
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Disila- and digermabenzenes†
Takahiro
Sasamori
 *
*
Division of Chemistry, Faculty of Pure and Applied Sciences, Tsukuba Research Center for Energy Materials Science (TREMS), University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan. E-mail: sasamori@chem.tsukuba.ac.jp
First published on 26th March 2021
Abstract
Reactions of isolable disilynes and digermynes with alkynes can result in the formation of the corresponding disila- (DSBs) and digermabenzenes (DGBs), wherein two carbon atoms of the benzene ring are replaced by silicon or germanium atoms. Detailed structural and spectroscopic analyses of these DSBs and DGBs have revealed that they exhibit considerable aromaticity, comparable to that of benzene. However, in contrast to the all-carbon system benzene, these DSBs and DGBs are highly reactive toward small molecules such as oxygen, hydrogen, 1,3-dienes, and water. During the investigation of their reactivity, we discovered that a 1,2-DGB works as a catalyst for the cyclotrimerization of arylalkynes, which provides access to the corresponding 1,2,4-triarylbenzenes. In this perspective article, our recent progress in the area of DSB and DGB chemistry is summarized.
1. Introduction
Multiple-bond compounds of heavier-group-14 elements represent the heavier homologues of unsaturated organic compounds.1 Traditionally, these compounds had been considered hard to isolate as stable monomeric compounds under ambient conditions. This notion was mostly due to their inherently high reactivity toward self-oligomerization and addition reactions with moisture and/or aerobic oxygen on account of their π-bonds, which are weak relative to the corresponding σ-bonds.1,2 However, it has since been demonstrated that the introduction of sterically demanding substituents on the heavier-group-14 elements can kinetically protect the corresponding π-bonds and thus render such compounds stable enough to be isolated under ambient conditions. Thus, several examples of kinetically stabilized aromatic compounds including a heavier-group-14 element using sterically demanding substituents have been reported,1–3 in addition to the already known heavy aromatic systems that bear a heavier-main-group element other than those from group 14 or a transition metal.4 With those examples in hand, the physical and chemical properties of the π-bonds between these heavier-group-14 elements have been thoroughly investigated experimentally and theoretically.2 It has been revealed that the bonding situation of these E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E double bonds are different from those of CC double bonds. For example, in contrast to the planar geometry of most CC double bonds, EE bonds (E: heavier-group-14 element) often exhibit a trans-pyramidalyzed geometry. Moreover, the extent of bond-shortening of the EE bond relative to the corresponding single bond is not as pronounced as for the CC bonds. These structural features could be feasibly interpreted in terms of a double donor–acceptor bond (based on valence-bond theory) or so-called second-order Jahn–Teller mixing of the π- and σ*-orbitals (Fig. 1).2,3 Thus, in case of heavier-group-14 elements, the term “π-bond” would be formally suitable, as it comprises np-orbitals and the mixing of orbitals, which would be different from the carbon systems. In this regard, it should be of great interest, to replace not only a HC moiety of benzene with an RE moiety (R = substituent; E = heavier-group-14 element), but also a HCCH moiety of benzene with an REER moiety.
E double bonds are different from those of CC double bonds. For example, in contrast to the planar geometry of most CC double bonds, EE bonds (E: heavier-group-14 element) often exhibit a trans-pyramidalyzed geometry. Moreover, the extent of bond-shortening of the EE bond relative to the corresponding single bond is not as pronounced as for the CC bonds. These structural features could be feasibly interpreted in terms of a double donor–acceptor bond (based on valence-bond theory) or so-called second-order Jahn–Teller mixing of the π- and σ*-orbitals (Fig. 1).2,3 Thus, in case of heavier-group-14 elements, the term “π-bond” would be formally suitable, as it comprises np-orbitals and the mixing of orbitals, which would be different from the carbon systems. In this regard, it should be of great interest, to replace not only a HC moiety of benzene with an RE moiety (R = substituent; E = heavier-group-14 element), but also a HCCH moiety of benzene with an REER moiety.
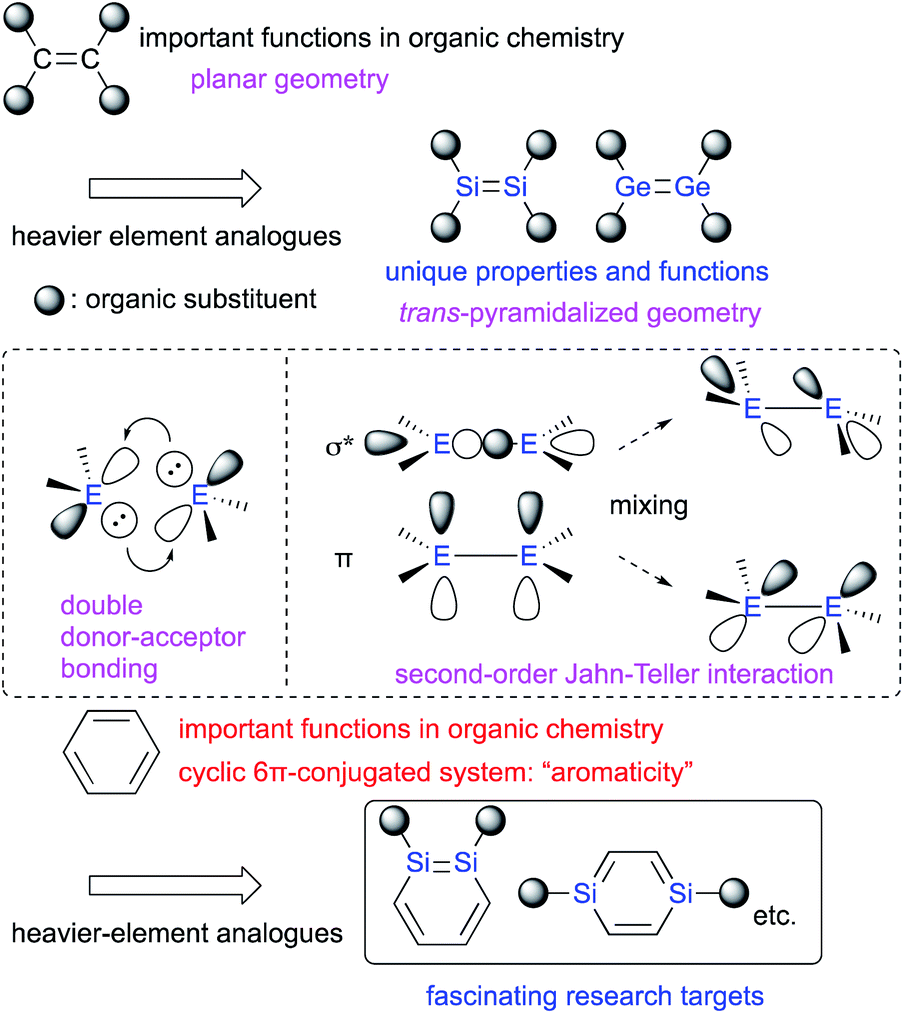 | ||
| Fig. 1 Heavier group-14 analogues of carbon-based π-bond compounds. | ||
The concept of π-bonds is closely associated with the concept of ‘π-conjugation’, and both are of pivotal importance for not only fundamental aspects of organic chemistry, but also for materials science in order to create e.g. organic optoelectronic materials.5 In contemporary organic chemistry, benzene has received particular attention due to its characteristic ‘aromaticity’, which arises from its cyclic π-conjugation system in combination with the delocalization of its six π-electrons (Fig. 1).6 Benzene analogues that incorporate heavier-group-14 elements have also garnered much interest as potential building blocks for optoelectronic materials, albeit that some difficulties associated with isolating these compounds due to their intrinsic reactivity and/or facile self-oligomerization remain to be addressed.6,7 Recently, the synthesis and isolation of stable metallabenzenes that contain heavier-group-14 elements have been achieved by using bulky substituents, which allows evaluating the aromaticity and reactivity of the heavier-group-14-element analogues of benzene relative to that of the all-carbon homologue. In this context, the recent developments of our research on disila- (DSBs) and digermabenzenes (DGBs) will be summarized.
2. Theoretical aspects
Among the benzene analogues that contain two silicon or germanium atoms, 1,2-disilabenzenes (1,2-DSBs) or 1,2-digermabenzenes (1,2-DGBs) represent some of the most fascinating research targets, especially with respect to the effect of replacing a CC unit in the 6π-electron aromatic system of benzene with an EE unit (E = Si or Ge), considering the unique differences of the π-electron character of the disilene (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) SiSi
SiSi![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ) moiety relative to the olefin moiety (CC).1–3,8 In other words, it should be very interesting to investigate which one of the two conceivable canonical structures of 1,2-dimetallabenzenes (I–Avs.I–B) provides the predominant contribution to the overall electronic structure. This is different to the 1,3- and 1,4-dimetallabenzenes, where the conceivable canonical resonance structures A and B should exhibit the same electronic structures (Fig. 2).
) moiety relative to the olefin moiety (CC).1–3,8 In other words, it should be very interesting to investigate which one of the two conceivable canonical structures of 1,2-dimetallabenzenes (I–Avs.I–B) provides the predominant contribution to the overall electronic structure. This is different to the 1,3- and 1,4-dimetallabenzenes, where the conceivable canonical resonance structures A and B should exhibit the same electronic structures (Fig. 2).
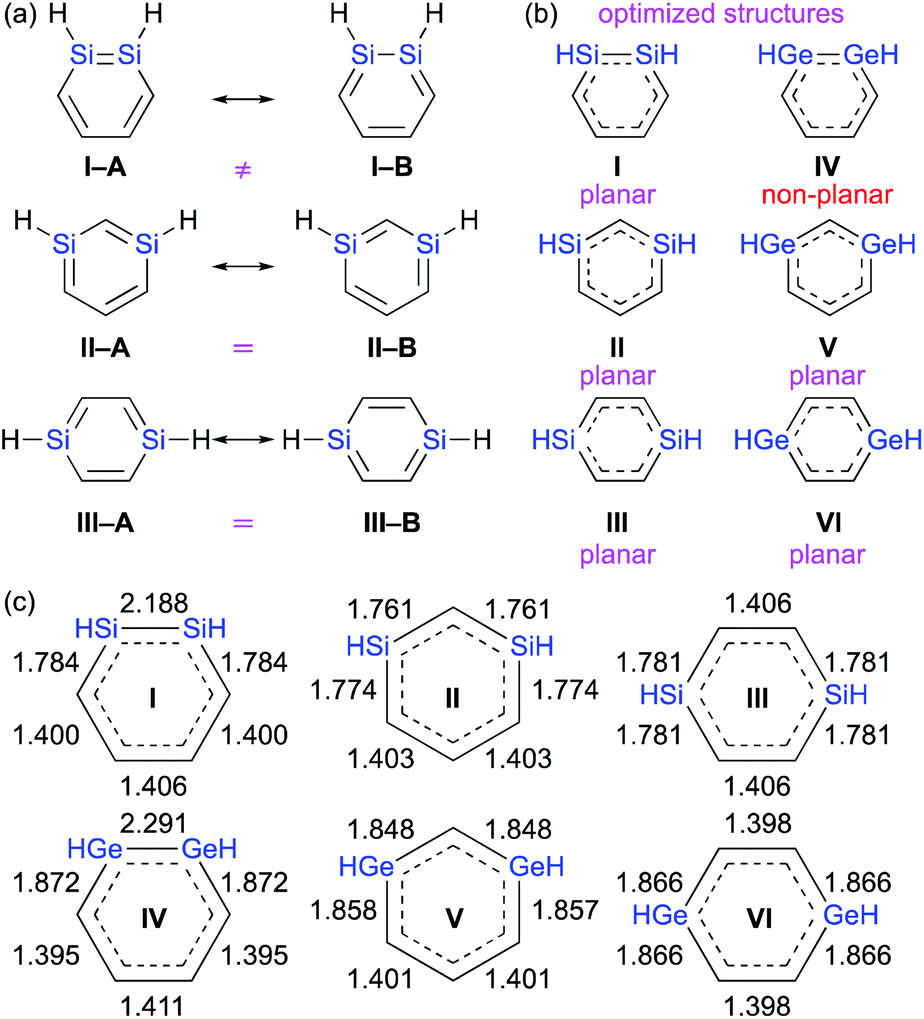 | ||
| Fig. 2 (a) Canonical resonance structures, (b) planarity of the optimized structures, and (c) optimized bond lengths of DSBs and DGBs, calculated at the MP2/6-311G(3d) level of theory. | ||
Moreover, the conceivable valence isomerizations of the dimetallabenzenes should also be very interesting (Fig. 3), given that the corresponding dimetalla-benzvalene, -prismane, and -Dewar-benzene isomers would exhibit unique cage structures. Considering that several theoretical investigations on dimetallabenzenes and their valence isomers have already been reported,9 we summarize only the most crucial results of our calculations in Table 1.10 Notably, 1,3-DSB is the most stable DSB, while 1,2-DGB is the most stable DGB. In addition, the relative energies of the valence isomers that do not contain an EE double bond are shown in Table 1. As in the case of the all-carbon systems, the corresponding benzvalene-, prismane-, and Dewar-type isomers are thermodynamically unstable relative to the dimetallabenzenes, albeit that the energy difference between the valence isomers and the dimetallabenzenes is smaller than those for the corresponding carbon cases. Against this theoretical background, it seems feasible to assume that the synthesis of dimetallabenzenes, in contrast to some isolated valence isomers,11 remained unsuccessful for a long time due to the lack of appropriate synthetic routes and sufficient kinetic stability to avoid self-oligomerization.12 The unexpected thermodynamic stability of dimetallabenzenes relative to the other valence isomers should most likely be interpreted in terms of their considerable aromatic stabilization energy and/or the severe ring strain in the valence isomers.
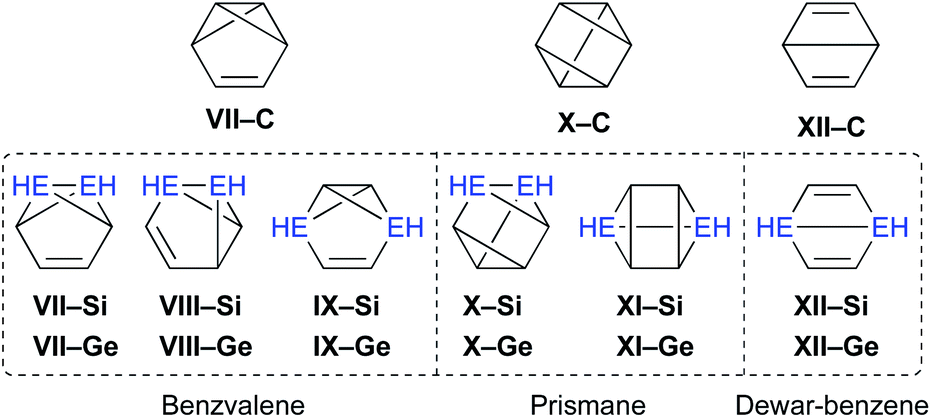 | ||
| Fig. 3 Possible valence isomers of benzene and dimetallabenzenes (E = C, Si, Ge) that bear no Si/Ge π-bond. | ||
| (a) Isomers | DSBs | DGBs | ||||
|---|---|---|---|---|---|---|
| 1,2-(I) | 1,3-(II) | 1,4-(III) | 1,2-(IV) | 1,3-(V) | 1,4-(VI) | |
| a Not the minimum structure. | ||||||
| Relative energy | 5.3 | 0.0 | 10.8 | 0.0 | 11.0 | 19.5 |
The theoretically optimized bond lengths of DSBs and DGBs should be indicative of their resonance structures (Fig. 2). Even though the C–C bond lengths in 1,2-DSBs are almost identical, those in 1,2-DGBs are slightly different (1.395 and 1.411 Å), which suggests nonnegligible bond-alternation and a GeGe character. Moreover, 1,2-DGB IV exhibits a non-planar geometry with a trans-bent structure at the GeGe moiety, which is similar to other isolable digermenes,8 and different to the completely planar structures of other DSBs and DGBs. In their entirety, the results of our theoretical calculations show that the π-electrons of the Ge–Ge moiety in a 1,2-DGB should be localized to at least some extent in the cyclic 6π-electrons conjugation system.
From a theoretical perspective, several indicators have been proposed for the evaluation of aromaticity.13 In order to compare the aromaticity of benzene, DSBs, and DGBs, the NICS(r) and NICSzz(r) values for H-substituted model compounds have been calculated at the GIAO-MP2/6-311G(3d)//MP2/6-311G(3d) (Fig. 4).14 The NICSzz(r) profile for benzene exhibits the highest absolute value (−32.3) at r = 1.0 Å above the center of the C6 plane, while the highest absolute value (−12.0) for the corresponding NICS(r) profile is observed at r = 0.7 Å. The profiles for DSBs I, II, and III as well as DGBs IV, V, and VI are similar in as far as they show considerable aromaticity, even though their absolute values are slightly lower than those of benzene (Fig. 1c). Based on the NICS(r) and NICSzz(r) profiles for both DSBs and DGBs, the 1,3-dimetallabenzenes exhibit the lowest degree of aromaticity among the corresponding dimetallabenzene derivatives. Based on the highest absolute NICS(r) and NICSzz(r) values, it can thus be concluded that the aromaticity in the DSBs and DGBs should be considerable, albeit lower than in benzene.
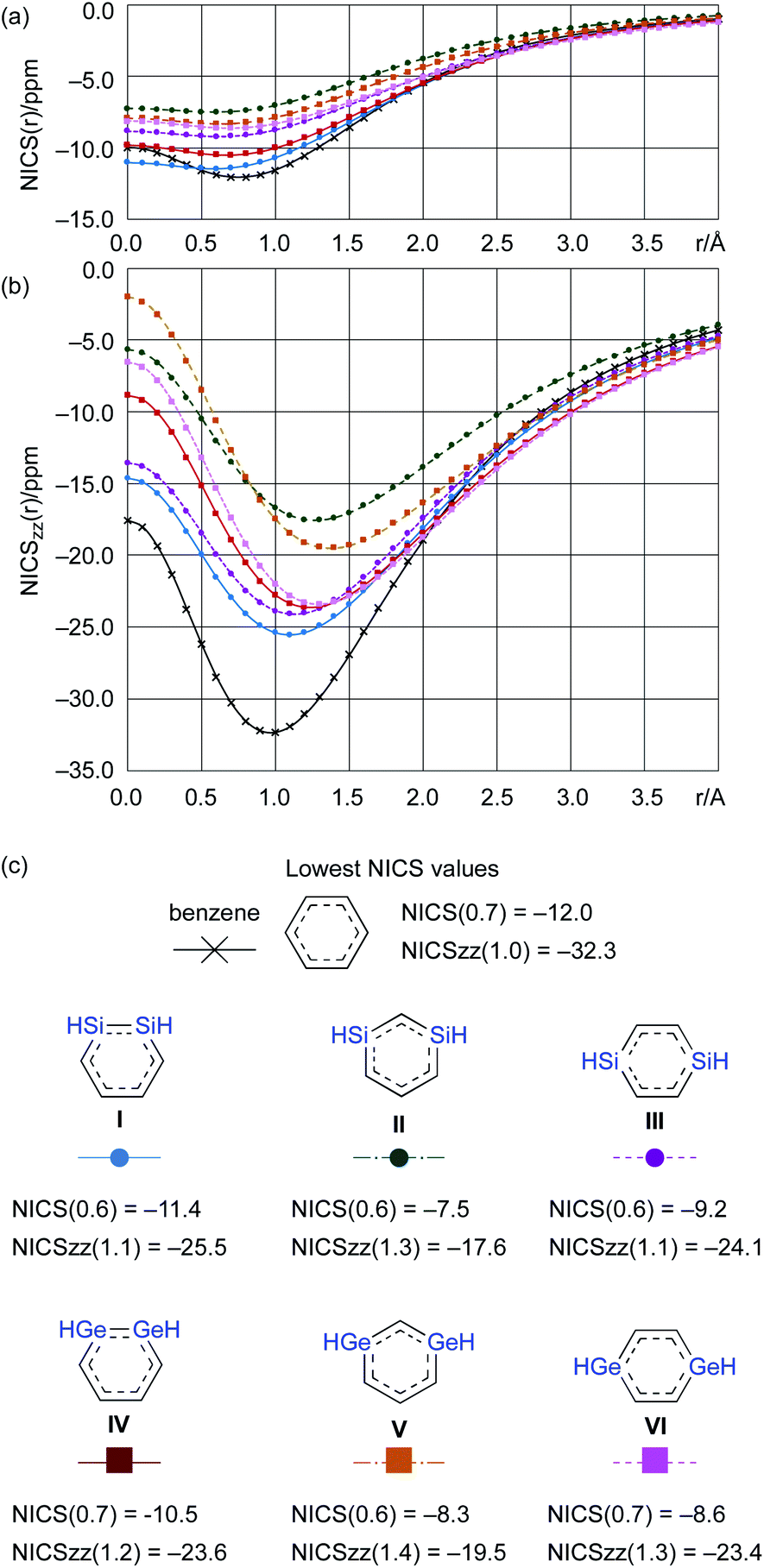 | ||
| Fig. 4 (a) NICS(r) plot, (b) NICSzz(r) plot, and (c) the lowest NICS values for benzene, DSBs (I, II, III), and DGBs (IV, V, VI), calculated at the GIAO-MP2/6-311G(3d)//MP2/6-311G(3d,p) level of theory. | ||
Considering these theoretical investigations, it should also be very interesting to examine the electronic structures of 1,2-DSBs and 1,2-DGBs, especially with respect to their resonance structures.
3. 1,2-Disila- and 1,2-digermabenzenes
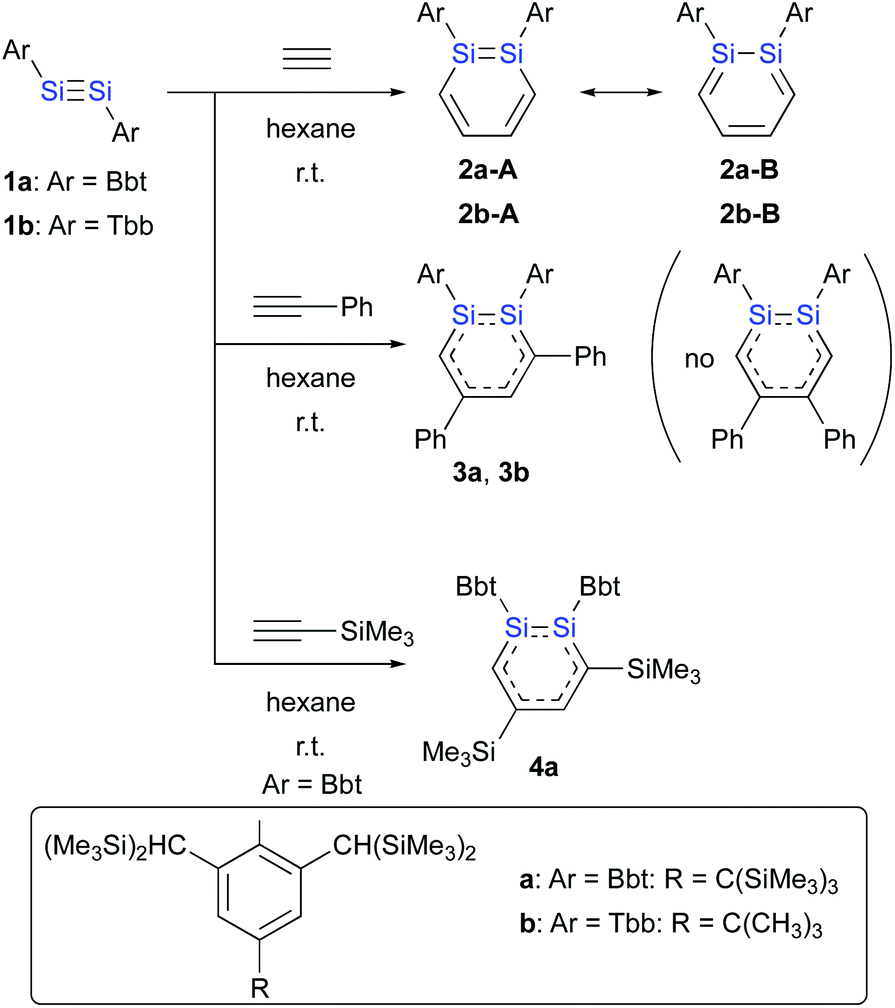
The only hitherto reported synthetic route to 1,2-DSBs proceeds via the reaction of an isolable disilyne (RSi![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiR)15 with acetylenes, i.e., a formal [2 + 2 + 2] cycloaddition (Scheme 1).16 Sekiguchi et al. have reported the first successful isolation of a stable 1,2-DSB from the reaction of the stable disilyne RSiSiSiRSi (1c; RSi = Si(iPr)[CH(SiMe3)2]2)15b with phenylacetylene.16a However, the separation and purification of the two isomers obtained, i.e., the 3,5-diphenyl- and 4,5-diphenyl-1,2-DSBs (3
SiR)15 with acetylenes, i.e., a formal [2 + 2 + 2] cycloaddition (Scheme 1).16 Sekiguchi et al. have reported the first successful isolation of a stable 1,2-DSB from the reaction of the stable disilyne RSiSiSiRSi (1c; RSi = Si(iPr)[CH(SiMe3)2]2)15b with phenylacetylene.16a However, the separation and purification of the two isomers obtained, i.e., the 3,5-diphenyl- and 4,5-diphenyl-1,2-DSBs (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio) were problematic due to the lability of the products toward air and moisture.
:2 ratio) were problematic due to the lability of the products toward air and moisture.
 | ||
| Scheme 1 Formation of 1,2-DSBs. | ||
Given that stable diaryldisilynes ArSiSiAr (1a: Ar = Bbt; 1b: Ar = Tbb; Bbt = 2,6-[(Me3Si)2CH]2-4-[(Me3Si)3C]-phenyl; Tbb = 4-t-butyl-2,6-[(Me3Si)2CH]2-phenyl) had already been reported by Tokitoh et al.,17,18 we wanted to use these as precursors for the synthesis of diaryl-substituted 1,2-DSBs.
The reactions of disilynes 1a and 1b with alkynes to give the corresponding 1,2-DSBs are shown in Scheme 1.16b,18 Treatment of hexane solutions of 1a and 1b with acetylene at r.t. afforded 1,2-Ar2-1,2-DSBs 2a (Ar = Bbt) and 2b (Ar = Tbb), respectively.16b Thus, the substituent effect of Bbt and Tbb during the formation of 1,2-DSBs is negligible.18 In contrast to the reaction of disilyldisilyne 1c,16a the reaction of disilyne 1a with phenylacetylene results in the selective formation of 3,5-diphenyl-1,2-DSB 3a together with an inseparable and unidentified by-product, which was not the corresponding 4,5-diphenyl-1,2-disilabenzene. Conversely, treatment of 1b with phenylacetylene in hexane at r.t. quantitatively afforded 3,5-diphenyl-1,2-DSB 3b as the sole product.
The difference with respect to the observed regioselectivity in the reaction of disilyldisilyne 1c and diaryldisilyne 1b with phenylacetylene is quite remarkable. The formation mechanism of a 1,2-DSB via the reaction of a disilyne with two molecules of acetylene has been theoretically investigated:18 a formal [2 + 2] cycloaddition between the SiSi moiety of the disilyne (VII) and the CC moiety of an acetylene results in the formation of the initial 1,2-disilacyclobutadiene intermediate (VIII), which can undergo a further reaction with another molecule of acetylene to give 1,2-disilabenzene IX (Scheme 2a). Based on this mechanism, the reaction of disilyne 1 with diphenylacetylene has been attempted in order to try and isolate the possible disilacyclobutene intermediate,19 similar to the case of the digermacyclobutadiene derivative (vide infra).20,21 When 1 was treated with diphenylacetylene, cyclic compound 6 was obtained quantitatively, suggesting the intermediate formation of 1,2-disilacyclobutadiene 5, which would readily undergo a [1,5]-sigmatropic hydrogen migration followed by an intramolecular [2 + 2] cycloaddition (Scheme 2). Thus, as shown in previously reported theoretical investigations, the initial formal [2 + 2] cycloadditions of disilynes 1b and 1c with phenylacetylene to give the corresponding 3-phenyl-1,2-disilacyclobutadienes Int-8b and Int-8c could occur via the electrophilic attack of the SiSi moieties at the CC moieties, followed by an intramolecular ring expansion in Int-7b and Int-7c (Scheme 3). Moreover, the potential energy surfaces (PESs) of the subsequent reactions of 1,2-disilabutadienes Int-8 with phenylacetylene to give the corresponding 1,2-DSBs have been theoretically investigated. Specifically, Pro-3 and Pro-13, have been theoretically calculated for Tbb- and RSi-substituted models (Scheme 3).18
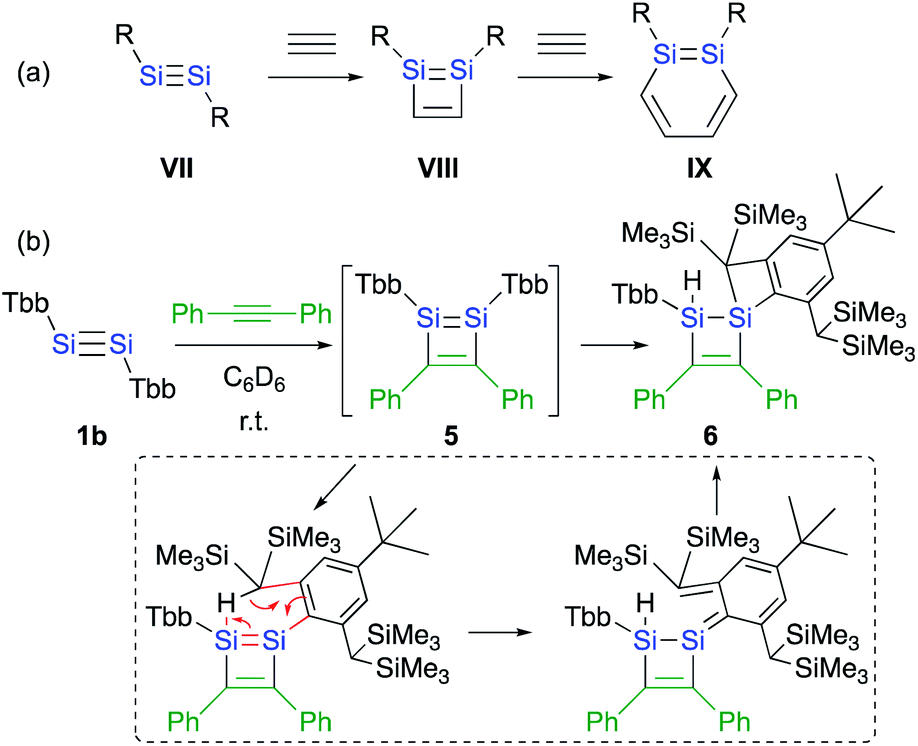 | ||
| Scheme 2 (a) A general scheme of a plausible formation mechanism for DSBs. (b) Reaction of disilyne 1b with diphenylacetylene. | ||
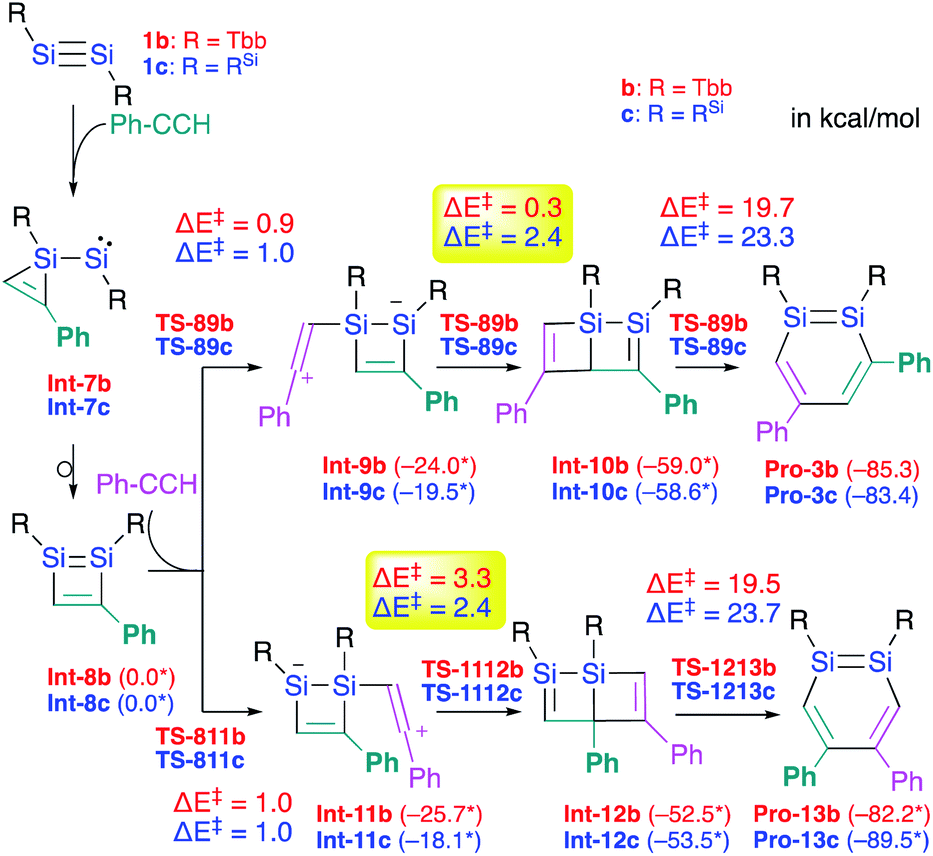 | ||
| Scheme 3 Theoretically investigated reaction mechanisms for the formation of 3,5-diphenyl- and 4,5-diphenyl-1,2-DSBs (Pro 3 and Pro-13) with Tbb and RSi substituents, together with the reaction barriers (in kcal mol−1) calculated at the B3PW91-D3(BJ)/lanl2dz+d (for Si) 6-31G(d) (for C,H) level of theory. *values in parentheses represent potential energies relative to that of Int-8.18 | ||
The insertion of the CC moiety of phenylacetylene into the Si–C bond of Int-8 should be crucial for the selectivity of the 1,2-DSBs. In both cases (b: R = Tbb; c: RSi), the insertion of phenylacetylene into the 1,4-Si–C bond of Int-8 would give 3,5-diphenyl-1,2-DSBs (Pro-3), while that into the 2,3-Si–C bond would lead to the formation of 4,5-diphenyl-1,2-DSBs (Pro-13). The entire process consists of (i) the addition of PhCCH (Int-8 → Int-9 or Int-11), (ii) a ring-closure (Int-9 → Int-10 or Int-11 → Int-12), and (iii) a ring-expansion (Int-10 → Pro-3 or Int-12 → Pro-13), whereby step (iii) should be the rate-determining step for the formation of Pro-3 or Pro-13. However, the selectivity cannot be readily rationalized exclusively in terms of the reaction barriers of the rate-determining steps, which are almost identical (b: R = Tbb, ΔE‡ = 19.7 kcal mol−1 for Pro-3b and 19.5 kcal mol−1 for Pro-13b; c: R = RSi, ΔE‡ = 23.3 kcal mol−1 for Pro-3c and 23.7 kcal mol−1 for Pro-13c). In step (ii), the barriers for Int-9c → Int-10c and Int-11c → Int-12c are almost identical (ΔE‡ = 2.4 kcal mol−1), while that for Int-9b → Int-10b (ΔE‡ = 0.3 kcal mol−1) is by 3.0 kcal mol−1 lower than that for Int-11b → Int-12b (ΔE‡ = 3.3 kcal mol−1), which would explain the selective formation of Pro-3b in the reaction of 1b with phenylacetylene in the Tbb-substituted case but not in the RSi-substituted case.
The successful isolation of stable 1,2-DSBs from the reaction of the corresponding disilyne with alkynes16,18 naturally prompted us to examine the reactions with Ge-analogues, i.e., the reaction of isolable digermynes with alkynes in the expectation to obtain the corresponding 1,2-DGBs.20,22 Digermyne TbbGeGeTbb (14) was prepared according to a previously reported synthetic procedure for stable digermynes.23 Exposure of a hexane solution of 14 to an acetylene atmosphere at r.t. afforded 1,2-DGB 15 (61% 1H NMR yield) as a pale yellow crystalline solid together with 1,4-digermabarrelene 16 (22% 1H NMR yield) (Scheme 4).20 Detailed theoretical calculations revealed that the formation of 1,2-DGB 15 should proceed according to a formation mechanism analogous to that for 1,2-DSBs 2a,b, via the 1,2-digermacyclobutadiene intermediate 17, which was experimentally supported by the formation of isolable 3,4-diphenyl-1,2-digermacyclobutadiene 18 in the reaction of 14 with diphenylacetylene.20,21 The formation of 16 could potentially be rationalized in terms of the intermediacy of 1,4-digermabenzene 19, which can be expected to readily undergo a subsequent [4 + 2] cycloaddition with one molecule of acetylene to give 16.
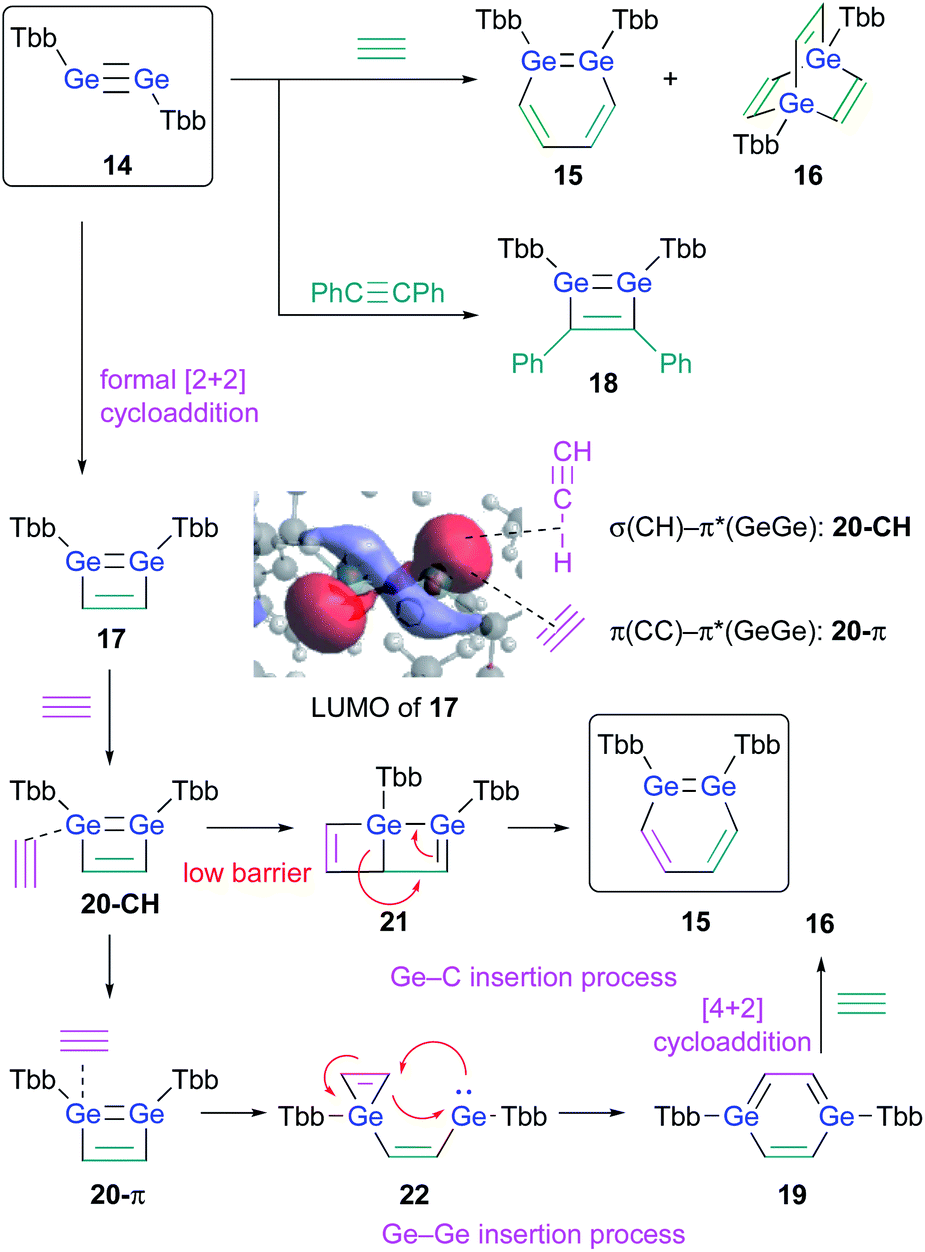 | ||
| Scheme 4 Formation of 1,2-DGB 15 and 1,4-digermabarrelene 16 in the reaction of digermyne 14 with acetylene. | ||
The potentially underlying reaction mechanism has been investigated using detailed DFT calculations.20 The key point for the selectivity between products 15 and 16 should be the interaction modes between 17 and acetylene. Given the characteristically low-lying π*(GeGe) orbital of 17, the σ orbital (CH) of acetylene would be approaching to form complex 20-CH as an initial intermediate. 20-CH would then be converted to complex 20-π, which exhibits an orbital interaction between the CC π orbital and the π*(GeGe) orbital, which is a mildly endothermic process. Conversely, with a slightly lower barrier relative to the conversion process to 20-π, 20-CH could undergo a pericyclic reaction to give 21, which would furnish 1,2-DGB 15. Complex 20-π could be converted into 1,4-digermabenzene 19via the process shown in Scheme 4, and 19 could undergo a facile [4 + 2] cycloaddition with another molecule of acetylene to furnish 16, which should be a thermodynamically very stable product, via a barrierless process. Thus, given all the reactions of 17 with acetylene, the pathways to the formation of 1,2-DGB 15 and 1,4-digermabarrelene 16 could be considered as kinetically and thermodynamically controlled processes, respectively.20
The molecular structures of 1,2-DSB 2b and 1,2-DGB 15 are shown in Fig. 5,18,22 and selected structural parameters are summarized in Table 2. In both cases, the E2C4 moiety exhibits crystallographic C2 symmetry. Interestingly, the sum of the interior angles of the E2C4 rings (Table 2) suggests that the Ge2C4 ring of 15 adopts a non-planar geometry, wherein the Ge–Ge axis comprises an angle of ca. 8.6° relative to the C4 plane, which stands in sharp contrast to the planarity observed for the Si2C4 ring in 2b.17,18 These structural features were expected considering the theoretical calculations shown in Fig. 2. The bond lengths in the E2C4 moieties (E–E′, E–C2, C2–C3, and C3–C3′) fall in between those of the corresponding single and double bonds. Thus, the six π electrons should be delocalized over the E2C4 rings, which suggests considerable aromaticity. However, the C3–C3′ bond lengths are slightly longer than the C2–C3 bonds in both cases, which indicates a predominant contribution from resonance structure A (Fig. 2) with E = E-double-bond character, rather than from resonance structure B with E–E single bond character.
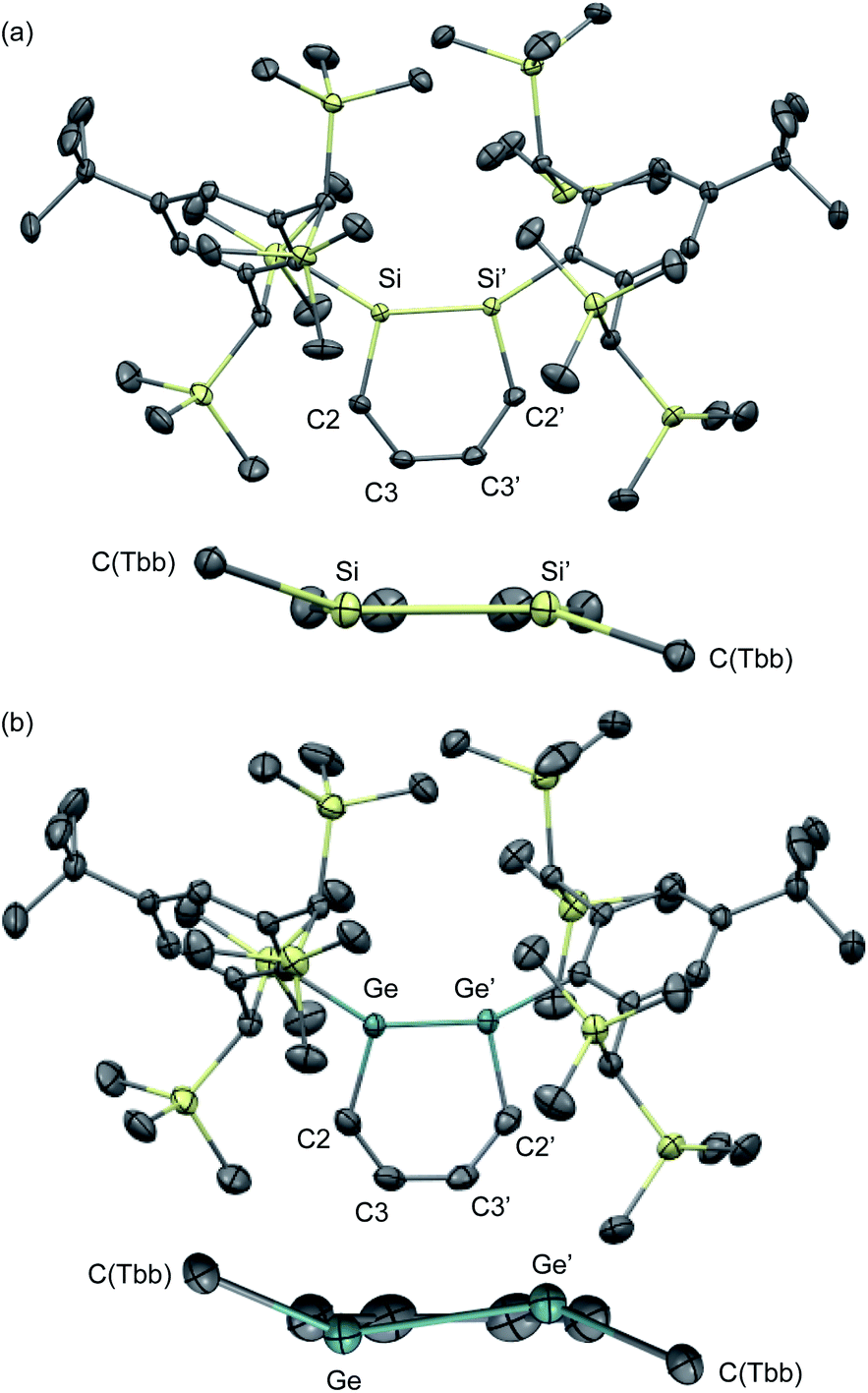 | ||
| Fig. 5 Molecular structures of (a) 1,2-DSB 2b and (b) 1,2-DGB 15 with thermal ellipsoids at 50% probability; hydrogen atoms are omitted for clarity. | ||
| Bond lengths/Å | ||||
|---|---|---|---|---|
| E–E′ | E–C2 | C2–C3 | C3–C3′ | |
| a Sum of the interior angles in the E2C4 moiety. | ||||
| 2b | 2.2101(8) | 1.8071(5) | 1.3787(7) | 1.4141(9) |
| 15 | 2.3118(4) | 1.897(3) | 1.359(4) | 1.417(4) |
| Bond angles/° | ||||
|---|---|---|---|---|
| E′–E–C2 | E–C2–C3 | C2–C3–C3′ | Σ(E2C4)a | |
| 2b | 103.44(3) | 130.10(4) | 126.38(5) | 719.8 |
| 15 | 101.34(9) | 129.3(2) | 128.1(3) | 717.5 |
As described above, the reaction of disilyne 1b with phenylacetylene selectively affords 3,5-diphenyl-1,2-DSB 3b.18 As expected, the reaction between digermyne 14 and 2 equiv. of phenylacetylene at r.t. in C6D6 afforded 3,5-diphenyl-1,2-DGB as the sole product in 68% isolated yield.24 However, in contrast to the silicon case, 1,2-DGB 23 is able to undergo a further reaction with phenylacetylene at r.t. to furnish tricyclic 24 (Scheme 5).24
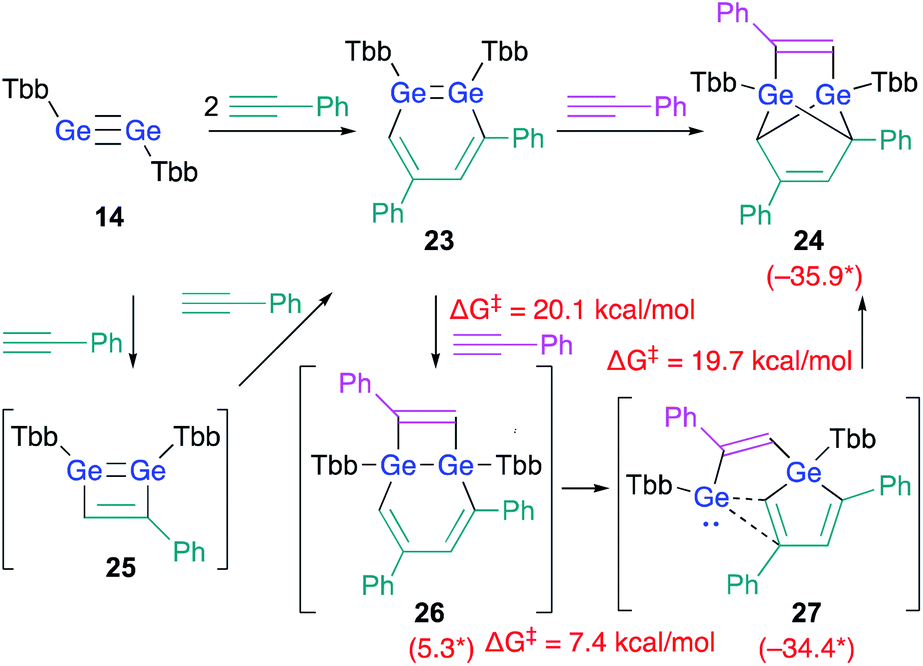 | ||
| Scheme 5 Reactions of digermyne 14 with phenylacetylene. *values in parenthesis represent free-energy values relative to that of 23 (in kcal mol−1); calculated at the TPSSTPSS-D3 level of theory.24 | ||
DFT calculations on the reaction mechanism of this reaction revealed the most reasonable pathway for the formation of 24 from digermyne 14. The formation of 3,5-diphenyl-1,2-DGB 23 from 14 could be explained reasonably using 1,2-digermacyclobutadiene intermediate 25 in analogy to the afore-mentioned cases (vide supra). A third molecule of phenylacetylene could attack the LUMO of 23, which should be predominantly localized around the GeGe moiety, to give 26via a formal [2 + 2] cycloaddition. Then, the ring-contraction of the Ge2C4 moiety could occur via a carbon migration from one Ge atom to the other Ge atom to form germole–germylene 27. Finally, an intramolecular [4 + 1] cycloaddition between the germole and the germylene moieties in 27 could afford 24. Based on theoretical calculations at the TPSSTPSS-D3 level of theory, the rate-determining step for the entire processes should be 23 → 26 (ΔG‡ = 20.1; ΔG = 5.3 kcal mol−1).
When 24 was further treated with phenylacetylene in C6D6 at 60 °C, phenylacetylene was selectively converted into 1,2,4-triphenylbenzene, while 24 was not consumed during the reaction. Given that simply heating phenylacetylene in the absence of any other compound under otherwise identical conditions did not show any reaction, it is feasible to conclude that 23 catalyzes the cyclotrimerization of phenylacetylene to give 1,2,4-triphenylbenzene. These experimental results demonstrate that the thermal reaction of arylacetylenes 28 in the presence of a catalytic amount of digermyne 14 smoothly furnishes the corresponding 1,2,4-triarylbenzenes (29) as the sole product in high yield (Scheme 6).24 Notably, the reaction proceeds with absolute stereoselectivity, i.e., other triarylbenzene isomers such as 30 were not obtained in these reactions. Compound 24 should be the resting state of the catalyst, considering that it is the only compound that was observed during the 1H NMR monitoring of the catalytic reactions. Thus, digermyne 14 acts as a pre-catalyst for the cyclotrimerization of arylacetylenes in the absence of a transition-metal catalyst,25 specifically for the perfectly regioselective cyclotrimerization of terminal arylacetylenes.
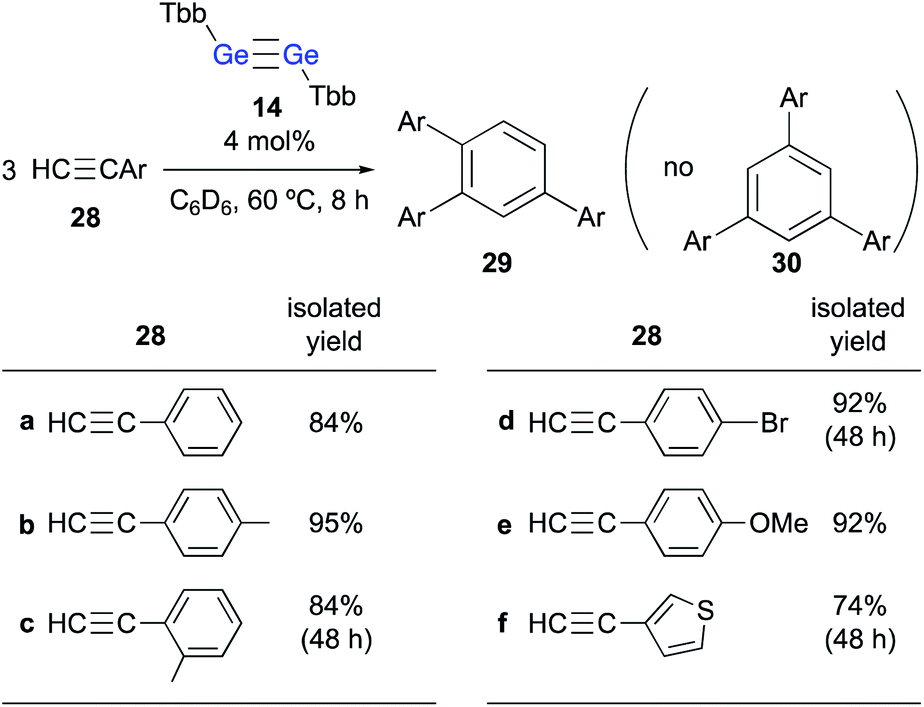 | ||
| Scheme 6 Cyclotrimerization of arylacetylenes 28 in the presence of a catalytic amount of digermyne 14. | ||
The experimental results, including crossover reactions, combined with the results of detailed theoretical calculations, have helped to reveal the reaction mechanism underlying the cyclotrimerization of phenylacetylene catalyzed by 23 (Scheme 7). The reaction barrier from 24 to 31 is the highest (ΔG‡ = 21.7, ΔG = 2.5 kcal mol; calculated at the TPSSTPSS-D3 level of theory) in the entire catalytic cycle shown in Scheme 7, which is consistent with the experimental result that only 24 was observed during the 1H NMR monitoring of the reactions.
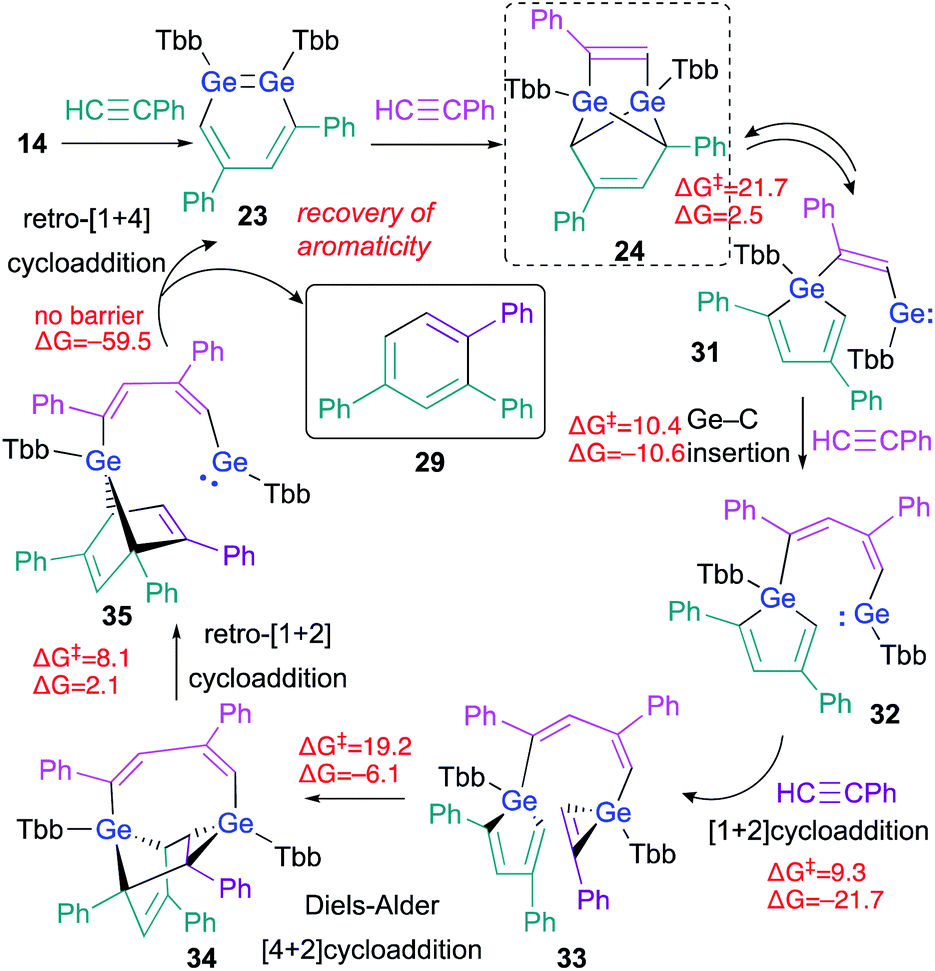 | ||
| Scheme 7 Plausible mechanism for the cyclotrimerization of phenylacetylene catalyzed by 1,2-DGB 23; free-energy values are shown in kcal mol−1. | ||
As described above, the tricyclic skeleton of 24 can be considered as the intramolecular [1 + 4] cycloadduct of a germole and a germylene. Thus, the retro [1 + 4] cycloaddition of 24 should give germole–germylene 31 in the equilibrium state. Then, the Ge–C insertion of phenylacetylene could occur to give 32, the germylene moiety of which could undergo a further [1 + 2] cycloaddition with phenylacetylene to give 33.26 The intramolecular Diels–Alder reaction of 33 can then be expected to smoothly afford 34. The ring-strain in 34 should be released via a retro [1 + 2] cycloaddition in the germirane moiety to furnish 35. The driving force for the recovery of the aromaticity of both triphenylbenzene 29 and 1,2-DGB 23 would promote the conversion of germanorbornadiene 35 to 23 with concomitant elimination of 29via a retro [1 + 4] cycloaddition. The key step for the stereoselectivity in these reactions should be 33 → 34, given the favorable orientations of the frontier orbital interactions, which are required for Diels–Alder reactions.27
In this section, we have discussed the synthesis of 1,2-DSBs and 1,2-DGBs, and plausible mechanisms of their formation have been postulated based on a combination of experimental and theoretical results. For the formation of both 1,2-DSBs and 1,2-DGBs, the key intermediates should be the corresponding 1,2-disila- and 1,2-digermacyclobutadienes. The effective orbital interaction between the σ(H–C) orbital and the low-lying LUMO of the 1,2-dimetallacyclobutadiene intermediates should promote the formation of the 1,2-dimetallabenzenes. Interestingly, digermyne 14 works as an effective pre-catalyst for the cyclotrimerization of arylacetylenes to selectively afford the corresponding 1,2,4-triarylbenzenes; in this process, the 1,2-DGB could be a key intermediate.
4. 1,4-Disila- and 1,4-digermabenzenes
Considering the aforementioned theoretical calculations on the reaction of digermyne 14 with acetylene (Scheme 4), which furnishes 1,2-digermabenzene 15 and 1,4-digermabarrelene 16, it has suggested that the orbital interaction between the π*(GeGe) orbital of 17 and the σ(H–C) orbital of an alkyne would lead to the formation of 1,2-DGB 15, while that between the π*(GeGe) orbital of 17 and the π(CC) orbital of an alkyne would lead to the formation of 1,4-DGB 19.20 Moreover, it has been reported that reactions of an amidinato-supported disilyne with diphenylacetylene afford the corresponding 1,4-disilabenzene derivatives, albeit that the isolated 1,4-disilabenzene derivatives contain tetra-coordinated silicon atoms due to the bidentate character of the amidinato ligand.28 These results prompted us to examine the reaction of 1,2-DSBs and 1,2-DGBs with internal alkynes, which do not contain a H–C moiety, with the objective to isolate the corresponding 1,4-DSBs and 1,4-DGBs.
In contrast to the Ge case, 1,2-disilacyclobutadiene 5 could not be isolated due to its lability on account of the intramolecular cyclization (Scheme 2b).19 We speculated that dialkylacetylenes, which exhibit higher nucleophilicity relative to diphenylacetylene, could potentially be more suitable for a further insertion reaction toward the transiently generated 1,2-disilacyclobutadiene to generate the corresponding 1,4-DSB without the intramolecular cyclization of the corresponding 1,2-disilacyclobutadiene intermediate as a side reaction. As expected, the reaction of disilyne 1b with 3-hexyne resulted in the competitive formation of 1,4-DSB 36 and disilabenzvalene 37 in a 1:2 ratio (Scheme 8).19 Unfortunately, 1,4-DSB 36 is difficult to isolate from the mixture due to its lability. Interestingly, we found that 36 is photochemically converted into 37. Although it would be feasible to think that a contamination of 37 in the reaction of disilyne 1b with 3-hexyne could be due to the photochemical isomerization of 36, the reaction of 1b with 3-hexyne in the dark under otherwise identical conditions was not successful, i.e., a mixture of 36 and 37 was obtained. Thus, a plausible reaction mechanism could be that shown in Scheme 8, where silirene–silylene 40, generated via the orbital interaction between the π*(SiSI) orbital of disilabutadiene intermediate 38 and the π(CC) orbital of 3-hexyne should be the mutual intermediate for the formation of 1,4-DSB 36 and disilabenzvalene 37.29 In other words, the competitive formation of 36 and 37 should most likely be interpreted in terms of an intramolecular Si–C insertion and the intramolecular [1 + 2] cycloaddition between the silylene and the silirene moieties in 40.
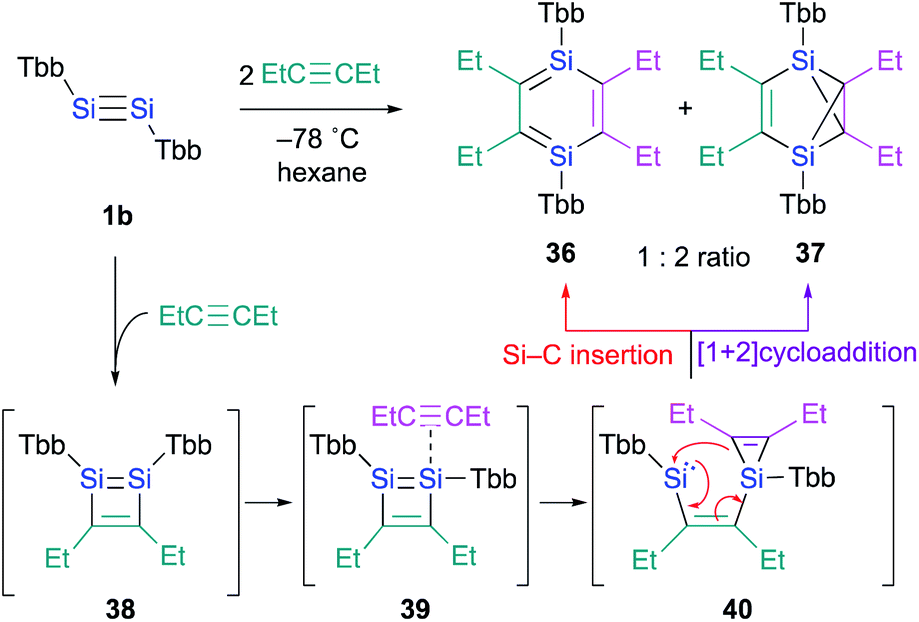 | ||
| Scheme 8 Formation of 1,4-DSB 36 in the reaction of disilyne 1b with 3-hexyne. | ||
Thus, we realized that the reaction of disilynes with internal alkynes, which do not contain a H–C moiety, can result in the formation of the corresponding 1,4-DSB via orbital interactions between the π*(SiSi) orbital of the 1,2-disilacyclobutadiene intermediate and the π(CC) orbitals.
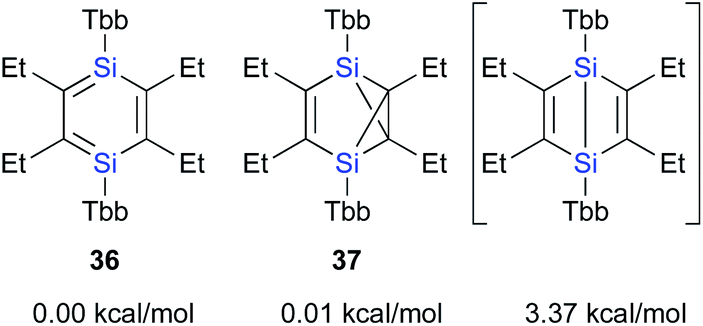 | ||
| Fig. 6 Relative energies of Tbb2Et4Si2C6 valence isomers. | ||
The calculated potential energies of 36 and 37 are almost identical at the B3PW91-D3(BJ)/6-311G(3df,p)//B3PW91/6-311G(3df)[Si],6-31G(d)[C,H] level of theory, while that of the imaginary Dewar-type isomer is slightly higher (3.37 kcal mol−1) relative to that of 1,4-DSBZ 36 (Fig. 6). In contrast to the cases of the relative energies of the corresponding H6Si2C4 valence isomers, the energy differences between the valence isomers of the ‘real’ models are very small, suggesting that the relative stability of these valence isomers should be effectively perturbed by the electronic/steric properties of the substituents.
With 3,4-diphenyl-1,2-digermacyclobutadiene 18 in hand, in contrast to the Si case, the reaction of 18 with a further molecule of diphenylacetylene could be attempted in the expectation of generating the corresponding 1,4-DGB. Heating 1,2-digermabutadiene 18 in the presence of an excess of diphenylacetylene at 60 °C afforded 1,4-DGB 41 (Scheme 9).30 1,4-DGB 41 should be formed via a mechanism similar to that for the formation of 19 (Scheme 2). Probably due to the steric demand of the Tbb and phenyl groups, a further [4 + 2] cycloaddition of 41 with diphenylacetylene does not occur, and, as a result, 41 can be isolated as a stable compound. Moreover, the reaction of digermyne 14 with 3-hexyne smoothly furnishes the corresponding 1,4-DGB 42 as the sole product, albeit that the formation of potential intermediates such as the corresponding 1,2-digermacyclobutadiene was not observed.30
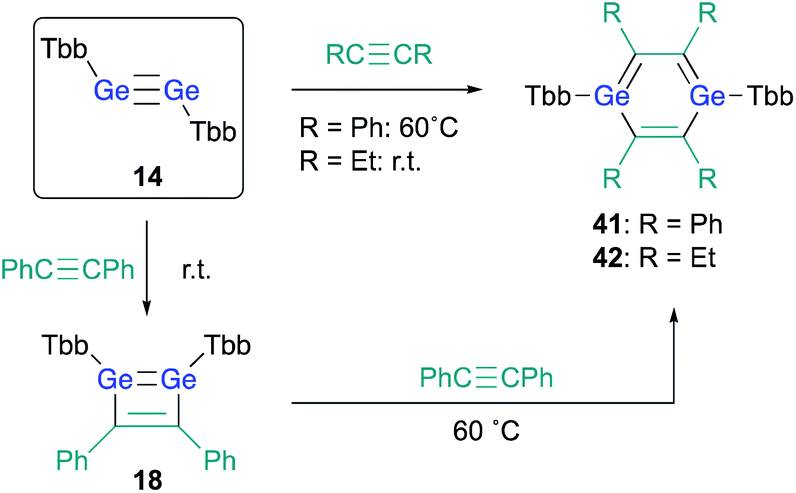 | ||
| Scheme 9 Synthesis of 1,4-DGBs 41 and 42. | ||
When 1,4-DGB 42 was treated with acetylene, the [4 + 2] cycloadduct, i.e., digermabarrelene 43 was obtained, which suggests a formation mechanism similar to that of 1,4-digermabarrelene 16 from the [4 + 2] cycloaddition of 19 with acetylene (Scheme 4). In addition, 42 was found to activate carbon dioxide (CO2) and molecular hydrogen (H2) at r.t. to give the corresponding syn-cycloadducts 44 and 45, respectively. Apart from the addition of CO2, the addition reactions of 42 with small molecules proceed smoothly and irreversibly at r.t. to give the 1,4-adducts (Scheme 10).30 Interestingly, heating of 44 at 60 °C in C6D6 gave 42 with concomitant elimination of CO2, suggesting that the cycloaddition of 1,4-DGB 42 with CO2 is reversible.
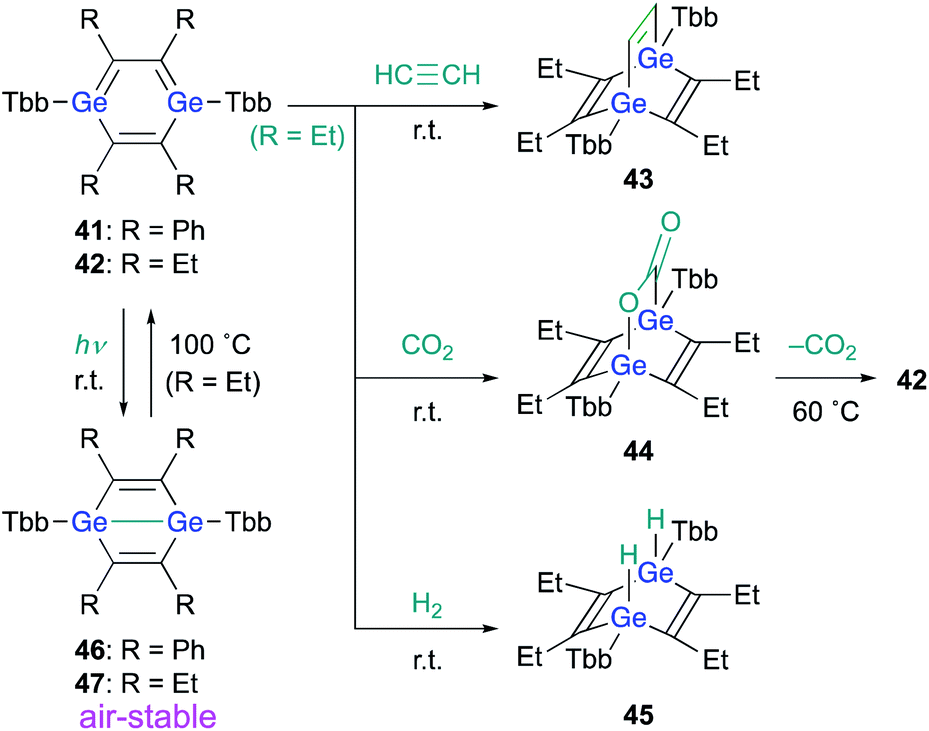 | ||
| Scheme 10 Reactions of 1,4-DGB 42 with small molecules. | ||
Notably, 1,4-digermabenzenes 41 and 42 are photo-responsive upon exposure to LED light (410–490 nm), resulting in the quantitative formation of the corresponding digerma-Dewar-benzenes 46 and 47 (Scheme 10). The photochemical isomerization of 1,4-DGBs 46 and 47 stands in sharp contrast to that of 1,4-DSB 36, which gave disilabenzvalene 37; the reason for the different types of photochemical isomerization of 1,4-DSBs and 1,4-DGBs remains unclear at present. Although the photochemical isomerization of tetraphenyl-1,4-DGB 41 to digerma-Dewar-benzene 46 is thermally irreversible, tetraethyl-1,4-DGB 42 can be thermally regenerated quantitatively by heating digerma-Dewar-benzene 47 to 100 °C for 2 h in toluene. The thermal conversion of 47 can be interpreted reasonably well in terms of the relative thermodynamic energies between 1,4-DGB 42 and digerma-Dewar-benzene 47. Theoretical calculations at the TPSSTPSS-D3(BJ)/6-311G(2d,p) level of theory showed that 47 is by 2.2 kcal mol−1 less stable than 42, while 46 is by 6.9 kcal mol−1 more stable than 41. It should be noted here that digerma-Dewar-benzenes 46 and 47 are inert toward air and moisture for at least two weeks, in contrast to 1,4-DGB 42, which is highly air- and moisture-sensitive. Considering the photochemical and chemical reactivity of 1,4-DGB 42, we attempted the activation of H2 with air-stable 1,4-digerma-Dewar-benzene 47. Treatment of 47 with H2 (1 atm, 100 °C, toluene, 2 h) resulted in the quantitative formation of hydrogen-adduct 45, suggesting a thermal isomerization of 47via intermediate 42, which can activate H2. Thus, 1,4-digerma-Dewar-benzene 47 can be considered as a shelf-stable main-group-element-based catalyst for small molecules that can be activated thermally.30
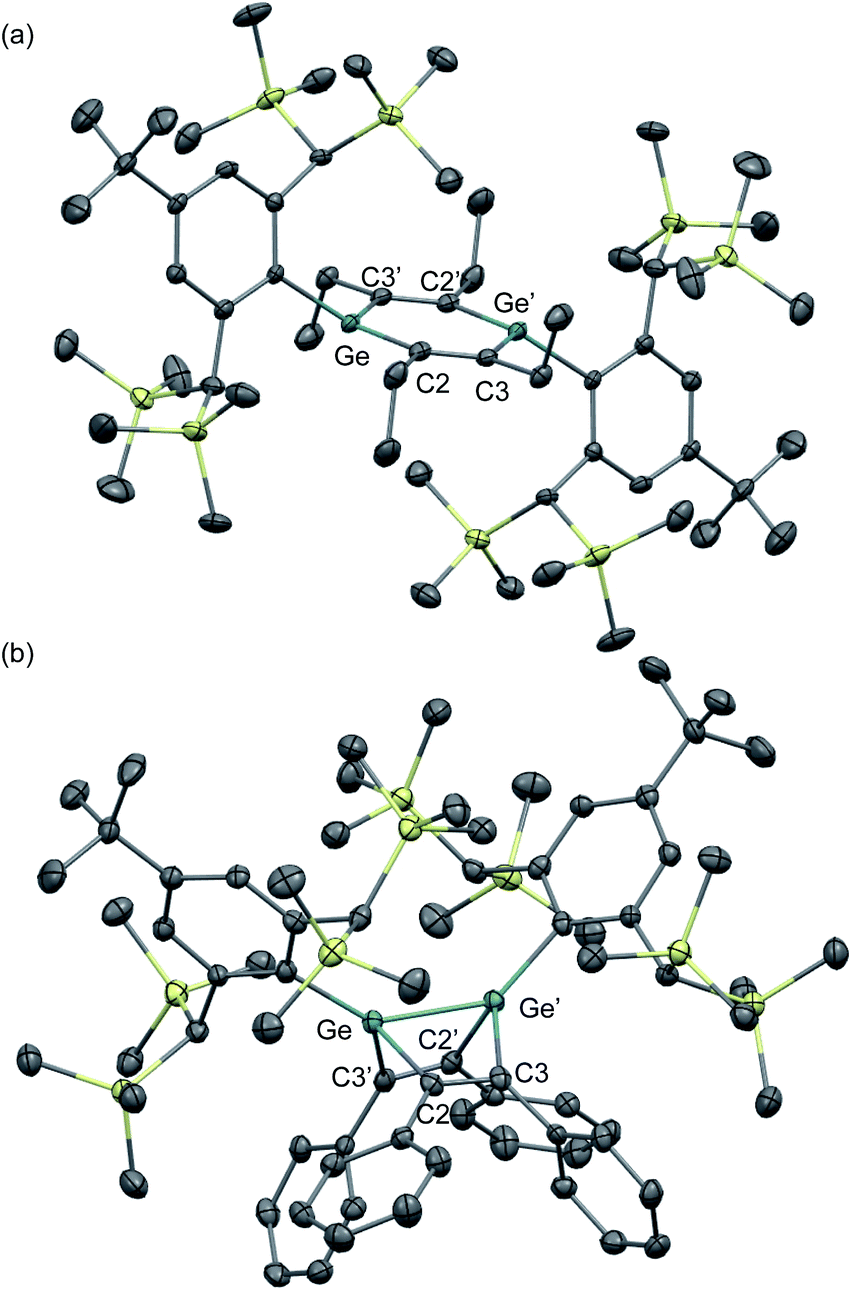 | ||
| Fig. 7 Molecular structures of (a) 1,4-DGB 42 and (b) digerma-Dewar-benzene 46 with thermal ellipsoids at 50% probability; hydrogen atoms are omitted for clarity. | ||
The molecular structures of the obtained 1,4-DGB 42 and digerma-Dewar-benzene 46 have been determined by single-crystal XRD analyses (Fig. 7). As shown in Fig. 7, 1,4-DGB 42 exhibits a slight deviation from ideal planarity of the Ge2C4 hexagon, i.e., the Ge2C4 hexagon adopts a chair-like geometry that includes a bent angle of ca. 10° between the Ge–C2–C3′ and C2–C3–C2′–C3′ planes. The Ge–C (1.888(3) Å) and C–C bond lengths (1.375(4) Å) in the Ge2C4 ring in 42 fall in between the corresponding double and single bond lengths, which suggests considerable π-electron delocalization over the aromatic Ge2C4 ring. In contrast, the Ge–Ge (2.3966(8) Å) and Ge–C bond lengths (2.049(4)/1.988(3) Å) in digerma-Dewar-benzene 46 are indicative of a typical Dewar-benzene structure with defined single-bond character.
In summary, as theoretically expected, the reactions of disilynes and digermynes with internal alkynes result in the formation of 1,4-DSBs and 1,4-DGBs, respectively. Interestingly, the 1,4-DSBs and 1,4-DGBs photochemically isomerize in different ways to give the corresponding disilabenzvalene and digerma-Dewar-benzene, respectively. Since the photochemical isomerization of the 1,4-DGBs is reversible upon heating, the digerma-Dewar-benzene works as a shelf-stable main-group-element-based activator for small molecules.
5. Conclusions
Stable 1,2-disilabenzenes (1,2-DSBs), 1,2-digermabenzenes (1,2-DGBs), and 1,4-digermabenzenes (1,4-DGBs) can be synthesized via the reaction of the corresponding dimetallynes with alkynes. Initially, our experimental and theoretical research was focused on a detailed understanding of the formation mechanism of the 1,2-dimetallabenzenes. The thus obtained results provided us with sufficient information to develop a strategy for the synthesis of 1,4-dimetallabenzenes. We discovered that the 1,2- and 1,4-dimetallabenzenes can be synthesized using appropriate synthetic methods, provided that sufficient kinetic stabilization is available. Their structural features suggest that these compounds exhibit considerable aromaticity, similar to that of benzene, which has been corroborated by theoretical calculations. Detailed electron-density analyses of these aromatic systems, which are currently in progress in our laboratory, can be expected to provide further important experimental insights into the aromaticity of these compounds. A fundamental investigation into the chemical properties of 1,2-DGBs revealed that a 1,2-DGB and a digermyne can work as Ge-based catalysts for the cyclotrimerization of terminal alkynes. We have thus demonstrated that a detailed fundamental investigation using a combination of experimental and theoretical techniques is able to find unique applications for the chemistry of dimetallabenzenes as main-group-element catalysts. These fundamental investigations, which combine experimental and theoretical aspects, should ultimately produce low-coordinated main-group-element compounds that serve as promising prospectives for transition-metal-free catalysts in C–C coupling reactions.Author contributions
The manuscript was written and checked by the author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to thank all of his past and present co-workers for their invaluable contributions (for individual names, please see the references). The author would like to express his gratitude especially to Dr Tomohiro Sugahara, Dr Joon Soo Han, and Dr Jing-Dong Guo for their continuous and dedicated effort during the experimental and theoretical works. The author would moreover like to thank Prof. Dr Norihiro Tokitoh and Prof. Dr Shigeru Nagase (Kyoto University), and Prof. Dr Rainer Streubel (Rheinische Friedrich-Wilhelms-Universität Bonn, Germany) for their kind assistance and helpful discussions. The author is also grateful to Prof. Dr Arturo Espinosa Ferao (Universidad De Murcia, Spain) for his kind collaboration on theoretical calculations. The author is also indebted to Dr Daisuke Hashizume (RIKEN) for his kind assistance with XRD experiments. For manufacturing the outstanding custom-tailored glassware used in the experimental work, the author would like to thank the glassblowers Mr Toshiaki Noda and Ms. Hideko Natsume. Furthermore, the author would like to thank Dr U. F. J. Mayer from Mayer Scientific Editing (http://www.mayerscientificediting.com/) for his assistance and the kind discussions during the preparation of this manuscript.Notes and references
- For selected reviews, see: (a) G. Raabe and J. Michl, Chem. Rev., 1985, 85, 419–509 CrossRef CAS; (b) T. Sasamori and N. Tokitoh, Encyclopedia of Inorganic Chemistry, ed. R. Bruce King, John Wiley & Sons, Chichester, 2nd edn, 2005, pp. 1698–1740 Search PubMed; (c) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed; (d) T. Sasamori and N. Tokitoh, Bull. Chem. Soc. Jpn., 2013, 86, 1005–1021 CrossRef CAS; (e) T. Matsuo and N. Hayakawa, Sci. Technol. Adv. Mater., 2018, 109, 108–129 CrossRef PubMed; (f) A. Rammo and D. Scheschkewitz, Chem.–Eur. J., 2018, 24, 6866–6885 CrossRef CAS PubMed.
- S. Nagase, Bull. Chem. Soc. Jpn., 2014, 87, 167–195 CrossRef CAS.
- (a) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed; (b) J.-D. Guo, D. J. Liptrot, S. Nagase and P. P. Power, Chem. Sci., 2015, 6, 6235–6244 RSC; (c) F. Hanusch, L. Groll and S. Inoue, Chem. Sci., 2021, 12, 2001–2015 RSC.
- For examples of isolable metallabenzenes that contain other elements than those from group 14, see: (a) J. R. Bleeke, Chem. Rev., 2001, 101, 1205–1227 CrossRef CAS PubMed; (b) A. J. Ashe III, S. Al-Ahmad and J. W. Kampf, Angew. Chem., Int. Ed. Engl., 1995, 34, 1357–1359 CrossRef; (c) T. Nakamura, K. Suzuki and M. Yamashita, J. Am. Chem. Soc., 2014, 136, 9276–9279 CrossRef CAS PubMed; (d) T. Ishii, K. Suzuki, T. Nakamura and M. Yamashita, J. Am. Chem. Soc., 2016, 138, 12787–12790 CrossRef CAS PubMed; (e) Z. Zhu, X. Wang, M. M. Olmstead and P. P. Power, Angew. Chem., Int. Ed., 2009, 121, 2061–2064 CrossRef.
- For selected examples, see: (a) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed; (b) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (c) Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- V. Y. Lee and A. Sekiguchi, Angew. Chem., Int. Ed., 2007, 46, 6596–6620 CrossRef CAS PubMed.
- (a) N. Tokitoh, Bull. Chem. Soc. Jpn., 2004, 77, 429–441 CrossRef CAS; (b) N. Tokitoh, Acc. Chem. Res., 2004, 37, 86–94 CrossRef CAS PubMed; (c) M. Saito, Coord. Chem. Rev., 2012, 256, 627–636 CrossRef CAS; (d) Y. Mizuhata, S. Fujimori, T. Sasamori and N. Tokitoh, Angew. Chem., Int. Ed., 2017, 56, 4588–4592 CrossRef CAS PubMed.
- (a) R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed; (b) T. Sasamori and N. Tokitoh, Bull. Chem. Soc. Jpn., 2013, 86, 1005–1021 CrossRef CAS; (c) V. Ya Lee and A. Sekiguchi, in Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds, Wiley & Sons, New York, 2010 CrossRef; (d) M. Kira and T. Iwamoto, Adv. Organomet. Chem., 2006, 54, 73–148 CrossRef CAS.
- For selected examples, see: (a) S.-Y. Kang, K. Yoshizawa, T. Yamabe, A. Naka and M. Ishikawa, J. Organomet. Chem., 2000, 611, 280–287 CrossRef CAS; (b) U. D. Priyakumar, M. Punnagai and G. N. Sastry, J. Organomet. Chem., 2004, 689, 1284–1287 CrossRef CAS; (c) D. M. Dhevi, U. D. Priyakumar and G. N. Sastry, J. Mol. Struct.: THEOCHEM, 2002, 618, 173–179 CrossRef CAS; (d) K. K. Baldridge, Organometallics, 2000, 19, 1477–1487 CrossRef CAS; (e) A. S. Ivanov and A. I. Boldyrev, J. Phys. Chem. A, 2012, 116, 9591–9598 CrossRef CAS PubMed; (f) S. Nagase, H. Teramae and T. Kudo, J. Chem. Phys., 1987, 86, 4513–4517 CrossRef CAS; (g) U. D. Priyakumar and G. N. Sastry, Organometallics, 2002, 21, 1493–1499 CrossRef CAS; (h) U. D. Priyakumar, D. Saravanan and G. N. Sastry, Organometallics, 2002, 21, 4823–4832 CrossRef CAS; (i) A. D. Zdetsis, J. Chem. Phys., 2007, 127, 214306 CrossRef PubMed; (j) K. Abersfelder, A. J. White, R. J. Berger, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2011, 50, 7936–7939 CrossRef CAS PubMed; (k) K. Abersfelder, A. J. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564–566 CrossRef CAS PubMed; (l) A. Sekiguchi, T. Yatabe, C. Kabuto and H. Sakurai, J. Am. Chem. Soc., 1993, 115, 5853–5854 CrossRef CAS; (m) A. Tsurusaki, C. Iizuka, K. Otsuka and S. Kyushin, J. Am. Chem. Soc., 2013, 135, 16340–16343 CrossRef CAS PubMed.
- All calculations were performed using the Gaussian 16 program.
- For examples, see: (a) Y. Kabe, K. Ohkubo, H. Ishikawa and W. Ando, J. Am. Chem. Soc., 2000, 122, 3775–3776 CrossRef CAS; (b) W. Ando, T. Shiba, T. Hidaka, K. Morihashi and O. Kikuchi, J. Am. Chem. Soc., 1997, 119, 3629–3630 CrossRef CAS; (c) N. Nakata, T. Oikawa, T. Matsumoto, Y. Kabe and A. Sekiguchi, Organometallics, 2005, 24, 3368–3370 CrossRef CAS; (d) K. M. Welsh, J. D. Rich and R. West, J. Organomet. Chem., 1987, 325, 105–115 CrossRef CAS; (e) J. D. Rich and R. West, J. Am. Chem. Soc., 1982, 104, 6884–6886 CrossRef CAS; (f) G. Maier, K. Schöttler and H. P. Reisenauer, Tetrahedron Lett., 1985, 26, 4079–4082 CrossRef CAS.
- (a) Considering not only the valence isomers of disilabenzenes, the theoretical global-minima search on C4Si2H6 isomers suggested the bicyclo[2.1.1]hexene silylene species should be the global minima, which is more stable relative to the corresponding 1,3-disilabenzene by 37 kJ mol−1 at CCSD(T)/CBS//B3LYP/6-311++G(d,p) level of theory, see: A. S. Ivanov and A. I. Boldyrev, J. Phys. Chem. A, 2012, 116, 9591–9598 CrossRef CAS PubMed; (b) The stable bicyclo[2.1.1]hexene silylene derivative has been isolated, see: C. R. W. Reinhold, Z. Dong, J. M. Winkler, H. Steinert, M. Schmidtmann and T. Müller, Chem.–Eur. J., 2018, 24, 848–854 CrossRef CAS PubMed.
- F. D. Proft and P. Geerlings, Chem. Rev., 2001, 101, 1451–1464 CrossRef PubMed.
- A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed.
- (a) N. Wiberg, W. Niedermayer, G. Fischer, H. Nöth and M. Suter, Eur. J. Inorg. Chem., 2002, 1066–1070 CrossRef CAS; (b) A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 315, 1755–1757 CrossRef PubMed.
- (a) R. Kinjo, M. Ichinohe, A. Sekiguchi, N. Takagi, M. Sumimoto and S. Nagase, J. Am. Chem. Soc., 2007, 129, 7766–7767 CrossRef CAS PubMed; (b) J. S. Han, T. Sasamori, Y. Mizuhata and N. Tokitoh, Dalton Trans., 2010, 39, 9238–9240 RSC.
- (a) T. Sasamori, K. Hironaka, Y. Sugiyama, N. Takagi, S. Nagase, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Am. Chem. Soc., 2008, 130, 13856–13857 CrossRef CAS PubMed; (b) T. Sasamori, J. S. Han, K. Hironaka, N. Takagi, S. Nagase and N. Tokitoh, Pure Appl. Chem., 2010, 82, 603–612 CAS.
- T. Sugahara, J.-D. Guo, D. Hashizume, T. Sasamori, S. Nagase and N. Tokitoh, Dalton Trans., 2018, 47, 13318–13322 RSC.
- T. Sugahara, T. Sasamori and N. Tokitoh, Dalton Trans., 2019, 48, 9053–9056 RSC.
- T. Sugahara, J.-D. Guo, T. Sasamori, Y. Karatsu, Y. Furukawa, A. Espinosa Ferao, S. Nagase and N. Tokitoh, Bull. Chem. Soc. Jpn., 2016, 89, 1375–1384 CrossRef CAS.
- (a) C. Cui, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 5062–5063 CrossRef CAS PubMed; (b) L. Zhao, C. Jones and G. Frenking, Chem.–Eur. J., 2015, 21, 12405–12413 CrossRef CAS PubMed; (c) N. Y. Tashkandi, L. C. Pavelka, C. A. Caputo, P. D. Boyle, P. P. Power and K. M. Baines, Dalton Trans., 2016, 45, 7226–7230 RSC.
- T. Sasamori, T. Sugahara, T. Agou, J.-D. Guo, S. Nagase, R. Streubel and N. Tokitoh, Organometallics, 2015, 34, 2106–2109 CrossRef CAS.
- (a) M. Stender, A. D. Phillips, R. J. Wright and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 1785–1787 CrossRef CAS; (b) L. Pu, A. D. Phillips, A. F. Richards, M. Stender, R. S. Simons, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2003, 125, 11626–11636 CrossRef CAS PubMed; (c) Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023–1031 CrossRef CAS PubMed.
- T. Sugahara, J.-D. Guo, T. Sasamori, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2018, 57, 3499–3503 CrossRef CAS PubMed.
- For examples of transition-metal-catalyzed cyclotrimerization of alkynes, see: (a) D. L. Broere and E. Ruijter, Synthesis, 2012, 44, 2639–2672 CrossRef CAS; (b) G. Domíngueza and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC; (c) B. R. Galan and T. Rovis, Angew. Chem., Int. Ed., 2009, 48, 2830–2834 CrossRef CAS PubMed; (d) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2915 CrossRef CAS PubMed; (e) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49–92 CrossRef CAS PubMed.
- M. Walewska, J. Hlina, J. Baumgartner, T. Müller and C. Marschner, Organometallics, 2016, 35, 2728–2737 CrossRef CAS PubMed.
- K. N. Houk, J. Am. Chem. Soc., 1973, 95, 4092–4094 CrossRef CAS.
- (a) S. S. Sen, H. W. Roesky, K. Meindl, D. Stern, J. Henn, A. C. Stückl and D. Stalke, Chem. Commun., 2010, 46, 5873–5875 RSC; (b) H.-X. Yeong, H.-W. Xi, K. H. Lim and C.-W. So, Chem.–Eur. J., 2010, 16, 12956–12961 CrossRef CAS PubMed.
- (a) J. D. Rich and R. West, J. Am. Chem. Soc., 1982, 104, 6884–6886 CrossRef CAS; (b) G. Maier, K. Schöttler and H. P. Reisenauer, Tetrahedron Lett., 1985, 26, 4079–4082 CrossRef CAS; (c) K. M. Welsh, J. D. Rich and R. West, J. Organomet. Chem., 1987, 325, 105–115 CrossRef CAS; (d) W. Ando, T. Shiba, T. Hidaka, K. Morihashi and O. Kikuchi, J. Am. Chem. Soc., 1997, 119, 3629–3630 CrossRef CAS; (e) Y. Kabe, K. Ohkubo, H. Ishikawa and W. Ando, J. Am. Chem. Soc., 2000, 122, 3775–3776 CrossRef CAS; (f) N. Nakata, T. Oikawa, T. Matsumoto, Y. Kabe and A. Sekiguchi, Organometallics, 2005, 24, 3368–3370 CrossRef CAS.
- T. Sugahara, J.-D. Guo, D. Hashizume, T. Sasamori and N. Tokitoh, J. Am. Chem. Soc., 2019, 141, 2263–2267 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Emeritus Professor Akira Sekiguchi on the occasion of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2021 |