
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to P-stereogenic compounds via desymmetrizing enantioselective bromination†‡
Qiu-Hong
Huang
a,
Qian-Yi
Zhou
a,
Chen
Yang
 a,
Li
Chen
a,
Jin-Pei
Cheng
ab and
Xin
Li
*a
a,
Li
Chen
a,
Jin-Pei
Cheng
ab and
Xin
Li
*a
aState Key Laboratory of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: xin_li@nankai.edu.cn
bCenter of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing 100084, China
First published on 12th February 2021
Abstract
A novel and efficient desymmetrizing asymmetric ortho-selective mono-bromination of bisphenol phosphine oxides under chiral squaramide catalysis was reported. Using this asymmetric ortho-bromination strategy, a wide range of chiral bisphenol phosphine oxides and bisphenol phosphinates were obtained with good to excellent yields (up to 92%) and enantioselectivities (up to 98.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 e.r.). The reaction could be scaled up, and the synthetic utility of the desired P-stereogenic compounds was proved by transformations and application in an asymmetric reaction.
:1.5 e.r.). The reaction could be scaled up, and the synthetic utility of the desired P-stereogenic compounds was proved by transformations and application in an asymmetric reaction.
P-Stereogenic compounds are a class of privileged structures, which have been widely present in natural products, drugs and biologically active molecules (Fig. 1a).1–4 In addition, they are also important chiral materials for the development of chiral catalysts and ligands (Fig. 1b), because the chirality of the phosphorus atom is closer to the catalytic center which can cause remarkable stereo-induction.5,6 Thus, the development of efficient methods for the synthesis of P-stereogenic compounds with novel structures and functional groups is very meaningful.5a Conventional syntheses of P-stereogenic compounds mainly depended on the resolution of diastereomeric mixtures and chiral-auxiliary-based approaches, in which stoichiometric amounts of chiral reagents are usually needed.7 By comparison, asymmetric catalytic strategies, including asymmetric desymmetric reactions of dialkynyl, dialkenyl, diaryl and bisphenol phosphine oxides,8–14 (dynamic) kinetic resolution of tertiary phosphine oxides,15 and asymmetric reactions of secondary phosphine oxides,16 can effectively solve the above-mentioned problems and have been considered as the most direct and efficient synthesis methods for constructing P-chiral phosphine oxides (Fig. 1c). Among them, organocatalytic asymmetric desymmetrization methods have been sporadic, in which the reaction sites were mainly limited to the hydroxyl group of bisphenol phosphine oxides that hindered their further transformation.8–11 It is worth mentioning that asymmetric desymmetrization methods, especially organocatalytic desymmetrization reactions, due to their unique advantages of mild reaction conditions and wide substrate scope, have become an important strategy for asymmetric synthesis. Accordingly, the development of efficient organocatalytic desymmetrization strategy for the synthesis of important functionalized P-stereogenic compounds which contain multiple conversion groups is very meaningful and highly desirable.
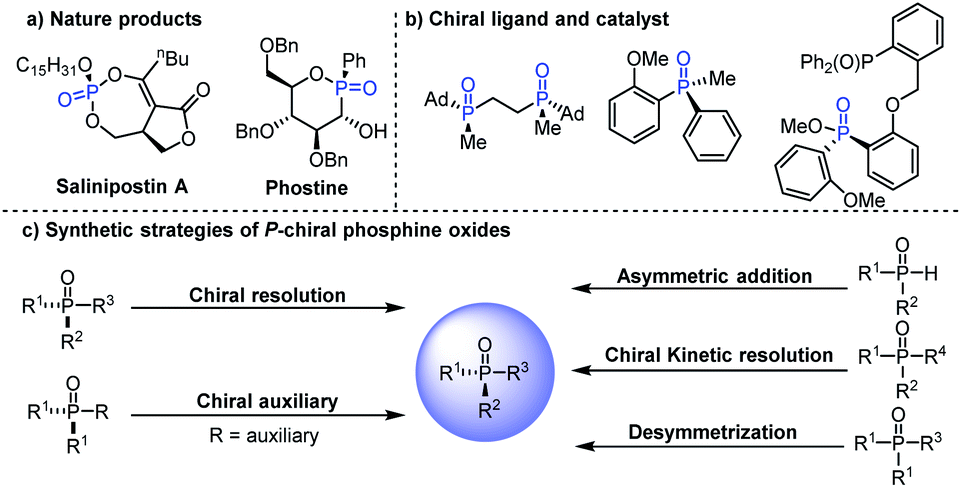 | ||
| Fig. 1 (a) Examples of natural products containing P-stereogenic centers. (b) P-Stereogenic compound type ligand and catalyst. (c) Typical P-stereogenic compounds' synthetic strategies. | ||
On the other hand, asymmetric bromination has been demonstrated to be one of the most attractive approaches for chiral compound syntheses.17 Since the pioneering work on peptide catalyzed asymmetric bromination for the construction of biaryl atropisomers,18a the reports on constructing axially biaryl atropisomers,18 C–N axially chiral compounds,19 atropisomeric benzamides,20 axially chiral isoquinoline N-oxides,21 and axially chiral N-aryl quinoids22 by electrophilic aromatic bromination have been well developed (Scheme 1a). In comparison, the desymmetrization of phenol through asymmetric bromination to construct central chirality remains a daunting task. Miller discovered a series of tailor made peptide catalyzed enantioselective desymmetrizations of diarylmethylamide through ortho-bromination (Scheme 1b).23 Recently, Yeung realized amino-urea catalyzed desymmetrizing asymmetric ortho-selective mono-bromination of phenol derivatives to fix a new class of potent privileged bisphenol catalyst cores with excellent yields and enantioselectivities (Scheme 1b).24 Despite this elegant work, there is no report on the synthesis of P-centered chiral compounds using the desymmetrizing asymmetric bromination strategy.
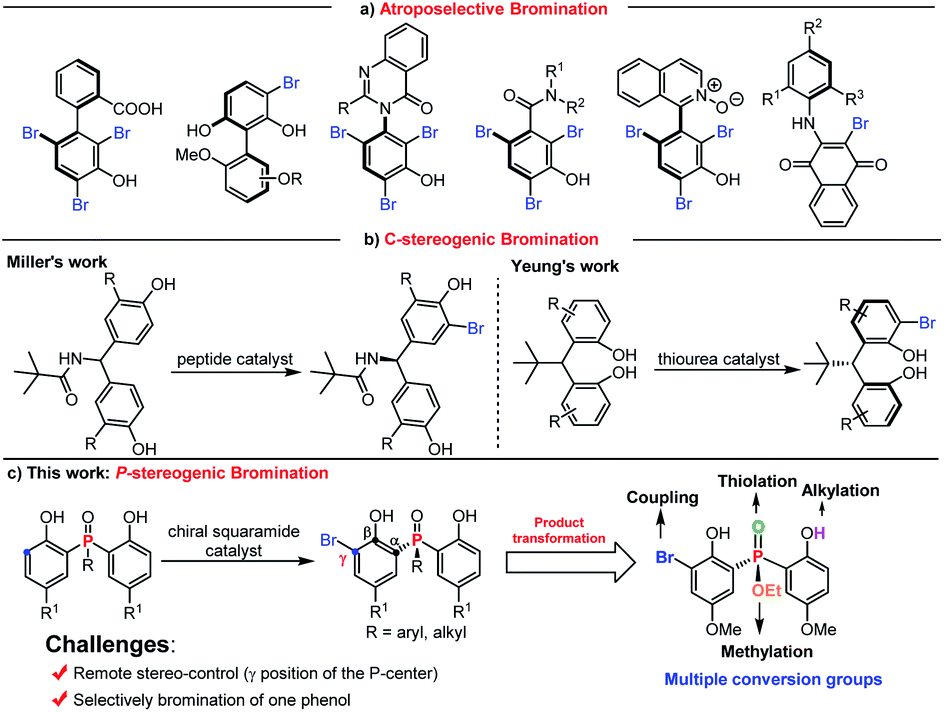 | ||
| Scheme 1 (a) Constructing axially chiral compounds by asymmetric bromination. (b) Known synthesis of central chiral compounds via asymmetric bromination. (c) This work: access to P-stereogenic compounds via desymmetrizing enantioselective bromination. | ||
Taking into account the above-mentioned consideration, we speculated that bisphenol phosphine oxides and bisphenol phosphinates are potential substrate candidates for desymmetrizing asymmetric bromination to construct P-stereogenic centers. The advantages of using bisphenol phosphine oxides and bisphenol phosphinates as substrates are shown in two aspects. First, the ortho-position of electron rich phenol is easy to take place electrophilic bromination reaction. Second, the corresponding bromination product structure contains abundant synthetic conversion groups, including bromine, hydroxyl group, alkoxy group and phosphoryl group. To achieve this goal, two challenges need to be overcome: (i) finding a suitable chiral catalyst for the desymmetrization process to induce enantiomeric control is troublesome, due to the remote distance between the prochiral phosphorus center and the enantiotopic site; (ii) selectively brominating one phenol to inhibit the formation of an achiral by-product is difficult. Herein, we report a chiral squaramide catalyzed asymmetric ortho-bromination strategy to construct a wide range of chiral bisphenol phosphine oxides and bisphenol phosphinates with good to excellent yields and enantioselectivities (Scheme 1c). It is worth mentioning that the obtained P-stereogenic compounds can be further transformed at multiple sites.
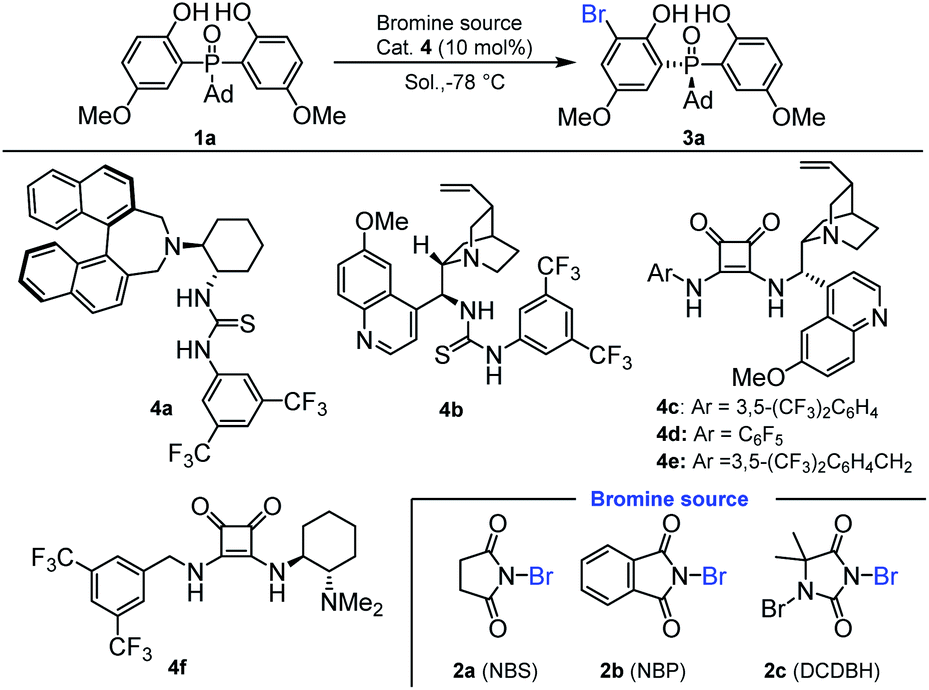
Our initial investigation was carried out with bis(2-hydroxyphenyl)phosphine oxide 1a and N-bromosuccinimide (NBS) 2a as the model substrates, 10 mol% chiral amino-thiourea 4a as the catalyst, and toluene as the solvent, which were stirred at −78 °C for 12 h. As a result, the reaction gave the desired desymmetrization product 3a in 65% yield with 56:44 e.r. (Table 1, entry 1). Then, thiourea 4b was tested, in which a little better result was obtained (Table 1, entry 2). To our delight, using the chiral squaramides 4c–4f as the catalysts, the enantiomeric ratios of the desymmetrization products had been significantly improved (Table 1, entries 3–6). Especially, when chiral squaramide catalyst 4c was applied to this reaction, the enantiomeric ratio of 3a was increased to 95:5 (Table 1, entry 3). To further improve the yield and enantioselectivity, we next optimized the reaction conditions by varying reaction media and additives. As shown in Table 1, the reaction was affected by the solvent dramatically. Product 3a was obtained with low yield and enantioselectivity in DCM (Table 1, entry 7). Also, when Et2O was used as the solvent, the yield and e.r. value of product 3a were all decreased (Table 1, entry 8). As a result, the initial used toluene was the optimal solvent. We also inspected the effect of different bromine sources, and found that the initially used NBS was the optimal one (Table 1, entries 3, 11 and 12). Fortunately, by adjusting the amount of bisphenol phosphine oxides to 1.5 equiv., the yield and the enantiomeric ratio of 3a were increased to 80% and 96.5:3.5, respectively (Table 1, entries 3, 13 and 14). Further increasing the amount of bisphenol phosphine oxides to 2.0 equiv. resulted in a reduced enantioselectivity (Table 1, entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Bromine source | Solvent | Yieldb (%) | e.r.c |
|
a Reaction conditions: a mixture of 1a (0.05 mmol), 2a (0.05 mmol) and cat. 4 (10 mol%) in the solvent (0.5 mL) was stirred at −78 °C for 12 h.
b Isolated yield.
c Determined by HPLC analysis.
d 3 Å MS (10.0 mg) was used as the additive.
e 4 Å MS (10.0 mg) was used as the additive.
f
1a:2a = 1.2:1.
g
1a:2a = 1.5:1.
h
1a:2a = 2.0:1.
|
|||||
| 1 | 4a | 2a | Toluene | 65 | 56:44 |
| 2 | 4b | 2a | Toluene | 49 | 68:32 |
| 3 | 4c | 2a | Toluene | 61 | 95:5 |
| 4 | 4d | 2a | Toluene | 41 | 75:25 |
| 5 | 4e | 2a | Toluene | 53 | 93:6 |
| 6 | 4f | 2a | Toluene | 39 | 61:39 |
| 7 | 4c | 2a | DCM | 47 | 89:11 |
| 8 | 4c | 2a | Et2O | 39 | 67:33 |
| 9d | 4c | 2a | Toluene | 69 | 94:6 |
| 10e | 4c | 2a | Toluene | 61 | 93:7 |
| 11 | 4c | 2b | Toluene | 63 | 94:6 |
| 12 | 4c | 2c | Toluene | 65 | 87:13 |
| 13f | 4c | 2a | Toluene | 75 | 95:5 |
| 14g | 4c | 2a | Toluene | 80 | 96.5:3.5 |
| 15h | 4c | 2a | Toluene | 79 | 95:5 |
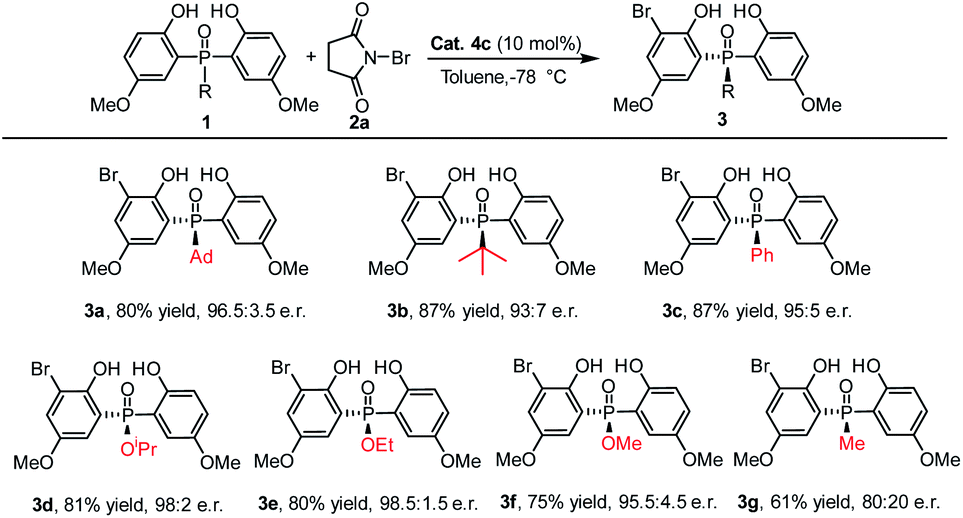
Under the optimized reaction conditions, the scope of the desymmetrizing asymmetric ortho-selective mono-bromination of phosphine oxides was examined. Firstly, the variation of the P-center substituted group was investigated. As shown in Table 2, a variety of P-aryl, P-alkyl substituted phosphine oxides and phosphinates (3a–3f) were well amenable to this reaction and the corresponding ortho-brominated products were obtained in good yield (up to 87%) with high enantiomeric ratios (up to 98.5:1.5 e.r.). Moreover, regardless of whether the R was a bulky group or a smaller one, the enantiomeric ratios of the products were maintained at excellent levels. Especially, when the P-center substituted group was ethoxyl (1e), the corresponding bromination product 3e was obtained in 80% yield with 98.5:1.5 e.r. When a P-methyl substituted phosphine oxide was used as the substrate, a moderate yield and enantiomeric ratio were obtained for 3g.
Next, using the ethoxyl substituted phosphinate as the template, a diversity of phosphinates with a 5-position substituent on the phenyl ring were examined (Table 3). To our delight, a range of phosphinates with different alkyl substituent on the phenyl ring was suitable for the currently studied reaction and the desired products 3h–3l were obtained with very good enantioselectivities (90.5:9.5–97.5:2.5 e.r.). Furthermore, substrates with aryl and alkoxy groups at the 5-position of the phenol moiety were also tolerated well under the reaction conditions, and gave the products 3m–3q with good to excellent yields (81–92%) and enantioselectivities (95:5–98.5:1.5 e.r.). Moreover, when a disubstituted phenol phosphinate substrate was used, the desired bromination product 3r was also delivered with a good yield and e.r. value.
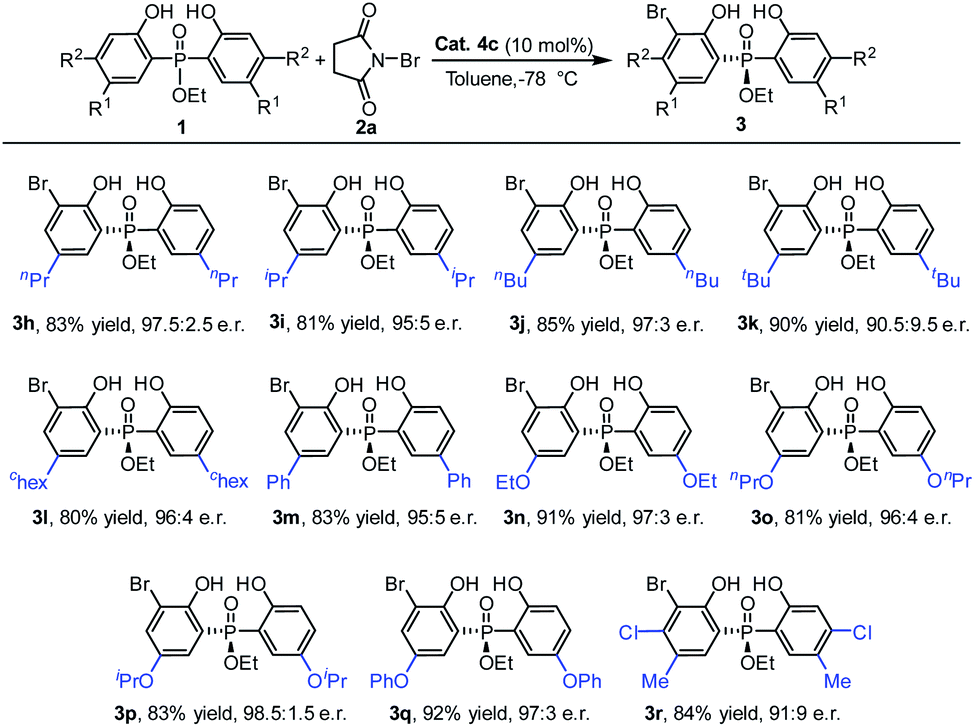
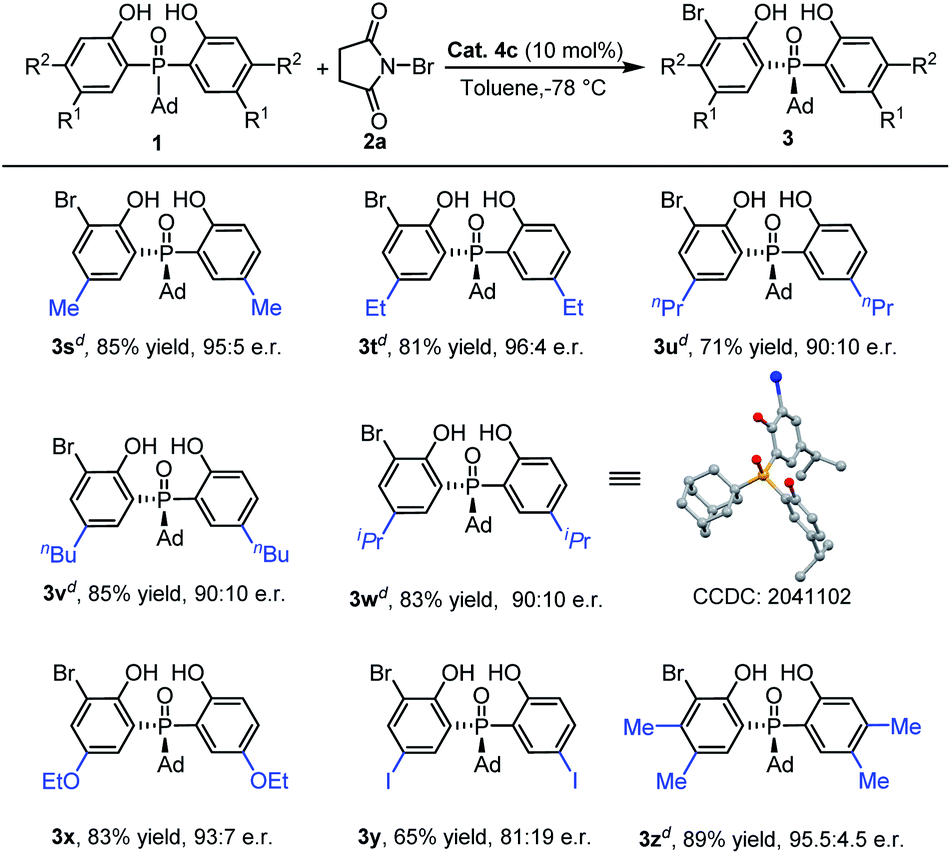
Then, we turned our attention to inspect the scope of ortho-bromination of P-adamantyl substituted phosphine oxides. As exhibited in Table 4, 5-methyl, 5-ethyl and 4,5-dimethyl aryl substituted phosphine oxides could be transformed into the corresponding products (3s, 3t and 3u) with excellent yields (81–89%) and enantioselectivities (95:5–96:4 e.r.). Upon increasing the size of the 5-position substituent on the phenyl ring of phosphine oxides, the enantioselectivities of the products 3v–3y had a little decreasing tendency (81:19–93:7 e.r.). The absolute configuration of 3v was determined by X-ray diffraction analysis and those of other products were assigned by analogy.25
To demonstrate the utility of this desymmetrizing asymmetric ortho-selective mono-bromination, the reaction was scaled up to 1.0 mmol, and the corresponding product 3a was obtained in 80% yield with 96.5:3.5 e.r. (98.5:1.5 e.r. after single recrystallization) (Scheme 2a). The encouraging results implied that this strategy had the potential for large-scale production. Additionally, the transformations of products 3a and 3e were also investigated (Scheme 2b). In the presence of Pd(OAc)2 and bulky electron-rich ligand S-Phos, 3a could react with phenylboronic acid effectively, in which the desired cross-coupling product 5 was generated in high yield with maintained enantioselectivity. In the presence of Lawesson's reagent, 3a could be transformed into thiophosphine oxide 6 with a high yield and e.r. value. Furthermore, 3e could react with methyl lithium to afford the DiPAMP analogue 3g in 85% yield with 98.5:1.5 e.r. And 3e could also be converted to chiral bidentate Lewis base 7 by a straightforward alkylation reaction. It was encouraging to find that 7 could be used as a catalyst for the asymmetric reaction between trans-chalcone and furfural, in which the desired product 8 was furnished with moderate stereoselectivity (Scheme 2c).26
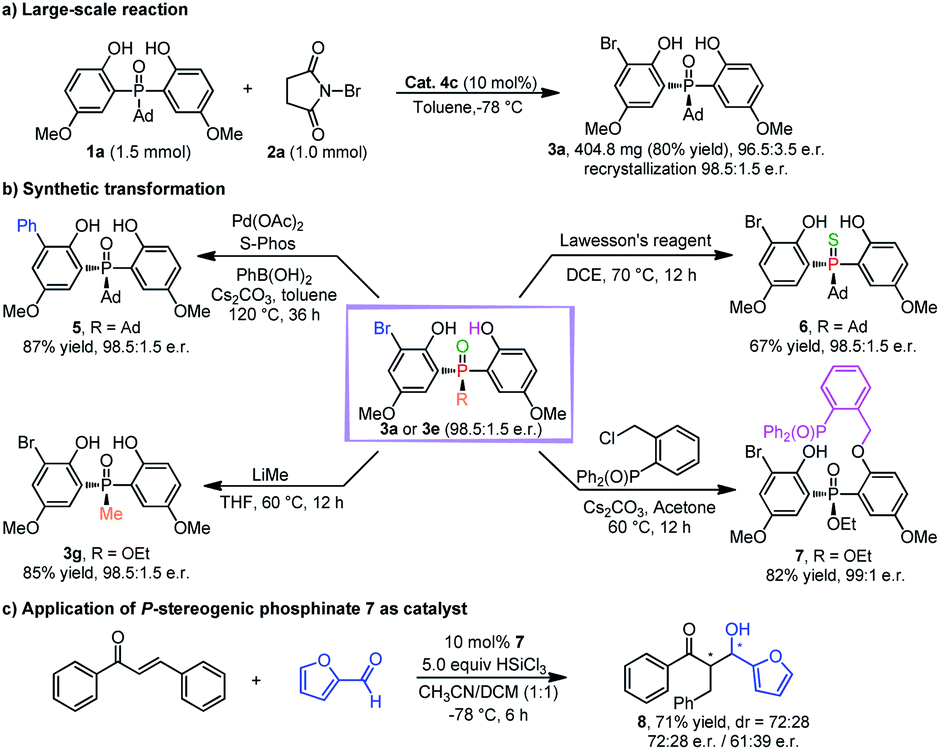 | ||
| Scheme 2 (a) Large-scale reaction. (b) Synthetic transformations. (c) Application of the transformed product. | ||
Since the mono-bromination product 3a could undergo further bromination to form the dibromo adduct, we wondered whether this second bromination is a kinetic resolution process. As shown in Scheme 3a, a racemic sample of 3a was subjected to the catalytic conditions ((±)-3a and 2a in a 2:1 molar ratio). Upon complete consumption of 2a (with the formation of a dibromo product in 49% yield), the mono-bromination product 3a was recovered in 51% yield with 99:1 e.r. This result indicated that the second bromination was indeed a kinetic resolution process and had a positive contribution to the enantioselectivity. Considering the excellent enantiomeric ratio of recovered 3a, we further investigated the reaction of rac-9 with 2a under kinetic resolution conditions (Scheme 3b). To our delight, the unreacted raw material 9 can be obtained in 51% yield with 99.5:0.5 e.r., and chiral dihalogenated product 10 can also be generated in 49% yield with 90:10 e.r.
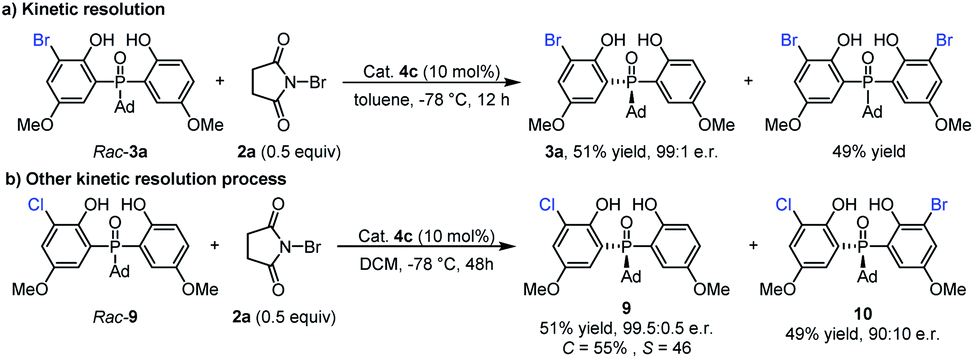 | ||
| Scheme 3 Kinetic resolution process. | ||
To investigate the mechanism, we performed some control experiments. First, a mono-methyl protected phosphine oxide substrate was prepared and subjected to ortho-bromination under the optimal conditions. As shown in Scheme 4a, the corresponding product 11 was obtained with 72.5:27.5 e.r. When the same reaction conditions were applied to the dimethyl protected phosphine oxide substrate, no reaction occurred (Scheme 4b). These results indicated that the phenol moieties of the substrate were essential for the bromination reaction. In fact, hydrogen bonds formed between the two phenolic hydroxyl groups and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O could be observed in the single crystal structure of the product 3w.25 Furthermore, when thiophosphine oxide, which had a weak hydrogen bond acceptor PS group, was prepared and tested in the reaction, the corresponding product 6 was obtained with a lower yield and enantioselectivity than that of 3a (Scheme 4c). This result suggested that the intramolecular hydrogen bonds of the substrate might be beneficial for both the reactivity and the enantioselectivity.27 In light of the control experiments and previous studies,24 two possible mechanisms were proposed (see the ESI‡).
O could be observed in the single crystal structure of the product 3w.25 Furthermore, when thiophosphine oxide, which had a weak hydrogen bond acceptor PS group, was prepared and tested in the reaction, the corresponding product 6 was obtained with a lower yield and enantioselectivity than that of 3a (Scheme 4c). This result suggested that the intramolecular hydrogen bonds of the substrate might be beneficial for both the reactivity and the enantioselectivity.27 In light of the control experiments and previous studies,24 two possible mechanisms were proposed (see the ESI‡).
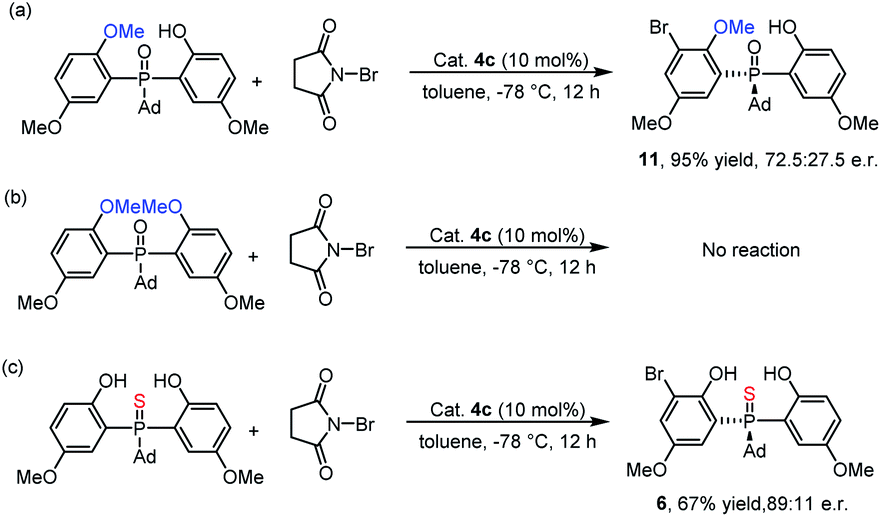 | ||
| Scheme 4 Control experiments: (a) mono-methyl protected phosphine oxide substrate was evaluated; (b) dimethyl protected phosphine oxide substrate was examined; (c) thiophosphine oxide substrate was investigated. | ||
In summary, a novel and efficient desymmetrizing asymmetric ortho-selective mono-bromination of bisphenol phosphine oxides under chiral squaramide catalysis was reported. Using this asymmetric ortho-bromination strategy, a wide range of chiral bisphenol phosphine oxides and bisphenol phosphinates were obtained with good to excellent yields and enantioselectivities. The reaction could be scaled up, and the synthetic utility of the desired P-stereogenic compounds was proved by transformations and application in an asymmetric reaction. Ongoing studies focus on the further mechanistic investigations and the potential applications of these chiral P-stereogenic compounds in other asymmetric transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (grant no. 21971120 and 21933008) and Natural Science Foundation of Tianjin (18JCYBJC28400) for financial support.Notes and references
- C. J. Schulze, G. Navarro, D. Ebert, J. DeRisi and R. G. Linington, J. Org. Chem., 2015, 80, 1312 CrossRef CAS.
- E. Cholongitas and G. V. Papatheodoridis, Ann. Gastroenterol., 2014, 27, 331 Search PubMed.
- L. Clarion, C. Jacquard, O. Sainte-Catherine, S. Loiseau, D. Filippini, M.-H. Hirlemann, J.-N. Volle, D. Virieux, M. Lecouvrey, J.-L. Pirat and N. Bakalara, J. Med. Chem., 2012, 55, 2196 CrossRef CAS.
- W. R. G. Dostmann, S. S. Taylor, H.-G. Genieser, B. Jastorff, S. O. Døskeland and D. Øgreid, J. Biol. Chem., 1990, 265, 10484 CrossRef CAS.
- For selected reviews, see: (a) R.-Y. Zhu, K. Liao, J.-S. Yu and J. Zhou, Acta Chim. Sin., 2020, 78, 193 CrossRef; (b) M. Matsukawa, H. Sugama and T. Imamoto, Tetrahedron Lett., 2000, 41, 6461 CrossRef.
- (a) K. Iseki, Y. Kuroki, M. Takahashi and Y. Kobayashi, Tetrahedron Lett., 1996, 37, 5149 CrossRef CAS; (b) K. Iseki, Y. Kuroki, M. Takahashi and S. Kishimoto, Tetrahedron, 1997, 53, 3513 CrossRef CAS.
- Selected examples for chiral resolution: (a) O. Korpiun, R. A. Lewis, J. Chickos and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4842 CrossRef CAS; for chiral auxiliaries: (b) O. Berger and J.-L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377 CrossRef CAS; (c) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J.-N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474 CrossRef CAS; (d) D. Gwon, D. Lee, J. Kim, S. Park and S. Chang, Chem.–Eur. J., 2014, 20, 12421 CrossRef CAS; for asymmetric oxidation of tertiary phosphines: (e) E. Bergin, C. T. O'Connor, S. B. Robinson, C. P. O'Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566 CrossRef CAS; (f) K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906 CrossRef CAS.
- Z. Huang, X. Huang, B. Li, C. Mou, S. Yang, B.-A. Song and Y. R. Chi, J. Am. Chem. Soc., 2016, 138, 7524 CrossRef CAS.
- G.-H. Yang, Y. Li, X. Li and J.-P. Cheng, Chem. Sci., 2019, 10, 4322 RSC.
- R.-Z. Guo, Z.-Q. Liu and X.-D. Zhao, CCS Chem., 2020, 2, 2617 CrossRef.
- Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734 CrossRef CAS.
- (a) G. Nishida, K. Noguchi, M. Hirano and K. Tanaka, Angew. Chem., Int. Ed., 2008, 47, 3410 CrossRef CAS; (b) Y.-K. Tahara, T. Sato, R. Matsubara, K. S. Kanyiva and T. Shibata, Heterocycles, 2016, 93, 685 CrossRef CAS; (c) Y. Zheng, L. Guo and W. Zi, Org. Lett., 2018, 20, 7039 CrossRef CAS; (d) Y. Zhang, F. Zhang, L. Chen, J. Xu, X. Liu and X. Feng, ACS Catal., 2019, 9, 4834 CrossRef CAS; (e) R. Y. Zhu, L. Chen, X. S. Hu, F. Zhou and J. Zhou, Chem. Sci., 2020, 11, 97 RSC.
- (a) J. S. Harvey, S. J. Malcolmson, K. S. Dunne, S. J. Meek, A. L. Thompson, R. R. Schrock, A. H. Hoveyda and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 762 CrossRef CAS; (b) Z. Wang and T. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 1702 CrossRef CAS.
- (a) D. Gwon, S. Park and S. Chang, Tetrahedron, 2015, 71, 4504 CrossRef CAS; (b) Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS; (c) Z.-Q. Lin, W.-Z. Wang, S.-B. Yan and W.-L. Duan, Angew. Chem., Int. Ed., 2015, 54, 6265 CrossRef CAS; (d) L. Liu, A.-A. Zhang, Y. Wang, F. Zhang, Z. Zuo, W.-X. Zhao, C.-L. Feng and W. Ma, Org. Lett., 2015, 17, 2046 CrossRef CAS; (e) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364 CrossRef CAS; (f) Y.-S. Jang, M. Dieckmann and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 15088 CrossRef CAS; (g) Y. Lin, W.-Y. Ma, Q.-Y. Sun, Y.-M. Cui and L.-W. Xu, Synlett, 2017, 28, 1432 CrossRef CAS; (h) Y.-S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS; (i) Z. Li, Z.-Q. Lin, C.-G. Yan and W.-L. Duan, Organometallics, 2019, 38, 3916 CrossRef CAS.
- (a) S. Liu, Z. F. Zhang, F. Xie, N. A. Butt, L. Sun and W. B. Zhang, Tetrahedron: Asymmetry, 2012, 23, 329 CrossRef CAS; (b) K. M.-H. Lim and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 8122 CrossRef CAS; (c) Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981 RSC.
- (a) X. Fu, W.-T. Loh, Y. Zhang, T. Chen, T. Ma, H. Liu, J. Wang and C.-H. Tan, Angew. Chem., Int. Ed., 2009, 48, 7387 CrossRef CAS; (b) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183 CrossRef CAS; (c) Y. Zhang, H. He, Q. Y. Wang and Q. Cai, Tetrahedron Lett., 2016, 57, 5308 CrossRef CAS; (d) Q. Dai, W.-B. Li, Z.-M. Li and J.-L. Zhang, J. Am. Chem. Soc., 2019, 141, 20556 CrossRef CAS; (e) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, J. Am. Chem. Soc., 2019, 141, 16584 CrossRef CAS; (f) H. Qiu, Q. Dai, J. He, W. Li and J. Zhang, Chem. Sci., 2020, 11, 9983 RSC.
- (a) S. Fujisaki, H. Eguchi, A. Omura, A. Okamoto and A. Nishida, Bull. Chem. Soc. Jpn., 1993, 66, 1576 CrossRef CAS; (b) X. Xiong and Y.-Y. Yeung, ACS Catal., 2018, 8, 4033 CrossRef CAS; (c) O. M. Beleh, E. Miller, F. D. Toste and S. J. Miller, J. Am. Chem. Soc., 2020, 142, 16461 CrossRef CAS.
- (a) J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS; (b) K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964 CrossRef CAS; (c) R. Miyaji, K. Asano and S. Matsubara, Chem.–Eur. J., 2017, 23, 9996 CrossRef CAS.
- (a) M. E. Diener, A. J. Metrano, S. Kusano and S. J. Miller, J. Am. Chem. Soc., 2015, 137, 12369 CrossRef CAS; (b) J. M. Crawford, E. A. Stone, A. J. Metrano, S. J. Miller and M. S. Sigman, J. Am. Chem. Soc., 2018, 140, 868 CrossRef CAS; (c) X. C. Yan, A. J. Metrano, M. J. Robertson, N. C. Abascal, J. Tirado-Rives, S. J. Miller and W. L. Jorgensen, ACS Catal., 2018, 8, 9968 CrossRef CAS; (d) A. J. Metrano and S. J. Miller, Acc. Chem. Res., 2019, 52, 199 CrossRef CAS.
- (a) K. T. Barrett and S. J. Miller, J. Am. Chem. Soc., 2013, 135, 2963 CrossRef CAS; (b) K. T. Barrett, A. J. Metrano, P. R. Rablen and S. J. Miller, Nature, 2014, 509, 71 CrossRef CAS.
- R. Miyaji, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 6766 CrossRef CAS.
- S. D. Vaidya, S. T. Toenjes, N. Yamamoto, S. M. Maddox and J. L. Gustafson, J. Am. Chem. Soc., 2020, 142, 2198 CrossRef CAS.
- (a) C. A. Lewis, A. Chiu, M. Kubryk, J. Balsells, D. Pollard, C. K. Esser, J. Murry, R. A. Reamer, K. B. Hansen and S. J. Miller, J. Am. Chem. Soc., 2006, 128, 16454 CrossRef CAS; (b) C. A. Lewis, J. L. Gustafson, A. Chiu, J. Balsells, D. Pollard, J. Murry, R. A. Reamer, K. B. Hansen and S. J. Miller, J. Am. Chem. Soc., 2008, 130, 16358 CrossRef CAS; (c) A. E. Hurtley, E. A. Stone, A. J. Metrano and S. J. Miller, J. Org. Chem., 2017, 82, 11326 CrossRef CAS.
- X. Xiong, T. Zheng, X. Wang, Y.-L. S. Tse and Y.-Y. Yeung, Chem, 2020, 6, 919 CAS.
- CCDC: 2041102, see the ESI‡ for more details.
- (a) M. Sugiura, N. Sato, Y. Sonoda, S. Kotani and M. Nakajima, Chem.–Asian J., 2010, 5, 478 CrossRef CAS; (b) M. Sugiura, N. Sato, S. Kotani and M. Nakajima, Chem. Commun., 2008, 44, 4309 RSC.
- We calculated the nucleophilic parameters of different substrates to illustrate that the intramolecular hydrogen bonding is possible. For details, see the ESI.‡.
Footnotes |
| † Dedicated to the 100th anniversary of Chemistry at Nankai University. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 2041102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc07008d |
| This journal is © The Royal Society of Chemistry 2021 |