
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular design of efficient yellow- to red-emissive alkynylgold(III) complexes for the realization of thermally activated delayed fluorescence (TADF) and their applications in solution-processed organic light-emitting devices†
Cathay Chai
Au-Yeung
 ,
Lok-Kwan
Li
,
Man-Chung
Tang
,
Shiu-Lun
Lai
,
Wai-Lung
Cheung
,
Maggie
Ng
,
Mei-Yee
Chan
* and
Vivian Wing-Wah
Yam
*
,
Lok-Kwan
Li
,
Man-Chung
Tang
,
Shiu-Lun
Lai
,
Wai-Lung
Cheung
,
Maggie
Ng
,
Mei-Yee
Chan
* and
Vivian Wing-Wah
Yam
*
Institute of Molecular Functional Materials and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: wwyam@hku.hk; chanmym@hku.hk; Fax: +86-852-2857-1586; Tel: +86-852-2859-2153
First published on 22nd June 2021
Abstract
Here, we report the design and synthesis of a new class of fused heterocyclic alkynyl ligand-containing gold(III) complexes, which show tunable emission colors spanning from the yellow to red region in the solid state and exhibit thermally activated delayed fluorescence (TADF) properties. These complexes display high photoluminescence quantum yields of up to 0.87 and short excited-state lifetimes in sub-microsecond timescales, yielding high radiative decay rate constants on the order of up to 106 s−1. The observation of the drastic enhancement in the emission intensity of the complexes with insignificant change in the excited-state lifetime upon increasing the temperature from 200 to 360 K indicates an increasing radiative decay rate. The experimentally estimated energy splitting between the lowest-lying singlet excited state (S1) and the lowest-lying triplet excited state (T1), ΔES1–T1, is found to be as small as ∼0.03 eV (250 cm−1), comparable to the value of ∼0.05 eV (435 cm−1) obtained from computational studies. The delicate choice of the cyclometalating ligand and the fused heterocyclic ligand is deemed the key to induce TADF through the control of the energy levels of the intraligand and the ligand-to-ligand charge transfer excited states. This work represents the realization of highly emissive yellow- to red-emitting gold(III) TADF complexes incorporated with fused heterocyclic alkynyl ligands and their applications in organic light-emitting devices.
Introduction
With the unprecedented success of the commercialization of ultrahigh-definition mobile phones and televisions on the market, organic light-emitting devices (OLEDs) are regarded as attractive candidates for cutting-edge flat-panel displays and solid-state lightings.1–4 Recent interest is also focused on the development of thermally activated delayed fluorescence (TADF) compounds as the emitting material for highly efficient OLEDs due to their capability to harvest 100% excitons.5–11 The TADF mechanism features an efficient reverse intersystem crossing (RISC) from the lowest-lying triplet excited state (T1) to the lowest-lying singlet excited state (S1) via a small energy splitting (ΔES1–T1), which can be generally achieved by reducing the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) overlap integral. Not only in the pure organic compounds, photoluminescence via the TADF mechanism has also been reported in four-coordinate N^N- and P^P-ligand-based copper(I) complexes,12–14 two-coordinate carbene-containing copper(I),14–18 gold(I)17–19 and silver(I)18,20 complexes and others.21–24 Particularly, a series of two-coordinate cyclic (alkyl)(amino)carbene (CAAC)–copper-amide complexes has been found to show photoluminescence quantum yields (PLQYs) of > 0.99.15 More recently, analogous gold(I) and silver(I) complexes have also been found to exhibit TADF properties and high PLQYs approaching unity.18 Such high radiance characteristics have demonstrated the high potential of metal-containing TADF complexes for OLED applications.Compared to the elaborate studies in the copper(I) TADF complexes, the TADF properties of gold(III) complexes have been relatively less explored. Since our research group’s report on the first room-temperature luminescent dialkyl- and diarylgold(III) diimine complexes25 and subsequently the diphenylpyridine (C^NPY^C)-containing gold(III) complexes and their application studies in OLEDs,26,27 developments in recent years have expanded the variety of luminescent gold(III) complexes, in which different moieties, such as pyridine, isoquinoline or triazine, were incorporated into the tridentate or tetradentate ligand scaffolds.28–34 In 2015, a class of pyrazine-based gold(III) complexes was first reported and shown to exhibit TADF properties.35 However, there was no report on the electroluminescence (EL) properties of the gold(III) TADF complexes until 2017 when several cyclometalated C^NPY^C gold(III) TADF emitters and their applications in OLEDs were reported.36 These gold(III) complexes were reported to show high PLQYs of up to 0.89 in solid-state thin films and short-lived excited states in the sub-microsecond regime. Such high radiative decay rate constants (kr) on the order of up to 105–106 s−1 implied that the emission origin is not only limited to the triplet parentage but also occurred with substantial mixing of the singlet character.36–38 The corresponding devices achieved high external quantum efficiencies (EQEs) of up to 25%.36–38 However, with emitters of the C^NPY^C ligand framework, the emission colors of the gold(III) TADF complexes and their corresponding fabricated devices are mainly in the green and yellow regions.36–38 Therefore, it would be motivating to demonstrate the use of versatile ligands to manipulate the HOMO–LUMO energy gaps to tune the emission color of the gold(III) TADF complexes over a wide range in the visible spectrum under the same pincer ligand framework.
To realize a broad emission range under the same tridentate ligand framework, our group has recently designed and synthesized a series of C^NPY^C-containing alkynylgold(III) emitters which features the σ-donating fused heterocyclic alkynyl ligands.31 The structural modification to the ligands, including incorporating the fluorene– and carbazole–triphenylamine hybrid to replace the unsubstituted triphenylamine moiety, can greatly affect the HOMO energies of the complexes, as reflected by their electrochemical studies.30–33 With the identical tridentate ligands, these complexes show a large spectral shift of approximately 110 nm (ca. 3310 cm−1) that covers green to red emission with high PLQYs of up to 0.8 in solid-state thin films.31 Furthermore, the fluorene– and carbazole–triphenylamine hybrid in the alkynyl ligands provide excellent carrier-transporting properties and sufficiently high triplet energies to ensure exothermic energy transfer from the host to the emitters.31,32 Based on these design strategies, solution-processed OLEDs employing these fused heterocyclic ligand-containing C^NPY^C alkynylgold(III) complexes have been shown to exhibit emission colors from sky-blue to near-infrared,30–33 and high EQEs of 14.5%, 13.5% and 9.3% for green-,32 yellow-,31 and orange-emitting31 devices, respectively.
Herein, we report a new series of cyclometalated alkynylgold(III) complexes 1–6 (Scheme 1) consisting of strong σ-donating fused heterocyclic alkynyl ligands to realize a systematic approach to tune the emission colors of the gold(III) complexes and to investigate the capabilities of the fused ligands to induce TADF behavior. Instead of the C^NPY^C-26,28,31 or diphenylisoquinoline (C^NISQ^C)30-containing cyclometalating ligand in the previous works, new diphenylthienopyridine (C^NTHPY^C)-containing cyclometalating ligands, i.e., 4-tBuC^NTHPY^CtBu-4 and 3,5-F2C^NTHPY^CF2-3,5 (THPY = thienopyridine), are employed to afford 1–3 and 4–6, respectively. The use of C^NPY^C ligands would limit the emission colors of the gold(III) complexes to the range from green to orange.26,28,31 However, the replacement of the C^NPY^C ligand by the C^NISQ^C ligand would lead to a much lower-lying LUMO energy and shift the emission to the near-infrared region with low PL quantum efficiencies.30 Therefore, here, we adjust the LUMO energy to achieve the yellow- to red-emitting gold(III) complexes by the use of C^NTHPY^C cyclometalating ligands. The incorporation of fluorene– and carbazole–triphenylamine hybrids onto the alkynyl ligands not only can effectively increase the HOMO energies of the complexes but also can enable a good spatial separation of the HOMO and the LUMO, as supported by the electrochemical and computational studies. As a result, a wide range of emission colors spanning from green to red have been observed in the 1,3-bis(N-carbazolyl)benzene (mCP) thin films of these complexes. The high PLQYs of up to 0.87, together with short excited-state lifetimes in sub-microsecond timescales, suggest a high kr on the order of up to 106 s−1. It is worth noting that some of these complexes show an exceptional increase in the emission intensity upon increasing the temperature. In particular, the solid-state emission of 3 shows a hypsochromic shift with increasing intensity and possesses a decreasing excited-state lifetime upon increasing the temperature from 200 K to 360 K, indicating an increasing radiative decay rate. By fitting the measured emission decay lifetimes to the full kinetic scheme developed previously,18 the ΔES1–T1 of 3 and 5 are estimated to be ∼0.03 eV (250 and 259 cm−1, respectively), comparable to the value of ∼0.05 eV (435 and 454 cm−1, respectively) obtained from time-dependent density functional theory (TDDFT) calculations, supporting the fact that the new class of alkynylgold(III) complexes display TADF at ambient temperature. More importantly, it is found that a relatively large energy difference between the intraligand (IL) and the charge transfer (CT) states is the key to facilitate the TADF behavior. The corresponding solution-processed OLEDs have achieved maximum EQEs of 8.0% and 5.3% for yellow- and red-emitting devices, respectively. This work represents the realization of highly emissive yellow- to red-emitting gold(III) TADF complexes and also the first demonstration on the application studies of red-emitting OLEDs based on TADF gold(III) emitters.
 | ||
| Scheme 1 Molecular structures of the cyclometalated alkynylgold(III) complexes 1–6. | ||
Results and discussion
Synthesis, characterization and thermal properties of alkynylgold(III) complexes
The cyclometalating ligands, 4-tBuC^NTHPY^CtBu-4 and 3,5-F2C^NTHPY^CF2-3,5, were synthesized by the Suzuki–Miyaura cross-coupling reaction with palladium-catalyzed C–C bond formation between 5,7-dichlorothieno[2,3-c]pyridine and 4-tert-butylphenylboronic acid or 3,5-difluorophenylboronic acid, respectively. The resulting cyclometalating ligands were then mercurated and transmetalated to the gold(III) metal center to afford the cyclometalated chlorogold(III) precursors. 1–6 were synthesized by treating the respective chlorogold(III) precursors with the corresponding alkynyl ligands in the presence of a catalytic amount of copper(I) iodide in triethylamine and tetrahydrofuran solution (Scheme S1†). After obtaining the crude product, these complexes were purified by column chromatography followed by recrystallization by diffusion of diethyl ether into a concentrated dichloromethane solution of the respective complexes (see SI for detailed procedures).30–33 All the complexes were characterized by 1H, 13C{1H} and 19F{1H} nuclear magnetic resonance (NMR) spectroscopy, high-resolution electrospray ionization (ESI) mass spectrometry (MS) and infrared (IR) spectroscopy. Thermogravimetric analysis has been conducted on representative complexes, in which high decomposition temperatures (Td) of above 330 °C have been found (Fig. S1 and Table S1†), demonstrating the high robustness of these alkynylgold(III) complexes.Electrochemistry
Cyclic voltammetric studies have been carried out on 1–6 in dichloromethane (0.1 M nBu4NPF6) at 298 K. Their electrochemical data and their corresponding HOMO and LUMO energies are summarized in Table S2,† while the cyclic voltammograms are shown in Fig. S2.† In general, 1–3 and 4–6 show a quasi-reversible reduction couple at ca. −1.45 V and −1.18 V vs. saturated calomel electrode (SCE), respectively, which are found to be insensitive to the choice of auxiliary ligands. With reference to the previously reported gold(III) complexes,30–33 these reduction couples could be attributed to the reduction of the C^NTHPY^C ligand. Considering the electron-withdrawing nature of the fluorine substituents in the cyclometalating ligands of 4–6, which leads to the stabilization of the π*(3,5-F2C^NTHPY^CF2-3,5) orbital, a less negative reduction potential is found for 4–6 (ca. −1.18 V vs. SCE) when compared to 1–3 (ca. −1.45 V vs. SCE). On the other hand, the potential for oxidation of 1–6 are sensitive to the nature of the heterocyclic core unit and could be ascribed to alkynyl ligand-centered oxidation.30,31 Apparently, with the incorporation of a fluorene–triphenylamine hybrid in the alkynyl ligand, 2 and 5 display a less positive potential for oxidation (ca. +0.80 V vs. SCE) than those incorporated with the conventional triphenylamine alkynyl ligand (ca. +0.86 V vs. SCE) in 1 and 4. The replacement of the fused fluorenyl to carbazolyl moieties in the alkynyl ligand of 3 and 6 results in a further less positive potential for oxidation (ca. +0.51 V vs. SCE). These suggest that the replacement of the conventional triphenylamine moiety with fluorene– and carbazole–triphenylamine hybrid can lead to a gradual destabilization of the π orbital of the corresponding alkynyl ligands,31 which further confirm the versatility of these fused alkynyl ligands in delicately controlling the HOMO–LUMO energy gap of these gold(III) complexes.Photophysical properties
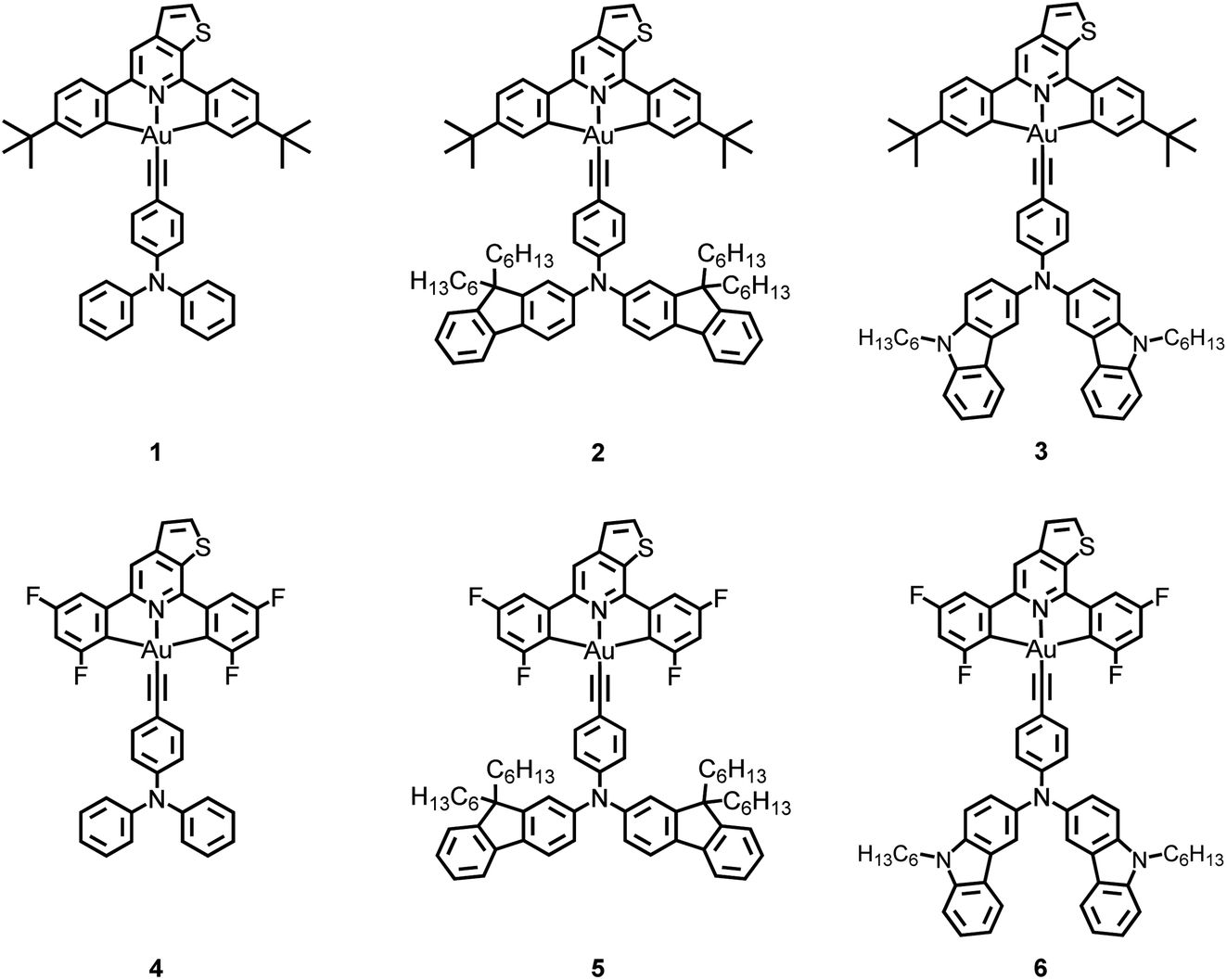
The UV-visible absorption spectra and normalized emission spectra of 1–6 in toluene at 298 K are shown in Fig. S3† and 1a, respectively. All the complexes show an intense absorption band at the wavelength (λ) of ca. 300–400 nm and a moderately intense vibronic-structured band at ca. 390–450 nm with the extinction coefficient (ε) of the order of 104 mol−1 dm3 cm−1, while 3–6 display an additional absorption tail extending to ca. 550–650 nm. The photophysical data of 1–6 are summarized in Table 1. The absorption bands at wavelengths shorter than 340 nm are mainly attributed to the spin-allowed IL [π→π*] transitions of the triphenylamine and/or carbazole moieties,30,31 while the intense absorption bands at ca. 340–400 nm of 2 and 5, respectively, are mainly attributed to IL [π→π*] transitions of the fluorene moieties.31–33 The less intense vibronic-structured absorption bands at ca. 390–450 nm are mainly attributed to the metal-perturbed IL [π→π*(C^NTHPY^C)] transitions.30,39 The additional absorption tail for 3–6 beyond 450 nm could be assigned to the ligand-to-ligand charge transfer (LLCT) [π(fused heterocyclic alkynyl)→π*(C^NTHPY^C)] transition.30–32 Apparently, 6 shows a relatively red-shifted absorption band compared to 5 [i.e., λ: 6 (517 nm) > 5 (485 nm)] due to the higher-lying HOMO level arising from the electron-donating carbazole-fused heterocyclic alkynyl ligand.31 With the same alkynyl ligand, 6 also exhibits a red-shifted absorption band than 3 [i.e., λ: 6 (517 nm) > 3 (464 nm)]. This could be attributed to the electron-withdrawing effect exerted by the fluorine substituents, leading to a lower-lying LUMO and therefore a narrower energy gap for the LLCT transition.30 These findings are in good agreement with the electrochemical and computational studies (see below).| Complex | Medium (T/K) | Absorption λmax/nm (εmax/dm3 mol−1 cm−1) | Emission λmax/nm (τo/μs) | Φ PL(sol)a | Φ PL(film)b | k r /s−1 | k nr /s−1 |
|---|---|---|---|---|---|---|---|
a The relative luminescence quantum yield, measured at room temperature using [Ru(bpy)3]Cl2 in degassed acetonitrile as the reference (excitation wavelength = 436 nm, Φlum = 0.060).
b Absolute luminescence quantum yield of thin films of the gold(III) complexes doped into 10% 1,3-bis(carbazol-9-yl)benzene (mCP) measured with 350 nm excitation.
c Radiative decay rate constant determined from the equation kr = Φem/τ; non-radiative decay rate constant determined from the equation knr = (1 − Φem)/τ.
d Measured in EtOH–MeOH–THF (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:1, v/v). :10:1, v/v).
|
|||||||
| 1 | Toluene (298) | 333 (39340), 398 (8360), 414 (10640), 436 (8360) |
524, 564, 606 (5.7) | 0.029 | 5.14 × 103 | 1.70 × 105 | |
| Solid (298) | 585 (0.1) | ||||||
| Solid (77) | 543, 585 (2.2) | ||||||
| Glass (77)d | 563, 604, 659 (141.6) | ||||||
| Thin film (298) | |||||||
| 5 wt% in mCP | 528, 568, 616 (96.1) | 0.57 | 5.93 × 103 | 4.47 × 103 | |||
| 10 wt% in mCP | 528, 568, 616 (91.9) | 0.48 | 5.22 × 103 | 5.66 × 103 | |||
| 15 wt% in mCP | 528, 568, 616 (82.1) | 0.54 | 6.58 × 103 | 5.60 × 103 | |||
| 20 wt% in mCP | 528, 568, 616 (74.1) | 0.46 | 6.21 × 103 | 7.29 × 103 | |||
| 2 | Toluene (298) | 320 (38040), 335 (55500), 366 (60660), 416 (14080), 436 (14530) |
525, 563, 609 (3.9) | 0.083 | 2.12 × 104 | 2.35 × 105 | |
| Solid (298) | 551 (3.5) | ||||||
| Solid (77) | 539, 582, 628 (101.6) | ||||||
| Glass (77)d | 561, 606, 659 (101.7) | ||||||
| Thin film (298) | |||||||
| 5 wt% in mCP | 528, 568, 616 (79.3) | 0.52 | 6.56 × 103 | 6.05 × 103 | |||
| 10 wt% in mCP | 528, 568, 616 (72.5) | 0.53 | 7.31 × 103 | 6.48 × 103 | |||
| 15 wt% in mCP | 528, 568, 616 (62.5) | 0.48 | 7.68 × 103 | 8.32 × 103 | |||
| 20 wt% in mCP | 528, 568, 616 (55.9) | 0.53 | 9.48 × 103 | 8.41 × 103 | |||
| 3 | Toluene (298) | 333 (60230), 395 (10190), 414 (12770), 437 (13110), 464 (6450) |
606 (0.3) | 0.066 | 2.19 × 105 | 3.11 × 106 | |
| Solid (298) | 646 (0.2) | ||||||
| Solid (77) | 642 (2.0) | ||||||
| Glass (77)d | 520, 637 (5.0) | ||||||
| Thin film (298) | |||||||
| 5 wt% in mCP | 562 (5.7) | 0.87 | 1.53 × 105 | 2.28 × 104 | |||
| 10 wt% in mCP | 569 (3.8) | 0.84 | 2.21 × 105 | 4.21 × 104 | |||
| 15 wt% in mCP | 575 (3.2) | 0.76 | 2.38 × 105 | 7.50 × 104 | |||
| 20 wt% in mCP | 583 (2.3) | 0.75 | 3.26 × 105 | 1.09 × 105 | |||
| 4 | Toluene (298) | 330 (41450), 378 (4740), 400 (5990), 421 (6960), 458 (4330) |
660 (<0.1) | 0.022 | 2.20 × 105 | 9.78 × 106 | |
| Solid (298) | 633 (0.7) | ||||||
| Solid (77) | 598 (5.7) | ||||||
| Glass (77)d | 523, 564, 610 (5.4) | ||||||
| Thin film (298) | |||||||
| 5 wt% in mCP | 559 (13.5) | 0.53 | 3.93 × 104 | 3.48 × 104 | |||
| 10 wt% in mCP | 562 (10.1) | 0.49 | 4.85 × 104 | 5.05 × 104 | |||
| 15 wt% in mCP | 565 (8.6) | 0.48 | 5.56 × 104 | 6.02 × 104 | |||
| 20 wt% in mCP | 569 (5.5) | 0.47 | 8.50 × 104 | 9.58 × 104 | |||
| 5 | Toluene (298) | 298 (38360), 360 (69724), 422 (8369), 485 (7000) |
656 (2.8) | 0.028 | 1.01 × 104 | 3.47 × 105 | |
| Solid (298) | 630 (0.6) | ||||||
| Solid (77) | 628 (1.9) | ||||||
| Glass (77)d | 607, 620 (1.2) | ||||||
| Thin film (298) | |||||||
| 5 wt% in mCP | 585 (1.3) | 0.70 | 5.38 × 105 | 2.31 × 105 | |||
| 10 wt% in mCP | 597 (0.9) | 0.60 | 6.59 × 105 | 4.40 × 105 | |||
| 15 wt% in mCP | 600 (0.6) | 0.61 | 9.68 × 105 | 6.19 × 105 | |||
| 20 wt% in mCP | 610 (0.5) | 0.52 | 1.06 × 106 | 9.80 × 105 | |||
| 6 | Toluene (298) | 299 (45760), 364 (22910), 399 (9900), 420 (7170), 517 (4520) |
740 (1.6) | 0.012 | 7.25 × 103 | 6.18 × 105 | |
| Solid (298) | 683 (0.1) | ||||||
| Solid (77) | 647, 687 (0.5) | ||||||
| Glass (77)d | 732 (0.3) | ||||||
| Thin film (298) | |||||||
| 5 wt% in mCP | 636 (0.4) | 0.40 | 1.00 × 106 | 1.50 × 106 | |||
| 10 wt% in mCP | 643 (0.3) | 0.35 | 1.16 × 106 | 2.17 × 106 | |||
| 15 wt% in mCP | 647 (0.3) | 0.24 | 8.28 × 105 | 2.62 × 106 | |||
| 20 wt% in mCP | 650 (0.2) | 0.29 | 1.45 × 106 | 3.55 × 106 | |||
Upon excitation at λ ≥ 400 nm, 1 and 2 show a vibronic-structured emission band peaking at ca. 524 nm with an emission decay of ca. 5.7 μs, which could be assigned as the metal-perturbed 3IL [π→π*(C^NTHPY^C)] excited state, possibly with the mixing of some CT character.30 On the other hand, 3–6 feature a broad structureless emission band with the emission maximum peaking at ca. 600–740 nm with the decay lifetime of ca. 0.1–2.8 μs (Fig. S4†), which could be assigned as originating from the LLCT [π(alkynyl)→π*(C^NTHPY^C)] excited state.31,32 To confirm the CT nature of the excited states of 3–6, solvent-dependent emission studies have been carried out on the representative complex 3. Fig. S5† depicts the different emission behaviors of 3 in various solvents at 298 K. Upon increasing the polarity of the solvent from toluene to ethyl acetate, a bathochromic shift of the LLCT band maxima from ca. 604 nm to 760 nm is observed. This, together with a slope of ca. 18500 cm−1 in the corresponding Lippert–Mataga plot (Fig. S6†), is in agreement with the charge-transfer character of the excited state and a better stabilization of the LLCT state in more polar solvents.
To gain further insights into the nature of the excited states, nanosecond transient absorption (TA) measurements have been performed on 1 and 3 in degassed toluene solution at 298 K. Their TA difference spectra determined at different delay times after a 355 nm laser pulse excitation are shown in Fig. S7.† The TA absorption profiles of 1 and 3 are unique with respect to each other, indicating that they do not share the same excited state origin. For 1, the TA spectrum features an intense broad band maximum at ca. 600 nm with ground-state bleaching at ca. 440 nm. The TA difference spectrum of 3 is characterized by a ground-state bleaching at ca. 300–370 nm and two intense positive absorption bands centered at 430 nm and beyond 600 nm. The bleaching at ca. 300–370 nm is ascribed to the depletion of the ground state upon excitation, corresponding to the IL [π–π*(C^NTHPY^C)] transition, while the higher-energy band centered at 430 nm is typical of the triplet absorption due to the C^NTHPY^C radical anion.28 The absorption beyond 600 nm is tentatively assigned as the (4-diphenylaminophenyl)ethynyl radical cation absorption, given that a similar TA band has also been observed for the radical cation of free 4-aminophenylacetylene and other 4-ethynyl-N,N-diphenylaniline-containing gold(III) complexes.28,39 The absorption decay lifetimes of 1 and 3 are found to be 7.8 μs and 0.3 μs, respectively, which are comparable to their emission decay lifetimes in toluene. This further supports the assignment of the TA bands, as derived from the IL [π→π*(C^NTHPY^C)] and LLCT [π(alkynyl)→π*(C^NTHPY^C)] states of 1 and 3, respectively.
The emission behaviors of these complexes in solid-state thin films have also been investigated. The normalized photoluminescence (PL) spectra of thin films of 10 wt% 1–6 doped in mCP at 298 K are shown in Fig. 1b. Apparently, 1 and 2 exhibit vibronic-structured emission bands peaking at ca. 528 nm, while 3–6 feature broad structureless emission bands peaking at ca. 560–640 nm. The emission bands of 1 and 2 are found to be independent of dopant concentrations (Table 1 and Fig. S8a and b†), suggesting a suppressed excimeric emission due to the incorporation of the bulky tert-butyl groups.40 In sharp contrast, the emission bands of 3–6 show a bathochromic shift upon increasing the dopant concentrations (Fig. S8c–f†), probably due to the presence of the hydrophobic alkyl chains and the large π surface in the fused heterocyclic alkynyl ligand in 3–6.31 Moreover, the concentration-dependent emission behavior could be a consequence of the change in the local polarity of the host–guest matrix that perturbs the relative contribution of the IL states and the CT states of the complexes.32 Prompt fluorescence was not observed in 1–6. This could be ascribed to the fast intersystem crossing (ISC) rate constants associated with the heavy gold metal center in these complexes.‡ It is worth noting that high PLQYs of up to 0.87 have been found in this class of gold(III) complexes. For 3–6, which exhibit structureless emission profiles in thin films, they generally show shorter decay lifetimes (0.2–5.7 μs), when compared to those of 1 and 2 (55.9–96.1 μs) (Fig. S9†). Hence, the kr of 3–6 in thin films (up to 1.45 × 106 s−1) are found to be much higher than those of 1 and 2 (up to 9.48 × 103 s−1). The larger kr of 3–6 are suggestive of the presence of emission origins that may not be limited only to the 3LLCT excited state.36,37 These results have prompted us to conduct the following variable-temperature experiments to further elucidate the excited state nature of these gold(III) complexes.
 | ||
| Fig. 1 (a) Normalized emission spectra of 1–6 in toluene at 298 K. (b) Normalized PL spectra of 10 wt% 1–6 doped in mCP thin films at 298 K. | ||
Variable-temperature studies
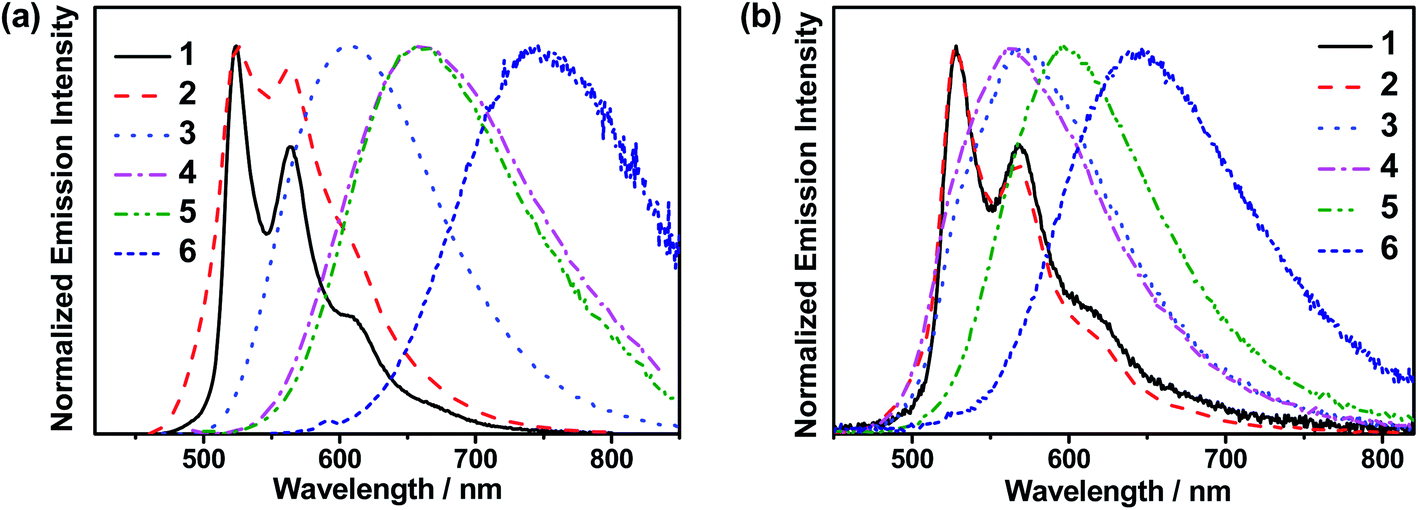
The emission spectra of 3 in toluene at various temperatures have been recorded, as shown in Fig. S10.† Upon increasing the temperature from 190 K to 300 K, the emission band maximum of 3 shows a hypsochromic shift from 633 nm to 603 nm with a gradual drop of the emission intensity. The decreasing emission intensity with temperature could be attributed to the enhanced non-radiative decay due to the molecular motions, which are more efficient in the solution state, while the hypsochromic shift may be due to several reasons. It is possible that the increase in temperature reduces the polarity of toluene, destabilizing the LLCT excited state of 3.41 Alternatively, such an observation may also suggest a thermally activated process, which involves another excited state that is of higher energy than the original emissive state.34,42To gain deeper insights into the excited state nature of these gold(III) complexes, the solid-state luminescence of selected complexes at different temperatures has been investigated, so that any solvent-related effects which could contribute to the changes in the emission can be excluded. The emission profiles of 1 in 5 wt% doped mCP thin film at 77 K, 200 K and 320 K are shown in Fig. 2a, while those of 3 in 5 wt% doped mCP thin film are shown in Fig. 2b. The vibronic-structured emission band of 1, typical of 3IL [π→π*(C^NTHPY^C)] emission origin, shows a drop in intensity upon increasing the temperature, attributed to the increased rate of non-radiative decay. In sharp contrast, the intensity of the emission band of 3 is found to be significantly enhanced upon increasing the temperature. A similar growth in emission intensity has also been found in 4–6 (Fig. S11†). Different from the broad and structureless emission band of 3, typical of LLCT emission, observed at 298 K, a small shoulder emerges at ca. 525 nm in the emission band of 3 at 77 K, indicative of the involvement of the 3IL [π→π*(C^NTHPY^C)] state.36 A small shoulder at ca. 520 nm is also found for 5 at 77 K. The presence of 3IL emission at 77 K is due to the subtle destabilization of the LLCT states upon lowering the temperature, such that the IL character is observed as the energy levels of the 3IL and the LLCT states become closer. Compared to 3, the structureless emission profile remains in 6 at both 298 K and 77 K. It is likely that replacing the cyclometalating ligand in 3 by the fluoro-substituted one in 6 would elevate the energy of the 3IL excited state and at the same time would lower the LLCT excited state, which widens the energy gap between the 3IL state and the lower-lying LLCT state, such that a structureless emission band with little involvement of the 3IL state, regardless of the temperature, is observed in 6 but not in 3. In contrast, the emission of 4 in mCP thin films becomes vibronic-structured at 77 K, indicating an 3IL emission origin and the strongest extent of 3IL excited state participation in the emission of 4 among 3–6. The extent of participation of the 3IL state can be reflected by the observed kr of the solid-state luminescence of the complexes, where a larger kr indicates a smaller extent of participation of the long-lived 3IL state, resulting from the larger energy difference between the higher-lying 3IL excited state and the lower-lying LLCT excited state. The observed kr of 3, 5 and 6 in mCP thin films are on the order of 105 to 106 s−1, higher than that of 4 which is on the order of 104 s−1 (Table 1).
 | ||
| Fig. 2 Emission spectra of (a) 1 and (b) 3 in 5 wt% doped mCP thin films at 77 K, 200 K and 320 K. (c) Emission spectra and (d) normalized emission spectra of 3 in 5 wt% doped mCP thin films upon increasing the temperature from 120 K to 360 K. | ||
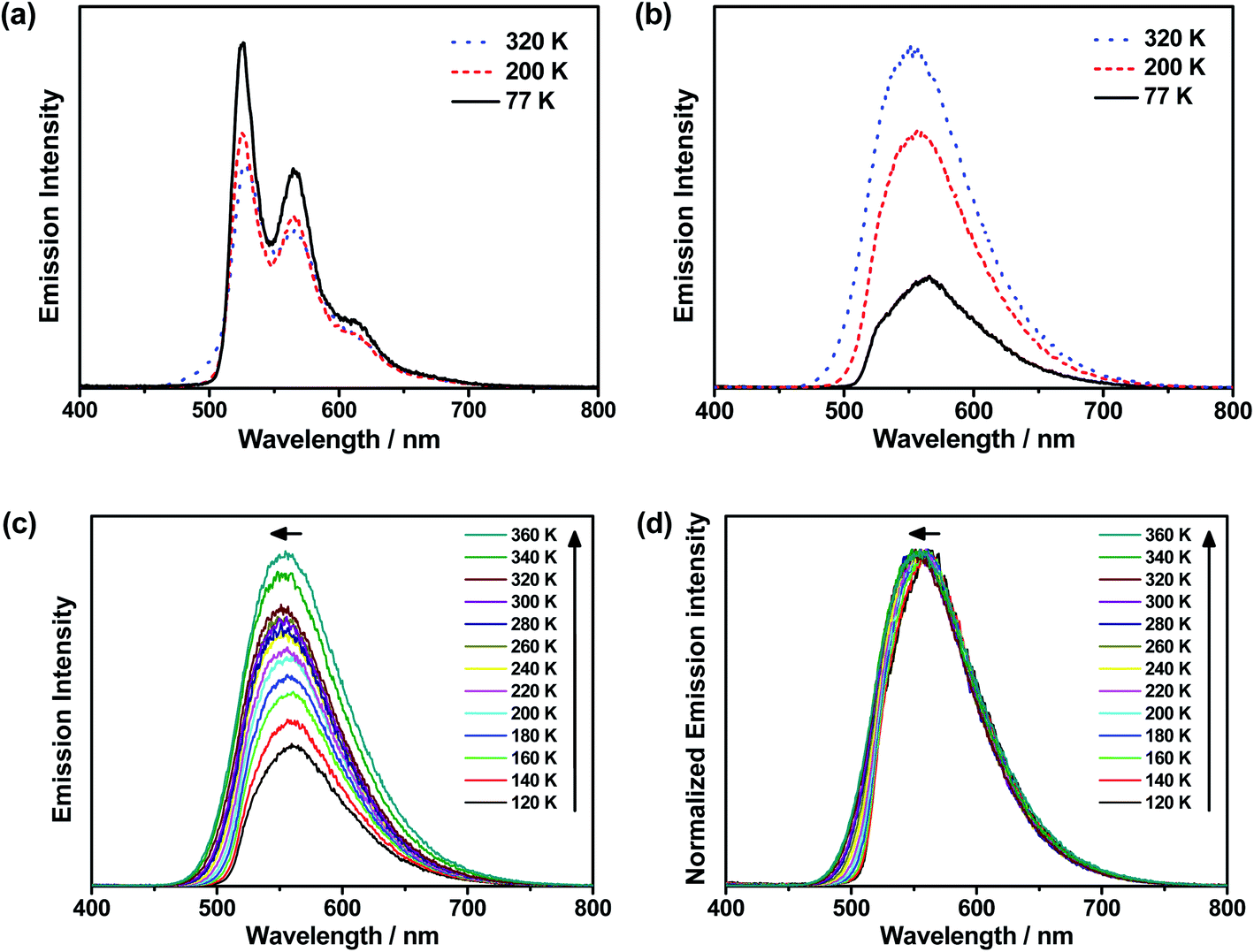
To further investigate the enhanced emission upon increasing the temperature in 3–6, the emission profiles and decay times of 3 in 5 wt% doped mCP thin films have been recorded upon increasing the temperature from 120 K to 360 K (Fig. 2c), in which a blue shift of the emission band maximum from 562 nm to 553 nm (∼290 cm−1) is observed. Notably, the excited state lifetime distinctly shortens from 34.05 μs at 77 K gradually to 3.41 μs at 360 K (Table S3†). Together with the increasing emission intensity, the significant increase in kr upon increasing the temperature is suggestive of the presence of a thermally activated emission process and the involvement of a higher-lying excited state with a higher decay rate.13,18,36 A possible emission origin involved could be the singlet states, which possess short decay lifetimes, with a growing contribution for emission with increasing temperature.18 As discussed earlier, the emission of 3 at low temperatures should involve not only the LLCT states but also the 3IL excited state, with a long emission lifetime of 34.05 μs and a structured band observed in the delayed emission spectra at 77 K (Fig. S12a†). When the temperature is increased to 200 K, the delayed emission profile turns structureless, indicating the decrease in the 3IL excited state character in the emission. In order to explore the possible involvement of the singlet state and to obtain an estimation of ΔES1–T1, a linear relation has been constructed in the 200–360 K temperature regime based on the full kinetic scheme for two level systems.18,43 An Arrhenius plot derived from the full kinetic scheme was used to estimate ΔES1–T1.§ In this fitting, only the data obtained above 200 K are taken into consideration in order to minimize the involvement of the 3IL excited state and to maximize the possible contribution of TADF in the emission.18 The Arrhenius plot of ln(kTADF) vs. 1/T, presenting a slope of ΔES1–T1/kB and an intercept of [ln(b)], which is a function of the S1 → T1 intersystem-crossing rate constant (kISC) and the radiative rate of the singlet state (kfl),17 is shown in Fig. 3a. Under this model, there are several assumptions, including the PLQY remaining continuously high so that kr ≫ knr, and the lifetime is being controlled principally by ΔES1–T1.18 As a result, a ΔES1–T1 value, by fitting to the equation,18
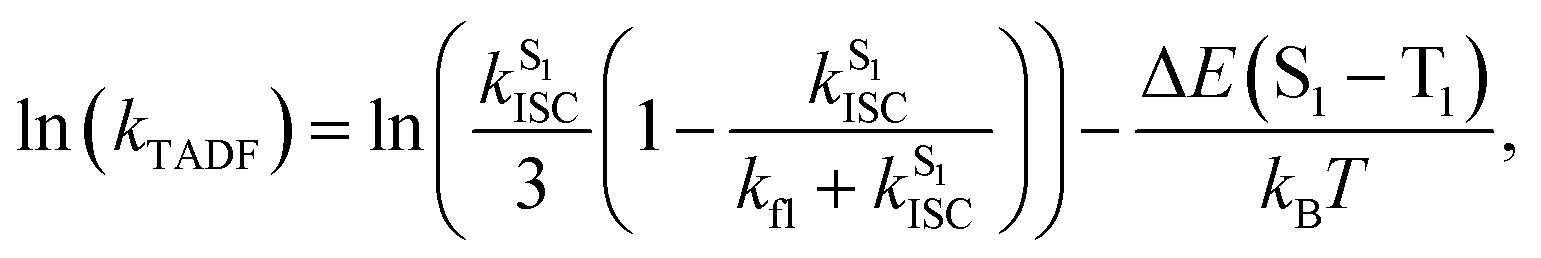 | (1) |
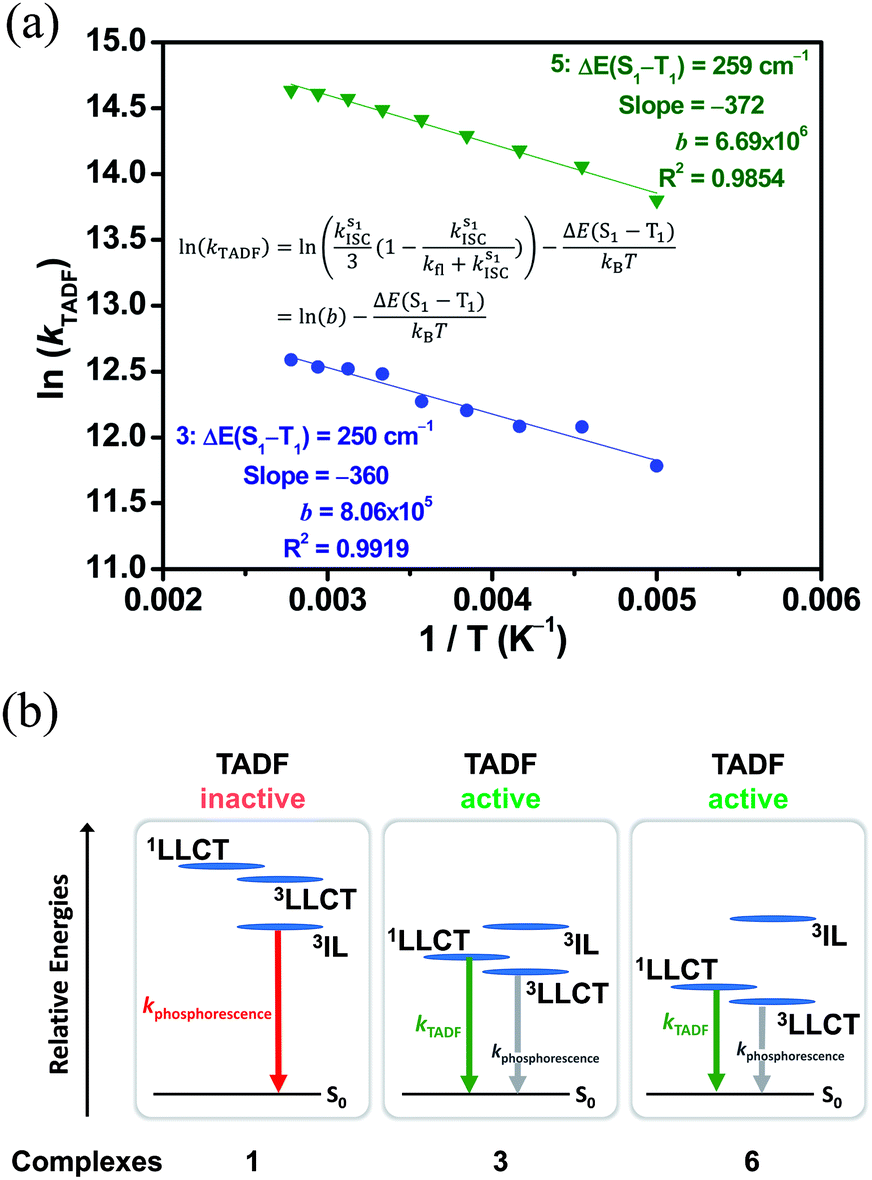 | ||
Fig. 3 (a) Plot of ln(kTADF) vs. 1/T of 3 ( ) and 5 ( ) and 5 ( ) in 5 w% doped mCP thin films monitored at 560 and 570 nm, respectively, and the fits of the temperature-dependent lifetime data from 200 to 360 K to eqn (1). (b) Qualitative state diagram depicting the relative energies of the 1,3LLCT states and the 3IL state of 1, 3 and 6 in the solid state at 298 K. The trend of the relative energies of the states is based on the results from the emission studies. ) in 5 w% doped mCP thin films monitored at 560 and 570 nm, respectively, and the fits of the temperature-dependent lifetime data from 200 to 360 K to eqn (1). (b) Qualitative state diagram depicting the relative energies of the 1,3LLCT states and the 3IL state of 1, 3 and 6 in the solid state at 298 K. The trend of the relative energies of the states is based on the results from the emission studies. | ||
The decay times of the selected complexes in 5 wt% doped mCP thin films from 77 K to 360 K have also been recorded (Table S3, Fig. S13 and S14†). Decreases in the excited state lifetime can be observed for all the four complexes (i.e., 3–6), which display enhanced luminescence upon increasing the temperature. The Arrhenius model was also used to fit the data of 5, which displays a comparably high PLQY among 3–6, in the temperature region (T = 200–360 K). By fitting to the model based on the full kinetic scheme, the ΔES1–T1 value is estimated to be 259 cm−1 (0.03 eV) for 5 (Fig. 3a), which also indicates a small energy gap for the singlet–triplet splitting.
These calculated results indicate a narrow energy splitting between the 1LLCT and 3LLCT states. It is also suggested that the enhanced emission upon increasing temperature observed in 3, as well as in 4–6, can be ascribed to the occurrence of TADF among the 1,3LLCT states. Notably, a large energy difference between the higher-lying 3IL excited state and the lower-lying LLCT excited states could restrict the participation of the rather longer-lived 3IL state in the emissions of the gold(III) complexes, leading to a larger observed kr in the solid-state TADF (Fig. 3b). Furthermore, the observation of the 3IL emission for 1 indicates that the 3IL excited state is at a lower energy than the LLCT excited state (Fig. 3b). The relative energies of the various excited states for complexes 1, 3 and 6 that account for the luminescence behavior are depicted in Fig. 3b. Despite the 3IL energies of 3 and 6 being similar, with 6 slightly higher in energy than 3, the lower 1,3LLCT energies of 6 than 3 with the same auxiliary ligand are in line with the lower-lying π* orbital energy of the fluoro-substituted C^NTHPY^C ligand due to the presence of the electronegative fluoro group, as revealed by the cyclic voltammetric studies. These observations illustrate the importance of the pairing of the cyclometalating ligands and the auxiliary ligands, which in turn controls the energy difference between the 3IL and LLCT states to facilitate the TADF behavior.
Computational studies
To gain a deeper insight into the electronic structures and the nature of the absorption and emission origins of these gold(III) complexes, density functional theory (DFT) and TDDFT calculations have been performed on selected complexes, i.e., 1, 3, 5 and 6. The optimized ground-state (S0) geometries and the selected structural parameters of the complexes are shown in Fig. S15.† It should be noted that the thienopyridine moiety and one of the phenyl rings on the cyclometalating ligand are coplanar to each other, with a dihedral angle close to 0°, in contrast to the previously reported isoquinoline-containing analogues.30 The first twenty-five singlet excited states of 1, 3, 5 and 6, computed by the TDDFT/conductor-like continuum model (CPCM) method, are summarized in Table S4,† and the simulated UV-vis spectra, generated by Multiwfn,44 are shown in Fig. S16–S19.† The selected molecular orbitals involved in the transitions of 3 is shown in Fig. 4, while those for other complexes are shown in Fig. S20–S22.† The low-energy absorption bands of 3, 5 and 6 computed at ca. 550–660 nm correspond to the HOMO → LUMO excitation, in which the HOMOs are the π orbitals localized on the alkynyl ligand and the LUMOs are the π* orbitals on the cyclometalating ligand. Hence, the low-energy absorption bands of 3, 5 and 6 can be assigned to the LLCT [π(alkynyl)→π*(C^NTHPY^C)] transition, which supports the experimental energy trend of the absorption tails and their spectral assignments. For all the complexes, the absorption bands computed at ca. 390–400 nm are predominantly contributed by the metal-perturbed IL [π→π*] transitions of the C^NTHPY^C ligand, which supports the experimental assignment of the moderately intense vibronic-structured bands at 400–470 nm. For 5, the intense absorption band computed at 375 nm mainly corresponds to the IL [π→π*] transition of the fluorene moiety. The orbital energy diagram showing the frontier molecular orbitals of 1, 3, 5 and 6 is depicted in Fig. S23.† Upon replacing the triphenylamine unit in 1 by the carbazole– or fluorene–triphenylamine hybrid in 3, 5 and 6, the HOMO localized on the alkynyl ligand is destabilized by ca. 0.2–0.4 eV. On the other hand, the replacement of the electron-donating tert-butyl groups by the electron-withdrawing fluorine atoms on the C^NTHPY^C ligand from 1 and 3 to 5 and 6 leads to the stabilization of the LUMO by ca. 0.3–0.4 eV. Overall, the trend of the HOMO and LUMO energies is in good agreement with their electrochemical data (Table S2†).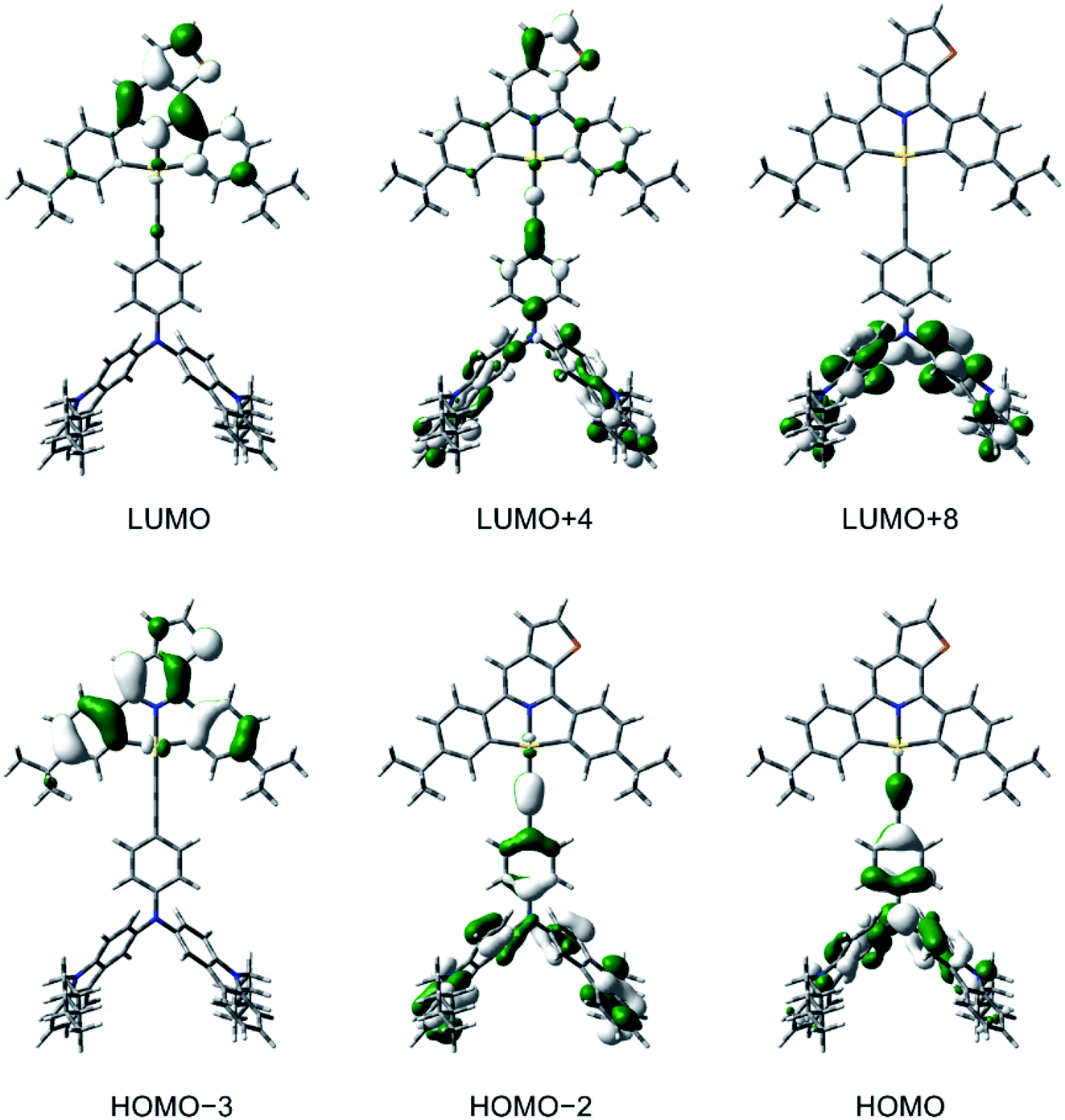 | ||
| Fig. 4 Spatial plots (isovalue = 0.03) of selected molecular orbitals of 3 at the optimized ground-state geometry. | ||
To confirm the nature of the emissive states, the geometries of T1 of 1, 3, 5 and 6 have been optimized with the unrestricted UPBE0 functional, with their plots of spin density shown in Fig. S24.† The spin density of 1 is predominantly localized on the C^NTHPY^C ligand, which supports the experimental assignment of the 3IL [π→π*(C^NTHPY^C)] character in the emissive state. On the other hand, the spin density of 3, 5 and 6 is localized on both the C^NTHPY^C and the alkynyl ligand. This supports the LLCT [π(fused heterocyclic alkynyl)→π*(C^NTHPY^C)] character in the emissive state. Similar bond angles and bond lengths were found in the optimized geometries of the S1 and T1 states of 3 (Fig. S25†), which could lead to a small reorganization energy, thus facilitating the process of TADF.35 The emission wavelengths of 1, 3, 5 and 6, approximated by the energy difference between the S0 and T1 states (ΔET1–S0), and those of 3, 5 and 6, approximated by the energy difference between the S0 and S1 states (ΔES1–S0) at their corresponding optimized geometries, are summarized in Table S5.† The calculated ΔET1–S0 shows a red shift on going from 1 (518 nm) to 3 and 5 (ca. 630 nm) to 6 (704 nm), which is generally in line with the experimental trend observed in the emission spectra. To gain further insights into the excited states involved in the TADF processes, the geometries of the S1 and the T1 states of 3 and 5 have been optimized using TDDFT calculations. The calculated ΔES1–S0 shows a red shift on going from 3 (575 nm) to 5 (625 nm) to 6 (699 nm), which is also in line with the experimental trend observed in the emission spectra. The computed ΔES1–T1 of 3 and 5 are 0.054 eV and 0.056 eV (i.e., 435 cm−1 and 454 cm−1), respectively, which are in fair agreement with the experimental value of 250 cm−1 and 259 cm−1 obtained by the full kinetic scheme.
OLED fabrication and characterization
Complexes 1–6 have been utilized to prepare solution-processed OLEDs to examine their EL properties. Solution-processed OLEDs with the configuration of indium tin oxide (ITO)/poly(ethylenedioxythiophene):poly(styrene sulfonic acid) (PEDOT:PSS; 40 nm)/emissive layer (30 nm)/tris(2,4,6-trimethyl-3-(pyridin-3-yl)phenyl)borane (3TPyMB; 5 nm)/1,3,5-tri[(3-pyridyl)phen-3-yl]benzene (TmPyPB; 30 nm)/LiF (0.8 nm)/Al (100 nm) were fabricated, in which the emissive layer was formed by mixing the respective gold(III) complex with mCP in chloroform via a spin-coating technique. Except for 6, which sparingly dissolves in chloroform, the EL spectra of the solution-processed OLEDs based on 1–5 are shown in Fig. 5a, S26–S30.† It is found that there is an insignificant red shift in the emissions for devices based on 1 and 2. Particularly, their EL spectra resemble their emission spectra in both solution and solid-state thin films. Upon increasing the dopant concentration, the EL spectra of devices made with 1 remain unchanged; while the emission intensity at λ = ∼580 nm is found to be increased in the devices made with 2. Both x and y values of the CIE coordinates for these devices are slightly varied (±0.02) even when the dopant concentration is increased from 5 wt% to 20 wt%. On the other hand, the EL spectra of devices made with 3, 4 and 5 are strongly dependent on the dopant concentrations. It is believed that the polarity of the gold(III) complexes is higher than that of the mCP host material, as reported in our recent publication,45 leading to a bathochromic shift in the EL spectra, in good agreement with the emission studies where the LLCT band maximum is red-shifted in more polar solvents.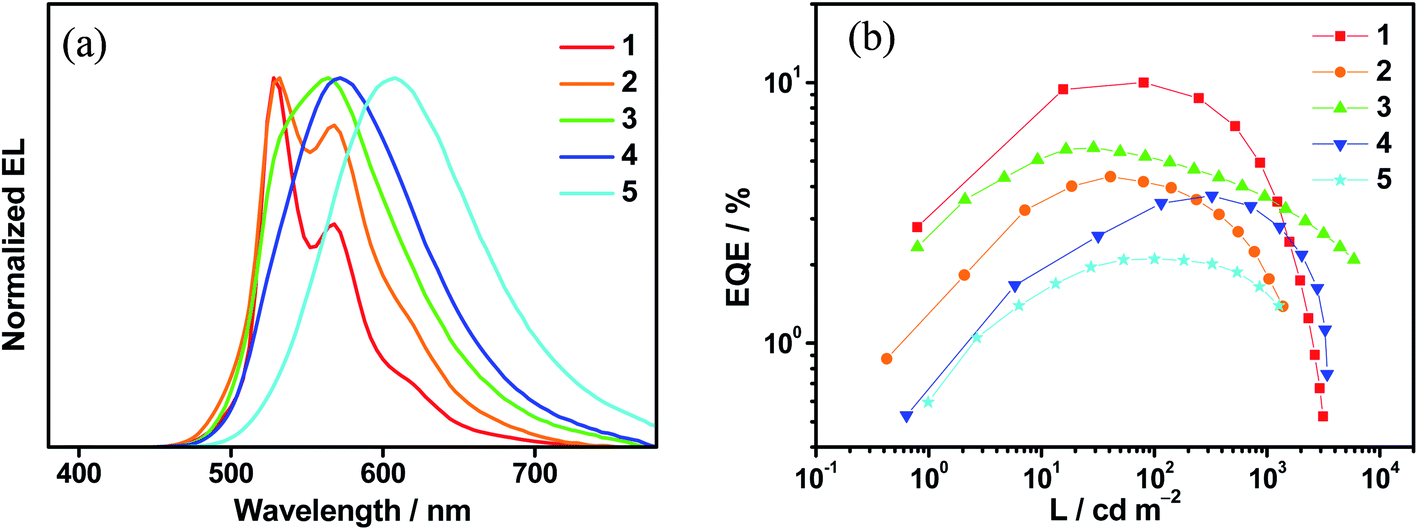 | ||
| Fig. 5 (a) Normalized EL spectra of solution-processed devices based on 10 wt% 1–5. (b) Plots of EQEs as a function of luminance of solution-processed devices based on 10 wt% 1–5. | ||
Satisfactory performance has been achieved for all the solution-processed devices. Table S22† summarizes their key device parameters and Fig. S31–S35† show their current density–luminance–voltage characteristics. Particularly, green-emitting devices based on 1 exhibit the highest current efficiencies of up to 37.0 cd A−1 and EQEs of up to 10.0%. On the other hand, yellow- to red-emitting devices based on 3–5 exhibit maximum EQEs of up to 8.0%. It is found that the presence of TADF properties in the gold(III) complexes can significantly reduce the efficiency roll-offs of the solution-processed devices. Fig. 5b shows the plots of EQEs of the solution-processed devices as a function of luminance. At a luminance of 1000 cd m−2, large efficiency roll-offs of ∼56.4% and ∼59.1% are noted for devices based on 1 and 2, respectively. In sharp contrast, the efficiency roll-offs are greatly reduced to ∼35.5%, ∼17.3% and ∼24.8% for devices made with 3, 4 and 5, respectively. The larger kr in 3–5, which are one to three magnitudes larger than those in 1 and 2, may be one of the factors governing the reduced efficiency roll-offs. Other factors including the molecular properties, destabilization of the HOMO and stabilization of the LUMO, can alter the position of the recombination zone and thus the efficiency roll-offs. Further work is needed to elucidate the underlying mechanism responsible for the reduced efficiency roll-off needs. This work demonstrates the first report of red-emitting devices based on gold(III) TADF emitters, with CIE coordinates of (0.60, 0.40) and a maximum EQE of 5.3%.
Conclusions
To summarize, a new class of fused heterocyclic ligand-containing alkynylgold(III) complexes has been designed and synthesized. These complexes have been found to exhibit tunable PL properties with high PLQYs of up to 0.87 in solid-state thin films. Through a judicious choice of the cyclometalating and alkynyl ligands, delicate control of the energy levels of the IL and LLCT states has been achieved. The larger energy difference between the higher-lying 3IL excited state and the lower-lying LLCT excited state of the complexes leads to a smaller participation of the long-lived 3IL state in the emission of 3–6. As a result, these complexes have shown enhanced emission intensity upon increasing the temperature, with kr on the order of up to 106 s−1, which can be ascribed to the occurrence of TADF. Solution-processed gold(III)-based OLEDs with maximum EQEs of up to 10.0% have been realized, in which the TADF gold(III) complexes can effectively reduce the efficiency roll-offs from ∼60% down to ∼17% at a luminance of 1000 cd m−2. This work further confirms the versatility of the fused heterocyclic alkynyl ligands in delicately controlling the HOMO–LUMO energy gap and demonstrates the realization of TADF alkynylgold(III) complexes and their application in solution-processed OLEDs, providing new insights for the design of gold(III) TADF complexes.Data availability
The datasets supporting this article have been uploaded as part of the supplementary material.Author contributions
V. W.-W. Y. initiated and designed the research. V. W.-W. Y., L.-K. L., M.-C. T. and C. C. A.-Y. designed the gold(III) complexes. C. C. A.-Y. conducted the synthesis, characterization, photophysical and electrochemical measurements of the gold(III) complexes. L.-K. L. and M. N. performed and analyzed the computational calculations. S.-L. L., W.-L. C. and M.-Y. C. carried out the OLED fabrication and characterizations. V. W.-W. Y. supervised the work. All authors discussed the results and contributed to the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
V. W.-W. Y. acknowledges UGC funding provided by the University of Hong Kong (HKU) for supporting the Electrospray Ionization Quadrupole Time-of-Flight Mass Spectrometry Facilities under the support for Interdisciplinary Research in Chemical Science, the HKU Development Fund, and the support from the University of Hong Kong under the Research Committee (URC) Strategically Oriented Research Theme on Functional Materials for Molecular Electronics. The work described in this paper was supported by a grant from Hong Kong Quantum AI Lab Ltd under the AIR@InnoHK administered by the Innovation and Technology Commission (ITC). C. C. A.-Y., L.-K. L. and W.-L. C. acknowledge the receipt of postgraduate studentships from HKU. L.-K. L. acknowledges the receipt of a University Postgraduate Fellowship from the University of Hong Kong. The computations were performed using the HKU ITS research computing facilities. Dr M.-Y. Leung is gratefully acknowledged for her assistance in variable-temperature studies.Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS
.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed
- M. Liu, Y. Seino, D. Chen, S. Inomata, S.-J. Su, H. Sasabe and J. Kido, Chem. Commun., 2015, 51, 16353–16356 RSC
- X.-K. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J.-L. Brédas, Adv. Mater., 2017, 29, 1702767 CrossRef PubMed
- D.-H. Kim, A. D'Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS
- Z. Han, X.-Y. Dong and S.-Q. Zang, Adv. Opt. Mater., 2021, 2100081 CrossRef
- L. Bergmann, G. J. Hedley, T. Baumann, S. Bräse and I. D. W. Samuel, Sci. Adv., 2016, 2, e1500889 CrossRef
- R. Czerwieniec, J. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed
- S. Shi, M. C. Jung, C. Coburn, A. Tadle, M. R. D. Sylvinson, P. I. Djurovich, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 3576–3588 CrossRef CAS PubMed
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed
- R. Hamze, S. Shi, S. C. Kapper, D. S. Muthiah Ravinson, L. Estergreen, M.-C. Jung, A. C. Tadle, R. Haiges, P. I. Djurovich, J. L. Peltier, R. Jazzar, G. Bertrand, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 8616–8626 CrossRef CAS PubMed
- P. J. Conaghan, S. M. Menke, A. S. Romanov, S. T. E. Jones, A. J. Pearson, E. W. Evans, M. Bochmann, N. C. Greenham and D. Credgington, Adv. Mater., 2018, 30, 1802285 CrossRef PubMed
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Inorg. Chem., 2017, 56, 13274–13285 CrossRef CAS PubMed
- M. Osawa, I. Kawata, R. Ishii, S. Igawa, M. Hashimoto and M. Hoshino, J. Mater. Chem. C, 2013, 1, 4375–4383 RSC
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed
- K.-T. Chan, T.-L. Lam, D. Yu, L. Du, D. L. Phillips, C.-L. Kwong, G. S. M. Tong, G. Cheng and C.-M. Che, Angew. Chem., Int. Ed., 2019, 58, 14896–14900 CrossRef CAS PubMed
- H. Mieno, R. Kabe, M. D. Allendorf and C. Adachi, Chem. Commun., 2018, 54, 631–634 RSC
- V. W.-W. Yam, S. W.-K. Choi, T.-F. Lai and W.-K. Lee, J. Chem. Soc., Dalton Trans., 1993, 1001–1002 RSC
- V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107–3110 CrossRef CAS PubMed
- K. M.-C. Wong, X. Zhu, L.-L. Hung, N. Zhu, V. W.-W. Yam and H.-S. Kwok, Chem. Commun., 2005, 2906–2908 RSC
- V. K.-M. Au, K. M.-C. Wong, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 14273–14278 CrossRef CAS PubMed
- B. Y.-W. Wong, H.-L. Wong, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2017, 56, 302–305 CrossRef CAS PubMed
- L.-K. Li, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M. Ng, C. K.-M. Chan, M.-Y. Chan and V. W.-W. Yam, Chem. Mater., 2019, 31, 6706–6714 CrossRef CAS
- M.-C. Tang, C.-H. Lee, M. Ng, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2018, 57, 5463–5466 CrossRef CAS PubMed
- M.-C. Tang, L. H.-Y. Lo, W.-L. Cheung, S.-L. Lai, M.-Y. Chan and V. W.-W. Yam, Chem. Commun., 2019, 55, 13844–13847 RSC
- C.-H. Lee, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2018, 9, 6228–6232 RSC
- M.-C. Tang, M.-Y. Leung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 13115–13124 CrossRef CAS PubMed
- J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold and M. Bochmann, Chem. Commun., 2015, 51, 16629–16632 RSC
- W.-P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 14036–14041 CrossRef CAS PubMed
- D. Zhou, W.-P. To, Y. Kwak, Y. Cho, G. Cheng, G. S. M. Tong and C.-M. Che, Adv. Sci., 2019, 6, 1802297 CrossRef CAS PubMed
- D. Zhou, W.-P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips and C.-M. Che, Angew. Chem., Int. Ed., 2020, 59, 6375–6382 CrossRef CAS PubMed
- V. K.-M. Au, D. P.-K. Tsang, K. M.-C. Wong, M.-Y. Chan, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2013, 52, 12713–12725 CrossRef CAS PubMed
- M.-C. Tang, C.-H. Lee, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 9341–9349 CrossRef CAS
- M. Aydemir, S. Xu, C. Chen, M. R. Bryce, Z. Chi and A. P. Monkman, J. Phys. Chem. C, 2017, 121, 17764–17772 CrossRef CAS
-
T. Palmeira and M. N. Berberan-Santos, in Highly Efficient OLEDs, ed. H. Yersin, Wiley-VCH, 2019, TADF Kinetics and Data Analysis in Photoluminescence and in Electroluminescence, pp. 229–255 Search PubMed
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed
- M.-Y. Leung, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2020, 142, 2448–2459 CrossRef CAS PubMed
- K. T. Chan, G. S. M. Tong, W.-P. To, C. Yang, L. Du, D. L. Phillips and C.-M. Che, Chem. Sci., 2017, 8, 2352–2364 RSC
- D. S. M. Ravinson and M. E. Thompson, Mater. Horiz., 2020, 7, 1210–1217 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic route for cyclometalated gold(III) complexes; thermogravimetric analysis; electrochemical studies; photophysical studies; computational studies; and electroluminescence studies (PDF). See DOI: 10.1039/d1sc02256c |
| ‡ It is worth noting that in this series of gold(III) complexes, prompt fluorescence is not readily observable. Unlike pure organic TADF molecules which usually exhibit distinct lifetimes for prompt (in ns range) and delayed (in μs range) emissions, with an intersystem crossing (ISC) rate constant kISC < 107 s−1, these gold(III) complexes exhibit a large spin–orbit coupling (SOC) constant associated with the heavy metal center, which would promote the rate of ISC from the S1 to the T1 state. ISC for TADF gold(III) complexes has been found to occur in the range of 0.2–13 ps.46 Such a high ISC rate constant (as high as 1010 s−1) is fast enough to outcompete the radiative decay of the S1 state, channeling the S1 state to the T1 state, which then undergoes facile RISC to give short-lifetime TADF and thus prompt fluorescence was not observed. Different from the pure organic TADF compounds in which the kISC and kRISC are merely governed by ΔE(S1–T1), the large SOC constant associated with a heavy gold metal center would also be a major factor that promotes the rate of ISC from the S1 to the T1 state. As both kISC and kRISC are fast and the ΔE(S1–T1) is small, a fast thermal equilibration between the S1 and T1 states will occur. This can be described by the equilibrium constant, Keq, and the ratio of kRISC to kISC.47 It has been found that different metal-containing TADF complexes can have very fast and similar kISC and kRISC values, but very different Keq values, in which a smaller kRISC-to-kISC ratio corresponds to a larger ΔE(S1–T1) value.47 Thus, the main contribution of the fast kRISC and kISC originates from the larger SOC constant with less significant contribution from the ΔE(S1–T1) value. |
| § The two-level Boltzmann model was not preferred as large uncertainties may be introduced since the fitting would be affected largely by the drastic increase in the lifetime at the low temperature region due to the involvement of the long-lived 3IL excited state. |
| This journal is © The Royal Society of Chemistry 2021 |