
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA unified strategy to prostaglandins: chemoenzymatic total synthesis of cloprostenol, bimatoprost, PGF2α, fluprostenol, and travoprost guided by biocatalytic retrosynthesis†
Kejie
Zhu
ab,
Meifen
Jiang
ab,
Baijun
Ye
 ab,
Guo-Tai
Zhang
c,
Weijian
Li
c,
Pei
Tang
c,
Zedu
Huang
*ab and
Fener
Chen
*abc
ab,
Guo-Tai
Zhang
c,
Weijian
Li
c,
Pei
Tang
c,
Zedu
Huang
*ab and
Fener
Chen
*abc
aEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China. E-mail: huangzedu@fudan.edu.cn; rfchen@fudan.edu.cn
bShanghai Engineering Research Center of Industrial Asymmetric Catalysis of Chiral Drugs, 220 Handan Road, Shanghai, 200433, P. R. China
cSichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu, 610041, P. R. China
First published on 1st July 2021
Abstract
Development of efficient and stereoselective synthesis of prostaglandins (PGs) is of utmost importance, owing to their valuable medicinal applications and unique chemical structures. We report here a unified synthesis of PGs cloprostenol, bimatoprost, PGF2α, fluprostenol, and travoprost from the readily available dichloro-containing bicyclic ketone 6a guided by biocatalytic retrosynthesis, in 11–12 steps with 3.8–8.4% overall yields. An unprecedented Baeyer–Villiger monooxygenase (BVMO)-catalyzed stereoselective oxidation of 6a (99% ee), and a ketoreductase (KRED)-catalyzed diastereoselective reduction of enones 12 (87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 to 99:1 dr) were utilized in combination for the first time to set the critical stereochemical configurations under mild conditions. Another key transformation was the copper(II)-catalyzed regioselective p-phenylbenzoylation of the secondary alcohol of diol 10 (9.3:1 rr). This study not only provides an alternative route to the highly stereoselective synthesis of PGs, but also showcases the usefulness and great potential of biocatalysis in construction of complex molecules.
:13 to 99:1 dr) were utilized in combination for the first time to set the critical stereochemical configurations under mild conditions. Another key transformation was the copper(II)-catalyzed regioselective p-phenylbenzoylation of the secondary alcohol of diol 10 (9.3:1 rr). This study not only provides an alternative route to the highly stereoselective synthesis of PGs, but also showcases the usefulness and great potential of biocatalysis in construction of complex molecules.
Introduction
Prostaglandins (PGs) are hormone-like lipid compounds often found in animals and human-beings,1 and they are shown to display a multitude of biological functions. To date, more than 20 PG analogs have been developed as marketed medicines, such as the veterinary drugs cloprostenol (1) and fluprostenol (4), and antiglaucoma drugs bimatoprost (2) and travoprost (5) (Fig. 1).1c In particular, bimatoprost has recently become a block-buster drug. Because of their valuable medicinal applications and unique chemical structures, tremendous efforts have been devoted to the efficient synthesis of PGs.1,2 In fact, since Corey's landmark synthesis of prostaglandin F2α (PGF2α, 3) in the late 1960s,3 PGs have become one touchstone of state-of-the-art synthetic methodologies,4 as exemplified in the elegant synthesis recently reported by the groups of Aggarwal,2,4a–c Hayashi,4d–h Grubbs,4i Fletcher,4j Nicolaou4k,l and Baran.4m Our group has developed an efficient and modular synthesis of PGs (Scheme S1†).5 Crucial to our success was the stereocontrolled synthesis of the key lactone intermediate 7avia a chiral spiro-phosphoric acid-catalyzed Baeyer–Villiger (B–V) oxidation of bicyclic ketone 6a (Scheme S1†).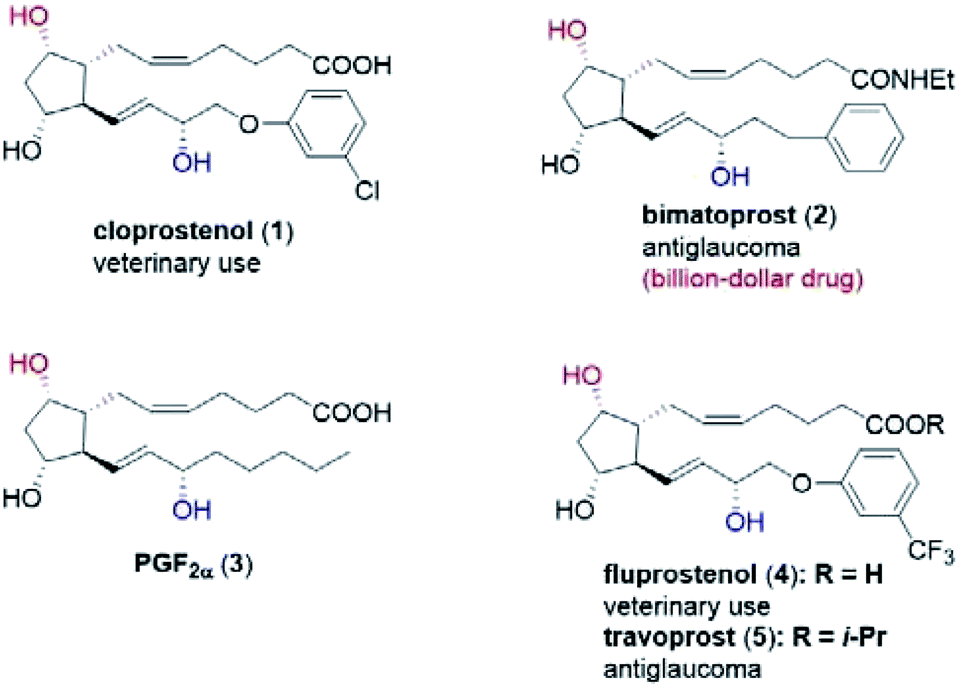 | ||
| Fig. 1 Structure of selected prostaglandins: cloprostenol (1), bimatoprost (2), PGF2α (3), fluprostenol (4), and travoprost (5). | ||
The past two decades have witnessed the rapid development of biocatalysis into a sophisticated technology, mainly thanks to the ever-increasing bioinformatics and protein engineering tools.6 Compared to chemocatalysis, biocatalysis usually offers unparalleled chemo-, regio-, and stereoselectivity pivotal for efficient synthesis of complex molecules like pharmaceuticals and bioactive natural products.6e,f,7 To harness the synthetic potential of these extraordinary selectivities and other advantageous properties inherent to biocatalysis (e.g. mild reaction conditions) more productively, Turner introduced the concept of biocatalytic retrosynthesis to the scientific community about seven years ago,6a which has since been widely and increasingly utilized when planning synthetic routes to target molecules.8 From a retrosynthetic viewpoint, a biocatalytic reaction can be adopted in two distinct ways: either being used as an alternative approach to replace an equivalent chemical step, or being employed to fulfill a disconnection which is impossible to realize by traditional chemical means.6a As an example in the former scenario, ketoreductase (KRED)-catalyzed stereoselective reduction was demonstrated to be a milder and more sustainable process compared to (−)-DIP-Cl-mediated reduction, in the synthesis of a key chiral alcohol intermediate to the active pharmaceutical ingredient (API) montelukast.9 So far, the latter scenario has been less exploited.6a Recently, a regio- and stereoselective transformation from prochiral ketoenones to 2,6-disubstituted piperidines, a formidable task for classical chemical methods, was accomplished via an ω-transaminase triggered intramolecular aza-Michael reaction and subsequent epimerization.8a In continuation of our interest in the development of efficient, stereoselective, and green synthesis of PGs, as well as application of biocatalysis to organic synthesis,10 herein we report a tractable, chemoenzymatic total synthesis of prostaglandins guided by biocatalytic retrosynthesis.
Results and discussion
In our biocatalytic retrosynthesis (Fig. 2), cloprostenol (1), bimatoprost (2), PGF2α (3), and fluprostenol (4), prostaglandins containing an allylic alcohol moiety on the ω-side-chain, could be synthesized from lactones 13a–13d through a three-step sequence of p-phenylbenzoyl (PPB) ester hydrolysis, DIBAL-H reduction, and Wittig olefination. We envisioned that the crucial stereogenic center of the ω-side-chain of 13a–13d could be installed via a KRED-catalyzed diastereoselective reduction of enones 12a–12d, which were prepared from C11-OH-PPB-protected Corey alcohol 11 (prostaglandin numbering) through primary alcohol oxidation, followed by Horner–Wadsworth–Emmons (HWE) olefination. It was hoped that 11 might be furnished via a catalyst-controlled regioselective p-phenylbenzoylation of the C11-OH of the known diol 10,11 which was synthesized from lactone 7avia a three-step sequence of dechlorination, the Prins reaction, and deformylation. At this stage, the key lactone 7a was envisioned to be accessible through a Baeyer–Villiger monooxygenase (BVMO)-catalyzed stereoselective oxidation of bicyclic ketone 6a. Finally, travoprost (5) can be synthesized from fluprostenol (4) via a late-stage i-propyl ester formation reaction.4b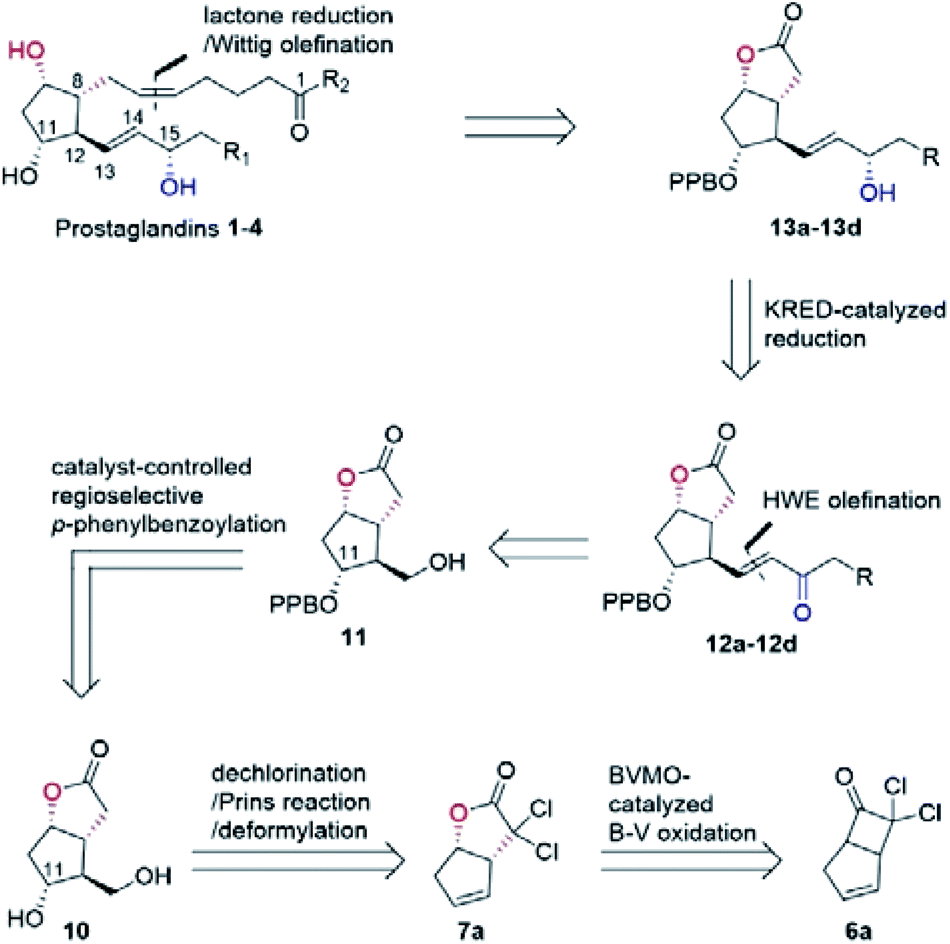 | ||
| Fig. 2 Biocatalytic retrosynthesis of prostaglandins 1–4. | ||
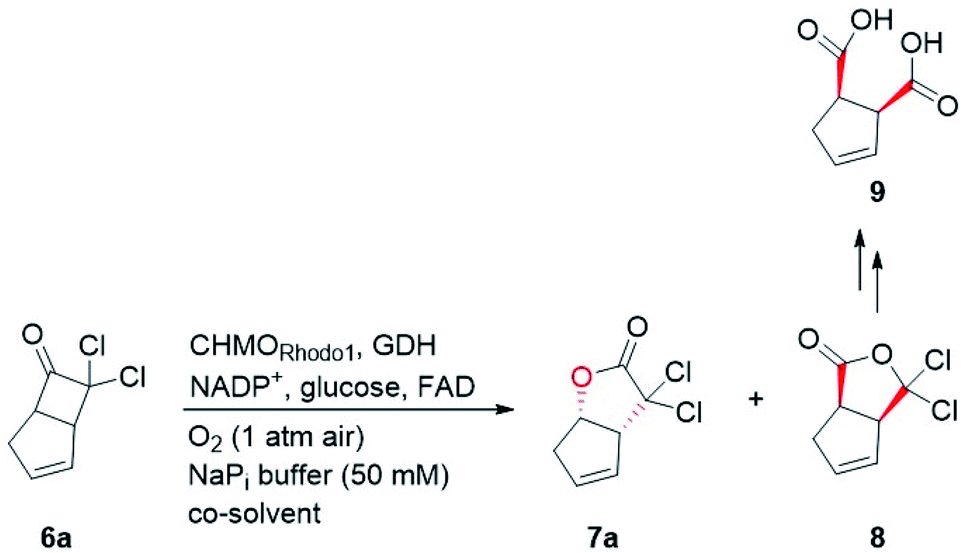
Bicyclo[3.2.0]hept-2-en-6-one (14), a bicyclic ketone lacking the dichloro functionality in comparison to 6a, was routinely employed as the model substrate in BVMO-catalyzed oxidation reactions (Scheme S2†).12 A regiodivergent conversion of 14 has been observed for most BVMOs, resulting in the formation of the enantioenriched normal lactone (NL) 15 and abnormal lactone (AL) 16 in similar amounts (Scheme S2†). Under such circumstances, the separation of 15 and 16 is troublesome. On the other hand, our previous study on chiral spiro-phosphoric acid-catalyzed B–V oxidation suggested that it was beneficial to introduce the dichloro functionality into the cyclobutanone ring of 6a. Firstly, formation of NL 7a becomes more favored because of the electron-withdrawing effect of the dichloro group (Scheme S1†). Secondly, the formed AL 8 could be readily converted to the easily removable cyclopentene dicarboxylic acid 9 during the reaction workup, thus facilitating the isolation of the desired 7a (Scheme S1†). Therefore, we were interested in the development of BVMO-catalyzed oxidation of dichloro-containing bicyclic ketone 6a to lactone 7a, which has not been reported, to the best of our knowledge. In the present study, three “regiodivergent” BVMOs, namely CHMORhodo1,13 CHMOArthro,13 and CHMOBrevi1,14 as well as the unique “regioselective” enzyme BVMO-MO14 originating from Rhodococcus jostii,15 are examined in the oxidation of 6a. On one hand, moderate to high conversion of 6a and formation of trace-amounts of the desired 7a (≤5%) were observed in the CHMOArthro-, CHMOBrevi1-, and BVMO-MO14-catalyzed reactions (Table S2†). On the other hand, 6a was completely consumed in the CHMORhodo1-catalyzed reaction, but the yield of 7a was only 25% (Table 1, entry 1, also see Table S2†), much lower than the 50% maximum theoretical yield for a resolution event. To account for this mass imbalance, we investigated the stability of 6a and racemic-7a. In the absence of BVMO, it was found that 6a and racemic-7a decomposed readily, with only 58% and 66% remaining, respectively, after incubation for 90 minutes (Fig. S5†). Presumably, the electron-withdrawing dichloro group makes the carbonyl more electron-deficient, hence rendering 6a and 7a more susceptible to hydrolysis as previously suggested.16 To improve the yield of 7a, CHMORhodo1-catalyzed oxidation reactions were carried out at different pHs (Table 1, entries 2 and 3) or at different temperatures (Table 1, entries 4 and 5). No significant improvement was achieved. Next, five organic solvents were screened in an effort to increase the product yield (Table 1, entries 6–10). To our delight, the use of methyl tert-butyl ether (MTBE) and 2-methoxyethanol (MME) significantly increased the yield of 7a to 38% and 36%, respectively. Not only the yield, but also the enantiomeric purity of 7a was revealed to be dependent on the co-solvents employed. While the use of MTBE resulted in an inferior enantiomeric purity (73% ee), no adverse effect was observed when using MME. Hence, the latter solvent was chosen for the remaining study. Gratifyingly, a semi-preparative-scale (0.5 mmol) oxidation of 6a under the above optimized conditions furnished the desired 7a in 38% isolated yield with 99% ee (Table 1, entry 11). Hence, we have realized the BVMO-catalyzed oxidation of 6a, which is prone to hydrolysis, to normal lactone 7a under buffer conditions in good yield and excellent stereoselectivity for the first time. The chemical structure and the stereochemical assignment of 7a were unambiguously established by X-ray crystallography (Fig. 3). Cyclopentene dicarboxylic acid 9, resulting from the hydrolysis of abnormal lactone 8, was isolated in 35% yield with 82% ee (determined upon derivatization, see Scheme S6† for details).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Co-solvent | pH | 6a remainingb (%) | Yield of 7ab (%) | ee of 7ac (%) |
| a Reaction conditions (1 mL): 6a (10 mM), glucose (60 mM), NADP+ (0.2 mM), FAD (0.05 mM), 0.79 mL of 20% (w/v) cell-free extract (CFE) of CHMORhodo1 in NaPi buffer (50 mM), 0.016 mL of 15% (w/v) CFE of glucose dehydrogenase (GDH) in NaPi buffer (50 mM, pH 7.0), and 0.1 mL co-solvent (if applicable) in NaPi buffer (50 mM). Reaction mixtures were incubated at 25 °C with 200 rpm shaking for 90 min. b Determined by GC analysis using undecane as the internal standard. c Determined by SFC analysis. d Not applicable (N.A.). e Not determined (N.D.). f The reaction temperature was 30 °C. g The reaction temperature was 35 °C. h The reaction volume was 50 mL (0.5 mmol scale). i Isolated yield. | |||||
| 1 | N.A.d | 7.5 | 0 | 25 | 99 |
| 2 | N.A. | 7.0 | 0 | 26 | N.D.e |
| 3 | N.A. | 6.5 | 6 | 26 | N.D. |
| 4f | N.A. | 7.5 | 0 | 22 | N.D. |
| 5g | N.A. | 7.5 | 0 | 22 | N.D. |
| 6 | Cyclohexane | 7.5 | 23 | 23 | N.D. |
| 7 | DMSO | 7.5 | 0 | 28 | N.D. |
| 8 | MTBE | 7.5 | 0 | 38 | 73 |
| 9 | Dioxane | 7.5 | 0 | 32 | 82 |
| 10 | MME | 7.5 | 0 | 36 | 99 |
| 11h | MME | 7.5 | 0 | 40 (38i) | 99 |
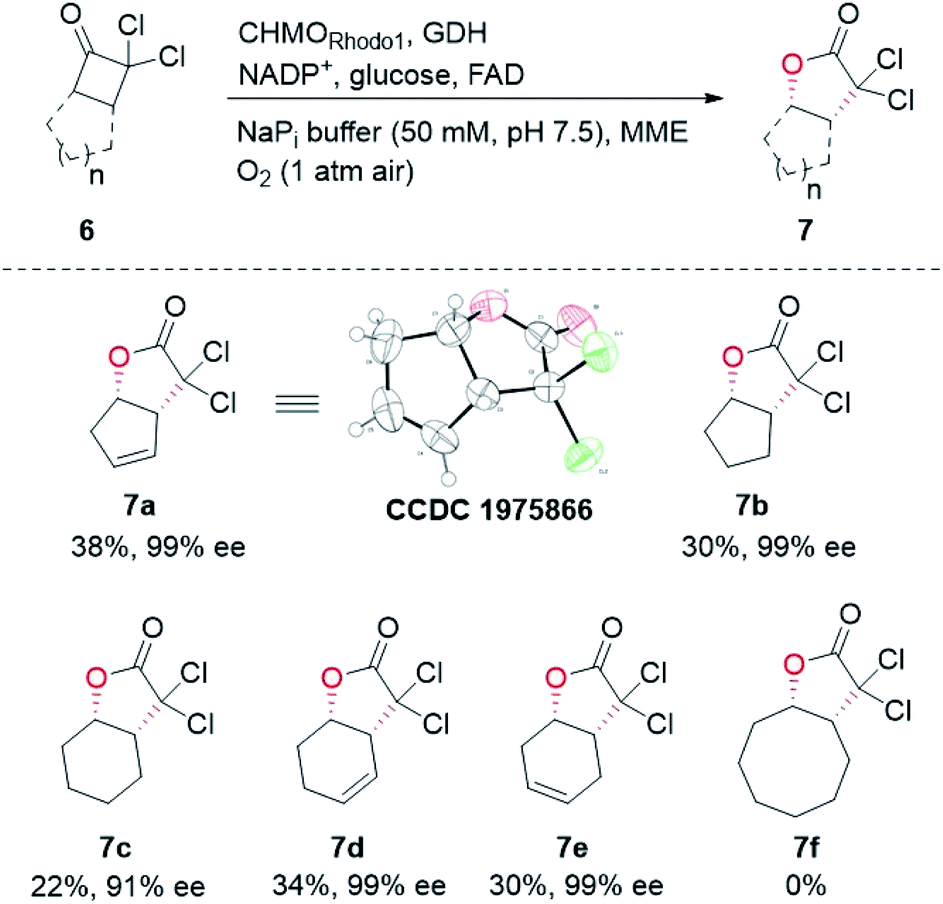 | ||
| Fig. 3 Substrate scope of CHMORhodo1-catalyzed B–V oxidation of bicyclic cyclobutanones. | ||
Other bicyclic cyclobutanones were employed as substrates to evaluate the feasibility of the CHMORhodo1-catalyzed oxidation reaction (Fig. 3). Firstly, ketone 6b without an olefin moiety was well recognized by CHMORhodo1, furnishing the desired normal lactone 7b in 30% isolated yield with 99% ee. Secondly, the size of the fused ring seemed to play an important role in this enzyme-catalyzed B–V oxidation process. For instance, the oxidation of substrates containing a 5-membered or 6-membered fused ring (6b–6e) occurred smoothly, affording the corresponding lactones 7b–7e in 22–34% isolated yield with 91–99% ee. However, ketone 6f with an 8-membered fused ring could not be converted by CHMORhodo1 and no desired lactone 7f was detected. It is possible that the overlarge size of this fused ring prevents a proper binding of 6f to CHMORhodo1.
The transformation of 7a into the known diol 10 was realized via a three-step sequence in flow chemistry, which was significantly more time-economical compared to batch reactions (Fig. 4).5 Firstly, a continuous flow dechlorination of 7a was accomplished in a packed bed reactor (Shenzhen E-Zheng Tech Co., Ltd) filled with zinc powder at 70 °C under 7 bar back-pressure with 10 min residence time, giving NL 15 in 90% yield. Then NL 15 was dissolved in a 10:1 HCOOH/H2SO4 solution containing pre-dissolved paraformaldehyde, and the mixture was pumped into a 0.5 mL PTFE reactor coil (i.d. = 0.8 mm) at 70 °C under 17 bar back-pressure with 15 min residence time to afford crude 17 as the major product with full conversion by the Prins reaction. After neutralization and removing the inorganic salt, the crude 17 in MeOH combined with NaOMe was pumped into a 0.5 mL PTFE reactor coil via a T-junction to complete deformylation followed by quenching with AcOH to give diol 10 smoothly in 81% yield over two steps (Fig. 4).
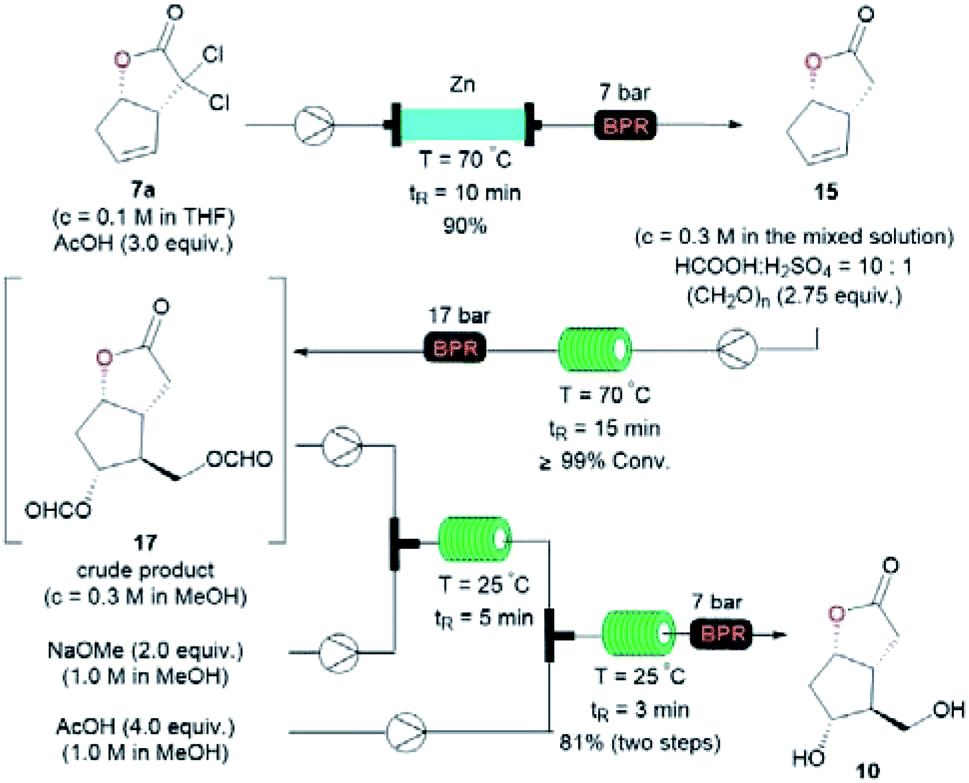 | ||
| Fig. 4 Conversion of lactone 7a into diol 10 in continuous flow. | ||
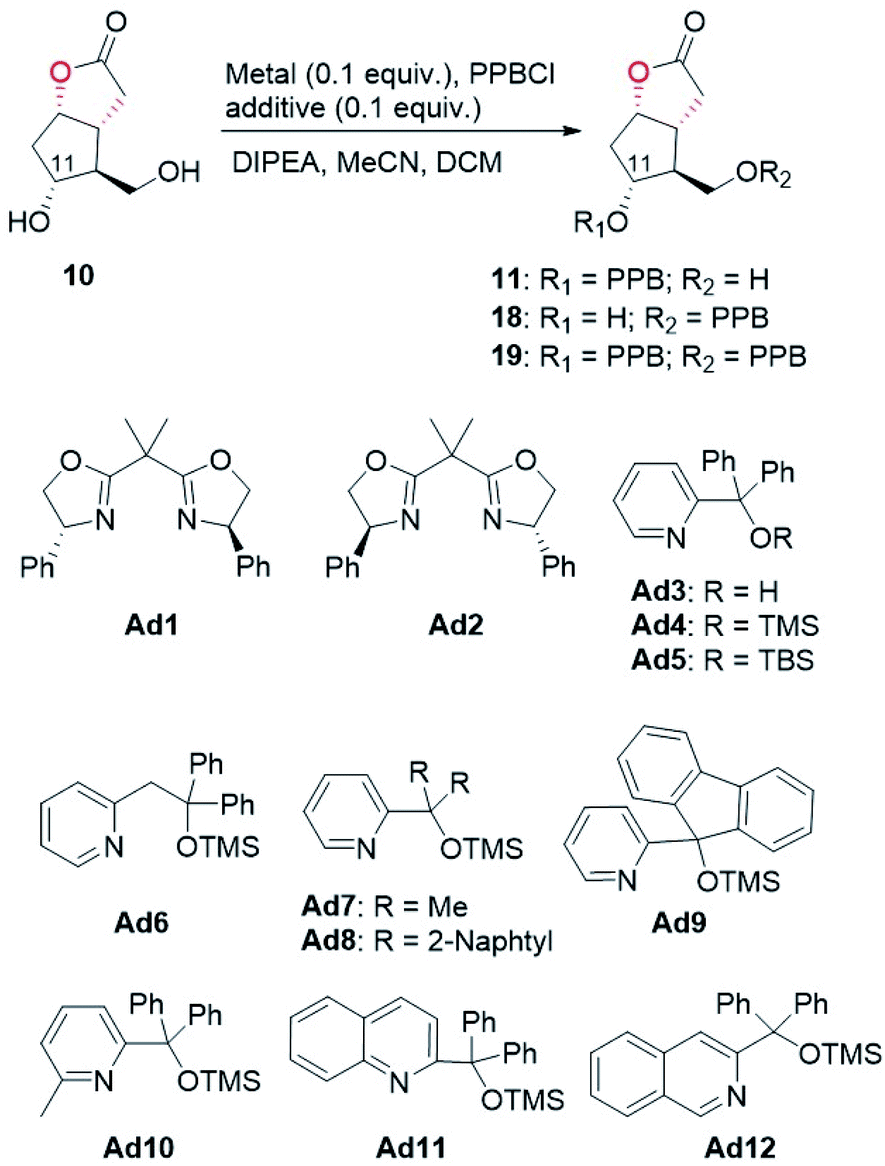
With access to diol 10, the site-selective protection of C11-OH could now commence. In the traditional total synthesis of PGs, the three-step choreography of protecting group manipulations of diol 10 would establish a C11-OH-protected Corey alcohol motif present within PGs, aiding purification and imparting chemoselectivity in subsequent transformations.17 However, this strategy is not step-economic for PG synthesis. To circumvent this limitation, we recently developed a directly regioselective oxidation of the primary alcohol of 10 by using a modified TEMPO/PhI(OAc)2 protocol to C11-OH non-masked Corey aldehyde for joining the ω-side-chain via Horner–Wadsworth–Emmons (HWE) olefination.5,18 While seemingly straightforward, successful implementation of our strategy would not be attractive and practical in the large-scale synthesis of prostaglandins, because most of the oil prostaglandin intermediates obtained in this protecting-group-free chemistry are unstable. Therefore, designing a one-step method for catalyst-controlled site-selective p-phenylbenzoylation of C11-OH in 10 would represent a valuable goal that promises streamlined access to the stable and nicely crystalline C11-OH-PPB-protected Corey alcohol 11 (Fig. 2).19
With this assumption in mind, the p-phenylbenzoylation of diol 10 was first conducted under typical acylation conditions as reported by the Dong group.20 As summarized in Table 2, monoesters 11 and 18 were both formed in 8% and 16% yields, respectively (entry 1). When the reaction temperature was lowered to −20 °C, only a trace amount of 18 (<5%) was obtained (entry 2). The use of copper salt and appropriate ligands/additives was previously demonstrated to promote regioselective acylation of substrates containing multiple hydroxyl groups.11 In the present study, we found that CuCl2 alone could accelerate the reaction, and importantly, the desired 11 was afforded in 48% yield as the major product (entry 3). To further improve the regioselectivity, several additives were tested. When chiral bisoxazoline compounds Ad1 and Ad2, the two commonly used ligands/additives previously in regioselective acylation reactions,11 were examined first, the undesired monoester 18 was predominantly generated (entries 4 and 5). To our delight, the use of additives Ad3–Ad6 containing a pyridine moiety favoured the formation of 11 (entries 6–9). In particular, the ratio of 11 to 18 was improved to 4.3:1 when Ad4 was attempted (entry 7, also see Fig. S6 and S7†). Encouraged by these results, we further designed and synthesized compounds Ad7–Ad12 based on the structure of Ad4. Upon screening (entries 10–15), a regioisomeric ratio of 6.4:1 was accomplished by using compound Ad9 containing a fluorene moiety as the additive (entry 12). Optimization of reaction conditions, including temperature, time, and the amount of DIPEA, further increased the regioisomeric ratio to 9.3:1, furnishing the desired monoester 11 in 73% isolated yield (entry 17). Taken together, by using a cheap copper(II) salt and an achiral additive Ad9, the regioselective p-phenylbenzoylation of diol 10 to the desired monoester 11 was realized in good yield and regioisomeric ratio. We believe that this regioselective p-phenylbenzoylation reaction will find wide application in the synthesis of prostaglandins. The detailed mechanism of this copper(II)-catalyzed regioselective acylation reaction warrants investigation in the future.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Metal | Additive | Yield of 11b (%) | Yield of 18b (%) | Yield of 19b (%) | Regioisomeric ratio (rr), 11:18 |
| a Reaction conditions (1.5 mL): 10 (0.1 mmol), PPBCl (0.1 mmol), DIPEA (0.1 mmol), CuCl2 (0.1 equiv., if applicable), additive (0.1 equiv., if applicable) in MeCN (1 mL) and DCM (0.5 mL). Reaction mixtures were stirred at −20 °C for 14 h. b Determined by 1H NMR analysis of the reaction crude using 1,3,5-trimethoxybenzene as the internal standard. c Reaction run at room temperature. d Not applicable (N.A.). e Stirred at −10 °C for 24 h. f Reaction scale (1 mmol), DIPEA (1.2 equiv.), stirred at −10 °C for 60 h. g Isolated yield. | ||||||
| 1c | N.A.d | N.A. | 8 | 16 | 1 | 1:2 |
| 2 | N.A. | N.A. | 0 | <5 | 0 | N.A. |
| 3 | CuCl2 | N.A. | 48 | 18 | 4 | 2.7:1 |
| 4 | CuCl2 | Ad1 | 13 | 63 | 0 | 1:4.8 |
| 5 | CuCl2 | Ad2 | 32 | 45 | 3 | 1:1.4 |
| 6 | CuCl2 | Ad3 | 54 | 14 | 2 | 4.0:1 |
| 7 | CuCl2 | Ad4 | 57 | 13 | 2 | 4.3:1 |
| 8 | CuCl2 | Ad5 | 40 | 18 | 2 | 2.2:1 |
| 9 | CuCl2 | Ad6 | 57 | 20 | 2 | 2.9:1 |
| 10 | CuCl2 | Ad7 | 59 | 19 | 2 | 3.0:1 |
| 11 | CuCl2 | Ad8 | 55 | 15 | 3 | 3.6:1 |
| 12 | CuCl2 | Ad9 | 58 | 9 | 2 | 6.4:1 |
| 13 | CuCl2 | Ad10 | 49 | 18 | 3 | 2.8:1 |
| 14 | CuCl2 | Ad11 | 52 | 22 | 2 | 2.4:1 |
| 15 | CuCl2 | Ad12 | 50 | 17 | 2 | 3.0:1 |
| 16e | CuCl2 | Ad9 | 66 | 8 | 2 | 8.1:1 |
| 17f | CuCl2 | Ad9 | 83 (73g) | 9 | 2 | 9.3:1 |
With the key C11-OH-PPB-protected Corey alcohol 11 in hand, we then turned our attention to efficiently install the ω-side-chain of PGs (Fig. 5). Oxidation of 11 with TEMPO and trichloroisocyanuric acid (TCCA) in ethyl acetate and dimethyl carbonate at 0 °C provided the desired crude C11-OH-PPB-protected Corey aldehyde20,21 and set the stage for the C13–C14 HWE olefination. Addition of the crude aldehyde 20 to the solutions of phosphonates 21a–21d in dichloromethane in the presence of 30% aq. NaOH at 0 °C afforded the corresponding enones 12a–12d in good isolated yields (71–75% over two steps) as a single geometric isomer with an (E)-configuration at the newly formed C13–C14 double bond judged by the 1H NMR spectral analysis.22
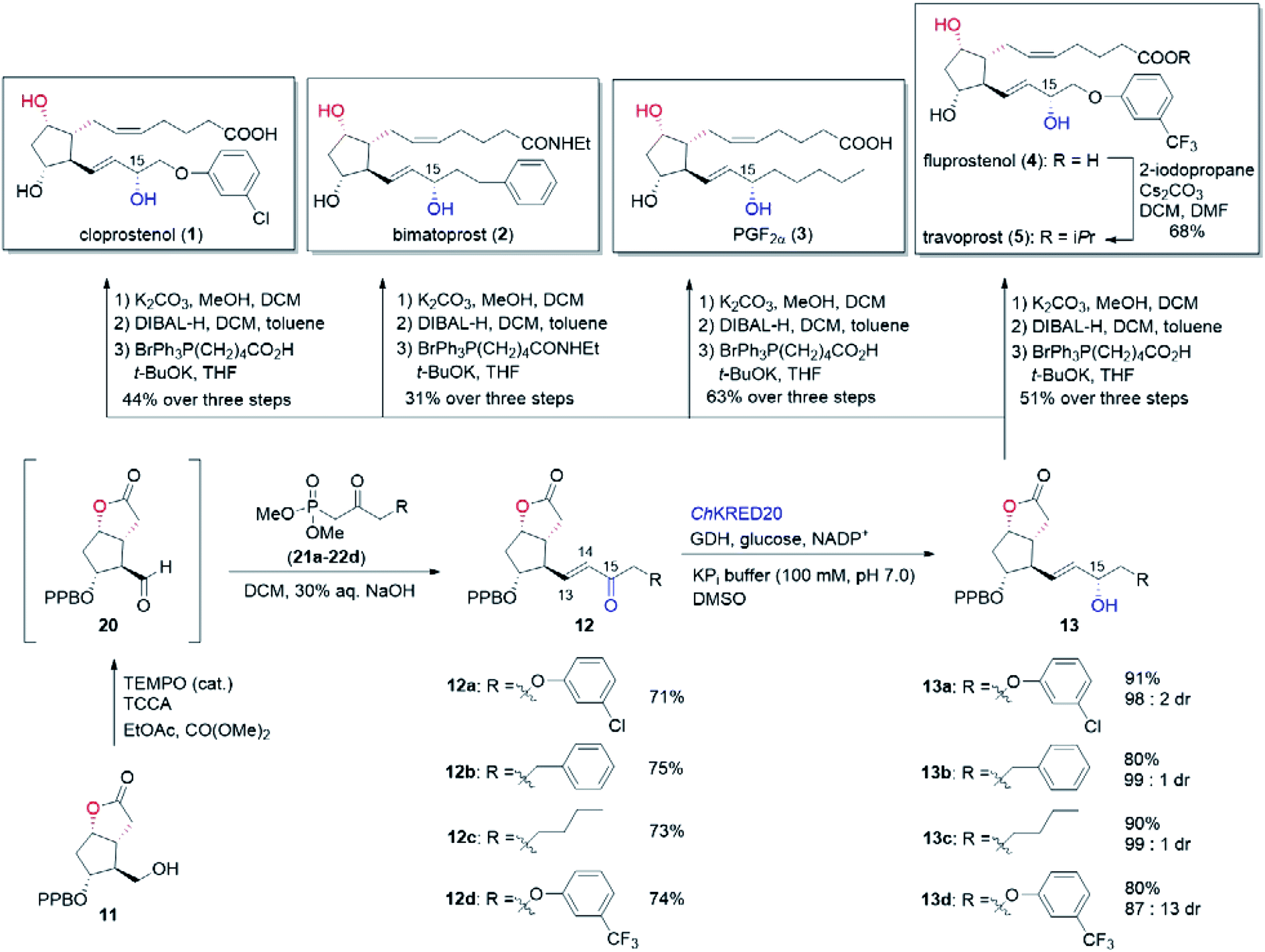 | ||
| Fig. 5 Completion of the chemoenzymatic total synthesis of cloprostenol (1), bimatoprost (2), PGF2α (3), fluprostenol (4), and travoprost (5). | ||
Stereoselective reduction of the keto functionality of the prostaglandin's ω-side-chain is dominated by chemical approaches, with biocatalytic reduction being rarely exploited. Recently, the Romano group reported an efficient and highly diastereoselective synthesis of a key allylic alcohol-containing intermediate to bimatoprost via the yeast Pichia anomala-mediated reduction.23 However, careful optimization of the biotransformation conditions was necessary in order to suppress the competing reduction of the carbon–carbon double bond by the enoate reductase present in the same yeast. To alleviate this inconvenience, use of KREDs recombinantly over-expressed in E. coli should be a viable option. Due to their excellent stereoselectivity, broad substrate spectrum, good stability, and high volumetric productivity, such over-expressed KREDs either in isolated form (purified enzyme or crude-cell lysate) or in whole-cell form, have been widely adopted in the synthesis of pharmaceuticals and bioactive molecules.9,10b,24 The Pietruszka group has reported an elegant chemoenzymatic total synthesis of travoprost.25 Ketoreductase was employed to stereoselectively prepare the chiral alcohol containing the ω-side-chain, which was then attached to the cyclopentenone ring via a three-component coupling strategy. Recombinantly over-expressed KRED-catalyzed stereoselective reduction of pre-assembled enone intermediates to prostaglandins, such as enones 12, has not been reported, to the best of our knowledge.
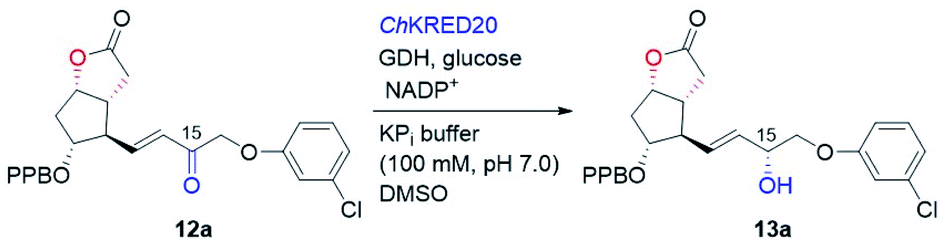
To this end, we examined the bioreduction of 12a using a small library of in-house preserved KREDs (Table S3†). Out of 15 enzymes tested, ten could not transform 12a at all. The conversion of the rest five enzymatic reactions was in the range of 4–43%, with ChKRED20 giving the best performance (Table 3, entry 1, also see Table S3†).26 It was noticed that the low conversion of 12a was to some extent due to its apparently poor solubility under current reaction conditions, which probably arises from its hydrophobic structure. To help solubilize 12a and improve the conversion, reactions with a larger amount of the co-solvent DMSO were attempted. Indeed, the conversions of 12a were increased to 58% and 71% (Table 3, entries 2 and 3), respectively, when 5% and 10% of DMSO were used. Although using 20% of DMSO further improved the conversion to 83%, an inferior diastereoselectivity (93.9:6.1 dr) was obtained (Table 3, entry 4). On the other hand, decrease of the substrate concentration from 10 mM to 5 mM boosted the reaction conversion to 90%, and importantly the excellent diastereoselectivity (97.9:2.1 dr) was retained (Table 3, entry 5). We believe that efficient reduction of the higher concentration of 12a would become possible by optimization of the reaction and protein engineering in the future.6d,27 Pleasingly, ChKRED20-catalyzed reduction of 12a under the optimized conditions at a semi-preparative-scale occurred smoothly, delivering the desired allylic alcohol 13a in 91% isolated yield with 97.9:2.1 dr (Table 3, entry 6). The configuration of the newly generated stereogenic center (C-15, prostaglandin numbering) was assigned as α by comparing to that of 13a prepared using (−)-DIP-Cl-mediated reduction.28 From 13a, hydrolysis of the PPB ester to alcohol, followed by the DIBAL-H mediated reduction of the lactone to the hemiacetal, and a final Wittig olefination furnished cloprostenol (1) in 44% over three steps (Fig. 5). The applicability of our developed route was further demonstrated by the synthesis of another four PGs: bimatoprost, PGF2α, fluprostenol, and travoprost (Fig. 5). When enones 12b–12d with different ω-side-chains were subjected to ChKRED20-catalyzed reduction, the desired allylic alcohols 13b–13d were isolated in 80–90% yields with 87:13 to 99:1 dr. Analogous to the above synthesis of cloprostenol (1), bimatoprost (2), PGF2α (3), and fluprostenol (4) were prepared from 13b, 13c, and 13d in a three-step sequence with 31%, 63%, and 51% yields, respectively. Finally, the transformation from fluprostenol (4) to travoprost (5) was accomplished in 68% yield by using 2-iodopropane and Cs2CO3 in the mixed solvent of DCM and DMF.4b Our chemoenzymatic synthesis of cloprostenol (1), bimatoprost (2), PGF2α (3), fluprostenol (4), and travoprost (5) was completed in 11–12 steps from bicyclic ketone 6a with 3.8–8.4% overall yields.
|
|
||||
|---|---|---|---|---|
| Entry | DMSO (v/v, %) | 12a (mM) | Conv.b (%) | dr (C-15, α:β)b |
| a Reaction conditions (0.5 mL): 12a (5 mM or 10 mM), glucose (2 equiv. relative to 12a), NADP+ (0.2 mM), ChKRED20 (1 mg mL−1), 0.1 mL of 15% (w/v) cell-free extract (CFE) of GDH, and DMSO (v/v, 3–20%) in KPi buffer (100 mM, pH 7.0). Reaction mixtures were incubated at 30 °C with 200 rpm shaking for 17 h. b Determined by SFC analysis. c The reaction volume was 40 mL and the reaction time was 33 h. d Isolated yield. | ||||
| 1 | 3 | 10 | 43 | 97.8:2.2 |
| 2 | 5 | 10 | 58 | 97.4:2.6 |
| 3 | 10 | 10 | 71 | 96.7:3.3 |
| 4 | 20 | 10 | 83 | 93.9:6.1 |
| 5 | 10 | 5 | 90 | 97.9:2.1 |
| 6c | 10 | 5 | 96 (91d) | 97.9:2.1 |
Conclusions
In summary, a unified, biocatalytic retrosynthesis-guided route has been developed for the synthesis of cloprostenol (1), bimatoprost (2), PGF2α (3), fluprostenol (4), and travoprost (5) from the readily available dichloro-containing bicyclic ketone 6a in 11–12 steps with 3.8–8.4% overall yields, featuring a BVMO-catalyzed stereoselective oxidation of 6a (99% ee), a time-economical three-step transformation of 7a into diol 10 using flow chemistry, a copper(II)-catalyzed regioselective p-phenylbenzoylation of the secondary alcohol of diol 10 (9.3:1 rr), and a KRED-catalyzed diastereoselective reduction of enones 12 (87:13 to 99:1 dr). Compared to chemocatalytic reactions, these two key enzymatic transformations were performed under milder conditions and meanwhile exhibited superior stereoselectivity. Our study not only provides an alternative route to the highly stereoselective synthesis of prostaglandins, but also showcases the usefulness and great potential of biocatalysis in construction of complex molecules.
Data availability
The experimental data is included in the ESI.Author contributions
Z. H. and F. C. conceived and directed the project and wrote the paper with assistance from K. Z. K. Z., M. J., B. Y., G. Z., W. L., and P. T. performed the experiments and analyzed the data. All authors discussed the results and commented on the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 22071033 and 21801047) and Shanghai Sailing Program (18YF1402100) is greatly appreciated.Notes and references
- (a) P. W. Collins and S. W. Djuric, Chem. Rev., 1993, 93, 1533–1564 CrossRef CAS; (b) S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Chem. Rev., 2007, 107, 3286–3337 CrossRef CAS; (c) H. Peng and F. Chen, Org. Biomol. Chem., 2017, 15, 6281–6301 RSC.
- S. H. Bennett, G. Coulthard and V. K. Aggarwal, Chem. Rec., 2020, 20, 936–947 CrossRef CAS PubMed.
- E. J. Corey, N. M. Weinshenker, T. K. Schaaf and W. Huber, J. Am. Chem. Soc., 1969, 91, 5675–5677 CrossRef CAS PubMed.
- (a) A. Pelss, N. Gandhamsetty, J. R. Smith, D. Mailhol, M. Silvi, A. Watson, I. Perez-Powell, S. Prévost, N. Schützenmeister, P. Moore and V. K. Aggarwal, Chem.–Eur. J., 2018, 24, 9542–9545 CrossRef CAS PubMed; (b) S. Prévost, K. Thai, N. Schgtzenmeister, G. Coulthard, W. Erb and V. K. Aggarwal, Org. Lett., 2015, 17, 504–507 CrossRef PubMed; (c) G. Coulthard, W. Erb and V. K. Aggarwal, Nature, 2012, 489, 278–281 CrossRef CAS PubMed; (d) N. Umekubo and Y. Hayashi, Org. Lett., 2020, 22, 9365–9370 CrossRef CAS PubMed; (e) N. Umekubo, Y. Suga and Y. Hayashi, Chem. Sci., 2020, 11, 1205–1209 RSC; (f) N. Umekubo and Y. Hayashi, Eur. J. Org. Chem., 2020, 6221–6227 CrossRef CAS; (g) G. Kawauchi, S. Umemiya, T. Taniguchi, K. Monde and Y. Hayashi, Chem.–Eur. J., 2018, 24, 8409–8414 CrossRef CAS PubMed; (h) Y. Hayashi and S. Umemiya, Angew. Chem., Int. Ed., 2013, 52, 3450–3452 CrossRef CAS PubMed; (i) J. Li, T. S. Ahmed, C. Xu, B. M. Stoltz and R. H. Grubbs, J. Am. Chem. Soc., 2019, 141, 154–158 CrossRef CAS PubMed; (j) R. Kučera, F. W. Goetzke and S. P. Fletcher, Org. Lett., 2020, 22, 2991–2994 CrossRef PubMed; (k) K. C. Nicolaou, K. K. Pulukuri, S. Rigol, P. Heretsch, R. Yu, C. I. Grove, C. R. H. Hale, A. ElMarrouni, V. Tetz, M. Brönstrup, M. Aujay, J. Sandoval and J. Gavrilyuk, J. Am. Chem. Soc., 2016, 138, 6550–6560 CrossRef CAS PubMed; (l) K. C. Nicolaou, P. Heretsch, A. ElMarrouni, C. R. H. Hale, K. K. Pulukuri, A. K. Kudva, V. Narayan and K. S. Prabhu, Angew. Chem., Int. Ed., 2014, 53, 10443–10447 CrossRef CAS PubMed; (m) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. Bao, F. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed; (n) J. Egger, S. Fischer, P. Bretscher, S. Freigang, M. Kopf and E. M. Carreira, Org. Lett., 2015, 17, 4340–4343 CrossRef CAS; (o) J. Egger, P. Bretscher, S. Freigang, M. Kopf and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 17382–17385 CrossRef CAS PubMed; (p) S. J. Danishefsky, M. P. Cabal and K. Chow, J. Am. Chem. Soc., 1989, 111, 3456–3457 CrossRef CAS; (q) G. Stork, P. M. Sher and H. L. Chen, J. Am. Chem. Soc., 1986, 108, 6384–6385 CrossRef CAS; (r) B. Achmatowicz, E. Baranowska, A. R. Daniewski, J. Pankowski and J. Wicha, Tetrahedron, 1988, 44, 4989–4998 CrossRef CAS; (s) E. J. Corey, Z. Arnold and J. Hutton, Tetrahedron Lett., 1970, 11, 307–310 CrossRef; (t) M. Suzuki, T. Kawagishi, T. Suzuki and R. Noyori, Tetrahedron Lett., 1982, 23, 4057–4060 CrossRef CAS; (u) M. Suzuki, T. Kawagishi and R. Noyori, Tetrahedron Lett., 1982, 23, 5563–5566 CrossRef CAS; (v) M. Suzuki, A. Yanagisawa and R. Noyori, J. Am. Chem. Soc., 1985, 107, 3348–3349 CrossRef CAS; (w) M. Suzuki, A. Yanagisawa and R. Noyori, J. Am. Chem. Soc., 1988, 110, 4718–4726 CrossRef CAS; (x) R. Noyori and M. Suzuki, Angew. Chem., Int. Ed., 1984, 23, 847–876 CrossRef; (y) G. L. Bundy, W. P. Schneider, F. H. Lincoln and J. E. Pike, J. Am. Chem. Soc., 1972, 94, 2123–2124 CrossRef CAS PubMed.
- K. Zhu, S. Hu, M. Liu, H. Peng and F. Chen, Angew. Chem., Int. Ed., 2019, 58, 9923–9927 CrossRef CAS PubMed.
- (a) N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed; (b) M. Hönig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942–8973 CrossRef PubMed; (c) R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem.–Eur. J., 2017, 23, 12040–12063 CrossRef CAS PubMed; (d) F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed; (e) R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC; (f) S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed; (g) U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- S. M. K. McKinnie, Z. D. Miles, P. A. Jordan, T. Awakawa, H. P. Pepper, L. A. M. Murray, J. H. George and B. S. Moore, J. Am. Chem. Soc., 2018, 140, 17840–17845 CrossRef CAS PubMed.
- (a) J. Ryan, M. Šiaučiulis, A. Gomm, B. Maciá, E. O'Reilly and E. Caprio, J. Am. Chem. Soc., 2016, 138, 15798–15800 CrossRef CAS PubMed; (b) H. Fu, J. Zhang, M. Saifuddin, G. Cruiming, P. G. Tepper and G. J. Poelarends, Nat. Catal., 2018, 1, 186–191 CrossRef CAS; (c) X. Zhang, E. King-Smith and H. Renata, Angew. Chem., Int. Ed., 2018, 57, 5037–5041 CrossRef CAS PubMed; (d) J. Xu, A. P. Green and N. J. Turner, Angew. Chem., Int. Ed., 2018, 57, 16760–16763 CrossRef CAS; (e) M. Lazzarotto, L. Hammerer, M. Hetmann, A. Borg, L. Schmermund, L. Steiner, P. Hartmann, F. Belaj, W. Kroutil, K. Gruber and M. Fuchs, Angew. Chem., Int. Ed., 2019, 58, 8226–8230 CrossRef CAS PubMed; (f) H. Eastman, J. Ryan, B. Maciá, V. Caprio and E. O'Reilly, ChemCatChem, 2019, 11, 3760–3762 CrossRef CAS; (g) F. Parmeggiani, A. R. Casamajo, D. Colombo, M. C. Ghezzi, J. L. Galman, R. A. Chica, E. Brenna and N. J. Turner, Green Chem., 2019, 21, 4368–4379 RSC; (h) J. Li, F. Li, E. King-Smith and H. Renata, Nat. Chem., 2020, 12, 173–179 CrossRef CAS PubMed; (i) F. Taday, J. Ryan, S. P. Argent, V. Caprio, B. Maciá and E. O'Reilly, Chem.–Eur. J., 2020, 26, 3729–3732 CrossRef CAS PubMed; (j) Z. Xu, P. Yao, X. Sheng, J. Li, J. Li, S. Yu, J. Feng, Q. Wu and D. Zhu, ACS Catal., 2020, 10, 8780–8787 CrossRef CAS; (k) L. T. Boulton, D. Brick, M. E. Fox, M. Jackson, I. C. Lennon, R. McCague, N. Parkin, D. Rhodes and G. Ruecroft, Org. Process Res. Dev., 2002, 6, 138–145 CrossRef CAS.
- J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. N. Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193–198 CrossRef CAS.
- (a) X. Wu, Z. Huang, Z. Wang, Z. Li, J. Wang, J. Lin and F. Chen, J. Org. Chem., 2020, 85, 5598–5614 CrossRef CAS PubMed; (b) C. Hu, M. Liu, X. Yue, Z. Huang and F. Chen, Org. Process Res. Dev., 2020, 24, 1700–1706 CrossRef CAS.
- (a) I.-H. Chen, K. G. M. Kou, D. N. Le, C. M. Rathbun and V. M. Dong, Chem.–Eur. J., 2014, 20, 5013–5018 CrossRef CAS PubMed; (b) Y. Matsumura, T. Maki, S. Murakami and O. Onomura, J. Am. Chem. Soc., 2003, 125, 2052–2053 CrossRef CAS PubMed; (c) C. Mazet, S. Roseblade, V. Köhler and A. Pfaltz, Org. Lett., 2006, 8, 1879–1882 CrossRef CAS PubMed; (d) C. L. Allen and S. J. Miller, Org. Lett., 2013, 15, 6178–6181 CrossRef CAS PubMed.
- (a) H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed; (b) M. D. Mihovilovic, Curr. Org. Chem., 2006, 10, 1265–1287 CrossRef CAS; (c) M. Bucko, P. Gemeiner, A. Schenkmayerova, T. Krajcovic, F. Rudroff and M. D. Mihovilovic, Appl. Microbiol. Biotechnol., 2016, 100, 6585–6599 CrossRef CAS PubMed; (d) M. J. L. J. Fürst, A. Gran-Scheuch, F. S. Aalbers and M. W. Fraaije, ACS Catal., 2019, 9, 11207–11241 CrossRef.
- P. C. Brzostowicz, D. M. Walters, S. M. Thomas, V. Nagarajan and P. E. Rouvière, Appl. Environ. Microbiol., 2003, 69, 334–342 CrossRef CAS PubMed.
- P. C. Brzostowicz, K. L. Gibson, S. M. Thomas, M. S. Blasko and P. E. Rouvière, J. Bacteriol., 2000, 182, 4241–4248 CrossRef CAS PubMed.
- B. D. Summers, M. Omar, T. O. Ronson, J. Cartwright, M. Lloyd and G. Grogan, Org. Biomol. Chem., 2015, 13, 1897–1903 RSC.
- (a) H. C. Stevens, D. A. Reich, D. R. Brandt, K. R. Fountain and E. J. Gaughan, J. Am. Chem. Soc., 1965, 87, 5257–5259 CrossRef CAS; (b) T. Asao, T. Machiguchi and Y. Kitahara, Bull. Chem. Soc. Jpn., 1970, 43, 2662 CrossRef CAS; (c) P. R. Brook and A. J. Duke, J. Chem. Soc. C, 1971, 1764–1769 RSC.
- (a) C. A. González-González, A. Fuentes-Benítez, E. Cuevas-Yáñez, D. Corona-Becerril, C. González-Romero and D. González-Calderón, Tetrahedron Lett., 2013, 54, 2726–2728 CrossRef; (b) D. González-Calderón, M. G. Mejía-Dionicio, M. A. Morales-Reza, J. G. Aguirre-de Paz, A. Ramírez-Villalva, M. Morales-Rodríguez, A. Fuentes-Benítes and C. González-Romero, Bioorg. Chem., 2016, 69, 1–6 CrossRef PubMed; (c) M. Jackson, V. H. Dahanukar, S. C. Joseph, V. Vardhana, V. R. Eda and S. K. Ramdas, US Pat., 2013/0184476A1, 2013 Search PubMed.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS.
- (a) E. J. Corey, S. M. Albonico, U. Koelliker, T. K. Schaaf and R. K. Varma, J. Am. Chem. Soc., 1971, 93, 1491–1493 CrossRef CAS PubMed; (b) E. J. Corey, R. K. Varma and K. B. Becker, J. Am. Chem. Soc., 1972, 94, 8616–8618 CrossRef CAS PubMed.
- J. Lv, J. Ge, T. Luo and H. Dong, Green Chem., 2018, 20, 1987–1991 RSC.
- (a) C. Ohta, S. Kuwabe, T. Shiraishi, I. Shinohara, H. Araki, S. Sakuyama, T. Makihara, Y. Kawanaka, S. Ohuchida and T. Seko, J. Org. Chem., 2009, 74, 8298–8308 CrossRef CAS PubMed; (b) L. De Luca, G. Giacomelli and A. Porcheddu, Org. Lett., 2001, 3, 3041–3043 CrossRef CAS PubMed; (c) G. Chambournier, A. Kornilov, H. M. Mahmoud, I. Vesely and S. D. Barrett, US Pat., 2012/0283451A1, 2012 Search PubMed; (d) B. Resul, J. Stjernschantz, K. No, C. Liljebris, G. Selén, M. Astin, M. Karlsson and L. Z. Bito, J. Med. Chem., 1993, 36, 243–248 CrossRef CAS PubMed.
- (a) G. A. Tolstikov, M. S. Miftakhov, M. E. Adler, N. G. Komissarova, O. M. Kuznetsov and N. S. Vostrikov, Synthesis, 1989, 940–942 CrossRef CAS; (b) A. Gutman, G. Nisnevich, M. Etinger, I. Zaltzman, L. Yudovich, B. Pertsikov and B. Tishin, US Pat., 2005/0209337A1, 2005 Search PubMed; (c) A. Gutman, I. Rukhman, B. Tishin, L. Yudovich, A. Vilensky, B. Pertsikov and G. Nisnevich, US Pat., 2009/0163596A1, 2009 Search PubMed.
- M. L. Contente, P. Zambelli, S. Galafassi, L. Tamborini, A. Pinto, P. Conti, F. Molinari and D. Romano, J. Mol. Catal. B: Enzym., 2015, 114, 7–12 CrossRef CAS.
- (a) S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC; (b) X. Gong, Z. Qin, F. Li, B. Zeng, G. Zheng and J. Xu, ACS Catal., 2019, 9, 147–153 CrossRef CAS; (c) B. Su, L. Xu, X. Xu, L. Wang, A. Li, J. Lin, L. Ye and H. Yu, Green Synth. Catal., 2020, 1, 150–159 CrossRef.
- C. Holec, D. Sandkuhl, D. Rother, W. Kroutil and J. Pietruszka, ChemCatChem, 2015, 7, 3125–3130 CrossRef CAS.
- (a) Y. Liu, T. Tang, X. Pei, C. Zhang and Z. Wu, J. Mol. Catal. B: Enzym., 2014, 102, 1–8 CrossRef CAS; (b) F. Zhao, Y. Jin, Z. Liu, C. Guo, T. Li, Z. Li, G. Wang and Z. Wu, Appl. Microbiol. Biotechnol., 2017, 101, 8395–8404 CrossRef CAS PubMed.
- G. Qu, A. Li, C. G. Acevedo-Rocha, Z. Sun and M. T. Reetz, Angew. Chem., Int. Ed., 2020, 59, 13204–13231 CrossRef CAS PubMed.
- Y. Chen, H. Yan, H. Chen, J. Weng and G. Lu, Chirality, 2015, 27, 392–396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1975866. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03237b |
| This journal is © The Royal Society of Chemistry 2021 |