
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative activating effects of metal ion and Brønsted acid on a metal oxo species†
Gui
Chen
 a,
Li
Ma
b,
Po-Kam
Lo
c,
Chi-Keung
Mak
*c,
Kai-Chung
Lau
*c and
Tai-Chu
Lau
*c
a,
Li
Ma
b,
Po-Kam
Lo
c,
Chi-Keung
Mak
*c,
Kai-Chung
Lau
*c and
Tai-Chu
Lau
*c
aDongguan Cleaner Production Technology Center, School of Environment and Civil Engineering, Dongguan University of Technology, Dongguan, Guangdong 523808, China
bDepartment of Chemistry, Jinan University, Guangzhou, 510632, China
cDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Hong Kong, China. E-mail: bhtclau@cityu.edu.hk; kaichung@cityu.edu.hk; chikmak6@cityu.edu.hk
First published on 21st October 2020
Abstract
Metal oxo (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) complexes are common oxidants in chemical and biological systems. The use of Lewis acids to activate metal oxo species has attracted great interest in recent years, especially after the discovery of the CaMn4O5 cluster in the oxygen-evolving centre of photosystem II. Strong Lewis acids such as Sc3+ and BF3, as well as strong Brønsted acids such as H2SO4 and CF3SO3H, are commonly used to activate metal oxo species. In this work, we demonstrate that relatively weak Lewis acids such as Ca2+ and other group 2 metal ions, as well as weak Brønsted acids such as CH3CO2H, can readily activate the stable RuO4− complex towards the oxidation of alkanes. Notably, the use of Ca2+ and CH3CO2H together produces a remarkable cooperative effect on RuO4−, resulting in a much more efficient oxidant. DFT calculations show that Ca2+ and CH3CO2H can bind to two oxo ligands to form a chelate ring. This results in substantial lowering of the barrier for hydrogen atom abstraction from cyclohexane.
O) complexes are common oxidants in chemical and biological systems. The use of Lewis acids to activate metal oxo species has attracted great interest in recent years, especially after the discovery of the CaMn4O5 cluster in the oxygen-evolving centre of photosystem II. Strong Lewis acids such as Sc3+ and BF3, as well as strong Brønsted acids such as H2SO4 and CF3SO3H, are commonly used to activate metal oxo species. In this work, we demonstrate that relatively weak Lewis acids such as Ca2+ and other group 2 metal ions, as well as weak Brønsted acids such as CH3CO2H, can readily activate the stable RuO4− complex towards the oxidation of alkanes. Notably, the use of Ca2+ and CH3CO2H together produces a remarkable cooperative effect on RuO4−, resulting in a much more efficient oxidant. DFT calculations show that Ca2+ and CH3CO2H can bind to two oxo ligands to form a chelate ring. This results in substantial lowering of the barrier for hydrogen atom abstraction from cyclohexane.
Introduction
High-valent metal oxo (MO) complexes are common oxidants in the chemical laboratory and in biological systems. Brønsted acids have long been used to increase the oxidizing power of MO via protonation of the oxo ligand (MO + HX → M−OH+ + X−). However, in recent years, the use of Lewis acids (LA) such as metal ions and boranes to activate MO has received tremendous attention (MO + LA → MO–LA).1–3 In particular, the interest in understanding the interaction of Lewis acids with metal oxos is stimulated by the discovery that the oxygen-evolving center (OEC) of photosystem II (PSII) is composed of a CaMn3O4 cubane and a dangling Mn linked via two μ-oxos.4–10 A possible role of Ca2+ is to function as a Lewis acid to modulate the redox reactivity of the manganese oxo complexes.
Strong Lewis acids such as BF3 and Sc3+ are usually used to activate metal oxo species. For example, we have reported that the oxidation of alkanes by MnO4− is accelerated by over seven orders of magnitude in the presence of BF3.11 The strong Lewis acid Sc3+ and the strong Brønsted acid CF3SO3H have been used to activate Fe(IV) and Mn(IV) oxo complexes,12,13 as well as metal superoxo species.14,15
There is also much interest in the use of the relatively weak Lewis acid, Ca2+, to activate MO because of its relevance to the CaMn4O5 cluster in Photosystem II; although as expected, its effect is usually much smaller than those of Sc3+ or BF3. For example, Agapie et al. reported that Ca2+ and other metal ions can increase the reduction potentials (E0) of manganese oxo clusters, and there is a linear correlation between E0 and pKa of the metal ions.16,17 Ca2+ can also enhance the catalytic water oxidation activity of manganese oxides18,19 and induce the oxidative release of O2 from a non-heme iron peroxo complex.20 We have also recently reported that Ca2+ can increase the rate of oxidation of alcohols by MnO4− (ref. 21) and induce intermolecular O–O coupling of FeO42− to give O2.22 So far, significant activating effects of Ca2+ are only reported on metal oxo complexes that are thermodynamically strong oxidants. On the other hand, there has been little or no report on the activation of metal oxo species by weak Brønsted acids such as alkanoic acids.
We have been investigating the effects of Lewis acids on the reactivity of simple metal oxo species bearing two or more oxos without bulky ancillary ligands.11,21–25 In this type of systems, more than one oxo sites are available for binding to two or more metal ions or Brønsted acids. We report herein the activation of a stable metal oxo complex, RuO4−, by weak Lewis acids such as Ca2+ and other group II ions, as well as by weak Brønsted acids such as CH3CO2H. Although iron oxo complexes are common oxidants in chemical and biological systems, they are usually relatively unstable. Hence, we choose to investigate a very stable ruthenium oxo species. Although in a high oxidation state of +VII, RuO4− is a mild oxidant that readily oxidizes alcohols but is inactive towards alkanes. However, in the presence of a few equiv. of group 2 metal ions or acetic acid, it readily oxidizes cyclohexane at ambient conditions. Notably, the use of Ca2+ and CH3CO2H together produces a remarkable cooperative effect on RuO4−, resulting in a much more efficient oxidant than the use of a strong Lewis acid in the presence or absence of a Brønsted acid.
Results and discussion
Effects of Lewis acids on oxidation of cyclohexane by RuVIIIO4 and RuVIIO4−
Tetraoxo complexes of ruthenium in oxidation states +VIII and +VII are known, with RuVIIIO4 being a much stronger oxidant than RuVIIO4−. As a comparison, we have initially chosen to study the effects of Brønsted and Lewis acids (metal ions and BF3) on the oxidation of alkanes by RuO4. RuO4 is a strong oxidant that is known to oxidize alkanes slowly at room temperature.26 In our hands, when RuO4 (0.01 M) was treated with an excess of cyclohexane (1.0 M) in CH3CN, 0.2 mol of cyclohexanone/mol of RuO4 (20 mol%) was produced after 5 h at 23 °C, as analysed by GC and GC/MS (Fig. 1a and Table S1†). No cyclohexanol could be detected. Since we previously reported that the oxidation of alkanes by MnO4− is greatly enhanced by just a few equiv. of BF3,11 we attempted to do the same with RuO4. However, when BF3 was added to RuO4, no enhancement in the rate of cyclohexane oxidation was observed; on the contrary, a lower yield of cyclohexanone (11 mol%) was obtained (Fig. 1a and Table S1†). We then examined if Ca2+ can activate RuO4, but virtually no effect on cyclohexane oxidation was observed when a few equiv. of Ca(OTf)2 (OTf is CF3SO3−) was added. We also tried to activate RuO4 with Brønsted acids such as CF3SO3H and CH3CO2H (AcOH), but again there were no effects. These results indicate that the oxo ligands in the highly electrophilic RuO4 have little or no affinity for CH3CO2H and various Lewis acids. | ||
| Fig. 1 Time traces for cyclohexane oxidation by (a) RuO4/Lewis acid and (b) RuO4−/Lewis acid in CH3CN. Conditions: RuO4 or [nPr4N][RuO4] (0.01 M), Lewis acid (0.04 M), cyclohexane (1.0 M) at 23 °C. | ||
On the other hand, RuO4− is a much weaker oxidant than RuO4; it is known to oxidize alcohols but not alkanes.27 When we treated [nPr4N][RuO4] with cyclohexane in CH3CN, no product could be detected after 5 days at 23 °C. Electrospray ionization mass spectrometry (ESI/MS) of the solution after 5 days shows that RuO4− and nPr4N+ are the only species present. The UV/Vis spectrum of the solution also remained unchanged after 5 days. However, upon adding 4 equiv. of BF3 to [nPr4N][RuO4] in CH3CN, 10 mol% of cyclohexanone was produced within 3 h at 23 °C (Fig. 1b and Table S2†). Again, no cyclohexanol product could be detected. More significantly, Ca(OTf)2 is also able to activate RuO4−, and a higher yield of 14 mol% of cyclohexanone was attained (Fig. 1b, S1 and Table S2†). As will be described below, the RuO4−/Ca2+ system functions as one-electron oxidant, and since the oxidation of cyclohexane to cyclohexanone is a four-electron process, the actual yield is 56%. Other group II metal ions were also found to activate RuO4−, with cyclohexanone production ranging from 11 to 15 mol% (Fig. 2, Table S2†). The relatively strong Lewis acid Sc(OTf)3 was also used and it gave the highest amount of 23 mol% of cyclohexanone. The rate and yield decrease in the order of Sc3+ (pKa = 4.3) > Mg2+ (11.2) > Ca2+ (12.7) > Sr2+ (13.2) ≫ Ba2+ (13.4); this trend correlates with their pKa values in H2O, which is a measure of their Lewis acidity.28 These results indicate that the oxo ligands in RuO4− are much more basic than those in RuO4, so they readily bind to Lewis acids. Electron-withdrawing by the Lewis acids via the oxo ligand enhances the oxidizing power of RuO4−.
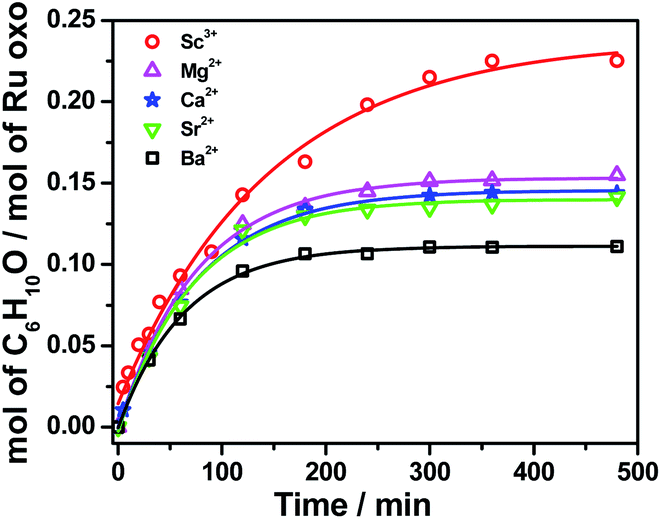 | ||
| Fig. 2 Effects of various Lewis acids on cyclohexane oxidation by RuO4− in CH3CN. Conditions: [nPr4N][RuO4] (0.01 M), Lewis acid (0.04 M), cyclohexane (1.0 M) at 23 °C. | ||
Kinetics of the oxidation of cyclohexane by RuO4−/Ca2+
The initial rate for cyclohexanone production by [nPr4N][RuO4] increases with [Ca2+] but eventually levels off at [Ca2+] > 5 mM (Fig. 3a). A plot of 1/(initial rate) versus 1/[Ca(OTf)2] is linear (Fig. 3b). In addition, when the concentration of RuO4− was doubled, the initial rate was also doubled. Such a kinetic behavior can be represented by eqn (1) and (2). The reacting calcium species is proposed to be Ca(OTf)+, as supported by results of DFT calculations described below. The initial rate of the reaction is shown in eqn (3). | (1) |
 | (2) |
 | (3) |
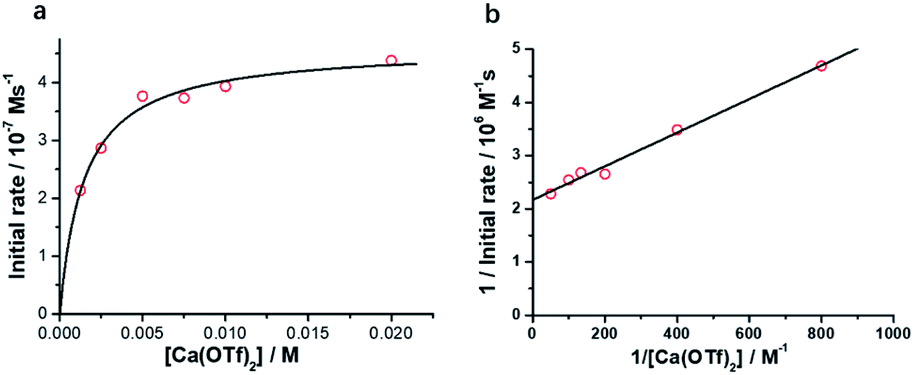 | ||
| Fig. 3 Effects of [Ca(OTf)2] on cyclohexane oxidation by RuO4− in CH3CN. Conditions: [nPr4N][RuO4] (0.01 M), cyclohexane (1.0 M) in CH3CN at 23 °C. (a) Plot of initial rate vs. [Ca(OTf)2]. (b) Plot of 1/initial rate vs. 1/[Ca(OTf)2] (slope = (3.16 ± 0.15) ×103, y-intercept = (2.16 ± 0.06) × 106, r = 0.988). | ||
From Fig. 3b, k = 1/(intercept [RuO4−]) = (4.63 ± 0.13) × 10−5 M−1 s−1 and K = intercept/slope = (6.83 ± 0.18) × 102 M−1 at 23 °C. The observed equilibrium constant K indicates relatively strong binding of Ca(OTf)+ to RuO4−, in accordance with the observed rate saturation behaviour.
Activation of RuO4− by Brønsted acids
The effects of Brønsted acids on cyclohexane oxidation by RuO4− were also investigated. Addition of 1 equiv. of the strong acid CF3SO3H (pKa = 0.23 in H2O) to [nPr4N][RuO4] (0.01 M) in CH3CN containing excess cyclohexane led to the formation of 15 mol% of cyclohexanone after 3 h at 23 °C. However, addition of ≥4 equiv. of CF3SO3H to [nPr4N][RuO4] resulted to rapid formation of a black precipitate with only a trace amount of cyclohexanone. Such a phenomenon is due to disproportionation of RuO4−, as represented by eqn (4):29| 4RuO4− + 4H+ → 3RuO4 + RuO2 + 2H2O E0 = 0.46 V | (4) |
Interestingly, [nPr4N][RuO4] is also readily activated by the relatively weak acid CH3CO2H (pKa = 4.76) to oxidize cyclohexane to cyclohexanone, with no evidence of disproportionation even in the presence of high concentrations of AcOH (>1 M). Upon addition of 12–48 equiv. of AcOH to [nPr4N][RuO4] in CH3CN containing excess cyclohexane, the brown solution gradually turned green, and 16 mol% of cyclohexanone was produced within 1–2 h at 23 °C. The oxidation state of the ruthenium product was determined to be +6 (see below, Fig. S2†), hence the actual yield is 64%. Although the yield is independent of [AcOH], the rate of oxidation increases with increasing [AcOH] (Fig. 4); a plot of the initial rate versus [AcOH] gives a straight line. The initial rate is also doubled when [RuO4−] is doubled. A proposed reaction scheme is shown in eqn (5)–(7). The first step involves protonation of an oxo ligand of RuO4− by AcOH, this step is supported by DFT calculations described below. The resulting [Ru(O)3(OH)] species is hydrogen-bonded to a second AcOH molecule to generate the active intermediate [AcOH·Ru(O)3(OH)] that oxidizes cyclohexane. The rate law is shown in eqn (8); at K′[CH3CO2H] ![[left double angle bracket]](https://www.rsc.org/images/entities/char_27ea.gif) 1, the rate law becomes that of eqn (9).
1, the rate law becomes that of eqn (9).
| RuO4− + CH3CO2H → [Ru(O)3(OH)] + CH3CO2− | (5) |
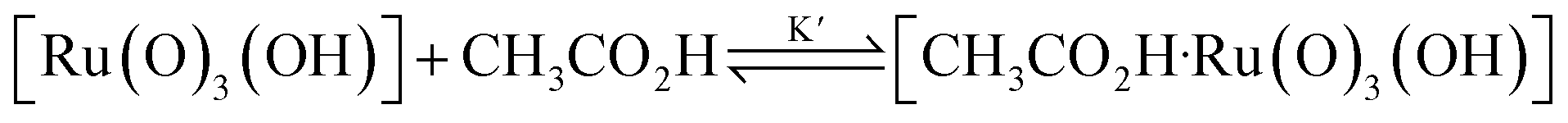 | (6) |
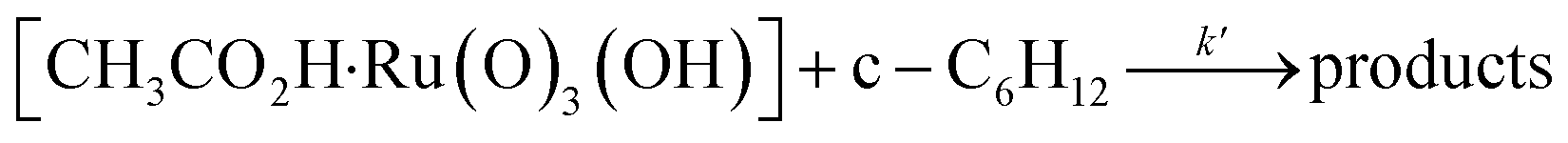 | (7) |
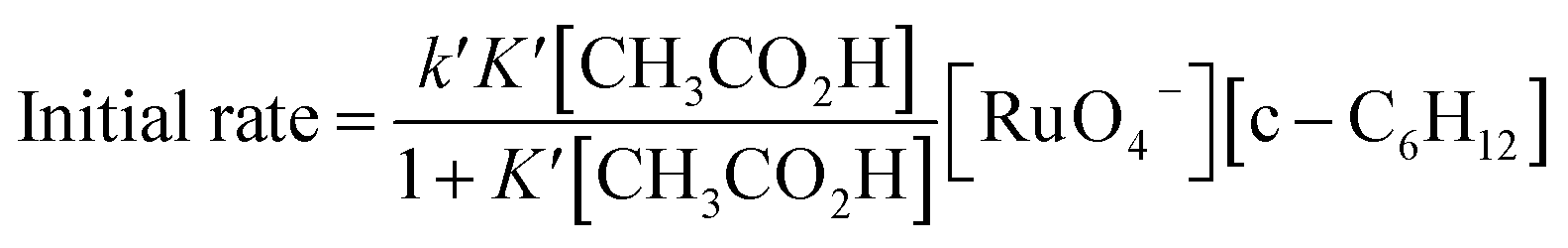 | (8) |
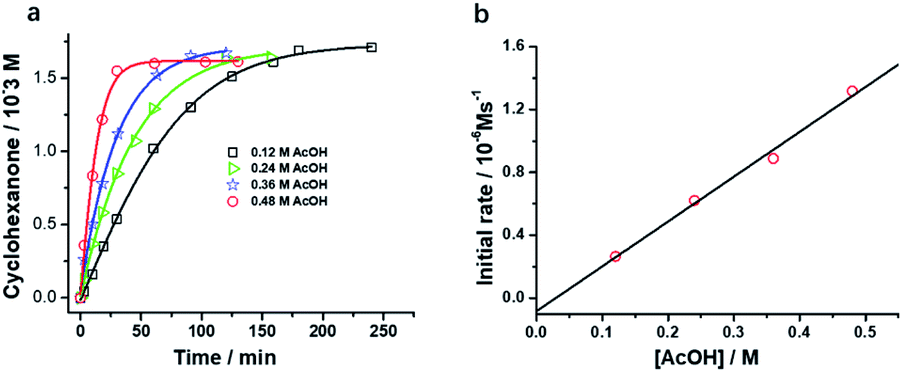 | ||
| Fig. 4 (a) Effects of CH3CO2H on cyclohexane oxidation by RuO4− in CH3CN. Conditions: [nPr4N][RuO4] (0.01 M), cyclohexane (1.0 M) in CH3CN at 23 °C. (b) Plot of initial rate versus [AcOH]. (slope = (2.72 ± 0.13) ×10−6, y-intercept = -(3.33 ± 3.82) × 10−8, r = 0.9908). | ||
At K′[CH3CO2H] 1
| Initial rate = k′K′[AcOH][RuO4−][c-C6H12] = kAcOH[AcOH][RuO4−][c-C6H12] | (9) |
From the slope of Fig. 4b and using [RuO4−] = 0.01 M and [c-C6H12] = 1.0 M, kAcOH is found to be (2.72 ± 0.13) ×10−4 M−2 s−1 at 23 °C.
Cooperative activating effects of metal ions and AcOH
Remarkably, when the oxidation of cyclohexane by RuO4− was carried out in the presence of Ca2+ and AcOH, both the rate and product yield were enhanced, as shown in Fig. 5. The amount of cyclohexanone produced by the RuO4−/Ca2+/AcOH system is 38 mol%, compared with ca. 16 mol% by both RuO4−/Ca2+ and RuO4−/AcOH systems. In this case, the oxidation state of the ruthenium product was found to be +5 (see below, Fig. S3†), so this system functions as a two-electron oxidant and the actual yield is 76%, higher than the 64% using Ca2+ or AcOH alone. The yield of cyclohexanone was increased to 92% when the amount of acetic acid was increased to 250 equiv. (CH3CN/AcOH: 6/1, Table 1). Note that in the absence of Ca(OTf)2, the yield of cyclohexanone did not increase with [AcOH], as illustrated in Fig. 4a. Similar cooperative effects of M2+ and AcOH were also found for other group 2 ions (Table 1), with a maximum yield of 99% observed for Sr2+/AcOH. However, no such cooperative effects were found for stronger Lewis acids such as BF3 and Sc(OTf)3, the yields remain the same in the absence or presence of AcOH. These results demonstrate the strong cooperative effects of relatively mild Brønsted and Lewis acid in activating a metal oxo species. Although the reaction rate is lower than that of BF3 or Sc(OTf)3, the Group 2 ion/AcOH combination is much more efficient in terms of product yield. Similar to the case of CF3SO3H discussed above, partial decomposition of RuO4− occurs in the presence of a strong Lewis acid, hence resulting in lower yields.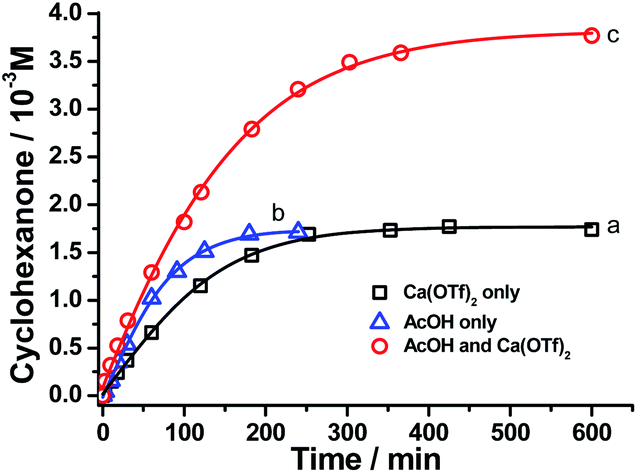 | ||
| Fig. 5 Time trace for cyclohexane oxidation by [nPr4N][RuO4] (0.01 M) in CH3CN at 23 °C in the presence of (a) 1 equiv. Ca2+, (b) 12 mol equiv. of AcOH and (c) 1 mol equiv. Ca2+ + 12 mol equiv. AcOH. | ||
| Entry a | Lewis acid | Yield of cyclohexanoned (%) | Time |
|---|---|---|---|
| a Conditions: [nPr4N][RuO4], 0.01 M; Lewis acid, 0.04 M; cyclohexane, 1.0 M; solvent, CH3CN/HOAc (6:1, v/v); at 23 °C. b Under argon. c In the presence of 10 equiv. of BrCCl3, only a trace amount of bromocyclohexane was detected. d Yield of cyclohexanone was calculated based on [nPr4N][RuO4] acting as two-electron oxidant. Yield = (mol of cyclohexanone)/(mol of [nPr4N][RuO4]) × 2 × 100%, no cyclohexanol was detected. | |||
| 1 | Ca(OTf)2 | 92 | 5 h |
| 2b | Ca(OTf)2 | 92 | 5 h |
| 3c | Ca(OTf)2 | 91 | 5 h |
| 4 | Sr(OTf)2 | 99 | 5 h |
| 5 | Mg(OTf)2 | 86 | 5 h |
| 6 | Ba(OTf)2 | 86 | 5 h |
| 7 | BF3 | 42 | 3 min |
| 8 | Sc(OTf)3 | 46 | 6 min |
The kinetics of cyclohexane oxidation by RuO4−/Ca2+/AcOH were investigated. Saturation kinetics were also observed when [Ca2+] was increased (Fig. 6). Based on Fig. 6 and the mechanisms proposed above for RuO4−/Ca2+ and RuO4−/AcOH, the mechanism for this system can be represented by the following equations.
| RuO4− + CH3CO2H → [Ru(O)3(OH)] + CH3CO2− | (10) |
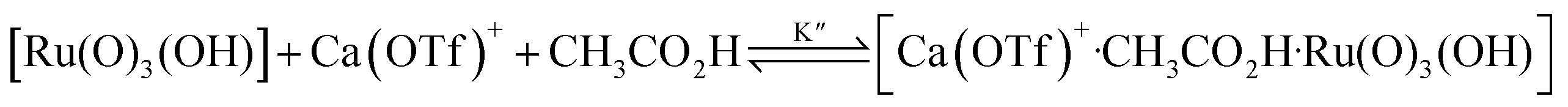 | (11) |
 | (12) |
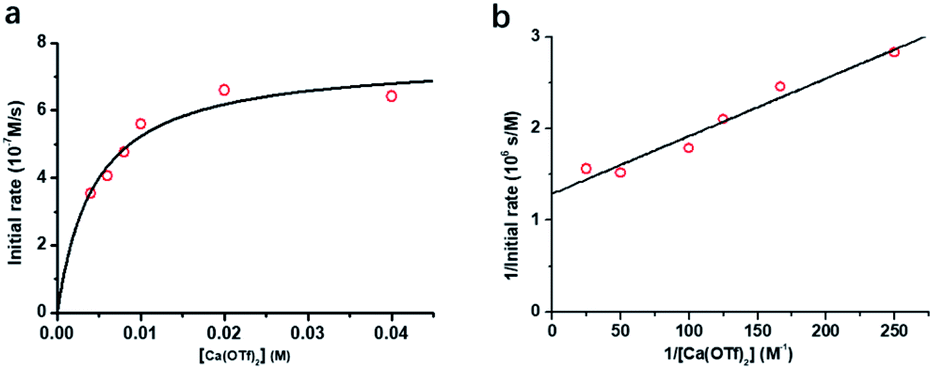 | ||
| Fig. 6 (a) Plot of initial rate vs. [Ca2+] for the Ca(OTf)2/AcOH activated oxidation of cyclohexane (0.10 M) by RuO4− (0.01 M) in CH3CN at 23.0 °C. (b) The corresponding plot of 1/initial rate vs. 1/[Ca2+]. Slope = (6.29 ± 0.64) ×103, y-intercept = (1.29 ± 0.09) × 106, r = 0.9504. Conditions: RuO4− (0.01 M); cyclohexane (1.0 M); AcOH (0.12 M); Ca(OTf)2 (0.002 M − 0.040 M). | ||
The rate-law is as shown in eqn (13):
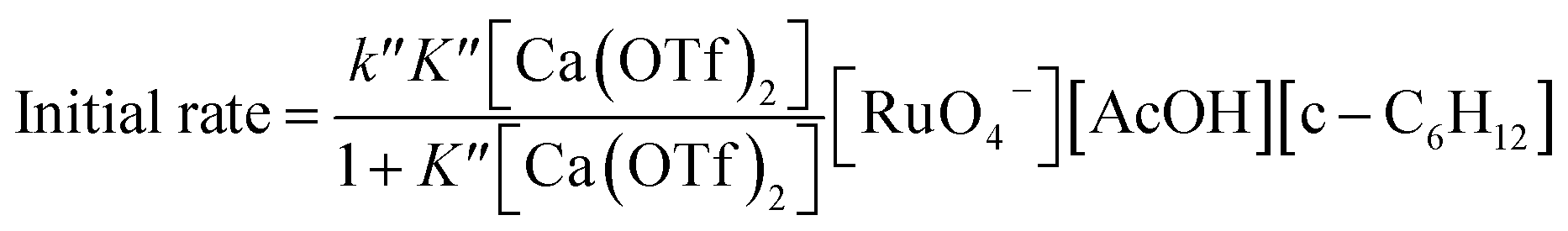 | (13) |
From eqn (11) and Fig. 6b, K′′ and k′′ are found to be (2.05 ± 0.31) × 102 M−1 and (6.49 ± 0.45) × 10−4 M−1 s−1, respectively.
A similar cooperative effect was also found for Mg2+/AcOH (Fig. S4†). However, no increase in rate and yield were found for Sc3+/AcOH (Fig. S5†).
Ruthenium intermediates and products
Electrospray ionization mass spectrometry (ESI/MS) was employed to detect any intermediate formed between RuO4− and Ca2+. The mass spectrum of [nPr4N][RuO4] in CH3CN exhibits a single peak at m/z 166.1 due to RuO4− (Fig. S6†). Upon addition of 0.25 equiv. of Ca(OTf)2, a new peak at m/z 653.9 appeared, which is assigned to [RuO4·Ca(CF3SO3)2·CF3SO3H]− (Fig. S7†). MS/MS of this ion (m/z 653.9) gives fragment peaks due to CF3SO3− (m/z 148.9) and RuO4− (m/z = 165.9) (Fig. S8†). This result provides evidence for the binding of Ca2+ to RuO4−.The brown color of the solution of [nPr4N][RuO4]/Ca(OTf)2 gradually lightened during cyclohexane oxidation, eventually a dark brown precipitate was observed and the solution became colorless. The dark precipitate, which is probably a Ca2+-bridged polymeric species, was dissolved in 0.1 M HNO3 and the solution was titrated spectrophotometrically with the strong oxidant (NH4)2Ce(NO3)6 (Ce(IV)). Upon addition of Ce(IV) to the ruthenium product solution, the characteristic vibronic-structured peaks at around 380 nm due to RuO4 appeared,26 and 2.3 ± 0.3 equiv. of Ce(IV) was consumed (Fig. S2†). This result indicates that the oxidation state of Ru in the dark brown product is +6 and hence the Ca2+/RuO4− system acts as one-electron oxidant in the reaction with cyclohexane.
In cyclohexane oxidation by [nPr4N][RuO4]/AcOH, the brown solution gradually turned dark green but no precipitate was observed. ESI/MS of the dark green solution shows the appearance of a peak at m/z 209 (Fig. S9†), which can be assigned to [RuVIO3(AcO)]−. When CH3CO2H was replaced by CD3CO2D, the m/z 209 peak was shifted to m/z 212, indicating that the m/z 209 peak consists of 1 AcO− ion. The assignment of +6 oxidation state to the ruthenium product is also supported by Ce(IV) titration, which consumes two equiv. of Ce(IV) to generate RuO4. Hence the RuO4−/AcOH system also functions a one-electron oxidant.
On the other hand, spectrophotometric titration of the product solution of Ca2+/AcOH/RuO4− after cyclohexane oxidation shows that it consumes 3.1 ± 0.2 equiv. of Ce(IV), hence in this case the oxidation state of the Ru product is +5 and the system is a two-electron oxidant towards cyclohexane (Fig. S3†).
Mechanistic studies
The same results were obtained for cyclohexane oxidation carried out under argon or air (Tables 1 and S2†). Also, the addition of BrCCl3, a radical scavenger, had little effects on the oxidation of cyclohexane, and only a trace amount of bromocyclohexane was detected (Tables 1 and S2†). These results indicate that no freely diffusing alkyl radicals are formed in the oxidation of cyclohexane by RuO4− in the presence of Ca2+ and/or AcOH.The kinetic isotope effects (KIE) for cyclohexane oxidation by RuO4− under various conditions were determined by competitive oxidation of an equimolar mixture of c-C6H12 and c-C6D12. The KIE for RuO4−/Ca2+, RuO4−/AcOH and RuO4−/Ca2+/AcOH were found to be 6.4 ± 0.2, 13.9 ± 0.4 and 6.5 ± 0.2, respectively. Such large KIEs are indicative of C–H bond cleavage in the rate-limiting step.
Based on the experimental results, the oxidation of alkane by RuO4− in the presence of Lewis acid (LA) appears to be consistent with a mechanism that involves the initial binding of LA to RuO4− to generate a precursor complex, which then reacts with alkane via a H-atom abstraction/O-rebound mechanism to generate the corresponding alcohol. Such a mechanism is commonly accepted for C–H bond activation by cytochrome P450 and various metal oxo species.30 However, since only ketones are detected in the present case, this suggests that the initially formed alcohol is rapidly oxidized to give the ketone. This is supported by a competitive experiment involving the oxidation of a mixture of cyclohexane and cyclopentanol (10:1) by RuO4−/Ca2+, which resulted in the rapid and exclusive formation of cyclopentanone (Fig. S10†). No alkene or products derived from its oxidation were observed in the oxidation of alkane by RuO4−/Ca2+, which rules out a dehydrogenation mechanism that has been shown to occur in alkane oxidation by non-heme iron(IV) oxo species.31 The binding of a Lewis acid to RuO4− enhances its oxidizing power, as observed in non-heme iron(IV) oxo complexes32 and manganese oxo clusters.16,17
Theoretical calculations
In order to obtain further insights into the activating effects of Ca(II) and AcOH on RuO4−, the reaction mechanisms for the oxidation of cyclohexane catalysed by RuO4− in the presence of Ca(OTf)2 and/or AcOH have been theoretically studied by density functional theory (DFT). As a comparison similar studies with RuO4 have also been carried out.In the oxidation of cyclohexane by [RuO4]− in CH3CN (Fig. 7), cyclohexane and [RuO4]− first form an intermediate, INT1(RuO4−), in which the two species are weakly attracted together ([RuO4⋯C6H12]−). HAT then occurs from C6H12 to RuO via a transition state TS1(RuO4−) to form a second intermediate, INT2(RuO4−). The reaction barrier (ΔG298‡) for the HAT is 26.8 kcal mol−1 in CH3CN. Such a large ΔG298‡ agrees with the experimental observation that RuO4− hardly reacts with cyclohexane at room temperature. The C1 of the cyclohexyl radical in INT2(RuO4−) bears −0.93 electrons, consistent with a HAT process. The cyclohexyl radical then binds to another oxo ligand to generate an alkoxo intermediate [RuO2(OH) (OC6H11)]−via TS2(RuO4−). It should be noted that the step after H-abstraction is not characterized as a rebound step, in contrast to cytochrome P450 and other mono-oxo species. Rather, another oxo group which is not used for H-atom abstraction combines with the carbon atom with a low barrier. Because of this reactivity pattern, a ruthenium-bound alkoxide instead of alcohol is formed as an intermediate. Then in the next step, proton transfer from Ru–OH to the alkoxide occurs via TS3(RuO4−) to generate the cyclohexanol product. Similar reaction pathways are observed for cyclohexane oxidation by RuO4, except in this case no radical intermediate (INT2) is formed (Fig. S11†). The reaction barrier (ΔG298‡) is 17.8 kcal mol−1, consistent with the experimental observation that RuO4 reacts readily with cyclohexane at room temperature.
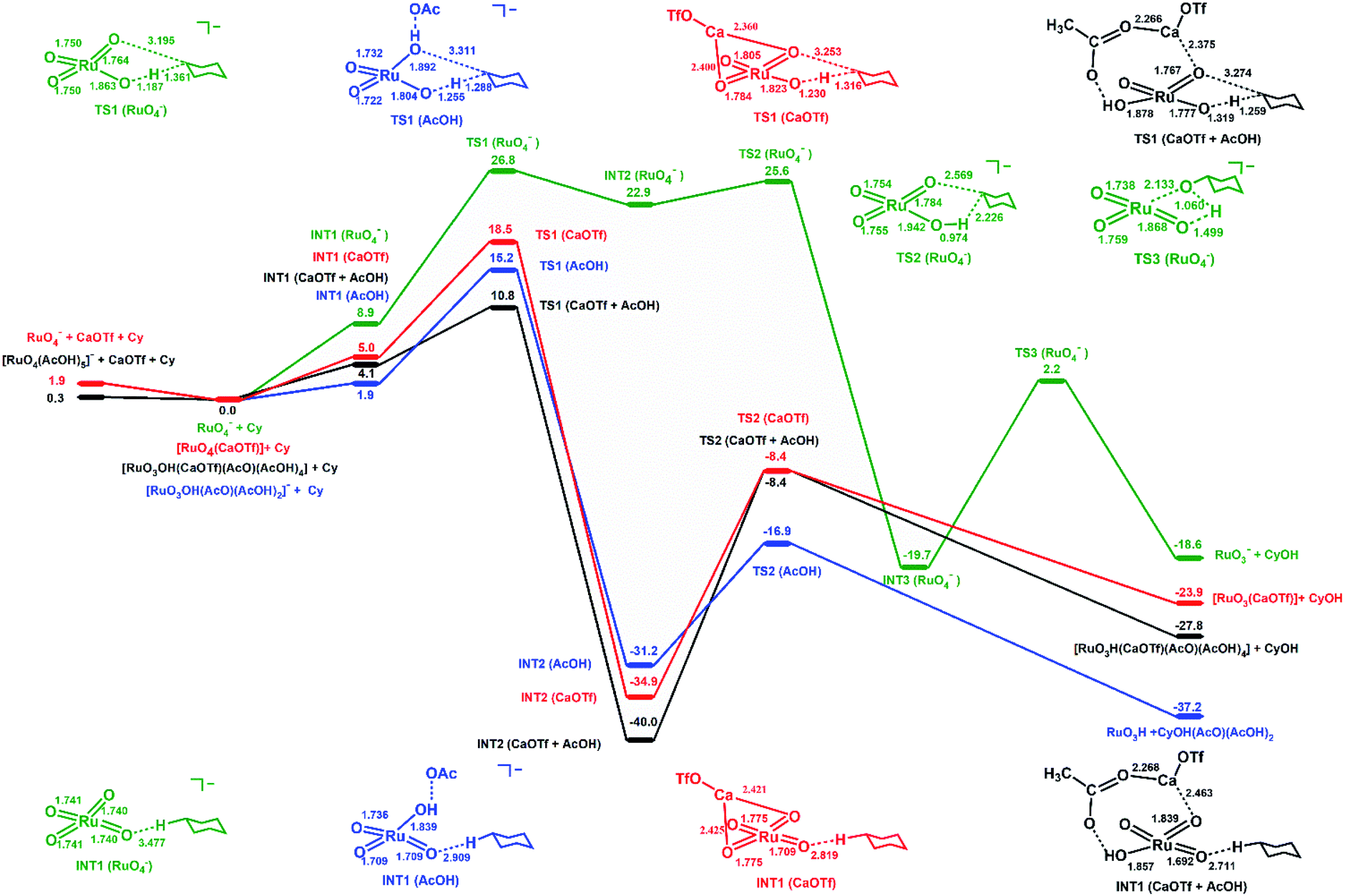 | ||
| Fig. 7 PESs for cyclohexane oxidation by [RuO4]−/[RuO4(AcOH)]−/[RuO4(CaOTf)]/[RuO4(AcOH)(CaOTf)] at the B3LYP level using LanL2TZ(f) basis set (Ru) and 6–311++G(d,p) basis set (non-metals). Relative 298 K Gibbs free energies in acetonitrile are given in kcal mol−1. Not all acetic acid molecules are shown for simplicity. | ||
In the oxidation of cyclohexane by RuO4− in the presence of [Ca(OTf)]+, the reaction mechanism is similar. [Ca(OTf)]+ forms an intermediate, INT1(CaOTf), with RuO4−; the Ca is bound to two oxo ligands. Due to the electron withdrawing effects of Ca(II) centre, the Ru–O bond lengths are changed from 1.740 (in RuO4−, Table S3†) to 1.775 (oxo bound to Ca) and 1.709 Å (free oxo) in INT1(CaOTf). HAT from C6H12 then occurs via the shorter and more electrophilic RuO bond. In this case there is no cyclohexyl radical intermediate, INT2(RuO4−); HAT and binding of cyclohexyl radical to a second oxo occur in a single step. The ΔG298‡ for the oxidation of cyclohexane by [RuO4(CaOTf)] (Fig. 7 and Table S3,† entry 3), via TS1(CaOTf), is 18.5 kcal mol−1. Such a lowering of 8.3 kcal mol−1 is in agreement with the observed accelerating effect of Ca(II). We have also found the ΔG298‡ for the oxidation of cyclohexane by [RuO4(Ca)]+ (Table S3,† entry 5) is higher than that by [RuO4(CaOTf)], so [RuO4(Ca)]+ should not be the active species in the oxidation of cyclohexane.
In the presence of acetic acid, RuO4− is protonated to give INT1(AcOH), [RuO3OH(AcO)⋯C6H12]. The AcO− is held by two additional AcOH molecules through hydrogen bonding (structures given in Table S3†). The Ru–OH bond distance is 1.858 Å; protonation results in shortening of two of the RuO from 1.740 Å (in RuO4−) to 1.709 Å. HAT by INT1(AcOH) occurs via one of the shorter and more electrophilic RuO; the resulting cyclohexyl radical then binds to Ru–OH⋯OAc to generate Ru bound cyclohexanol in the same step, INT2(AcOH). The ΔG298‡ for HAT from C6H12 to INT1(AcOH) via TS1(AcOH) is 15.2 kcal mol−1, which is significantly lower than the ΔG298‡ for RuO4− alone by 11.6 kcal mol−1, in accordance with the experimentally observed accelerating effects of AcOH on RuO4−.
In the presence of both [CaOTf]+ and AcOH, the intermediate with RuO4−, INT1(CaOTf + AcOH), consists of AcO− and Ca forming a chelate ring with RuO and Ru–OH, as well as three H-bonded AcOH molecules (Fig. 7 and Table S3,† entry 4). The free RuO bonds are further shortened to 1.692 Å. Accordingly the ΔG298‡ for HAT from cyclohexane via TS1(CaOTf + AcOH) is lowered to 10.8 kcal mol−1, which is smaller than the value of 18.5 kcal mol−1 and 15.2 kcal mol−1, respectively, with Ca(OTf)+ or AcOH alone. This is in agreement with the observed cooperative activating effects of AcOH and Ca(II). HAT and binding of the resulting cyclohexyl radical to a Ca-bound oxo ligand occur in one step. Protonation by Ru–OH to the alkoxide then occurs to generate cyclohexanol. The potential energy surfaces (PES) for RuO4/Ca(OTf)+, RuO4/AcOH and RuO4/Ca(OTf)+/AcOH are shown in Fig. S11.† The ΔG298‡ for HAT by RuO4 alone is 17.8 kcal mol−1, consistent with the experimental observation that RuO4 is able to oxidize cyclohexane at ambient conditions. There are little or no changes in the RuO distances of RuO4 upon binding to Ca(OTf)+ and/or AcOH, and there are only small changes in the reaction barriers, in agreement with experimental observations. This is in accordance with the RuO bonds being highly electrophilic and non-basic, hence there is little affinity for Lewis acids.
Conclusions
Our results demonstrate a remarkable cooperative effect of a weak Brønsted acid and a weak Lewis acid on the activation of a metal oxo species. RuO4−, although in high oxidation state of +VII, is a weak oxidant due to stabilization by the four oxo ligands. However, it can be readily activated by a mild Lewis acid such as Ca2+ or other group II metal ions, as well as a weak Brønsted acid such as CH3CO2H. The addition of both Ca2+ and CH3CO2H generates a highly efficient system that can oxidize unactivated C–H bonds with much higher yields than the use of strong Lewis acids such as Sc3+ or BF3, with or without CH3CO2H. Such an observation may provide insights into the design of active oxidants based on metal oxo species in combination with relatively weak Brønsted and Lewis acids, especially if the metal oxo or the substrate is sensitive to strong acids. Our studies may also be relevant to oxidation by metal oxo species in biological systems, where only mild Brønsted acids such as alkanoic or amino acids, and mild Lewis acids such as Zn2+ or Ca2+, are present in cells. So may be highly efficient oxidizing systems can be generated in biological systems using this strategy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hong Kong Research Grants Council (CityU 11336816), the National Science Foundation of China (21975043) and Guangdong Provincial Key Platforms and Major Scientific Research Projects for Colleges and Universities (2018KTSCX227). We thank Dr Hajime Hirao for very helpful discussions.Notes and references
- T. Devi, Y.-M. Lee, W. Nam and S. Fukuzumi, Coord. Chem. Rev., 2020, 410, 213219 CrossRef CAS.
- Y. Liu and T.-C. Lau, J. Am. Chem. Soc., 2019, 141, 3755–3766 CrossRef CAS.
- S. Fukuzumi, Coord. Chem. Rev., 2013, 257, 1564–1575 CrossRef CAS.
- D. J. Vinyard, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577–606 CrossRef CAS.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS.
- K. M. Davis, B. T. Sullivan, M. C. Palenik, L. Yan, V. Purohit, G. Robison, I. Kosheleva, R. W. Henning, G. T. Seidler and Y. Pushkar, Phys. Rev. X, 2018, 8, 041014 CAS.
- Y. Pushkar, K. M. Davis and M. C. Palenik, J. Phys. Chem. Lett., 2018, 9, 3525–3531 CrossRef CAS.
- W. W. Y. Lam, S.-M. Yiu, J. M. N. Lee, S. K.-Y. Yau, H.-K. Kwong, T.-C. Lau, D. Liu and Z. Lin, J. Am. Chem. Soc., 2006, 128, 2851–2858 CrossRef CAS.
- Y. Morimoto, H. Kotani, J. Park, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 403–405 CrossRef CAS.
- H. Yoon, H. Yoon, Y.-M. Lee, X. Wu, K.-B. Cho, R. Sarangi, W. Nam and F. S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 9186–9194 CrossRef CAS.
- T. Devi, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2020, 142, 365–372 CrossRef CAS.
- T. Devi, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2018, 140, 8372–8375 CrossRef CAS.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS.
- E. Y. Tsui and T. Agapie, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 10084–10088 CrossRef CAS.
- M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233–2237 CrossRef CAS.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330–2339 RSC.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef CAS.
- H. Du, Po-K. Lo, Z. Hu, H. Liang, K.-C. Lau, Y.-N. Wang, W. W. Y. Lam and T.-C. Lau, Chem. Commun., 2011, 47, 7143–7145 RSC.
- L. Ma, W. W. Y. Lam, P.-K. Lo, K.-C. Lau and T.-C. Lau, Angew. Chem., Int. Ed., 2016, 55, 3012–3016 CrossRef CAS.
- T.-C. Lau and C.-K. Mak, J. Chem. Soc., Chem. Commun., 1993, 766–767 RSC.
- S.-M. Yiu, Z.-B. Wu, C.-K. Mak and T.-C. Lau, J. Am. Chem. Soc., 2004, 126, 14921–14929 CrossRef CAS.
- S.-M. Yiu, W.-L. Man, X. Wang, W. W. Y. Lam, S.-M. Ng, H.-K. Kwong, K.-C. Lau and T.-C. Lau, Chem. Commun., 2011, 47, 4159–4161 RSC.
- W. P. Griffith, Ruthenium Oxidation Complexes: Their Uses as Homogenous Organic Catalysts, Springer, 2011 Search PubMed.
- W. P. Griffith, S. V. Ley, G. P. Whitcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 1625–1627 RSC.
- S. J. Hawkes, J. Chem. Educ., 1996, 73, 516 CrossRef CAS.
- E. A. Seddon and K. R. Seddon, The Chemistry of Ruthenium, Elsevier, Amsterdam, 1984, vol. 52 Search PubMed.
- J. L. McLain, J. Lee and J. T. Groves, Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Imperial College Press, London, 2000 Search PubMed.
- K.-B. Cho, X. Wu, Y.-M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222–20225 CrossRef CAS.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04069j |
| This journal is © The Royal Society of Chemistry 2021 |