
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymers with advanced structural and supramolecular features synthesized through topochemical polymerization
Kuntrapakam
Hema†‡
 ,
Arthi
Ravi‡
,
Cijil
Raju‡
and
Kana M.
Sureshan
*
,
Arthi
Ravi‡
,
Cijil
Raju‡
and
Kana M.
Sureshan
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Maruthamala, Vithura, Thiruvananthapuram-695551, India. E-mail: kms@iisertvm.ac.in
First published on 23rd February 2021
Abstract
Polymers are an integral part of our daily life. Hence, there are constant efforts towards synthesizing novel polymers with unique properties. As the composition and packing of polymer chains influence polymer's properties, sophisticated control over the molecular and supramolecular structure of the polymer helps tailor its properties as desired. However, such precise control via conventional solution-state synthesis is challenging. Topochemical polymerization (TP), a solvent- and catalyst-free reaction that occurs under the confinement of a crystal lattice, offers profound control over the molecular structure and supramolecular architecture of a polymer and usually results in ordered polymers. In particular, single-crystal-to-single-crystal (SCSC) TP is advantageous as we can correlate the structure and packing of polymer chains with their properties. By designing molecules appended with suitable reactive moieties and utilizing the principles of supramolecular chemistry to align them in a reactive orientation, the synthesis of higher-dimensional polymers and divergent topologies has been achieved via TP. Though there are a few reviews on TP in the literature, an exclusive review showcasing the topochemical synthesis of polymers with advanced structural features is not available. In this perspective, we present selected examples of the topochemical synthesis of organic polymers with sophisticated structures like ladders, tubular polymers, alternating copolymers, polymer blends, and other interesting topologies. We also detail some strategies adopted for obtaining distinct polymers from the same monomer. Finally, we highlight the main challenges and prospects for developing advanced polymers via TP and inspire future directions in this area.
 Kuntrapakam Hema | Kuntrapakam Hema received her master's degree from Sri Venkateswara University, Tirupati in 2011. Later she obtained her PhD from the Indian Institute of Science Education and Research Thiruvananthapuram (IISER TVM), India, under the supervision of Prof. Kana M. Sureshan in 2019. Currently, she is a postdoctoral fellow at the Weizmann Institute of Science under the supervision of Prof. Rafal Klajn. Her research interests include designing and executing chemical reactions in confined media. |
 Arthi Ravi | Arthi Ravi received her BSc degree in Chemistry from Holy Cross College (Autonomous), Tiruchirappalli, Tamil Nadu. She is currently pursuing her integrated PhD in Chemistry under the guidance of Prof. Kana M. Sureshan, at the Indian Institute of Science Education and Research Thiruvananthapuram (IISER TVM), India. Her research involves developing methodologies and solid-state reaction strategies and investigating the supramolecular aspects of sugar mimics. |
 Cijil Raju | Cijil Raju received his BSc and MSc in Chemistry from Newman College Thodupuzha (M. G. University, Kottayam, Kerala) in 2013 and 2015, respectively. He is currently a PhD student under the supervision of Prof. Kana M. Sureshan, at the Indian Institute of Science Education and Research Thiruvananthapuram (IISER TVM), India. His research includes the design and synthesis of polymers via the topochemical approach. |
 Kana M. Sureshan | Prof. Kana M. Sureshan obtained his PhD from the National Chemical Laboratory, Pune, India. He was a postdoctoral researcher at Ehime University, Japan (2002–2004), the University of Bath, UK (2004–2006) and the Max-Planck Institute for Molecular Physiology, Dortmund, Germany (2006–2008). In 2008, he joined the Institute of Life Sciences, Hyderabad as a Senior Scientist. In 2009, he joined IISER Thiruvananthapuram, as an Assistant Professor. Currently he is a full professor of chemistry. He is a fellow of the Royal Society of Chemistry and the Indian Academy of Sciences. His research area includes supramolecular chemistry, topochemistry, crystal engineering, organic synthesis, etc. |
1. Introduction
The bulk properties of polymers depend on various factors such as the nature of the linkage, tacticity, molecular weight, conformation and packing. Control over the molecular and supramolecular structure is essential for tuning the properties of polymers. While there are several strategies for solution-phase synthesis to control the polymer structure,1 controlling the supramolecular features (e.g. packing) of such solution-synthesized polymers is challenging. On the other hand, topochemical polymerization (TP), the solid-state polymerization of a monomer that occurs under the stringent control of a crystal lattice, is an efficient method that offers control over both the molecular and supramolecular structures of polymers.2 One of the attractive features of TP is that it yields ordered or crystalline polymers and in many cases, such a reaction occurs in a SCSC manner. The pre-organization of monomer molecules in the crystal lattice determines the topochemical reactivity and the structure of the resulting polymer. Followed by the seminal work of Schmidt on the photodimerization of cinnamic acid in crystals,3 many topochemical reactions were invented and have been extensively exploited for polymer synthesis. Topochemical [2+2] cycloaddition polymerization of diolefins,2c,4 [4+4] cycloaddition polymerization of anthracene-based monomers,5 [3+2] cycloaddition polymerization of azide- and alkyne-functionalized monomers,2e,6 1,4-addition polymerization of diynes2d,f,7 and dienes,2b,8 and 1,6-addition polymerizations of trienes,9 triynes,10 and quinodimethanes11 are some of the processes used for polymer synthesis. While there are a few reviews highlighting the design of monomers for TP and their topochemical reactivity,2b–g a review demonstrating the power of TP in producing polymers with advanced structural and supramolecular features is lacking. In this perspective, we have compiled selected examples of TP emphasizing its application in synthesizing organic polymers having an advanced structure and packing.2. Ladder polymers
In a ladder polymer, two parallelly running polymer chains connect to each other periodically by a covalent linker (Fig. 1). Ladder polymers are attractive as they possess enhanced thermal or mechanical stabilities compared to common single-stranded linear chain polymers.12 But achieving ladder polymers through solution-phase synthesis is strenuous. However, such polymers can be made by TP of a monomer having two polymerizable moieties separated by a linker.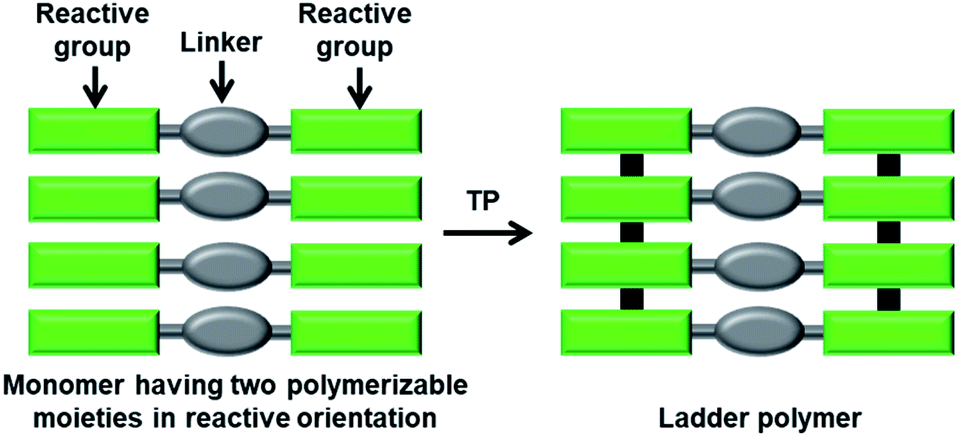 | ||
| Fig. 1 General scheme for the topochemical synthesis of a ladder polymer. | ||
TP of diacetylenes (DAs) which yields polydiacetylene (PDA) is the most commonly used method for the synthesis of ladder polymers (Fig. 2a). Diacetylene monomers that slip-stack with a stacking distance (ds) of about 4.9 Å, an orientation angle of 45° and a contact distance of 3.5 Å between the reactive ends (dCC) of the DA units of adjacent monomers, in crystals, undergo heat/light-induced topochemical 1,4-addition polymerization to form a PDA.7a,13 PDAs having 1D π-conjugated structures are attractive scaffolds with interesting electrical, chromic and non-linear optical properties.14 PDA-based ladder polymers are expected to show improved conductivity and/or non-linear optical properties as they possess more than one PDA chain in their structure.
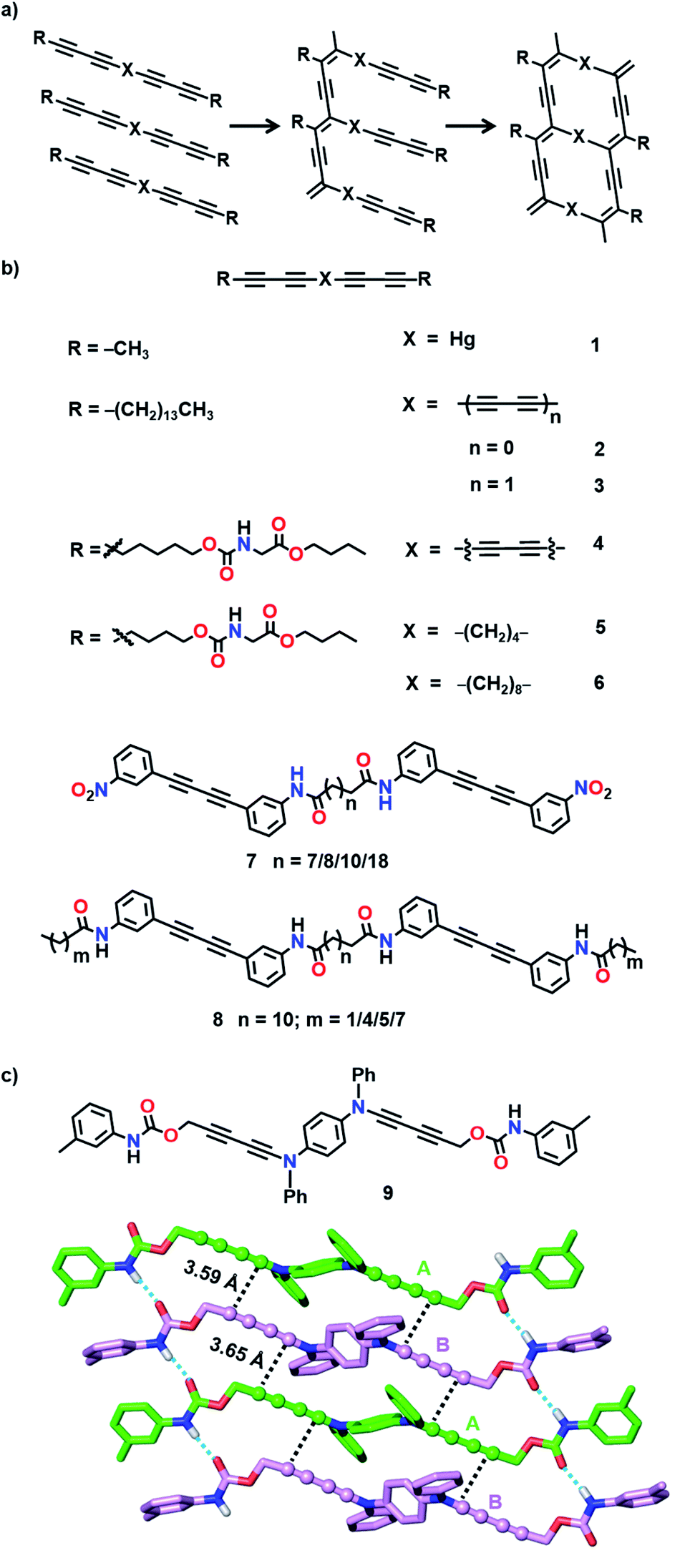 | ||
| Fig. 2 (a) Two-step topochemical 1,4-polymerization of bis(diacetylene)-based monomers to form ladder-PDAs. (b) Chemical structures of the bis(diacetylene)-based monomers 1–8. (c) Favourable packing of monomer 9 in crystals for the formation of a ladder-PDA upon TP. In the crystal packing of 9, the DA units are represented as a ball-and-stick model and H atoms are omitted for clarity, except for N–H. | ||
Wegner and co-workers synthesized the first PDA-based ladder through TP of bis(1,3-pentadiynyl)mercury (1, Fig. 2b).15 When the crystals were heated or irradiated with UV light, both the DA units in 1 underwent 1,4-addition polymerization. The authors proposed the formation of a ladder structure in the polymer, with two PDA strands running parallel to each other (Fig. 2a).
Hayamizu and co-workers synthesized the first conjugated fused-ring ladder polymer via the TP of an alkyl-substituted tetraacetylene-based monomer (2, Fig. 2b).16 The tetraacetylene-monomer underwent two-step topochemical 1,4-addition polymerization. First, UV irradiation of the crystals resulted in the formation of a PDA with diacetylene side groups. Further thermal treatment resulted in the 1,4-addition polymerization of the side group diacetylenes forming a ladder polymer having two conjugated PDA chains (Fig. 2a). Similarly, using a hexaacetylene-based monomer (3, Fig. 2b), the authors obtained a ladder-PDA wherein two PDA chains are conjugated through butadiynylene linkers.17 Probable reorganization of the PDA structure to form allene-type structures was proposed. In the examples mentioned above, although the polymers maintained their structural regularity, they were amorphous.
To preserve the crystallinity, Matsuda and co-workers attempted the synthesis of a PDA-ladder from a hexaacetylene-based monomer (4, Fig. 2b) with urethane side chains.18 The ladder formation occurred only to a partial extent owing to the restricted motion of molecules attributable to the intermolecular hydrogen bonding between the urethane units. Nevertheless, the crystallinity of the polymer was maintained.
Nakanishi and co-workers designed bis(diacetylene)-based monomers (5 and 6, Fig. 2b) having methylene linkers between the diacetylene units and urethane units in the side groups.19 While the methylene linkers offer required flexibility for TP, intermolecular hydrogen bonding through urethane units preserves the packing and maintains the crystallinity of the polymer. Upon γ-irradiation, monomers having four or more methylene units as spacers polymerized in a two-step fashion (Fig. 2a) to form ladder structures. The photopolymerization of 6 having an octamethylene linker resulted in the formation of a crystalline ladder polymer, whereas 5 having a tetramethylene linker resulted in an amorphous ladder polymer. The presence of a more flexible methylene linker would have provided the necessary flexibility for the shearing motion of diacetylene units in 6 and allowed the polymerization to take place without disturbing the crystal packing. Thus, the length of the spacers plays a vital role in the facile TP. These ladder-PDAs exhibited enhanced non-linear optical susceptibilities.19c
Takaragi and co-workers designed diphenylbutadiyne-based monomers having amide groups for the synthesis of ladder polymers.20 Amide groups could participate in intermolecular hydrogen bonding and align the molecules suitably for topochemical 1,4-addition polymerization. The authors prepared two sets of monomers having two diphenylbutadiyne units (7 and 8, Fig. 2b): one set of monomers having two amide groups (7) and another set of monomers having four amide groups (8) and compared their solid-state reactivities. In monomers having two amide groups, only one of the DA units polymerized upon photoirradiation to form a single-PDA, whereas the other diacetylene remained inactive. In monomers having four amide groups, both the DA units polymerized to form ladder polymers. It is assumed that in monomers containing only two amide units, molecular movement during the first polymerization step would have changed the alignment of the remaining diacetylene and rendered it unsuitable for further polymerization. In monomers having four amide groups, the multiple hydrogen bonds would have restricted the molecular movement to a certain extent, thus maintaining the other DA also in a reactive position for the formation of ladder polymers. Accordingly, it is important to note that a correct balance between flexibility and rigidity is necessary for favouring the polymerization of both the DA units in a monomer.
The urethane motif has been successfully employed to design suitable monomers for the synthesis of ladder polymers.18,19,21 Okuno and co-workers designed a monomer with two diacetylene units linked by a 1,4-phenylenediamine linker and aryl urethanes as side groups (9, Fig. 2c).21b The authors justified their choice of the arylamine linker in view of its effectiveness in cushioning the stress generated during the polymerization. The designed monomer 9 crystallized with two molecules (A and B) in the asymmetric unit (Fig. 2c). The arylurethane side groups participate in the intermolecular hydrogen bonding, thereby stacking the diyne units in an appropriate orientation for the TP (ds = 4.67 Å and 4.92 Å, dCC = 3.59 Å and 3.65 Å, and θ = 49° and 47°). The heating of the crystals resulted in a crystalline ladder polymer, having a small optical band gap and better conductivity than the regular PDA.
1,3-Diene-based molecules are another interesting category of monomers that are efficiently explored for the topochemical synthesis of ladder polymers. 1,3-diene monomers undergo topochemical 1,4-polymerization if the diene units align with a stacking distance of (ds) of about 5 Å, a distance of 4 Å between the reactive ends (dCC) of the diene units of neighbouring monomers and an orientation angle (θ) of 30-60°.2b Matsumoto and co-workers utilized p-xylylene diammonium disorbate (10, Fig. 3a), a monomer with two reactive sorbate functionalities separated by a diammonium xylylene linker, for the topochemical synthesis of a supramolecular ladder polymer.22 In the crystals, molecules stack to form 1D columns via π⋯π and C–H⋯π interactions between the xylylene groups. In addition, the ammonium and carboxylate groups attract each other by coulombic forces and participate in H-bonding to form robust 2D sheets. Along the 1D columns, the diene moieties stack suitably for the TP (Fig. 3a, ds = 4.99 Å, dCC = 5.47 Å, and θ = 60°). Photoirradiation resulted in the polymerization of both the sorbate units forming a crystalline supramolecular ladder polymer with an erythro-diisotactic-2,5-trans structure. However, unlike the step-wise ladder formation in diacetylene-based systems, both the sorbate units in 10 react simultaneously. The ladder structure of the polymer bestows enhanced thermal stability compared to the regular single-stranded diene polymer. While the above example detailed the formation of a supramolecular ladder, Chu and co-workers synthesized covalent ladder polymers from diene-based monomers.23 They synthesized monomers wherein two sorbate units are connected by an unreactive 1,4-phenylene diester linker. In the crystals of monomer 11, molecules stack to form a 1D columnar arrangement via π⋯π interactions between the phenyl units (Fig. 3b). The distance between the reactive carbons of adjacent diene units along the 1D columns is 3.66 Å and the stacking distance is 5.60 Å. The packing is suitable for topochemical 1,4-addition polymerization. The photoirradiation of the crystals resulted in the formation of a crystalline ladder polymer. The obtained ladder is stable even when treated with strong acids and bases.
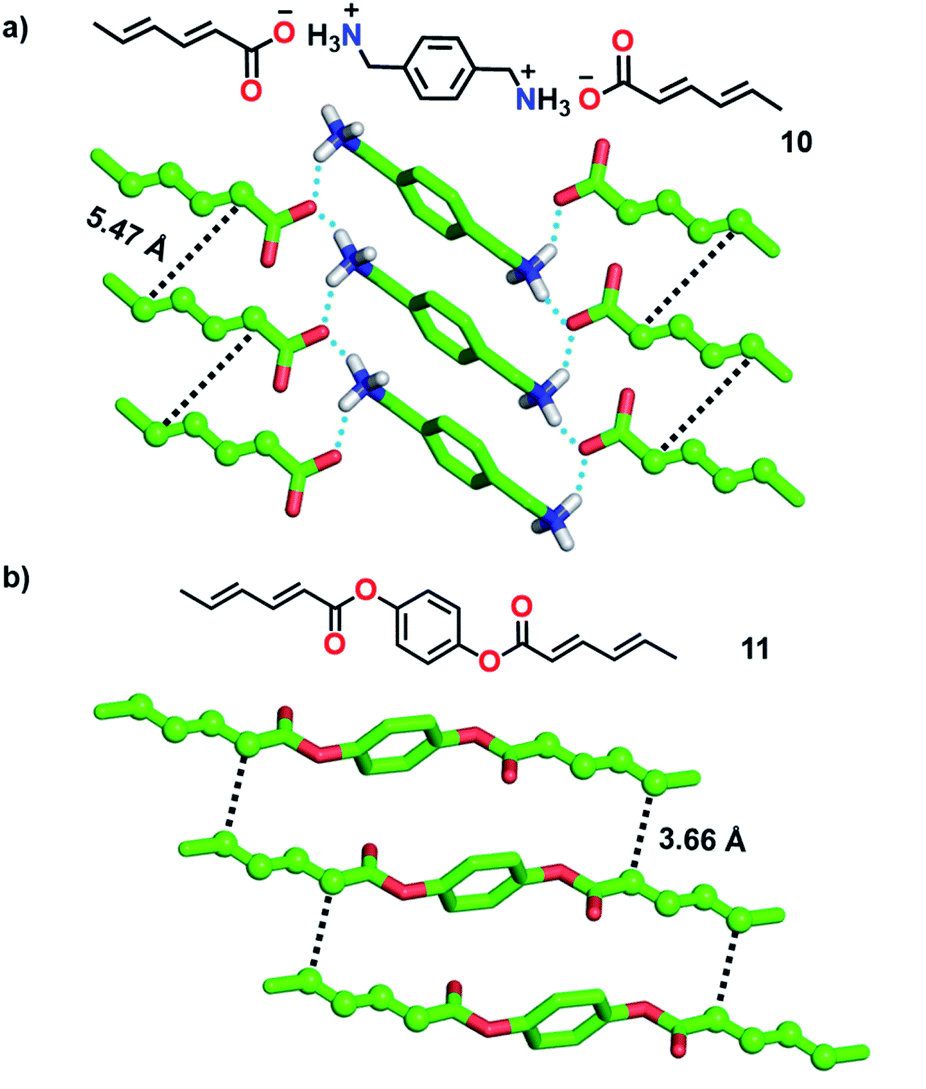 | ||
| Fig. 3 Favourable molecular packing of the diene-based monomers (a) 10 and (b) 11 for the formation of supramolecular and covalent polymer ladders, respectively. The diene units are depicted in a ball-and-stick model. H-atoms, except N–H, are omitted for clarity. | ||
While the synthesis of ladder polymers employs monomers with two reactive moieties, Morin and co-workers demonstrated the synthesis of conjugated nanowires by the TP of a star-shaped monomer with three DA units.24a Thus, further extension of the concept to monomers with more than two reactive moieties could result in polymers with unique structures.24b
3. Tubular polymers
The bottom-up synthesis of fully organic polymers with tubular architectures that mimic carbon nanotubes is of great interest.25 They find potential applications in transport, sensing, gas absorption, etc.26 One method for achieving a tubular architecture is the self-assembly of cyclic monomers into a supramolecular tube followed by their covalent crosslinking (Fig. 4). Cyclic monomers containing multiple diacetylene units (macrocyclic diacetylenes, MCDA) are established as excellent precursors for the topochemical synthesis of PDA-based organic tubular polymers that possess nanometre-sized interior cavities.2d,27 Suitably functionalized MCDAs containing multiple DA units self-stack into a columnar assembly through non-covalent interactions,28 sometimes aided by templates,29 forming a supramolecular nanotube. The subsequent 1,4-addition polymerization of the DA units along the columns results in nanotubular polymers, bearing PDA side-walls. The presence of PDAs imparts additional chromogenic and conductive properties to the tubular polymers.30 | ||
| Fig. 4 General scheme for the topochemical synthesis of a tubular polymer. | ||
Yee and co-workers made the first attempt towards the synthesis and characterization of tubular polymers.27a,b They synthesized a MCDA containing four DA units 12 (Fig. 5a) and obtained its crystals from different solvents. Among the various solvatomorphs of the cyclic tetramer 12, only chloroform solvate crystals underwent topochemical 1,4-addition polymerization. The chloroform molecules incorporated in the crystals are vital for maintaining the packing suitable for 1,4-addition polymerization of adjacent DA units. When the crystals were irradiated with 60Co γ-rays, four-fold topochemical 1,4-addition polymerization of all the DA units proceeded and produced a tubular polymer. The polymer crystals are red-brown, highly dichroic and insoluble in common organic solvents. IR spectral analysis revealed a tubular polymer reinforced with the PDA chains of a cumulene-type backbone. The tubular polymer is thermally stable up to 400 °C, which is higher than several conventional PDA polymers.
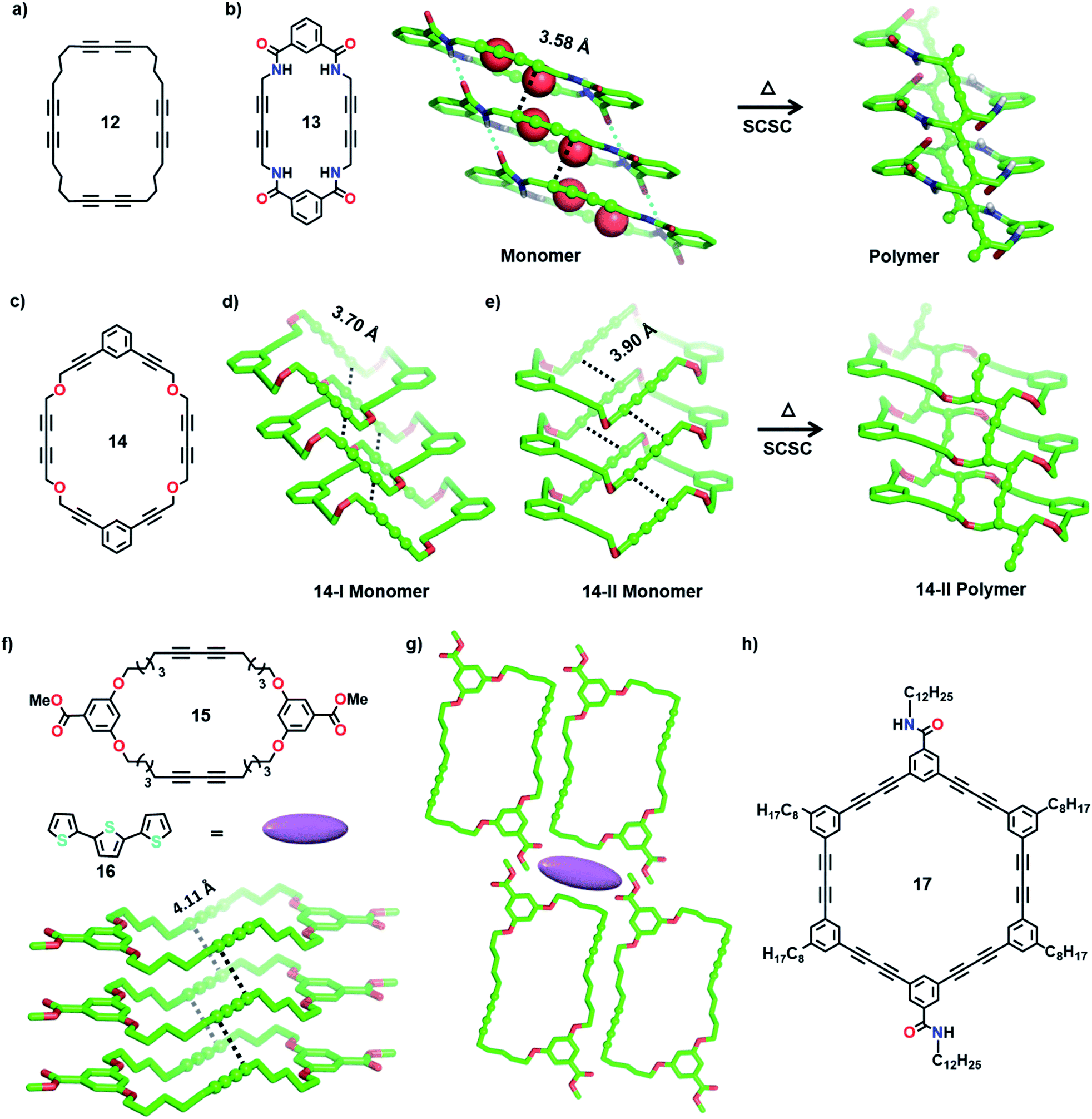 | ||
| Fig. 5 (a) Chemical structure of MCDA 12. (b) Structure and SCSC polymerization of MCDA 13 yielding a tubular polymer. Water molecules are depicted as space-filling models. (c) Chemical structure of monomer 14 and (d) favourable packing of MCDA 14 in polymorph 14-I for TP. (e) SCSC polymerization of monomer 14 in polymorph 14-II resulting in the corresponding tubular polymer. (f) Chemical structures of MCDA 15 and terthiophene 16, and favourable packing of MCDA 15 in their cocrystal for the formation of a tubular polymer. (g) Crystal packing of the cocrystal 15·16 showing the presence of terthiophene 16 in the space between four supramolecular columns of MCDA 15. (h) Chemical structure of amide-based MCDA 17. The DA units are represented as a ball-and-stick model. H-atoms except N–H are omitted for clarity. | ||
Rubin and co-workers studied the packing of several dehydro[24]annulene derivatives with four sets of DA units in crystals and liquid/solid interfaces and demonstrated their suitable alignment for TP.31 Additionally, MCDAs with two DA units functionalized with amide groups or aromatic units have proved very facile for constructing tubular polymers.2d In a remarkable effort, Shimizu and co-workers achieved tubular polymers of an amide-functionalized MCDA 13 containing two DA units (Fig. 5b).28a,32 In the dihydrate form of MCDA 13 (space group = P21/c), the macrocycles stack into tubular columns through N–H⋯O hydrogen bonds. The columns host water molecules in their cavity, and these water molecules hydrogen-bond to the carbonyl oxygens. Along the column, both sets of DA units align favourably for TP (dCC = 3.58 Å, ds = 4.99 Å, and θ = 42°). Upon heating in an inert atmosphere, 1,4-addition polymerization occurred along the columns, resulting in tubular polymers where each nanotube contains two parallel PDA chains. Water molecules are lost from the crystals during the reaction. During TP, the methylene carbons act as pivots, and the relatively planar macrocycles transform into a folded conformation. It is noteworthy that though significant molecular movement occurs in the crystal, the reaction proceeds in an SCSC fashion. This microporous PDA nanotube absorbs iodine.
In another interesting report, Lauher and co-workers synthesized an ether-linked MCDA 14 (Fig. 5c) containing two parallelly placed DA units to achieve a covalent organic nanotube.28b They obtained two different polymorphs of 14 from its dichloromethane solution at different temperatures: 14-I (monoclinic, P21/c) and 14-II (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). In both polymorphs, the macrocycles stack through π⋯π interactions to form tubular structures (Fig. 5d and e), with the DA units aligned parallelly for TP (14-I: dCC = 3.70 Å, ds = 5.09 Å, and θ = 46°; 14-II: dCC = 3.90 Å, ds = 4.84 Å, and θ = 52°). The monoclinic 14-I yielded an amorphous polymer when left at room temperature for 30 days or heated. In contrast, the form 14-II underwent SCSC polymerization upon annealing, yielding a PDA nanotube (Fig. 5e). The polymerization was accompanied by tilting of the macrocycle, forming a tubular architecture with a circular cross section of 1 nm, similar to a carbon nanotube.
). In both polymorphs, the macrocycles stack through π⋯π interactions to form tubular structures (Fig. 5d and e), with the DA units aligned parallelly for TP (14-I: dCC = 3.70 Å, ds = 5.09 Å, and θ = 46°; 14-II: dCC = 3.90 Å, ds = 4.84 Å, and θ = 52°). The monoclinic 14-I yielded an amorphous polymer when left at room temperature for 30 days or heated. In contrast, the form 14-II underwent SCSC polymerization upon annealing, yielding a PDA nanotube (Fig. 5e). The polymerization was accompanied by tilting of the macrocycle, forming a tubular architecture with a circular cross section of 1 nm, similar to a carbon nanotube.
Following these reports, Kim and co-workers synthesized several ether-linked MCDAs for obtaining tubular polymers. They systematically varied the length of alkyl spacers between DA units and aromatic linkers in the MCDAs and investigated their TP in crystals.30a The resulting tubular polymers displayed interesting solvatochromic and thermochromic properties. In a subsequent study, they attempted to access the porous channels of an ether-linked MCDA-15 (Fig. 5f) for complexing guest molecules. They investigated the optical properties of a host–guest cocrystal before and after DA polymerization.33 As it is known that PDAs generally absorb in the visible region, the authors chose terthiophene (16, Fig. 5f, emits in the visible range) as a guest to facilitate energy transfer between the guest and PDA. In the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystal of 15·16, the molecules of MCDA-15 adopt a chair-like conformation and align into columnar assemblies through π⋯π interactions. Along the 1D columns, the DA units align appropriately for TP (Fig. 5g, dCC = 4.11 Å, ds = 4.71 Å, and θ = 57°). The terthiophene guest molecules are proposed to be present in the voids between four parallelly packed columns in a linear array (Fig. 5g). This MCDA-15·16 cocrystal displays strong blue fluorescence from the terthiophene. Upon UV irradiation, TP occurs along the columns, resulting in a conjugated polymer (PDA nanotube). This polymerization resulted in the quenching of terthiophene fluorescence due to the effective energy transfer between the fluorescent terthiophene (donor) and PDA (acceptor). This suggests that a PDA-based donor–acceptor system can be applied in optoelectronics.
:1 cocrystal of 15·16, the molecules of MCDA-15 adopt a chair-like conformation and align into columnar assemblies through π⋯π interactions. Along the 1D columns, the DA units align appropriately for TP (Fig. 5g, dCC = 4.11 Å, ds = 4.71 Å, and θ = 57°). The terthiophene guest molecules are proposed to be present in the voids between four parallelly packed columns in a linear array (Fig. 5g). This MCDA-15·16 cocrystal displays strong blue fluorescence from the terthiophene. Upon UV irradiation, TP occurs along the columns, resulting in a conjugated polymer (PDA nanotube). This polymerization resulted in the quenching of terthiophene fluorescence due to the effective energy transfer between the fluorescent terthiophene (donor) and PDA (acceptor). This suggests that a PDA-based donor–acceptor system can be applied in optoelectronics.
Morin and co-workers recently reported tubular polymers of several phenylacetylene- and phenylene butadiynylene-based MCDAs in the gel/xerogel state.34 Increasing the number of polymer chains forming the backbone of the tubular polymer is expected to enhance their properties, including stability. With the intention of preparing larger, rigid and robust PDA-walled nanotubes, the authors designed a phenylene butadiynylene-based amide-functionalized MCDA 17 containing six DA units (Fig. 5h). The molecules of 17 self-assemble to form an organogel in cyclohexane. The PXRD spectrum of the gel suggested that the macrocycles stack via π⋯π interactions. UV irradiation of the xerogel of MCDA 17 resulted in polymerization forming a dark-blue PDA. Raman spectral analysis revealed that all six DA units underwent 1,4-addition polymerization. The formation of nanotubes was evident from the high-resolution transmission electron microscopy (HRTEM) image of a single polymer nanotube with a hollow interior cavity.34a In a later study, the authors have demonstrated facile TP of several MCDAs in higher yields by applying a pressure of 1 GPa in a sapphire-anvil cell at 390 K.34b
4. Higher-dimensional polymers
There is huge interest in fully organic 2D polymers that mimic graphene, as they find extensive applications in gas storage, filtration, separation of materials, catalysis, nonlinear optics, photovoltaics and production of materials with a range of conductivities for energy storage devices.35 The topochemical approach for 2D polymer synthesis is advantageous over solution-phase synthesis as it enables thorough structural elucidation and gives ordered 2D polymers.36 For the topochemical synthesis of 2D polymers, monomers with multiple reactive units are required. The successful 2D TP necessitates the alignment of these reactive moieties in favourable orientation for TP in two dimensions.37One common strategy for the synthesis of a 2D polymer is the design of monomers having three reactive units (Fig. 6).24b,38 Ozaki and co-workers designed a monomer 1,15,17,31-dotriacontatetrayne (18, Fig. 7a) with two different reactive units: an internal diacetylene and two terminal alkynes, and obtained its monolayer on highly oriented pyrolytic graphite as the substrate.39 The authors monitored the photopolymerization of 18 using Penning ionization electron spectroscopy (PIES)39a at various stages. Later, imaging through scanning tunneling microscopy (STM) provided direct evidence for the formation of a 2D polymer.39b The molecules of 18 self-assemble into columns and such columns align adjacent to each other. Under UV light, 1,4-addition polymerization of DA units proceeded along the column, and 1,2-addition polymerization of terminal acetylenes occurred between the molecules on adjacent columns. The resulting 2D polymer constitutes alternately placed polydiacetylene and polyacetylene chains connected periodically by alkyl chains (Fig. 7a). This is the first synthesis of a covalently-linked 2D polymer.
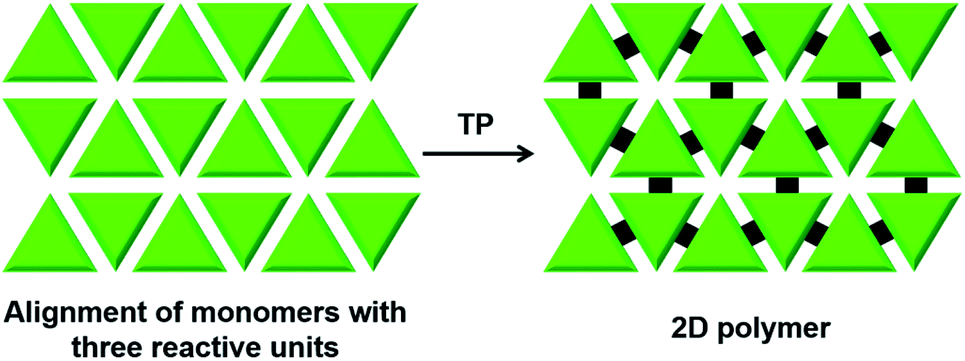 | ||
| Fig. 6 General scheme for the topochemical synthesis of a 2D polymer from a monomer having three reactive units. | ||
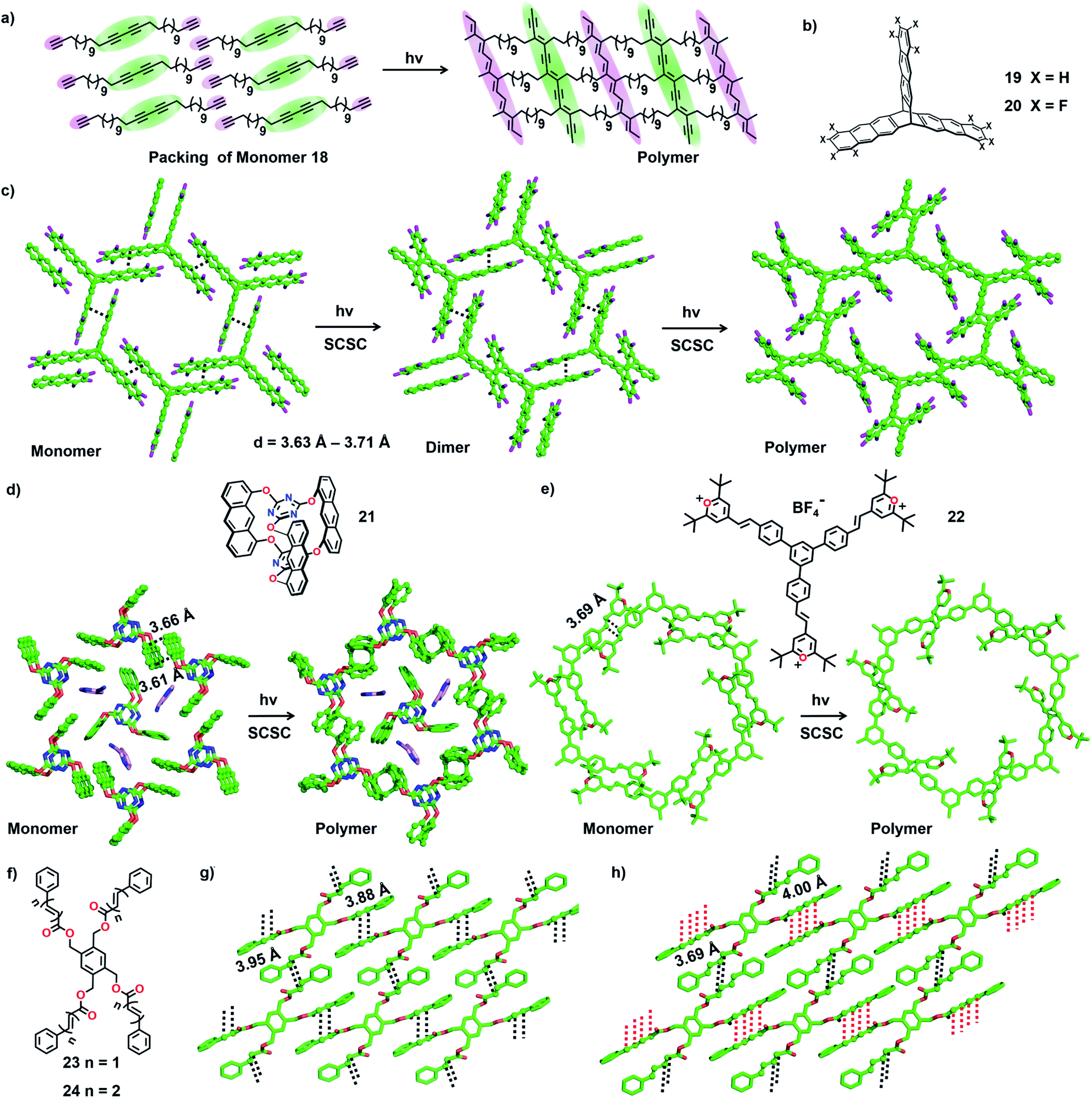 | ||
| Fig. 7 (a) Schematic representation of the 2D polymerization of monomer 18. (b) Chemical structures of triptycene monomers 19 and 20. (c) Two-step SCSC polymerization of monomer 20 to a 2D polymer. (d) SCSC 2D polymerization of anthraphane 21. (e) SCSC 2D polymerization of monomer 22. In the packing diagram, the disordered portions are omitted for clarity. (f) Chemical structures of monomers 23 and 24. (g) Favourable packing of monomer 23 for the formation of a 2D polymer. (h) Favourable packing of monomer 24 for the formation of a 2D polymer. The reactive anthracene units and all olefin units are depicted as a ball-and-stick model. H atoms are omitted for clarity. | ||
In 2012, Sakamoto and co-workers successfully carried out topochemical 2D polymerization via a [4+2] cycloaddition reaction of a monomer constituting three 1,8-diethynylanthrylene units linked to benzo-1,3,5-triate unit via terphenylene linkers.40 Soon after, King and co-workers introduced a triptycene-based monomer 19 (Fig. 7b), having three anthracene blades.5b,41 The monomers pack in a lamellar fashion such that each of the three anthracene blades stacks with an anthracene unit of a neighbouring monomer. Upon irradiation, polymerization occurred resulting in a crystalline 2D polymer. However, during the course of the reaction, the crystals cracked, leading to the loss of single crystal nature.41 Hence, the authors introduced the well-known arene–perfluoroarene interaction to improve the stacking between the anthracene blades (compound 20, Fig. 7b).5b The molecules of 20 also adopted a face-to-face (ftf) antiparallel stacking between the anthracene units of adjacent monomers (d = 3.63 Å–3.71 Å, Fig. 7c). The authors observed a two-step SCSC conversion from monomer 20 to a dimer and then to a polymer via a topochemical [4+4] cycloaddition reaction under blue light (460 nm). The polymer underwent depolymerization via retrocycloaddition to a monomer when heated (150 °C). Heating the polymer crystals at 50 °C in 1-methyl-2-pyrrolidone (NMP) resulted in the exfoliation of the 2D polymer to thin layers without any depolymerization.
Later, Schlüter and co-workers introduced anthraphane 21 for the synthesis of a 2D polymer.5c In the crystal packing, molecules of 21 showed an interesting dual role. One-third of the molecules act as a template, around which the remaining two-third of the molecules adopt a hexagonal arrangement via ftf π⋯π stacking (Fig. 7d). Upon photoirradiation (465 nm), the hexagonally arranged molecules underwent SCSC topochemical [4+4] cycloaddition polymerization, leaving the template unreacted. Interestingly, the thermal (180 °C) depolymerization to a monomer also occurred in a SCSC manner. Thin layers of this 2D-polymer could be exfoliated by heating these crystals in NMP at 50 °C for 2–3 weeks.36b Subsequently, the authors investigated the structural transformations during photopolymerization in detail. They recorded SCXRD data for the crystals of 21 at different reaction intervals and proposed that the 2D polymerization occurs in a self-impeding fashion.42
Monomers containing multiple olefin units pre-organized in a reactive orientation are also known to undergo TP to yield 2D polymers through [2+2] cycloaddition.43,44 Monomer 22 possesses three styrylpyrylium arms (Fig. 7e). Each olefin-containing arm stacks antiparallelly (d = 3.93 Å) with an olefin unit of a nearby monomer forming a supramolecular 2D layer (Fig. 7f).44 In each layer, a set of six monomer units together form a honeycomb like lattice. Photoirradiation (λ = 530 nm; T = 4 °C) of the crystals results in [2+2] cycloaddition forming a 2D polymer in a SCSC fashion. Thermal depolymerization to a monomer via retro-cycloaddition occurs at 150 °C. Heating a dispersion of polymer crystals in γ-butyrolactone at 80 °C for six days exfoliates a few layers.
Chu and co-workers designed tetrapodal monomers with reactive ene (23) or diene (24) units in the arms and studied their TP in crystals.43 The symmetric four-armed monomer 23, functionalized with an olefin unit in each arm, packs in a 2D layer via π⋯π stacking of the arms of neighbouring monomers. This arrangement brings the reactive olefins to a distance suitable for their [2+2] cycloaddition reaction (d = 3.88–3.95 Å). Similarly, the monomer 24 functionalized with conjugated dienes in each of the four arms crystallizes with a layered packing via π⋯π stacking between the arms of adjacent molecules (Fig. 7g). The packing favours two of the diene arms in an orientation (d = 4.0 Å) to form ladderane (Fig. 7h, distances marked with red lines) between them and the remaining two arms in an orientation (d = 3.69 Å) to form a cyclobutane ring (Fig. 7h, distances marked with black lines) between them, upon a topochemical reaction. As expected, upon irradiation (UV or sunlight) of the powdered crystals, 23 results in a cyclobutane-linked 2D polymer, while 24 yields a 2D polymer containing both ladderane and cyclobutane linkages.
Can one make 3D polymers via TP? In this context, a recent report by Thomas and co-workers is very promising. They synthesized a 2D COF (25) by a condensation reaction between trimethyltriazine and terephthalaldehyde (Fig. 8).45 Powley refinement of PXRD data combined with crystal structure modelling showed eclipsed stacking of COF layers with a stacking distance of 3.4 Å. This packing is suitable for a topochemical [2+2] cycloaddition reaction between parallelly stacked olefin units. Upon irradiation, the 2D COF showed a colour change from yellow to colourless. This is due to the formation of cyclobutane linkages via [2+2] cycloaddition between the 2D COF layers to yield a 3D COF, which disrupt the conjugation in the 2D COF, leading to the colour change. Unfortunately, the crystallinity was lost during polymerization, giving a 3D COF as an amorphous powder. Later, Perepichka and co-workers achieved the 3D COF of 25, with the retention of crystallinity, by tuning the polarity of the dispersion media as well as by varying the irradiation source.46 They also added a naphthalene-based crystalline 3D COF (26) to the library via TP between the layers of 2D COF 26 (Fig. 8). The formation of cyclobutane linkages was confirmed by FT-IR and 13C NMR spectroscopic techniques. Heating the 3D COFs at 200 °C induced partial regeneration of the 2D COFs.
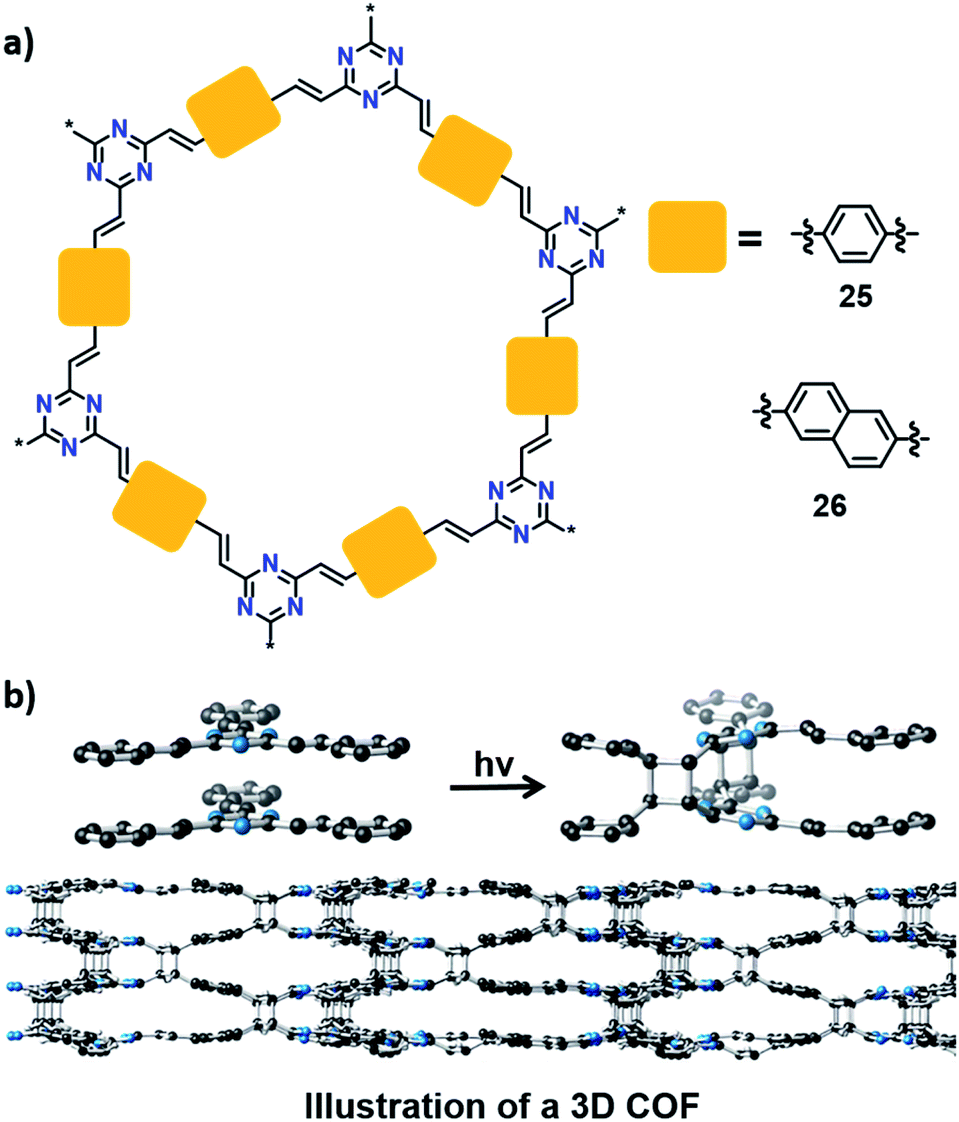 | ||
| Fig. 8 (a) Representation of 2D COFs 25 and 26. (b) Transformation of a 2D into a 3D COF via a topochemical [2+2] cycloaddition reaction between olefin units in the adjacent layers of COF 25. | ||
5. Copolymers
Copolymers, polymers made from more than one type of monomer unit, are attractive materials showing interesting properties. Statistical (random) copolymers and alternating copolymers, in which the different monomers are connected alternatively, are two important types of copolymers, the latter being synthetically challenging, especially when the monomers are of similar nature and reactivity. TP offers a solution for this problem; a judiciously designed crystal having more than one type of monomer co-aligned alternately in reactive geometry would undergo TP to give alternating copolymers. While TP using a mixed crystal of two or more isomorphous monomers would give a random copolymer, the use of cocrystals having a stoichiometric ratio of all comonomers results in a sequence-defined alternating copolymer (Fig. 9).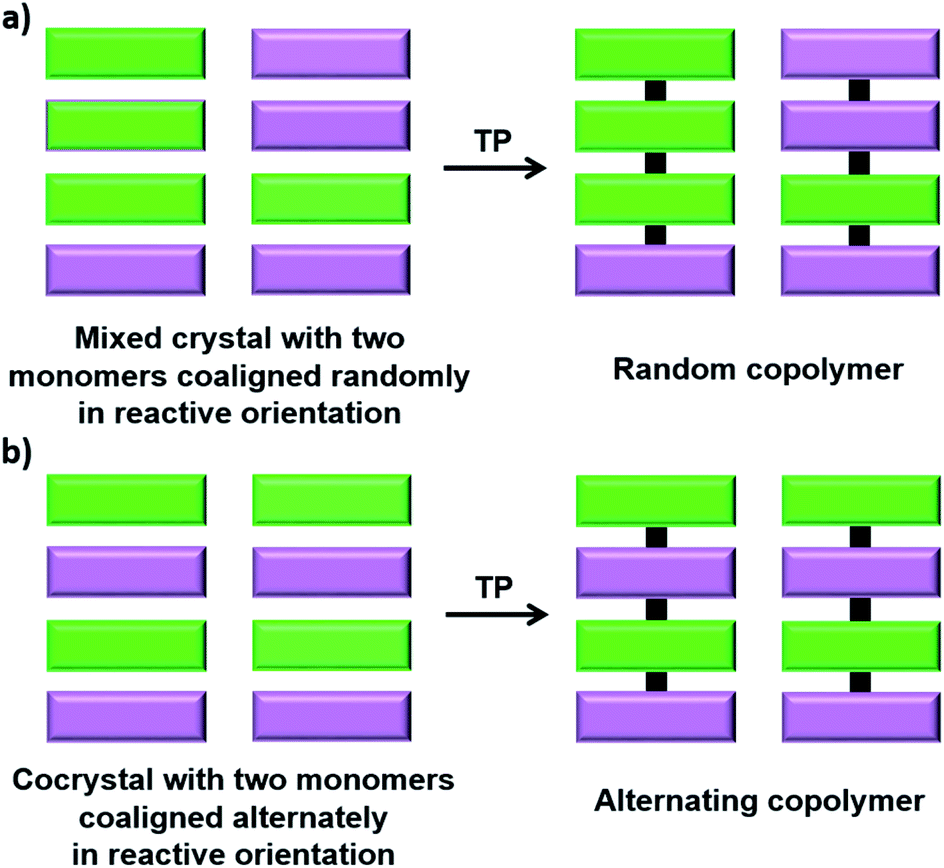 | ||
| Fig. 9 General scheme for the topochemical synthesis of (a) a random copolymer and (b) an alternating copolymer. | ||
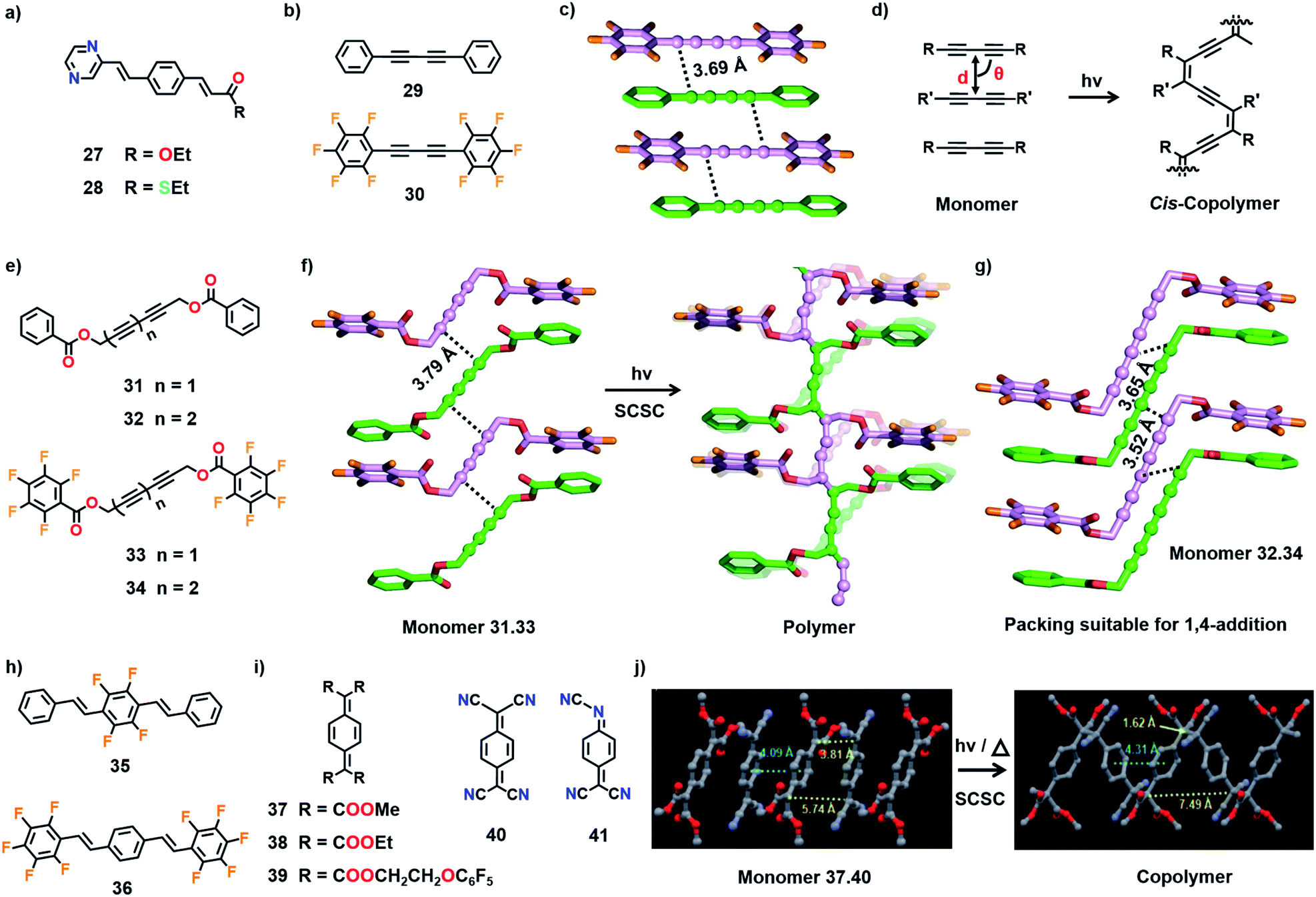
Ester (27) and thioester (28) derivatives (Fig. 10a) are isomorphous and both compounds crystallize in a packing suitable for [2+2] cycloaddition polymerization.47 Molecules 27 and 28 react in their individual crystals to form the corresponding crystalline polymers upon photoirradiation (UV light). Hasegawa and co-workers exploited the isomorphous nature of these molecules to prepare their mixed crystals. Due to the isomorphous nature of 27 and 28, they could form mixed crystals with similar favourable alignment for TP as in the individual crystals and form copolymers upon photopolymerization. Both 27 and 28 readily form mixed crystals either by crystallization from methanol or by grinding both the compounds together. The mixed crystal formed with a nonstoichiometric ratio of 27 and 28 (45:55). In the crystals, both the molecules are present in a random sequence (solid solution) and the resulting copolymer contains a random arrangement. This is the first report validating the formation of a copolymer through TP. Implementing a similar approach, recently Okada and co-workers prepared a PDA copolymer from the mixed crystals of diacetylene monomers (solid solutions).48 However, in these cases, a detailed analysis of the crystal structures and (or) molecular packing of the mixed crystals (monomers) and the corresponding copolymers is not feasible, as the molecules are usually present in a random sequence.
 | ||
| Fig. 10 Chemical structures of the (a) isomorphous monomers 27 and 28 and (b) the complementary monomers 29–30. (c) Suitable molecular packing in the cocrystal 29·30 for the formation of an alternating cis-copolymer. (d) General schematic for the cis-copolymer formation from a diacetylene-based cocrystal. (e) Chemical structures of 31–34. (f) SCSC polymerization of the cocrystal 31·33 to the corresponding alternating copolymer. In the polymer structure, the disordered distribution of monomer (faded) and polymer parts are superposed. (g) Alternate alignment of comonomers in the cocrystal 32·34 in a packing suitable for 1,4-addition TP. (h) Chemical structures of diolefin-based monomers 35 and 36. (i) Chemical structures of monomers 37–41. (j) SCSC 1,6-polymerization of the cocrystal 37·40. The DAs and PDA polymer backbone in (c)–(g) are depicted in a ball-and-stick model. H atoms are omitted for clarity. (j) is adapted from ref. 11b with permission from John Wiley and Sons, copyright 2011. | ||
A cocrystal comprising a 1:1 ratio of two reactive molecules is proven to be the most suitable system for the rational synthesis of copolymers through the topochemical method. Molecules exhibiting complementary interactions have been explored effectively for the preparation of 1:1 cocrystals. For example, benzene and perfluorobenzene rings have quadrupole moments with a similar magnitude but opposite in sign.49 This aromatic pair is known to stack alternately in cocrystals and is a reliable supramolecular synthon for the rational design of monomers for TP.
Grubbs and co-workers implemented phenyl-perfluorophenyl interactions for achieving 1:1 cocrystals of two diyne-based monomers (Fig. 10b) diphenylbutadiyne (29) and decafluorodiphenylbutadiyne (30).50 Both the molecules stack alternately to form columns (Fig. 10c). The distance between the diyne carbons is 3.69 Å and the mean angle between the stacking axis and the molecular axis is 75°. The packing of monomers in the cocrystal (29·30) suggests the probable formation of an alternating copolymer with cis-PDA configuration (Fig. 10d). Interestingly, while the individual monomers did not undergo effective TP, the cocrystal underwent TP to form a copolymer upon UV irradiation. However, the crystals shattered during polymerization and only the smaller oligomers were observed in the mass spectrum. Although the crystal packing suggests the possibility of cis-specific copolymerization, the structure of the polymer could not be determined.
Frauenrath and co-workers prepared the cocrystals of symmetrical diacetylene and triacetylene monomers by employing phenyl–perfluorophenyl stacking as a supramolecular synthon (Fig. 10e).51 However, these molecules possess ester linkers connecting the diacetylene or triacetylene units with aromatic groups and hence are more flexible than the rigid arylbutadiynes 29 and 30. Whereas the individual molecules did not polymerize effectively, their cocrystals (31·33 and 32·34) reacted to form alternating copolymers. In the cocrystals of 31·33, both the diacetylenes situate alternately to form columns and stack the phenyl and perfluorophenyl groups in a face-to-face manner. The diacetylenes exhibit appropriate packing for TP (dCC = 3.79 Å, ds = 4.80/5.10 Å, and θ = 46°/53°, Fig. 10f). When the crystals were irradiated with UV light, 1,4-addition polymerization occurred as expected, yielding an alternating copolymer. During the polymerization, ester linkages between the diacetylene and aromatic units act as hinges and assist the shearing movement of diacetylene without causing drastic movement in the aromatic side groups. The single crystalline nature of the copolymer is preserved after the reaction. This is the first PDA copolymer whose structure is proved unambiguously. The triacetylene-based cocrystals 32·34 also adopted a similar packing as in 31·33 (Fig. 10g). The triacetylenes 32 and 34 stack alternatively through perfluorophenyl–phenyl interactions (Fig. 10g). Like diacetylenes, triacetylenes also underwent 1,4-addition polymerization in the cocrystals, yielding a copolymer with a poly(trans-ene-yne) backbone conjugated to acetylene groups in the side chain.52
Phenyl/perfluorophenyl stacking has also been exploited for the cocrystallization of olefin-based monomers for achieving the corresponding copolymers via TP.53 Grubbs and co-workers obtained a 1:1 cocrystal of diolefins 35 and 36 (Fig. 10h) and achieved their copolymer formation through [2+2] cycloaddition polymerization.53a
Another class of molecular pairs that form 1:1 cocrystals with alternate alignment is 7,7,8,8-tetrakis(alkoxycarbonyl)quinodimethanes (37–39) and 7,7,8,8-tetracyanoquinodimethane (40), due to their ability to form charge-transfer type complexation (Fig. 10i). For instance, 7,7,8,8-tetrakis(methoxycarbonyl)quinodimethane (37) forms a charge-transfer complex with the electron-accepting 40 in the solution state (acetonitrile) and polymerizes spontaneously to form an alternating copolymer. Exploiting this charge-transfer complexation behaviour, Itoh and co-workers prepared the cocrystals of 37 and 40.11b In the cocrystals (37 and 40), both the molecules stack alternately with a torsion angle of 56.4° to form 1D columns. Their centres of gravity are separated by 4.09 Å and aligned in a straight line. In the crystals, two distances are present between the reactive exomethylene carbons, 3.81 Å and 5.74 Å (Fig. 10j). When the crystals were either irradiated with UV light or heated, TP occurred by connecting the molecules along the stacks through the proximal exomethylene carbons to produce an alternating copolymer with syn-orientation of the aromatic rings. As the centres of gravity of the molecules are aligned in a straight line, polymerization occurred with less molecular movement and the single crystal nature of the copolymer is preserved.
Subsequently, the concept has been extended to other tetrakis(alkoxycarbonyl)quinodimethanes (38 and 39) for the synthesis of the corresponding alternating copolymers with 40.11c,54 However, when the unsymmetrical N,7,7-tricyanoquinone methide (41, Fig. 10i) was utilized as an electron-accepting analogue for cocrystallization with tetrakis(alkoxycarbonyl)quinodimethanes, cocrystals underwent TP to produce amorphous alternating copolymers. The centres of gravity in the cocrystals containing 41 are not aligned in a straight line, necessitating a large movement of molecules during polymerization, leading to the loss of crystallinity.
It is clear from the above examples that the cocrystallization of two monomers followed by their TP is a powerful and successful strategy for making alternating copolymers. Interestingly, there are cases of topochemical copolymerization wherein one of the monomers diffuses into the other monomer crystal during polymerization. Matsumoto and co-workers observed that crystals of octadecyl sorbate procured from ethanol (42) react with oxygen resulting in an alternating copolymer with peroxy linkages (Fig. 11a).55 Later, a few alkyl sorbates and N-substituted sorbamides were also found to react in the solid state in the presence of oxygen, forming alternating copolymers with oxygen.56
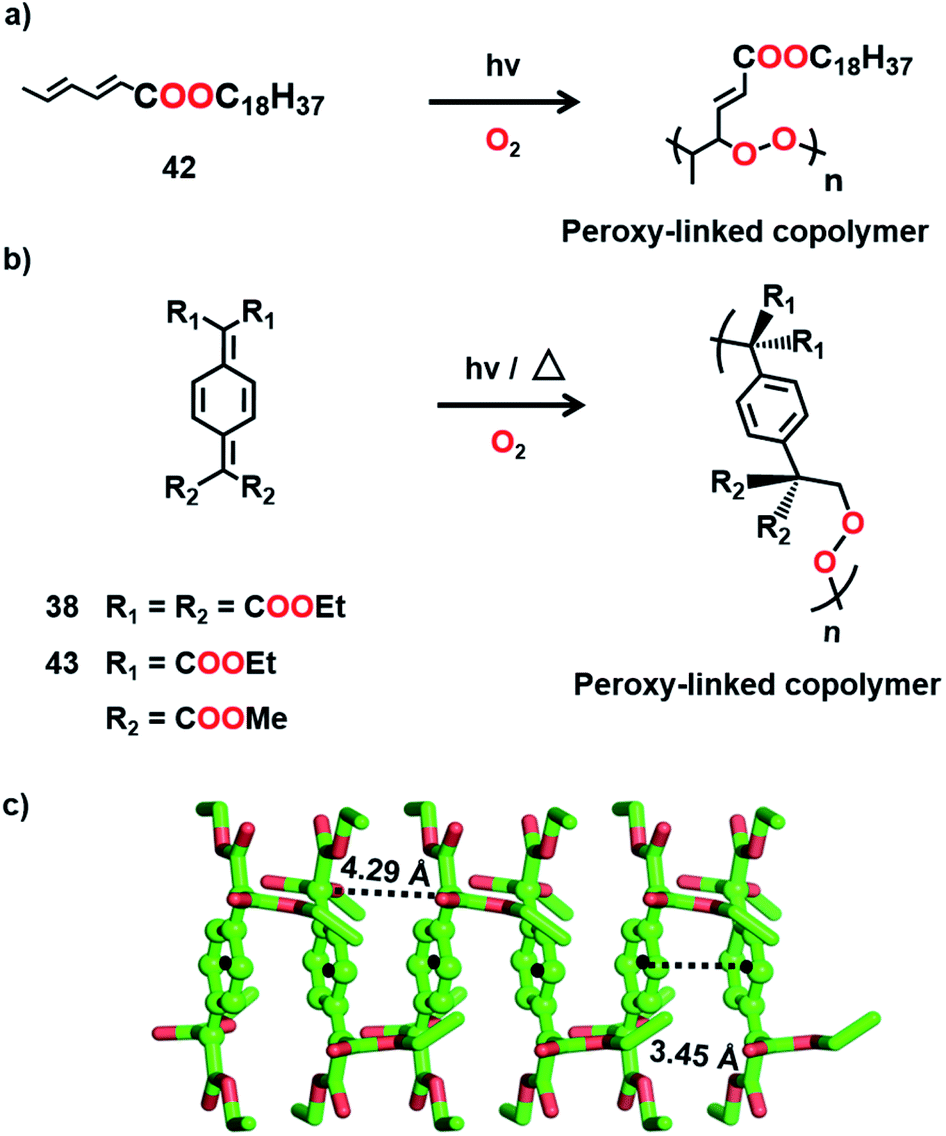 | ||
| Fig. 11 Topochemical copolymerization of (a) 42, (b) 38 and 43 with oxygen to form copolymers with peroxy linkages. (c) A suitable molecular packing of 38 for the insertion of oxygen. The quinodimethane units are depicted in a ball-and-stick model. H atoms are omitted for clarity. | ||
Itoh and co-workers observed that the crystals of 7,7,8,8-tetrakis-(ethoxycarbonyl)quinodimethane (38), upon UV irradiation or heating, polymerize in the presence of air or oxygen to form a crystalline alternating copolymer with peroxy linkages (Fig. 11b).57 But it does not react in a vacuum. The crystal structure reveals that, although the molecules of 38 stack in a columnar arrangement (Fig. 11c), the molecular packing is not appropriate for homopolymerization through 1,6-addition. However, the voids present in the crystal can permit the diffusion of oxygen, which can link the adjacent molecules of 38 by reacting at the exomethylene carbons. It is regarded that the copolymerization with oxygen commences at the surface of the crystals and further the diffusion of oxygen into the crystal lattice helps in the progress of the reaction along the 1D stacks. A similar derivative, 7,7-bis(ethoxycarbonyl)-8,8-bis(methoxycarbonyl) quinodimethane (43, Fig. 11b), also copolymerizes with oxygen to form an alternating copolymer.58 However, the copolymer obtained from 43 is amorphous. A larger stacking distance (5.11 Å) and/or a larger distance between the reactive exomethylene carbons (5.11 Å) could have necessitated significant molecular movement and resulted in the loss of crystallinity.
6. Polymer blends
A polymer blend is a mixture of at least two different polymers. While there are several interesting approaches for achieving a polymer blend in the solution state, the polymers are generally randomly aligned. Thus, it would be interesting to achieve polymer blends via the topochemical approach, as it results in different polymers to form a blend in an ordered fashion. However, achieving two different polymers in a crystal forming a blend is challenging as it requires two differently reacting monomers to be self-sorted in the same crystal (Fig. 12).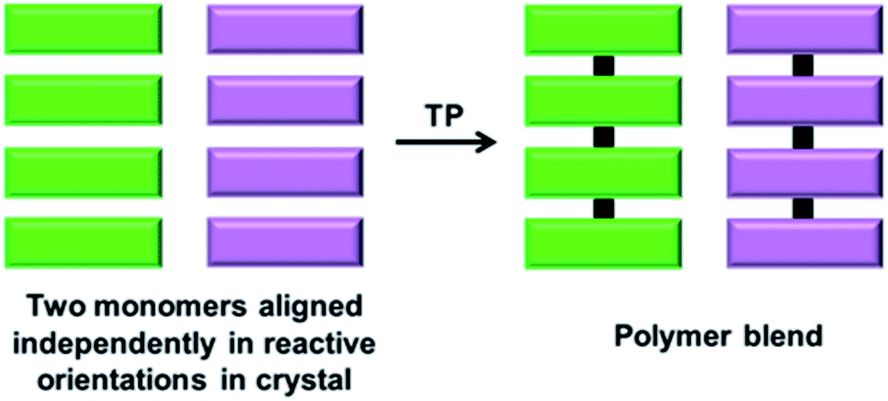 | ||
| Fig. 12 Schematic for the topochemical synthesis of a polymer blend from a crystal composed of two self-sorted reactive molecules. | ||
Matsumoto and co-workers attempted to synthesize two different polymers in a single crystal by the TP of a salt having two reactive moieties: diyne and diene (Fig. 13a). Upon polymerization, two polymers with diverse structures and properties are anticipated to co-exist in a single crystal. For achieving this, the authors prepared several diynediammonium dienecarboxylates as monomers.59 Among the various derivatives prepared, 4,4′-butadiynedibenzylammonium disorbate (44) adopted an appropriate alignment for the reaction. In the crystals of 44, diene and diyne units stack to form a 1D-columnar arrangement assisted by the stacking of phenyl rings. When the crystals were irradiated, the diene units underwent smooth polymerization in a SCSC fashion, whereas the diyne units were intact in the polymer single crystals. However, a low conversion of diynes to PDA was assumed based on the change in the colour of the monomer crystals.
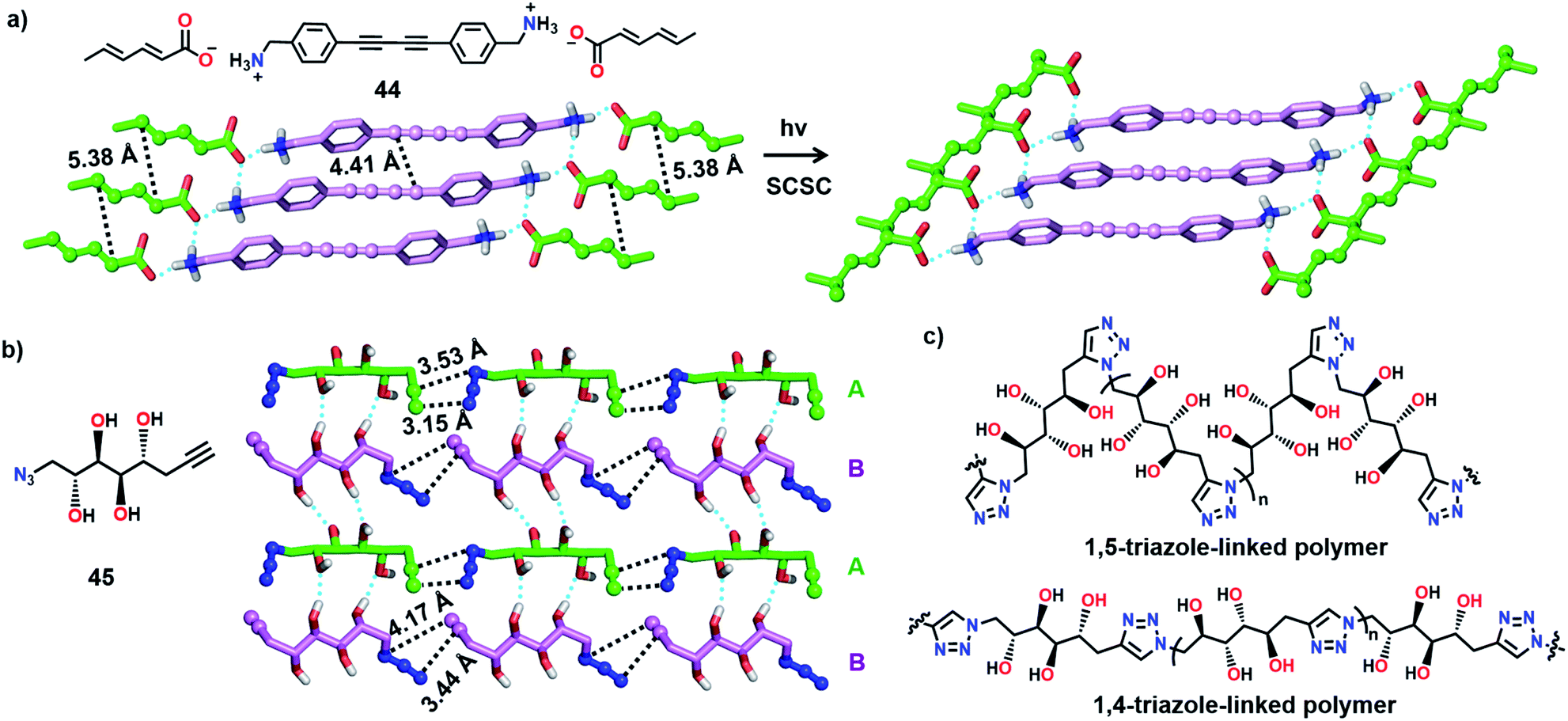 | ||
| Fig. 13 (a) SCSC TP of 44. While the diene moieties polymerized, diyne units remained intact. (b) Self-sorted head-to-tail alignment of symmetry-independent conformers A and B in monomer 45, a favourable alignment for the formation of a polymer blend. (c) Chemical structures of 1,5-and 1,4-triazole-linked polymers of 45. The diyne, diene, azide and alkyne units are represented in a ball-and-stick model. H atoms, except for O–H and N–H, are omitted for clarity. | ||
Recently, we have shown the synthesis of two different polymers from a monomer having one type of reactive unit (azide and alkyne pair).60 Depending on the pre-organization of azide and alkyne groups in a monomer, a topochemical azide–alkyne cycloaddition (TAAC) reaction generally produces polymers with one of the two possible linkages, i.e., either 1,4- or 1,5-triazolyl linkages. Remarkably, the monomer 45 attained a crystal packing such that two different polymers, one with 1,4- and the other with 1,5-triazole-linkages, could be formed upon TP (Fig. 13b). In the crystal structure, the molecules of 45 exist as two symmetry independent conformers A and B in a 1:1 ratio. Both the conformers self-sort to form head-to-tail aligned chains. Hence, the azide and alkyne units of each conformer are proximally aligned for the TAAC reaction with the conformers of the same type. Remarkably, one of the conformers (A) adopts an alignment suitable for the formation of the 1,5-triazole-linked polymer, and the other conformer (B) attains an orientation suitable for the formation of the 1,4-triazole-linked polymer. Such a molecular packing would result in the formation of an alternate miscible blend of 1,4- and 1,5-triazole-linked polymers upon the TAAC reaction (Fig. 13c). As anticipated, when the crystals of 45 were heated at 75 °C, a 1:1 polymer blend containing both 1,4- and 1,5-triazole-linked polymers was obtained. The interesting aspect of the solid-state synthesis of a polymer blend is that both the polymer chains co-exist in an ordered fashion and contain effective supramolecular interactions between the polymer chains.
7. Novel topologies
The properties of polymers depend not only on their molecular structure but also on their topology.61 The packing of polymer chains into distinct architectures results in polymers with intriguing properties and applications. By TP, it is possible to obtain polymers with unique topologies predefined by the packing of monomers.We have achieved a pseudoprotein with a helical-sheet topology that resembles the cross-α-packing (β-sheets formed via lateral hydrogen bonding of helices) of amyloid proteins via the SCSC TAAC polymerization of a dipeptide (46, Fig. 14a and b).62 In its crystal, the molecules of 46 align as β-sheets by means of N–H⋯O hydrogen bonds (Fig. 14c). Orthogonal to this β-sheet-arrangement, molecules adopt a head-to-tail alignment. Upon heating the crystals at 60 °C for nine days, a regiospecific TAAC reaction between the monomers in alternate layers resulted solely in a 1,4-triazole linked pseudoprotein with Gly–Phe–Gly repeats. As the reaction occurred between the monomers of alternate layers, the resulting pseudoprotein possesses a helical (left-handed) topology that retained the β-sheet packing in the lateral direction.
 | ||
| Fig. 14 (a) Chemical structure of dipeptide 46. (b) Schematic representation of a covalent helical-sheet. (c) SCSC TAAC polymerization of monomer 46 to a pseudoprotein with helical sheet packing. (d) Chemical structures of monomers di- and tripeptides 47 and 48. (e) Schematic representation of cross-laminated arrangement. (f) SCSC TAAC polymerization of dipeptide 47 to a cross-laminated polymer. (g) Chemical structure of dipeptide 49 and schematic representation of the twist-stacked topology. (h) SCSC TAAC polymerization of dipeptide 47 to a polymer in a twist-stacked topology. The azide, alkyne and triazole units are represented by a ball-and-stick model. H atoms except N–H are omitted for clarity. | ||
Very recently, we reported two topologically unique cross-laminated polymers synthesized via TAAC polymerization. The designed Gly-based dipeptide 47 and the tripeptide 48 (Fig. 14d) crystallize with a layered packing of molecules in their respective crystals. Each layer consists of parallelly packed 1D supramolecular chains that are formed by the head-to-tail arrangement of monomers. This arrangement places the azide and alkyne of adjacent molecules proximally preorganized in a reactive geometry in each supramolecular chain. Interestingly, molecules in adjacent layers orient in a criss-cross fashion assisted by H-bonding. Upon polymerization, these criss-cross packed monomers would transform into polymers with a cross-laminated topology (Fig. 14d and e).63 Both the monomers 47 and 48 evolve spontaneously to the respective polymers via TAAC polymerization (Fig. 14f) in a SCSC fashion. As expected, the pre-defined cross-laminated topology of the monomers was retained in the polymer crystals too. The crystals of the polymer displayed enhanced mechanical strength and thermal stability compared to the respective amorphous polymers made by dissolving the crystalline polymers in hexafluoroisopropanol.
Encouraged by the cross-laminated polymers described above, we have designed the topochemical synthesis of a more complex topologically defined polymer.61f We have designed an Ile-Leu-based dipeptide (49, Fig. 14g) that adopts a twist-stacked layered packing by virtue of its six-fold screw axis (P65 space group). The head-to-tail arrangement of molecules form supramolecular 1D-chains, and parallel packing of such chains forms a layer. These layers stack through hydrogen bonding between the molecules of adjacent layers with a twist of 60°. Heating the crystals at 75 °C effected SCSC polymerization to the corresponding polymer in a twist-stacked topology (Fig. 14h). These examples exemplify the importance of TP in the design of polymers with unique topologies by transcribing packing information from the monomer to the polymer.
8. Polymorphism for producing polymers with different structural or supramolecular features
The impact of polymorphism on topochemical reactivity was established way back in the 1960s through the photodimerization of cinnamic acid, wherein different polymorphs dimerize (or react) in different ways.64 Thus, the crystallization of a monomer into different reactive polymorphic forms and exploiting these polymorphs for polymerization could be interesting. Different polymorphs could polymerize to result in polymers with different molecular structures55 or the same polymer with different packing28b,65 (Fig. 15).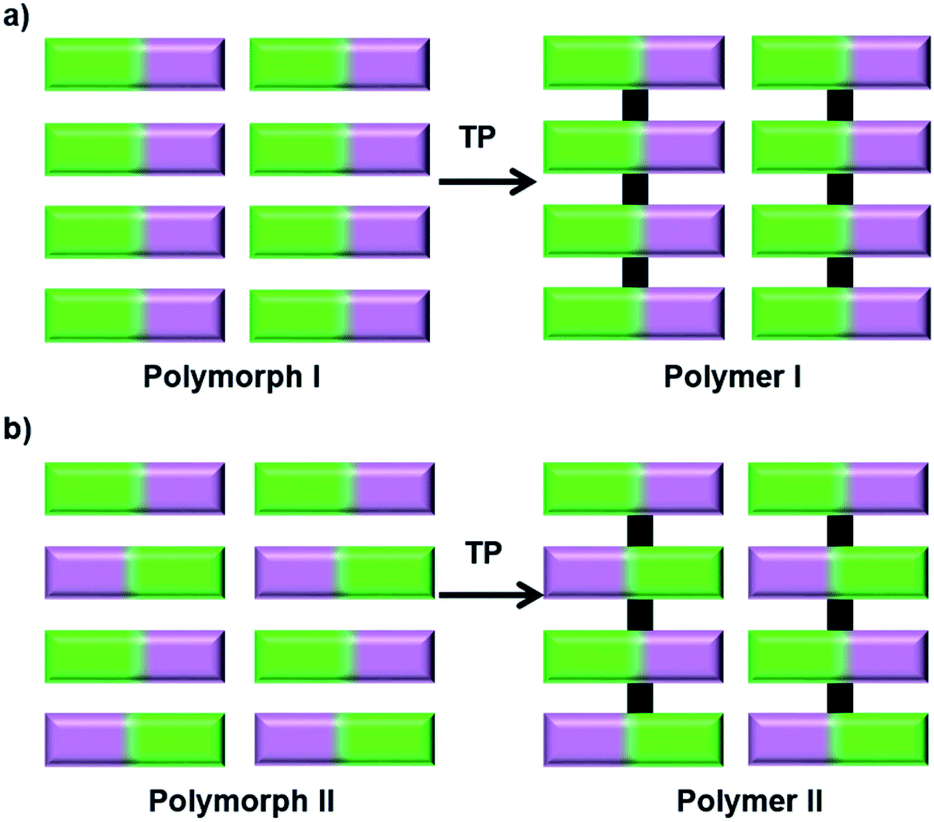 | ||
| Fig. 15 Schematic representation for the synthesis of polymers with different structures or packing via TP of different polymorphs of a monomer. | ||
8.1 Polymorphs of a monomer giving different polymers
The TP of polymorphs of a monomer to produce polymers with different primary structures has been observed by Yokoi and co-workers. The authors reported two distinct kinds of polymerization with two different polymorphs of octadecyl trans,trans-hexadienoate (42) in the crystalline state (Fig. 16a).55 The polymorph obtained from ethanol (form 42-I), upon irradiation, underwent copolymerization with O2 (from air) and formed a peroxy-linked alternating copolymer. On the other hand, polymorphs obtained from hexane or chloroform (form 42-II) when irradiated in the crystalline state undergo addition polymerization, resulting in a mixture comprising a trans-2,5-linked homopolymer as the major product and a peroxy-linked alternating copolymer as a minor product (Fig. 16a). This report showing the formation of structurally different polymers from two different polymorphs of a monomer is very promising.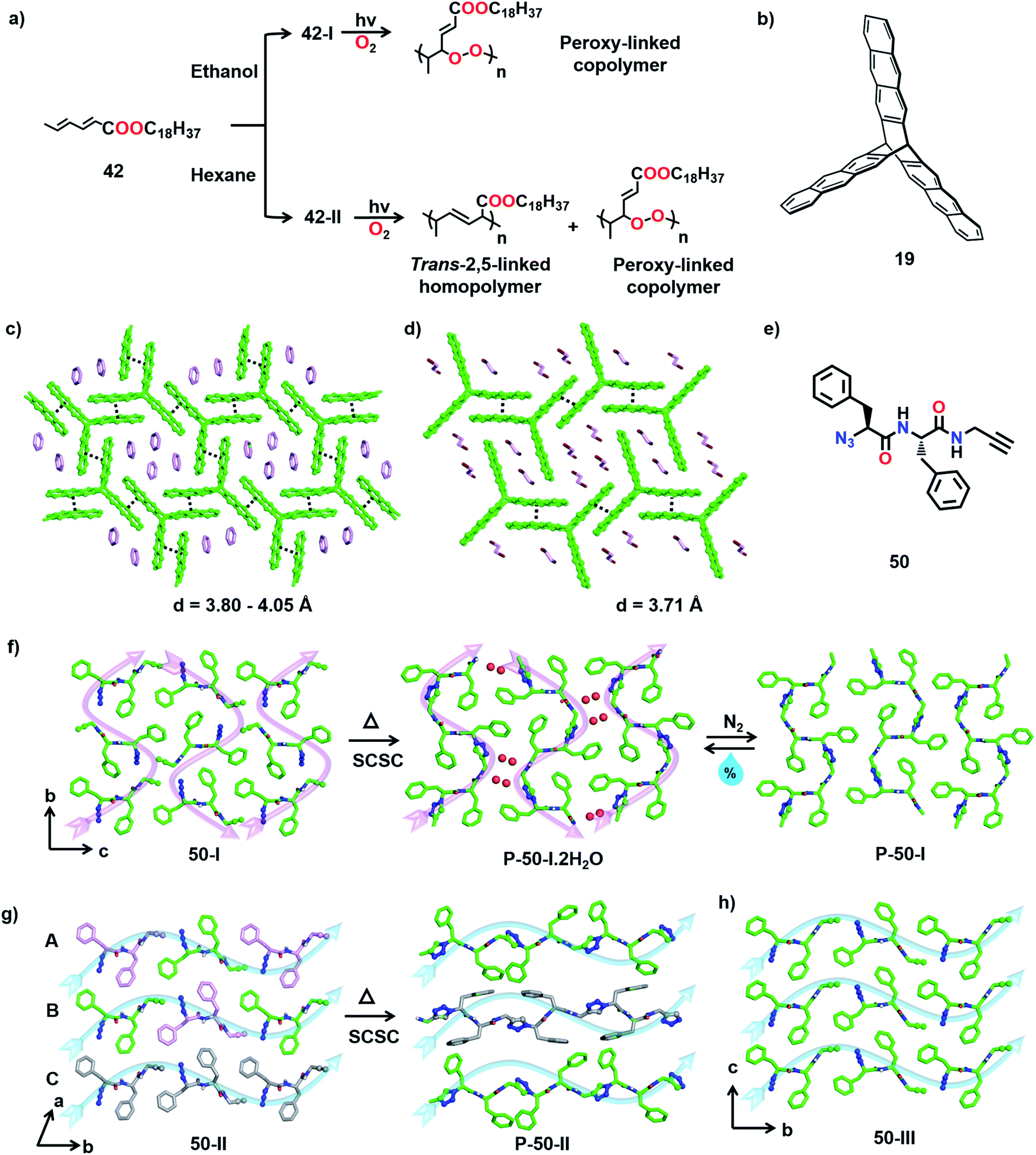 | ||
| Fig. 16 (a) Topochemical synthesis of two different polymers of monomer 42 arising from polymorphs 42-I and 42-II, respectively. (b) Chemical structure of triptycene-based monomer 19. (c) Favourable crystal packing of monomer 19 in benzene solvate for the formation of a 2D polymer. (d) Crystal packing of monomer 19 in 1,2-dibromoethane solvate suitable for 1D polymer formation. (e) Chemical structure of monomer 50. (f) SCSC TAAC polymerization of the 50-I polymorph to a hydrated pseudoprotein and its reversible dehydration. (g) SCSC TAAC polymerization of polymorph 50-II. (h) Crystal packing of monomer 50 in polymorph 50-III. The anthracene, azide, alkyne and triazole units are represented by a ball-and-stick model. H atoms are omitted for clarity except N–H. | ||
Similar to polymorphs, different solvatomorphs of the same monomer can have different molecular arrangements and thus can give different polymers. For example, the trifunctionalized triptycene monomer 19 (Fig. 16b) is known to form different solvates. The molecular arrangement in its benzene solvate is suitable for the formation of a 2D polymer (Fig. 16c), while the packing in dibromoethane solvate is suitable for the formation of a 1D polymer (Fig. 16d).41 While the authors have executed the topochemical 2D polymerization successfully, the polymerization of the dibromoethane solvate was not reported.
8.2 Polymorphs of a monomer giving polymorphs of a polymer
Diamond, graphite, carbon nanotubes, fullerenes, etc., are different forms of carbon with very different properties and fundamentally, this difference originates from their difference in packing. Likewise, one way to tune the properties of a polymer is to pack it in different ways.66 However, post-synthetic crystallization of a polymer is challenging and this poses obstacles in achieving such differently packed polymers. However, TP offers an effective solution to this problem. Thus, the crystallization of a monomer into different reactive polymorphic forms and TP of these different polymorphs represent an emerging method to make different forms of the same polymer. There were consistent efforts to obtain different crystalline forms of a polymer by topochemical reactions. Though there is success in getting two reactive polymorphs, often only one of the forms polymerized to give a crystalline polymer while the other gave an amorphous polymer.28b,65 Recently, we achieved trimorphs of a crystalline polymer using the topochemical approach.67We designed a diphenylalanine-derived monomer (50, Fig. 16e) for TAAC polymerization. We obtained three different crystal forms of the monomer 50 under different conditions. In the polymorph 50-I (orthorhombic, P212121) obtained from a methanol–toluene mixture, the molecules self-assemble into parallel β-sheets through N–H⋯O hydrogen bonding and orthogonal to the β-sheets, the molecules orient in a head-to-tail manner along ‘b’ and ‘c’ directions (Fig. 16f). Two other polymorphs (50-II and 50-III; monoclinic P21) of the dipeptide were obtained via gel-to-crystal transitions from toluene and xylene gels of compound 50, respectively. A similar β-sheet hydrogen bonding prevails in the crystals of the forms 50-II and 50-III, and the molecules align head-to-tail along the ‘b’-direction (Fig. 16g and h). While 50-II contains three molecules in its asymmetric unit, 50-III crystallizes with one molecule in its asymmetric unit. Interestingly, in the crystal structure of 50-II, the three symmetry independent monomer units (A, B, and C) align such that A and B arrange alternately in a head-to-tail fashion forming one type of chain while C forms another self-sorted head-to-tail chain in the crystal (Fig. 16g). In all the three polymorphs, the azide and alkyne units do not initially align proximally for the TAAC reaction. However, thermal annealing at 60 °C causes rotation of azide and alkyne units to a reactive alignment and facilitates TAAC polymerization in all the polymorphs. The polymerization proceeded in a regiospecific fashion resulting in trimorphs of the 1,4-triazolyl-linked polymer of 50.
In the case of polymorph 50-I, the molecules underwent TAAC polymerization in a SCSC manner to yield a 1,4-triazolyl-linked pseudoprotein (P-50-I). The polymer chains pack antiparallelly along the ‘c’ direction (Fig. 16f, indicated by pink arrows). Surprisingly, each asymmetric unit of the polymer (P-50-I) crystal structure contains two water molecules along with the repeating unit of the pseudoprotein. Upon heating the monomer 50-I crystal, a rearrangement of the molecules generates 1D columnar channels in the crystal lattice and the reacting crystal absorbs water molecules into these channels from the surroundings (Fig. 16f). Interestingly, hydrated P-50-I undergoes dehydration by passing an inert gas or by mild heating, in a SCSC fashion suggesting that the water molecules are loosely bound in the channels. The release of water molecules is accompanied by the sliding of polymer chains, thus closing the channels. However, upon exposure to air, the dehydrated polymer crystals re-hydrate in a SCSC fashion and this hydration–dehydration cycle can be repeated multiple times.68 This interesting feature of the pseudoprotein makes it a prospective material for atmospheric water harvesting and as a desiccant.69
Upon heating the crystals of 50-II and 50-III at 60 °C, TAAC polymerization proceeded along the ‘b’ direction (along blue arrows, Fig. 16g and h), resulting in 1,4-triazolyl-linked pseudoproteins P-50-II and P-50-III, respectively. In both P-50-II and P-50-III crystals, the polymer chains are packed parallelly along the ‘c’ direction. P-50-II contains two symmetry independent polymer chains: one resulting from a reaction between A and B (A–B–A–B) and one between the molecules of C along the chain. It is to be noted that though the polymerization was accompanied by a huge reorientation of the phenyl rings, the reaction still proceeded in a SCSC fashion. However, in the case of 50-III, TP resulted in a loss of quality of single crystals. Thus, three different polymorphs of the 1,4-triazolyl-linked diphenylalanine-based pseudoproteins are obtained: (a) P-50-I with an antiparallel arrangement of polymer chains, (b) P-50-II containing a blend of two conformationally different polymer chains arranged parallelly and (c) P-50-III with a parallel arrangement of adjacent polymer chains.
Owing to the differences in the packing of polymer chains, the three polymorphs exhibited distinct properties. While P-50-I can absorb and desorb water reversibly, P-50-II and P-50-III do not absorb water. The polymers also exhibited different thermal stabilities; P-50-III decomposes at 315 °C compared to P-50-I and P-50-II, which decompose at 300 °C. There are also substantial differences in the solubilities of the three polymers in DMSO, with P-50-I being insoluble in DMSO while the other polymorphs are soluble. The mechanical properties are also expected to vary. Thus, by adopting the topochemical approach, we achieved the synthesis of trimorphs of a 1,4-triazolyl-linked polymer having different packing and properties.
9. Different polymers by changing the comonomer guest
In an interesting recent report, Ke and co-workers synthesized copolymers with distinct topologies from the same host-monomer by varying the type of guest comonomer. The authors designed two olefin-appended monomers (51 and 52) containing a tetraphenylethylene core connected to four melamine units (Fig. 17a). In their respective crystals, the molecules form a 3D hydrogen-bonded porous organic framework via 20 N–H⋯N hydrogen bonds between the melamine units of each monomer with six surrounding monomers. When these porous host-crystals were suspended in a solution of the guest, dithiol comonomer, guest encapsulation occurred. The guest-encapsulated crystals upon UV irradiation gave the copolymer via a topochemical thiol–ene reaction.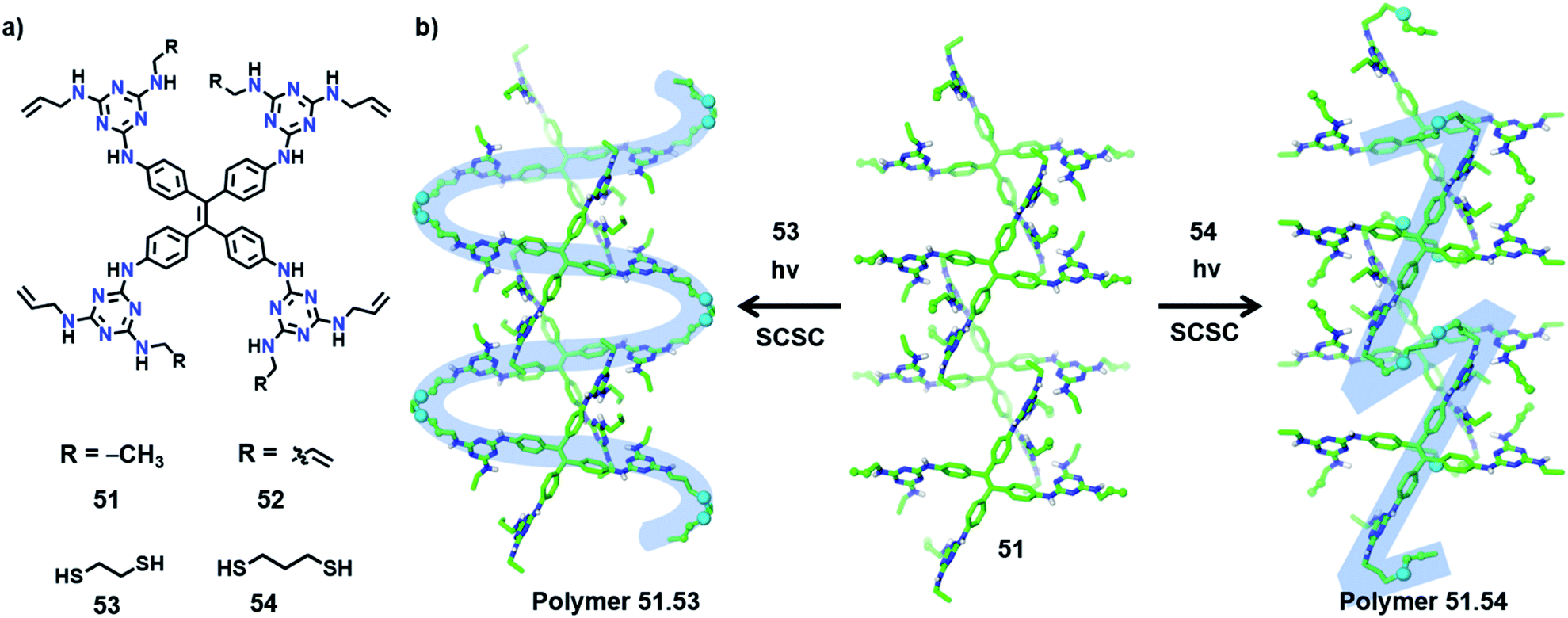 | ||
| Fig. 17 (a) Chemical structures of monomers 51–54. (b) SCSC topochemical synthesis of 51·53 (S-shaped polymer) and 51·54 (Z-shaped polymer) from monomer 51via a thiol–ene reaction with comonomers 53 and 54, respectively. | ||
By introducing alkyl dithiol guests of varying alkyl lengths, and subsequent thiol–ene photopolymerization, the authors could obtain polymers with distinct topologies from the same monomer (51 or 52).70 In some cases, SCSC polymerization was realized, which provided unequivocal proof for the divergent topologies dictated by the type of guest molecule. In the case of monomer 51, the molecules in two adjacent layers are positioned relative to each other at an angle of 120° (Fig. 17b). The incorporation of ethanedithiol (53) as a comonomer and subsequent irradiation result in an S-shaped linear polymer and two neighbouring S-shaped chains inter-weave. Here, the thiol–ene reaction proceeds between molecules on the adjacent layers (angled at 120°) in a SCSC fashion, with the molecules of 53 as a bridging linker.
On the other hand, when a longer linker (propanedithiol, 54) was used, SCSC thiol–ene polymerization occurred between molecules on the same layer yielding Z-shaped linear chains (Fig. 17b). Such linear chains are aligned parallelly in the polymer crystal. In either case, only 50% of the olefin units participated in the topochemical thiol–ene reaction. Similarly, in the case of monomer 52, a 3D interconnected polymer network or cross-linked 2D polymer network can be procured by applying comonomers 53 and 54, respectively. The authors further observed the elastic expansion of the polymer crystals via guest (iodine) inclusion, by a visible change in the colour and dimensions of the crystals (>200%), which revealed the flexibility of the framework. The polymers obtained from monomer 52 display a record iodine uptake, ∼3 g of iodine per gram of polymer. Also, different topologies of the polymers showed distinct iodine uptake capacities as well as morphology changes. This example demonstrating the structural and topological tuning of polymers by varying the guest monomers shows a new and exciting dimension of TP.
10. Conclusions and outlook
Ever since the development of crystal engineering as a tool for the rational design of crystals with a desired molecular orientation, the topochemical method has become a powerful strategy for polymer synthesis. In this perspective, we summarized the selected reports on the implementation of the topochemical approach for the otherwise challenging synthesis of polymers with advanced molecular and supramolecular architectures. These advanced polymers are attractive in terms of their unique properties by virtue of their molecular structure or packing.Unarguably, TP demonstrated its power in synthesizing structurally interesting ladder polymers, tubular polymers, polymer blends, copolymers, 2D polymers and other ordered polymers. While these demonstrations were made by exploiting only limited types of topochemical reactions, there is much potential to extend these strategies for other types of reactions. While DA-based monomers have been used extensively for the topochemical synthesis of tubular polymers, it would be interesting to synthesize organic nanotubes with different backbones via other categories of TP. While TP has proven to be successful in providing 2D polymers with an attractive structure and properties, mainly, monomers having only one type of reactive group have been explored for their synthesis. An advancement in this area would be to integrate different types of reactive units in the same molecule so as to achieve novel polymers with distinct polymer backbones and even multiple functions. Though a preliminary report of polymerization of 2D polymers to 3D is encouraging, by adopting a prudent monomer design, it would be exciting to explore the direct synthesis of a 3D polymer through TP. The diffusion of comonomers such as gaseous oxygen and dithiols into the crystals of other monomers leading to their topochemical copolymerization is intriguing. By judicious crystal engineering approaches, the design of monomers that allow the diffusion of other gaseous or liquid monomers into their crystals followed by their topochemical co-polymerization may be a possible reality in the future. One could also think of achieving challenging block copolymers or hyperbranched polymer architectures. The limited examples of exploiting polymorphism for accessing polymers of different structures and packing show the impact of this strategy and demand further research in these areas.
The topochemical synthesis of polymers with sophisticated molecular structures necessitates multiple reactive moieties in a monomer or co-existence of multiple monomers, in reactive orientation in the crystal. While the primary challenge is to design monomers in such a way that all the reactive units present in the monomer could attain favourable orientation, it is also important to have structural and packing features that permit the progress of the reaction without drastic changes in packing. The molecular motion/packing change accompanying the reaction could cause unfavourable packing of part of the reactive motifs making them unreactive. Nevertheless, such complications have been successfully overcome by persistent efforts and prudent design of the monomers. A suitable functionalization of the monomer or usage of templates to direct the self-assembly of monomers in favourable orientation and incorporating the linkers that provide suitable molecular flexibility during the reaction have substantial roles in successful TP. While significant developments have been made regarding the synthesis of polymers with sophisticated molecular features, tuning the supramolecular architecture of polymers is still in the initial stage. The challenge is to design a monomer that could efficiently orient and polymerize uniformly to form such complex architectures. One of the plausible ways to access such advanced polymer architectures could be by integrating TP with other supramolecular systems. A recent report on engineering rotaxane-based architectures through topochemical cross-linking of monomers present in inclusion complexes inspires probable research in this direction.71
In our lab, we are actively pursuing the implementation of TAAC for achieving polymers with sophisticated structural features. We have demonstrated the efficient synthesis of polymer blends, polymers with novel supramolecular architectures, interesting topologies, and polymorphic forms of a polymer. Still, there are a lot of exciting polymer architectures to be explored. Living in an era where polymers are an integral part of everyday life, tailoring the properties of polymers is an important aspect. We believe that this perspective would inspire researchers to perceive the topochemical approach as a versatile strategy for the synthesis of polymers with advanced structures and attractive properties.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
KMS thanks the Science and Engineering Research Board, Department of Science and Technology, Govt. of India for the financial support.References
-
(a) G. Pasparakis, N. Krasnogor, L. Cronin, B. G. Davis and C. Alexander, Chem. Soc. Rev., 2010, 39, 286–300 RSC
; (b) K. L. Kaligian and M. M. Sprachman, Mol. Syst. Des. Eng., 2019, 4, 144–161 RSC
-
(a) A. Matsumoto, S. Nagahama and T. Odani, J. Am. Chem. Soc., 2000, 122, 9109–9119 CrossRef CAS
-
(a) M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC
- Q.-H. Guo, M. Jia, Z. Liu, Y. Qiu, H. Chen, D. Shen, X. Zhang, Q. Tu, M. R. Ryder, H. Chen, P. Li, Y. Xu, P. Li, Z. Chen, G. S. Shekhawat, V. P. Dravid, R. Q. Snurr, D. Philp, A. C. H. Sue, O. K. Farha, M. Rolandi and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 6180–6187 CrossRef PubMed
-
(a) R. O. Al-Kaysi, R. J. Dillon, J. M. Kaiser, L. J. Mueller, G. Guirado and C. J. Bardeen, Macromolecules, 2007, 40, 9040–9044 CrossRef CAS
-
(a) A. Pathigoolla and K. M. Sureshan, Angew. Chem., Int. Ed., 2013, 52, 8671–8675 CrossRef CAS PubMed
-
(a) R. H. Baughman, J. Polym. Sci., Polym. Phys. Ed., 1974, 12, 1511–1535 CrossRef CAS
- A. Matsumoto, Prog. React. Kinet. Mech., 2001, 26, 59–109 CrossRef CAS
- T. Hoang, J. W. Lauher and F. W. Fowler, J. Am. Chem. Soc., 2002, 124, 10656–10657 CrossRef CAS PubMed
- J. Xiao, M. Yang, J. W. Lauher and F. W. Fowler, Angew. Chem., Int. Ed., 2000, 39, 2132–2135 CrossRef CAS PubMed
-
(a) T. Itoh, S. Nomura, T. Uno, M. Kubo, K. Sada and M. Miyata, Angew. Chem., Int. Ed., 2002, 41, 4306–4309 CrossRef CAS
-
(a) J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC
- G. Wegner, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1969, 24, 824–832 CrossRef CAS
-
(a) S. Okada, K. Hayamizu, H. Matsuda, A. Masaki and H. Nakanishi, Bull. Chem. Soc. Jpn., 1991, 64, 857–863 CrossRef CAS
- M. Steinbach and G. Wegner, Makromol. Chem., 1977, 178, 1671–1677 CrossRef CAS
- S. Okada, H. Matsuda, A. Masaki, H. Nakanishi and K. Hayamizu, Chem. Lett., 1990, 2213–2216 CrossRef CAS
- S. Okada, K. Hayamizu, H. Matsuda, A. Masaki, N. Minami and H. Nakanishi, Chem. Lett., 1992, 301–304 CrossRef CAS
- H. Matsuzawa, S. Okada, H. Matsuda and H. Nakanishi, Proc. SPIE, 1996, 2851, 14–25 CrossRef CAS
-
(a) H. Matsuzawa, S. Okada, H. Matsuda and H. Nakanishi, Chem. Lett., 1997, 26, 1105–1106 CrossRef
- H. Matsuo, S. Okada, H. Nakanishi, H. Matsuda and S. Takaragi, Polym. J., 2002, 34, 825–834 CrossRef CAS
-
(a) S. Inayama, Y. Tatewaki and S. Okada, Polym. J., 2010, 42, 201–207 CrossRef CAS
- S. Nagahama and A. Matsumoto, J. Am. Chem. Soc., 2001, 123, 12176–12181 CrossRef CAS
- X. Hou, Z. Wang, J. Lee, E. Wysocki, C. Oian, J. Schlak and Q. R. Chu, Chem. Commun., 2014, 50, 1218–1220 RSC
-
(a) J. R. Néabo, S. Rondeau-Gagné, C. Vigier-Carrière and J.-F. Morin, Langmuir, 2013, 29, 3446–3452 CrossRef PubMed
-
(a)
M. A. Balbo Block, C. Kaiser, A. Khan and S. Hecht, in Functional Molecular Nanostructures, ed. A. D. Schlüter, Springer, Berlin, Heidelberg, 2005, vol. 201, pp. 89–150 Search PubMed
-
(a) D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS
-
(a) A. Banerjie, J. B. Lando, K. C. Yee and R. H. Baughman, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 655–662 CrossRef CAS
-
(a) Y. Xu, M. D. Smith, M. F. Geer, P. J. Pellechia, J. C. Brown, A. C. Wibowo and L. S. Shimizu, J. Am. Chem. Soc., 2010, 132, 5334–5335 CrossRef CAS
-
(a) G. Shin, M. I. Khazi and J.-M. Kim, Macromolecules, 2019, 53, 149–157 CrossRef
-
(a) J.-M. Heo, Y. Kim, S. Han, J. F. Joung, S.-h. Lee, S. Han, J. Noh, J. Kim, S. Park, H. Lee, Y. M. Choi, Y.-S. Jung and J.-M. Kim, Macromolecules, 2017, 50, 900–913 CrossRef CAS
-
(a) M. Suzuki, A. Comito, S. I. Khan and Y. Rubin, Org. Lett., 2010, 12, 2346–2349 CrossRef CAS PubMed
- W. L. Xu, M. D. Smith, J. A. Krause, A. B. Greytak, S. Ma, C. M. Read and L. S. Shimizu, Cryst. Growth Des., 2014, 14, 993–1002 CrossRef CAS
- K. Bae, J.-M. Heo, M. I. Khazi, J. F. Joung, S. Park, Y. Kim and J.-M. Kim, Cryst. Growth Des., 2020, 20, 434–441 CrossRef CAS
-
(a) S. Rondeau-Gagné, J. R. Néabo, M. Desroches, I. Levesque, M. Daigle, K. Cantin and J.-F. Morin, Chem. Commun., 2013, 49, 9546–9548 RSC
-
(a) Z. Xiang, D. Cao and L. Dai, Polym. Chem., 2015, 6, 1896–1911 RSC
-
(a) A. D. Schlüter, T. Weber and G. Hofer, Chem. Soc. Rev., 2020, 49, 5140–5158 RSC
-
(a) M. Servalli, M. Solar, N. Trapp, M. Wörle and A. D. Schlüter, Cryst. Growth Des., 2017, 17, 6510–6522 CrossRef CAS
-
(a) J. Sakamoto, J. van Heijst, O. Lukin and A. D. Schlüter, Angew. Chem., Int. Ed., 2009, 48, 1030–1069 CrossRef CAS PubMed
-
(a) H. Ozaki, M. Kasuga, T. Tsuchiya, T. Funaki, Y. Mazaki, M. Aoki, S. Masuda and Y. Harada, J. Chem. Phys., 1995, 103, 1226–1228 CrossRef CAS
- P. Kissel, R. Erni, W. V. Schweizer, M. D. Rossell, B. T. King, T. Bauer, S. Götzinger, A. D. Schlüter and J. Sakamoto, Nat. Chem., 2012, 4, 287–291 CrossRef CAS PubMed
- R. Bhola, P. Payamyar, D. J. Murray, B. Kumar, A. J. Teator, M. U. Schmidt, S. M. Hammer, A. Saha, J. Sakamoto, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2013, 135, 14134–14141 CrossRef CAS PubMed
- G. Hofer, F. Grieder, M. Kröger, A. D. Schlüter and T. Weber, J. Appl. Crystallogr., 2018, 51, 481–497 CrossRef CAS
- Z. Wang, K. Randazzo, X. Hou, J. Simpson, J. Struppe, A. Ugrinov, B. Kastern, E. Wysocki and Q. R. Chu, Macromolecules, 2015, 48, 2894–2900 CrossRef CAS
- R. Z. Lange, G. Hofer, T. Weber and A. D. Schlüter, J. Am. Chem. Soc., 2017, 139, 2053–2059 CrossRef CAS
- A. Acharjya, P. Pachfule, J. Roeser, F.-J. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS
- T. Jadhav, Y. Fang, W. Patterson, C.-H. Liu, E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2019, 58, 13753–13757 CrossRef CAS PubMed
- Y. Maekawa, P.-J. Lim, K. Saigo and M. Hasegawa, Macromolecules, 1991, 24, 5722–5755 Search PubMed
- K. Sasamura and S. Okada, Polym. Bull., 2019, 76, 1675–1683 CrossRef CAS
- C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CrossRef CAS
- G. W. Coates, A. R. Dunn, L. M. Henling, D. A. Dougherty and R. H. Grubbs, Angew. Chem., Int. Ed., 1997, 36, 248–251 CrossRef CAS
- R. Xu, V. Gramlich and H. Frauenrath, J. Am. Chem. Soc., 2006, 128, 5541–5547 CrossRef CAS PubMed
- R. Xu, W. B. Schweizer and H. Frauenrath, Chem.–Eur. J., 2009, 15, 9105–9116 CrossRef CAS PubMed
-
(a) G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef CAS
- T. Fukushima, T. Uno, M. Kubo, T. Itoh, N. Tohnai and M. Miyata, Aust. J. Basic Appl. Sci., 2014, 8, 516–520 CAS
- A. Matsumoto, Y. Ishizu and K. Yokoi, Macromol. Chem. Phys., 1998, 199, 2511–2516 CrossRef CAS
-
(a) A. Matsumoto and A. Higashi, Macromolecules, 2000, 33, 1651–1655 CrossRef CAS
- S. Nomura, T. Itoh, M. Ohtake, T. Uno, M. Kubo, A. Kajiwara, K. Sada and M. Miyata, Angew. Chem., Int. Ed., 2003, 42, 5468–5472 CrossRef CAS PubMed
- T. Itoh, S. Nomura, M. Ohtake, T. Yoshida, T. Uno, M. Kubo, A. Kajiwara, K. Sada and M. Miyata, Macromolecules, 2004, 37, 8230–8238 CrossRef CAS
-
(a) T. Odani, S. Okada, C. Kabuto, T. Kimura, H. Matsuda, A. Matsumoto and H. Nakanishi, Chem. Lett., 2004, 33, 1312–1313 CrossRef CAS
- K. Hema and K. M. Sureshan, Angew. Chem., Int. Ed., 2019, 58, 2754–2759 CrossRef CAS PubMed
-
(a) X. Zou, H. Ren and G. Zhu, Chem. Commun., 2013, 49, 3925–3936 RSC
- K. Hema and K. M. Sureshan, Angew. Chem., Int. Ed., 2020, 59, 8854–8859 CrossRef CAS
- V. Athiyarath and K. M. Sureshan, Angew. Chem., Int. Ed., 2018, 58, 612–617 CrossRef
-
(a) M. D. Cohen and G. M. J. Schmidt, J. Chem. Soc., 1964, 1996–2000 RSC
- S. Nomura, T. Itoh, H. Nakasho, T. Uno, M. Kubo, K. Sada, K. Inoue and M. Miyata, J. Am. Chem. Soc., 2004, 126, 2035–2041 CrossRef CAS PubMed
-
(a) M. Yoshio, T. Kagata, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2006, 128, 5570–5577 CrossRef CAS PubMed
- R. Mohanrao, K. Hema and K. M. Sureshan, Nat. Commun., 2020, 11, 865 CrossRef CAS PubMed
- R. Mohanrao and K. M. Sureshan, Angew. Chem., Int. Ed., 2018, 57, 12435–12439 CrossRef CAS PubMed
- R. Mohanrao, K. Hema and K. M. Sureshan, ACS Appl. Polym. Mater., 2020, 2(11), 4985–4992 CrossRef CAS
- X. Jiang, X. Cui, A. J. E. Duncan, L. Li, R. P. Hughes, R. J. Staples, E. V. Alexandrov, D. M. Proserpio, Y. Wu and C. Ke, J. Am. Chem. Soc., 2019, 141, 10915–10923 CrossRef CAS PubMed
- M. Zhu, L. Yin, Y. Zhou, H. Wu and L. Zhu, Macromolecules, 2018, 51, 746–754 CrossRef CAS
Footnotes |
| † Current address: Department of Organic Chemistry, Weizmann Institute of Science, Rehovot-7610001, Israel. |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |