
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTug-of-war: molecular dynamometers against living cells for analyzing sub-piconewton interaction of a specific protein with the cell membrane†
Huipu
Liu‡
,
Yunlong
Chen‡
 ,
Jiawei
Wang
,
Yuanjiao
Yang
and
Huangxian
Ju
*
,
Jiawei
Wang
,
Yuanjiao
Yang
and
Huangxian
Ju
*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: hxju@nju.edu.cn
First published on 2nd September 2021
Abstract
Protein–membrane interactions play important roles in signal transductions and functional regulation of membrane proteins. Here, we design a molecular dynamometer (MDM) for analyzing protein–membrane interaction on living cells. The MDM is constructed by assembling an artificial lipid bilayer and alkylated Cy3-DNA azide (azide-Cy3-Cx) on a silica bubble. After a functional aptamer is covalently anchored onto the corresponding target protein on a living cell through UV irradiation, azide-Cy3-Cx is conjugated with the aptamer through a click reaction to produce a “tug-of-war” between the MDM and the cell due to the buoyancy of the silica bubble. This induces the detachment of the protein from the cell membrane or the alkane terminal from the MDM enabling sub-piconewton embedding force measurement by changing the alkane chain length and simple fluorescence analysis. The successful analysis of embedding force variation of a protein on the cell membrane upon post-translational modifications demonstrates the practicability and expansibility of this method for mechanics-related research in biological systems.
Introduction
The plasma membrane is constructed with a lipid bilayer along with rich proteins embedded in the bilayer.1 The interactions between the membrane and proteins can impact their configurations and activities by shaping the conformational energy landscape and determining their topology,2,3 which can be attributed to different interaction forces between the membrane and proteins. Generally, the interaction forces between biomolecules are transient in biochemical processes and tend to be lower than 100 pN, which is sufficient to drive their conformational changes, but insufficient to dissociate the general covalent bonds.4 Moreover, the interaction forces of the membrane with different kinds of membrane proteins vary in a huge range. For instance, the interaction force between integrin conjugated on the cytoskeleton5 and the membrane is tens of piconewtons (pN),6 while some proteins like mucins, which are embedded on the cell membrane only through hydrophobic interaction, have much lower interaction forces than those with the support of the cytoskeleton, although the exact level is unknown.Up to now some methods including atomic force microscopy7 and optical tweezers8 have been developed to study the modulation of dietary fatty acids to Piezo1 mechanical response,9 analyze the pulling force during the unfolding and extracting processes of a membrane protein from an artificial lipid bilayer,10,11 reveal the insertion and folding behavior of a single membrane protein,12 and explore the role of caveolae in membrane-mediated mechanical responses.13 Magnetic tweezers can be used to measure a wide range of forces (10 pN to 10 nN)14 and maneuver small magnetic probes inside living cells.15 However, most of these techniques are performed in artificial biological environments or the cells need to be fixed, leading to certain deviation from the real situation. Besides, these methods mainly apply to case analysis of a single protein, but cannot provide the statistical data of abundant proteins on the cell surface, which are critical for practical biological application. Moreover, their resolution or sensitivity is insufficient to detect the interaction forces of a few piconewtons.
Several methods based on fluorescence imaging have been proposed to visualize the cellular traction force by stretching the straight stem–loop DNA hairpin flanked with a fluorophore–quencher pair16 or a flexible linker tagged with a fluorophore-labeled specific recombinant peptide and a corresponding acceptor17 to produce the fluorescence signal of the traction force, which is sensitive to the forces of a few pN. However, the mechanistic difference between traction of the membrane and folding of the DNA hairpin or pulling of the flexible linker greatly affects the quantitative results. Grashoff et al.18 developed single-molecule fluorescence force spectroscopy with a genetically encoded vinculin tension sensor by combining confocal scanning fluorescence microscopy with optical tweezers. This technique achieved single piconewton sensitivity, but could not still respond to the hydrophobic embedding interaction.
To analyze the weak mucin–membrane hydrophobic interaction on living cells in a simple, convenient and universal way, this work constructed a “tug-of-war” mode with a newly designed MDM. The MDM was prepared by assembling a 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) bilayer19 and alkylated cyanine3 (Cy3)-DNA azide (azide-Cy3-Cx, x is the length of the alkane) on silica bubbles (Fig. 1A). Meanwhile, a dibenzocyclooctyne (DBCO) functionalized carboxyfluorescein (FAM)–aptamer (FA) was covalently cross-linked to the target protein embedded on the living cell membrane by diazirine (Dia) mediated photo-crosslinking (Fig. 1B).20,21 Both the contact of the azide group with the DBCO group for their binding and the “tug-of-war” of MDMs against the cells could be driven by the buoyancy of MDMs through inverting the cell seeded dishes. Owing to the hollow structure of silica bubbles, the MDM could float to approach the FA-labeled cells that were seeded on an inverted dish for binding azide with DBCO groups through a copper-free cycloaddition reaction,22 which triggered the “tug-of-war” for visualization of the protein–membrane interaction after inverting the dish back (Fig. 1C) via monitoring the change of FAM fluorescence, which depended on the amount of target protein left on the membrane. When the alkane–DSPE interaction on the silica bubbles was stronger than the protein–membrane interaction, the proteins could be pulled out of the membrane. The length of the alkane in azide-Cy3-Cx decided the winner of the “tug-of-war” and thus the amount of remaining target protein. The developed strategy achieved the semi-quantitative measurement of the protein embedding force at the sub-piconewton level. The successful analysis of the minor changes in the protein embedding force caused by palmitoylation or O-glycosylation, two important post-translational modifications to modulate the activity and function of proteins,23 demonstrated the practicability of the MDM and the proposed method. This work provides a convenient and sensitive protocol for revealing the biological mechanisms related to weak protein–membrane interactions.
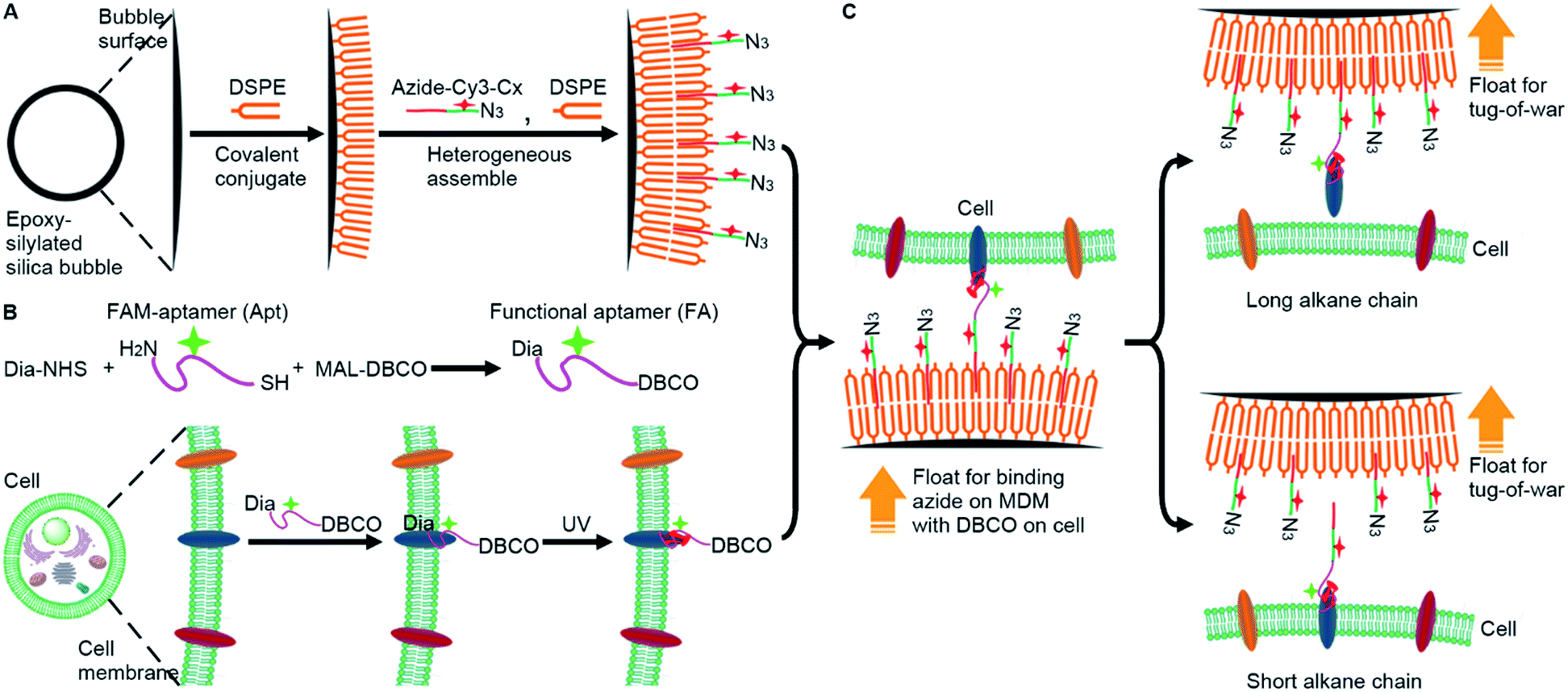 | ||
| Fig. 1 Schematic illustration of the molecular dynamometer (MDM), functional aptamer (FA), and “tug-of-war” tests. (A) Preparation of the MDM. (B) Preparation and conjugation of the FA with the target protein on the cell membrane. (C) Binding of the MDM with the FA-conjugated cells for “tug-of-war” tests. | ||
Results and discussion
Assembly and characterization of the MDM
Silica bubbles with a diameter of around 30 μm were first treated with 5 M H2SO4 to introduce epoxy groups onto the surface with methyldimethoxysilane, which resulted in greatly increased contact angle (Fig. 2A). After DSPE was assembled on the epoxy-modified bubbles by covalent conjugation of amine and epoxy groups, the contact angle slightly increased, indicating similar hydrophobicity. Upon the assembly of azide-Cy3-Cx on DSPE-modified bubbles and the formation of the second layer of DSPE by hydrophobic interaction, the contact angle of the thick film of the obtained bubbles further increased to 84.7° (x = 12), and obvious fluorescence of Cy3 could be observed around the bubbles (Fig. S1A† for x = 3), indicating the successful assembly of the MDM. Moreover, the change of the alkane length from 3 to 12 did not affect the assembly of azide-Cy3-Cx on the DSPE modified surface (Fig. 2B). However, the assembly sequence of azide-Cy3-Cx and the second layer of DSPE greatly affected the coverage of azide-Cy3-Cx on the MDM. If the second layer of DSPE was first formed, less azide-Cy3-Cx could be assembled on the bilayer, which led to almost negligible Cy3 fluorescence (Fig. S1A†). This could be attributed to the presence of 3-phosphoethanolamine at the end of the bilayer, which was unfavorable to the approach of azide-Cy3-Cx to the modified bubble surface for hydrophobic insertion (Fig. S1B†). After incubating the MDMs in 1640 medium for 30 min, negligible change of Cy3 fluorescence was observed on all MDMs (Fig. S2†), which indicated the excellent stability of the assembled azide-Cy3-Cx on MDM surfaces. The amounts of azide-Cy3-Cx on the MDM were obtained by measuring the Cy3 fluorescence of the remaining assembly solutions of azide-Cy3-Cx and DSPE-modified bubbles with the corresponding calibration curves (Fig. S3†). The average amounts of azide-Cy3-Cx molecules on each MDM were calculated to be 1.10 × 107 (C3-MDM), 1.21 × 107 (C6-MDM), 1.65 × 107 (C9-MDM) and 2.05 × 107 (C12-MDM), respectively.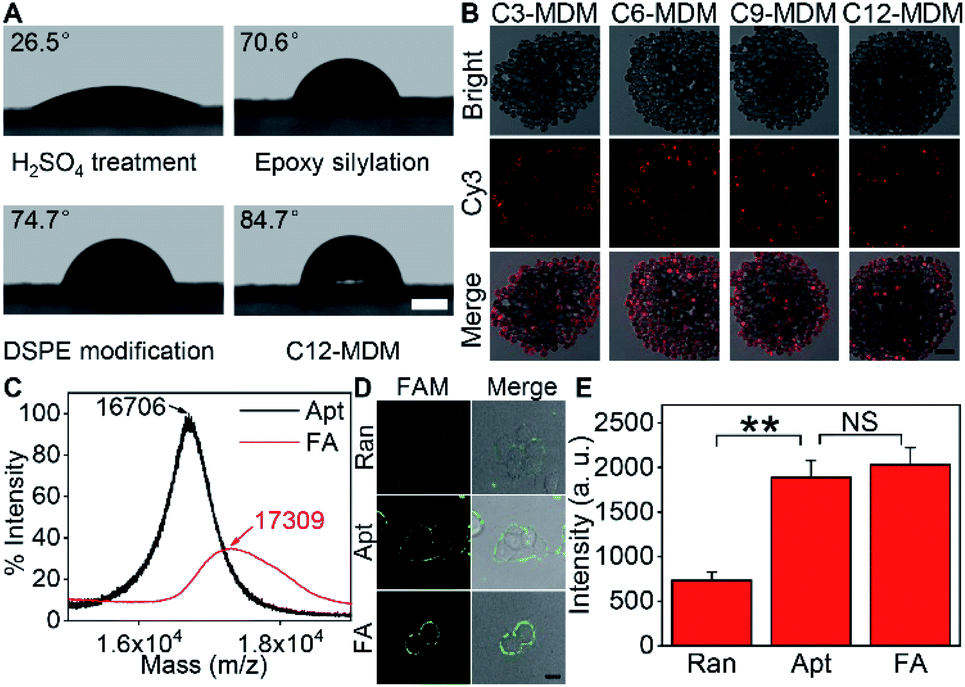 | ||
| Fig. 2 Characterization of the FA and MDM. (A) Contact angles of the thick films of H2SO4-treated, epoxy-silylated and DSPE-modified bubbles, and the MDM assembled with azide-Cy3-C12 (C12-MDM). Scale bar, 500 μm. (B) CLSM images of the MDM assembled with azide-Cy3-Cx. Scale bar, 100 μm. (C) Mass spectra of the FAM-aptamer (Apt) and FA for MUC1. (D) Bright field image, fluorescence image from FAM and overlay image of cells after incubation with random DNA (Ran), Apt and FA at 37 °C for 30 min. Scale bar, 15 μm. (E) Statistical analysis of the fluorescence intensities of FAM from (D), which was performed with the t test (**p < 0.005; NS, not significant). Error bars represent s.d. (n = 3 independent experiments). | ||
Synthesis of the FA and labeling of the target protein on cells
For proof-of-concept, mucin 1 protein (MUC1) was chosen as the target protein, which participates in the regulation of several genes related to cell proliferation and differentiation, and mediates adhesion events by binding to adhesion molecules.24 Human breast cancer MCF-7 cells were chosen as the cell model. The amount of MUC1 on each MCF-7 cell is around 1.3 × 105,25 which is much smaller than the coverage of azide-Cy3-Cx on each MDM, guaranteeing the sufficient binding of the MDMs to DBCO groups labelled on the cell surface. The aptamer of MUC1 (ref. 26) was customized by inserting a FAM label in the extended thymine sequence and terminating with a sulfhydryl group at the 3′ end and an amino group at the 5′ end, which was abbreviated as FAM-aptamer or Apt. The FA was prepared by one-pot incubation of Apt with dibenzocyclooctyne-PEG4-maleimide (MAL-DBCO) and succinimidyl 4,4′-azipentanoate (Dia-NHS) (Fig. S4†), and then purified by ultrafiltration. The mass spectrum of the obtained FA exhibited an obvious m/z increase from the 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 706 (estimated m/z: 16711.20) of Apt to 17309 (estimated m/z: 17269.93) (Fig. 2C), which indicated the successful conjugation of DBCO and Dia groups with Apt.
706 (estimated m/z: 16711.20) of Apt to 17309 (estimated m/z: 17269.93) (Fig. 2C), which indicated the successful conjugation of DBCO and Dia groups with Apt.
The recognizing ability of FA towards MUC1 was investigated with confocal laser scanning microscopy (CLSM) imaging. A FAM-labeled random DNA sequence (Ran) was used as the negative control. Ran-treated cells exhibited negligible fluorescence signal on the cell surface, while both Apt- and FA-treated cells exhibited equally strong fluorescence of FAM on the cell surface (Fig. 2D and E). Thus, the conjugation of DBCO and Dia did not affect the recognition ability of Apt towards MUC1 proteins on the cell surface.
The click reaction between DBCO in FA and azide was verified with gel electrophoresis analysis (Fig. S5†). After functionalizing Apt with MAL-DBCO and Dia-NHS, the molecular weight became larger due to the formation of FA (lanes 1 and 2). After the mixture of azide-Cy3-C18 and FA was incubated for 30 min, a new band occurred at the molecular weight larger than those of both azide-Cy3-C18 and FA (lanes 2, 3, and 5), indicating the efficient binding between azide-Cy3-C18 and FA. As expected, azide-Cy3-C18 did not react with Apt (lane 4). The efficient binding could also be achieved on the cell surface. After MCF-7 cells were incubated with Apt or FA and then with azide-Cy3-C18, the Apt-treated cells showed the fluorescence of only FAM, while the FA-treated cells exhibited the strong fluorescence of both FAM and Cy3 around the cell surface (Fig. S6†), which demonstrated the successful conjugation between azide-Cy3-Cx and FA through the click reaction on living cells. Besides, the fluorescence intensity of FAM did not decrease after conjugation of azide-Cy3-Cx, indicating that there was no Förster resonance energy transfer between FAM and Cy3, which may be attributed to the long distance of the two dyes (Fig. S7†).
To achieve “tug-of-war” of the MDM against the cells for embedding force measurement of the target protein on the cell surface, the FA should be first anchored onto the protein (Fig. 1B). The covalent anchor of the FA could be demonstrated by the displacement of the FA with a DNA sequence complementary to the aptamer. After the FA-treated MCF-7 cells were incubated with the DNA sequence, the strong fluorescence of FAM on the cell surface disappeared (Fig. S8†), indicating that the FA departed the surface due to the displacement hybridization, which peeled off the aptamer from MUC1.27 However, the exposure of FA-treated MCF-7 cells to UV irradiation retained the FAM fluorescence (Fig. S8†). Thus the FA was covalently anchored onto the target protein during UV exposure.20,21 To further confirm the UV irradiation-mediated covalent anchor between Dia and the protein, the MCF-7 cells were first incubated with random FAM-DNA (Ran) or Dia-modified Ran (Dia-Ran), which was obtained by conjugating Dia-NHS to the amino group in Ran. After direct UV irradiation without washing, Dia-Ran treated cells exhibited obvious FAM fluorescence, while the washing led to negligible FAM fluorescence of the treated cells even after UV irradiation (Fig. S9†), demonstrating the Dia-mediated photo-crosslinking and tiny nonspecific adsorption of the DNA probes. Therefore, UV irradiation induced the covalent anchor of the FA onto the target protein to obtain the FA labeled MCF-7 cells.
“Tug-of-war” test of the MDM against living cells
To measure the embedding force of MUC1 on the living cell membrane, MDM dispersions with the same amount of silica bubbles were added to the dishes seeded with FA-labeled MCF-7 cells, respectively. Upon the inversion of the dishes, the MDM approached the cells by buoyancy, which led to the binding of the azide group on the MDM with the DBCO group in the FA through a copper-free cycloaddition reaction22 (Fig. 1C). Therefore, after the dishes were inverted back to the normal position, the buoyancy led to a competition between the alkane on the MDM surface and the Dia-anchored MUC1 on the cell membrane to create a “tug-of-war” of the MDM against living cells. As designed, the MDMs with different alkane chain lengths led to the difference of FAM and Cy3 fluorescence intensities on the tested cell surface (Fig. 3A). C3-MDM and C6-MDM treated cells showed obvious FAM and Cy3 fluorescence, while the FAM and Cy3 fluorescence greatly weakened on C9-MD and C12-MD treated cells (Fig. 3A), which did not show significant difference (Fig. 3B). The high colocalization degree and the similar changing tendency of Cy3 fluorescence to that of FAM fluorescence demonstrated similar click reaction activity on MDMs and cells. The significant change of fluorescence intensity from C6-MDM to C9-MDM (Fig. 3B) indicated the departure of FA linked MUC1 from the cell membrane due to the stronger hydrophobic interaction of C9 and C12 with the DSPE bilayer on MDMs than the embedding force of MUC1 on the living cell membrane, and the embedding force was stronger than the hydrophobic interaction of C3 and C6 with the DSPE bilayer. Similarly, flow cytometric analysis with 5000 cells showed the same “tug-of-war” results (Fig. 3C). Thus, it was reasonable to conclude that the embedding force of MUC1 on the living cell membrane was between the hydrophobic interactions of C6 and C9 with the DSPE bilayer. | ||
| Fig. 3 Measurement of embedding force of MUC1. (A) CLSM images of FA-labeled MCF-7 cells after treatment with different MDMs. Scale bar, 15 μm. (B) FAM (black) and Cy3 (red) fluorescence intensities of FA-labeled cells treated with different MDMs. (C) Flow cytometric analysis of FAM fluorescence intensity on FA-labeled MCF-7 cells after treatment with C3/6/9/12-MDM. Error bars represent s.d. (n=3 independent experiments). (*p < 0.05; NS, not significant). | ||
Quantitation of the embedding force
To estimate the embedding force of MUC1 on the cell membrane, DSPE bilayer modified 10 μm silica microspheres (SM10) and azide-Cy3-Cx modified 20 μm silica microspheres (SM20) were designed to obtain a “ruler” via optical tweezers analysis (Fig. 4A). The SM20 with different alkane chain lengths showed strong Cy3 fluorescence (Fig. S10†). The contact angle tests demonstrated the presence of the DSPE bilayer on SM10 (Fig. S11†). By using an optical trap to fix SM10 and another optical trap to manipulate the movement of SM20 forward and backward for the contact of Cx with the DSPE bilayer, their hydrophobic interaction could be quantified by detecting the tiny displacement of SM10 during the contact, which ranged at the sub-piconewton level (Fig. S12†). The different kinds of alkane chains showed integer multiple values of force, which indicated the simultaneous insertion of multiple alkane chains. Therefore, the module values attributing to single alkane insertion could be collected (Fig. 4B), which were defined as a “ruler” of the exact mechanics values (Fig. 4C), and the embedding force of MUC1 on the living MCF-7 cell membrane could be inferred as 0.16–0.24 pN (Table S1†).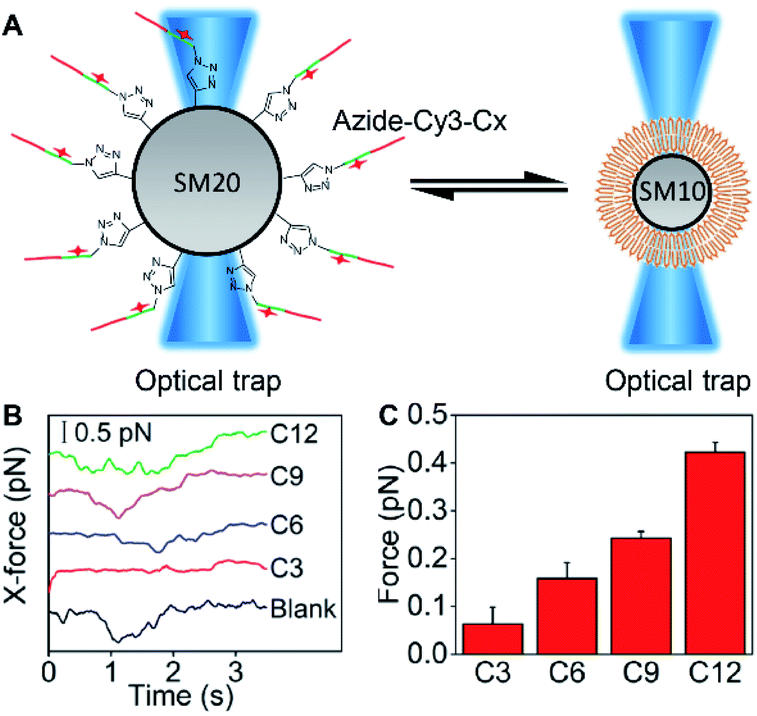 | ||
| Fig. 4 Quantification of hydrophobic interaction force. (A) Schematic illustration for quantifying the hydrophobic interaction forces between different alkane chains and the DSPE bilayer by moving the alkane chain modified SM20 right and left with an optical trap to contact the DSPE bilayer modified SM10 fixed with another optical trap. (B) X-direction forces for the alkane chain modified SM20 or unmodified SM20 (blank) acting on the DSPE bilayer modified SM10, which are measured with optical tweezers. (C) Hydrophobic interaction forces between the alkane chains and DSPE bilayer after deducting the blank. Error bars represent s.d. (n = 3 independent experiments). | ||
Embedding force variation of MUC1 upon post-translational modification
Palmitoylation and glycosylation are two important post-translational modifications of proteins, and play essential roles in the regulation of protein functions.28,29 MUC1 can be dually palmitoylated at the boundary of the transmembrane and the cytoplasmic domain,30 and abundantly O-glycosylated at serine or threonine sites.31 The membrane tethering of mucins can be effectively affected by the palmitoylation-mediated hydrophobicity32 or glycosylation-mediated hydrophilicity.33 To examine the practicability of the designed MDM, their effects on the protein embedding force were investigated by using 2-bromopalmitic acid (2-BP)34 and benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (BAG)35 to inhibit the global palmitoylation and O-glycosylation of proteins on MCF-7 cells, respectively.To visualize the inhibition of palmitoylation by 2-BP, the palmitoyl group on cells was metabolically labeled with palmitic acid (15-yne)36 and then with Alexa Fluor 555 azide through click chemistry. In comparison to the uninhibited cells, the inhibited cells showed an obvious decrease of Alexa Fluor 555 fluorescence around cells (Fig. S13†), indicating the decrease of the palmitoyl group on cells and thus the successful inhibition of palmitoylation by 2-BP. The 2-BP treated cells could also be labeled with the FA (Fig. S14†), which ensured the feasibility for the following “tug-of-war” test. After the 2-BP treated cells were labeled with the FA and then treated with MDMs to bind the azide group on the MDM with the DBCO group in the FA, the “tug-of-war” of MDMs against the living cells led to the decrease of both FAM and Cy3 fluorescence (Fig. 5A), and the decline of fluorescence intensity was significant between C3-MDM and C6-MDM treated cells (Fig. 5B), which indicated the decrease of MUC1 embedding force on 2-BP treated MCF-7, which should be in the range of 0.06–0.16 pN. Thus, the palmitoylation level was positively correlated to the MUC1 embedding force on the MCF-7 cell surface.
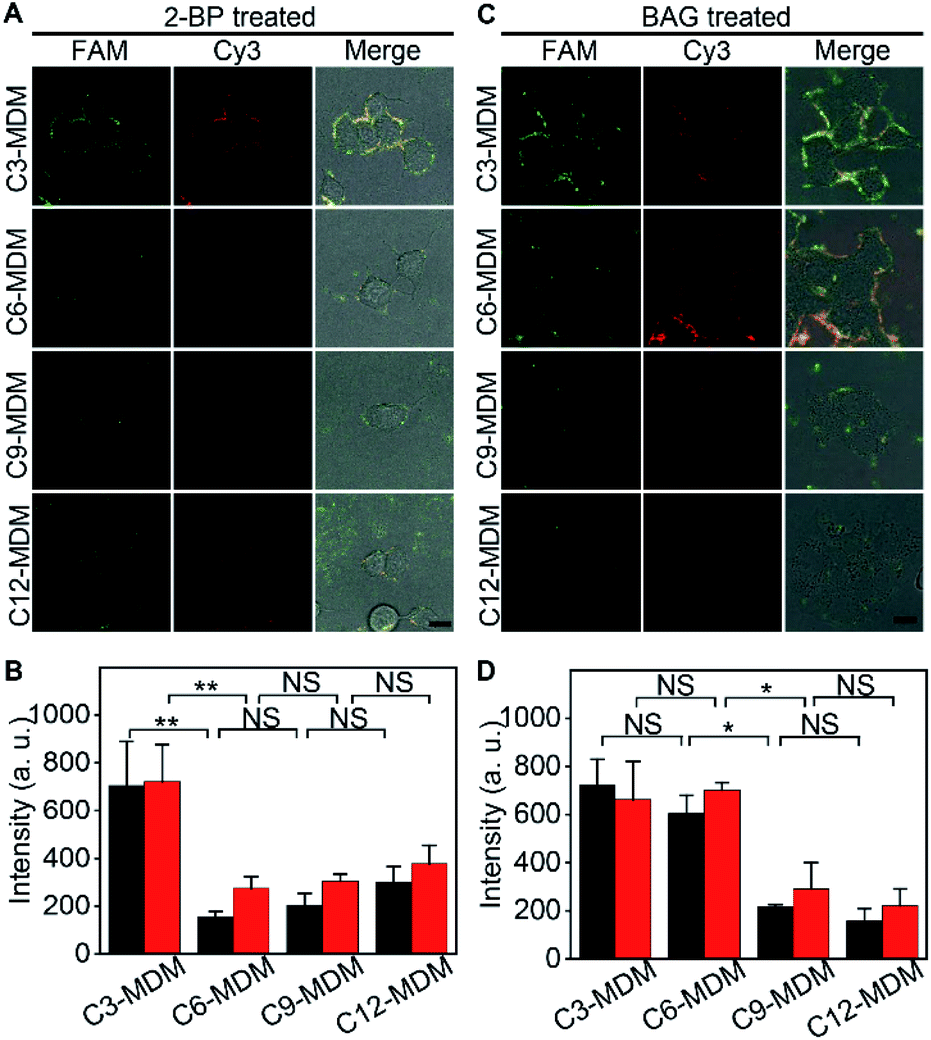 | ||
| Fig. 5 Variation of embedding force upon inhibition of post-translational modification of MUC1 on the cell membrane. (A) CLSM images and (B) the corresponding FAM (black) and Cy3 (red) fluorescence intensities of MCF-7 cells after being treated with 2-BP, labeled with the FA, and incubated with different types of MDMs. (C) CLSM images and (D) the corresponding FAM (black) and Cy3 (red) fluorescence intensities of MCF-7 cells after being treated with BAG, labeled with the FA, and incubated with different types of MDMs. Scale bar, 15 μm. Error bars represent s.d. (n = 3 independent experiments). (**p < 0.005; *p < 0.05; NS, not significant). | ||
The inhibition of O-glycosylation by BAG was demonstrated by labelling the BAG-treated cells with FITC conjugated soybean agglutinin (SBA), which could specifically bind to N-acetylgalactosamine (GalNAc) groups on cells.37 Compared to the uninhibited cells, the inhibition by BAG led to an obvious decrease of FITC fluorescence around cells (Fig. S15†). Compared to the cells without the treatment of BAG, similar fluorescence intensity of FAM was observed on the BAG-treated cells after labeling with the FA (Fig. S16†), which was attributed to the fact that the aptamer only recognizes the tandem repeat peptide region of MUC1.26 The “tug-of-war” tests on BAG-treated cells with different MDMs showed significant change of fluorescence intensity between C6-MDM and C9-MDM treated cells (Fig. 5C and D), indicating a MUC1 embedding force of 0.16–0.24 pN on BAG-treated MCF-7 cells, which was the same as that on uninhibited cells (Fig. 3B). Thus, the O-glycosylation level of MUC1 had tiny influence on MUC1 embedding force on the MCF-7 cell surface. The different effects of palmitoylation and glycosylation on protein embedding force could be attributed to their different modification sites on MUC1. The palmitoylation site of MUC1 is located at the boundary of the transmembrane and cytoplasmic domains,28 while the O-glycosylation site of MUC1 is located at the extracellular domain.31 Thus the palmitoylation of MUC1 increased its hydrophobic interaction with the cell membrane, and thus the embedding force on the MCF-7 cell surface.
Conclusions
In conclusion, a convenient and sensitive method has been developed for the quantitation of protein embedding force at the sub-piconewton level on the living cell membrane, which is suitable for proteins embedding only through hydrophobic interaction and without the support of the cytoskeleton. This method is simply performed with a fluorescence “tug-of-war” test along with a designed “ruler” obtained by the optical tweezers analysis. The “tug-of-war” is achieved between newly constructed MDMs with azide-Cy3-Cx assembled in the DSPE bilayer against living cells with the target protein. The “ruler” provides a universal way to semi-quantitatively transduce the fluorescence signals to the embedding force at the sub-piconewton level. The practicability of the developed method has been verified by assessing the effects of post-translational modifications on the embedding force of MUC1 on the MCF-7 cell membrane. This method provides an innovative idea to transduce the complicated biological interaction events to simplified artificial models for quantitative analysis, but is limited to the availability of suitable protein targets and precise quantitation references. This proposed protocol can be extended to other biological molecules on the plasma membrane by changing the corresponding recognizing components, and thus possesses great prospects for mechanics-related research in biological systems.Data availability
All relevant data is presented in the paper and ESI.† Raw data is available upon request by email to the corresponding author.Author contributions
H. J. initiated the projects. H. L. and Y. C. conceived the experiments. H. L., Y. C., J. W. and Y. Y. performed the experiments. H. L., Y. C. and H. J. analyzed the data and wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21635005, 21827812, 21890741, 21974063), National Key Research and Development Program of China (2018YFC1004704), and Fundamental Research Funds for the Central Universities (14380200).Notes and references
- P. Sengupta, T. Jovanovic-Talisman, D. Skoko, M. Renz, S. L. Veatch and J. Lippincott-Schwartz, Nat. Methods, 2011, 8, 969–975 CrossRef CAS PubMed.
- A. N. Bondar and M. J. Lemieux, Chem. Rev., 2019, 119, 6162–6183 CrossRef CAS PubMed.
- F. J. de Meyer, M. Venturoli and B. Smit, Biophys. J., 2008, 95, 1851–1865 CrossRef PubMed.
- V. P.-Y. Ma, Y. Liu, K. Yehl, K. Galior, Y. Zhang and K. Salaita, Angew. Chem., Int. Ed., 2016, 55, 5488–5492 CrossRef CAS PubMed.
- P. Kanchanawong, G. Shtengel, A. M. Pasapera, R. B. Ramko, M. W. Davidson, H. F. Hess and C. M. Waterman, Nature, 2010, 468, 580–584 CrossRef CAS PubMed.
- X. F. Wang and T. Ha, Science, 2013, 340, 991–994 CrossRef CAS PubMed.
- M. B. Viani, T. E. Schaffer, A. Chand, M. Rief, H. E. Gaub and P. K. Hansma, J. Appl. Phys., 1999, 86, 2258–2262 CrossRef CAS.
- K. Svoboda, C. F. Schmidt, B. J. Schnapp and S. M. Block, Nature, 1993, 365, 721–727 CrossRef CAS PubMed.
- L. O. Romero, A. E. Massey, A. D. Mata-Daboin, F. J. Sierra-Valdez, S. C. Chauhan, J. F. Cordero-Morales and V. Vásquezet, Nat. Commun., 2019, 10, 1200 CrossRef PubMed.
- V. Swaminathan, K. Mythreye, E. T. O'Brien, A. Berchuck, G. C. Blobe and R. Superfine, Cancer Res., 2011, 71, 5075–5080 CrossRef CAS PubMed.
- F. Oesterhelt, D. Oesterhelt, M. Pfeiffer, A. Engel, H. E. Gaub and D. J. Müller, Science, 2000, 288, 143–146 CrossRef CAS PubMed.
- D. N. Ganchev, D. T. Rijkers, M. M. Snel, J. A. Killian and B. de Kruijff, Biochemistry, 2004, 43, 14987–14993 CrossRef CAS PubMed.
- T. Serdiuk, A. Steudle, S. A. Mari, S. Manioglu, H. R. Kaback, A. Kuhn and D. J. Müller, Sci. Adv., 2019, 5, eaau6824 CrossRef PubMed.
- B. Sinha, D. Köster, R. Ruez, P. Gonnord, M. Bastiani, D. Abankwa, R. V. Stan, G. Butler-Browne, B. Vedie, L. Johannes, N. Morone, R. G. Parton, G. Raposo, P. Sens, C. Lamaze and P. Nassoy, Cell, 2011, 144, 402–413 CrossRef CAS PubMed.
- A. H. B. de Vries, B. E. Krenn, R. van Driel and J. S. Kanger, Biophys. J., 2005, 88, 2137–2144 CrossRef CAS PubMed.
- Y. Liu, L. Blanchfield, V. P.-Y. Ma, R. Andargachew, K. Galior, Z. Liu, B. Evavold and K. Salaita, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5610–5615 CrossRef CAS PubMed.
- Y. Chang, Z. Liu, Y. Zhang, K. Galior, J. Yang and K. Salaita, J. Am. Chem. Soc., 2016, 138, 2901–2904 CrossRef CAS PubMed.
- C. Grashoff, B. D. Hoffman, M. D. Brenner, R. Zhou, M. Parsons, M. T. Yang, M. A. McLean, S. G. Sligar, C. S. Chen, T. Ha and M. A. Schwartz, Nature, 2010, 466, 263–266 CrossRef CAS PubMed.
- C. Steinem, A. Janshoff, W. P. Ulrich, M. Sieber and H. J. Galla, Biochim. Biophys. Acta, 1996, 1279, 169–180 CrossRef.
- D. Wang, S. Du, A. Cazenave-Gassiot, J. Ge, J. S. Lee, M. R. Wenk and S. Q. Yao, Angew. Chem., Int. Ed., 2017, 56, 5829–5833 CrossRef CAS PubMed.
- D. Höglinger, A. Nadler, P. Haberkant, J. Kirkpatrick, M. Schifferer, F. Stein, S. Hauke, F. D. Portere and C. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1566–1571 CrossRef PubMed.
- X. H. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed.
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255–261 CrossRef CAS PubMed.
- M. A. Hollingsworth and B. J. Swanson, Nat. Rev. Cancer, 2004, 4, 45–60 CrossRef CAS PubMed.
- T. Gao, B. Wang, L. Shi, X. Zhu, Y. Xiang, J. Anzai and G. Li, Anal. Chem., 2017, 89, 10776–10782 CrossRef CAS PubMed.
- C. S. M. Ferreira, C. S. Matthews and S. Missailidis, Tumor Biol., 2006, 27, 289–301 CrossRef CAS PubMed.
- X. Chen, L. Qiu, R. Cai, C. Cui, L. Li, J. Jiang and W. Tan, ACS Appl. Mater. Interfaces, 2020, 12, 37845–37850 CrossRef CAS PubMed.
- Y. Yang, J. M. Hsu, L. Sun, L. C. Chan, C. W. Li, J. L. Hsu, Y. Wei, W. Xia, J. Hou, Y. Qiu and M.-C. Hung, Cell Res., 2019, 29, 83–86 CrossRef PubMed.
- C.-W. Li, S.-O. Lim, W. Xia, H.-H. Lee, L.-C. Chan, C.-W. Kuo, K.-H. Khoo, S.-S. Chang, J.-H. Cha, T. Kim, J. L. Hsu, Y. Wu, J.-M. Hsu, H. Yamaguchi, Q. Ding, Y. Wang, J. Yao, C.-C. Lee, H.-J. Wu, A. A. Sahin, J. P. Allison, D. Yu, G. N. Hortobagyi and M.-C. Hung, Nat. Commun., 2016, 7, 12632 CrossRef CAS PubMed.
- C. L. Kinlough, R. J. McMahan, P. A. Poland, J. B. Bruns, K. L. Harkleroad, R. J. Stremple, O. B. Kashlan, K. M. Weixel, O. A. Weisz and R. P. Hughey, J. Biol. Chem., 2006, 281, 12112–12122 CrossRef CAS PubMed.
- N. Martínez-Sáez, J. M. Peregrina and F. Corzana, Chem. Soc. Rev., 2017, 46, 7154–7175 RSC.
- R. N. Hannoush and J. Sun, Nat. Chem. Biol., 2010, 6, 498–506 CrossRef CAS PubMed.
- M. A. Longo and D. Combes, J. Biotechnol., 1997, 58, 21–32 CrossRef CAS.
- X. X. Gao and R. N. Hannoush, Nat. Chem. Biol., 2014, 10, 61–68 CrossRef CAS PubMed.
- A. Paszkiewicz-Gadek, H. Porowska, D. Lemancewicz, S. Wolczynski and A. Gindzienski, Int. J. Mol. Med., 2006, 17, 669–674 CAS.
- R. N. Hannoush and N. Arenas-Ramirez, ACS Chem. Biol., 2009, 4, 581–587 CrossRef CAS PubMed.
- H. Lis, B. A. Sela, L. Sachs and N. Sharon, Biochim. Biophys. Acta, 1970, 211, 582–585 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, supporting figures and table. See DOI: 10.1039/d1sc03059k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |