
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent probes for visualizing ROS-associated proteins in disease
Hui
Wang
a,
Xin
Wang
a,
Ping
Li
 *a,
Mingyan
Dong
a,
Shao Q.
Yao
*b and
Bo
Tang
*a
*a,
Mingyan
Dong
a,
Shao Q.
Yao
*b and
Bo
Tang
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Institute of Biomedical Sciences, Shandong Normal University, Jinan 250014, P. R. China. E-mail: lip@sdnu.edu.cn; tangb@sdnu.edu.cn
bDepartment of Chemistry, National University of Singapore, Singapore 117543, Singapore. E-mail: chmyaosq@nus.edu.sg
First published on 24th August 2021
Abstract
Abnormal expression of proteins, including catalytic and expression dysfunction, is directly related to the development of various diseases in living organisms. Reactive oxygen species (ROS) could regulate protein expression by redox modification or cellular signal pathway and thus influence the development of disease. Determining the expression level and activity of these ROS-associated proteins is of considerable importance in early-stage disease diagnosis and the identification of new drug targets. Fluorescence imaging technology has emerged as a powerful tool for specific in situ imaging of target proteins by virtue of its non-invasiveness, high sensitivity and good spatiotemporal resolution. In this review, we summarize advances made in the past decade for the design of fluorescent probes that have contributed to tracking ROS-associated proteins in disease. We envision that this review will attract significant attention from a wide range of researchers in their utilization of fluorescent probes for in situ investigation of pathological processes synergistically regulated by both ROS and proteins.
 Hui Wang | Hui Wang received her PhD degree from Nankai University in 2013. She is an associate professor in the College of Chemistry at Shandong Normal University. Her current research interests include design and synthesis of dual-mode fluorescent probes for enzymes and fluorescence imaging of Golgi biomarkers in disease. |
 Xin Wang | Xin Wang received her PhD degree at the college of Chemistry, Chemical Engineering and Materials Science in Shandong Normal University in 2020. Her research interests are focused on two-photon fluorescence imaging of depression related active molecules in vivo. |
 Ping Li | Ping Li received her PhD degree in 2008 from Shandong Normal University. In 1998, she joined the faculty at Shandong Normal University, where she is currently a professor of the College of Chemistry, Chemical Engineering and Materials Science. She is Taishan Distinguished Professor and the leader of the Changjiang Scholars and Innovative Research Team in University. Her research interests include synthesis and bioimaging application of fluorescent probes for biologically active molecules. |
 Mingyan Dong | Mingyan Dong is currently pursuing a masters degree at the college of Chemistry, Chemical Engineering and Materials Science in Shandong Normal University. Her research interests are focused on two-photon fluorescence imaging of intracellular microenvironment. |
 Shao Q. Yao | Shao Q. Yao received his PhD from Purdue University (1998) and post-doctoral training (1998–2001) from University of California, Berkeley and The Scripps Research Institute. He joined National University of Singapore (NUS) in 2001 and is currently a Full Professor at the Department of Chemistry, NUS. His current research interest is in catalomics – proteome-wide interrogation of enzymes and their associated molecules by using techniques in chemistry, biology and materials science. He is currently on the scientific advisory board of numerous international journals. |
 Bo Tang | Bo Tang is now a professor of chemistry at Shandong Normal University. He received his PhD degree in 1994 from Nankai University. He began his independent career as a professor of chemistry at Shandong Normal University in 1994. He obtained the National Fund for Outstanding Young Scientists in 2007, and was a Chief Scientist for the 973 Program in 2012. His research interests include the synthesis of molecular and nano probes and the application in biological imaging, green chemical production, synthesis of fluorescent materials, solar chemical conversion and storage and so on. He has contributed more than 300 journal articles, as well as 21 invited book chapters and reviews, and obtained 39 granted patents. |
1. Introduction
All cells depend on preserving protein homeostasis to maintain the correct biological functions. In response to pathological stimuli, hundreds of proteins are aberrantly expressed, accompanied by abnormal changes in the activities of numerous enzymes in cells. Abundant reports have suggested that neurodegenerative disorders, such as Alzheimer's disease (AD) and prion disease, share similar cellular and molecular mechanisms, including deviant protein aggregation and deposition.1,2 The development and metastasis of cancer is also directly related to abnormal expression levels of proteins including up/down-regulated activities of enzymes.3–5 Take cathepsins for example: many of these enzymes exhibit high proteinase activity in tumour cells and participate in angiogenesis and cell invasion.6 The activity of matrix metalloproteinases, another class of well-known enzymes, is also elevated in many tumour cells to degrade extracellular matrix proteins and promote tumour metastasis.7A series of oxygen metabolites, including superoxide anions (O2˙−), hydrogen peroxide (H2O2), hydroxyl radicals (˙OH), organic peroxides and so on, collectively known as reactive oxygen species (ROS), are present in mammalian cells.8,9 Intracellular ROS are constantly generated at low concentrations, which can regulate protein expression by redox modification or cellular signal pathways.10,11 When cells undergo oxidative stress, amino acid residues in many susceptible proteins may be damaged by oxidative outburst. For instance, ROS can convert the side chains of certain amino acid residues (i.e., Pro, Arg and Lys) into glutamic semialdehydes and aminoadipic semialdehydes by iron-catalyzed oxidation.12 ROS also mediate the formation of 3-nitrotyrosine in proteins by nitration of tyrosine residues. These oxidative modifications might destroy the targeted protein structures and affect their functions, consequently affecting metabolism, protein folding and degradation of the false proteins. Eventually, various serious diseases may occur as a result.
Therefore, it is vitally necessary to accurately detect the expression and activity of ROS-associated proteins in disease in vivo. This will enable identification of damaged metabolic networks at early stages of diseases, as well as their pathogenesis, leading to acceleration of early diagnosis of major diseases and their drug development.
Although several approaches have been employed to assess the level of protein expression, such as two-dimensional electrophoresis (2-DE), immunoblotting and immunofluorescence techniques, they have severe limitations. (1) In immunoblotting and 2-DE, the protein of interest (POI) needs to be extracted from cells or tissue homogenates, which may degrade due to temperature, pH and protease, leading to inaccurate test results. (2) Immunofluorescence requires specific binding of fluorophore labelled antibodies to their antigens and measures the expression levels of proteins based on fluorescence signals. Although protein extraction is avoided in immunofluorescence, this technique can only be performed on dead cells or fixed tissue slices, which could not be implemented for noninvasive studies of cells and intact organisms. In summary, these methods could not be applied for accurate in situ protein detection in vivo.
Recently, fluorescent probes have been widely used in conjunction with fluorescence imaging techniques to track molecular events in living cells. Fluorescence imaging is a noninvasive approach for in situ detection of various biomolecules.13,14 So, by using fluorescence-based probes, the expression level and catalytic activity of ROS-associated proteins could be continuously monitored in situ in living cells on a real-time scale, which demonstrates broad application prospects compared with traditional methods. In this review, to provide strategies used in the design of fluorescence probes for ROS-relevant proteins and their application to the detection of disease, we summarize recently published fluorescent probes for the optical detection of ROS-associated proteins in disease. Considering proteins with and without catalytic activity, we clarify the difference between design strategies of fluorescent probes for them. Subsequently, according to different diseases, recent examples and applications of fluorescent probes for the measurement of ROS-associated proteins in the past decade will be introduced. Finally, with the aim of exploiting more powerful imaging tools in this area, we will give a perspective on further challenges and opportunities in this field.
2. Classification of proteins and probe design strategies
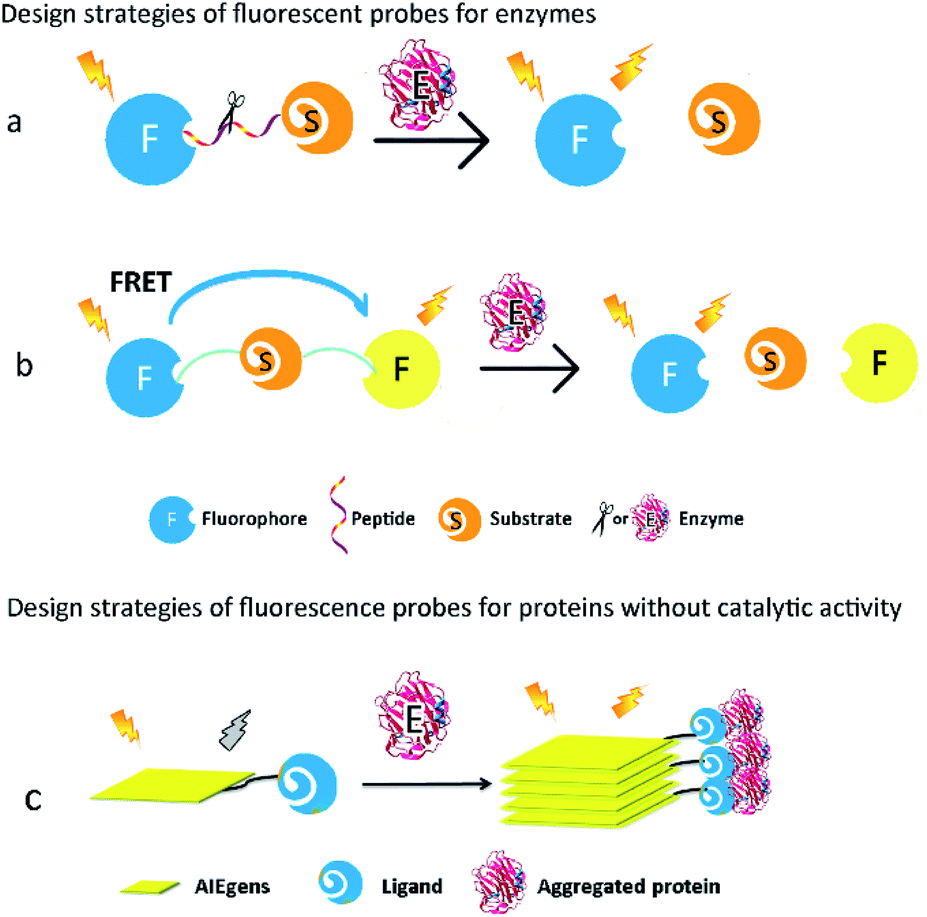
Proteins are major targets of ROS. Some ROS-induced protein modifications can result in unfolding or alteration of protein structure and activity. For example, ROS-mediated S-glutathionylation, nitrosation, and carbonylation play significant roles in the modulation of protein function in cells. The overall biology of oxidative protein modifications however remains complicated and undefined. Thus, development of fluorescent probes for detection of ROS-associated proteins could be quite beneficial for a better understanding of oxidative-damaged proteins in various diseases. Typically, proteins can be broadly divided into two groups, one with catalytic activity (the so-called enzymes) and the other without catalytic activity. According to their different characterizations, feasible strategies should be proposed for the design of their corresponding fluorescent probes.For enzymatic activity-based fluorescent probes, the covalent bond is cleaved/formed or the functional group transformed into a new group under specific enzyme-catalysed conditions. These reactions will change the optical properties of the probe, such as different fluorescence intensities or emission wavelengths, and consequently report the activity of the enzyme.15 However, the chemical structures of the probes would not be altered when they encounter proteins without catalytic activity, but the microenvironmental changes strongly affect the photophysical properties or the aggregated states of the probes. These alterations are accompanied by changes in the fluorescence intensity of the probe, leading to detection and monitoring of the protein expression level.
2.1. Design strategies of fluorescent probes for proteins with catalytic activity
Most ROS-associated enzymes could be classified into different classes according to the different chemical reactions they catalyse, such as oxido-reductases, hydrolases, and transferases. Oxido-reductases can catalyse the transfer of electrons between metabolites in order to maintain redox homeostasis. Numerous oxido-reductases exhibit direct relationships with various human diseases. For instance, monoamine oxidases are overexpressed in Alzheimer's disease. In addition, nitroreductases play essential roles in tumour metastasis. Hydrolases, including lipases, nucleosidases, and peptidases, are a class of enzymes that catalyse the hydrolysis of some chemical bonds, and are widely used as disease biomarkers. The activities of caspases, metallomembrane proteases, and aminopeptidases, all of which are sub-classes of peptidases, are common indicators of cancer. Transferases can transfer specific functional groups from the donor to acceptor substances, and among them, glycosyl transferases are involved in transferring monosaccharides in metabolisms.Since enzymes show high specificity toward substrates in catalytic reactions, specific chemical structures or chemical bonds could be recognized. For example, glucosidases just hydrolyse the glucose-formed glucosidic bond. On the basis of enzymes' features, various fluorescent probes have been synthesized for the assessment of different catalytic activities in numerous biological processes in the past decade, especially those relevant to major human diseases.
How to regulate optical properties of a fluorescent probe before and after it is catalysed by the target enzyme is the key factor in the probe design. In general, fluorescence properties of the probes are tuned by intermolecular charge transfer (ICT), intermolecular photoinduced electron transfer (PeT), fluorescence resonance energy transfer (FRET), and so on (Fig. 1). When enzymatic reactions cause the cleavage of covalent bonds or the alteration of functional groups in the probes, fluorescence signals emitted by the probes can be subsequently changed, leading to eventual qualitative or quantitative detection of the target enzymes.
 | ||
| Fig. 1 Design strategies of fluorescent probes for proteins with or without catalytic activity. | ||
2.2. Design strategies of fluorescent probes for proteins without catalytic activity
Numerous proteins without catalytic activity participate in cellular metabolism, and are also closely associated with various diseases caused by aggregation of proteins such as β-amyloid (Aβ) and tau. The expression levels of these proteins fluctuate during the course of pathology development, and thus have emerged as potential diagnostic biomarkers of these diseases. Since these proteins do not possess any catalytic activity and cannot catalyse the cleavage or formation of covalent bonds, they can't alter the molecular structures of a fluorescent probe. Thus, to image these proteins, the design of the corresponding fluorescent probes requires very different strategies from those for the enzymatic probes. Fluorescent probes based on the aggregation-induced emission (AIE) mechanism and environmental-sensitive strategies have been developed in the past decade.16,17AIE-based fluorescent probes are composed of two parts, namely, AIE luminogens (AIEgens) and a specific ligand of target proteins (Fig. 1c). The connection between AIEgens and the ligand is classified into either a covalent bond or noncovalent binding. The latter usually connects AIEgens and the ligand by hydrogen bonding, ionic bonding, and hydrophobic interaction. A ligand with high affinity and specificity forces AIEgens of the fluorescent probes to accumulate at the proximity of aggregated proteins. AIEgens are nonemissive in the dispersed state as earlier mentioned, due to their ability to consume the excited-state energy through intramolecular motion upon being photo-excited. In their aggregated state on the other hand, AIEgens emit intense fluorescence because the intramolecular motion is spatially restricted.18 Therefore, AIE-based fluorescent probes can afford useful imaging information of aggregated peptides or proteins, especially those commonly found in neurodegenerative diseases, such as Aβ and tau.19,20
For proteins whose expression levels are clearly affected by their surrounding environment, such as polarity and viscosity, environment-sensitive fluorescent probes are often used to detect them.21 These probes contain either a donor–acceptor or donor–acceptor–donor, which is connected by a conjugated π-bridge. Depending on whether intramolecular rotation is limited, the probe's coplanarity differs in the excited state upon successful binding to the target protein. Better coplanarity of the fluorescent probe causes a strong fluorescence change or red-shift of the emission wavelength, which can be effectively correlated with the expression level of the target protein.22 Because the aggregation of Aβ and tau dramatically alters these proteins' surrounding polarity and viscosity in cells, many environment-sensitive fluorescent probes, designed for imaging of these proteins based on changes in their intramolecular rotation, have been reported in the past decade.23–25
3. Fluorescent probes for ROS-associated proteins in diseases
Many post-translational modifications of proteins could be controlled or caused by ROS. For instance, the sulfhydryl group in cysteine residues of proteins can be oxidized to form an intramolecular disulphide bridge by H2O2 or sulphur reactive radicals. Various diseases, including cancer (e.g., hepatic carcinoma), cerebral disease (e.g., depression), cardiovascular diseases (e.g., ischemia reperfusion), fibrotic diseases (e.g., pulmonary fibrosis), and inflammations (e.g., arthritis), have been reported to possess direct relevance to the expression level or the activities of critical ROS-associated proteins.263.1. Cancer
ROS can promote tumour development and progression in various types of cancers. In cancer cells, high levels of ROS are generated as a result of increased enzymatic activities in oxidases, cyclooxygenases, and lipoxygenases. The excess ROS overwhelms the antioxidant systems (e.g., coenzyme Q, ferritin, glutathione, superoxide dismutase, thioredoxins and peroxidases), leading to oxidative stress.27,28 In order to maintain the delicate balance of intracellular ROS levels, the expression level of antioxidant proteins (such as thioredoxins and peroxidases) in tumour cells is elevated. Moreover, many proteins involved in cell proliferation, differentiation, biomacromolecule synthesis, tissue invasion and metastasis, are also known to be ROS-sensitive in various cancers.29DEVD (Asp–Glu–Val–Asp) is the most commonly used peptide substrate to be specifically hydrolyzed by caspase-3,39 but DEVD is also the optimum substrate for caspase-7. Therefore, the fluorescence probes that are designed based on DEVD as the substrate to detect caspase-3/7 cannot selectively respond to caspase-3 or caspase-7. Nevertheless, several fluorescence probes were still developed by introducing DEVD into the probe to detect caspase-3.
Several small-molecule fluorescence probes for detection of caspases have been reported.40,41 Kim et al. described a water-soluble fluorescent probe (1, Ac-DEVD-NH-CV) responsive to caspase-3 (Fig. 2). After being cleaved by caspase-3, the probe (1) was converted into hydrophobic CV-NH2, which would aggregate and emit strong fluorescence. This probe was an attractive tool to study caspase-3 in vitro and in vivo.42
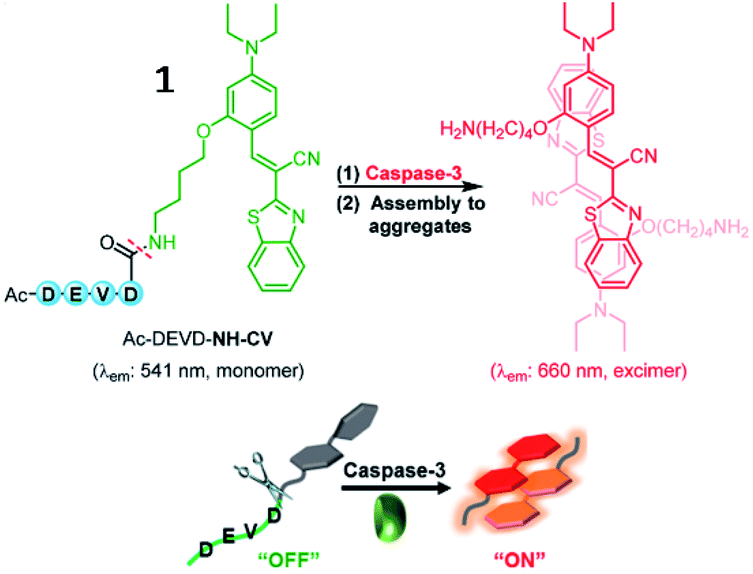 | ||
| Fig. 2 Proposed turn-on sensing of probe 1 for caspase-3. Reprinted from ref. 42 with permission. Copyright 2017 American Chemical Society. | ||
Jiang et al. developed a small-molecule probe (2) for fluorescence imaging of caspase-8 activity through a thiol-ene click reaction (Fig. 3).43 Upon reaction with caspase-8, the probe (2) was cleaved at the peptide bond between cysteine and the caspase-8-specific substrate, releasing the fluorophore. Due to the intramolecular hydrogen bonds, probe 2 locally aggregated and emitted bright fluorescence at 520 nm. This is the first fluorescent probe based on a thiol-Michael addition click reaction for bioimaging of caspase.
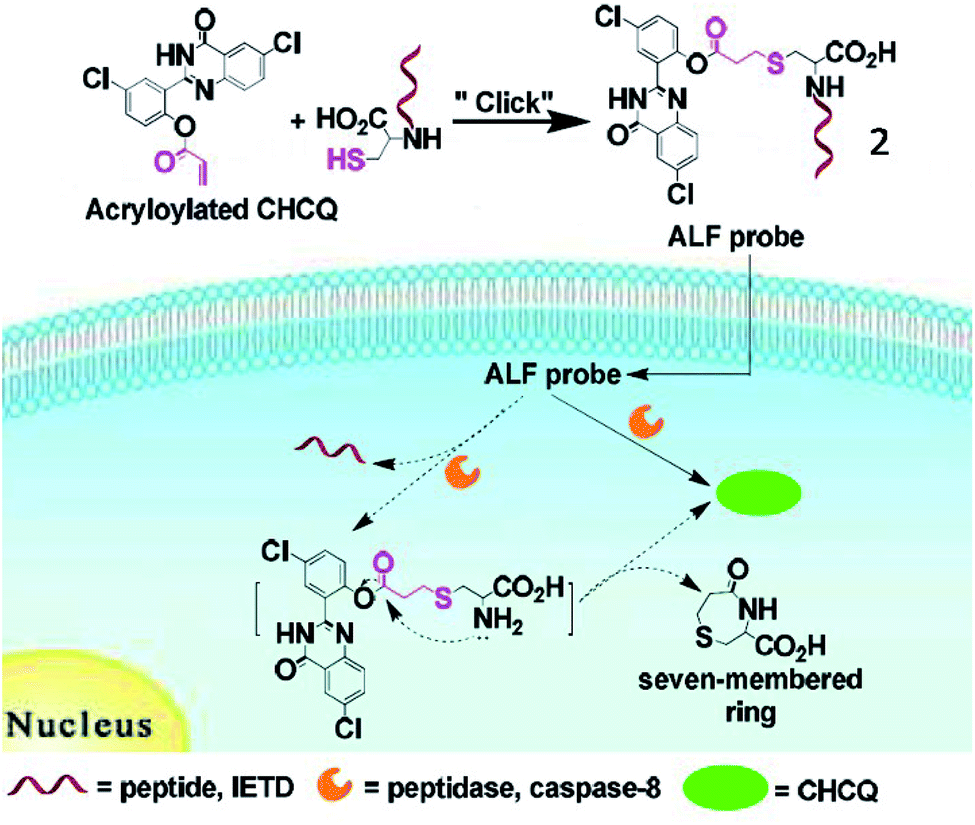 | ||
| Fig. 3 Illustration of the activity localization fluorescence peptide probe 2 for peptidase imaging. Reprinted from ref. 43 with permission. Copyright 2016 American Chemical Society. | ||
Based on the cyclization reaction between free cysteine and 2-cyano-6-hydroxyquinoline (CHQ), Rao and Ye et al. explored a self-assembled fluorescent small-molecule probe (3, C-SNAF) for the detection of caspase-3/7 (Fig. 4). The cyclization reaction between free cysteine and CHQ is a biocompatible, kinetically fast intramolecular condensation (6 × 10−3 s−1).37 After reaction with caspase-3/7 and GSH, the amino and thiol groups of the D-cysteine masked by DEVD and disulfide bonds are released, then, C-SNAF undergoes macrocyclization and in situ nanoaggregation, which leads to long residence time and enhanced fluorescence. The caspase-3/7-triggered nanoaggregation of probe 3 enables effective imaging of caspase-3/7 activities in vivo and can be used to monitor tumour therapy responses. This is a novel strategy for directing the in situ self-assembly of synthetic small molecules into nanoaggregates in living systems.
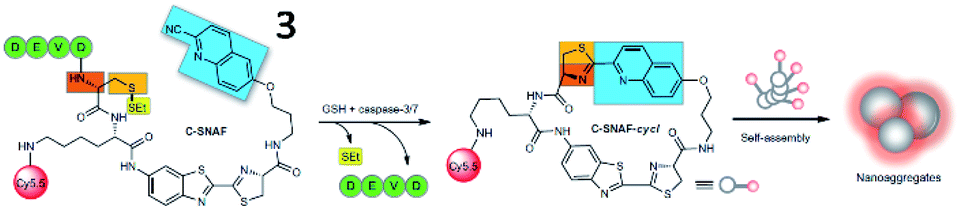 | ||
| Fig. 4 Proposed caspase-3/7 and reduction-controlled conversion of C-SNAF into C-SNAF-cycl through the bioorthogonal intramolecular cyclization reaction, followed by self-assembly into nanoaggregates in situ. Reprinted from ref. 37 with permission. Copyright 2014 Macmillan Publishers Limited. | ||
Recently, Ye et al. reported a photoacoustic (PA) probe (4, 1-RGD) for caspase-3 detection based on the same self-assembly strategy (Fig. 5).44 The probe (4) consists of a CHQ and a D-cysteine residue, a caspase-3-cleavable peptide substrate DEVD, a glutathione (GSH)-reducible disulfide bond, a tumour tissue-targetable cyclic peptide (c-RGD) and a clinically used near-infrared (NIR) dye (indocyanine green, ICG). Probe 4 can target tumor tissues, since c-RGD targets the αvβ3 integrin receptor which is overexpressed in tumor cells. Upon interaction with GSH and caspase-3, free Cys and CHQ underwent fast intramolecular condensation to form a cyclized product, subsequently inducing stronger intermolecular interactions to promote molecular self-assembly into nanoparticles. Thus, probe 4 could realize both PA and fluorescence imaging of caspase-3 activity and distribution within tumour tissues. This work reports the first smart PA probe for real-time monitoring of caspase-3 activity in deep tissues.
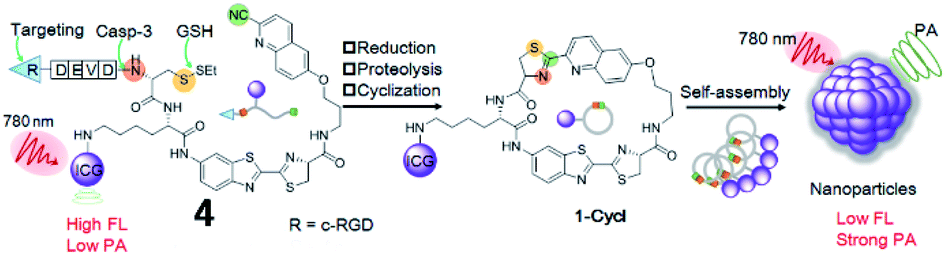 | ||
| Fig. 5 Development of a PA imaging probe 4 for detecting caspase-3 activity. Reprinted from ref. 44 with permission. Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Several AIE-based fluorescent “turn-on” probes for caspase-3 detection have been reported by Tang and co-workers (Fig. 6). In 2012, probe 5 consisting of a DEVD peptide and an AIEgen (TPE, tetraphenylethene) was synthesized to monitor caspase-3/-7 activities from mammalian cells in real time.45 In the presence of caspase-3/-7, the peptide was cleaved from TPE, resulting in fluorescence recovery of TPE due to the AIE effect. This work provides an effective strategy for real-time imaging of live-cell apoptosis. However, its short emission wavelength limited its application in imaging of apoptosis in vivo.
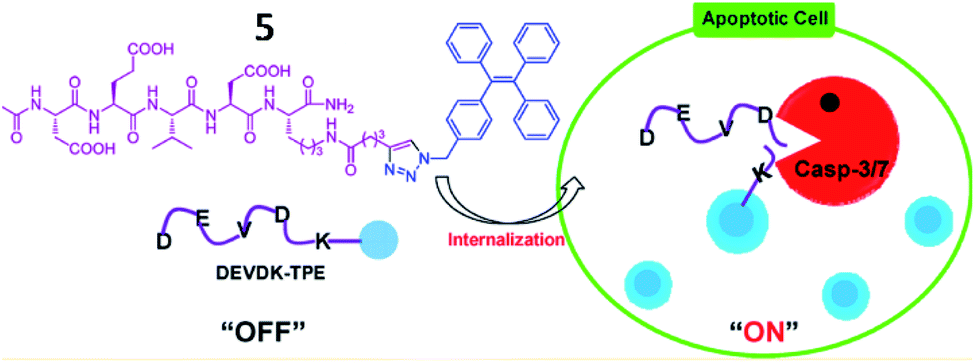 | ||
| Fig. 6 Illustration of probe 5 for caspase-3/-7 activities. Reprinted from ref. 45 with permission. Copyright 2012 American Chemical Society. | ||
In 2016, another FRET- and AIE-based fluorescent probe (6) for caspase-3 detection was developed (Fig. 7).46 Coumarin chromophore was used as the energy donor and linked with an AIEgen (as an energy acceptor) through the DEVD sequence. Upon encountering caspase-3, the AIEgen was enzymatically released and subsequently aggregated, inducing the recovery of coumarin fluorescence and AIEgen fluorescence. This probe provides “turn-on” of two fluorescence signals during analyte identification, which was used for caspase-3-related drug screening.
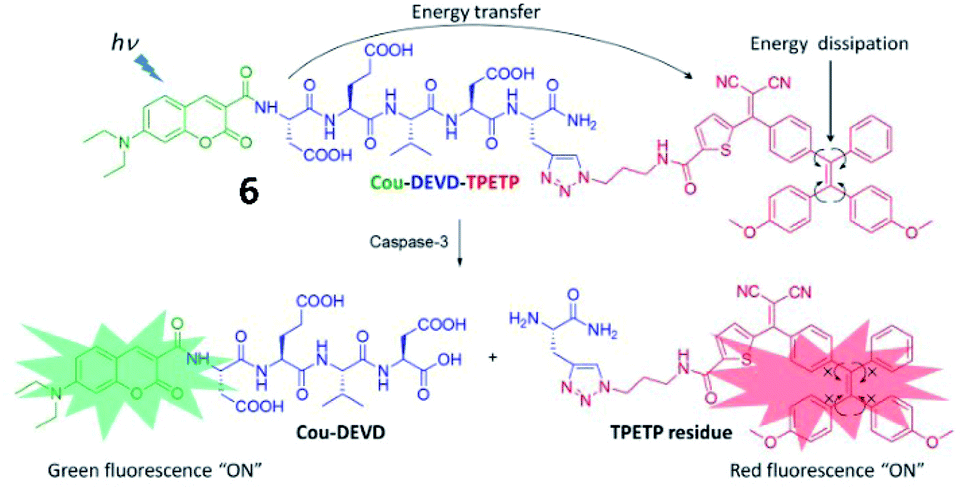 | ||
| Fig. 7 Schematic illustration of the FRET probe using AIEgen as an energy quencher with dual signal output for self-validated caspase-3 detection. Reprinted from ref. 46 with permission. Copyright 2016 The Royal Society of Chemistry. | ||
Tang et al. synthesized a multicolour fluorescent nanoprobe (7) for the imaging of caspase-3, -8 and -9 simultaneously during cell apoptosis (Fig. 8a). By attaching caspase peptide substrates labelled with three different fluorophores onto gold nanoparticles through Au–Se bonds, nanoprobe 7 could synchronously visualize the activations of caspase-3, -8, and -9. Utilizing this nanoprobe, the authors revealed that caspase-8 and caspase-9 could activate caspase-3 during the cell apoptosis process. This nanoprobe is a promising tool for real-time monitoring of caspase cascade activations.47 Based on this work, the Tang group also developed another Au–Se-bonded nanoprobe (8) for fluorescence imaging of caspase-3 and cathepsin B (Fig. 8b). In the presence of caspase-3 and cathepsin B, the peptide chains with fluorophores were specifically cleaved by caspase-3 or cathepsin B, leading to fluorescence recovery. This work confirmed that cathepsin B and caspase-3 were sequentially activated in cancer cells during apoptosis. Both probes 7 and 8 could be effective tools for understanding the cell apoptotic mechanisms.48
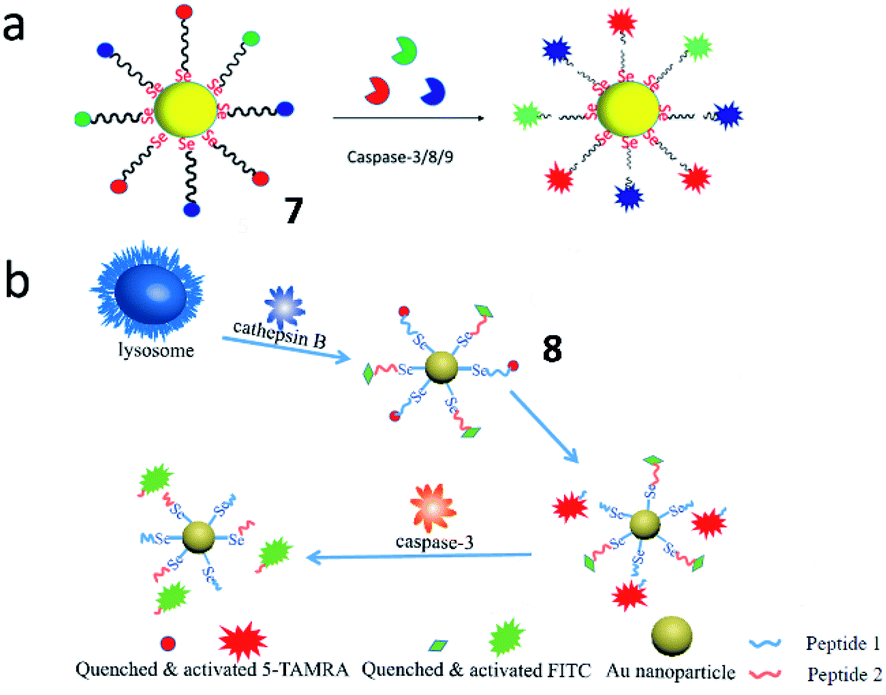 | ||
| Fig. 8 (a) The structure of probe 7 for monitoring the activities of caspase-3, -8 and -9 simultaneously. Reprinted from ref. 47 with permission. Copyright 2019 American Chemical Society. (b) The structure of probe 8 for monitoring the activities of caspase-3 and cathepsin B. Reprinted from ref. 48 with permission. Copyright 2019 Elsevier Ltd. | ||
Li et al.55 reported three ratiometric fluorescent probes 9–11 for detecting the activities of APN (Fig. 9). They chose 1,8-naphthalimide, a prototype ICT fluorophore as a reporter, and alanine and norvaline as the recognition moieties. After the cleavage of the amide bond between the amino acids and the fluorophores by APN, 1,8-naphthalimide can be released and the ICT effect is enhanced.
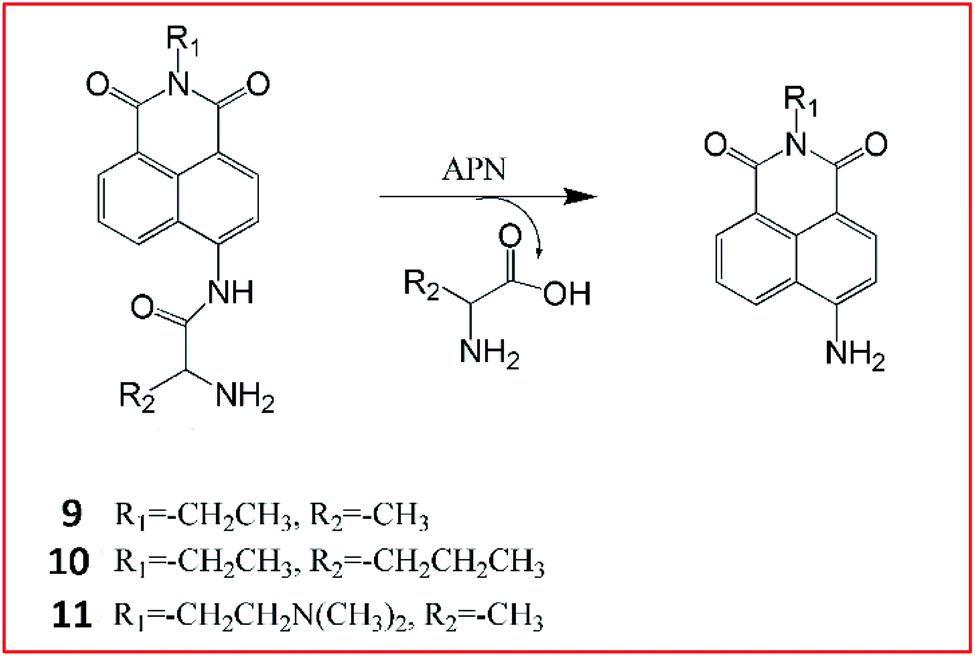 | ||
| Fig. 9 Detection mechanism of probes 9–11 for detecting the activities of APN. | ||
The maximum fluorescence emission redshifts from blue fluorescence to a strong bright green fluorescence.55
Ma et al. developed an ultrasensitive ratiometric fluorescent probe (12, CVN) to visualize APN (Fig. 10a).56 The amino substitution by alanine caused a distinct blue-shift of the fluorescence peak from 626 to 575 nm. After reaction with APN, the fluorescence intensity of the probe at 575 nm decreased and that at 626 nm increased gradually. The detection limit of CVN was determined to be 33 pg mL−1, suggesting that 12 was an ultrasensitive probe for detecting APN, making it the most sensitive APN-detecting fluorescent probe known. The same group further explored another NIR fluorescent probe (13, HCAN) for detection of APN in 2017 (Fig. 10b).57 The NIR dye hemicyanine was used as the fluorescence-reporting moiety and an alanine residue was taken as the recognition group. The amino substitution by alanine efficiently quenches the fluorescence of hemicyanine, since the electron-withdrawing effect of the amide bond disturbs the conjugation of the fluorophore. After reaction with APN, the amide bond is cleaved to release the fluorophore and emit strong fluorescence. Probe 13 was the first NIR-based fluorescent probe for imaging of APN with high selectivity and sensitivity.
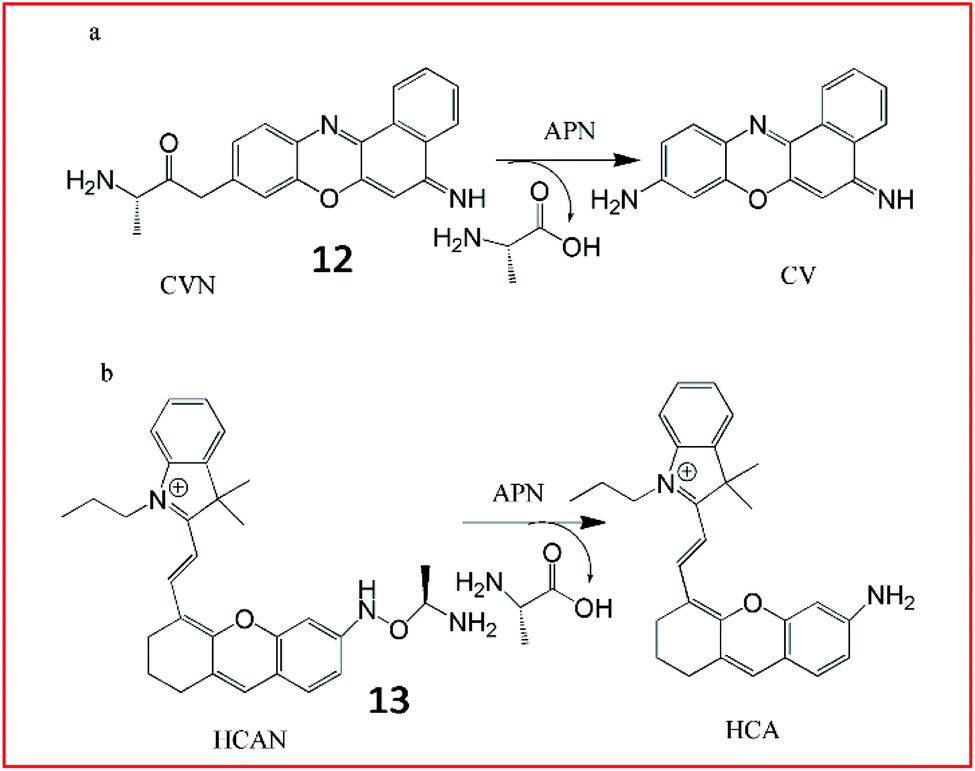 | ||
| Fig. 10 Structure of probes 12 and 13 and detection mechanism with APN. | ||
For tracking metastatic cancer and imaging tumours during surgery, Peng and Yoon et al. developed an APN-activatable fluorescent probe (14, YH-APN; Fig. 11a).58 The probe was constructed with a dicyanoisophorone as the fluorescence-reporting group and L-alanine as the recognition group for APN, which emitted strong NIR fluorescence upon APN cleavage. The probe (14) was able to monitor the activity of APN in cancer cells with ultrasensitivity and excellent selectivity. This probe could also be successfully lighted up in tumour cells, exhibiting great potential for image-guided surgery.
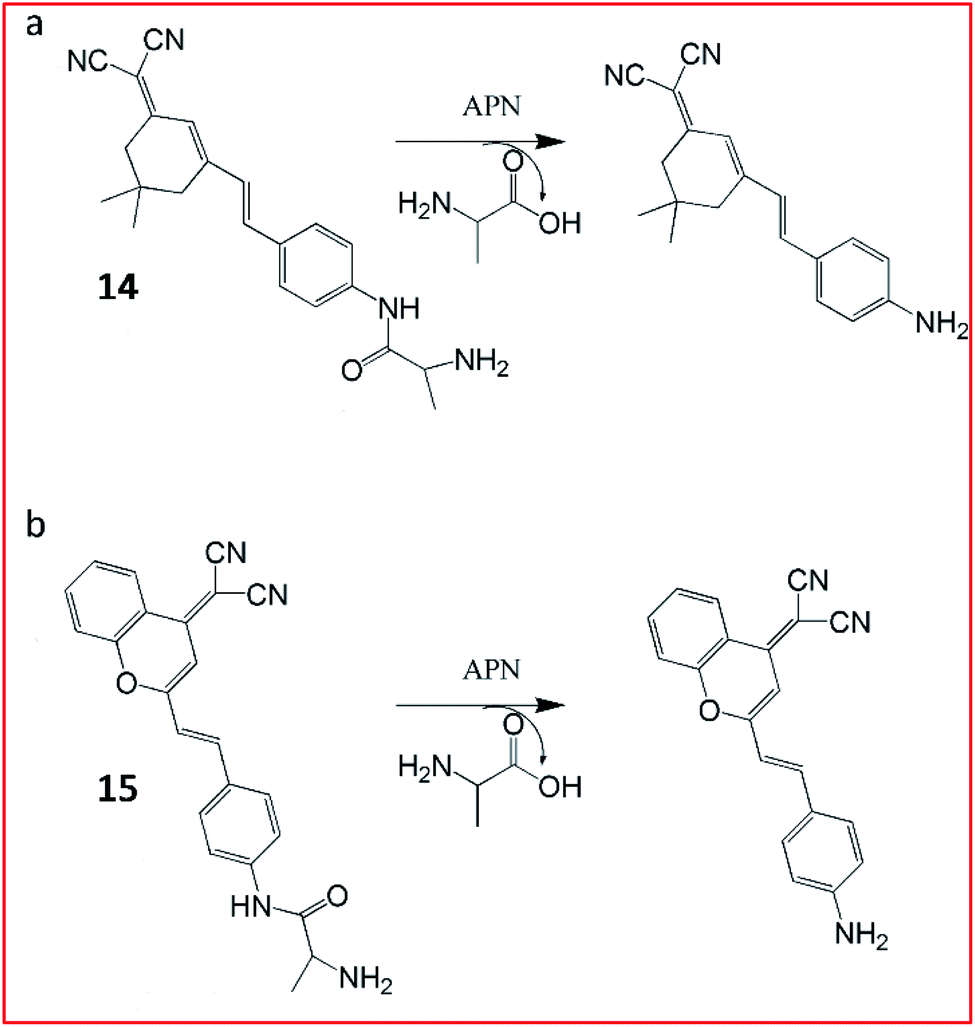 | ||
| Fig. 11 Structure of probes 14 and 15 and their detection mechanism with APN. | ||
Peng et al. reported a two-photon NIR fluorescence probe (15, DCM-APN) to track APN in vivo (Fig. 11b).59 The probe contained an L-alanine as the substrate of APN and a dicyanomethylene-benzopyran (DCM) as the chromophore. Probe 15 had been successfully employed to distinguish normal cells (LO2 cells) from cancer cells (HepG-2 and B16/BL6). Furthermore, the probe could be used to investigate the activity of APN in hepatocellular carcinoma tissues in situ. The NIR excitation and emission wavelengths provided the deep-tissue imaging opportunity for the probe.
Consequently, several ROS-associated MMPs are considered potential diagnostic and prognostic biomarkers for cancers.64–66
Until now, a large number of MMP-responsive fluorescent probes for imaging have been successfully developed, all of which were designed by incorporating chromophore-modified substrates and a quencher.67 Youn et al. developed a soluble probe, 16, for detecting the activity of MMP-3 from serum to monitor the progress of rheumatoid arthritis (Fig. 12).68 This MMP-3 probe was constructed with a peptide (GVPLSLTMGK) modified with a dark quencher 3 (BHQ-3) and an NIR fluorophore (Cy5.5) and glycol chitosan (GC). After being specifically cleaved by MMP-3, Cy5.5 was released and the NIR fluorescence signal in the probe was restored. This MMP-3 probe was used to selectively detect the activity of MMP-3 in diluted serum from CIA mice.
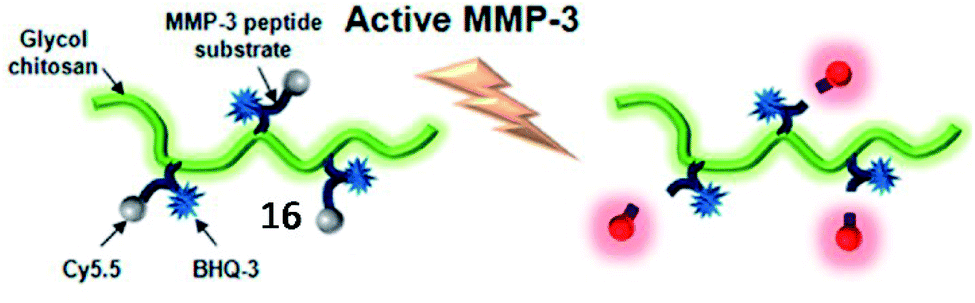 | ||
| Fig. 12 Schematic diagram of the MMP-3 probe 16. When MMP-3 encounters the specific substrate recognition site of the probe, a pronounced NIR fluorescence signal recovery is observed. Reprinted from ref. 68 with permission. Copyright 2013 American Chemical Society. | ||
Chen et al. developed an MMP-13 probe (17, D-MMP-P12) to image MMP-13 activity in vivo. D-MMP-P12 contained a Cy5.5 dye, an MMP substrate (GPLGVRGKGG), a quencher BHQ-3, and a polyethylene glycol (PEG)-12 chain (Fig. 13).69 Specific cleavage of the peptidase substrate in D-MMP-P12 by MMP-13 generated strong fluorescence enhancement of Cy5.5. This probe could be used to visualize MMP-13 activity in squamous cell carcinoma (SCC)-7 tumour-bearing mice. A similar strategy was employed by Urano and coworkers, who designed a FRET-based NIR fluorescent probe 18 for the determination of MMP activities (Fig. 14).70 Upon being cleaved by MMPs, the fluorescent dye was cleaved off the probe, departed from the quencher and emitted NIR fluorescence. This probe could detect MMP activity in vitro, in cultured cells and in tumour-bearing mice.
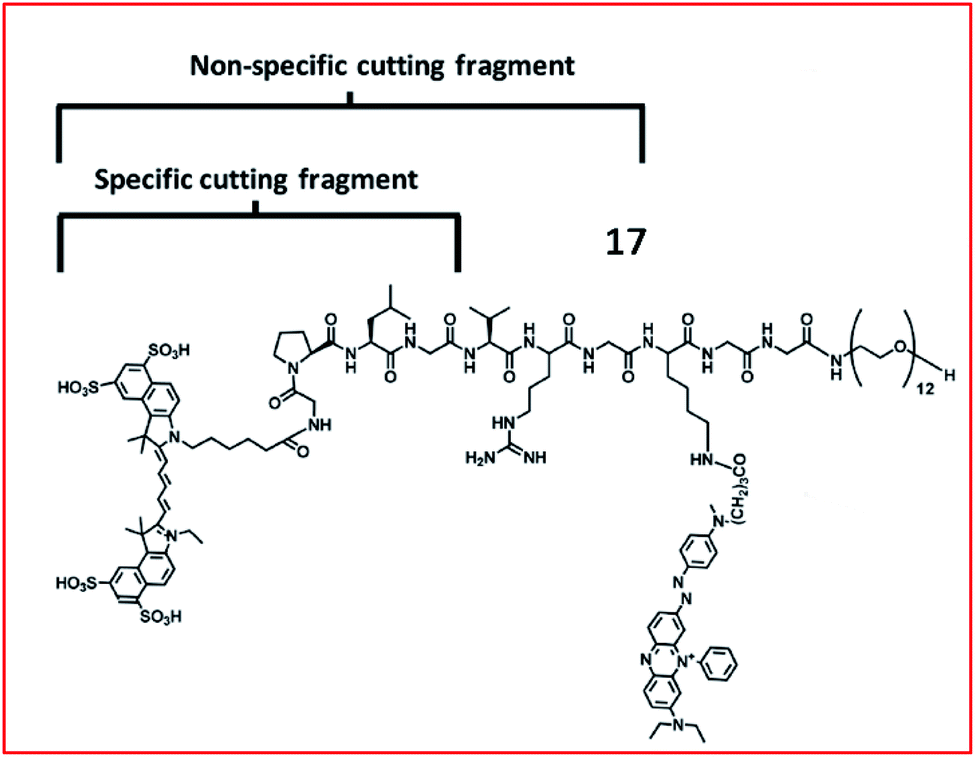 | ||
| Fig. 13 Structure of MMP probe 17. Reprinted from ref. 69 with permission. Copyright 2013 American Chemical Society. | ||
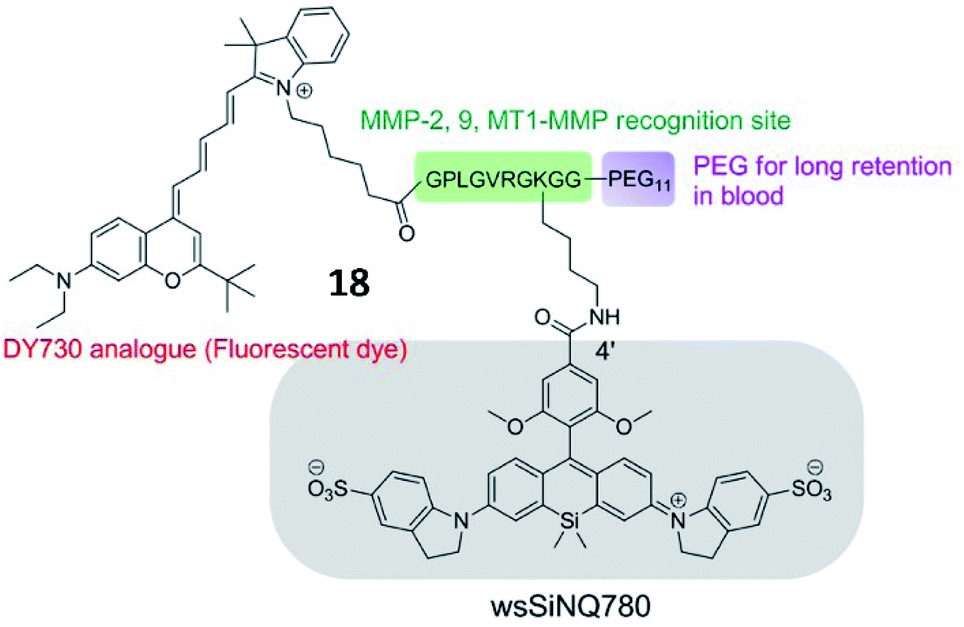 | ||
| Fig. 14 Chemical structure of MMP probe 18. Reprinted from ref. 70 with permission. Copyright 2015 American Chemical Society. | ||
Tang et al. developed an Au–Se bond based nanoprobe for detection of MMP activities with anti-interference ability for glutathione (Fig. 15).71 Although the Au–Se bond and Au–S bond possessed similar physical and chemical properties, the Au–Se bond showed higher stability than the Au–S bond. The probe (19) was an easily assembled nanosensor, which was coupled with a selenol- and FITC-modified Gly–Pro–Leu–Gly–Val–Arg–Gly (the specific substrate of MMP-2) peptide onto the surface of AuNPs. Compared to the commonly used Au–S probes, the Au–Se probes display high thermal stability and good anti-interference ability towards glutathione.
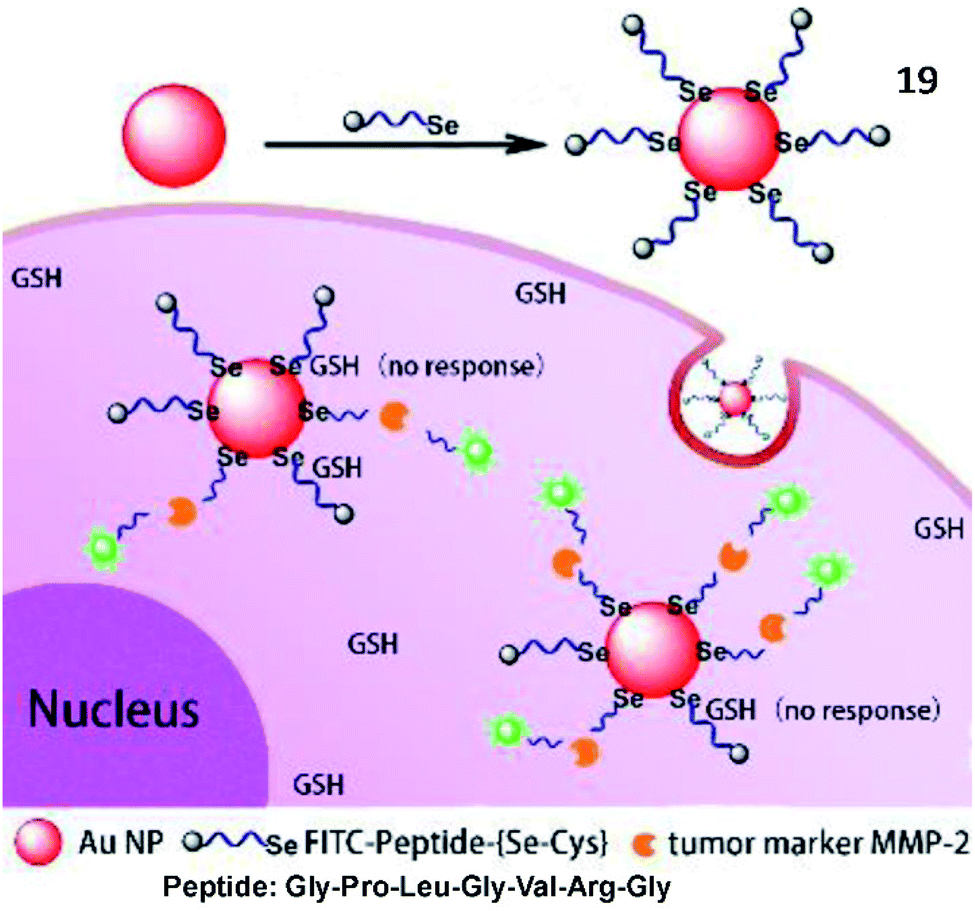 | ||
| Fig. 15 Structure of probe 19 and detection mechanism with MMP-2. Reprinted from ref. 71 with permission. Copyright 2018 American Chemical Society. | ||
In addition, Zhang et al. developed a multi-FRET fluorescent probe (20, Mc-probe) for simultaneous imaging of MMP-2 and caspase-3 activities (Fig. 16).72 Mc-probe was constructed with an MMP-2 sensitive peptide (GPLGVRG), a caspase-3 specifically responsive peptide (SDEVDS), and fluorophores (5(6)-carboxyfluorescein (FAM)/tetramethylrhodamine (TAMRA)/Dabcyl). The fluorescence of FAM and TAMRA was initially quenched due to the FRET process. After being cleaved by MMP-2, FRET between TAMRA and Dabcyl was blocked, restoring TAMRA fluorescence. After further cleavage by caspase-3, FAM fluorescence was recovered due to the blockade of FRET between FAM and TAMRA. These kinds of fluorescent probes designed for simultaneous imaging of different enzymes have great potential for precise disease diagnosis and therapeutic efficacy evaluation.
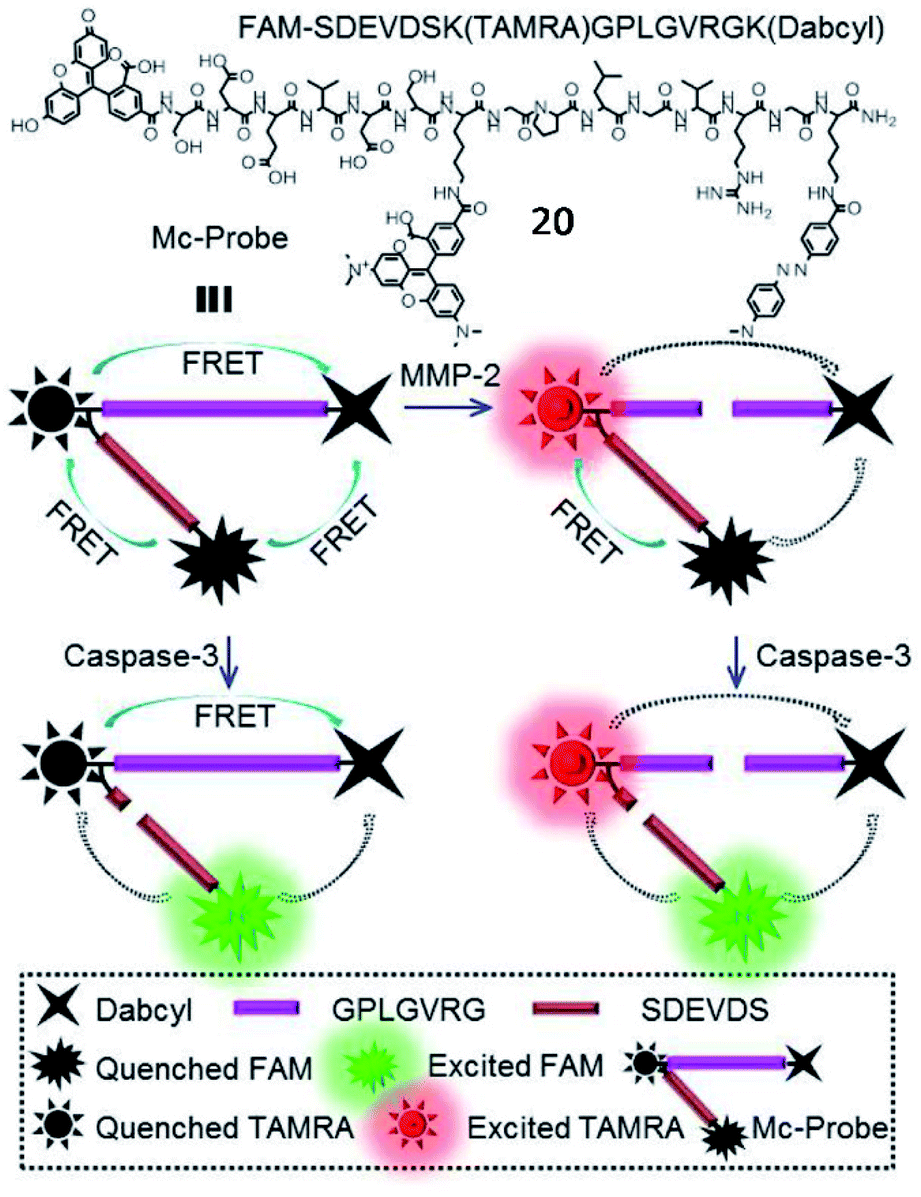 | ||
| Fig. 16 Chemical structure and the proposed processes of probe 20 for spatiotemporal MMP-2 and caspase-3 imaging. Reprinted from ref. 72 with permission. Copyright 2017 American Chemical Society. | ||
Moreover, an MMP-2-responsive PA and fluorescent probe (21, QC) was developed by Gao and coworkers (Fig. 17).73 The probe was established by the covalent linkage of an NIR dye (Cy5.5) and a quencher (QSY21) with the MMP-2 cleavable peptide (GPLGVRGY). Probe 21 self-assembled into nanoparticles in aqueous solution due to the presence of both a hydrophilic group (Cy5.5) and hydrophobic group (QSY21). When probe 21 was cleaved by MMP-2, the fluorophore Cy5.5 was released, emitting NIR fluorescence and a PA signal. Utilizing NIR/PA dual imaging, the fluorescence probe 21 could quantitatively detect MMP-2 with deep-penetration in tumour tissues from 4T1 tumour-bearing nude mice.
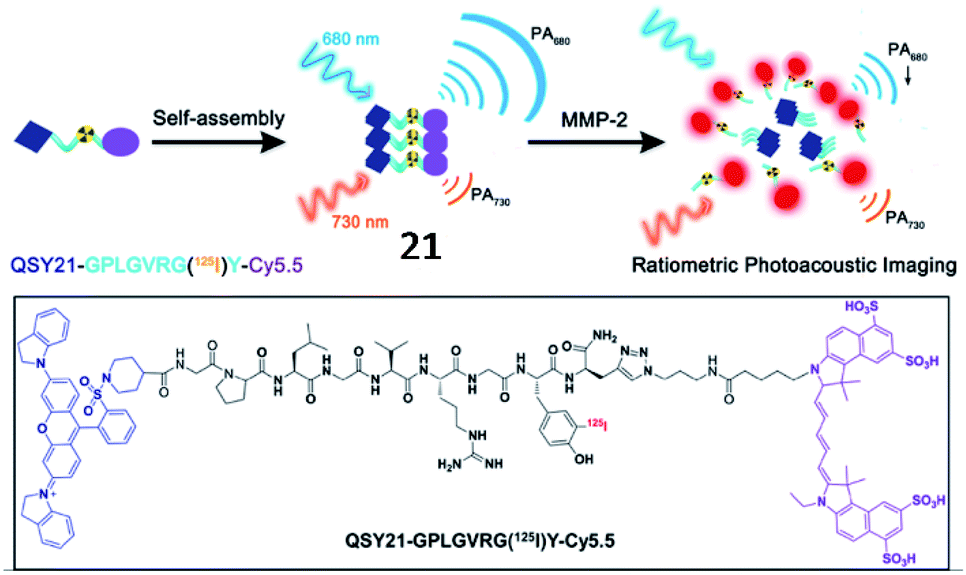 | ||
| Fig. 17 Probe 21 for non-invasive detection of MMP-2 activity through fluorescence and photoacoustic imaging. Reprinted from ref. 73 with permission. Copyright 2019 American Chemical Society. | ||
Xia et al. developed an MMP-2 activatable probe (22, DOX-FCPPs-PyTPE, DFP) to image MMP-2 activity and deliver drugs in vivo (Fig. 18).74 Probe 22 contained a TPE derivative (PyTPE) as the AIE-active unit, a functionalized cell penetrating peptide (FCPPs, CRRRRRRRRRP), a short MMP-2 specific substrate (LGLAG), and a therapeutic unit (doxorubicin, DOX). After being cleaved by MMP-2, the released hydrophobic PyTPE aggregated, generating strong yellow fluorescence. Then, the hydrophilic FCPPs-DOX could quickly permeate into MMP-2 overexpressed cells. This probe could distinguish different cell types which possessed varied MMP-2 activities, and also has high cytotoxicity to MMP-2 over-expressed cells. This work could be a milestone in MMP-2 related theranostics.
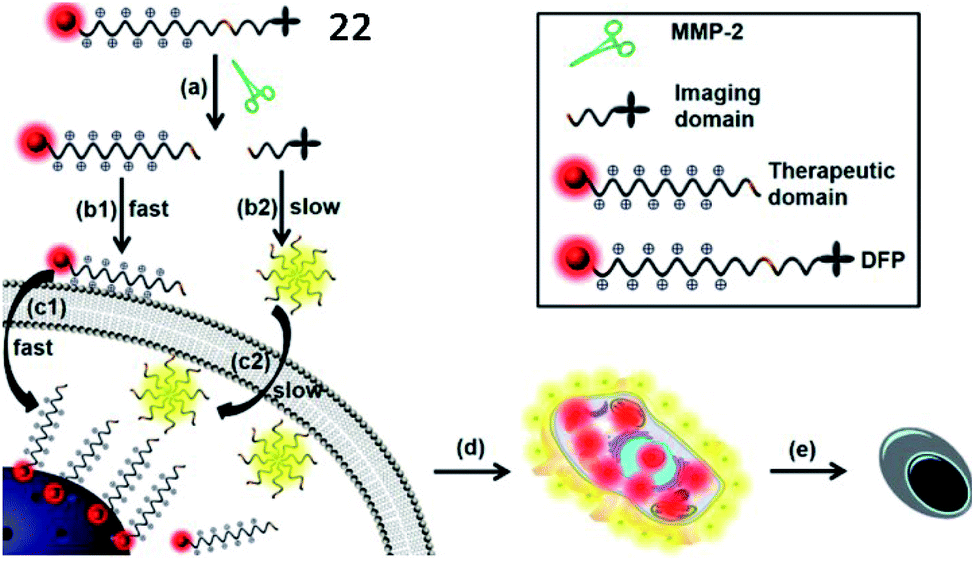 | ||
| Fig. 18 Probe 22 for rapid drug delivery and release tracking in MMP-2 over-expression living cells. Reprinted from ref. 74 with permission. Copyright 2016 American Chemical Society. | ||
Several cathepsin activatable small-molecule probes have been created for fluorescence imaging in vivo. Chen et al. developed a lysosome-targeting probe (23) to sense cathepsin B activity in living cancer cells (Fig. 19).79 The probe was incorporated by a cathepsin B-recognitive peptide substrate Cbz–Lys–Lys–p-aminobenzyl alcohol (Cbz–Lys–Lys–PABA) and a lysosome locating group morpholine. The fluorescence intensity of the probe increased 73-fold upon cathepsin B-mediated cleavage of the substrate, suggesting that the probe possessed high sensitivity toward cathepsin B.
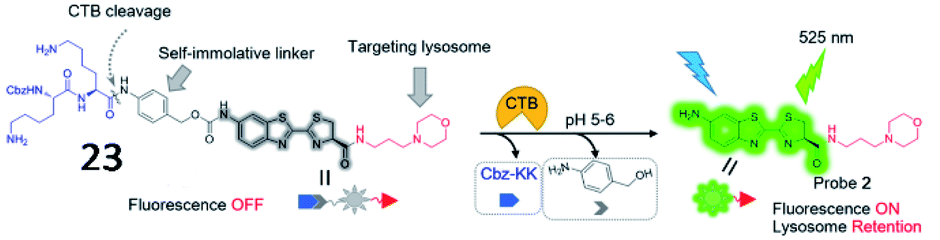 | ||
| Fig. 19 The chemical structure of probe 23 and the proposed chemical conversion in response to cathepsin B. Reprinted from ref. 79 with permission. Copyright 2016 American Chemical Society. | ||
Kim et al. explored a fluorescent probe (24, RR-S-Ac3ManNAz) for the detection of cathepsin B activity by incorporating a cathepsin B-specific substrate (Lys–Gly–Arg–Arg, KGRR) into a tri-acetylated N-azidoacetyl-D-mannosamine (Fig. 20).80 After being cleaved by cathepsin B in cells, the released S-Ac3ManNAz could be hydrolysed to produce Ac3ManNAz, which was eventually labelled by a dibenzylcyclooctyne-modified NIR dye (Cy5.5) through bioorthogonal click chemistry. This strategy showed promise for the inspection of enzyme activities in vitro and in vivo.
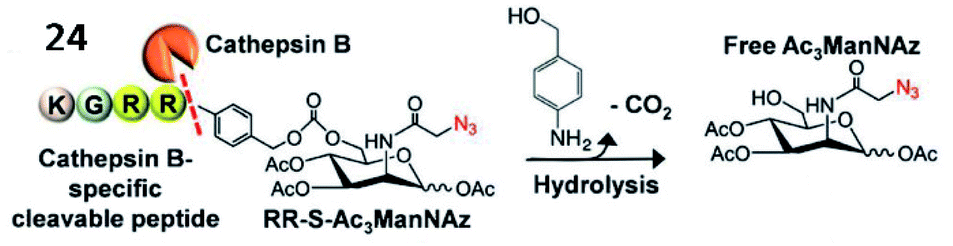 | ||
| Fig. 20 Probe 24 for rapid drug delivery and release tracking in MMP-2 over-expression living cells. Reprinted from ref. 80 with permission. Copyright 2016 American Chemical Society. | ||
In addition, several fluorescent probes for simultaneous imaging and treatment of cancer have been reported. Yoon et al. described a cathepsin B-activated fluorescent probe (25, CyA-P-CyB) for detecting cathepsin B activity and investigated its application in imaging tumour tissues in vivo (Fig. 21).81 The probe (25) consisted of two cyanine moieties (CyA and CyB), linked by a cathepsin B-activated peptide (Gly–Phe–Leu–Gly). After being cleaved by cathepsin B, the probe released two chromophores, evoking two fluorescence signals. This probe also exhibited great phototoxicity to tumour cells in mice, thus providing great potential for tumour treatment.
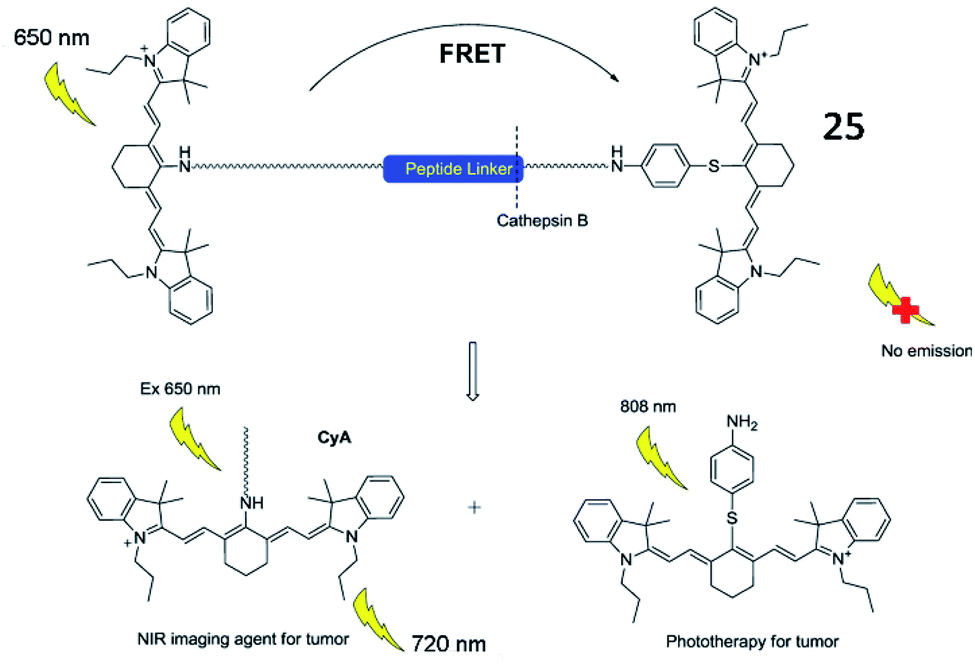 | ||
| Fig. 21 Illustration of near-infrared imaging and phototherapy of cathepsin B using the probe 25. Reprinted from ref. 81 with permission. Copyright 2017 Elsevier Ltd. | ||
Liu and Tang et al. described a cathepsin B-activatable probe (26) based on an AIEgen, which could simultaneously image and treat cancer though photodynamic therapy (Fig. 22).82 The probe is composed of four parts: (1) an AIE fluorogen as an imaging reagent and photosensitizer, (2) a peptide substrate of cathepsin B, (3) a hydrophilic linker, and (4) a cRGD-targeting moiety. After cancer-cellular uptake, the fluorescence of the probe (26) was dramatically enhanced due to the AIE effect upon cleavage of the GFLG peptide by cathepsin B, and the PDT was activated. Probe 26 could be used for image-guided PDT in cancer cells. These studies provide valuable strategies for the development of probes for cancer diagnosis and treatments.
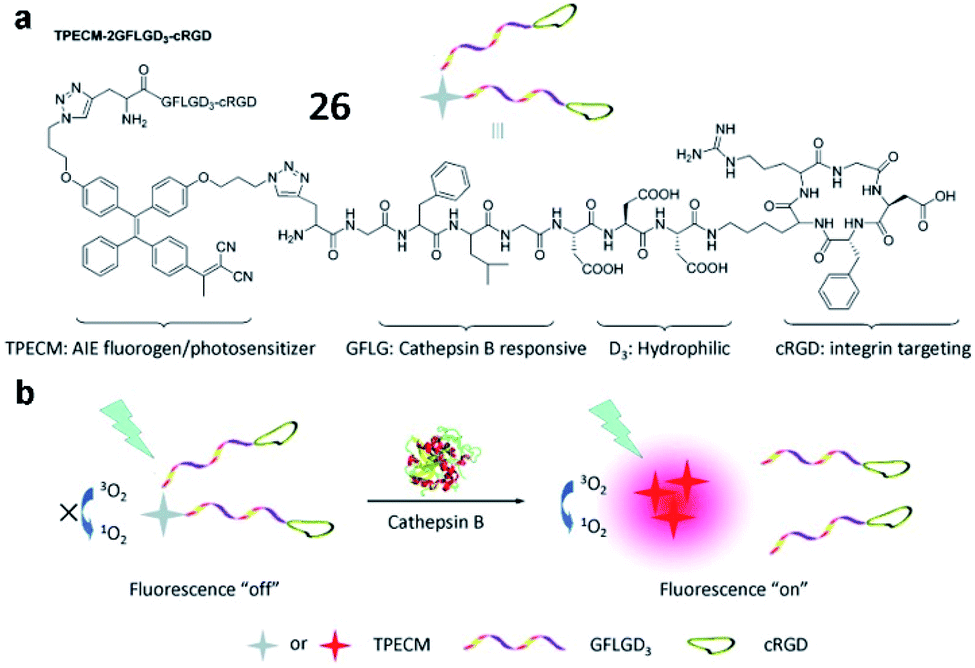 | ||
| Fig. 22 The chemical structure of probe 26 and the proposed chemical conversion in response to cathepsin B. Reprinted from ref. 82 with permission. Copyright 2016 American Chemical Society. | ||
Many tyrosinase activity-based fluorescent probes with a tyrosinase substrate and fluorophore have been constructed to image tyrosinase activity in vivo. Ma et al. synthesized a small-molecule “off–on” fluorescent probe (27, Mela-TYR) to monitor tyrosinase in cells (Fig. 23a). This probe was engineered by engineering 4-aminophenol-derived urea and morpholine into a naphthalimide scaffold, and morpholine was used as a melanosome-targeting group. Upon the enzymatic cleavage of 27, the naphthalimide fluorescence recovered. This probe provides a practical tool to investigate the activity of tyrosinase in cells.89
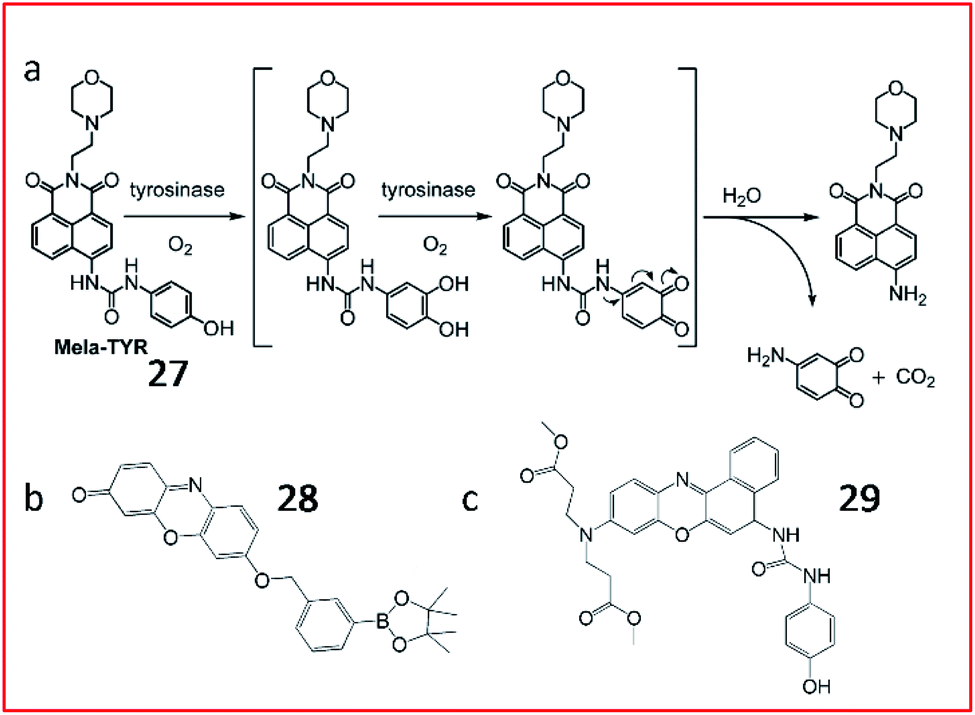 | ||
| Fig. 23 (a) Probe 27 reaction with tyrosinase. Reprinted from ref. 89 with permission. Copyright 2016 American Chemical Society. (b) Structure of probe 28. (c) Structure of probe 29. | ||
Subsequently, several fluorescent probes were developed following the above strategy. Zhang et al. developed a resorufin-based fluorescent probe (28) for the detection of tyrosinase (Fig. 23b). m-Tolylboronic acid pinacol ester was used as a substrate for tyrosinase; thus, resorufin was released in the presence of tyrosinase. This probe exhibits high selectivity and sensitivity towards tyrosinase, suggesting its use as a promising tool for the detection of tyrosinase in biosystems.90
Wu et al. developed a fluorescent probe (29, NBR-AP) to sense tyrosinase by modifying a phenoxazine derivative with a hydroxyphenyl urea group (substrate for tyrosinase) (Fig. 23c). In the presence of tyrosinase, the carbamide bond broke, and the fluorescent dye was released, emitting strong fluorescence. Probe 29 could selectively detect tyrosinase activity in cells and living zebrafish. Moreover, based on changes in tyrosinase activity, the probe could indicate melanoma metastasis in a mouse model.91
A PeT-based two-photon fluorescent probe (30, Tyro-1) was designed by Bhuniya and coworkers to track the activity of intracellular tyrosinase (Fig. 24). BODIPY was chosen as the chromophore due to its photophysical stability, large Stokes shift and high quantum yield. The tyrosinase activity in various cell types was detected by probe 30. Moreover, utilizing the probe, the authors disclosed that H2O2 can promote tyrosinase activity in cells.86
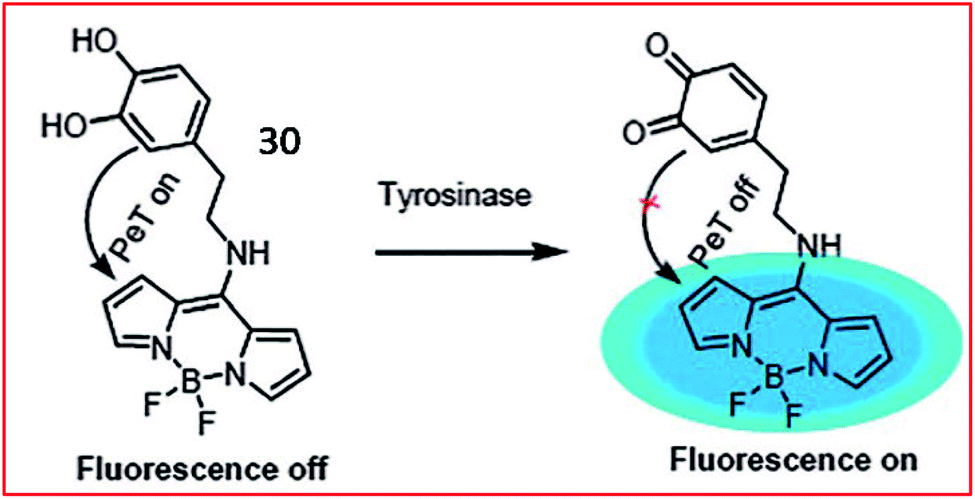 | ||
| Fig. 24 Fluorescence changing mechanism of probe 30 in the presence of tyrosinase. Reprinted from ref. 86 with permission. Copyright 2017 The Royal Society of Chemistry. | ||
Since previously reported fluorescent probes for tyrosinase showed similar responses to both tyrosinase and some reactive oxygen species (ROS), thereby suffering from the ROS interferences, Ma et al. proposed a new tyrosinase-recognition moiety, 3-hydroxybenzyloxy, in which the presence of the unique 3-hydroxy (instead of 4-hydroxy) group facilitates the hydroxylation at the 4-position vacancy by tyrosinase but not by ROS, and the resulting hydroxylation unit would be spontaneously removed by the subsequent 1,6-rearrangement–elimination (Fig. 25a).92 The probe (31) can specifically identify tyrosinase instead of ROS. The probe consisted of a 3-hydroxybenzyloxy moiety as a new recognition unit and a stable hemicyanine group as a fluorescent reporter, which can be used to detect endogenous tyrosinase activity in live cells and zebrafish.
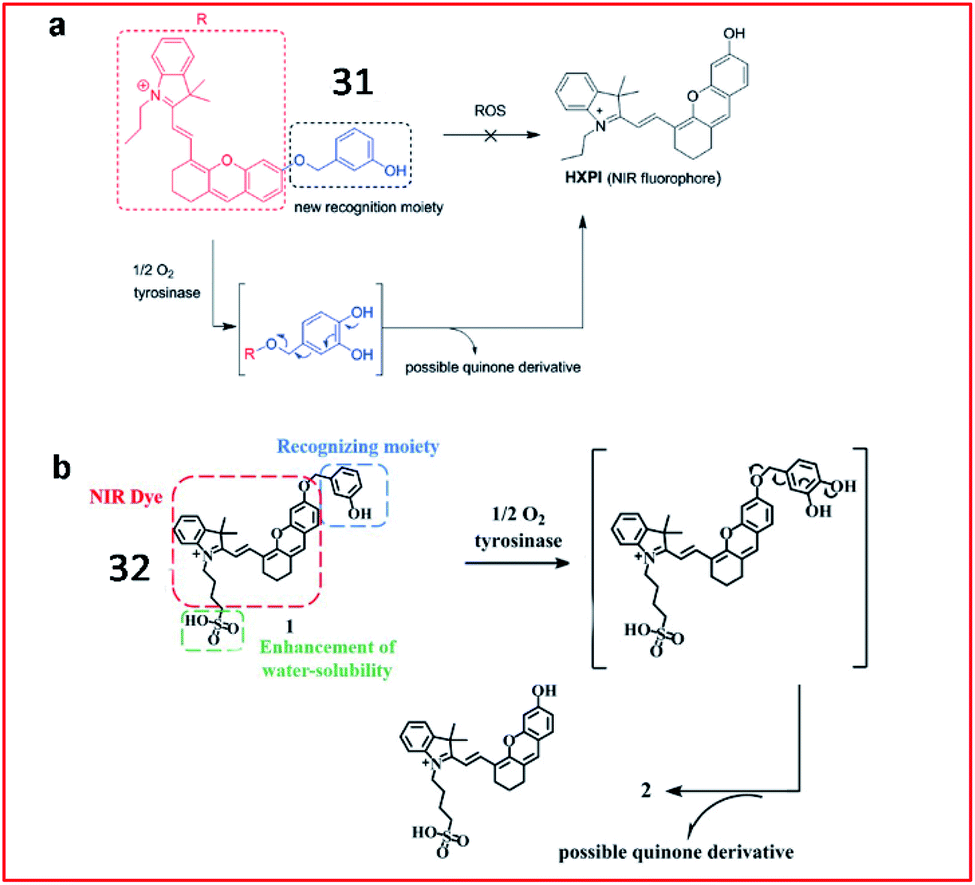 | ||
| Fig. 25 (a) The proposed reaction mechanisms of probe 31 with tyrosinase and ROS. Reprinted from ref. 92 with permission. Copyright 2016 Wiley-VCH. (b) Probe 32 reaction with tyrosinase. Reprinted from ref. 93 with permission. Copyright 2019 The Royal Society of Chemistry. | ||
Recently, Ding et al. reported a hydrosoluble NIR fluorescent probe (32) to image tyrosinase activity by using 3-hydroxybenzyloxy as the substrate of tyrosinase too (Fig. 25b). In the presence of tyrosinase, the maximum absorption wavelength of the probe was red-shifted from 600 nm to 670 nm, and the fluorescence intensity at 708 nm was significantly enhanced. This probe (32) was successfully used to monitor tyrosinase activity in cells and mice.93
Many other fluorescent probes have also been developed for the fluorescence imaging of tyrosinase activity.94–97 Su et al. designed a ratiometric fluorescence probe, containing red-emissive QDs and green-emissive QDs as fluorophores to provide ratiometric signals. Dopamine and QDs were conjugated to the surface of silica nanoparticles. Upon reaction with tyrosinase, dopamine transformed into dopamine quinine, thereby quenching the fluorescence of the green QDs. By monitoring the green and red fluorescence intensities, tyrosinase activity could be quantified.98 In 2018, Ouyang et al. reported a PeT-based NIR fluorescent probe (HB-NP) with a large Stokes shift (195 nm) for the detection of tyrosinase.99 HB-NP showed high selectivity towards tyrosinase and successfully realized the quantitative detection of tyrosinase in diverse living cells, especially in melanoma cells. In 2011, a BODIPY-based “turn-on” fluorescent probe to assess the activity of tyrosinase and the effect of tyrosinase inhibitors was presented by the Kim group.100
Over the past decade, several fluorescent probes have been reported to detect the activity of GGT. Kim et al. reported a mitochondria-targeted ratiometric probe (33, RP-Glu); the acid moiety of AP-Glu was replaced with an ester group, and an amide group was directly conjugated to indocyanine (Fig. 26). Probe 33 initially showed weak luminescence in a shorter wavelength region because of its amide functionality. When exposed to γ-GGT, the probe displayed strong fluorescence at the far-green region of the spectra through the enzyme-mediated amide bond cleavage reaction. This probe presented a ratiometric fluorescence response in cellular mitochondria and could detect cancer in mouse colons by imaging mitochondrial GGT activity.108
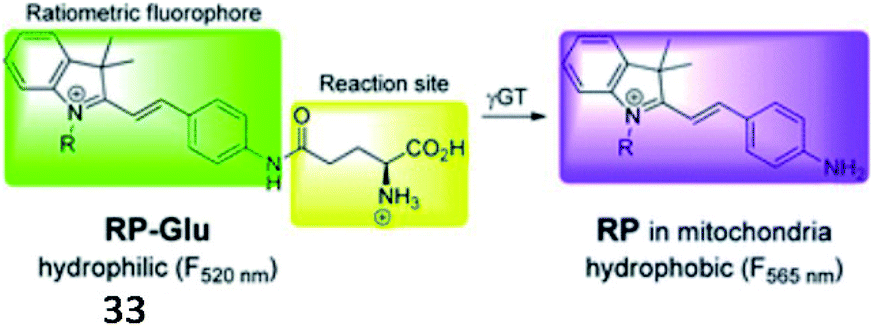 | ||
| Fig. 26 Proposed mechanism of probe 33 with γ-GGT. Reprinted from ref. 108 with permission. Copyright 2016 The Royal Society of Chemistry. | ||
Li and Xu et al. reported a ratiometric GGT-responsive sensor (34, Py-GSH) with high sensitivity. The probe consisted of a GSH group as the GGT-responsive moiety and pyronin B as the fluorescent group (Fig. 27). After reaction with GGT, the GSH group was cleaved, and electron rearrangement of Py-CG occurred, yielding strong green fluorescence at 540 nm. Probe 34 could rapidly and selectively track GGT activity in GGT-positive small peritoneal metastatic tumours (∼1 mm in diameter) in a mouse model. Moreover, this probe could detect microscopic cancer nodules in clinical tumour resection.109
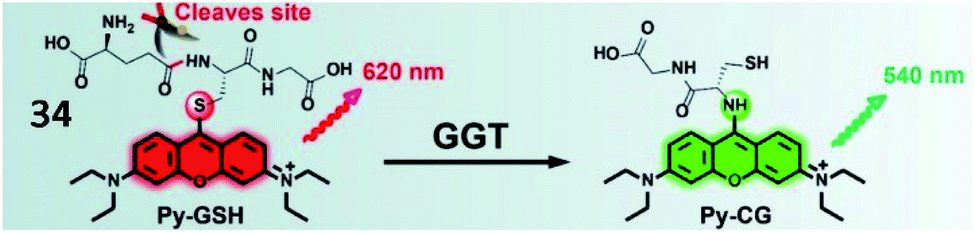 | ||
| Fig. 27 The presence of GGT leads to γ-glutamyl cleavage of 34. Reprinted from ref. 109 with permission. Copyright 2021 Ivyspring International Publisher. | ||
In addition, several NIR fluorescent probes were also developed for the detection of γ-GGT. Li et al. synthesized a zero cross-talk ratiometric NIR fluorescent probe (35, Cy-GSH) consisting of cyanine as a chromophore and GSH as the GGT-recognition unit to sense the activity of GGT (Fig. 28).110 Upon reaction with GGT, the γ-glutamyl moiety of probe 35 was removed, and the maximum emission wavelength was blue-shifted from 805 nm to 640 nm. Probe 35 could detect GGT in the blood, cells, and tumour tissues with high sensitivity and a low detection limit. Chen et al. also developed an NIR fluorescence sensor (36) constructed with hemicyanine dye as a signalling moiety, p-aminobenzyl alcohol as a self-immolative linker, γ-Glu as a substrate and c-RGD as the tumour-targeting group.111 Upon GGT catalysis against the self-immolative linker of probe 36, the free NIR dye was released, producing a strong fluorescence signal. This probe successfully targeted tumour cells in living mice in a non-invasive manner by αvβ3 receptor-mediated endocytosis. In the same year, another light-up NIR fluorescent probe (37, DCDHF-Glu) was developed by the Kong group.112 The probe 37 contained a dicyanomethylene dihydrofuran derivative as the signal reporter unit and a γ-glutamyl group as the enzyme-active trigger moiety. In the presence of GGT, the recognized substrate of GGT was removed, accompanied by a “turn-on” fluorescence response based on ICT.
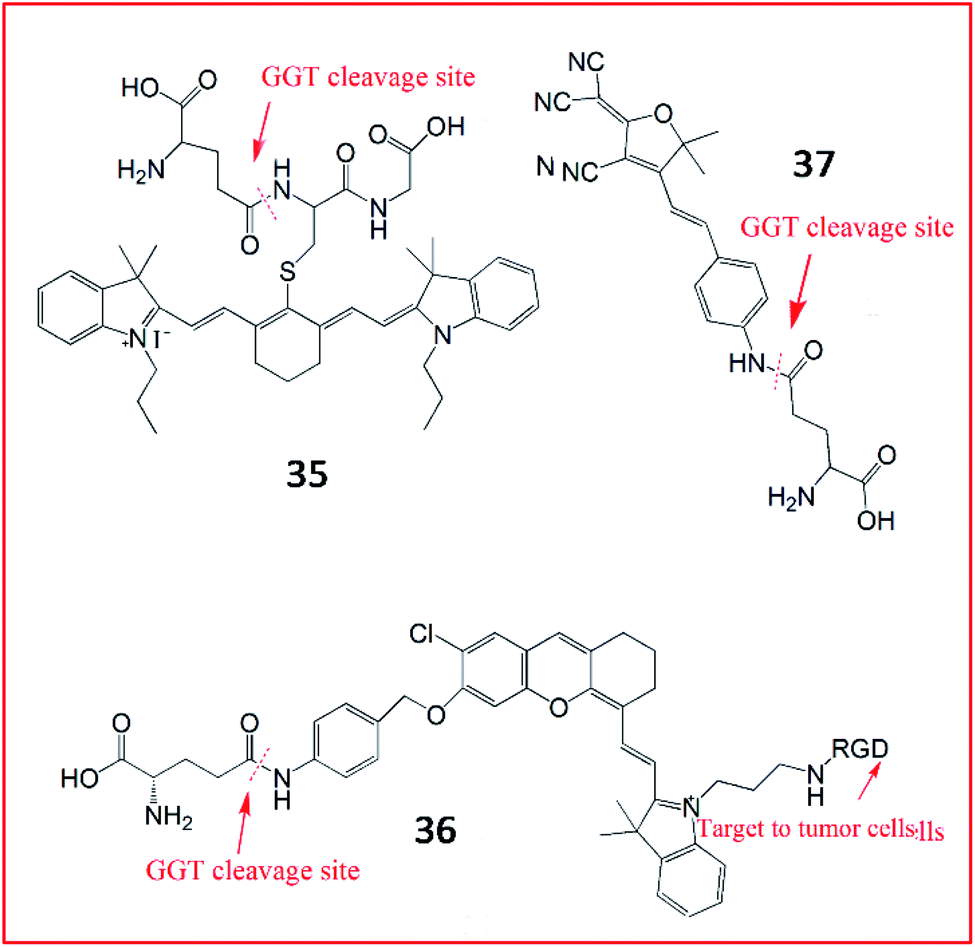 | ||
| Fig. 28 The structures of probes 35–37. These probes were used to detect the activity of GGT. | ||
In addition, many other fluorescent probes have been developed over the past decade to sense the activity of GGT.113–116 For instance, Wang et al. reported the fluorescent probe NM-GSH, which was composed of a γ-glutamyl group as a substrate of GGT and 1,8-naphthalimide as a chromophore. This probe could selectively image ovarian cancer cells.117 Peng et al. explored a two-photon fluorescent probe, TCF-GGT, by modifying a γ-GGT enzyme-specific substrate to 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran derivatives.118 Specific cleavage of the amide bond in the probe by γ-GGT released an electron-donating amine moiety to generate TCF-NH2, inducing remarkable fluorescence enhancement. This probe was applied for the identification of ovarian cancer cells from normal cells and real-time tracking of γ-GGT activity in a tumour xenotransplantation model in mice. Moreover, Urano et al. synthesized several fluorescent probes for the detection of GGT. The highly sensitive fluorescence probe gGlu-HMRG and the rhodamine-based fluorescent probe gGlu-HMJCR were manufactured to detect lysosomal GGT activities in ovarian cancer. Furthermore, the authors designed the fluorescent probe gGlu-HMJSiR for real-time monitoring of GGT activity in cells.119–121
In 2011, Urano and Nagano et al. created a fluorescent probe (38, HMDER-βGal) in which a β-galactopyranoside group was incorporated into the hydroxymethyl group (HMDER) of the scaffold for the detection of β-galactosidase activity (Fig. 29a).125 The fluorescence intensity of probe 38 obviously increased upon reaction with β-galactosidase. This is the first fluorescent probe in which spirocyclization of a hydroxymethyl group was utilized to control fluorescence emission upon reaction with the enzyme at physiological pH. In 2015, the authors developed a new fluorescent probe (39, HMRef-βGal) that exhibited bright fluorescence (>1400-fold fluorescence enhancement) upon the addition of β-galactosidase due to its optimized intramolecular spirocyclic effect (Fig. 29b).126 Probe 39 was successfully applied to diagnose cancer and mark tumour metastases in vivo with high sensitivity.
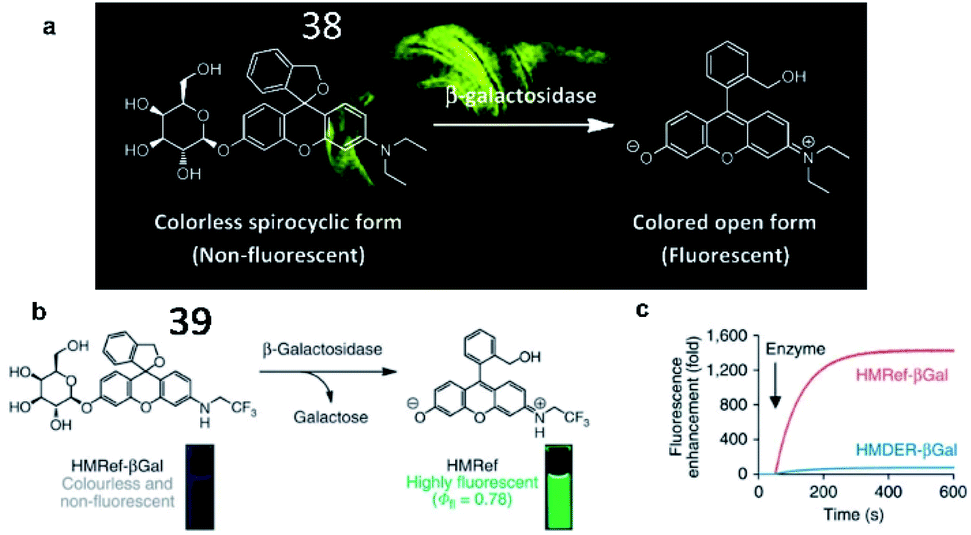 | ||
| Fig. 29 (a) The proposed reaction mechanisms of probe 38 with β-galactosidase. Reprinted from ref. 125 with permission. Copyright 2011 American Chemical Society. (b) Activation of 39 on enzymatic reaction with β-galactosidase. (c) Time course of the enzymatic reaction of 39 with β-galactosidase. (b) and (c) Reprinted from ref. 126 with permission. Copyright 2015 Macmillan Publishers Limited. | ||
Several activatable ratiometric fluorescent probes have been designed to quantitate β-galactosidase activity. Lin et al. designed an ICT-FRET-based ratiometric fluorescent probe (40, CG) to detect β-galactosidase activity (Fig. 30). In probe 40, 7-diethylamino coumarin was incorporated as a fluorescent donor, and 4-hydroxy-1,8-naphthalimide was incorporated as an acceptor in a FRET pair. In the presence of β-galactosidase, the specific substrate β-D-galactopyranoside was hydrolysed and released from the probe, which was accompanied by the restoration of fluorescence based on the ICT effect. Probe 40 exhibited a fast response rate (less than 20 s) and a low detection limit (0.081 U mL−1) for β-galactosidase. This probe displays great potential for revealing the function of β-galactosidase in biological organisms.127
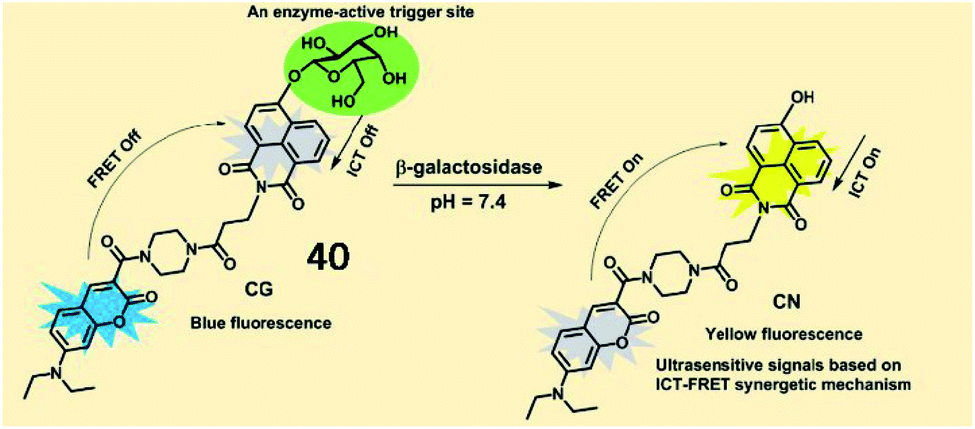 | ||
| Fig. 30 The proposed reaction mechanisms of probe 40 with β-galactosidase. Reprinted from ref. 127 with permission. Copyright 2019 American Chemical Society. | ||
To achieve deeper tissue penetration, a ratiometric NIR fluorescent probe (41, DCM-βgal) to quantify the activity of β-galactosidase was reported by Zhu and coworkers (Fig. 31).128 The chromophore dicyanomethylene-4H-pyran was utilized as a fluorescent reporter, and a cleavable β-galactosidase moiety was used as an enzyme-active trigger. The emission peak at 685 nm was enhanced upon incubation with β-galactosidase. The probe could be used for the ratiometric tracking of endogenously overexpressed β-galactosidase distribution in living 293T cells. This work provides a potential tool for in vivo real-time tracking of enzyme activity in preclinical applications.
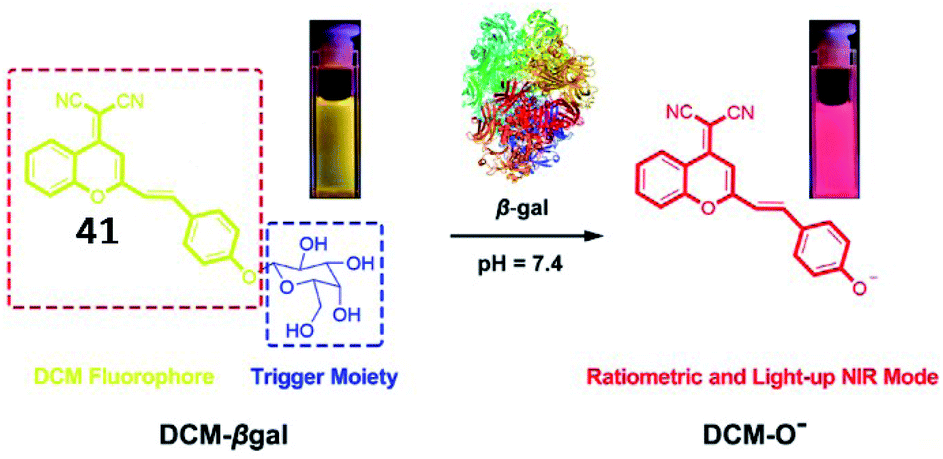 | ||
| Fig. 31 Proposed sensing mechanism for β-galactosidase enzymatic activation of 41. Reprinted from ref. 128 with permission. Copyright 2016 American Chemical Society. | ||
Kim et al. developed another ratiometric NIR fluorescent probe (42, DCDHF-βgal) for non-invasive imaging of β-galactosidase (Fig. 32).129 The maximum emission band was red-shifted from 615 nm to 665 nm upon the activation of probe 42 by β-galactosidase, indicating that the activity of β-galactosidase could be ratiometrically monitored by this probe. Furthermore, probe 42 could detect hepatocellular carcinoma with high selectivity and sensitivity by sensing β-galactosidase overexpression.
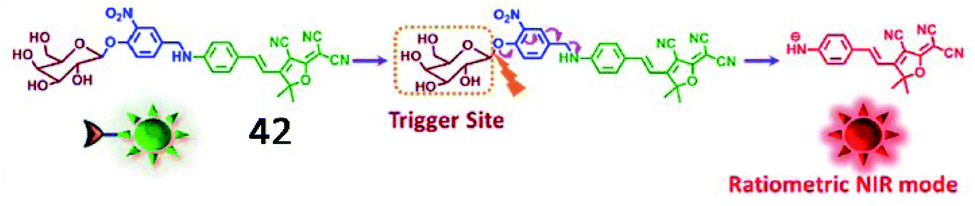 | ||
| Fig. 32 Conceptual scheme of 42 when encountering β-galactosidase. Reprinted from ref. 129 with permission. Copyright 2017 Elsevier Ltd. | ||
Subsequently, several fluorescent probes were developed based on the same strategy shown in Fig. 32. Liu et al. reported a lysosome-targeting NIR fluorescent probe (43, Lyso-Gal) for the visualization of lysosomal β-galactosidase in vivo (Fig. 33). Probe 43 consisted of hemicyanine as the fluorophore and galactose as the recognition group. Upon reaction with β-galactosidase, Lyso-OH, which emitted bright fluorescence, was produced. With its beneficial high sensitivity and ultra-fast response (∼1 min) to β-galactosidase, probe 43 could successfully visualize endogenous β-galactosidase in ovarian cancer cells.130 Compared with other reported probes, probe 43 showed the fastest response to β-galactosidase to date.
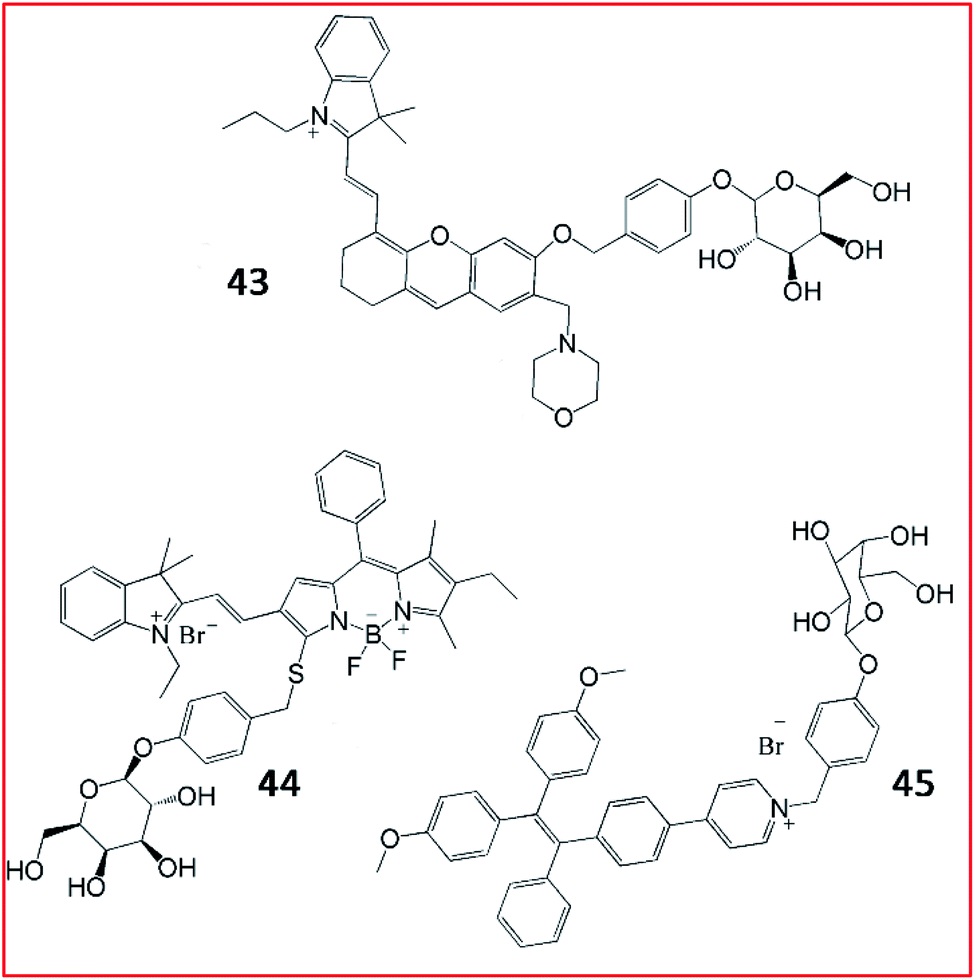 | ||
| Fig. 33 The structures of fluorescent probes 43–45. These probes were developed for the detection of β-galactosidase. | ||
As reported in a recent study, Gu et al. developed an NIR-II fluorescent probe (44, BOD-M-β-gal) for the detection of β-galactosidase (Fig. 33). Probe 44 showed bright fluorescence after cleavage by β-galactosidase. Overexpression of β-galactosidase was visualized by probe 44 with NIR-II emission at 900–1300 nm. Probe 44 could be applied to detect and bio-image SOVK3 cancer cells and ovarian tumour tissues in nude mice.131 Until now, probe 44 is the only reported NIR-II fluorescent probe for the detection of β-galactosidase activity.
Tang et al. devised an AIE-based bioimaging probe (45, TPE-Gal) to monitor the activity of β-galactosidase in living cells (Fig. 33). To form this tetraphenylethylene (AIEgen)-based light-up fluorescent probe, a D-galactose residue was applied as the substrate of β-galactosidase in probe 45. Upon activation by the enzyme, the D-galactose residue left, promoting the 1, 6-elimination of p-quinone-methide, releasing the tetraphenylethylene. Consequently, aggregated tetraphenyl ethylenes emitted a distinct fluorescence signal. In general, probe (45) displayed high specificity, good cell permeability, and high biocompatibility for the imaging of β-galactosidase in cells.132
3.2. Cardiovascular disease
Cardiovascular disease (CVD), a chronic disease responsible for the largest global disease burden in the world, is closely associated with age. CVD is also associated with a number of different disorders including ischaemia/reperfusion injury, ischemic heart disease, hypertension and diabetes. Many reports have indicated that overproduction of ROS is a major factor contributing to the pathogenesis of CVD due to overloading of the capacity of antioxidants and antioxidant enzymes in vivo.133,134Creatine kinase (CK), which catalyses the transfer of a phosphoryl group from adenosine triphosphate (ATP) to adenosine diphosphate (ADP), is closely associated with many bioenergetic processes. CK is widely expressed in many organs, such as smooth muscle, cardiac muscle, the kidney, the brain and skeletal tissue. Previous studies reported that CK expression in cells could protect against ischaemia/reperfusion injury.135 In addition, the CK isoenzyme, CK-B is used as a biomarker of myocardial infarction. Zhang et al. reported a water-soluble AIE-based fluorescent probe (46, TPEMA) to sense the activity of CK-B (Fig. 34).136 The inhibitor ethylmalonic acid was used as the targeting moiety to deliver the probe into the cavity of CK-B, and TPE was used as the fluorophore. When probe 46 was inserted into the cavity of CK-B, the molecular motion of TPE was restricted, and TPE emitted strong fluorescence. Altogether, probe 46 displayed excellent selectivity and high sensitivity towards the CK-B subunit both in cells and in vitro.
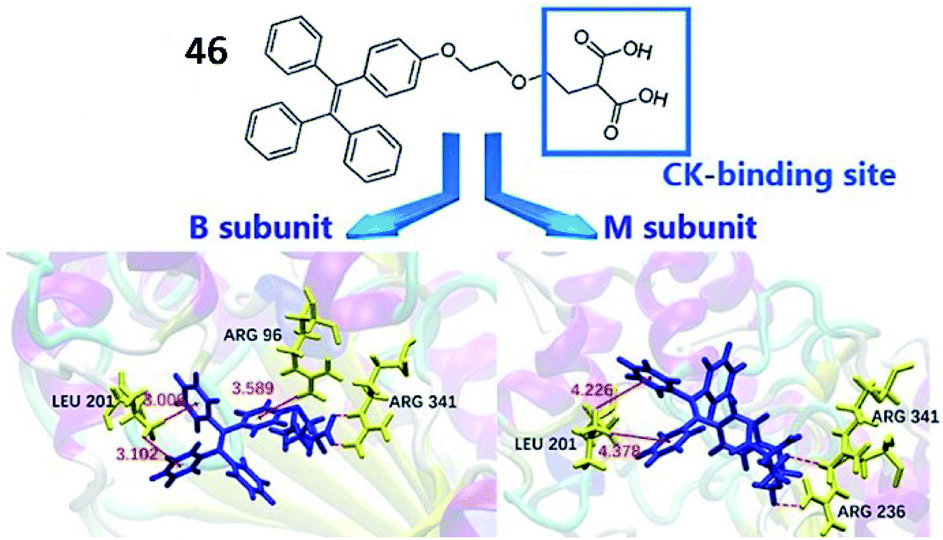 | ||
| Fig. 34 The structure of probe 46 (TPEMA). Reprinted from ref. 136 with permission. Copyright 2019 Wiley-VCH. | ||
Fang et al. developed a naphthalimide-based TrxR-activatable fluorescent probe (47, TRFS-green) in 2014 (Fig. 35a).137 The probe showed green fluorescence after its cyclic disulfide scaffold was cleaved by TrxR in the presence of NADPH. 47 could be used to visualize TrxR activity in living cells and screen inhibitors of TrxR. Then, a red-emission fluorescent probe (48, TRFS-red) was reported by the same group (Fig. 35b).138 The probe contained Nile blue derivative as the fluorophore and 1,2-dithiolan-4-ol as the TrxR recognition group. The maximum absorption wavelength of probe 48 was red-shifted from 530 nm to 615 nm upon activation by TrxR. Compared with the authors' previously reported probe 47 (35-fold), probe 48 possessed higher sensitivity (90-fold fluorescence enhancement) towards TrxR. Recently, the Fang group reported another new fluorescent probe (49, Fast-TRFS) to detect the activity of TrxR fast and specifically (Fig. 35a).139 Based on previous work, a superfast probe was optimized, synthesized and demonstrated to exhibit 150-fold enhanced fluorescence after incubation with TrxR. More importantly, 49 displays better selectivity to TrxR than do 47 and 48. Further mechanistic studies reveal that switching on the fluorescence of 49 is achieved by the reduction of the disulfide bond only and the process of cyclization-driven release (CDR) is not necessary, which is different from the mechanism underlying the activation of 47 or 48. This work provides a high-throughput assay to screen for inhibitors of TrxR.
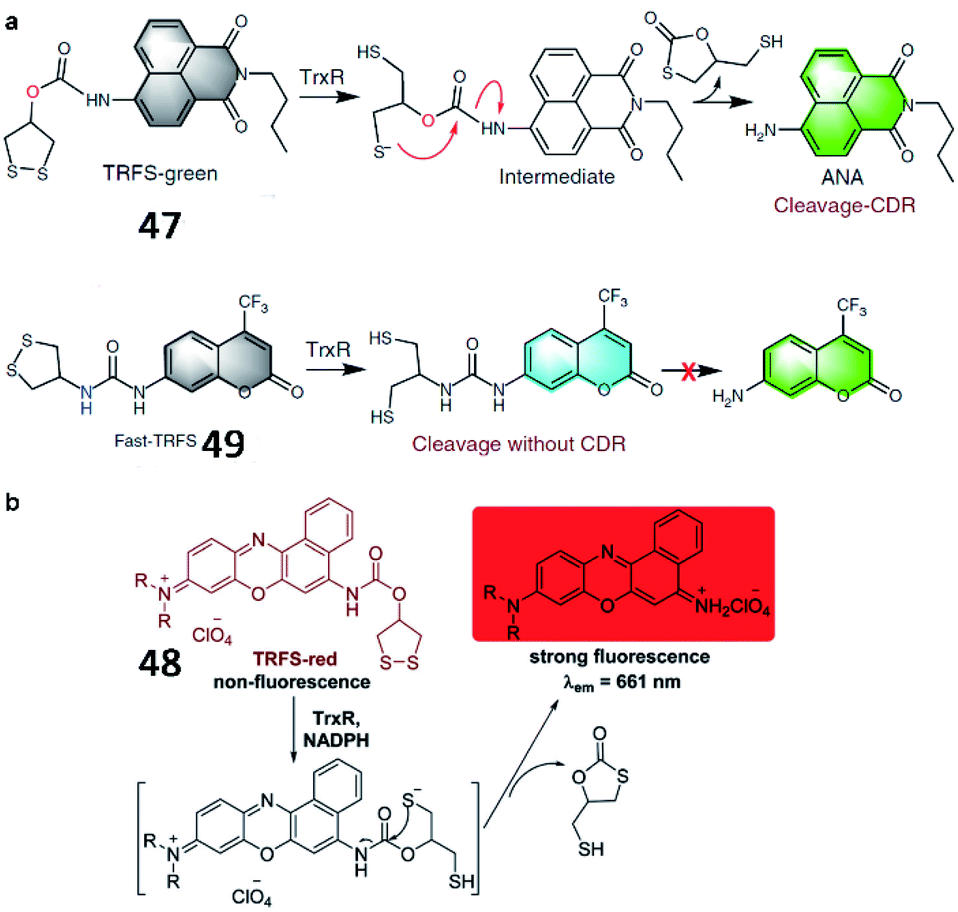 | ||
| Fig. 35 (a) Reaction mechanism of 47 and 49. Reprinted from ref. 139 with permission. Copyright 2019 Springer Nature. (b) Proposed mechanism for the activation of 48 by TrxR/NADPH. Reprinted from ref. 138 with permission. Copyright 2016 The Royal Society of Chemistry. | ||
3.3. Neurodegenerative disease
Neurodegenerative diseases are a heterogeneous group of disorders caused by progressive damage to the central nervous system. Common neurodegenerative diseases include AD, PD and Huntington's disease (HD).140 In most neurodegenerative diseases, abundant abnormal proteins (Aβ, tau) aggregate in neural cells and extracellular compartments, affecting post-translation processing, protein solubility, and fibril formation. These aggregated proteins cannot be degraded by proteases, autophagy or the ubiquitin–proteasome system. In addition, ROS induced oxidative stress has been implicated in the exacerbation of neurodegenerative diseases.141,142 Therefore, the development of fluorescent probes to detect ROS and ROS-associated proteins is necessary for a comprehensive understanding of the biological mechanism of ROS in neurodegenerative diseases.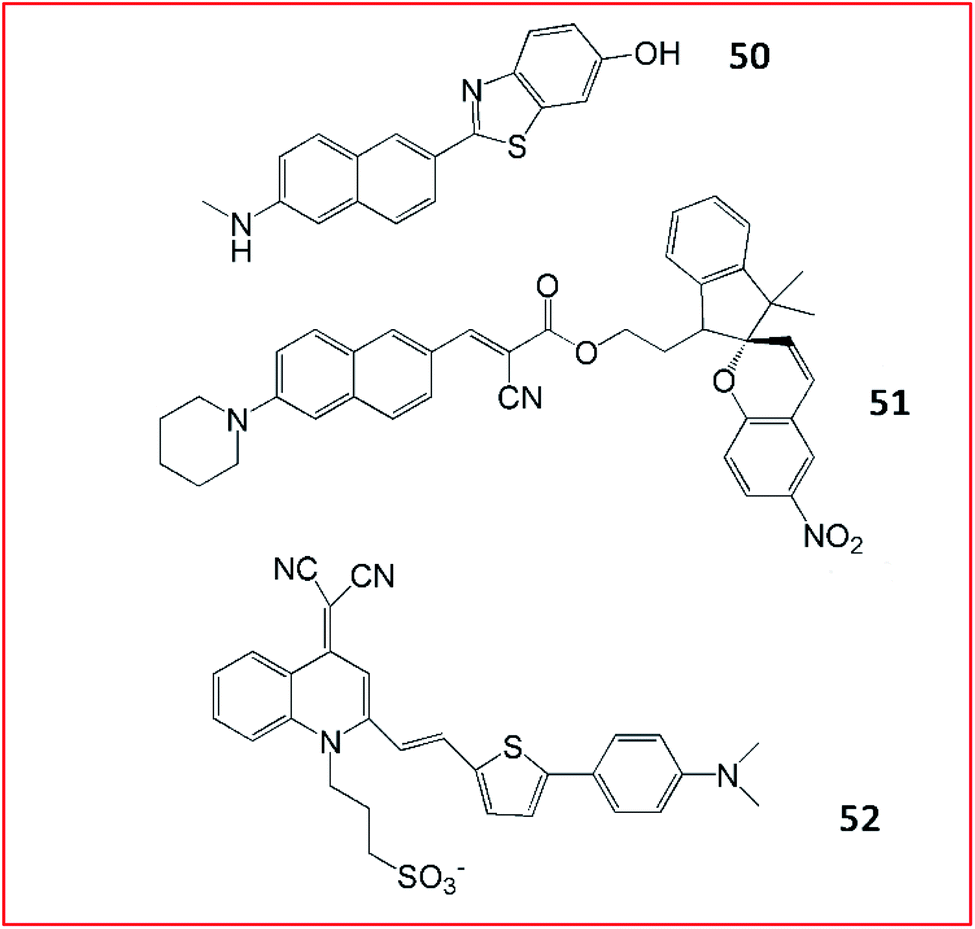 | ||
| Fig. 36 The structures of probes 50–52. These probes were developed for the detection of Aβ. | ||
Yi et al. reported several fluorescent probes for the detection of Aβ in AD (Fig. 36). In 2016, a spiropyran-based fluorescent probe (51, AN-SP) with an amino naphthalenyl-2-cyano-acrylate constituent as the targeting group for the imaging of Aβ oligomers with high affinity was developed.146 The fluorescence of probe 51 visibly changed in the presence of Aβ oligomers. The probe could detect Aβ oligomers in both brain sections and living AD mouse models.
Many AIE-based fluorescent probes have been developed to explore aggregated Aβ. Zhu et al. developed an NIR AIE-based fluorescent probe (52, QM-FN-SO3) for the visualization of Aβ plaques (Fig. 36).147 This probe 52 was ultrasensitive towards Aβ plaques, owing to its remarkable binding affinity for Aβ plaques. Moreover, probe 52 could traverse the blood–brain barrier with excellent photostability. Aβ plaques in living mice were successfully visualized by probe 52.
Tian et al. reported a ratiometric AIE-based fluorescent probe (53) for the imaging of Aβ and fibrils (Fig. 37). A silole-based AIEgen and fluorescent glycoprobes joined by supramolecular assembly constitute the probe (AIE-GNP). In the presence of Aβ, since the interaction between the AIEgen (DES) and the hydrophobic region of Aβ was stronger, Aβ could competitively bind DES and block FRET between DES and glycoprobes (Dks). The FRET efficiency between AIEgen and the glycoprobes accurately indicated the levels of Aβ and fibrils.148
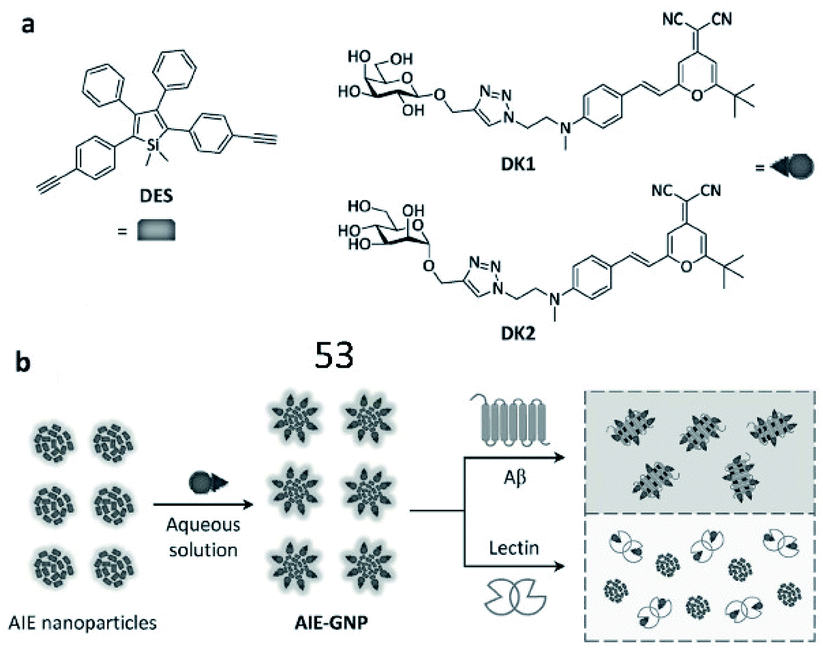 | ||
| Fig. 37 (a) Structures of the AIEgen (DES) and glycoprobes (DK1 and DK2). (b) Schematic illustration of probe 53 for detection of Aβ. Reprinted from ref. 148 with permission. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Several fluorescent probes have been reported to specifically bind and label tau in vivo. Schmidt et al. synthesized a set of trimethine cyanine derivative-based compounds to screen fluorescent probes for the selective imaging of tau fibrils (Fig. 38). Probes 54 could be used to visualize tau fibrils in the human AD brain and olfactory epithelium sections. These results suggest that probe 54 can target tau fibrils with a high binding rate, good aqueous solubility and low cytotoxicity.152
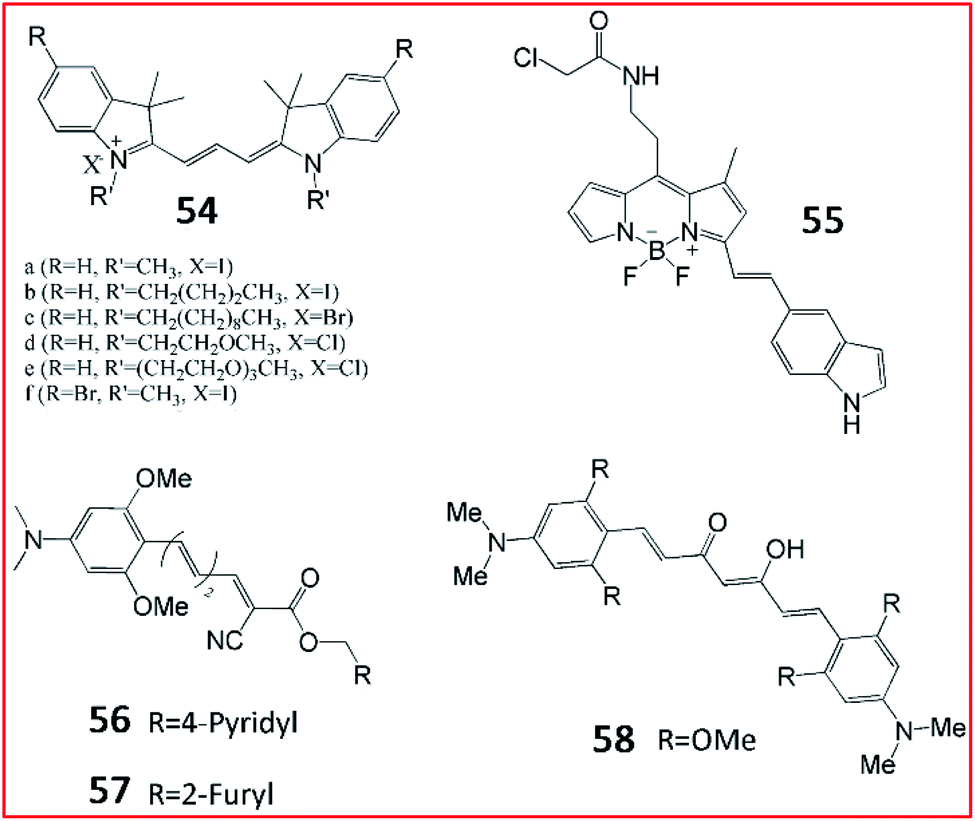 | ||
| Fig. 38 The structures of probes 54–58. These probes can image tau aggregates selectively. | ||
Kim et al. designed a BODIPY-based fluorescent probe (55, BD-tau) to observe aggregated tau in cells (Fig. 38). Probe 55 exhibited a 3.2-fold increase in fluorescence intensity in tau aggregates. Probe 55 could selectively image tau aggregates without interference from tau pre-aggregates or BSA, suggesting its high selectivity. Probe 55 could be used to detect pathological tau aggregates in live hippocampal neurons, indicating its potential for screening anti-tau aggregate drugs.153
NIR fluorescent probes may possess wider application in fluorescence imaging due to their deep penetration. Chong et al. developed NIR fluorescent probes 56 (3 g) and 57 (3 h) to identify tau fibrils in cells (Fig. 38).154 The fluorophore curcumin was used as a fluorescent reporter in a series of probes with a novel donor–π–acceptor architecture. The fluorescence properties of these probes were evaluated. Upon mixing with tau, the probe 56 and 57 exhibited large Stoke shifts with emission at 650 nm. In addition, probes 56 and 57 evoked brighter fluorescence towards tau aggregates over Aβ fibrils, suggesting that probes 56 and 57 possessed high selectivity.74 The authors also reported a curcumin-based NIR fluorescent probe (58) for the imaging of tau fibrils in 2015.155 A curcumin derivative was chosen as the fluorescent reporter and tau fibril-specific binding group. Intense fluorescence was observed upon mixing with tau fibrils, confirming its higher binding affinity for pre-aggregated tau.
Inspired by the characteristic structures of MAO-A inhibitors, Ma et al. reported a strategy to design and synthesize a fluorescent probe (59) for the specific imaging of MAO-A (Fig. 39). Based on this strategy, the structure (i.e. phenol with different substituents) of clorgyline (the specific inhibitor of MAO-A) was combined with propylamine, a new specific recognition unit for MAO-A, connected to the fluorophore resorufin.159 In the presence of MAO-A, the amino group in the probe was oxidized to imine and underwent deamination to form an aldehyde. Then, the propionaldehyde was removed by a β-elimination reaction, and the substituted phenol was removed by 1,6-rearrangement elimination, ultimately releasing resorufin. Altogether, probe 59 possesses the highest specificity for the detection of MAO-A and can distinguish between MAO-A and MAO-B.
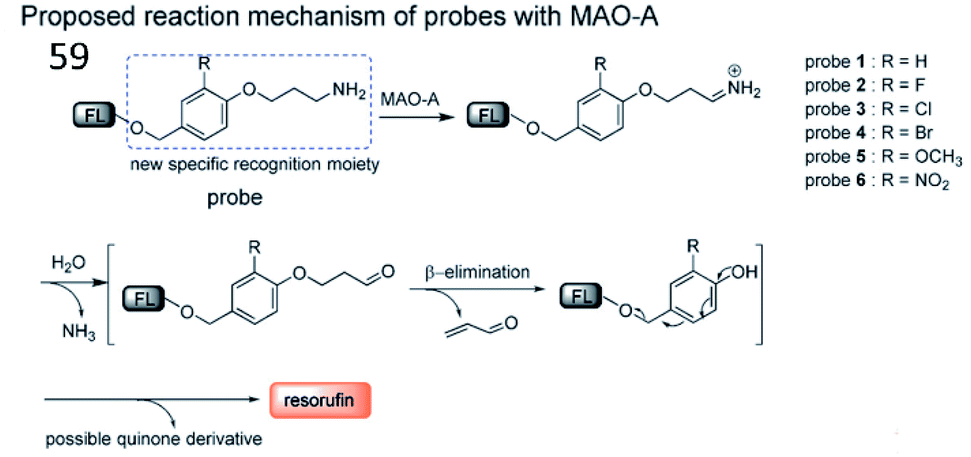 | ||
| Fig. 39 The proposed detection mechanism of probe 59 with MAO-A. Reprinted from ref. 159 with permission. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Many two-photon fluorescent probes have been reported to detect the activities of MAOs. Kim et al. developed a two-photon probe (60, PCP-1) for the specific targeting and imaging of MAO-A (Fig. 40). This probe consisted of a rhodamine derivative as a two-photon fluorescent group and an MAO-A-selective inhibitor (moclobemide) as a specific recognition. The probe targeted and bound MAO-A through a selective inhibitor of MAO-A and could monitor MAO-A in cancer cells. In addition, probe 60 prevented the proliferation and metastasis of cancer cells by inhibiting the activity of MAO-A.160
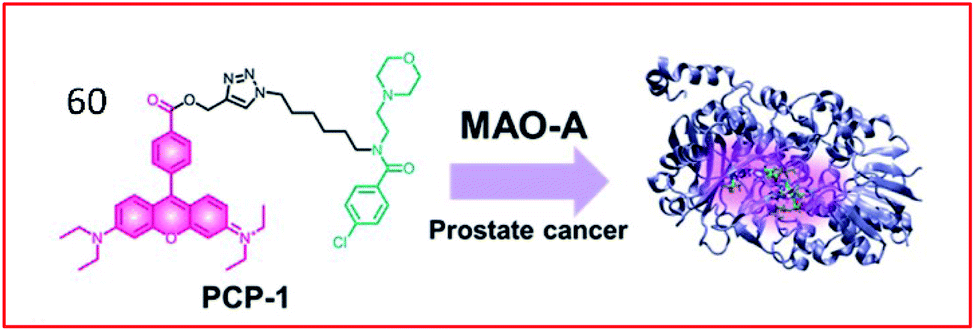 | ||
| Fig. 40 Design of probe 60 targeting MAO-A for the detection of prostate cancer. Reprinted from ref. 160 with permission. Copyright 2019 The Royal Society of Chemistry. | ||
Yao et al. reported the first two-photon enzyme probe (61, U1) for the specific detection of MAO-B activity (Fig. 41a).161 Probe 97 contained a 2-methylamino-6-acetylnaphthalene (acedan) moiety as the fluorophore and a propylamine moiety as a reactive group specific for MAO-B. Initially, due to the PeT process between acedan and the adjacent carbamate, the fluorescence of acedan was quenched. In the presence of MAO-B, the amino group was oxidized to an aldehyde group (via an imine intermediate), and propionaldehyde and CO2 were spontaneously released through β-elimination. Then, free Flu1 was released, emitting strong fluorescence based on blockade of the PeT effect. This probe could detect the activity of MAO-B in biological samples with high sensitivity and specificity. In 2015, by combining the fluorophore of probe U1 with the proteome profiling handle of the previously reported proteome-profiling-enabled probe P3, the authors further described a new activity-based dual-purpose probe (62, M2) for the detection of MAO-B activity (Fig. 41b).162 Probe 62 can detect endogenous MAO-B activity via in situ proteome profiling and live-cell imaging. Furthermore, this probe could detect the activity of MAO-B in established PD models.
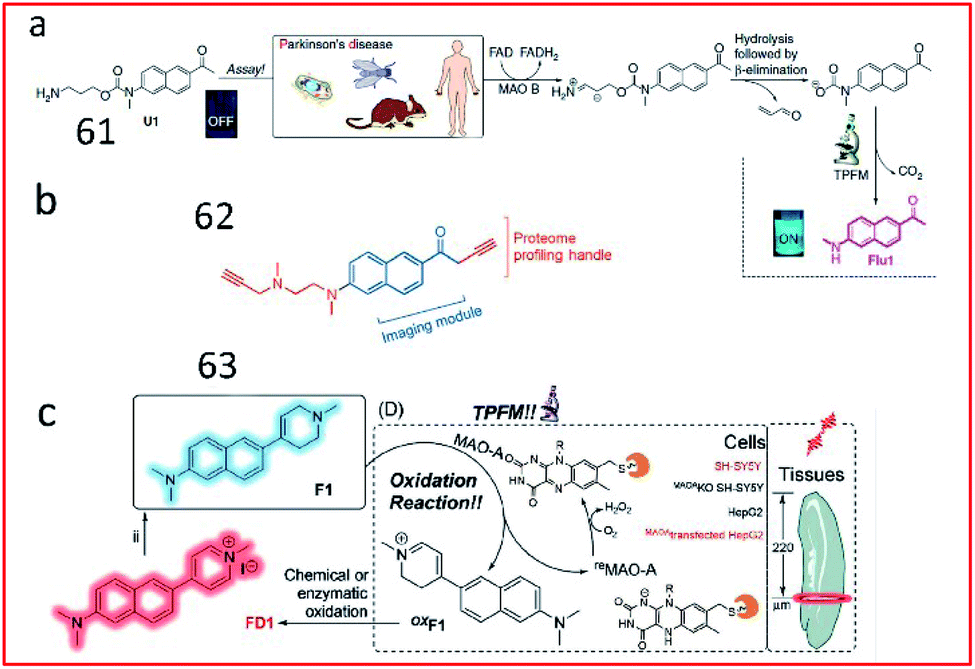 | ||
| Fig. 41 Overview of the design and synthesis of MAO-specific probes. (a) 61. Reprinted from ref. 161 with permission. Copyright 2014 Macmillan Publishers Limited. (b) 62. Reprinted from ref. 162 with permission. Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) 63. Reprinted from ref. 163 with permission. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Based on this work, the structure of naphthalene in probe U1 was retained by exchanging the positions of the propylamine warhead and fluorophore, and combined with N-alkylated tetrahydropyridine to obtain the first MAO-A-specific two-photon fluorescent probe (63, F1) (Fig. 41c).163 In the presence of MAO-A, the probe was oxidized to oxF1, and then transformed to FD1 through chemical or enzymatic oxidation in the system. Subsequently, the tetrahydropyridine group in probe 63 was oxidized to pyridine, which had strong electron-withdrawing ability, leading to increased fluorescence based on an enhanced push–pull electron effect. Probe 63 was successfully used to detect MAO-A activity in fresh mouse/human brain and tumour tissues. Probes 61–63 reported by Yao161–163 and co-workers possess excellent selectivity towards MAO-B or MAO-A and are powerful tools for PD diagnosis.
Several NIR MAO-activatable fluorescent probes were designed. Qin et al. created four mitochondrial-targeted dihydroxanthene (DH)-derived NIR fluorescent probes, DHMP1–4, for the detection of MAO-A; among these probes, probe 64 (DHMP2) was the most efficacious (Fig. 42). Probe 64 contained a dihydroxanthene group as the fluorophore, a propylamine group as the MAO-A reaction unit, and a tetramethyl-indole cation group as a mitochondrial targeting group. Upon cleavage by MAO-A, the hydroxyl group of the probe was exposed, restoring fluorescence. Probe 64 was used to monitor MAO-A activity in cellular mitochondria, zebrafish and tumour-bearing nude mice.164
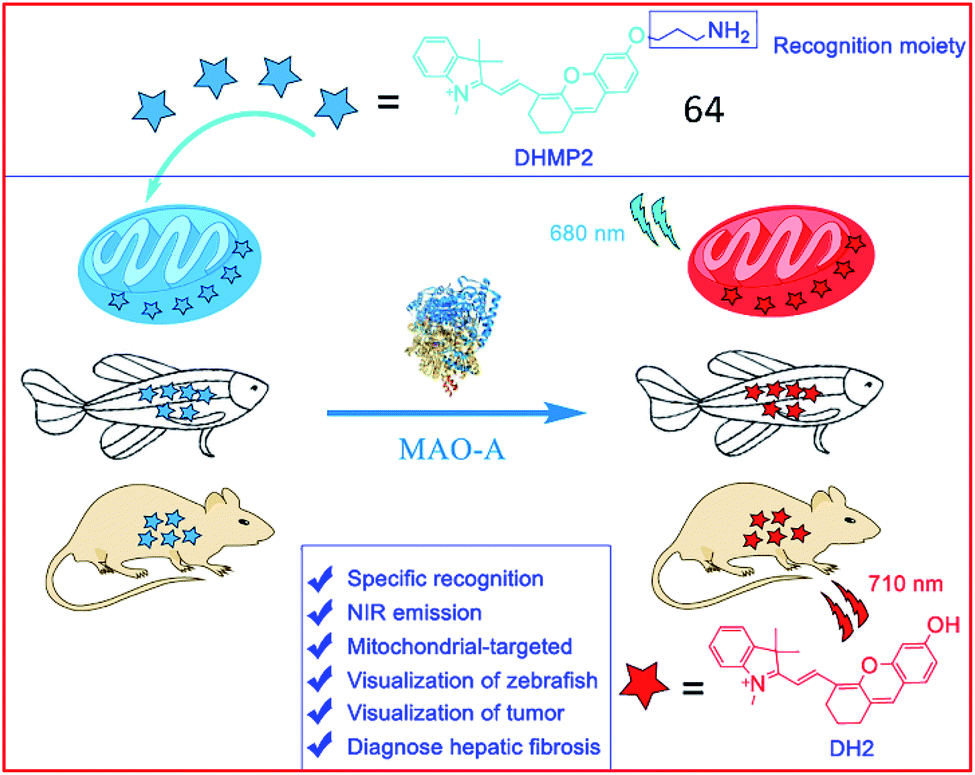 | ||
| Fig. 42 The structure of probe 64. Reprinted from ref. 164 with permission. Copyright 2020 American Chemical Society. | ||
To simultaneously image the activity of MAO-B and the generation of ROS, Chen and co-workers developed two NIR ratiometric fluorescent probes, probes 65 (MitoHCy-NH2) and 66 (MitoCy-NH2) (Fig. 43). In the presence of MAO-B, conversion of the amino group to an imine intermediate catalysed by MAO-B was accompanied by the generation of ROS, and the reduced fluorophore in probe 65 was oxidized by ROS to restore fluorescence. Subsequently, the absence of the propylamine group caused β-elimination, leading to blue-shifting of the fluorescence maximum emission wavelength of the probe. Therefore, probes 65 and 66 can be used for the synchronous detection of MAO-B and ROS in cell and aging mouse models.165
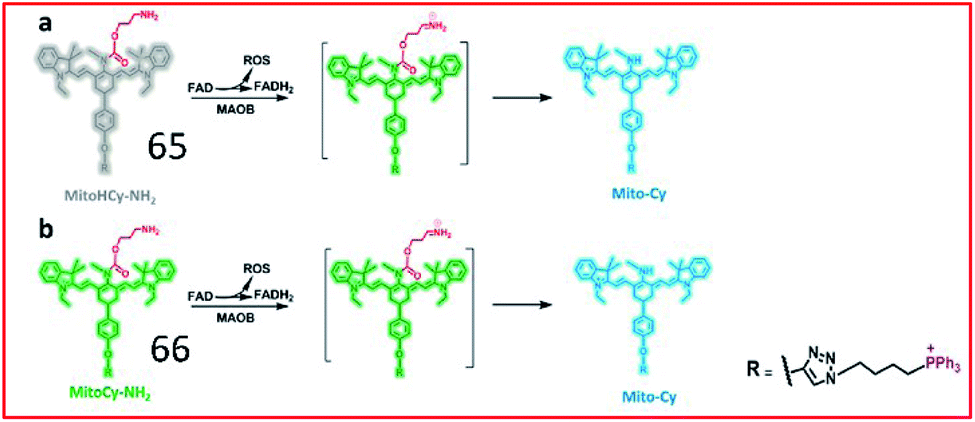 | ||
| Fig. 43 Molecular structures and the proposed detection mechanism of 65 and 66. Reprinted from ref. 165 with permission. Copyright 2018 American Chemical Society. | ||
Recently, a double-locked strategy was used to design a fluorescent probe. Zhang et al. designed a double-locked probe (67, NML) for the detection of LAP and MAO activities (Fig. 44). Probe 67 contained leucine, propylamine and an NIR fluorophore (NF). The leucine was connected to the amino group of propylamine through a pseudo-peptide bond, and the other end of the propylamine was incorporated into NF with an elimination-type spacer (para-hydroxybenzyl group). In the presence of LAP and MAO, the pseudo-peptide bond between the leucine and propylamine groups was cleaved by LAP, and the exposed amino group was oxidized by MAO, which further caused a β-elimination reaction and released the NF. Probe 67 could distinguish normal, drug liver injury and cirrhotic mice by detecting the activities of LAP and MAO in the serum.166
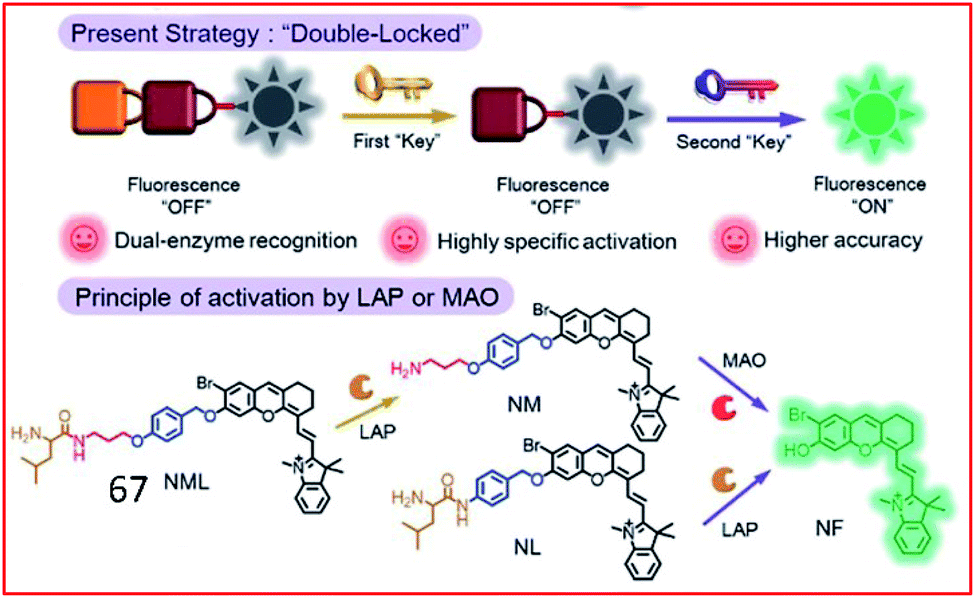 | ||
| Fig. 44 Design strategy of the enzyme-activated molecular probe 67. Reprinted from ref. 166 with permission. Copyright 2019 The Royal Society of Chemistry. | ||
Tang et al. synthesized a two-photon fluorescence probe (68, MCYN) that contained merocyanine as a fluorophore and dimethyl carbamate as an enzymatic recognition unit for the real-time detection of AChE activity (Fig. 45).168 When AChE catalysed the cleavage of the dimethyl carbamate group in probe 68, MCYO was produced, accompanied by an enhanced push–pull electron effect, causing the emission of bright fluorescence. Probe 68 was successfully employed to sensitively and selectively monitor AChE activity in living cells and in the brains of mice with depression. This work also suggested that the O2˙− level is positively correlated with AChE activity in depression.
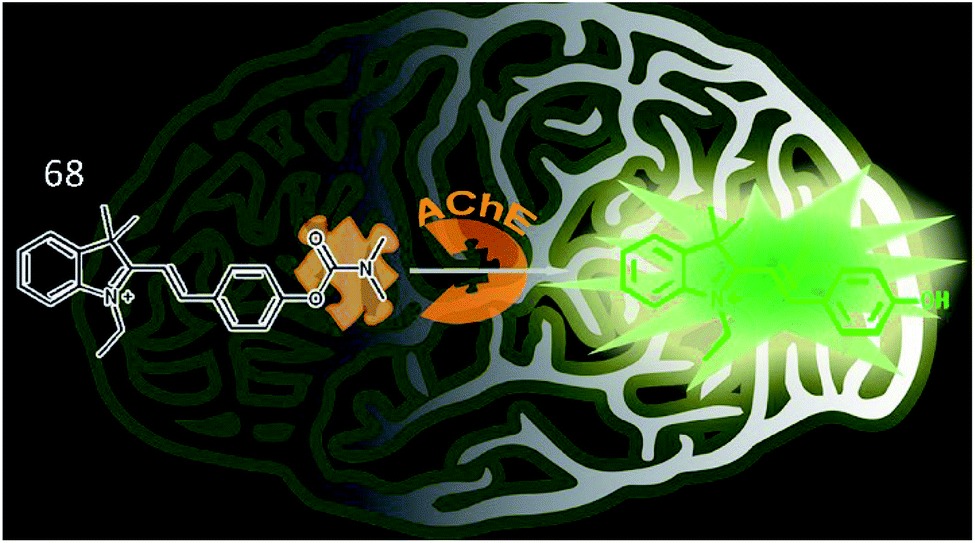 | ||
| Fig. 45 Recognition mechanism of probe 68 and structure of MCYO. Reprinted from ref. 168 with permission. Copyright 2019 American Chemical Society. | ||
Based on this work, Guo et al. developed an NIR “turn-on” fluorescent sensor (69, CyN) consisting of an N,N-dimethyl carbamyl moiety as the AChE recognition substrate and hemicyanine as the fluorescent reporter for the highly selective detection of the AChE (Fig. 46).169 Upon cleavage of the carbamyl group by AChE, probe 69 was transformed into a hemicyanine dye (CyOH). The fluorescence intensity of the probe was distinctly enhanced due to the push–pull electron effect in CyOH. This probe was used to monitor changes in AChE activity in cells and in zebrafish.
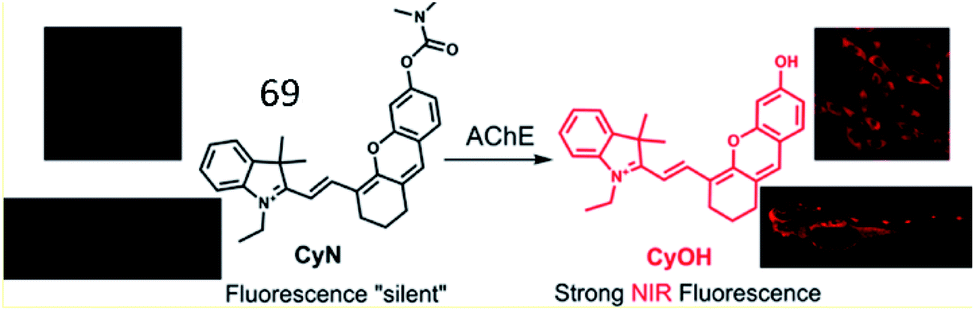 | ||
| Fig. 46 Proposed recognition mechanism for 69 sensing AChE. Reprinted from ref. 169 with permission. Copyright 2019 American Chemical Society. | ||
Furthermore, many other NIR fluorescence sensors and FRET-based nano probes were also developed to sense the activity of AChE.170,171 For instance, Qian et al. assembled a fluorescence nanoprobe, polycytosine-templated silver nanoclusters (dC12–Ag NCs), for the ultrasensitive tracking of AChE activity.172 Hu et al. exploited a carbon dot (CD)-based ratiometric fluorescence sensor to track AChE and butyrylcholinesterase (BChE) activities.173
3.4. Fibrotic disease
Fibrosis is frequently associated with many chronic inflammatory diseases, such as cardiomyopathy, diabetes, obesity, pneumonia and liver diseases. The excessive accumulation of fibrous connective tissues around damaged tissue leads to scarring, organ dysfunction, death, and diseases including renal fibrosis, idiopathic pulmonary fibrosis (IPF) and liver fibrosis.174 ROS play an essential role in fibrosis by their involvement in the TGF-β1 signalling pathway and post-transcriptional and epigenetic mechanisms. Although many immunological and molecular mechanisms contribute to the progression of fibrotic disease, specific biomarkers to identify disease and evaluate the effect of therapy are lacking.175 Therefore, it is highly important to develop fast, specific fluorescent probes to sense biomarkers of fibrotic diseases in real time.Li et al. reported a bioluminogenic probe (70) for the imaging of FAP in vivo and screening of FAP inhibitors in vitro.181N-Carbobenzyloxy–Gly–Pro–OH (Cbz–Gly–Pro–OH) served as the FAP substrate in this probe (Fig. 47). When the substrate of firefly luciferase, aminoluciferin, was caged by Cbz–Gly–Pro–OH, the bioluminescence of probe 70 was undetectable. After being released by FAP, the probe generated bioluminescence via the reaction between aminoluciferin and firefly luciferase. Probe 70 showed a 185-fold increase in bioluminescence upon the addition of 500 ng mL−1 FAP. With an extremely low detection limit of 0.254 ng mL−1, probe 70 was used to evaluate the activity of FAP with high selectivity and sensitivity in living mice.
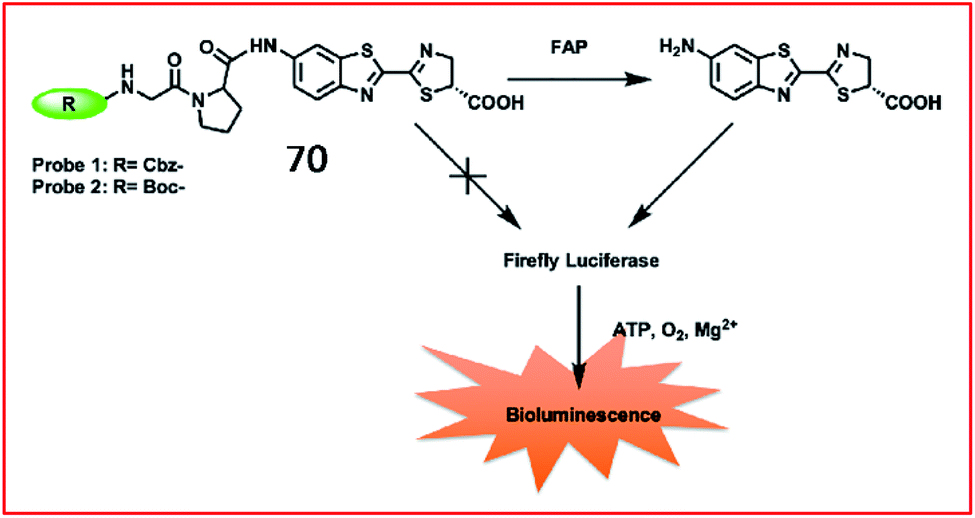 | ||
| Fig. 47 Design strategy for the probes 70 of FAP. Reprinted from ref. 181 with permission. Copyright 2019 American Chemical Society. | ||
Recently, a series of NIR fluorescent probes for the detection of FAP were developed. Pu et al. reported an NIR fluorescent probe (71, FNP1) that could be activated by FAPa to image keloid and skin cells (Fig. 48).182 This probe consisted of a CyOH as the fluorophore and a carbobenzyloxy–Gly–ProOH (Cbz–Gly–Pro) as a reactive group specific for FAPa. These two moieties were linked by a carbamate-based self-immolative moiety. With the addition of FAPa, the substrate and the self-immolative moiety were cleaved; therefore, CyOH, which contained a free hydroxyl group, emitted bright fluorescence. Due to its beneficial high selectivity and sensitivity towards FAPa, probe 71 can distinguish keloid cells from other normal skin cells, allowing it to detect abnormal scar fibrous lesions at an early stage.
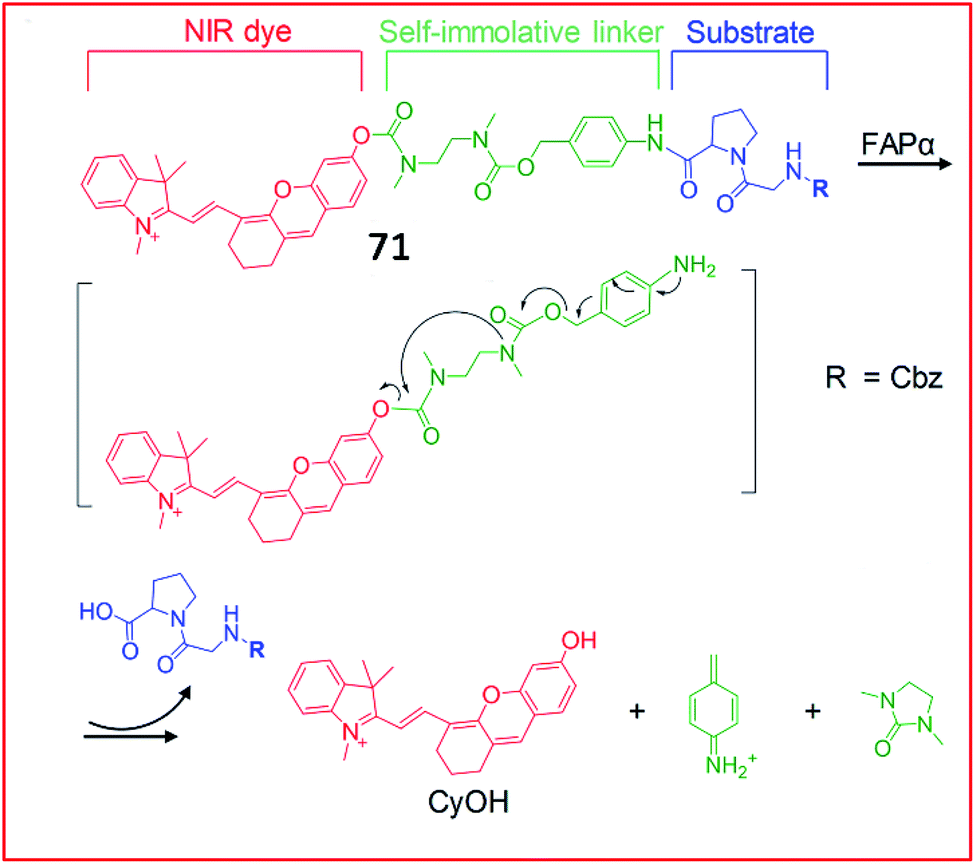 | ||
| Fig. 48 Design and mechanism of 71 for FAP imaging. Reprinted from ref. 182 with permission. Copyright 2018 Wiley-VCH. | ||
Wu et al. developed a hemicyanine-based FAP-activatable NIR fluorescent probe (72, HCFP) consisting of hemicyanine as the fluorophore and a dipeptide derivative as the recognition group for FAP (Fig. 49).183 In the presence of FAP, the amide bond was cleaved, releasing the amino fluorophore, which exhibited strong fluorescence. Probe 72 possesses good selectivity and sensitivity for cancer cell detection via in vitro and in vivo imaging.
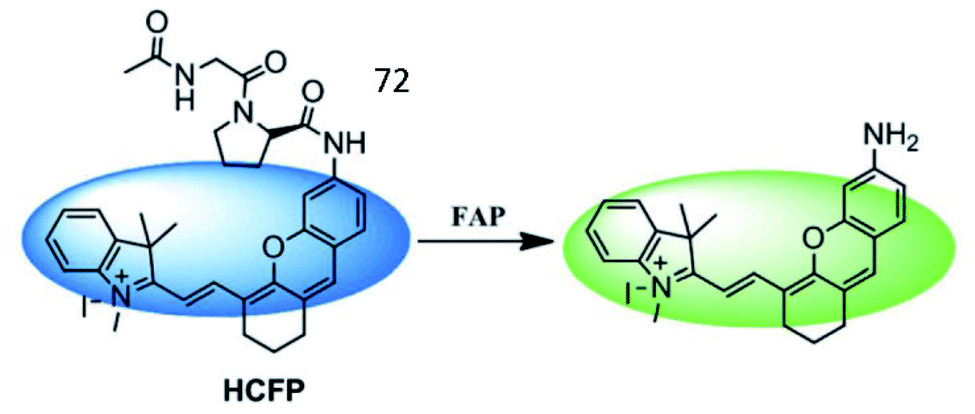 | ||
| Fig. 49 Mechanism of the probes 72 for FAP imaging. Reprinted from ref. 183 with permission. Copyright 2018 The Royal Society of Chemistry. | ||
Cheng et al. explored a FRET-based NIR fluorescence probe (73, APNFAP) for the detection of FAPα in vivo (Fig. 50). Probe 73 consisted of an NIR dye (Cy5.5) and a quenching dye (QSY21) linked by a FAPα-specific peptide substrate (KGPGPNQC). Due to the FRET effect between Cy5.5 and QSY21, the fluorescence of probe 73 was initially quenched. Upon cleavage by FAPα, the specific peptide bond was broken, causing the release of Cy5.5, which emitted bright fluorescence. Probe 73 was applied to label FAPα-positive tumour cells with high selectivity in vivo.184
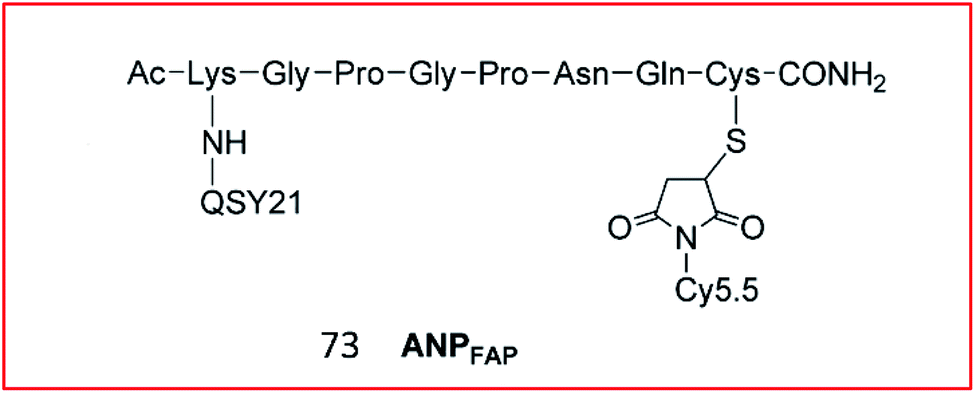 | ||
| Fig. 50 Structure of probe 73 for FAP imaging. Reprinted from ref. 184 with permission. Copyright 2012 American Chemical Society. | ||
Nagano et al. designed and synthesized the first GST fluorescent probe 74.187 The probe was composed of a xanthene group as a fluorophore and 3,4-dinitrobenzanilide (NNBA) as an enzyme-recognition site (Fig. 51). Probe 74 contained a hydrophilic carboxyl group that could detect GST activity in recombinant cells and lysates in vitro. To improve permeability, a lipophilic methyl group was introduced into probe 75. Therefore, 75 allowed good optical characterization to detect GST in living cells. In the presence of GST, GST catalysed GSH to replace the nitro group on the 5-position carbon in NNBA, enhancing fluorescence via blockade of the PeT process. Therefore, probe 74 can be used for high-throughput screening of GST inhibitors, and probe 75 can be applied to detect GST activity in live cells with high spatiotemporal resolution.
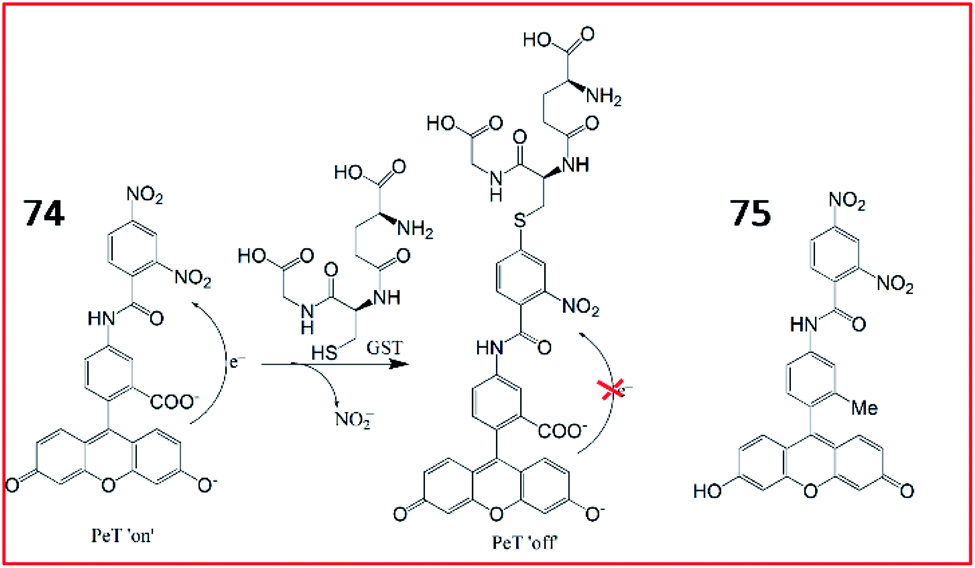 | ||
| Fig. 51 The structures of probe 74 and 75, and GST-catalyzed glutathionylation of 74. | ||
Lv et al. developed an “off–on” switch NIR fluorescent probe (76, Cy-GST) for the detection of GSTs (Fig. 52). Cy-GST contained a specific substrate of human GST (3,4-dinitrobenzanilide moiety) as the specific response group, and a benzoheptamethine cyanine dye as the fluorescent reporter group. The fluorescence of the probe was quenched due to PeT between the fluorophore and the strong electron-withdrawing 3,4-dinitrobenzanilide moiety. In the presence of GSTs, GSTs catalysed the binding of glutathione to probe 76, resulting in the sulfhydryl substitution derivative Cy-GST-GSH. Glutathionylation of probe 76 blocked the PeT process and restored the fluorescence. Probe 76 was used to detect GST in cells and IPF mouse models with high selectivity.188
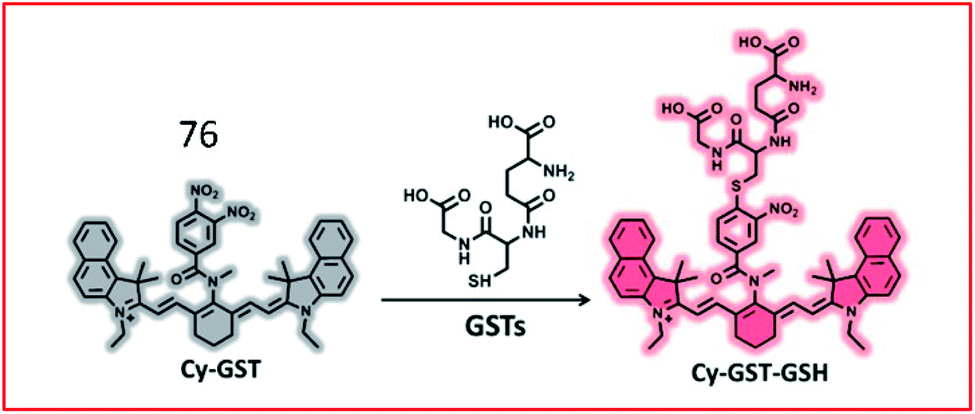 | ||
| Fig. 52 Proposed detection mechanism of the probe 76 against GSTs. Reprinted from ref. 188 with permission. Copyright 2019 American Chemical Society. | ||
3.5. Infectious diseases and inflammation diseases
Viruses, bacteria, and parasites comprise a vast group of aetiological agents that cause acute or chronic diseases and inflammation in humans. ROS have been implicated in several infectious diseases by inducing the assembly and activation of inflammasomes, which are multiprotein cytoplasmic complexes involved in innate immune signaling pathways in response to various pathogens and injury. Numerous proteins participate in intracellular immune-related signaling pathways and have the potential to be biomarkers or drug targets in the future. Recently, many fluorescent probes to sense the expression levels or activities of inflammatory proteins have been reported.189The leukotriene A4 hydrolase (LTA4H, EC 3.3.2.6) is a cytosolic dual-function enzyme that catalyses the hydrolysis of leukotriene A4 (LTA4) to form the proinflammatory mediator leukotriene B4 (LTB4). As a chemoattractant and stimulator that induces ROS generation in macrophages, neutrophils, T-cells, and other fibroblasts, LTB4 is involved in many human pathologies, such as chronic obstructive pulmonary disease, inflammatory bowel disease, rheumatoid arthritis, asthma, and connective tissue disease.190,191 Therefore, LTA4H is believed to participate in inflammatory progression by the biosynthesis of LTB4, which suggests that the development of fluorescent probes targeting LTA4H may contribute to drug discovery.
In 2018, Tang et al. discovered a small molecular fluorescent probe (77, ASPC) for the detection of LTA4H. This novel two-photon fluorescent probe (77) incorporated the substrate of LTA4H (unnatural amino acid L-AspBzl) into the fluorophore 7-amino-4-methylcoumarin (AMC) (Fig. 53).192 The amide bond between AMC and L-AspBzl in probe 77 could be recognized and broken by LTA4H, inducing bright green fluorescence under two-photon excitation at 690 nm. Probe 77 was capable of monitoring the activity of LTA4H in lipopolysaccharide-stimulated cells by one-photon and two-photon fluorescence imaging. Moreover, this probe could identify inflamed lung tissues by imaging LTA4H.
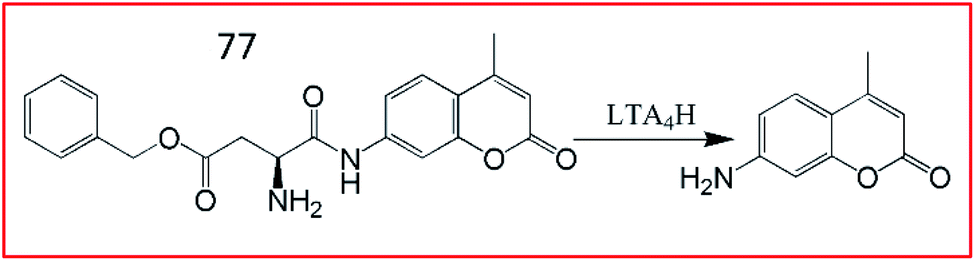 | ||
| Fig. 53 Schematic illustration of probe 77 for the detection of LTA4H. | ||
To obtain deeper tissue penetration, a fluorescent and photoacoustic dual-mode imaging probe (78, Cy-ASP) was developed by the same group in 2019 (Fig. 54).193 The NIR fluorophore hemicyanine (Cy-NH2) was chosen as the fluorescent and photoacoustic signal reporter, and L-AspBzl was used as the recognition site for LTA4H. Probe 78 could visualize LTA4H at inflammatory sites without dissection via fluorescence and photoacoustic dual-model imaging.
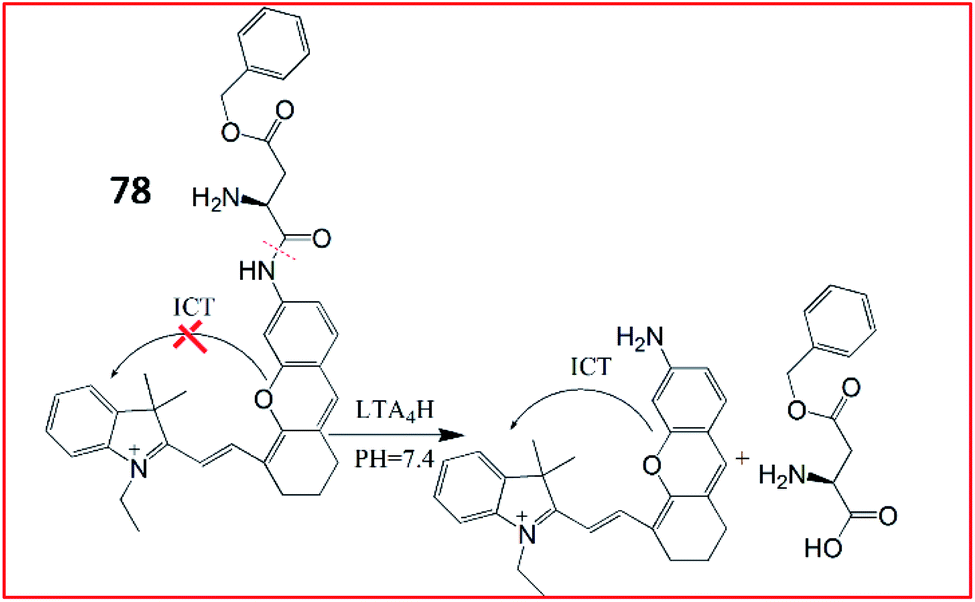 | ||
| Fig. 54 Sensing mechanism of probe 78 for the detection of LTA4H. | ||
Human neutrophil elastase (NE) is a single chain glycoprotein consisting of 267 amino acids belonging to the serine protease family.
When inflammation occurs, neutrophils are recruited to the inflammatory site, where they release the toxic molecules NE and ROS, leading to tissue destruction at the inflammatory site. Many studies have shown that a rise in NE levels is related to a variety of inflammatory diseases, especially chronic obstructive pulmonary disease, and acute lung injury. Yang et al. reported several fluorescent sensors to detect the activity of NE. The first non-peptide-based fluorescent probe that they developed (79) contained a 7-amino-4-trifluoromethyl coumarin group as the fluorophore and a pentafluoro-propionyl group as the quencher (Fig. 55a).194 The amide bond between the fluorophore and quencher was the reaction site for human NE. Specific cleavage of the amide bond in the probe by NE strongly enhances fluorescence. In 2019, the authors further presented an NIR fluorescent probe (80, NEP), which was based on a small-molecule substrate, for the detection of NE (Fig. 55b). Probe 80 consisted of a pentafluoroethyl moiety as the recognition unit and a hemicyanine dye moiety as the fluorophore. In the presence of NE, cleavage of the amide bond by NE restored the ICT process, inducing the strong NIR fluorescence of hemicyanine dye. Probe 80 was applied for the real-time monitoring of NE activity in an acute kidney injury (ALI) mouse model.195
 | ||
| Fig. 55 (a) The structure of probe 79. Reprinted from ref. 194 with permission. Copyright 2013 American Chemical Society. (b) Sensing mechanism of probe 80 for NE. Reprinted from ref. 195 with permission. Copyright 2019 American Chemical Society. | ||
4. Conclusions
The expression levels and activities of proteins are closely related to numerous metabolic processes, which are accompanied by oxidative stress to varying degrees, in living organisms. The cooperation between proteins and ROS affects the occurrence and development of diseases via either ROS-regulating proteins or ROS control by proteins. Thus, a thorough understanding of the expression levels and activities of ROS-associated proteins may promote defining the pathogenesis of diseases and accelerate drug development. In this review, we illustrate the strategies of fluorescent probe design and sensing mechanisms of fluorescent probes to identify these proteins with or without catalytic activity and summarize the advances in the imaging of these proteins in different diseases with typical probes made over the past decade. Even so, as some challenges for the accurate in situ detection of ROS-associated proteins remain, the following considerations are suggested:(1) Highly sensitive fluorescent probes for in situ monitoring of the dynamic ROS modification of proteins should be created. To date, ROS modification patterns and protein sites are usually investigated by mass spectrometry in vitro. However, this method cannot accurately evaluate the concentrations of ROS and proteins in living cells and neglects upstream and downstream regulation. Given that native conditions are absent, this in vitro method can only partially indicate the interaction between ROS and proteins. Therefore, one major issue that needs to be addressed is how to exploit novel fluorescent probes to examine the ROS modification patterns of proteins in real time.
(2) New probes with deeper signal penetration are needed. Optical penetration depths limit the clinical application of existing fluorescent probes, such as NIR probes whose penetration is still shallow at approximately 1 mm. The following strategies are selected as possible solutions. First, NIR fluorescent probes whose emission wavelengths are 1500–1700 nm may be improved tools due to their deep penetration (10 mm), low level of photo-scattering, and nearly absent autofluorescence.196 Second, PAI probes can provide images at a high spatial resolution from deep tissues through the combination of optical and ultrasonic signals. Third, CRET-based NIR fluorescent probes without an additional light source are promising for deep tissue imaging applications.197
(3) The probe response time should be improved. Low substrate specificity and the high degree of steric hindrance due to the probe seriously impede enzymatic reactions, leading to extended probe response times towards the target enzyme. To raise the reaction rate, screening novel specific substrates with low Michaelis constant might be helpful.
(4) The simultaneous detection of multiple proteins is needed. In living organisms, multiple proteins synchronously regulate cellular processes; however, traditional approaches still require extensive efforts to verify the undefined roles of some proteins in signalling networks. Using new probes with high selectivity and sensitivity, multicolour in situ fluorescence imaging will provide convincing evidence to reveal how various ROS-associated proteins are implicated in diseases.
(5) Diagnosis and therapy should be integrated. Based on the roles of ROS-associated proteins in diseases, multifunctional fluorescent probes could not only diagnose various diseases in real time by fluorescence imaging but also facilitate the use of suitable treatments.198 To further improve efficiency, theranostic agents might have other applications, such as MRI, drug delivery, and PDT/PTT.
Author contributions
Hui Wang was the main author of the review, and completed the collection and analysis of relevant literature and the writing of the first draft of the paper. Xin Wang and Mingyan Dong participated in the analysis and collation of literature; Ping Li, Shao Q. Yao, Bo Tang were the originators and principals of the review, who directed the writing of the paper. The final text was read and agreed upon by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding supports from the National Natural Science Foundation of China (91753111, 21675105, 21535004, 21927811, 21405097, 22174088), and the Key Research and Development Program of Shandong Province (2018YFJH0502) from China, the GSK-EDB Trust Fund (R-143-000-688-592) and the Synthetic Biology Research & Development Programme (SBP) of National Research Foundation (SBP-P4 and SBP-P8) of Singapore, are acknowledged.Notes and references
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10(Suppl), S10–S17 CrossRef.
- J. P. Taylor, J. Hardy and K. H. Fischbeck, Science, 2002, 296, 1991–1995 CrossRef CAS PubMed.
- H. W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X. B. Zhang and W. Tan, Chem. Soc. Rev., 2018, 47, 7140–7180 RSC.
- J. Zhang, X. Chai, X. P. He, H. J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683–722 RSC.
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS.
- O. C. Olson and J. A. Joyce, Nat. Rev. Cancer, 2015, 15, 712–729 CrossRef CAS PubMed.
- L. M. Coussens, B. Fingleton and L. M. Matrisian, Science, 2002, 295, 2387–2392 CrossRef CAS.
- A. Martinez, M. Portero-Otin, R. Pamplona and I. Ferrer, Brain Pathol., 2010, 20, 281–297 CrossRef CAS.
- H. Xiao, W. Zhang, P. Li, W. Zhang, X. Wang and B. Tang, Angew Chem. Int. Ed. Engl., 2020, 59, 4216–4230 CrossRef CAS.
- K. Brieger, S. Schiavone, F. J. Miller and K. H. Krause, Swiss Med. Wkly., 2012, 142, w13659 CAS.
- A. Popa-Wagner, S. Mitran, S. Sivanesan, E. Chang and A. M. Buga, Oxid. Med. Cell. Longevity, 2013, 2013, 963520 Search PubMed.
- D. H. F. H. B, Biomarkers, 1997, 2, 117–123 Search PubMed.
- X. Zhang, Y. Xiao and X. Qian, Angew Chem. Int. Ed. Engl., 2008, 47, 8025–8029 CrossRef CAS.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- L. Qian, L. Li and S. Q. Yao, Acc. Chem. Res., 2016, 49, 626–634 CrossRef CAS.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. D. Zhang, J. Mei, X. L. Hu, X. P. He and H. Tian, Small, 2016, 12, 6562–6567 CrossRef CAS PubMed.
- H. Shi, J. Liu, J. Geng, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 9569–9572 CrossRef CAS.
- H. Tong, K. Lou and W. Wang, Acta Pharm. Sin. B, 2015, 5, 25–33 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- W. M. Chang, M. Dakanali, C. C. Capule, C. J. Sigurdson, J. Yang and E. A. Theodorakis, ACS Chem. Neurosci., 2011, 2, 249–255 CrossRef CAS PubMed.
- C. H. Heo, K. H. Kim, H. J. Kim, S. H. Baik, H. Song, Y. S. Kim, J. Lee, I. Mook-jung and H. M. Kim, Chem. Commun., 2013, 49, 1303–1305 RSC.
- C. H. Heo, A. R. Sarkar, S. H. Baik, T. S. Jung, J. J. Kim, H. Kang, I. Mook-Jung and H. M. Kim, Chem. Sci., 2016, 7, 4600–4606 RSC.
- J. Zhang, X. Wang, V. Vikash, Q. Ye, D. Wu, Y. Liu and W. Dong, Oxid. Med. Cell. Longevity, 2016, 2016, 4350965 Search PubMed.
- L. Tochhawng, S. Deng, S. Pervaiz and C. T. Yap, Mitochondrion, 2013, 13, 246–253 CrossRef CAS PubMed.
- V. Sosa, T. Moline, R. Somoza, R. Paciucci, H. Kondoh and M. E. LLeonart, Ageing Res. Rev., 2013, 12, 376–390 CrossRef CAS PubMed.
- G. Y. Liou and P. Storz, Free Radic. Res., 2010, 44, 479–496 CrossRef CAS.
- M. Poreba, A. Strozyk, G. S. Salvesen and M. Drag, Cold Spring Harbor Perspect. Biol., 2013, 5, a008680 Search PubMed.
- Z. Su, Z. Yang, Y. Xu, Y. Chen and Q. Yu, Mol. Canc., 2015, 14, 48 CrossRef PubMed.
- S. J. Riedl and Y. Shi, Nat. Rev. Mol. Cell Biol., 2004, 5, 897–907 CrossRef CAS.
- R. S. Hotchkiss and D. W. Nicholson, Nat. Rev. Immunol., 2006, 6, 813–822 CrossRef CAS.
- X. H. Cao, A. H. Wang, C. L. Wang, D. Z. Mao, M. F. Lu, Y. Q. Cui and R. Z. Jiao, Chem. Biol. Interact., 2010, 183, 357–362 CrossRef CAS.
- J. H. Kim, S. Y. Lee, S. Y. Oh, S. I. Han, H. G. Park, M. A. Yoo and H. S. Kang, Oncol. Rep., 2004, 12, 1233–1238 CAS.
- T. I. Kim, H. Jin, J. Bae and Y. Kim, Anal. Chem., 2017, 89, 10565–10569 CrossRef CAS.
- D. Ye, A. J. Shuhendler, L. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. Rao, Nat. Chem., 2014, 6, 519–526 CrossRef CAS.
- M. Hu, L. Li, H. Wu, Y. Su, P. Y. Yang, M. Uttamchandani, Q. H. Xu and S. Q. Yao, J. Am. Chem. Soc., 2011, 133, 12009–12020 CrossRef CAS PubMed.
- O. Julien and J. A. Wells, Cell Death Differ., 2017, 24, 1380–1389 CrossRef CAS.
- S. Y. Li, L. H. Liu, H. Cheng, B. Li, W. X. Qiu and X. Z. Zhang, Chem. Commun., 2015, 51, 14520–14523 RSC.
- L. E. Edgington, A. B. Berger, G. Blum, V. E. Albrow, M. G. Paulick, N. Lineberry and M. Bogyo, Nat. Med., 2009, 15, 967–973 CrossRef CAS.
- T. I. Kim, H. Jin, J. Bae and Y. Kim, Anal. Chem., 2017, 89, 10565–10569 CrossRef CAS.
- W. Liu, S. J. Liu, Y. Q. Kuang, F. Y. Luo and J. H. Jiang, Anal. Chem., 2016, 88, 7867–7872 CrossRef CAS PubMed.
- Y. Wang, X. Hu, J. Weng, J. Li, Q. Fan, Y. Zhang and D. Ye, Angew Chem. Int. Ed. Engl., 2019, 58, 4886–4890 CrossRef CAS PubMed.
- H. Shi, R. T. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS PubMed.
- Y. Yuan, R. Zhang, X. Cheng, S. Xu and B. Liu, Chem. Sci., 2016, 7, 4245–4250 RSC.
- X. Liu, X. Song, D. Luan, B. Hu, K. Xu and B. Tang, Anal. Chem., 2019, 91, 5994–6002 CrossRef CAS PubMed.
- X. Gao, J. Li, M. Luan, Y. Li, W. Pan, N. Li and B. Tang, Biosens. Bioelectron., 2020, 147, 111755 CrossRef CAS PubMed.
- B. Bauvois and D. Dauzonne, Med. Res. Rev., 2006, 26, 88–130 CrossRef CAS.
- P. Mina-Osorio, Trends Mol. Med., 2008, 14, 361–371 CrossRef CAS PubMed.
- C. Santiago, G. Mudgal, J. Reguera, R. Recacha, S. Albrecht, L. Enjuanes and J. M. Casasnovas, Sci. Rep., 2017, 7, 46045 CrossRef CAS PubMed.
- Y. Wang, B. Pang, R. Zhang, Y. Fu and Q. Pang, Drug Des., Dev. Ther., 2019, 13, 3217–3228 CrossRef CAS.
- T. Kessler, A. Baumeier, C. Brand, M. Grau, L. Angenendt, S. Harrach, U. Stalmann, L. H. Schmidt, G. Gosheger, J. Hardes, D. Andreou, J. Dreischaluck, G. Lenz, E. Wardelmann, R. M. Mesters, C. Schwoppe, W. E. Berdel, W. Hartmann and C. Schliemann, Clin. Transl. Oncol., 2018, 11, 1271–1282 CrossRef.
- Y. Luan and W. Xu, Curr. Med. Chem., 2007, 14, 639–647 CrossRef CAS PubMed.
- L. Chen, W. Sun, J. Li, Z. Liu, Z. Ma, W. Zhang, L. Du, W. Xu, H. Fang and M. Li, Org. Biomol. Chem., 2013, 11, 378–382 RSC.
- X. He, Y. Xu, W. Shi and H. Ma, Anal. Chem., 2017, 89, 3217–3221 CrossRef CAS.
- X. He, Y. Hu, W. Shi, X. Li and H. Ma, Chem. Commun., 2017, 53, 9438–9441 RSC.
- H. Li, Q. Yao, W. Sun, K. Shao, Y. Lu, J. Chung, D. Kim, J. Fan, S. Long, J. Du, Y. Li, J. Wang, J. Yoon and X. Peng, J. Am. Chem. Soc., 2020, 142, 6381–6389 CrossRef CAS PubMed.
- H. Li, Y. Li, Q. Yao, J. Fan, W. Sun, S. Long, K. Shao, J. Du, J. Wang and X. Peng, Chem. Sci., 2019, 10, 1619–1625 RSC.
- T. Shiomi, V. Lemaitre, J. D'Armiento and Y. Okada, Pathol. Int., 2010, 60, 477–496 CrossRef CAS.
- S. T. Alamgeer, U. H. Hasan, A. M. Uttra, S. Qasim, J. Ikram, M. Saleem and Z. R. Niazi, Phytomedicine, 2020, 66, 153134 CrossRef.
- G. H. Lee, S. W. Jin, S. J. Kim, T. H. Pham, J. H. Choi and H. G. Jeong, Toxicol. Res., 2019, 35, 93–101 CrossRef CAS.
- D. E. Lee, M. Y. Chung, T. G. Lim, W. B. Huh, H. J. Lee and K. W. Lee, J. Food Sci., 2013, 78, H1464–H1469 CrossRef CAS.
- C. Muvva, S. Patra and S. Venkatesan, PLoS One, 2016, 11, e0159321 CrossRef PubMed.
- C. Gialeli, A. D. Theocharis and N. K. Karamanos, FEBS J., 2011, 278, 16–27 CrossRef CAS.
- S. Quintero-Fabian, R. Arreola, E. Becerril-Villanueva, J. C. Torres-Romero, V. Arana-Argaez, J. Lara-Riegos, M. A. Ramirez-Camacho and M. E. Alvarez-Sanchez, Front. Oncol., 2019, 9, 1370 CrossRef.
- R. Lee, S. J. Choi, K. C. Moon, J. W. Park, K. Kim, S. Y. Yoon and I. Youn, ACS Biomater. Sci. Eng., 2019, 5, 3039–3048 CrossRef CAS PubMed.
- A. Lee, S. J. Choi, K. Park, J. W. Park, K. Kim, K. Choi, S. Y. Yoon and I. Youn, Bioconjugate Chem., 2013, 24, 1068–1074 CrossRef CAS PubMed.
- L. Zhu, Y. Ma, D. O. Kiesewetter, Y. Wang, L. Lang, S. Lee, G. Niu and X. Chen, ACS Chem. Biol., 2014, 9, 510–516 CrossRef CAS.
- T. Myochin, K. Hanaoka, S. Iwaki, T. Ueno, T. Komatsu, T. Terai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 4759–4765 CrossRef CAS PubMed.
- X. Gao, L. Jiang, B. Hu, F. Kong, X. Liu, K. Xu and B. Tang, Anal. Chem., 2018, 90, 4719–4724 CrossRef CAS PubMed.
- H. Cheng, S. Y. Li, H. R. Zheng, C. X. Li, B. R. Xie, K. W. Chen, B. Li and X. Z. Zhang, Anal. Chem., 2017, 89, 4349–4354 CrossRef CAS PubMed.
- L. Yin, H. Sun, H. Zhang, L. He, L. Qiu, J. Lin, H. Xia, Y. Zhang, S. Ji, H. Shi and M. Gao, J. Am. Chem. Soc., 2019, 141, 3265–3273 CrossRef CAS.
- Y. Cheng, F. Huang, X. Min, P. Gao, T. Zhang, X. Li, B. Liu, Y. Hong, X. Lou and F. Xia, Anal. Chem., 2016, 88, 8913–8919 CrossRef CAS PubMed.
- J. Ni, Z. Wu, V. Stoka, J. Meng, Y. Hayashi, C. Peters, H. Qing, V. Turk and H. Nakanishi, Aging Cell, 2019, 18, e12856 CrossRef.
- S. U. Seo, S. M. Woo, M. W. Kim, H. S. Lee, S. H. Kim, S. C. Kang, E. W. Lee, K. J. Min and T. K. Kwon, Redox Biol., 2020, 30, 101422 CrossRef CAS PubMed.
- A. M. Szpaderska and A. Frankfater, Cancer Res., 2001, 61, 3493–3500 CAS.
- V. Turk, V. Stoka, O. Vasiljeva, M. Renko, T. Sun, B. Turk and D. Turk, Biochim. Biophys. Acta, 2012, 1824, 68–88 CrossRef CAS PubMed.
- Y. Wang, J. Li, L. Feng, J. Yu, Y. Zhang, D. Ye and H. Y. Chen, Anal. Chem., 2016, 88, 12403–12410 CrossRef CAS PubMed.
- M. K. Shim, H. Y. Yoon, J. H. Ryu, H. Koo, S. Lee, J. H. Park, J. H. Kim, S. Lee, M. G. Pomper, I. C. Kwon and K. Kim, Angew Chem. Int. Ed. Engl., 2016, 55, 14698–14703 CrossRef CAS.
- X. Chen, D. Lee, S. Yu, G. Kim, S. Lee, Y. Cho, H. Jeong, K. T. Nam and J. Yoon, Biomaterials, 2017, 122, 130–140 CrossRef CAS.
- Y. Yuan, C. J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew Chem. Int. Ed. Engl., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077–4098 RSC.
- R. Freeman, J. Elbaz, R. Gill, M. Zayats and I. Willner, Chemistry, 2007, 13, 7288–7293 CrossRef CAS.
- M. Perluigi, F. De Marco, C. Foppoli, R. Coccia, C. Blarzino, M. L. Marcante and C. Cini, Biochem. Biophys. Res. Commun., 2003, 305, 250–256 CrossRef CAS.
- B. K. Naidu, M. Won, I. Shim, N. Velusamy, Z. Yang, J. Qu, J. S. Kim and S. Bhuniya, Chem. Commun., 2017, 53, 11213–11216 RSC.
- I. Tessari, M. Bisaglia, F. Valle, B. Samori, E. Bergantino, S. Mammi and L. Bubacco, J. Biol. Chem., 2008, 283, 16808–16817 CrossRef CAS.
- K. Cho, C. S. Ryu, S. Jeong and Y. Kim, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2020, 228, 108655 CAS.
- J. Zhou, W. Shi, L. Li, Q. Gong, X. Wu, X. Li and H. Ma, Anal. Chem., 2016, 88, 4557–4564 CrossRef CAS PubMed.
- H. Li, W. Liu, F. Zhang, X. Zhu, L. Huang and H. Zhang, Anal. Chem., 2018, 90, 855–858 CrossRef CAS.
- C. Zhan, J. Cheng, B. Li, S. Huang, F. Zeng and S. Wu, Anal. Chem., 2018, 90, 8807–8815 CrossRef CAS.
- X. Wu, L. Li, W. Shi, Q. Gong and H. Ma, Angew Chem. Int. Ed. Engl., 2016, 55, 14728–14732 CrossRef CAS.
- J. Zhang, Z. Li, X. Tian and N. Ding, Chem. Commun., 2019, 55, 9463–9466 RSC.
- Z. Li, Y. F. Wang, C. Zeng, L. Hu and X. Liang, J. Anal. Chem., 2018, 90, 3666–3669 CAS.
- R. Gill, R. Freeman, J. P. Xu, I. Willner, S. Winograd, I. Shweky and U. Banin, J. Am. Chem. Soc., 2006, 128, 15376–15377 CrossRef CAS.
- M. Wang, J. L. Xie, J. Li, Y. Y. Fan, X. Deng, H. L. Duan and Z. Q. Zhang, ACS Sens., 2020, 5, 1634–1640 CrossRef CAS PubMed.
- Y. Chen, Anal. Biochem., 2020, 594, 113614 CrossRef CAS.
- X. Yan, H. Li, W. Zheng and X. Su, Anal. Chem., 2015, 87, 8904–8909 CrossRef CAS.
- M. Peng, Y. Wang, Q. Fu, F. Sun, N. Na and J. Ouyang, Anal. Chem., 2018, 90, 6206–6213 CrossRef CAS.
- T. I. Kim, J. Park, S. Park, Y. Choi and Y. Kim, Chem. Commun., 2011, 47, 12640–12642 RSC.
- R. Drozdz, C. Parmentier, H. Hachad, P. Leroy, G. Siest and M. Wellman, Free Radic. Biol. Med., 1998, 25, 786–792 CrossRef CAS.
- M. J. Accaoui, M. Enoiu, M. Mergny, C. Masson, S. Dominici, M. Wellman and A. Visvikis, Biochem. Biophys. Res. Commun., 2000, 276, 1062–1067 CrossRef CAS PubMed.
- M. W. Lieberman, A. L. Wiseman, Z. Z. Shi, B. Z. Carter, R. Barrios, C. N. Ou, P. Chevez-Barrios, Y. Wang, G. M. Habib, J. C. Goodman, S. L. Huang, R. M. Lebovitz and M. M. Matzuk, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 7923–7926 CrossRef CAS.
- S. S. Terzyan, A. W. Burgett, A. Heroux, C. A. Smith, B. H. Mooers and M. H. Hanigan, J. Biol. Chem., 2015, 290, 17576–17586 CrossRef CAS.
- M. B. West, Y. Chen, S. Wickham, A. Heroux, K. Cahill, M. H. Hanigan and B. H. Mooers, J. Biol. Chem., 2013, 288, 31902–31913 CrossRef CAS.
- A. Paolicchi, A. Pompella, P. Tonarelli, A. Gadducci, A. R. Genazzani, F. Zunino, G. Pratesi and R. Tongiani, Anticancer Res., 1996, 16, 3053–3058 CAS.
- H. Ma, L. Zhang, B. Tang, Y. Wang, R. Chen, B. Zhang, Y. Chen, N. Ge, Y. Wang, Y. Gan, S. Ye and Z. Ren, Ann. Surg Oncol., 2014, 21, 3084–3089 CrossRef PubMed.
- S. Park, D. J. Bae, Y. M. Ryu, S. Y. Kim, S. J. Myung and H. J. Kim, Chem. Commun., 2016, 52, 10400–10402 RSC.
- X. Zhou, Y. Liu, Q. Liu, L. Yan, M. Xue, W. Yuan, M. Shi, W. Feng, C. Xu and F. Li, Theranostics, 2019, 9, 4597–4607 CrossRef CAS.
- J. Ou-Yang, Y. Li, W. L. Jiang, S. Y. He, H. W. Liu and C. Y. Li, Anal. Chem., 2019, 91, 1056–1063 CrossRef CAS PubMed.
- Z. Luo, Z. Huang, K. Li, Y. Sun, J. Lin, D. Ye and H. Y. Chen, Anal. Chem., 2018, 90, 2875–2883 CrossRef CAS.
- F. Liu, Z. Wang, W. Wang, J. G. Luo and L. Kong, Anal. Chem., 2018, 90, 7467–7473 CrossRef CAS PubMed.
- H. Zhang, K. Wang, X. Xuan, Q. Lv, Y. Nie and H. Guo, Chem. Commun., 2016, 52, 6308–6311 RSC.
- L. Li, W. Shi, Z. Wang, Q. Gong and H. Ma, Anal. Chem., 2015, 87, 8353–8359 CrossRef CAS.
- J. Liu, S. Zhang, B. Zhao, C. Shen, X. Zhang and G. Yang, Biosens. Bioelectron., 2019, 142, 111497 CrossRef CAS PubMed.
- T. Fujii, M. Kamiya and Y. Urano, Bioconjugate Chem., 2014, 25, 1838–1846 CrossRef CAS.
- H. Tong, Y. Zheng, L. Zhou, X. Li, R. Qian, R. Wang, J. Zhao, K. Lou and W. Wang, Anal. Chem., 2016, 88, 10816–10820 CrossRef CAS.
- H. Li, Q. Yao, F. Xu, N. Xu, R. Duan, S. Long, J. Fan, J. Du, J. Wang and X. Peng, Biomaterials, 2018, 179, 1–14 CrossRef CAS PubMed.
- Y. Urano, M. Sakabe, N. Kosaka, M. Ogawa, M. Mitsunaga, D. Asanuma, M. Kamiya, M. R. Young, T. Nagano, P. L. Choyke and H. Kobayashi, Sci. Transl. Med., 2011, 3, 110ra119 Search PubMed.
- R. J. Iwatate, M. Kamiya and Y. Urano, Chemistry, 2016, 22, 1696–1703 CrossRef CAS.
- R. J. Iwatate, M. Kamiya, K. Umezawa, H. Kashima, M. Nakadate, R. Kojima and Y. Urano, Bioconjugate Chem., 2018, 29, 241–244 CrossRef CAS PubMed.
- Q. Husain, Crit. Rev. Biotechnol., 2010, 30, 41–62 CrossRef CAS.
- G. P. Dimri, X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E. E. Medrano, M. Linskens, I. Rubelj, O. Pereira-Smith and A. Et, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9363–9367 CrossRef CAS.
- J. Zhang, P. Cheng and K. Pu, Bioconjugate Chem., 2019, 30, 2089–2101 CrossRef PubMed.
- M. Kamiya, D. Asanuma, E. Kuranaga, A. Takeishi, M. Sakabe, M. Miura, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2011, 133, 12960–12963 CrossRef CAS.
- D. Asanuma, M. Sakabe, M. Kamiya, K. Yamamoto, J. Hiratake, M. Ogawa, N. Kosaka, P. L. Choyke, T. Nagano, H. Kobayashi and Y. Urano, Nat. Commun., 2015, 6, 6463 CrossRef CAS PubMed.
- X. Kong, M. Li, B. Dong, Y. Yin, W. Song and W. Lin, Anal. Chem., 2019, 91, 15591–15598 CrossRef CAS PubMed.
- K. Gu, Y. Xu, H. Li, Z. Guo, S. Zhu, S. Zhu, P. Shi, T. D. James, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2016, 138, 5334–5340 CrossRef CAS.
- E. J. Kim, R. Kumar, A. Sharma, B. Yoon, H. M. Kim, H. Lee, K. S. Hong and J. S. Kim, Biomaterials, 2017, 122, 83–90 CrossRef CAS PubMed.
- X. Li, Y. Pan, H. Chen, Y. Duan, S. Zhou, W. Wu, S. Wang and B. Liu, Anal. Chem., 2020, 92, 5772–5779 CrossRef CAS PubMed.
- J. A. Chen, H. Pan, Z. Wang, J. Gao, J. Tan, Z. Ouyang, W. Guo and X. Gu, Chem. Commun., 2020, 56, 2731–2734 RSC.
- G. Jiang, G. Zeng, W. Zhu, Y. Li, X. Dong, G. Zhang, X. Fan, J. Wang, Y. Wu and B. Z. Tang, Chem. Commun., 2017, 53, 4505–4508 RSC.
- Y. Kayama, U. Raaz, A. Jagger, M. Adam, I. N. Schellinger, M. Sakamoto, H. Suzuki, K. Toyama, J. M. Spin and P. S. Tsao, Int. J. Mol. Sci., 2015, 16, 25234–25263 CrossRef CAS PubMed.
- H. Pei, Y. Yang, H. Zhao, X. Li, D. Yang, D. Li and Y. Yang, Oxid. Med. Cell. Longevity, 2016, 2016, 5470457 Search PubMed.
- C. A. Lygate, S. Bohl, H. M. Ten, K. M. Faller, P. J. Ostrowski, S. Zervou, D. J. Medway, D. Aksentijevic, L. Sebag-Montefiore, J. Wallis, K. Clarke, H. Watkins, J. E. Schneider and S. Neubauer, Cardiovasc. Res., 2012, 96, 466–475 CrossRef CAS PubMed.
- T. Zang, Y. Xie, S. Su, F. Liu, Q. Chen, J. Jing, R. Zhang, G. Niu and X. Zhang, Angew Chem. Int. Ed. Engl., 2020, 59, 10003–10007 CrossRef CAS PubMed.
- L. Zhang, D. Duan, Y. Liu, C. Ge, X. Cui, J. Sun and J. Fang, J. Am. Chem. Soc., 2014, 136, 226–233 CrossRef CAS PubMed.
- H. Ma, J. Zhang, Z. Zhang, Y. Liu and J. Fang, Chem. Commun., 2016, 52, 12060–12063 RSC.
- X. Li, B. Zhang, C. Yan, J. Li, S. Wang, X. Wei, X. Jiang, P. Zhou and J. Fang, Nat. Commun., 2019, 10, 2745 CrossRef PubMed.
- M. Hamer and Y. Chida, Psychol. Med., 2009, 39, 3–11 CrossRef CAS PubMed.
- H. Quan, E. Koltai, K. Suzuki, A. J. Aguiar, R. Pinho, I. Boldogh, I. Berkes and Z. Radak, Biochim. Biophys. Acta, Mol. Basis Dis., 2020, 1866, 165778 CrossRef CAS.
- Z. Liu, T. Zhou, A. C. Ziegler, P. Dimitrion and L. Zuo, Oxid. Med. Cell. Longevity, 2017, 2017, 2525967 Search PubMed.
- H. Fu, M. Cui, P. Tu, Z. Pan and B. Liu, Chem. Commun., 2014, 50, 11875–11878 RSC.
- X. Zhang, Y. Tian, Z. Li, X. Tian, H. Sun, H. Liu, A. Moore and C. Ran, J. Am. Chem. Soc., 2013, 135, 16397–16409 CrossRef CAS.
- C. H. Heo, K. H. Kim, H. J. Kim, S. H. Baik, H. Song, Y. S. Kim, J. Lee, I. Mook-jung and H. M. Kim, Chem. Commun., 2013, 49, 1303–1305 RSC.
- G. Lv, A. Sun, P. Wei, N. Zhang, H. Lan and T. Yi, Chem. Commun., 2016, 52, 8865–8868 RSC.
- W. Fu, C. Yan, Z. Guo, J. Zhang, H. Zhang, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2019, 141, 3171–3177 CrossRef CAS.
- J. D. Zhang, J. Mei, X. L. Hu, X. P. He and H. Tian, Small, 2016, 12, 6562–6567 CrossRef CAS.
- M. Goedert and M. G. Spillantini, Science, 2006, 314, 777–781 CrossRef CAS PubMed.
- W. H. Stoothoff and G. V. Johnson, Biochim. Biophys. Acta, 2005, 1739, 280–297 CrossRef CAS.
- N. Esteras, J. D. Rohrer, J. Hardy, S. Wray and A. Y. Abramov, Redox Biol., 2017, 12, 410–422 CrossRef CAS.
- J. Gu, U. R. Anumala, H. R. Heyny-von, J. Holzer, V. Goetschy-Meyer, G. Mall, I. Hilger, C. Czech and B. Schmidt, ChemMedChem, 2013, 8, 891–897 CrossRef CAS.
- S. Lim, M. M. Haque, D. Su, D. Kim, J. S. Lee, Y. T. Chang and Y. K. Kim, Chem. Commun., 2017, 53, 1607–1610 RSC.
- Y. Seo, K. S. Park, T. Ha, M. K. Kim, Y. J. Hwang, J. Lee, H. Ryu, H. Choo and Y. Chong, ACS Chem. Neurosci., 2016, 7, 1474–1481 CrossRef CAS PubMed.
- K. S. Park, Y. Seo, M. K. Kim, K. Kim, Y. K. Kim, H. Choo and Y. Chong, Org. Biomol. Chem., 2015, 13, 11194–11199 RSC.
- E. Vandermarliere, B. Ghesquiere, V. Jonckheere, K. Gevaert and L. Martens, Proteomics, 2014, 14, 1990–1998 CrossRef CAS PubMed.
- L. Lai, J. Sun, S. Tarafdar, C. Liu, E. Murphy, G. Kim and R. L. Levine, Free Radic. Biol. Med., 2019, 145, 374–384 CrossRef CAS.
- M. L. Circu and T. Y. Aw, Free Radic. Biol. Med., 2010, 48, 749–762 CrossRef CAS.
- X. Wu, W. Shi, X. Li and H. Ma, Angew Chem. Int. Ed. Engl., 2017, 56, 15319–15323 CrossRef CAS.
- W. Y. Kim, M. Won, A. Salimi, A. Sharma, J. H. Lim, S. H. Kwon, J. Y. Jeon, J. Y. Lee and J. S. Kim, Chem. Commun., 2019, 55, 13267–13270 RSC.
- L. Li, C. W. Zhang, G. Y. Chen, B. Zhu, C. Chai, Q. H. Xu, E. K. Tan, Q. Zhu, K. L. Lim and S. Q. Yao, Nat. Commun., 2014, 5, 3276 CrossRef PubMed.
- L. Li, C. W. Zhang, J. Ge, L. Qian, B. H. Chai, Q. Zhu, J. S. Lee, K. L. Lim and S. Q. Yao, Angew Chem. Int. Ed. Engl., 2015, 54, 10821–10825 CrossRef CAS.
- H. Fang, H. Zhang, L. Li, Y. Ni, R. Shi, Z. Li, X. Yang, B. Ma, C. Zhang, Q. Wu, C. Yu, N. Yang, S. Q. Yao and W. Huang, Angew Chem. Int. Ed. Engl., 2020, 59, 7536–7541 CrossRef CAS PubMed.
- Z. M. Yang, Q. Y. Mo, J. M. He, D. L. Mo, J. Li, H. Chen, S. L. Zhao and J. K. Qin, ACS Sens., 2020, 5, 943–951 CrossRef CAS PubMed.
- R. Wang, X. Han, J. You, F. Yu and L. Chen, Anal. Chem., 2018, 90, 4054–4061 CrossRef CAS.
- Y. Liu, L. Teng, C. Xu, H. W. Liu, S. Xu, H. Guo, L. Yuan and X. B. Zhang, Chem. Sci., 2019, 10, 10931–10936 RSC.
- W. Li, W. Li, Y. Hu, Y. Xia, Q. Shen, Z. Nie, Y. Huang and S. Yao, Biosens. Bioelectron., 2013, 47, 345–349 CrossRef CAS PubMed.
- X. Wang, P. Li, Q. Ding, C. Wu, W. Zhang and B. Tang, J. Am. Chem. Soc., 2019, 141, 2061–2068 CrossRef CAS.
- J. Ma, T. Si, C. Yan, Y. Li, Q. Li, X. Lu and Y. Guo, ACS Sens., 2020, 5, 83–92 CrossRef CAS PubMed.
- Y. Chen, W. Liu, B. Zhang, Z. Suo, F. Xing and L. Feng, Anal. Biochem., 2020, 607, 113835 CrossRef CAS.
- X. Xu, Y. Cen, G. Xu, F. Wei, M. Shi and Q. Hu, Biosens. Bioelectron., 2019, 131, 232–236 CrossRef CAS PubMed.
- Y. Zhang, Y. Cai, Z. Qi, L. Lu and Y. Qian, Anal. Chem., 2013, 85, 8455–8461 CrossRef CAS.
- X. Xu, Y. Cen, G. Xu, F. Wei, M. Shi and Q. Hu, Biosens. Bioelectron., 2019, 131, 232–236 CrossRef CAS.
- K. Kis, X. Liu and J. S. Hagood, Expert Rev. Mol. Med., 2011, 13, e27 CrossRef PubMed.
- R. Kramann, D. P. DiRocco and B. D. Humphreys, J. Pathol., 2013, 231, 273–289 CrossRef CAS.
- M. T. Levy, G. W. McCaughan, C. A. Abbott, J. E. Park, A. M. Cunningham, E. Muller, W. J. Rettig and M. D. Gorrell, Hepatology, 1999, 29, 1768–1778 CrossRef CAS.
- A. Simkova, P. Busek, A. Sedo and J. Konvalinka, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140409 CrossRef CAS.
- N. Sampson, E. Brunner, A. Weber, M. Puhr, G. Schafer, C. Szyndralewiez and H. Klocker, Int. J. Cancer, 2018, 143, 383–395 CrossRef CAS PubMed.
- J. Xing, Q. Gong, R. Zou, Z. Li, Y. Xia, Z. Yu, Y. Ye, L. Xiang and A. Wu, J. Mater. Chem. B, 2018, 6, 1449–1451 RSC.
- A. Simkova, P. Busek, A. Sedo and J. Konvalinka, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140409 CrossRef CAS.
- Y. Lin, Z. Ma, Z. Li, Y. Gao, X. Qin, Z. Zhang, G. Wang, L. Du and M. Li, Anal. Chem., 2019, 91, 14873–14878 CrossRef CAS PubMed.
- Q. Miao, D. C. Yeo, C. Wiraja, J. Zhang, X. Ning, C. Xu and K. Pu, Angew Chem. Int. Ed. Engl., 2018, 57, 1256–1260 CrossRef CAS.
- J. Xing, Q. Gong, R. Zou, Z. Li, Y. Xia, Z. Yu, Y. Ye, L. Xiang and A. Wu, J. Mater. Chem. B, 2018, 6, 1449–1451 RSC.
- J. Li, K. Chen, H. Liu, K. Cheng, M. Yang, J. Zhang, J. D. Cheng, Y. Zhang and Z. Cheng, Bioconjugate Chem., 2012, 23, 1704–1711 CrossRef CAS PubMed.
- H. Raza, M. A. Robin, J. K. Fang and N. G. Avadhani, Biochem. J., 2002, 366, 45–55 CrossRef CAS.
- A. Oakley, Drug Metab. Rev., 2011, 43, 138–151 CrossRef CAS.
- Y. Fujikawa, Y. Urano, T. Komatsu, K. Hanaoka, H. Kojima, T. Terai, H. Inoue and T. Nagano, J. Am. Chem. Soc., 2008, 130, 14533–14543 CrossRef CAS PubMed.
- N. He, S. Bai, Y. Huang, Y. Xing, L. Chen, F. Yu and C. Lv, Anal. Chem., 2019, 91, 5424–5432 CrossRef CAS PubMed.
- F. Martinon, Eur. J. Immunol., 2010, 40, 616–619 CrossRef CAS.
- K. J. Cho, J. M. Seo and J. H. Kim, Mol. Cells, 2011, 32, 1–5 CrossRef CAS PubMed.
- X. Jiang, L. Zhou, D. Wei, H. Meng, Y. Liu and L. Lai, Bioorg. Med. Chem. Lett., 2008, 18, 6549–6552 CrossRef CAS.
- H. Wang, K. Xue, P. Li, Y. Yang, Z. He, W. Zhang, W. Zhang and B. Tang, Anal. Chem., 2018, 90, 6020–6027 CrossRef CAS.
- H. Wang, K. Xue, Z. Duan, Y. Yang, Z. He, C. Wu, W. Zhang, W. Zhang, P. Li and B. Tang, Sens. Actuators, B, 2019, 286, 243–249 CrossRef CAS.
- Q. Sun, J. Li, W. N. Liu, Q. J. Dong, W. C. Yang and G. F. Yang, Anal. Chem., 2013, 85, 11304–11311 CrossRef CAS PubMed.
- S. Y. Liu, H. Xiong, R. R. Li, W. C. Yang and G. F. Yang, Anal. Chem., 2019, 91, 3877–3884 CrossRef CAS.
- J. A. Chen, H. Pan, Z. Wang, J. Gao, J. Tan, Z. Ouyang, W. Guo and X. Gu, Chem. Commun., 2020, 56, 2731–2734 RSC.
- X. Jie, M. Wu, H. Yang and W. Wei, Anal. Chem., 2019, 91, 13174–13182 CrossRef CAS PubMed.
- Y. Cheng, F. Huang, X. Min, P. Gao, T. Zhang, X. Li, B. Liu, Y. Hong, X. Lou and F. Xia, Anal. Chem., 2016, 88, 8913–8919 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |