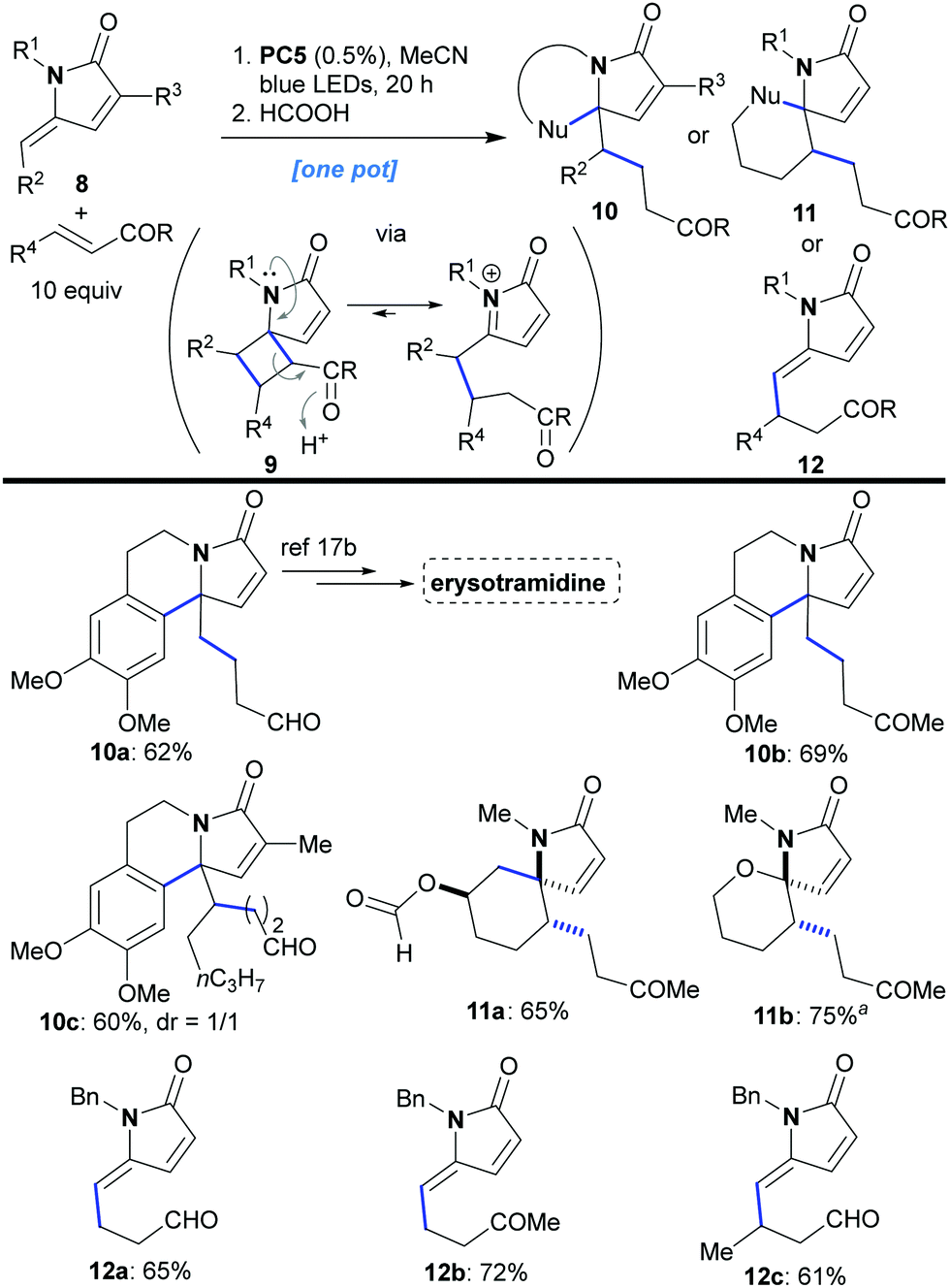 | ||
| Scheme 5 Functionalization of γ-alkylidene–γ-lactams based on photocatalytic [2 + 2]-cyclization followed by cyclobutane ring opening. aThe second step was performed using PTSA·H2O (2 equiv.) in CH2Cl2. | ||
Finally, we wanted to investigate the reaction of γ-alkylidene–γ-lactams with 2,3-dimethylbuta-1,3-diene. 1,3-Dienes have previously been utilized in photocycloadditions, producing either vinylcyclobutans or cyclohexenes depending on which mechanism is in operation (EnT or ET).18 When the optimized conditions were applied to conjugated lactams of type 8 in the presence of 2,3-dimethylbuta-1,3-diene (4 equiv., Scheme 6), spirocyclic compounds of type 13 were formed as the sole products (13a–13c, 70–74% yield, Scheme 6). Monitoring the reaction by 1H NMR, we observed the formation of the vinylcyclobutane 9h (Scheme 6) during the early stages of the process. This intermediate subsequently disappeared and spirocyclic product 13 was formed. This observation implies two sequential steps; an initial [2 + 2]-cycloaddition yielding 9h with subsequent rearrangement to 13 to give overall a [4 + 2]-transformation.
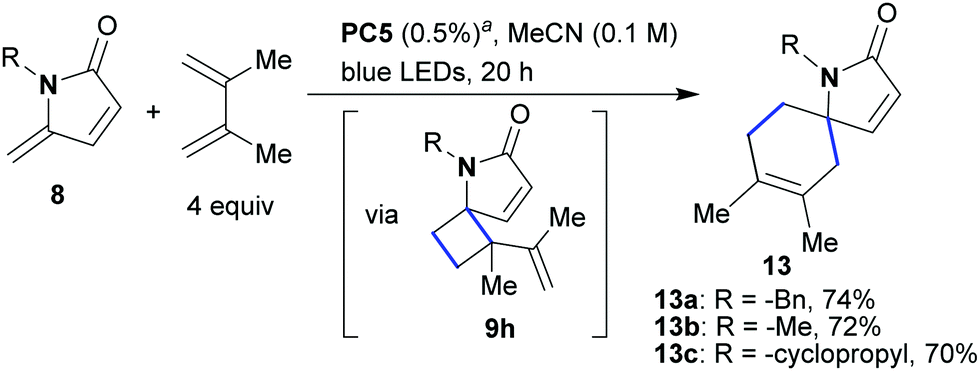 | ||
| Scheme 6 Photocatalytic cyclization of unsaturated γ-alkylidene–γ-lactams with 2,3-dimethylbuta-1,3-diene. aThe reactions occurred similarly using 0.5% of PC4. | ||
Stern–Volmer quenching and voltammetry studies were undertaken (see, ESI‡) and the results were consistent with an energy transfer from the excited state of the photocatalyst to the γ-alkylidene–γ-lactam being the mechanistic mode operating in these [2 + 2] cycloadditions. More precisely, compound 8a quenches the excited state of PC5 at a significantly higher rate than methylvinyl ketone or 2,3-dimethylbuta-1,3-diene and the redox potentials of PC4 and PC5 are not appropriate for initiation of an electron transfer pathway with 4a (see ESI‡).
Overall, a series of mild and highly efficient methodologies to access a diverse range of unusual rigid sp3-rich spirocycles, complex alkaloid frameworks or chain homologated products have been developed. Synthesis of the latter two groups was achieved by incorporating ring strain relieving cyclobutane opening into highly effective one pot cascade reaction sequences. The methodologies all rely initially on a novel photocatalyzed (visible light + PC) [2 + 2]-cycloaddition between γ-alkylidene–γ-lactams and an unsaturated partner which occurs via an energy transfer mechanism.
This research has been co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code:T2EDK-02364). We thank the Mass Spectrometry Facility of the University of Crete for obtaining the HRMS data. We are grateful to Prof. Athanassios G. Coutsolelos and Dr Georgios Charalambidis for help with voltammetry and Stern–Volmer studies.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-L. Reymond, Acc. Chem. Res., 2015, 48, 722 CrossRef CAS PubMed.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (b) P. A. Clemons, N. E. Bodycombe, H. A. Carrinski, J. A. Wilson, A. F. Shamji, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18787 CrossRef CAS PubMed.
- (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed; (b) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673 CrossRef CAS PubMed; (c) K. Hiesinger, D. Dar’in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150 CrossRef CAS PubMed.
- (a) D. Kalaitzakis, A. Bosveli, K. Sfakianaki, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2021, 60, 4335 CrossRef CAS PubMed; (b) S. Hoxha, D. Kalaitzakis, A. Bosveli, T. Montagnon and G. Vassilikogiannakis, Org. Lett., 2021, 23, 5354 CrossRef CAS; (c) T. Montagnon, D. Kalaitzakis, M. Sofiadis and G. Vassilikogiannakis, Org. Biomol. Chem., 2020, 18, 180 RSC; (d) G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, ChemPhotoChem, 2018, 2, 860 CrossRef CAS PubMed; (e) D. Kalaitzakis, M. Triantafyllakis, G. I. Ioannou and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2017, 56, 4020 CrossRef CAS; (f) D. Kalaitzakis, A. Kouridaki, D. Noutsias, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2015, 54, 6283 CrossRef CAS PubMed.
- (a) R. Bonnett, J. E. Davies and M. B. Hursthouse, Nature, 1976, 262, 327 CrossRef CAS PubMed; (b) K. Mreihil, A. F. McDonagh, B. Nakstad and T. W. R. Hansen, Pediatr. Res., 2010, 67, 656 CrossRef CAS.
- (a) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136 RSC; (b) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254 RSC.
- J. D. Winkler, C. M. Bowen and F. Liotta, Chem. Rev., 1995, 95, 2003 CrossRef CAS.
- (a) M. R. Fructos and A. Prieto, Tetrahedron, 2016, 72, 355 CrossRef CAS; (b) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918 CrossRef CAS PubMed.
- (a) N. Hoffmann, Chem. Rev., 2008, 108, 1052 CrossRef CAS PubMed; (b) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (c) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed; (d) B. Cox, K. I. Booker-Milburn, L. D. Elliott, M. Robertson-Ralph and V. Zdorichenko, ACS Med. Chem. Lett., 2019, 10, 1512 CrossRef CAS PubMed.
- For selected reviews see: (a) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190 RSC; (b) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed ; For selected non-enantioselective examples see: ; (c) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329 CrossRef CAS PubMed; (d) Y.-Q. Zou, S.-W. Duan, X.-G. Meng, X.-Q. Hua, S. Gao, J.-R. Chen and W.-J. Xiao, Tetrahedron, 2012, 68, 6914 CrossRef CAS; (e) A. E. Hurtley, Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 8991 CrossRef CAS PubMed; (f) V. Mojr, E. Svobodová, K. Straková, T. Neveselý, J. Chudoba, H. Dvořáková and R. Cibulka, Chem. Commun., 2015, 51, 12036 RSC; (g) J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, J. Am. Chem. Soc., 2017, 139, 9807 CrossRef CAS PubMed; (h) M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624 CrossRef CAS PubMed; (i) R. F. Higgins, S. M. Fatur, N. H. Damrauer, E. M. Ferreira, A. K. Rappé and M. P. Shores, ACS Catal., 2018, 8, 9216 CrossRef CAS; (j) F. M. Hörmann, T. S. Chung, E. Rodriguez, M. Jakob and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 827 CrossRef PubMed; (k) T. Neveselý, C. G. Daniliuc and R. Gilmour, Org. Lett., 2019, 21, 9724 CrossRef PubMed; (l) L. D. Elliott, S. Kayal, M. W. George and K. Booker-Milburn, J. Am. Chem. Soc., 2020, 142, 14947 CrossRef CAS PubMed.
- For selected examples see: (a) N. L. Bauld, Tetrahedron, 1989, 45, 5307 CrossRef CAS; (b) M. A. Miranda and H. García, Chem. Rev., 1994, 94, 1063 CrossRef CAS; (c) T. P. Yoon, ACS Catal., 2013, 3, 895 CrossRef CAS PubMed; (d) M. Riener and D. A. Nicewicz, Chem. Sci., 2013, 4, 2625 RSC.
- For relative reviews see: (a) Y. Yokoyama, Chem. Rev., 2000, 100, 1717 CrossRef CAS PubMed; (b) J. B. Metternich and R. Gilmour, Synlett, 2016, 2541 CAS; (c) J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 13654 CrossRef CAS PubMed.
- (a) V. M. Dembitsky, J. Nat. Med., 2008, 62, 1 CAS; (b) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559 CrossRef CAS PubMed; (c) A. Sergeiko, V. V. Poroikov, L. O. Hanuš and V. M. Dembitsky, Open J. Med. Chem., 2008, 2, 26 CrossRef CAS PubMed.
- For the triplet energy of the current applied photosensitizers see: (a) For PC1: D. P. Rillema, G. Allen, T. J. Meyer and D. Conrad, Inorg. Chem., 1983, 22, 1617 CrossRef CAS; (b) For PC2, PC3 and MB: T. Shen, Z.-G. Zhao, Q. Yu and H.-J. Xu, J. Photochem. Photobiol., A, 1989, 47, 203 CrossRef CAS; (c) For PC4: J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763 CrossRef CAS PubMed; (d) For PC5: A. Singh, K. Teegardin, M. Kelly, K. S. Prasad, S. Krishnan and J. D. Weaver, J. Organomet. Chem., 2015, 776, 51 CrossRef CAS.
- (a) E. A. Bell, M. Y. Qureshi, R. J. Pryce, D. H. Janzen, P. Lemke and J. Clardy, J. Am. Chem. Soc., 1980, 102, 1409 CrossRef CAS; (b) C. V. Stevens, G. Smagghe, T. Rammeloo and N. De Kimpe, J. Agric. Food Chem., 2005, 53, 1945 CrossRef CAS PubMed.
- B. Cox, J. Duffy, V. Zhdorichenko, C. Bellanger, J. Hurcum, B. Laleu, K. I. Booker-Milburn, L. D. Elliot, M. Robertson-Ralph, C. J. Swain, S. J. Bishop, I. Hallyburton and M. Anderson, ACS Med. Chem. Lett., 2020, 11, 2497 CrossRef CAS PubMed.
- (a) M. E. Amer, M. Shamma and A. J. Freyer, J. Nat. Prod., 1991, 54, 329 CrossRef CAS; (b) A. J. Blake, C. Gill, D. A. Greenhalgh, N. S. Simpkins and F. Zhang, Synthesis, 2005, 3287 CAS; (c) S. M. Weinreb, Chem. Rev., 2006, 106, 2531 CrossRef CAS PubMed; (d) C. Boonlarppradab, C. A. Kauffman, P. R. Jensen and W. Fenical, Org. Lett., 2008, 10, 5505 CrossRef CAS PubMed.
- Energy transfer based cyclizations with 1,3-dienes afford vinylcyclobutanes. For examples see: (a) Ref. 10c, e, j; (b) F. Tang, L. Tang, Z. Guan and Y.-H. He, Tetrahedron, 2018, 74, 6694 CrossRef CAS; (c) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391 CrossRef CAS PubMed; (d) Recently (ref. 10i), using isoprene as the 1,3-diene an overall-[4 + 2] reaction was observed.
Footnotes |
| † Dedicated to Prof. Gerasimos J. Karabatsos for his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: [DETAILS]. See DOI: https://doi.org/10.1039/d2cc03009h |
| § These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |