
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydride state accumulation in native [FeFe]-hydrogenase with the physiological reductant H2 supports its catalytic relevance†
Moritz
Senger
 *a,
Tobias
Kernmayr‡
b,
Marco
Lorenzi
b,
Holly J.
Redman
b and
Gustav
Berggren
*b
*a,
Tobias
Kernmayr‡
b,
Marco
Lorenzi
b,
Holly J.
Redman
b and
Gustav
Berggren
*b
aDepartment of Chemistry, Physical Chemistry, Uppsala University, 75120 Uppsala, Sweden. E-mail: moritz.senger@kemi.uu.se
bDepartment of Chemistry, Molecular Biomimetics, Uppsala University, 75120 Uppsala, Sweden. E-mail: gustav.berggren@kemi.uu.se
First published on 30th May 2022
Abstract
Small molecules in solution may interfere with mechanistic investigations, as they can affect the stability of catalytic states and produce off-cycle states that can be mistaken for catalytically relevant species. Here we show that the hydride state (Hhyd), a proposed central intermediate in the catalytic cycle of [FeFe]-hydrogenase, can be formed in wild-type [FeFe]-hydrogenases treated with H2 in absence of other, non-biological, reductants. Moreover, we reveal a new state with unclear role in catalysis induced by common low pH buffers.
Hydrogenases are redox enzymes catalysing molecular hydrogen (H2) uptake and production. The high H2 evolution frequency of prototypical [FeFe]-hydrogenases motivates basic research on the reaction mechanism and inspires catalyst design for a hydrogen economy.1–3 In these enzymes, hydrogen catalysis takes place at a hexa-iron cofactor, called the H-cluster. It is composed of a [4Fe4S]-cluster linked via a cysteine residue to a unique diiron site ([2Fe]) that binds two cyanide (CN−) and three carbonyl (CO) ligands. These ligands serve as intrinsic probes sensitive to infrared spectroscopy and report on reduction and protonation events during catalysis. The diiron site is bridged by an azadithiolate ligand (−SCH2NHCH2S−, ADT) that is proposed to shuttle protons via its amine group to the apical vacancy, the site of hydrogen catalysis, located at the Fe ion (Fed) distal to the [4Fe4S] cluster (Fig. 1). The most studied group of [FeFe]-hydrogenases (Group A) transfers protons to the active site via a conserved Proton Transfer Pathway (PTP) composed of amino acid residues and water molecules.4–6 Recently described [FeFe]-hydrogenases in group D lack this PTP.7,8 Several redox and protonation states have been characterised and proposed as catalytic intermediates.1,2 The starting point of catalysis is the oxidised state Hox. Under inert atmosphere conditions at low pH values and in presence of chemical reductant a blue-shifted variant of Hox is formed, commonly denoted HoxH.9,10 One electron reduction of Hox results in population of two singly reduced states: Hred’ or Hred. Hred’ features a reduced [4Fe4S] cluster while the diiron site is still in an oxidised configuration. Formation of the diiron site reduced state Hred is coupled to a proton uptake event, although the site of protonation is under discussion.11,12 A second reduction step yields the super-reduced state (Hsred) or a hydride state (Hhyd). Hsred is characterised by a reduction of both the [4Fe4S] cluster and the diiron site.13 In contrast, Hhyd features a terminal hydride at Fed, yielding a formally oxidized diiron site (Fig. 1).14–16 As the last step in hydrogen turnover, the terminal hydride of Hhyd is proposed to combine with a proton delivered via the PTP to generate molecular hydrogen.1–3 A hydride state similar to Hhyd but with an additionally protonated ADT bridge has been proposed as the last intermediate in hydrogen evolution.17–20
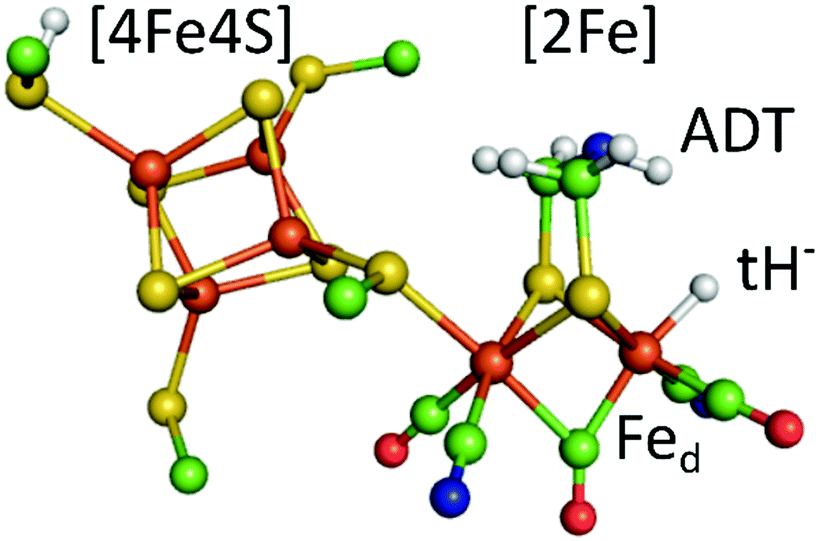 | ||
| Fig. 1 The calculated hydride state structure of the H-cluster in [FeFe]-hydrogenases. The H-cluster consists of a protein bound [4Fe4S] cluster that is covalently linked via a cysteine thiolate ligand to the diiron site ([2Fe]), further ligated by three carbonyl (CO) and two cyanide (CN−) ligands. A terminal hydride (tH−) at the distal iron ion is proposed to combine with a proton delivered by the ADT ligand to yield molecular hydrogen in the last step of hydrogen catalysis. Colour code: orange-iron, yellow-sulphur, green-carbon, red-oxygen, blue-nitrogen, white-hydrogen. The structure is drawn after the PDB coordinates 4XDC optimized for the Hhyd state by DFT calculations. (ref. 33). | ||
A terminal-hydride state of the H-cluster, considered to reflect the Hhyd state, can be stabilised by impairing the enzyme's catalytic function via amino acid variations in the PTP or cofactor alteration. This has enabled its characterization using a range of techniques, including NMR, NRVS, Mössbauer, EPR and FTIR spectroscopy.14–16,18,21,22 In native, fully functional, [FeFe]-hydrogenases the accumulation of a highly similar Hhyd state is commonly achieved by exposure to H2 at low pH values in the presence of the non-physiological reductant sodium dithionite (NaDT, Table S1, ESI†) (with exceptions19,22–24).16,17,19,22,25,26 Based on chronoamperometry experiments it was recently proposed that [FeFe]-hydrogenases are inhibited by NaDT at low pH values (or rather SO2, one of its oxidized by-products), i.e. conditions similar to Hhyd accumulation.27 Considering its in-depth spectroscopic characterization, and proposed central importance in the catalytic cycle, the possibility that the to-date characterized Hhyd-state reflects an inhibited “artefact-state” would represent a significant setback in our mechanistic understanding of [FeFe]-hydrogenase.
Here, we selectively enrich the double reduced states, Hhyd and Hsred, by exposure to molecular hydrogen and follow the respective absorbance changes of cofactor ligand bands by Attenuated Total Reflection Fourier-transform Infrared (ATR-FTIR) Spectroscopy. Data is collected at different pH values and buffers. This study reveals that small carboxylic acids, often used in low pH buffers in enzyme electrochemistry experiments, are non-innocent in formation of a new H-cluster species similar to Hox. At present it is not clear if this is an artefactual off-cycle state, or in fact a critical intermediate that previously have escaped detection. Moreover, we show the accumulation of Hhyd in native [FeFe]-hydrogenase, HydA1 from Chlamydomonas reinhardtii, at mildly basic and low pH values regardless of buffer choice and more importantly in the absence of NaDT. Our findings are well in accordance with the previously characterized Hhyd-state as a reaction intermediate rather than an experimental artefact.
A solution of the HydA1 [FeFe]-hydrogenase was applied to the crystal surface of the ATR-FTIR setup, following removal of any NaDT contaminations by chromatography and buffer exchange. The enzyme solution was subsequently dried and rehydrated under N2 atmosphere to form an auto-oxidised protein film.28,29 The hydrogenase adopted the expected oxidised active-ready state Hox, and was subsequently exposed to H2 gas. The resulting difference spectra are displayed in Fig. 2. As a consequence of H2 uptake, a mixture of the hydride state Hhyd (blue positive bands, 2081, 2066, 1979, 1960, 1860 cm−1) and the super-reduced state Hsred (red positive bands, 2070, 2025, 1954, 1918, 1882 cm−1) was populated while the oxidised state Hox (grey negative bands, 2089, 2071, 1965, 1940, 1802 cm−1) was depopulated. This holds true for different pH values, in a range of different buffers. Fig. 2 displays the ATR-FTIR difference spectra of HydA1 samples adjusted to pH 8 or pH 4 and subsequently exposed to H2 (absolute spectra and pH adjustment in Fig. S1, ESI†). In each spectrum the signature of Hhyd (blue positive bands) is clearly detectable. A larger fraction of Hsred is formed at pH 8 (100 mM Tris buffer) while a larger fraction of Hhyd is populated at pH 4 (100 mM propionate buffer), in both cases a concomitant loss of Hox is observed in contrast to earlier results where a depopulation of HoxH was observed at pH 4 due to the presence of NaDT.9,10,16 However, the preference for Hhyd accumulation at low pH is clearly independent of the presence of NaDT. As Hhyd and Hsred are generally considered to reflect two different tautomers, their relative stability as a function of pH value is arguably a consequence of the protonation state of the H-cluster surroundings. The reaction of auto-oxidised HydA1 films with H2 gas proceeds within one-two seconds while auto-oxidation, observed when H2 was replaced by N2, occurs over 10–20 seconds (compare Fig. S2, ESI†). Both kinetics report indirectly on the gas accessibility and hydration of the protein film in the ATR-FTIR setup. Dehydrated [FeFe]-hydrogenase samples were reported to be protected from exposure to gases.30
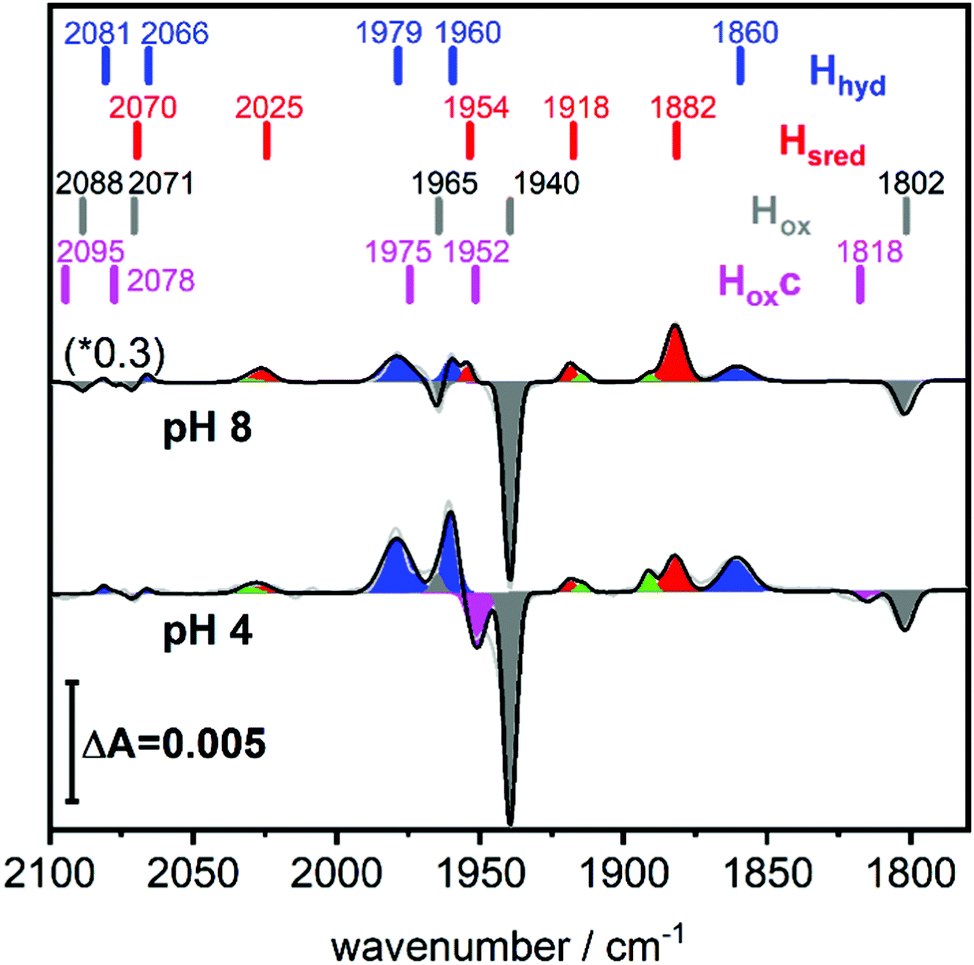 | ||
| Fig. 2 Hydrogen uptake induced ATR-FTIR difference spectra of the CO/CN ligands of HydA1 Upon exposure to H2 the oxidised state Hox (grey negative bands) is depopulated while double reduced redox states Hsred (red positive bands) and Hhyd (blue positive bands) accumulate. For both pH values, pH 8 (top, 100 mM Tris buffer, scaled by 0.3) and pH 4 (bottom, 100 mM propionate buffer), the formation of Hhyd (blue) is observed in the absence of NaDT. Magenta negative bands at 1952 and 1815 cm−1 indicate the newly observed, unknown H-cluster species denoted Hoxc. Green bands belong to Hred. The band positions are indicated by bars. | ||
Fig. 2 (bottom) shows that H2 exposure causes Hhyd accumulation in the absence of NaDT preferentially at low pH values (compare Fig. 2 top). This trend had been reported by several groups before, however always involving NaDT.16,17,19,25 Apart from the recent proposed binding of NaDT (or one of its oxidized products) to the H-cluster, protonation has been suggested to stabilize the reduction at the [4Fe4S] cluster for Hred’9,10,31 and a similar stabilisation effect was reported for Hhyd from both purely computational and spectroscopic approaches.32,33 Our findings verify that NaDT is not required to stabilize Hhyd. The observed pH dependent shift between Hsred and Hhyd instead supports a model of a protonatable site, either near the reduced [4Fe4S] cluster as previously proposed,3,31,34 or e.g. in the PTP. Residual traces of NaDT have been shown to lead to population of HoxH (2089, 2079, 1970, 1945, 1810 cm−1),27 which is absent in our spectra. The negative bands in the pH 4 difference spectrum (Fig. 2 bottom) at 1952 and 1815 cm−1 belong to an unknown band pattern significantly different from HoxH (full spectroscopic signature Fig. 3), and we denote this new state as Hoxc.
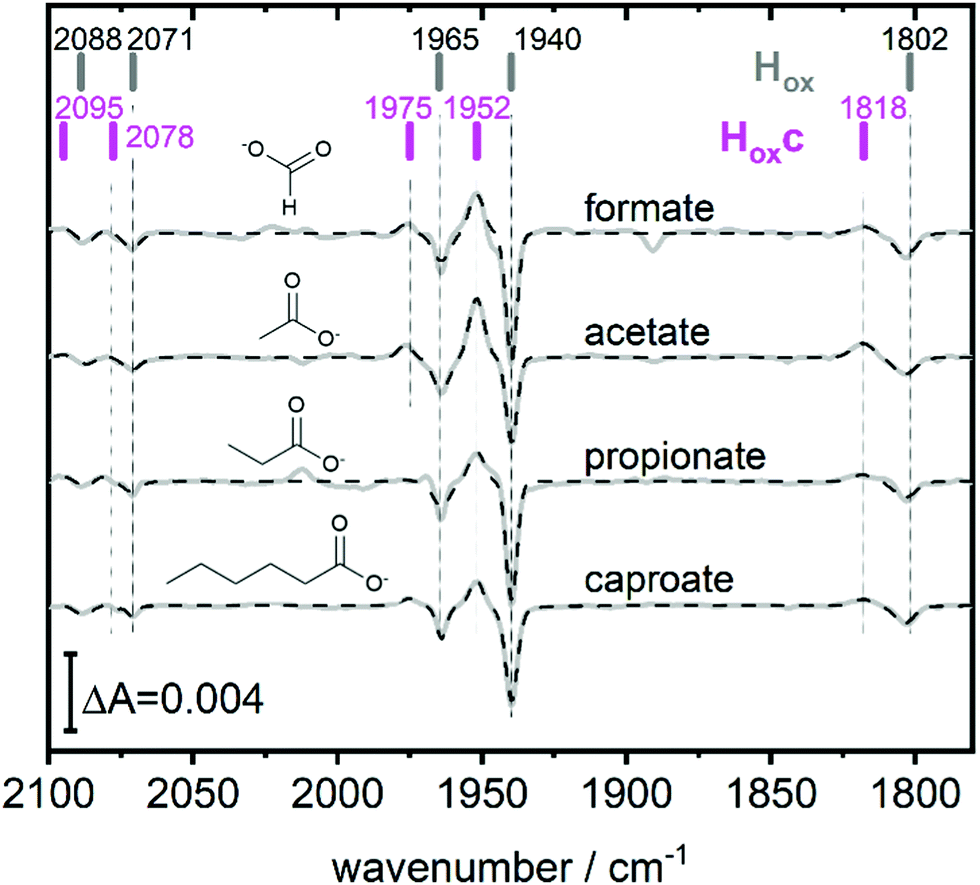 | ||
| Fig. 3 The full spectroscopic signature of Hoxc. Difference spectra highlighting the full band pattern of Hoxc (positive bands) for transitions from pH 8 (10 mM Tris, negative bands) to pH 4 with, from top to bottom, 100 mM formate, 100 mM acetate, 100 mM propionate and 100 mM caproate buffer. Data presented in grey and fit in dashed black. The band positions of Hox and Hoxc are indicated by bars. Acetate and propionate spectrum scaled by 2 and 0.5 respectively. | ||
This blue shifted Hox population was observable with pH 4 buffers composed of monocarboxylic acids, i.e. formic acid (pKa1 = 3.8), acetic acid (pKa = 4.8), propionic acid (pKa1 = 4.9) and caproic acid (pKa1 = 4.9) (Fig. 3). Conversely, di- and tricarboxylic acids such as succinic acid (pKa1 = 4.2), oxalic acid (pKa2 = 4.14) or citric acid (pKa1 = 3.1) did not induce the new species (Fig. S3, ESI†). Thus, stabilizations of Hoxc appear to be determined by the number of the carboxylic acid groups, rather than molecular weight of the acid.
Compared to Hox the overall band pattern of Hoxc is retained and in analogy to Hred’ (red shifted) and HoxH (blue shifted) we propose that the H-cluster geometry and electronic structure is highly similar. Moreover, the requirement for low pH in forming the state implies that the buffer molecule interacts in its protonated, uncharged, form (Fig. S4, ESI†). A complete characterisation of this new H-cluster state is beyond the scope of the current study, but we note that it could be of high relevance in particular as e.g. acetic acid (acetate) is commonly employed in protein film electrochemistry studies of [FeFe]-hydrogenase.27,35–45 Formaldehyde has previously been observed to act as an inhibitor of the H-cluster under reducing conditions.46,47 In the latter case, binding of formaldehyde on the [2Fe] subsite via a Fe–C bond was proposed from a combination of ENDOR spectroscopy and DFT calculations,48 and further supported by studies of model complexes.49 Whether a similar binding model can be applied also to carboxylic acids remains to be verified. The extent of the new state (Hoxc) formed in electrochemistry setups, its role in catalysis and its implication on catalytic currents, especially at low pH, remain unclear. In contrast to the Hox state, the same Hhyd state forms regardless of the nature of the buffer, rendering a stabilizing role of the buffer or other additives unlikely (Fig. 2 and Fig. S1, ESI†). Our data shows clearly that Hhyd can be accumulated in the absence of NaDT and without introducing loss-of-function mutations. In closing, this underscores the flat energy landscape of the H-cluster during catalysis, and it is tempting to conclude that the previously characterized Hhyd state indeed represents one of the final intermediates in hydrogen evolution catalysis of [FeFe]-hydrogenases.
Investigation by M. S. and T. K., M. L. (enzyme purification), H. J. R. (cofactor synthesis), writing (original draft) by M. S., writing (review & editing) M. S., T. K., M. L., H. J. R. and G. B.
The authors would like to thank Leif Hammarström for critically reviewing the manuscript. Open access funding was provided by Uppsala University. The European Research Council (ERC, to GB, Contract No. 714102) as well as the European Union's Horizon 2020 research and innovation program (Marie Skłodowska Curie Grant No. 897555 to M.S.) are gratefully acknowledged for funding.
Conflicts of interest
There are no conflicts to declare.References
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U. P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS.
- F. Wittkamp, M. Senger, S. T. Stripp and U. P. Apfel, Chem. Commun., 2018, 54, 5934–5942 RSC.
- M. Winkler, J. Esselborn and T. Happe, Biochim. Biophys. Acta, 2013, 1827, 974–985 CrossRef CAS PubMed.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U. P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Nat. Commun., 2018, 9, 4726 CrossRef PubMed.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knor, U. P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS PubMed.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Nemeth, N. Polidori, L. S. Meszaros, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 12789–12801 RSC.
- H. Land, P. Ceccaldi, L. S. Meszaros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2019, 10, 9941–9948 RSC.
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U. P. Apfel and S. T. Stripp, Angew. Chem., Int. Ed., 2017, 56, 16503–16506 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U. P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- S. Mebs, M. Senger, J. Duan, F. Wittkamp, U. P. Apfel, T. Happe, M. Winkler, S. T. Stripp and M. Haumann, J. Am. Chem. Soc., 2017, 139, 12157–12160 CrossRef CAS PubMed.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rudiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- A. Silakov, C. Kamp, E. Reijerse, T. Happe and W. Lubitz, Biochemistry-Us, 2009, 48, 7780–7786 CrossRef CAS PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS PubMed.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U. P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed.
- D. W. Mulder, M. W. Ratzloff, E. M. Shepard, A. S. Byer, S. M. Noone, J. W. Peters, J. B. Broderick and P. W. King, J. Am. Chem. Soc., 2013, 135, 6921–6929 CrossRef CAS PubMed.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed.
- L. S. Meszaros, P. Ceccaldi, M. Lorenzi, H. J. Redman, E. Pfitzner, J. Heberle, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 4608–4617 RSC.
- A. Adamska, A. Silakov, C. Lambertz, O. Rudiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef CAS PubMed.
- P. Knorzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, J. Am. Chem. Soc., 2018, 140, 7623–7628 CrossRef CAS PubMed.
- S. Morra, J. Duan, M. Winkler, P. A. Ash, T. Happe and K. A. Vincent, Dalton Trans., 2021, 50, 12655–12663 RSC.
- P. S. Corrigan, J. L. Tirsch and A. Silakov, J. Am. Chem. Soc., 2020, 142, 12409–12419 CrossRef CAS PubMed.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U. P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS PubMed.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 16894–16902 CrossRef CAS PubMed.
- M. A. Martini, O. Rudiger, N. Breuer, B. Noring, S. DeBeer, P. Rodriguez-Macia and J. A. Birrell, J. Am. Chem. Soc., 2021, 143, 18159–18171 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, F. Wittkamp, U. P. Apfel, J. Heberle, M. Haumann and S. T. Stripp, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8454–8459 CrossRef CAS PubMed.
- S. T. Stripp, ACS Catal., 2021, 11, 7845–7862 CrossRef CAS.
- J. Noth, R. Kositzki, K. Klein, M. Winkler, M. Haumann and T. Happe, Sci. Rep., 2015, 5, 13978 CrossRef CAS PubMed.
- K. Laun, I. Baranova, J. Duan, L. Kertess, F. Wittkamp, U. P. Apfel, T. Happe, M. Senger and S. T. Stripp, Dalton Trans., 2021, 50, 3641–3650 RSC.
- P. E. M. Siegbahn and R. Z. Liao, J. Phys. Chem. A, 2020, 124, 10540–10549 CrossRef CAS PubMed.
- S. Mebs, R. Kositzki, J. Duan, L. Kertess, M. Senger, F. Wittkamp, U. P. Apfel, T. Happe, S. T. Stripp, M. Winkler and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 28–41 CrossRef CAS PubMed.
- M. Haumann and S. T. Stripp, Acc. Chem. Res., 2018, 51, 1755–1763 CrossRef CAS PubMed.
- P. Rodríguez-Maciá, N. Breuer, S. DeBeer and J. A. Birrell, ACS Catal., 2020, 10, 13084–13095 CrossRef.
- P. Rodriguez-Macia, L. Kertess, J. Burnik, J. A. Birrell, E. Hofmann, W. Lubitz, T. Happe and O. Rudiger, J. Am. Chem. Soc., 2019, 141, 472–481 CrossRef CAS PubMed.
- O. Lampret, A. Adamska-Venkatesh, H. Konegger, F. Wittkamp, U. P. Apfel, E. J. Reijerse, W. Lubitz, O. Rudiger, T. Happe and M. Winkler, J. Am. Chem. Soc., 2017, 139, 18222–18230 CrossRef CAS PubMed.
- L. Kertess, F. Wittkamp, C. Sommer, J. Esselborn, O. Rudiger, E. J. Reijerse, E. Hofmann, W. Lubitz, M. Winkler, T. Happe and U. P. Apfel, Dalton Trans., 2017, 46, 16947–16958 RSC.
- L. Kertess, A. Adamska-Venkatesh, P. Rodriguez-Macia, O. Rudiger, W. Lubitz and T. Happe, Chem. Sci., 2017, 8, 8127–8137 RSC.
- O. Lampret, J. Duan, E. Hofmann, M. Winkler, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20520–20529 CrossRef CAS PubMed.
- V. Hajj, C. Baffert, K. Sybirna, I. Meynial-Salles, P. Soucaille, H. Bottin, V. Fourmond and C. Leger, Energy Environ. Sci., 2014, 7, 715–719 RSC.
- M. Del Barrio, M. Sensi, L. Fradale, M. Bruschi, C. Greco, L. de Gioia, L. Bertini, V. Fourmond and C. Leger, J. Am. Chem. Soc., 2018, 140, 5485–5492 CrossRef CAS PubMed.
- M. Winkler, J. Duan, A. Rutz, C. Felbek, L. Scholtysek, O. Lampret, J. Jaenecke, U. P. Apfel, G. Gilardi, F. Valetti, V. Fourmond, E. Hofmann, C. Leger and T. Happe, Nat. Commun., 2021, 12, 756 CrossRef CAS PubMed.
- S. Hardt, S. Stapf, D. T. Filmon, J. A. Birrell, O. Rudiger, V. Fourmond, C. Leger and N. Plumere, Nat. Catal., 2021, 4, 251–258 CrossRef CAS PubMed.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Leger, J. Am. Chem. Soc., 2021, 143, 20320–20325 CrossRef CAS PubMed.
- A. F. Wait, C. Brandmayr, S. T. Stripp, C. Cavazza, J. C. Fontecilla-Camps, T. Happe and F. A. Armstrong, J. Am. Chem. Soc., 2011, 133, 1282–1285 CrossRef CAS PubMed.
- C. E. Foster, T. Kramer, A. F. Wait, A. Parkin, D. P. Jennings, T. Happe, J. E. McGrady and F. A. Armstrong, J. Am. Chem. Soc., 2012, 134, 7553–7557 CrossRef CAS PubMed.
- A. Bachmeier, J. Esselborn, S. V. Hexter, T. Kramer, K. Klein, T. Happe, J. E. McGrady, W. K. Myers and F. A. Armstrong, J. Am. Chem. Soc., 2015, 137, 5381–5389 CrossRef CAS PubMed.
- F. Zhang, T. J. Woods, L. Zhu and T. B. Rauchfuss, Chem. Sci., 2021, 12, 15673–15681 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, FTIR spectra, Gas interaction kinetics. See DOI: https://doi.org/10.1039/d2cc00671e |
| ‡ Current address: Department of Chemistry, Ludwig-Maximilians-Universität MÜnchen, 81377 MÜnchen, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |