
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalysed ortho-alkynylation of nitroarenes†
Eric
Tan
ab,
Marc
Montesinos-Magraner
 a,
Cristina
García-Morales
ab,
Joan Guillem
Mayans
ab and
Antonio M.
Echavarren
*ab
a,
Cristina
García-Morales
ab,
Joan Guillem
Mayans
ab and
Antonio M.
Echavarren
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology (BIST), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: aechavarren@iciq.es
bDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, C/ Marcel·lí Domingo s/n, 43007 Tarragona, Spain
First published on 11th October 2021
Abstract
The ortho-alkynylation of nitro-(hetero)arenes takes place in the presence of a Rh(III) catalyst to deliver a wide variety of alkynylated nitroarenes regioselectively. These interesting products could be further derivatized by selective reduction of the nitro group or palladium-catalysed couplings. Experimental and computational mechanistic studies demonstrate that the reaction proceeds via a turnover-limiting electrophilic C–H metalation ortho to the strongly electron-withdrawing nitro group.
Introduction
Nitrobenzenes are among the most important bulk chemicals, used in a range of applications such as dyes, organic materials, solvents and perfumes.1 With a price comparable to benzene, nitrobenzene serves as precursor to most of the functionalized aromatic building blocks. Therefore, the development of methods for the functionalization of nitrobenzenes is of high interest.Nitrobenzenes can be functionalized at the ipso-position by reduction to an aniline and formation of the diazonium salt, allowing access to versatile aryl halides (Sandmeyer reaction).2 More recently, the functionalization of nitrobenzenes gained momentum with the discovery that rhodium,3 copper,4 and palladium5 complexes can undergo oxidative addition to nitrobenzenes, opening the door to their use as coupling partners in transition-metal catalysis. Nitrobenzenes are usually functionalized at the meta-position via electrophilic aromatic substitution, although functionalization at the ortho- and para-positions is also possible via the so-called vicarious nucleophilic substitution (Scheme 1a),1,6 where an α-halo-carbanion generated from an active methylene compound adds to the ortho- and/or para-position. However, this method is mainly limited to alkylation-type functionalization and often requires electronically activated nitrobenzenes to achieve synthetically useful yields and good selectivity. Therefore, the selective ortho-functionalization of unbiased nitrobenzene derivatives is an important and yet underdeveloped transformation in organic chemistry.
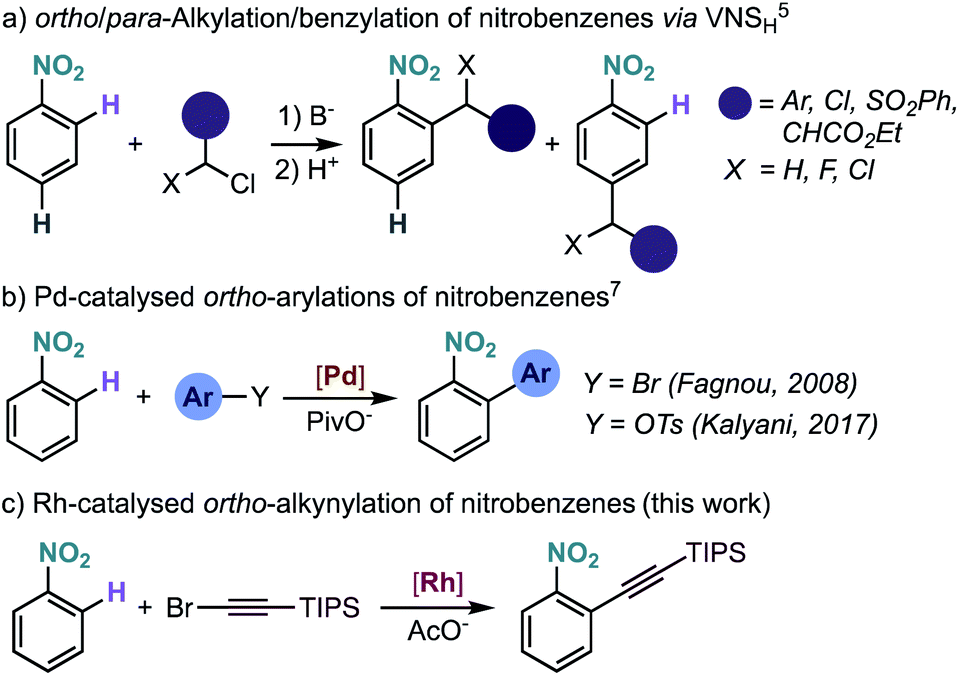 | ||
| Scheme 1 Intermolecular ortho-functionalization of nitrobenzenes and present studies. VNSH = vicarious nucleophilic substitution of hydrogen. | ||
The development of metal-catalysed directed C–H functionalization reactions has become an efficient method for the synthesis of functionalized arenes. Even though the ability of the ubiquitous nitro group to coordinate to metals is known,7 its use as directing group in catalysis is rare and limited to direct arylation.8 Our group observed the intramolecular Pd-catalysed ortho C–H arylation of nitrobenzene derivatives, although in that reaction the nitro group was not acting as a regiodirecting substituent.9a The first genuine intermolecular Pd-catalysed direct arylation of nitrobenzenes was achieved using aryl halides8a and tosylates,8b although an excess of nitrobenzene was required (Scheme 1b). The ortho C–H arylation of nitro-substituted nitrogen heteroarenes has been developed to a greater extent, but, in these examples, the site-selectivity is a consequence of the heteroarene electronics and not of the regiodirecting effect of the nitro group.10
Here we report the Rh-catalysed nitro-directed C–H alkynylation of nitrobenzenes, which tolerates a broad range of functional groups (Scheme 1c).11 Our experimental and computational mechanistic investigations are consistent with a turnover limiting electrophilic C–H activation step, followed by alkyne insertion and bromide elimination. We also disclose preliminary results on a related C–H iodination reaction, which may pave the way for other types of ortho-functionalization via sequential Rh/Pd catalysis.
Results and discussion
As part of our research program on the selective C–H alkynylation of functionalized molecules, we recently found that the combination of a Cp*Rh(III) catalyst and bromo-alkyne 2a is a highly active system for the alkynylation of a broad-range of C–H bonds.12 Our initial attempts to extend this reactivity to 2-methylnitrobenzene (1a) at 50 °C proved unsuccessful (Table 1, entry 1). Interestingly, formation of the corresponding alkynylated product 3a could be observed at 80 °C as the only product (Table 1, entry 2). A further increase of the temperature to 110 °C led to the formation of 3a in 95% yield (Table 1, entry 3). Control experiments showed the essential role of all reaction components (Table 1, entries 4–7). Other catalysts frequently used in C–H functionalization, such as MnBr(CO)5, Cp*Co(CO)I2, Pd(OAc)2, [RuCl2(p-cymene)]2 or [Cp*IrCl2]2 were inactive (Table 1, entry 8). The use of different silver salts (Table 1, entry 9), solvents (Table 1, entry 10), or carboxylate salts (Table 1, entry 11) led to unreactive catalytic systems. Regarding the alkyne counterpart, switching bromine for chlorine (2b) did not affect the reaction outcome (Table 1, entry 12). However, using iodo-alkyne 2c led to the formation of 3a in low yield (Table 1, entry 13).|
|
||
|---|---|---|
| Entry | Deviation from optimized conditions | Yield 3aa (%) |
| a Yield determined by 1H NMR with an internal standard. b Isolated yield. | ||
| 1 | 50 °C | 0 |
| 2 | 80 °C | 35b |
| 3 | None | 95b |
| 4 | Without [Cp*RhCl2]2 | 0 |
| 5 | Without Ag2CO3 | 0 |
| 6 | Without LiOAc | 0 |
| 7 | Without AgSbF6 | 0 |
| 8 | MnBr(CO)5, Cp*Co(CO)I2, Pd(OAc)2, [Cp*IrCl2] or [RuCl2(p-cymene)]2 instead of [Cp*RhCl2]2 | 0 |
| 9 | With AgNO3 or Ag2O instead of Ag2CO3 | 0 |
| 10 | THF or tert-amyl alcohol instead of DCE | 0 |
| 11 | NaOPiv instead of LiOAc | 0 |
| 12 | TIPS-Cl-acetylene (2b) instead of 2a | 96 |
| 13 | TIPS-I-acetylene (2c) instead of 2a | 10 |

The scope of the Rh-catalysed alkynylation of nitrobenzenes was explored using the optimized conditions for the synthesis of 3a (Scheme 2). Although the reactions were typically performed overnight for convenience, reaction times can be often reduced to just a few hours. Alkynylation of nitrobenzene was performed under the optimised reaction conditions to obtain a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of mono- and dialkynylated product in 75% yield. This ratio can be reversed by increasing the amount of bromoalkyne 2a, which results in the obtention of dialkynylated 3b′ in excellent yield. Functionalities such as alkyl (3a, 3c), aryl (3d, 3j), ether (3e), aldehyde (3f) and halides (3g–i), groups at the ortho position were well tolerated, leading to 3a–j in 40–95% yield and complete ortho-selectivity. In the case of meta-substituted nitrobenzenes, the alkynylation occurred exclusively at the least hindered site leading to methyl, dimethyl amine or vinyl substituted compounds (3k–m) groups, whereas fluoro and methoxy substituents led to a 2:1 mixture of mono- and dialkynylated products, favoring the formation of 1,2,3-trisubstituted nitrobenzenes (3n,o). Regarding para-substituted derivatives, mixtures of mono- and dialkynylated products (3p,q) with complete ortho-selectivity were also obtained. Polysubstituted substrates could also be efficiently alkynylated leading to (3r–ag) in 32–95% yield with perfect regioselectivity. In this context, substituents such as alkyl, aryl, ether, halide, and even trifluoromethyl were employed in different substitution patterns. We followed our investigations with polyaromatic nitrobenzenes, which gave the corresponding products (3ah, 3ai, 3an) in moderate to excellent yields. The formation of dialkynylated compound 3an might arise from a subsequent electrophilic alkynylation directed by the electron-donating MeO group. Importantly, alkynylated 1-nitropyrene 3ai was obtained in 90% yield at 1 mmol scale in only 4 hours. Next, we applied our reaction conditions to the alkynylation of nitro-heteroarenes. Expectedly, π-deficient heteroarenes, such as 3-nitropyridine, were unreactive.13 On the other hand, when π-excessive nitro-thiophene (1aj) and differently substituted nitro-indoles (1ak–am) were added in slightly excess (2 equiv.), the corresponding alkynylated products 3ak–am were obtained in moderate to good yields. Additionally, when 2 equivalents of 2a were used with 5-nitroindole 1am, double alkynylated 3am′ was produced selectively. Interestingly, other bromoalkynes bearing different bulky silyl protecting groups could be used as alkynylating reagents affording products 3ap–3as in moderate to good yields. However, when smaller silyl groups were tried, the desired products could not be observed (3at, 3au). Careful analysis of the crude reaction mixtures revealed that these silyl alkynes react preferentially with AgSbF6 yielding the corresponding trialkylsilyl fluorides. In all cases, the alkyne dimer and the chloroalkyne could also be detected. Similarly, carbon-substituted bromo-alkynes could not be used as alkynylating reagents (3av–3az).
:1 mixture of mono- and dialkynylated product in 75% yield. This ratio can be reversed by increasing the amount of bromoalkyne 2a, which results in the obtention of dialkynylated 3b′ in excellent yield. Functionalities such as alkyl (3a, 3c), aryl (3d, 3j), ether (3e), aldehyde (3f) and halides (3g–i), groups at the ortho position were well tolerated, leading to 3a–j in 40–95% yield and complete ortho-selectivity. In the case of meta-substituted nitrobenzenes, the alkynylation occurred exclusively at the least hindered site leading to methyl, dimethyl amine or vinyl substituted compounds (3k–m) groups, whereas fluoro and methoxy substituents led to a 2:1 mixture of mono- and dialkynylated products, favoring the formation of 1,2,3-trisubstituted nitrobenzenes (3n,o). Regarding para-substituted derivatives, mixtures of mono- and dialkynylated products (3p,q) with complete ortho-selectivity were also obtained. Polysubstituted substrates could also be efficiently alkynylated leading to (3r–ag) in 32–95% yield with perfect regioselectivity. In this context, substituents such as alkyl, aryl, ether, halide, and even trifluoromethyl were employed in different substitution patterns. We followed our investigations with polyaromatic nitrobenzenes, which gave the corresponding products (3ah, 3ai, 3an) in moderate to excellent yields. The formation of dialkynylated compound 3an might arise from a subsequent electrophilic alkynylation directed by the electron-donating MeO group. Importantly, alkynylated 1-nitropyrene 3ai was obtained in 90% yield at 1 mmol scale in only 4 hours. Next, we applied our reaction conditions to the alkynylation of nitro-heteroarenes. Expectedly, π-deficient heteroarenes, such as 3-nitropyridine, were unreactive.13 On the other hand, when π-excessive nitro-thiophene (1aj) and differently substituted nitro-indoles (1ak–am) were added in slightly excess (2 equiv.), the corresponding alkynylated products 3ak–am were obtained in moderate to good yields. Additionally, when 2 equivalents of 2a were used with 5-nitroindole 1am, double alkynylated 3am′ was produced selectively. Interestingly, other bromoalkynes bearing different bulky silyl protecting groups could be used as alkynylating reagents affording products 3ap–3as in moderate to good yields. However, when smaller silyl groups were tried, the desired products could not be observed (3at, 3au). Careful analysis of the crude reaction mixtures revealed that these silyl alkynes react preferentially with AgSbF6 yielding the corresponding trialkylsilyl fluorides. In all cases, the alkyne dimer and the chloroalkyne could also be detected. Similarly, carbon-substituted bromo-alkynes could not be used as alkynylating reagents (3av–3az).
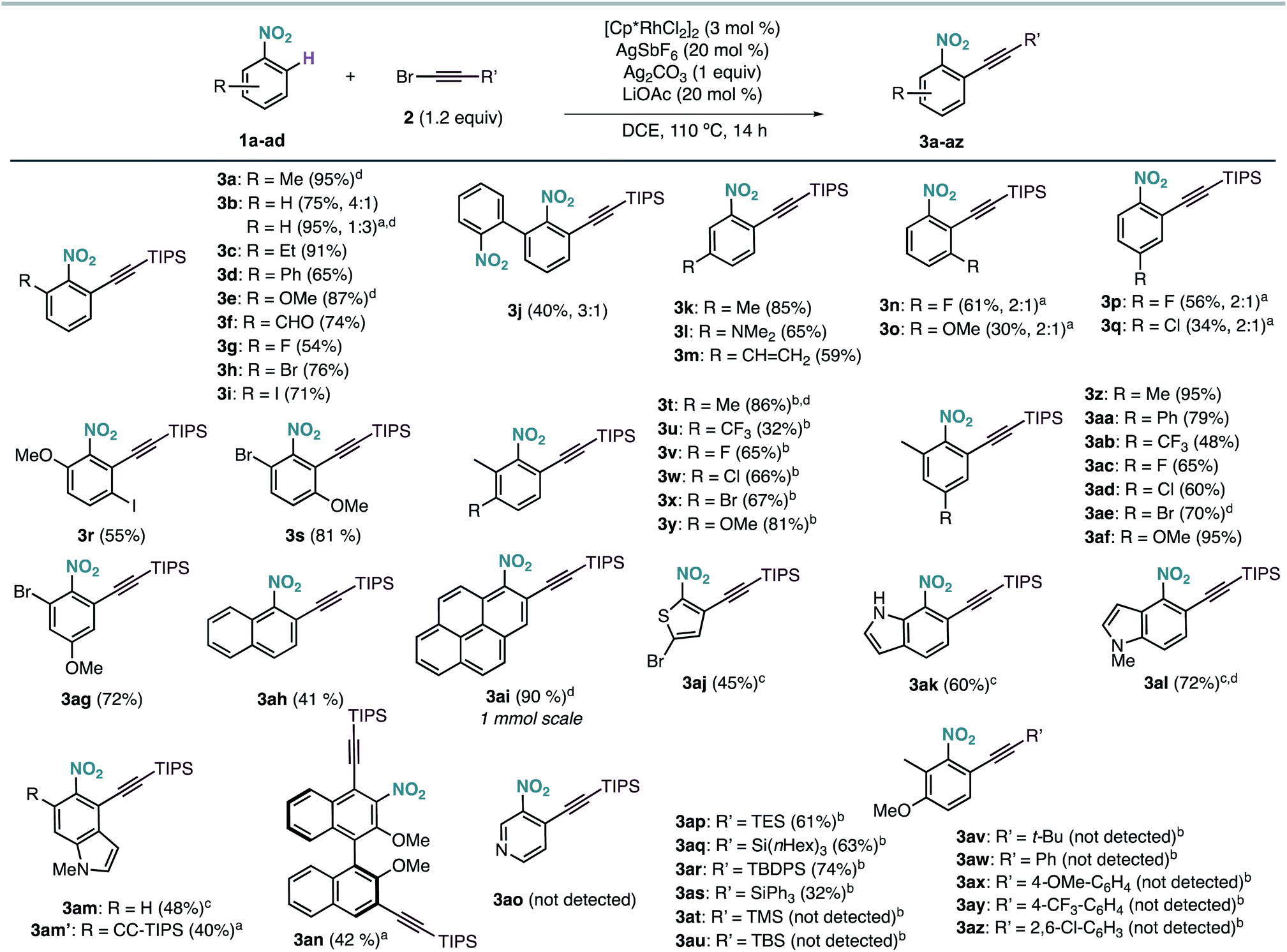 | ||
| Scheme 2 Rh-catalyzed ortho-alkynylation of nitrobenzenes. Yields of isolated products in parentheses. In cases in which dialkynylated products were also formed, mono- vs. dialkynylation selectivity is shown in parentheses. aWith 2 equiv. of bromo-alkyne. bAt 100 °C. cWith 2 equiv. of nitro-heteroarene. d4 h reaction time. | ||
Terminal alkyne 4 was easily accessed by treatment of 3b with a solution of TBAF in THF (Scheme 3). Selective reduction of the nitro group with Fe under acidic conditions delivered aniline 5 in excellent yield, with the triple bond untouched. We derivatized this compound further towards the synthesis of 2-(triisopropyl)silylindole (6) via Au(I)-catalysed cyclisation. This new protocol of alkynylation/reduction/cyclisation allows for the efficient synthesis of important indoles from nitroarene feedstocks. More complex anilines, such as chiral 7, can also be prepared by denitrative Pd-catalysed C–N coupling, following the procedure reported by Wu and coworkers.5g
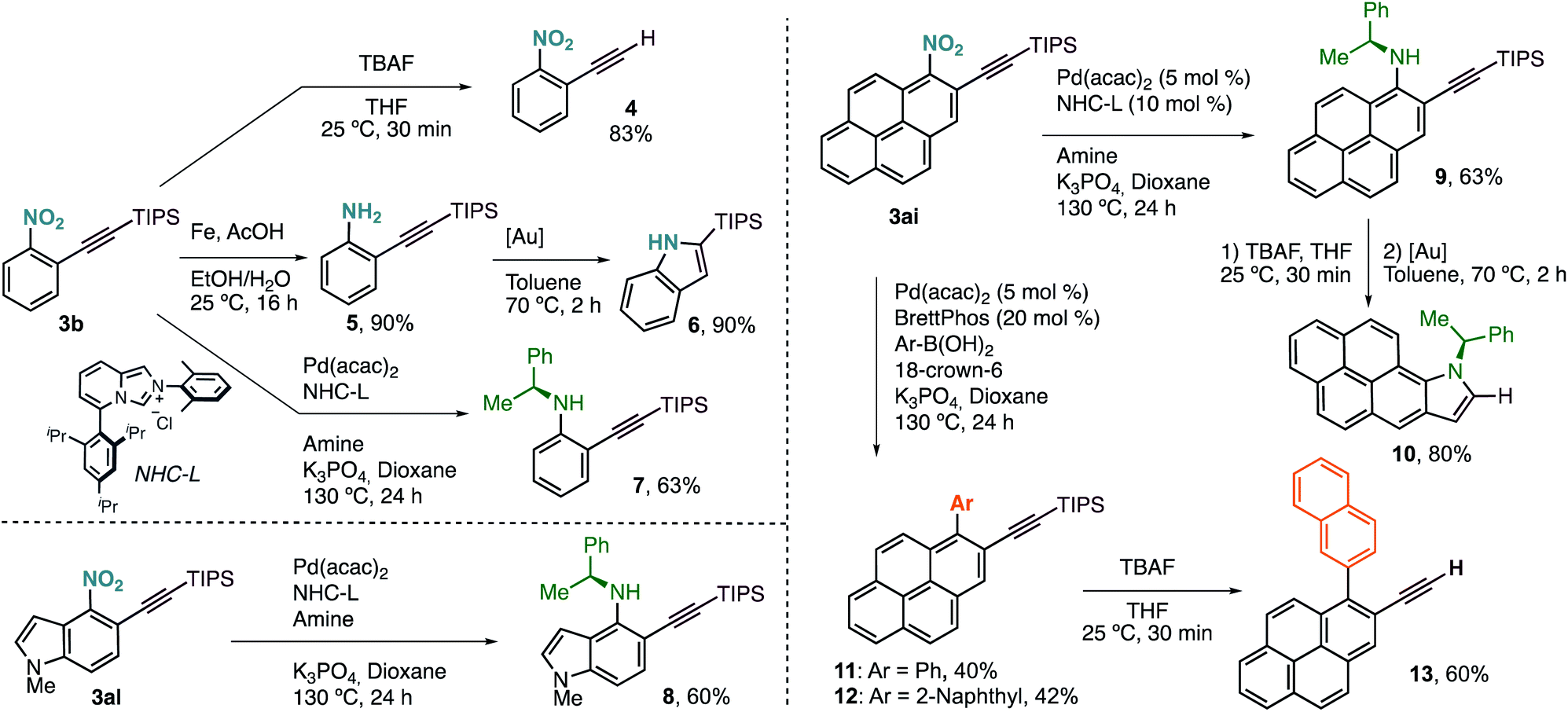 | ||
| Scheme 3 Synthetic transformations. [Au] = [(JohnPhos)Au(MeCN)]SbF6. See ESI† for further experimental details. | ||
This protocol can also be applied to the synthesis of more complex structures such as chiral aminoalkynylated indole 8, prepared in just two steps from simple nitroindole 1al.
Our alkynylation protocol can also be of high interest in the synthesis and modification of π-extended systems, which are attracting attention from the synthetic community due to their interesting properties.14 In this context, we performed the alkynylation of 1-nitropyrene, easily accessed by electrophilic nitration, in a 1 mmol scale with an excellent result (Scheme 2). The corresponding alkynylated nitropyrene 3ai was then subjected to the C–N coupling conditions to form chiral secondary aniline 9, again in good yield (Scheme 3). Deprotection followed by Au(I)-catalysed cyclisation delivered chiral π-extended heteroarene 10 in excellent overall yield. Alternatively, denitrative Suzuki–Miyaura-type coupling was possible by slightly modifying the reported reaction conditions.5a Different aromatic boronic acids were successfully coupled with 3ai to obtain the desired biaryl systems 11 and 12. The corresponding terminal alkynes are also accessible by TBAF deprotection. Thus, as an example of functionalized polycyclic aromatic hydrocarbon, 2-ethynyl-1-(naphthalen-2-yl)pyrene (13) can be easily obtained in just four steps regioselectively from pyrene by nitration, o-alkynylation, cross-coupling, and deprotection.
Interestingly, under our optimised reaction conditions, the alkynylation of nitrendipine, a commercialized antihypertensive agent, leads to alkynylated product 14 in 40% yield with concomitant oxidation of the 1,4-dihydropyridine ring (Scheme 4).
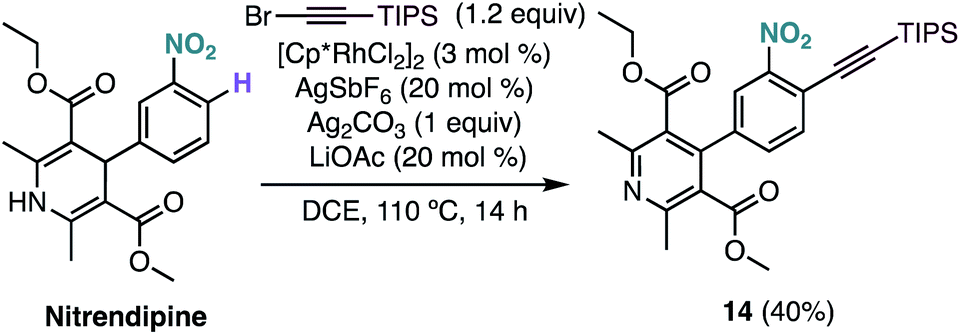 | ||
| Scheme 4 Functionalization of nitrendipine via Rh-catalysed C–H alkynylation. | ||
The site selective Rh-catalysed ortho-functionalization could also be extended to the iodination of nitrobenzenes using N-iodosuccinimide (Scheme 5a).15 Interestingly, an overall regioselective ortho-arylation of nitrobenzene could be performed by a sequential Rh-catalysed ortho-iodination/Pd-catalysed Suzuki–Miyaura coupling leading to biaryl 16 in 50% yield (Scheme 5b).
 | ||
| Scheme 5 Rh-catalysed ortho-iodination of 2-methylnitrobenzene and one-pot ortho-arylation via sequential Rh/Pd catalysis. Conditions A: [Cp*RhCl2]2 (3 mol%), AgSbF6 (15 mol%), LiOAc (20 mol%), NIS (1.2 equiv.), DCE, 120 °C, 14 h. Conditions B: Pd(PPh3)4 (5 mol%), DCE, K2CO3, 60 °C, 14 h. | ||
To understand the mechanism of this unprecedented nitro-directed Rh-catalysed ortho-alkynylation, we performed a series of studies encompassing experimental and computational approaches. The computational calculations were carried out using DFT at ωB97XD/6-31G(d)(H, C, O, N, F, Cl) + LANL2DZ(Rh, Ag, Br)//6-311G++(d,p)(H, C, O, N, F, Cl) + LANL2DZ(Br) + LANL2TZ(Rh, Ag) level of theory, taking into account the solvent effect (SMD = 1,2-dichloroethane).16
First, we studied computationally the complete mechanism of the Rh-catalysed alkynylation of nitrobenzene 1a.17 According to our studies, after several dissociative ligand events, intermediate II undergoes a turnover limiting C–H bond cleavage (ΔG‡ = 25.1 kcal mol−1, energy span) which proceeds through a concerted six-membered cyclic transition state with intramolecular acetate-assistance (TSII-III) (Fig. 1, violet section).18 Dissociative substitution of acetic acid by bromoalkyne 2a gives (η2-alkyne)Rh intermediate IV, which undergoes alkyne insertion through a low energy transition state (TSIV-V, ΔG‡ = 16.1 kcal mol−1) (Fig. 1, pink section). The final step consists in an almost barrierless AgOAc-assisted β-debromination (ΔG‡ = 5.8 kcal mol−1) to form o-alkynylated nitrobenzene 3a in an overall exergonic reaction (ΔG = −40.4 kcal mol−1) (Fig. 1, grey sections). The C–H cleavage transition state (TSII-III) features a slightly elongated C1–H1 (1.277 Å) bond, whereas C1–Rh (2.248 Å) and H1–O1 (1.359 Å) bond distances are considerably contracted compared to the previous intermediate II (Table in Fig. 1). The NBO analysis of TSII-III reveals two main electronic interactions not present in intermediate II related to the C–H bond cleavage event.13
 | ||
| Fig. 1 Simplification of the computed free energy profile for the Rh-catalysed alkynylation of 2-methylnitrobenzene (1a) and relevant structural information of stationary points II, TSII-III and III. Free energies in kcal mol−1 at 25 °C. | ||
First, a lone pair on O1 delocalized over H1 (ηO1, 86.3% O1 and 7.2% H1), which highlights the role of the acetate in the abstraction of H1 during TSII-III (Fig. S6†). Second, a NLMO associated to σ-C1–H1 bond which is delocalized over Rh (ΩC1–H1, 65.4% C, 20.6% H and 8.6% Rh) indicating the formation of the C1–Rh bond during TSII-III (Fig. S7†). All in all, the indicated bond distances and the NBO analysis suggest that TSII-III is considerably asynchronous with a greater extent of C1–Rh and O1–H1 bonds forming than C1–H1 bond cleavage.
Experimentally, we found a significant kinetic isotope effect (KIE = 4.0) (Scheme 6a), which supports the computational finding that the C–H bond cleavage corresponds to the turnover determining step of the catalytic cycle. Moreover, the computed KIE at 110 °C using Cp*Rh was 4.2 and accurately reproduces the experimental results (Scheme 6b).19 Interestingly, the computed KIE using a model system bearing Cp ligand was significantly smaller (KIE = 2.7).
 | ||
| Scheme 6 (a) Experimental and (b) computational KIE for the Rh-catalysed ortho-alkynylation of nitrobenzenes 1b and 1b-d5. Free energies in kcal mol−1 at 110 °C. | ||
Next, to understand whether or not a positive charge could build-up ortho to the nitro group, we performed initial rates measurements of meta-substituted 2-methylnitrobenzenes 1a,t-y (Fig. 2a). A Hammett correlation was found (R2 = 0.95 using σp) with a negative ρ value (ρ = −3.6), suggesting a decrease of electron density at the aryl ring in the C–H activation step (Fig. 2c, dark blue data, triangles). A computational Hammett correlation was also found for a series of meta-substituted nitrobenzenes and the σp parameter (ρ = −2.7, R2 = 0.81) (Fig. 2c, clear blue data, circles).20,21 The computed negative ρ value is in agreement with the experimental results. To understand the charge distribution in the transition state of the turnover-limiting step, we assessed the charge accumulation (Δδ+) over the meta-substituted nitrobenzene fragment in the concerted transition states by NBO analysis (Fig. 2b and c, pink data). There is a slight positive charge built up on the substituted nitrobenzenes (Δδ+ = 0.06–0.14), which agrees with the trends observed by Hammett analysis. In fact, a straight regression line was obtained by plotting Δδ+vs. σp parameters (R2 = 0.89) (Fig. S13†). Both the Hammett and NBO analysis suggest that the Rh-catalysed C–H activation corresponds to an electrophilic concerted metalation deprotonation process,12 in which both an electrophilic metal and a basic ligand cooperate in the cleavage of the C–H bond and subsequent rhodacycle formation.
 | ||
| Fig. 2 Experimental and computational Hammett correlations. The charge accumulation (Δδ+, represented as pink bars) has been calculated as the sum of the NPA on all the atoms of the nitrobenzene fragment on the TSRCp*II-III for all the different substituents specified in the graph (pink area in Fig. 2b). | ||
Despite our previous observation of this mechanism for the alkynylation of esters, ethers and ketones,12a the engagement of nitrobenzenes in acetate-assisted internal electrophilic substitution is rather remarkable, considering the strong electron-withdrawing character and reduced coordination ability of the nitro group. An inspection of the structural features in intermediate II (Table 2) shows an elongated O–Rh bond for the nitro (2.253 Å) when compared to the other directing groups (2.178–2.209 Å), showcasing the limited coordination offered by the –NO2 group (Table 2). Moreover, TSII-III exhibits a shorter C1–H1 bond and longer C1–Rh and O–Rh distances for nitrobenzene.
|
|
||||
|---|---|---|---|---|
| Structure | Directing group | C1–H1 (Å) | C1–Rh (Å) | O2(DG)–Rh (Å) |
| II | NO2 | 1.085 | 3.378 | 2.253 |
| CO2Me | 1.084 | 3.684 | 2.187 | |
| CH2OMe | 1.087 | 3.510 | 2.209 | |
| C(O)Me | 1.086 | 3.340 | 2.178 | |
| TSII-III | NO2 | 1.272 | 2.251 | 2.212 |
| CO2Me | 1.304 | 2.228 | 2.191 | |
| CH2OMe | 1.306 | 2.186 | 2.191 | |
| C(O)Me | 1.308 | 2.214 | 2.169 | |
| III | NO2 | 2.923 | 2.008 | 2.152 |
| CO2Me | 2.273 | 2.023 | 2.165 | |
| CH2OMe | 2.066 | 1.022 | 2.170 | |
| C(O)Me | 2.798 | 2.012 | 2.132 | |

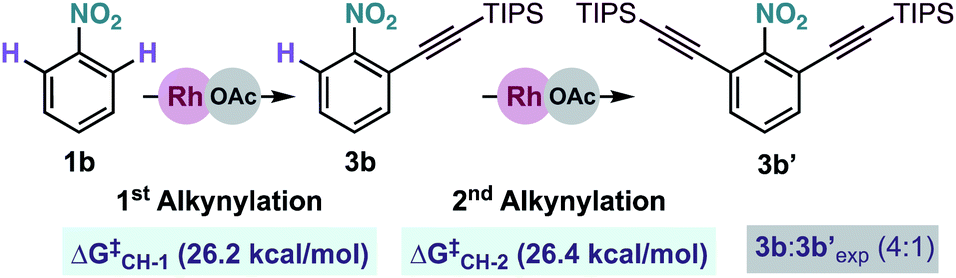
We also examined theoretically the formation of dialkynylated nitrobenzene 3b′ (Scheme 7). Structurally similar transition states were found both for the C–H cleavage of 1b and the subsequent C–H activation step of mono-alkynylated nitrobenzene 3b.13 The small differences between the activation barriers calculated for the first (ΔG‡CH-1 = 26.2 kcal mol−1) and the second (ΔG‡CH-2 = 26.4 kcal mol−1) ortho-C–H cleavages of nitrobenzene 1j explains the moderate mono/di-alkynylation selectivity observed experimentally under the optimal conditions 
 | ||
| Scheme 7 Energy comparison for the Rh-catalysed mono and di-alkynylation of nitrobenzene 1b. Free energies at 25 °C. | ||
Regarding nitro-heteroarenes, we found that the activation energy for the C–H metalation step is energetically more demanding for thiophene (1aj) and 3-nitropyridine (1ao) than for nitroarenes which make the overall ortho-alkynylation more challenging.13
Surprisingly, our calculations show that the alkyne insertion/Ag-assisted-β-debromination sequence is energetically feasible for the experimentally unsuccessful alkyne counterparts 2c, 2d, and 2f,13 which suggests that alkyne 2a has the correct steric and electronic balance to prevent scenarios in which the alkyne is unproductively consumed leading to secondary products prior to its insertion into the C–Rh bond. Particularly intrigued by the low activation barrier for the β-debromination for non-Si containing bromo phenylacetylene (2f) (ΔG‡ = 4.3 kcal mol−1), we examined the hyperconjugative interactions on the previous intermediates to the bromo elimination V and V′ for alkynes 2a and 2f, respectively (Fig. 3).22 For V, the NBO analysis revealed a strong interaction between σ(Rh-Cα) and σ*(Br-Cβ) orbitals (Eij(2) = 22.7 kcal mol−1) (Fig. 3, left), while no interaction was found between the σ(Si-Cα) and σ*(Br-Cβ) orbitals. The same scenario was encountered for V′, in which the σ(Rh-Cα) and σ*(Br-Cβ) orbitals also interact significantly (Eij(2) = 17.4 kcal mol−1) (Fig. 3, right). These results indicate that the Ag-assisted debromination is facilitated by the σ–σ-conjugation arising from the so-called “β-Rh effect”,17a,b rather than by β-Si effect.23
 | ||
| Fig. 3 Plot of the donor–acceptor NBO interaction for the σ–σ-conjugation across alkenyl-Rh for V and V′. Cutoff: 0.05. | ||
Conclusions
We have developed the first general o-alkynylation of abundant nitroarenes, which proceeds in a rather straightforward manner under catalytic conditions. Despite the strongly electron-withdrawing nature of the nitro group, our experimental and computational results demonstrate that the rhodium-catalysed o-alkynylation of nitroarenes takes place by an electrophilic concerted metalation/deprotonation pathway. The resulting o-alkynyl nitroarenes are precursors of indoles by reduction to the corresponding anilines and cyclization24 and have also been used as synthons for the preparation of other valuable products.25 In this context, the nitro group can be viewed as a “super pseudo-halogen”, that participates in a manifold of cross-coupling reactions,3–5 while allowing also the introduction of highly versatile alkynyl groups at the ortho position under catalytic conditions.Data availability
All experimental and computational data associated with the article are incorporated into the ESI.†Author contributions
Conceptualization: A. M. E. and E. T.; formal analysis: C. G.-M.; funding acquisition: A. M. E.; investigation: E. T., M. M.-M., C. G.-M., and J. G. M; methodology: E. T., and C. G.-M; project administration: A. M. E.; supervision: A. M. E.; visualization: E. T., M. M.-M., C. G.-M., and J. G. M.; writing – original draft: E. T., C. G.-M., and A. M. E.; writing – review & editing: E. T., M. M.-M., C. G.-M., J. G. M., and A. M. E.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the MCIN/AEI/10.13039/501100011033 (PID2019104815GB-I00 and Severo Ochoa Excellence Accreditation 2020–2023 CEX2019-000925-S), the AGAUR (2017 SGR 1257), and CERCA Program/Generalitat de Catalunya for financial support. M. M.-M. thanks Ministerio de Ciencia e Innovación for a Juan de la Cierva contract (IJC2019-040181-I).Notes and references
-
(a)
Nitro Compounds, Aromatic, in Ullmann's Encyclopedia of Industrial Chemistry, G. Booth, Wiley-VCH, Weinheim, 6th edn, 2000 Search PubMed
; (b) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, 2001 CrossRef
-
M. B. Smith and J. March, March's Advanced Organic Chemistry, John Wiley & Sons, Inc., New York, 5th edn, 2001 Search PubMed
- X. Zheng, J. Ding, J. Chen, W. Gao, M. Liu and H. Wu, Org. Lett., 2011, 13, 1726–1729 CrossRef CAS PubMed
-
(a) J. Zhang, J. Chen, M. Liu, X. Zheng, J. Ding and H. Wu, Green Chem., 2012, 14, 912–916 RSC
-
(a) M. R. Yadav, M. Nagaoka, M. Kashihara, R.-L. Zhong, T. Miyazaki, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2017, 139, 9423–9426 CrossRef CAS PubMed
-
(a) M. Makosza and J. Winiarski, Acc. Chem. Res., 1987, 20, 282–289 CrossRef CAS
-
(a) X. Zhang, M. Kanzelberger, T. J. Emge and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13192–13193 CrossRef CAS PubMed
-
(a) L. Caron, L.-C. Campeau and K. Fagnou, Org. Lett., 2008, 10, 4533–4536 CrossRef CAS PubMed
- Pd-catalysed C–H arylation of nitrobenzenes
in which nitro group is not acting as directing group:
(a) J. J. Gonzalez, N. Garcia, B. Gomez-Lor and A. M. Echavarren, J. Org. Chem., 1997, 62, 1286–1291 CrossRef CAS
-
(a) P. Guo, J. M. Joo, S. Rakshit and D. Sames, J. Am. Chem. Soc., 2011, 133, 16338–16341 CrossRef CAS PubMed
- For references in C(sp2)–H alkynylation:
(a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed
-
(a) E. Tan, O. Quinonero, M. E. De Orbe and A. M. Echavarren, ACS Catal., 2018, 8, 2166–2172 CrossRef CAS PubMed
- See ESI for further details.†.
-
(a) W. Yang, J. H. S. K. Monteiro, A. de Bettencourt-Dias, V. J. Catalano and W. A. Chalifoux, Angew. Chem., Int. Ed., 2016, 55, 10427–10430 CrossRef CAS PubMed
- For references in Rh-catalysed C–H halogenations, see:
(a) N. Schröder, J. Wencel-Delord and F. Glorius, J. Am. Chem. Soc., 2012, 134, 8298–8301 CrossRef PubMed
- A dataset collection of computational results is available in the ioChem-BD repository and can be accessed through https://doi.org/10.19061/iochem-bd-1-158, M. Álvarez-Moreno, C. Graaf, N. Lopez, F. Maseras, J. M. Poblet and C. Bo, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef PubMed
-
(a) B. E. Haines, R. Sarpong and D. G. Musaev, J. Am. Chem. Soc., 2018, 33, 10612–10618 CrossRef PubMed
- The alternative 4-membered cyclic transition state (TSCH-4, ΔG‡ = 38.7 kcal mol−1) and the intermolecular acetate-assisted transition state (TSCH-inter, ΔG‡ = 45.4 kcal mol−1) were found to be much higher in energy..
- The KIE was calculated based on the free energy difference between [Cp*RhAlkyneOAc] (ICp*) and the corresponding TSH/DCp*II-III (T = 110 °C).
- The Hammett plot was calculated based on the free energy difference between [Cp*RhAlkyneOAc] (ICp*) and the corresponding TSRCp*II-III. A better-fitting correlation was found using potential energies (ΔE‡) (ρ = −2.57, R2 = 0.91) (Fig. S11†).
- For an explanation on the plausible pitfalls using free activation energies to run comparisons between similar structures: H. Ryu, J. Park, H. K. Kim, J. Y. Park, S.-T. Kim and M.-H. Baik, Organometallics, 2018, 19, 3228–3239 CrossRef
- Similar results were obtained for VI and VI′ and no interaction was found between Br and Ag. See Fig. SX.†.
-
(a) Z. X. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 3172–3176 CrossRef CAS PubMed
-
(a) W.-M. Dai, L.-P. Sun and D.-S. Guo, Tetrahedron Lett., 2002, 43, 7699–7702 CrossRef CAS
- Lead references on other uses of o-alkynyl nitroarenes as synthons:
(a) M. S. Maier, K. Huell, M. Reynders, B. S. Matsuura, P. Leippe, T. Ko, L. Schaeffer and D. Trauner, J. Am. Chem. Soc., 2019, 43, 17295–17304 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental figures, synthetic and computational details, spectra for synthesized compounds. See DOI: 10.1039/d1sc04527j |
| This journal is © The Royal Society of Chemistry 2021 |