
Enhancing scanning electrochemical microscopy's potential to probe dynamic co-culture systems via hyperspectral assisted-imaging†
Sondrica
Goines
 a,
Mingchu
Deng
a,
Matthew W.
Glasscott‡
a,
Justin W. C.
Leung
b and
Jeffrey E.
Dick
*ac
a,
Mingchu
Deng
a,
Matthew W.
Glasscott‡
a,
Justin W. C.
Leung
b and
Jeffrey E.
Dick
*ac
aDepartment of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: jedick@email.unc.edu
bDepartment of Radiation Oncology, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA
cLineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
First published on 5th April 2022
Abstract
Precise determination of boundaries in co-culture systems is difficult to achieve with scanning electrochemical microscopy alone. Thus, biological scanning electrochemical microscope platforms generally consist of a scanning electrochemical microscope positioner mounted on the stage of an inverted microscope for correlated electrochemical and optical imaging. Use of a fluorescence microscope allows for site-specific fluorescence labeling to obtain more clearly resolved spatial and electrochemical data. Here, we construct a unique hyperspectral assisted-biological scanning electrochemical microscope platform to widen the scope of biological imaging. Specifically, we incorporate a variable fluorescence bandpass source into a biological scanning electrochemical microscope platform for simultaneous optical, spectral, and electrochemical imaging. Not only does this platform serve as a cost-effective alternative to white light laser imaging, but additionally it provides multi-functional analysis of biological samples. Here, we demonstrate the efficacy of our platform to discern the electrochemical contribution of site-specific cells by optically and spectroscopically resolving boundaries as well as cell types within a complex biological system.
Introduction
The study of dynamics within human tissue and among cells is based on mapping biochemical responses as a function of location; changes in cellular dynamics based on the local environment of cells may vary based on many cell traits such as location (i.e., the distance between cellular boundaries), topography, membrane permeability, or gene expression, with each characteristic directly contributing to changes in cell metabolism.1 The complexity of human tissue has directed bioanalytical chemistry towards single-cell analysis (i.e., the study of single-cell behavior both within a population or isolated) to reveal heterogeneities that cannot be observed when measuring over a large number of cells.2 Considering cancer, one of the deadliest diseases among us, begins with a mutation among the genetic code of just one rogue cell, single-cell studies are worth pursuing. Furthermore, the heterogeneity among cancer cells of the same tissue type makes personalized treatment plans based on single-cell analysis more valuable.3 While single-cell sequencing techniques have gained wide use due to the convenience of the amplification of DNA or RNA,4 few analytical tools are available to study single-cell reactivity, metabolomics, and proteomics of live-cells. Various techniques have been created based on mass spectrometry5,6 and fluorescence spectroscopy;7,8 however, these techniques suffer in terms of signal generated by a single cell (i.e., there is not much material within picoliter, 10−12 L, volumes to be analyzed leading to low signal-to-noise). Furthermore, typical mass spectrometry techniques require cell lysis or fixation to make a measurement, severely limiting our understanding of the reactivity of a live cell. Thus, analytical tools to study single-cell longitudinal behavior are necessary to elucidate dynamic changes – such as differences in a cell's life cycle, the differentiation of a stem cell, or real-time, single-cell carcinogenesis – and their dependence on the local environment.Biological applications of scanning electrochemical microscopy were apparent from the initial construction of the technique as it was non-invasive, non-destructive, and easily coupled with other analytical techniques for in vitro analysis of live-cells.9 Experiments involving scanning electrochemical microscopy have been used to image the redox activity of living, single cells with respect to cellular membrane transport, intracellular reactive oxygen and nitrogen species, and neurotransmission.10–16 Targeted scanning electrochemical microscope experiments may use redox processes to differentiate between cells within a sample. For example, quinone redox mediators have been used to differentiate between metastatic and non-transformed cells in electrochemical images; non-transformed cells generally displayed higher redox activity and apparent heterogeneous rate constants due to a higher number of available redox centers.17–20 Moreover, a nanoelectrode may be used instead of a typical microelectrode tip to achieve nanometer spatial resolution when differentiating between single cells.21,22 These experiments have allowed quantitative analysis at the single-cell level and have minimized perturbation to cellular homeostasis, encouraging the non-invasive study of living cells. In many of these experiments, however, a two-dimensional monoculture is used to model biological systems in vitro. Though monocultures allow for controlled in vitro analysis, they lack the complexity of in vivo systems. To better understand in vivo events, two-dimensional co-cultures, where two or more different populations of cells are grown in close proximity to one another, may be used to elucidate cellular activity based on intercellular communication.23,24 The difficulty lies in the robust, unambiguous determination of cell location, cell type, and intra- and extra-cellular boundaries.25 Here, we outline the combination of scanning electrochemical microscopy and variable fluorescence bandpass hyperspectral imaging as a means to overcome these challenges and observe dynamic changes.
Hyperspectral imaging widens the scope of research with the use of tunable filters, which allow users to increase spectral discrimination between wavelengths in comparison to the use of standard gratings. Spectral imaging has been used to understand cellular dynamics with respect to pharmacological responses,26 single-cell viability,27,28 and carcinogenesis.28–32 While most methods depend on labeling the analyte of interest, others have used spectral imaging to characterize analytes with known spectral properties, such as the experimental drug doxorubicin which has known fluorescence properties.26 Hyperspectral imaging is momentous to the field of biological imaging as it allows for the differentiation of biochemical complexes that are known to display unique spectral signatures.33
Hyperspectral imaging systems are characterized by their ability to collect hundreds of spectral bands; the temporal and spatial resolution of a hyperspectral imaging system are based on the limitations of the system's optical and mechanical components.33 Within our system, two tunable filter changers are coupled to a biological scanning electrochemical microscope platform (Scheme 1) to allow the user to select specific excitation and emission wavelengths with 1 nm resolution, depending on the filters in use, for spectral imaging. Whereas a standard fluorescence microscope allows one to follow a single wavelength range over time, our hyperspectral imaging system allows one to obtain multiple spectral bands by collecting a stack of two-dimensional images as a function of wavelength over time. Moreover, a spectrum is being recorded at each pixel within an image using a single channel of the microscope system. Thus, our system allows for richer insight into dynamic systems of interest compared to conventional microscope platforms. For example, the system outlined here could be used to detect spectral shifts that occur when chromophores encounter a phase within a cell that is very different from continuous water (e.g., liquid droplets34,35). Use of our hyperspectral imaging system in coordination with biological scanning electrochemical microscopy allows for differentiation of cells based on cell location, cell type, and cellular reactivity given use of an appropriate fluorescence label (or substance) and a redox mediator. While fluorescence imaging is often used in biological investigations, there is literature precedent for changes in redox activity (i.e., phototoxic effects) due to incident light,36 therefore the combination of hyperspectral imaging and electrochemical imaging would allow users to directly probe these effects. This analysis is necessary to resolve the biological redox mechanisms that give rise to the electrochemical signal.
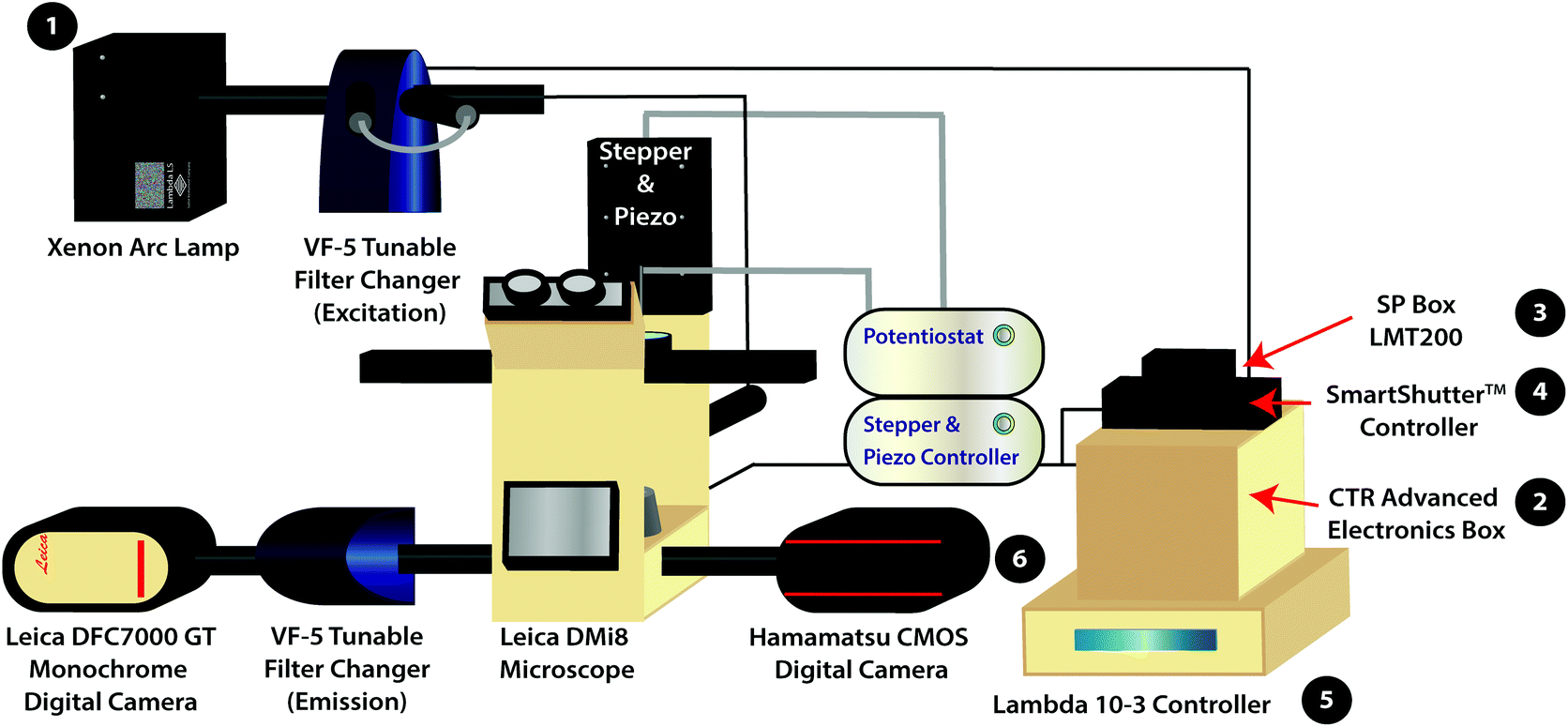 | ||
| Scheme 1 Schematic of the variable fluorescence bandpass imaging platform with numbered equipment for the power on mechanism. The (1) Xenon arc lamp functions as the white light source. The (2) CTR Advanced Electronics Box and (3) SP Box LMT200 are used to operate the Leica DMi8 Microscope and the Leica DFC7000 GT Monochrome Digital Camera. The (4) SmartShutter™ Controller operates the shutter for the light source. The (5) Lambda 10-3 Controller programs external VF-5 Tunable Filter Changers housing tunable filters. The (6) Hamamatsu CMOS Digital Camera is used to collect fluorescence images using the conventional filter cubes housed within the Leica DMi8 Microscope (i.e., GFP, Y5, DAPI, and TXR filter cubes). The Leica DFC7000 GT Monochrome Digital Camera is used to collect hyperspectral images. | ||
While hyperspectral imaging is feasible with white light laser confocal systems, these systems are quite expensive. For example, Leica Stellaris white light laser confocal systems cost upwards of $225![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (without a scanning electrochemical microscope). This article outlines a cost-effective, robust alternative to white light laser hyperspectral imaging: variable fluorescence bandpass hyperspectral imaging paired with scanning electrochemical microscopy for live cell microscopy. The combined system costs roughly $75000 less than the aforementioned system. A detailed technical motivation and user instructions are given in the ESI.† Here, we demonstrate the efficacy of our system by providing a proof-of-concept analysis where we differentiate between cell types by imaging a two-dimensional co-culture of hepatocarcinoma (Hep G2) and osteosarcoma (U2OS) cells using our uniquely designed hyperspectral assisted-scanning electrochemical microscope platform.
000 (without a scanning electrochemical microscope). This article outlines a cost-effective, robust alternative to white light laser hyperspectral imaging: variable fluorescence bandpass hyperspectral imaging paired with scanning electrochemical microscopy for live cell microscopy. The combined system costs roughly $75000 less than the aforementioned system. A detailed technical motivation and user instructions are given in the ESI.† Here, we demonstrate the efficacy of our system by providing a proof-of-concept analysis where we differentiate between cell types by imaging a two-dimensional co-culture of hepatocarcinoma (Hep G2) and osteosarcoma (U2OS) cells using our uniquely designed hyperspectral assisted-scanning electrochemical microscope platform.
Experimental
Reagents and materials
Blue-green (430/465), yellow-green (505/515), and red (580/605) Invitrogen FluoSpheres™ were purchased from Thermo Fisher Scientific. Hoechst stain solution (10 mg mL−1) was stored in the dark in a −20 °C freezer. Dulbecco's Modified Eagle's Medium (DMEM) – high glucose (with 4500 mg L−1 glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate) along with penicillin–streptomycin were purchased from Sigma-Aldrich. Gibco™ Dulbecco's Phosphate-Buffered Saline (DPBS, 1X, pH 7.4) and Gibco™ TrypLE™ Express were purchased from Thermo Fisher Scientific. Premium grade 100% fetal bovine serum, 1 M HEPES (sterile, pH 7.3), and 3.5 cm poly-L-lysine treated tissue culture dishes were purchased from VWR International, LLC. Hepatocarcinoma (Hep G2) cells were obtained from the University of North Carolina at Chapel Hill and RFP-LC8 transfected, osteosarcoma (U2OS) cells were obtained from the University of Arkansas for Medical Sciences. Hydroxymethylferrocene, 97% (ferrocenemethanol) was purchased from Alfa Aesar. All reagents were used without further purification.Instrumentation
The variable fluorescence bandpass hyperspectral imaging system was designed using a conventional inverted fluorescence microscope platform and a commercially available tunable filter system. The system is also generalizable to a wide library of cell types that go beyond those presented in this study.The hyperspectral imaging system was composed of a Lambda LS Xenon Arc Lamp (Sutter Instrument Company, Novato, CA), a Leica CTR Advanced Electronics Box (Leica Microsystems, Germany), a Leica SP Box LMT200 (Leica Microsystems, Germany), a conventional Leica DMi8 Inverted Microscope (Leica Microsystems, Germany), a Leica DFC7000 GT Monochrome Digital Camera (Leica Microsystems, Germany), a Lambda SC SmartShutter™ Controller (Sutter Instrument Company, Novato, CA), an ORCA-Flash4.0 V3 Digital CMOS Camera (Hamamatsu Photonics K. K., Hamamatsu City, Japan), two Lambda VF-5™ Tunable Filter Changers (Sutter Instrument Company, Novato, CA), and a Lambda 10-3 Controller (Sutter Instrument Company, Novato, CA). The tunable filter changers were each equipped with a combination of five VersaChrome® tunable filters produced by Semrock that allow excitation and emission wavelengths ranging from 380 nm to 700 nm. One must note that any combination of available filters may be used to amend the ranges permitted by the tunable filter changers. The first tunable filter changer was fitted against the xenon arc lamp to function as an excitation source, while the second was fitted between the Leica DMi8 Inverted Microscope and the Leica DFC7000 GT Monochrome Digital Camera to capture light emitted from (or transmitted through) the sample. In addition, the Leica DMi8 Inverted Microscope is equipped with conventional GFP, Y5, TXR, and DAPI filter cubes for standard fluorescence imaging as well as an 80/20 beam splitter for hyperspectral/fluorescence imaging using the tunable filter changers.
For correlated electrochemical analysis, the typical bright field light source and condenser of the inverted microscope were replaced with a stepper and piezo positioner/controller (CH Instruments, Inc., Austin, TX). The positioner/controller was mobilized by a 920D bipotentiostat (CH Instruments, Inc., Austin, TX), allowing simultaneous scanning electrochemical microscope analysis. Platinum microelectrode tips (r = 5 μm, 1 ≤ RG ≤ 7) and Ag/AgCl (1M KCl) electrodes were purchased from CH Instruments to be used as working and reference electrodes, respectively. A thin glassy carbon rod (r = 1.5 mm) was used as the counter electrode. While we recognize that platinum nanoelectrodes could be used to increase the resolution of electrochemical images, the purpose of this manuscript is to provide a proof-of-concept experiment.
Variable fluorescence bandpass hyperspectral imaging of Invitrogen FluoSpheres™
To demonstrate the capabilities and reproducibility of the hyperspectral imaging system, three types of Invitrogen FluoSpheres™ were imaged and their corresponding spectra were obtained. For sample preparation, 10 μL aliquots of blue-green (430/465), yellow-green (505/515), and red (580/605) fluorescent polystyrene microspheres were combined in a 3.5 cm poly-L-lysine treated tissue culture dish with approximately 2 mL Gibco™ Dulbecco's Phosphate-Buffered Saline (DPBS, 1X, pH 7.4).Leica LAS X imaging software was used to image the fluorescent polystyrene microspheres. Beads were focused in bright field using a standard halogen lamp and the 40× objective lens equipped with adaptive focus control as well as real-time control for optimum biological imaging. The Lambda 10-3 optical filter changer control system was programmed for the excitation and emission of each bead to set tunable filters to the appropriate wavelengths, and three separate images were obtained using the 80/20 beam splitter and the Leica digital camera. The resulting fluorescence images were overlaid. To obtain corresponding emission spectra, the Lambda 10-3 optical filter changer control system was programed to maintain an excitation wavelength of 425 nm while stepping through emission wavelengths from 450 nm to 700 nm with a step size of 10 nm. Leica LAS X time lapse imaging software was used to capture images at each emission wavelength using an emission based TTL trigger. Following image acquisition, a two-dimensional stack profile of the images was rendered to produce emission spectra.
Variable fluorescence bandpass hyperspectral imaging of Hep G2 cells with correlated scanning electrochemical microscopy
To demonstrate the bioanalytical utility of the system, Hep G2 cells were imaged with correlated electrochemical and spectral analysis.Hep G2 cells were cultured in a 3.5 cm poly-L-lysine treated tissue culture dish using DMEM – high glucose supplemented with 10% (v/v) fetal bovine serum, 2.5% (v/v) HEPES buffer, and 1% (v/v) penicillin–streptomycin (i.e., full growth media). Cells were incubated at 37 °C, 5% CO2, and 10% O2 until they reached 65 to 85% confluence. Hoechst stain solution (10 mg mL−1) was thawed and diluted to 10 μg mL−1 in DPBS (1X, pH 7.4). Spent media in the 3.5 cm dish was replaced with 1 mL 10 μg mL−1 Hoechst stain solution, following a DPBS (1X, pH 7.4) wash. The dish was covered in foil and placed on a rotator for 10 minutes at 10 rpm at room temperature. Following an additional DPBS (1X, pH 7.4) wash, the stain solution was replaced with a 2 mL solution of ferrocenemethanol in DPBS (1X, pH 7.4) for scanning electrochemical microscopy.
Cells were brought into focus using a standard halogen lamp and the 40× objective lens, and an initial bright field image was taken. Hep G2 cells were approximately 20 μm in diameter, but cell shape and size varied throughout the sample. A typical fluorescence image was captured using a conventional DAPI filter cube and the Hamamatsu digital camera (i.e., bypass mode, which is addressed in the ESI†). The variable fluorescence bandpass system was used to capture hyperspectral images; these additional fluorescence images were captured using the 80/20 beam splitter and the Leica digital camera. To obtain emission spectra, the Lambda 10-3 optical filter changer control system was programmed to maintain an excitation wavelength of 400 nm and step through emission wavelengths of 440 nm to 700 nm with a step size of 10 nm. A two-dimensional stack profile of the images captured at each emission wavelength was rendered to produce emission spectra of Hep G2 nuclei.
For subsequent scanning electrochemical microscopy, a Pt microelectrode tip (r = 5 μm) was placed in a 3D printed holder connected to the piezo positioner/controller above the Leica DMi8 stage. A thin glassy carbon rod and a Ag/AgCl electrode were used as the counter electrode and reference electrode, respectively. The cells and reference electrode were separated by a salt bridge to prevent silver leakage into cell media. An initial cyclic voltammogram was taken at the surface of the ferrocenemethanol solution, sufficiently far enough from the surface of the Hep G2 cells, to observe the typical faradaic response of ferrocenemethanol in DPBS (1X, pH 7.4) (Fig. S1†). For additional analysis, the current at +0.5 V vs. Ag/AgCl is used as the limiting current (i.e., iT,∞). The Pt microelectrode tip was then used to approach the surface of cells within the culture dish in the z-direction while poising the electrode sufficiently positive to oxidize ferrocenemethanol (i.e., +0.5 V vs. Ag/AgCl). The bipotentiostat simultaneously measured current versus the distance traveled by the electrode. The approach was concurrently monitored using bright field microscopy. Once a feedback response was observed near the surface of a Hep G2 cell membrane, the Pt microelectrode tip was retracted approximately 5 to 10 μm to avoid potential tip–sample crashes associated with constant-height imaging, while remaining at an appropriate working distance (i.e., z ≤ 2a, where z is the working distance and a is the electrode radius). This method of tip placement in 2D cell cultures has been previously validated.15,27,28 Next, the electrode tip was biased at +0.5 V vs. Ag/AgCl and used to scan an area of cells in the xy plane. Simultaneously measuring current at the electrode tip via amperometry resulted in an image of the cells based on their feedback (i.e., current) response. Electrochemical images were captured over a period of 5 to 7 minutes. Concurrently, the Pt microelectrode tip was monitored using a 40× objective lens with a resolution of 559 nm in the xy plane. No visual evidence of the tip contacting the cells or cellular perturbation was observed. Minimal changes in cell morphology indicated insignificant changes in cell viability. For time lapse imaging over longer periods, a stage top incubator equipped with a silicon inlet from Tokai Hit® may be used for probing cellular reactivity. For electrochemical data analysis, current scales were normalized by iT,∞ to display current response relative to the bulk solution.
Variable fluorescence bandpass hyperspectral imaging of co-cultured cells with correlated scanning electrochemical microscopy
Hoechst stained Hep G2 cells were cultured in a 3.5 cm dish with U2OS cells. U2OS cells were previously transfected to express RFP-LC8. Cells were incubated in full growth media at 37 °C, 5% CO2, and 10% O2 until they reached 65 to 85% confluence. Prior to imaging, spent media was removed from the sample dish and cells were washed with DPBS (1X, pH 7.4). Following this wash step, approximately 2 mL of a ferrocenemethanol in DPBS (1X, pH 7.4) solution was added to the dish for scanning electrochemical microscopy.Co-cultured cells were brought into focus using a standard halogen lamp, and an initial bright field image was taken. To capture RFP emission from U2OS cells, a conventional TXR filter cube and the Hamamatsu digital camera were used. To capture Hoechst emission from Hep G2 cells, a conventional DAPI filter cube and the Hamamatsu digital camera were used. The resulting fluorescence images were overlaid. Compared to Hep G2 cells, U2OS cells were typically elongated with a width between 15 to 20 μm. Spectral imaging was completed using an 80/20 beam splitter and the Leica digital camera. To obtain emission spectra of RFP-LC8 modified U2OS cells, the Lambda 10-3 optical filter changer control system was programmed to maintain an excitation wavelength of 580 nm and step through emission wavelengths of 610 nm to 700 nm with a step size of 10 nm. A two-dimensional stack profile of the images captured at each emission wavelength was rendered to produce emission spectra of RFP-LC8 in U2OS cells. Subsequent hyperspectral imaging of Hep G2 nuclei and electrochemical imaging of the sample were completed as previously described.
Results and discussion
To display the hyperspectral imaging capabilities of the variable fluorescence bandpass platform, we obtained a lambda scan of three fluorescent microspheres (r = 7.5 μm) (Fig. 1a). The use of standard overlap tunable filters while imaging over a wide spectral field resulted in minimal overlap in Fig. 1b. A common excitation wavelength and sufficient variation in emission wavelengths made subsequent imaging of each bead during a lambda scan feasible. Ten successive lambda scans (Fig. 1b) resulted in standard deviations of mean emission intensities no greater than 4% RSD. By selecting a region of interest within each bead type in the Leica LAS X imaging software, we retrieved the lambda scan of an individual bead (i.e., the spectra representative of the two-dimensional stack profile of the images captured at each emission wavelength, Fig. 1c), a feature necessary to capture dynamic correlated hyperspectral data within a single cell. As a control, this result implies our potential to optically visualize a stained organelle.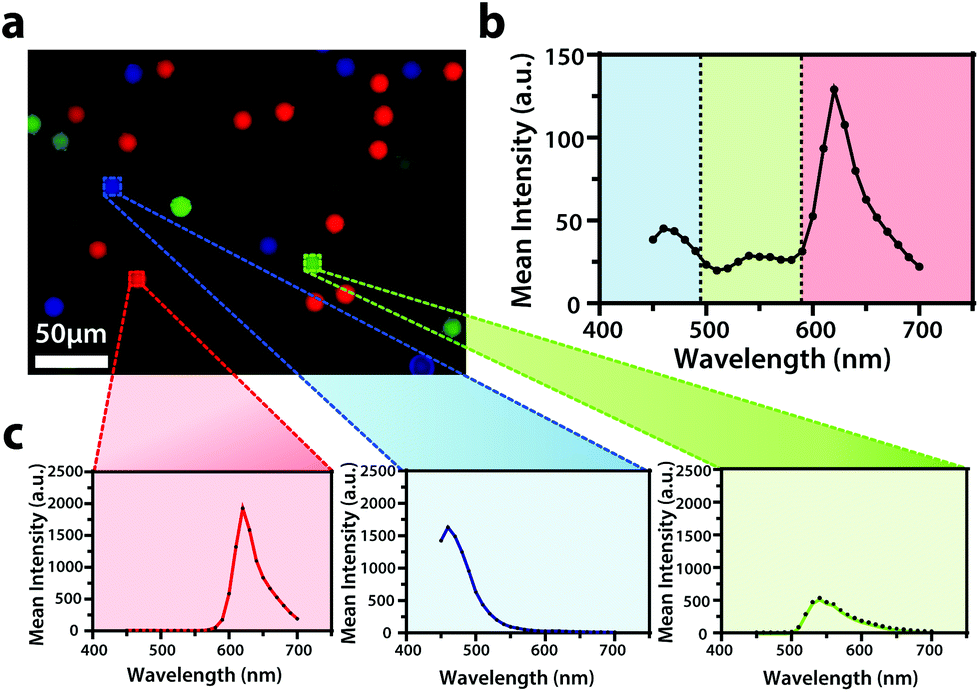 | ||
| Fig. 1 (a) An overlay of fluorescence images of blue-green (λex/λem,435/465 nm), yellow-green (λex/λem, 505/530 nm), and red (λex/λem, 580/605 nm) Invitrogen FluoSpheres™ combined in a 3.5 cm dish submerged in DPBS (1X, pH 7.4) with (b) an average emission spectrum of the sample (N = 10). (c) The average spectrum is accompanied by an individual average spectrum of each microsphere type within the sample (N = 10). The overlay of fluorescence images was false colored for visual representation. Conventional fluorescence images are presented using the bandpass wavelengths provided, while hyperspectral images are presented using the spectra provided. | ||
For electrochemical imaging of Hep G2 cells, initially, a cyclic voltammogram (Fig. S1†), in the polarographic convention, of ferrocenemethanol in DPBS (1X, pH 7.4) was obtained at the surface of the solution above the cells with a Pt microelectrode tip (r = 5 μm) as the working electrode. Next, an area of cells was approached, while driving the oxidation of ferrocenemethanol at +0.5 V vs. Ag/AgCl at the electrode surface, to observe a decrease in oxidative current in the polarographic convention. The decrease in current is a result of hindered diffusion of ferrocenemethanol to the Pt microelectrode tip as the cells were approached. An increase in current is measured just above the surface of the cells due to redox activity at the cell membrane (Fig. 2a). Simultaneous optical microscopy of the approach indicated close proximity to the area of cells, allowing us to retract the tip 5 to 10 μm above the surface of the cells using the piezoelectric positioner (Fig. 2b). This is often necessary when performing constant-height imaging with large aspect-ratio samples, such as two-dimensional cell cultures, to avoid tip–sample crashes.37 Fig. S2† is an example of an optical image following an approach to an area of cells. After performing these steps, subsequent xy scans at +0.5 V vs. Ag/AgCl produced an electrochemical image of the cells (Fig. 2c). In Fig. 2c, cell reactivity provides an increase in feedback relative to the dish surface. Correlated bright field images showed the typical morphology of low density Hep G2 cells. Complementary bright field/fluorescence overlaid images of the same position were obtained to locate the nuclei of cells that had been previously incubated in a Hoechst nuclear stain solution (Fig. 2d). When compared, these images (Fig. 2c and d) allowed us to spatially locate and differentiate cells on a single-cell basis. These images are essential for the unambiguous determination of cellular boundaries, which are often difficult to discern when using bright field and electrochemical imaging alone. Additionally, Fig. S3† demonstrates how electrochemical images may be used to address cell viability.
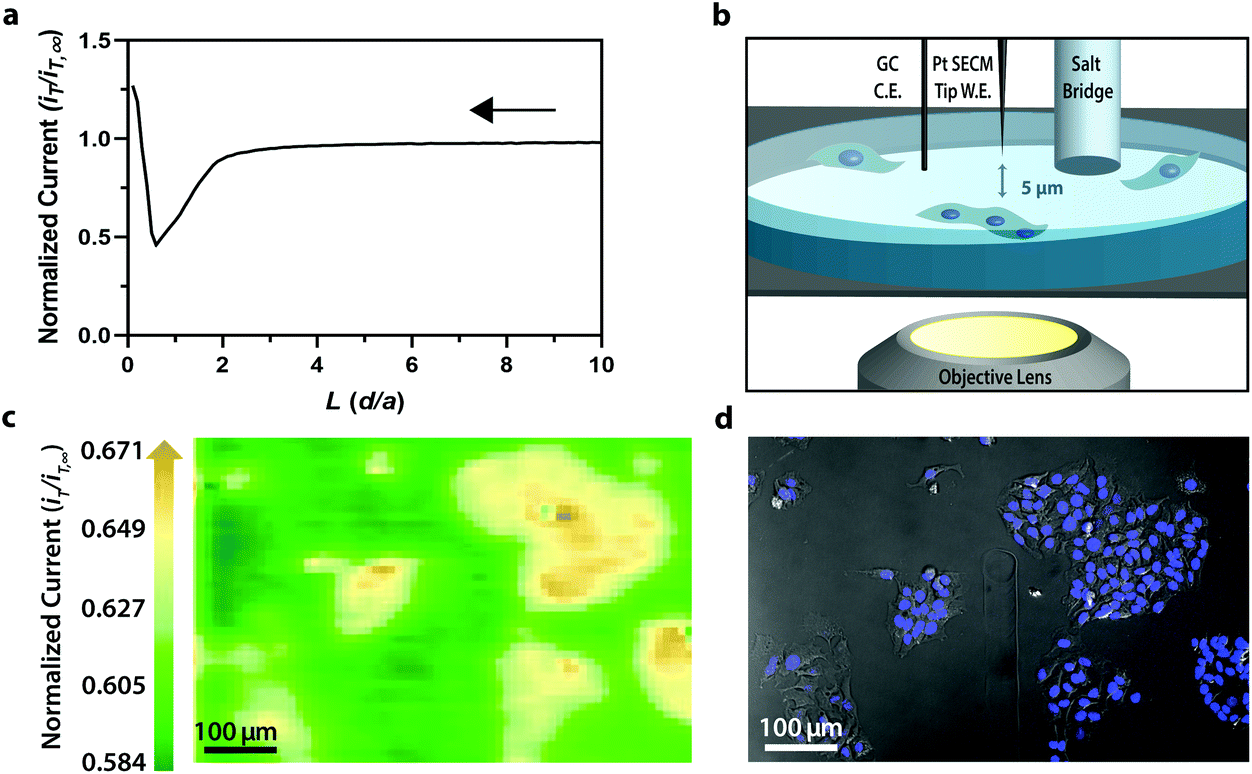 | ||
| Fig. 2 (a) Polarographic feedback response of a Pt microelectrode tip (r = 5 μm) vs. Ag/AgCl scanned in the z-direction over an insulated tissue culture dish with Hep G2 cells in 70 μM ferrocenemethanol in DPBS (1X, pH 7.4). (b) Schematic of tip-to-cell distance approximation. (c) Correlated polarographic electrochemical image and (d) bright field/fluorescence overlay of Hep G2 cells. The fluorescence image was false colored for visual representation. Cell nuclei stained with Hoechst 33342 (λex/λem, 400/497 nm). | ||
While the fluorescence microscopy detailed above may be achieved using a conventional fluorescence microscope, the variable fluorescence bandpass system described here allows multicolor imaging, enabling multi-fluorophore detection within dynamic systems.
The VersaChrome® filters used are capable of high transmission, ideal for spectroscopy, and out-of-band blocking. Additionally, Semrock designed these particular VersaChrome® tunable thin-film filters with steep edges to increase spectral discrimination compared to standard gratings while also providing more bandwidth control.
Though the bandpass resolution of our filters ranges from 13 nm to 16 nm, these filters were incorporated to decrease spectral distortion associated with the angle of incident light making 1 nm spectral resolution possible if a spectrum is obtained within the bandpass resolution of a single VersaChrome® filter (Fig. S4†). In addition, the Sutter Instrument® Lambda 10-3 optical filter changer control system used to operate our set up allows us to specify wavelengths in increments as low as 1 nm; lambda scans obtained using this novel combination of technology would allow one to discern features within dynamic spectral and optical data otherwise unrecognized by scans obtained with conventional filter cubes (i.e., one could observe slight shifts in the excitation of a site-specific fluorophore with nanometer resolution as fluorophore polarization may vary with respect to dynamic interactions38). Moreover, being coupled to a scanning electrochemical microscope, this system may be used to observe site-specific electrochemical activity while obtaining spectral and optical data to locate and differentiate between each cell (Fig. S4†). This is novel within biological imaging because different biochemical species display different spectral signatures.33 Moreover, hyperspectral assisted-electrochemical imaging allows one to investigate dynamic changes in cell metabolism using three independently, valuable methods: optical microscopy, spectroscopy, and electrochemistry. Fig. 3 serves as a model data set produced by our unique hyperspectral assisted-scanning electrochemical microscope system. Here, the correlated optical images (Fig. 3b) allow one to differentiate between cellular boundaries, enhancing spatial resolution when compared to the SECM image (Fig. 3a) obtained with a 10 μm diameter tip. In addition, hyperspectral imaging is used to yield a distinct fluorescence signal for Hep G2 nuclei (Fig. 3c). The novelty lies in the system's ability to capture a distinct emission spectra as spectral shifts are often indicative of changes in the intracellular environment.34,35
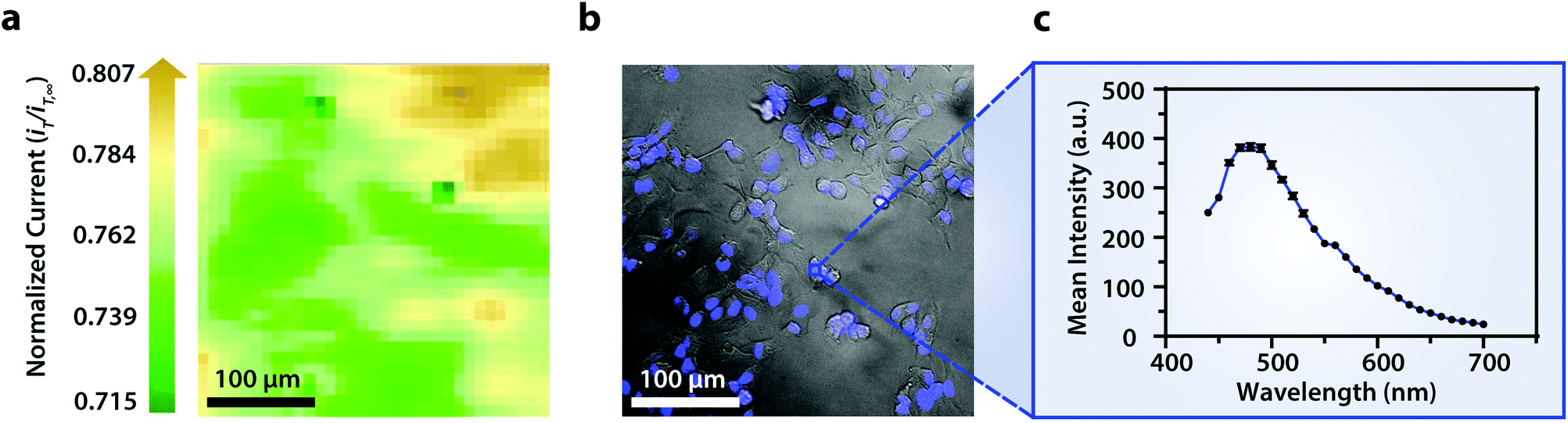 | ||
| Fig. 3 (a) Correlated polarographic electrochemical image and (b) bright field/fluorescence overlay of Hep G2 cells with a (c) correlated spectrum of Hoechst 33342 (λex/λem, 400/497 nm) for a single nucleus. The electrochemical image was obtained with a Pt microelectrode tip (r = 5 μm) vs. Ag/AgCl in 0.60 mM ferrocenemethanol in DPBS (1X, pH 7.4). The fluorescence image was false colored for visual representation. Cell nuclei stained with Hoechst 33342 (λex/λem, 400/497 nm). Conventional fluorescence images are presented using the bandpass wavelengths provided, while hyperspectral images are presented using the spectra provided. | ||
To establish the system's ability to distinguish between cell types via multicolor imaging, correlated electrochemical and optical data was obtained using a two-dimensional co-culture of Hep G2 and U2OS cells. Hep G2 cells were previously stained with the Hoechst nuclear stain solution, while U2OS cells were previously transfected with RFP-LC8; LC8 is a eukaryotic protein localized in the cytoplasm and the nucleus of cells.39 Here, both cell types exhibited an increase in their feedback response relative to the insulating dish (Fig. 4a); variation in the magnitude of the feedback response could indicate variation in cell height, oxidative stress, redox mediator permeability, or cell communication since cells within dynamic co-culture systems often display varying reactivity based on cell type.40
 | ||
| Fig. 4 (a) Correlated polarographic electrochemical image, (b) bright field image, and (c) fluorescence image of a two-dimensional co-culture of Hep G2 and U2OS cells. The electrochemical image was obtained with a Pt microelectrode tip (r = 5 μm) vs. Ag/AgCl in 0.35 mM ferrocenemethanol in DPBS (1X, pH 7.4). The fluorescence image was false colored for visual representation. Hep G2 nuclei stained with Hoechst 33342 (λex/λem, 400/497 nm). U2OS cells transfected with RFP-LC8 (λex/λem, 580/630 nm). | ||
Within this two-dimensional co-culture system, cell type cannot be clearly distinguished based on the electrochemical (Fig. 4a) and bright field (Fig. 4b) images alone. Thus, correlated fluorescence (Fig. 4c) and hyperspectral (Fig. 5b and c) images obtained using our unique variable fluorescence bandpass imaging platform were necessary to discern between cell types as well as cellular boundaries.
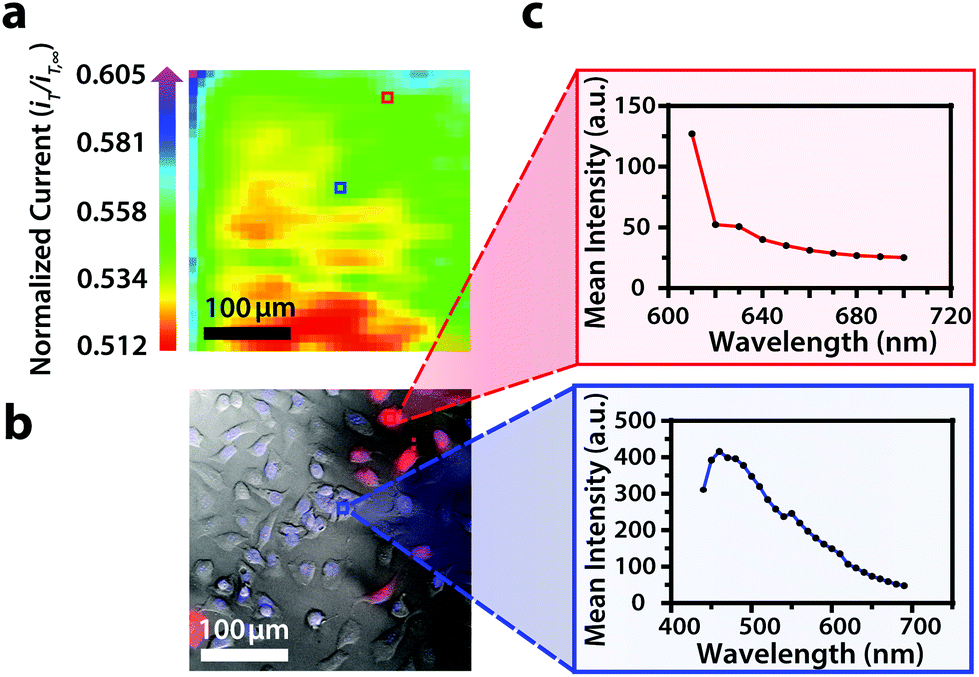 | ||
| Fig. 5 (a) Correlated polarographic electrochemical image, (b) bright field/fluorescence overlay, and (c) spectra for U2OS RFP-LC8 and Hoechst 33342 of two individual cells within a two-dimensional co-culture of Hep G2 and U2OS cells. The electrochemical image was obtained with a Pt microelectrode tip (r = 5 μm) vs. Ag/AgCl in 0.80 mM ferrocenemethanol in DPBS (1X, pH 7.4). The fluorescence image was false colored for visual representation. Hep G2 nuclei stained with Hoechst 33342 (λex/λem, 400/497 nm). U2OS cells transfected with RFP-LC8 (λex/λem, 580/630 nm). Conventional fluorescence images are presented using the bandpass wavelengths provided, while hyperspectral images are presented using the spectra provided. | ||
By correlating fluorescence images with electrochemical images, we differentiated between the electrochemical feedback of U2OS and Hep G2 cells within the co-culture system. In Fig. 4a, a cluster of Hep G2 cells near the bottom right of the image exhibited positive feedback based on the normalized current, while U2OS cells typically displayed less feedback relative to the insulating dish in comparison. Although this trend is evident in Fig. 4, we also observed that U2OS cells have the potential to exhibit similar feedback to Hep G2 cells with respect to the insulating dish in Fig. 5. These are preliminary, qualitative assessments since variation in the feedback response may be due to differences in cellular metabolism based on cell type, cell morphology, and diffusion layer overlap due to cell aggregates. Further hyperspectral analysis, similar to that shown in Fig. 5, may elucidate variations due to cellular metabolism if a redox fluorophore is used. Fig. S4† demonstrates the use of a redox indicator with a fluorescence signal.
Here, we recognize that the resolving power of this novel system can be improved through the use of nanoelectrode tips to achieve nanometer resolution during electrochemical imaging and through the use of redox indicative fluorophores to differentiate between the metabolic activity of each cell type. Additionally, the temporal resolution may be improved if an alternative to electrochemical mapping is used to assess redox activity, for example amperometric approaches have been used to determine heterogeneous rate constants above living cells18 and electrochemiluminescence has been used to image cell membranes.41 Here, electrochemical mapping was used to correlate cell location between electrochemical and fluorescence responses. Specifically, we demonstrate the use of a cost-effective, hyperspectral assisted-scanning electrochemical microscope system. Future investigations will be geared towards investigating cellular dynamics with nanometer spatial resolution and additional electrochemical techniques to push the resolving power of the system presented here.
Conclusions
As nanoelectrochemistry progresses further into single-cell analyses, the integration of optical techniques is essential to differentiate between cellular boundaries. In this manuscript, we describe the construction of a hyperspectral assisted-scanning electrochemical microscope capable of discerning cell location, cell type, and cellular boundaries within two-dimensional cell cultures. While typical scanning electrochemical microscope experiments involve imaging cells within monocultures, our innovative design is capable of robust evaluation of two-dimensional co-cultures, which are often used to model complex in vivo systems. A hallmark of the technique presented here is that it is apathetic to the chosen fluorophore, and multicolor imaging (i.e., the use of several fluorophores) is permitted. With this system serving as a foundation for correlated single-cell electrochemical and spectral studies, we hope to spark numerous investigations to determine which aspects of the biological system give rise to the redox response at the single-cell level. Additionally, spatial resolution can be enhanced with the use of nanoelectrode tips to understand dynamic interactions within single cells.Author contributions
Sondrica Goines, Matthew W. Glasscott, and Jeffrey E. Dick conceived and designed hyperspectral assisted-scanning electrochemical microscopy. Sondrica Goines cultured cells and performed the reported investigations. Mingchu Deng and Justin W. C. Leung cultured and transfected cells, respectively. Sondrica Goines drafted the manuscript; Jeffrey E. Dick edited and revised the manuscript. Jeffrey E. Dick managed all aspects of the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1650116. We acknowledge the generous support from the National Institutes of Health under grant number 1-R35-GM138133-01. U2OS cells were transfected by Dr. Justin W. C. Leung with the support of the National Institutes of Health (NCI: R01CA244261 and NIGMS: R35GM137798). Additionally, we gratefully acknowledge the University of North Carolina at Chapel Hill for initial start-up funds, which supported this work. We would also like to acknowledge Ross Warrington and Chris Murphy (Leica Microsystems) and Chris Ballard (Sutter Instrument Company) for their assistance in constructing this imaging platform. Additionally, we would like to thank Dr. Silvia Voci, Dr. Koun Lim, and Dr. Moinul H. Choudhury for helpful discussions.References
- P. Weiss, Rev. Mod. Phys., 1959, 31, 11–20 CrossRef CAS.
- E. Lubeck and L. Cai, Nat. Methods, 2012, 9, 743–U159 CrossRef CAS PubMed.
- I. Dagogo-Jack and A. T. Shaw, Nat. Rev. Clin. Oncol., 2018, 15, 81–94 CrossRef CAS PubMed.
- B. Hwang, J. H. Lee and D. Bang, Exp. Mol. Med., 2018, 50, 1–14 CrossRef CAS PubMed.
- A. Ali, Y. Abouleila, Y. Shimizu, E. Hiyama, S. Emara, A. Mashaghi and T. Hankemeier, Trac-Trends in Analytical Chemistry, 2019, p. 120 Search PubMed.
- L. Yin, Z. Zhang, Y. Z. Liu, Y. Gao and J. K. Gu, Analyst, 2019, 144, 824–845 RSC.
- X. A. Cambronne, M. L. Stewart, D. Kim, A. M. Jones-Brunette, R. K. Morgan, D. L. Farrens, M. S. Cohen and R. H. Goodman, Science, 2016, 352, 1474–1477 CrossRef CAS PubMed.
- G. W. Li and X. S. Xie, Nature, 2011, 475, 308–315 CrossRef CAS PubMed.
- D. Polcari, P. Dauphin-Ducharme and J. Mauzeroll, Chem. Rev., 2016, 116, 13234–13278 CrossRef CAS PubMed.
- B. Zhang, K. L. Adams, S. J. Luber, D. J. Eves, M. L. Heien and A. G. Ewing, Anal. Chem., 2008, 80, 1394–1400 CrossRef CAS PubMed.
- K. T. Kawagoe, J. A. Jankowski and R. M. Wightman, Anal. Chem., 1991, 63, 1589–1594 CrossRef CAS PubMed.
- R. M. Wightman, Science, 2006, 311, 1570–1574 CrossRef CAS PubMed.
- R. T. Kurulugama, D. O. Wipf, S. A. Takacs, S. Pongmayteegul, P. A. Garris and J. E. Baur, Anal. Chem., 2005, 77, 1111–1117 CrossRef CAS PubMed.
- J. G. Roberts, M. A. Voinov, A. C. Schmidt, T. I. Smirnova and L. A. Sombers, J. Am. Chem. Soc., 2016, 138, 2516–2519 CrossRef CAS PubMed.
- J. G. Roberts, B. P. Moody, G. S. McCarty and L. A. Sombers, Langmuir, 2010, 26, 9116–9122 CrossRef CAS PubMed.
- F. P. Filice and Z. F. Ding, Analyst, 2019, 144, 738–752 RSC.
- B. Liu, S. A. Rotenberg and M. V. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9855–9860 CrossRef CAS PubMed.
- B. Liu, S. A. Rogenberg and M. V. Mirkin, Anal. Chem., 2002, 74, 6340–6348 CrossRef CAS PubMed.
- S. A. Rotenberg and M. V. Mirkin, J. Mammary Gland Biol. Neoplasia, 2004, 9, 375–382 CrossRef PubMed.
- S. E. Salamifar and R. Y. Lai, Anal. Chem., 2013, 85, 9417–9421 CrossRef CAS PubMed.
- Y. Y. T. Sun and M. V. Mirkin, Anal. Chem., 2016, 88, 11758–11766 CrossRef PubMed.
- P. Elsamadisi, Y. X. Wang, J. Velmurugan and M. V. Mirkin, Anal. Chem., 2011, 83, 671–673 CrossRef CAS PubMed.
- M. Pandurangan and I. Hwang, Appl. Microbiol. Biotechnol., 2014, 98, 7359–7364 CrossRef CAS PubMed.
- L. Goers, P. Freemont and K. M. Polizzi, J. R. Soc., Interface, 2014, 11, 20140065 CrossRef PubMed.
- K. G. Battiston, J. W. C. Cheung, D. Jain and J. P. Santerre, Biomaterials, 2014, 35, 4465–4476 CrossRef CAS PubMed.
- Q. A. Alshammari, R. Pala, N. Katzir and S. M. Nauli, Sci. Rep., 2021, 11, 2703 CrossRef CAS PubMed.
- H. Matsuoka, Y. Kosai, M. Saito, N. Takeyama and H. Suto, J. Biotechnol., 2002, 94, 299–308 CrossRef CAS.
- K. Asai, Y. Sumiyama, M. Watanabe and K. Aizawa, Surg. Today, 2006, 36, 1075–1084 CrossRef PubMed.
- S. J. Leavesley, M. Walters, C. Lopez, T. Baker, P. F. Favreau, T. C. Rich, P. F. Rider and C. W. Boudreaux, J. Biomed. Opt., 2016, 21, 10 CrossRef.
- J. Moreau, P. Bouzy, J. Guillard, V. Untereiner, R. Garnotel, A. Marchal, C. Gobinet, C. Terryn, G. D. Sockalingum and G. Thiefin, Molecules, 2020, 25 Search PubMed.
- G. Thomas, J. van Voskuilen, H. Truong, J. Y. Song, H. C. Gerritsen and H. Sterenborg, Biomed. Opt. Express, 2014, 5, 4281–4299 CrossRef.
- A. Travo, O. Piot, R. Wolthuis, C. Gobinet, M. Manfait, J. Bara, M. E. Forgue-Lafitte and P. Jeannesson, Histopathology, 2010, 56, 921–931 CrossRef PubMed.
- Q. L. Li, X. F. He, Y. T. Wang, H. Y. Liu, D. R. Xu and F. M. Guo, J. Biomed. Opt., 2013, 18, 100901 CrossRef PubMed.
- R. Oliva, S. K. Mukherjee, Z. Fetahaj, S. Mobitz and R. Winter, Chem. Commun., 2020, 56, 11577–11580 RSC.
- M. Hazawa, S. Amemori, Y. Nishiyama, Y. Iga, Y. Iwashima, A. Kobayashi, H. Nagatani, M. Mizuno, K. Takahashi and R. W. Wong, Iscience, 2021, 24, 102865 CrossRef CAS PubMed.
- J. B. Robertson, C. R. Davis and C. H. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 21130–21135 CrossRef CAS.
- I. Beaulieu, S. Kuss, J. Mauzeroll and M. Geissler, Anal. Chem., 2011, 83, 1485–1492 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science+Business Media, LLC, 3rd edn., 2006 Search PubMed.
- K. L. West, J. L. Kelliher, Z. Z. Xu, L. W. An, M. R. Reed, R. L. Eoff, J. D. Wang, M. S. Y. Huen and J. W. C. Leung, Nucleic Acids Res., 2019, 47, 6236–6249 CrossRef CAS PubMed.
- S. Kuss, D. Polcari, M. Geissler, D. Brassard and J. Mauzeroll, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9249 CrossRef CAS PubMed.
- S. Voci, B. Goudeau, G. Valenti, A. Lesch, M. Jović, S. Rapino, F. Paolucci, S. Arbault and N. Sojic, J. Am. Chem. Soc., 2018, 140, 14753–14760 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an00319h |
| ‡ The present address of this author is U.S. Army Corps of Engineers, Engineer Research and Development Center, Vicksburg, MS 39180, USA. |
| This journal is © The Royal Society of Chemistry 2022 |