
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer defect engineering – conductive 2D organic platelets from precise thiophene-doped polyethylene†
Oksana
Suraeva
a,
Beomjin
Jeong
a,
Kamal
Asadi
a,
Katharina
Landfester
a,
Frederik R.
Wurm
 *ab and
Ingo
Lieberwirth
*a
*ab and
Ingo
Lieberwirth
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: lieberwirth@mpip-mainz.mpg.de
bSustainable Polymer Chemistry Group, MESA+ Institute for Nanotechnology, Faculty of Science and Technology, Universiteit Twente, PO Box 217, 7500 AE Enschede, The Netherlands. E-mail: frederik.wurm@utwente.nl
First published on 2nd March 2021
Abstract
We developed a simple way to create 2D conductive nanostructures with dielectric cores and conductive surfaces based on polyethylene with in-chain thiophene groups. Generally, thiophene-based polymers show great conductive properties, but exhibit a poor processability. Here, we use the crystallization of a polyethylene chain with precisely distributed thiophene groups as the platform for a self-organization of a lamellar structure. During crystallization, thiophene groups are expelled to the crystal surface. Subsequent copolymerization with 3,4-ethylenedioxythiophene (EDOT) molecules finally yields 2D platelets with a conductive surface. The electric properties of the surface are demonstrated by conductivity measurements. Given the molecular structure of the polymer, it can be assumed that the conductive layer consists of only one monoatomic layer of polymerized thiophene. We thus show a new way to create an ultra-thin, conductive surface on a polymer surface in just a few steps. Hence, the method presented here opens up a wide range of possibilities to produce complex, nanoscale electronic structures for microelectronic applications.
Introduction
Conductive polymers combine the mechanical advantages of polymers with the electronic properties of metals and semiconductors, which makes them valuable in the electronics and optical industries.1–3 Some of the most common organic molecules to obtain conductive materials are thiophene and its derivatives, because of their good electrical and chemical properties and environmental stability (both to oxygen and to moisture).4 However, the conjugated backbone of un-substituted polythiophenes is typically rigid and results in poor processability.5 To obtain control over the nanostructure and to increase the processability of thiophene containing materials, two different approaches have been described in the literature: the first approach utilizes the self-assembly of a polymer system to form a nanoscopic template.6 Subsequently, a conductive polymer like polythiophene is grafted upon this template. The resulting nanostructures thus formed have a dielectric core, but the surfaces show only a partial conductivity. The other approach aims to generate a polymeric material with bulk conductivity first.7 Subsequently, this conductive polymer is attached to a dielectric layer, which involves several additional steps.Here, we demonstrate a unique method to generate 2D polymer nanoplatelets with conductive surfaces. To achieve this, we utilize the crystallization driven self-assembly of a precisely synthesized polyethylene-based polymer with in-chain thiophene groups. We use acyclic diene metathesis (ADMET) polymerization8 to synthesize the necessary polymer. This approach allows us to generate an equidistant distribution of functional groups along the polymer backbone.9,10 We designed the polymer architecture in a way, so that the crystallization of this polymer automatically yields the desired 2D nanoplatelets with the thiophene groups arranged exclusively on their surface. Although the thiophene groups are arranged with crystal perfection on the surface, their intramolecular distance is too large to allow for charge carrier transport. Hence, to achieve the final surface conductivity of these nanoplatelets, we copolymerize the surface protruding thiophene groups with 3,4-ethylenedioxythiophene (EDOT). The resulting 2D nano platelets thus designed show a significant increase of conductivity after this copolymerization.
Experimental
Solvents were purchased from Acros Organics and Sigma Aldrich and used as received, unless otherwise stated. 3,4-Dibromothiophene, 11-bromo-1-undecene, 18-bromo-1-octadecene, 3,4-ethylenedioxythiophene (EDOT), (1,3-dppp)NiCl2, Mg, I2, CaCl2, MgSO4, HCl, and ethyl vinyl ether were used as received from Acros Organics and Sigma Aldrich. The 1st generation Grubbs catalyst was purchased from Sigma-Aldrich and stored under an argon atmosphere. Deuterated solvents were purchased from Acros Organics and Carl Roth Chemicals.Instrumentation
Thin layer chromatography (TLC) was performed using Merck aluminium-foil baked plates coated with Kieselgel 60 F245. The products were visualized using UV fluorescence (254 nm) or potassium permanganate stain. Flash column chromatography was performed over Merck silica gel C60 (40–60 μm) using eluent systems as described for each experiment.1H and 13C{H} NMR spectra were acquired on a 300 MHz Bruker system. The temperature was kept at 298.3 K and calibrated with a standard 1H methanol NMR sample using Topspin 3.0 (Bruker). 13C{H} NMR spectra were referenced internally to solvent signals. The 13C{H} NMR (101 MHz) measurements were performed with a 1H power gate decoupling method using 30° flip angle. All spectra were measured in CDCl3 at 298 K. The spectra were calibrated against the solvent signal and analyzed using MestReNova 14.1.0. (Mestrelab Research S.L).
Mass spectrum acquisitions were conducted on an Advion expressionL compact mass spectrometer (CMS) by using the atmospheric solid analysis probe (ASAP) technique. All spectra were acquired in the positive ion reflectron mode, m/z range from 10 to 2000 m/z, and acquisition speed 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m/z units per s. The obtained spectra were analyzed by using Advion CheMS Express software version 5.1.0.2.
000 m/z units per s. The obtained spectra were analyzed by using Advion CheMS Express software version 5.1.0.2.
Wide-angle X-ray diffraction WAXD patterns of the precision PEs were obtained on a Philips PW 1820 diffractometer (scattering angle 2° ≤ 2θ ≤ 60°, 0.02° step size), using Cu Kα radiation (λ = 1.5418 Å) as the X-ray source.
Gel-permeation chromatography (GPC) measurements were carried out in THF, with 1 g L−1 sample concentration. Sample injection was performed using a 717 plus auto sampler (Waters) at 30 °C (THF). Flow was 1 mL min−1. Three SDV columns (PSS) with the dimensions 300 × 80 mm, 10 μm particle size and pore sizes of 106, 104, and 500 Å were employed. Detection was accomplished with a DRI Shodex RI-101 detector (ERC) and a UV–vis S-3702 detector (Soma). Calibration was carried out using polystyrene standards provided by Polymer Standards Service.
Thermal analysis by differential scanning calorimetry (DSC) was carried out using a Mettler-Toledo DSC 822. Three scanning cycles of heating/cooling were performed under a nitrogen atmosphere (30 mL min−1) with a heating and cooling rate of 10 °C min−1 in a temperature range between −50 and 250 °C for polymers TH20, TH20-H, TH38, TH38-H and between −120 and 250 °C for all samples before and after oxidative coupling.
Thermal gravimetric analysis (TGA) was performed on a Mettler Toledo ThermoSTAR TGA/SDTA 851-Thermowaage under a nitrogen atmosphere. The heating rate was 10 °C min−1 in a temperature range between 25 and 900 °C.
For transmission electron microscopy (TEM) measurements, a FEI Tecnai F20 transmission electron microscope operated at an acceleration voltage of 200 kV was used to determine the crystal morphology and crystal structure. Bright field (BF) and parallel beam nano-electron diffraction (NBED) were used for measurements.
Conductivity measurement: glass substrates were sequentially cleaned with detergent, acetone, and 2-propanol by sonication for 5 min each and then dried. Polymers TH38-H, B and C (Scheme 2) were crystallized on the substrate from hot ethyl acetate, and dispersion of polymer A (Scheme 2) was applied on the substrate without additional preparation. Au electrodes were deposited on the dried films by thermal evaporation through shadow masks with a rate of 1 Å s−1 under a high vacuum of 10−7 mbar. The spacing between the electrodes amounted to 20 μm, and the electrode width was 10000 μm. Current (I)–voltage (V) measurement was performed using a Keithley 4200A semiconductor parameter analyzer in a vacuum probe station under 10−5 mbar.
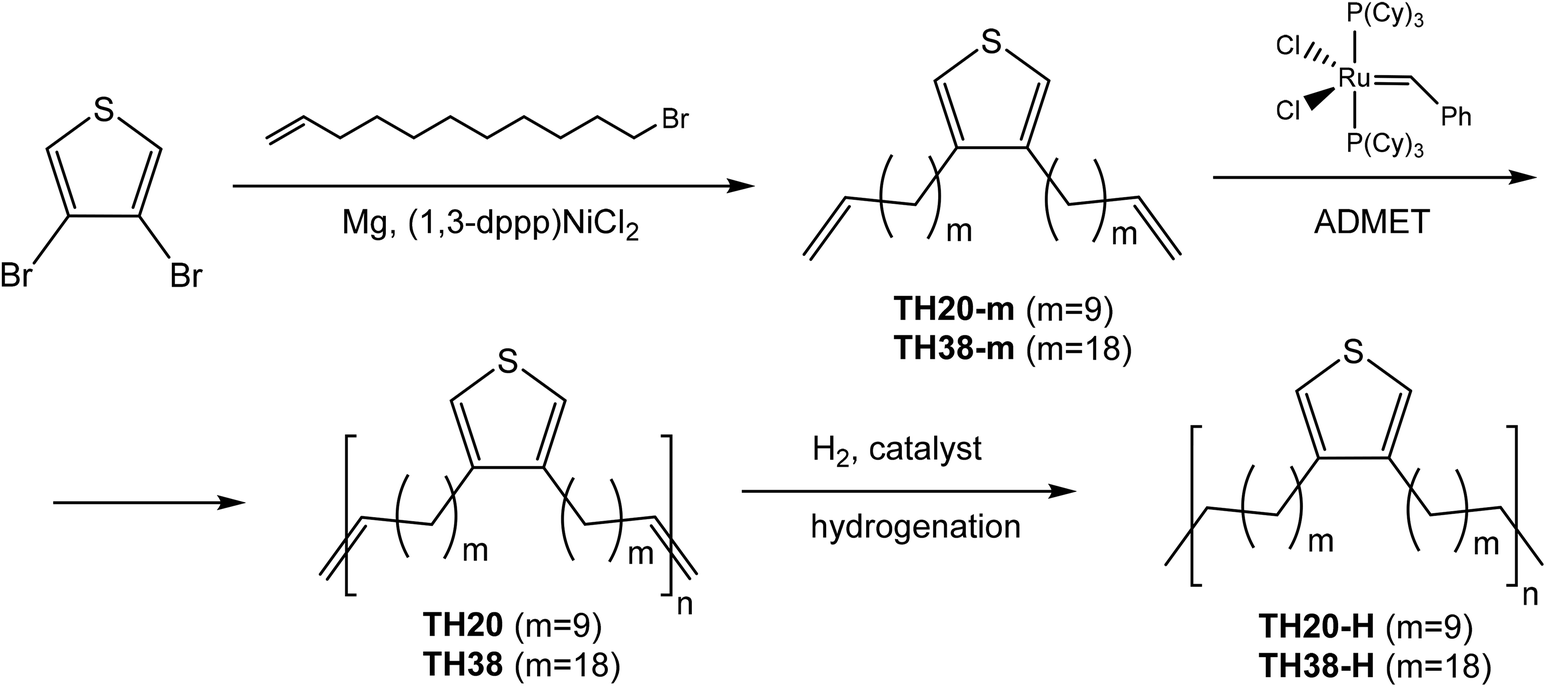 | ||
| Scheme 1 Synthesis of thiophene diene monomers with different lengths of the aliphatic spacers and the ADMET polymerization and hydrogenation. | ||
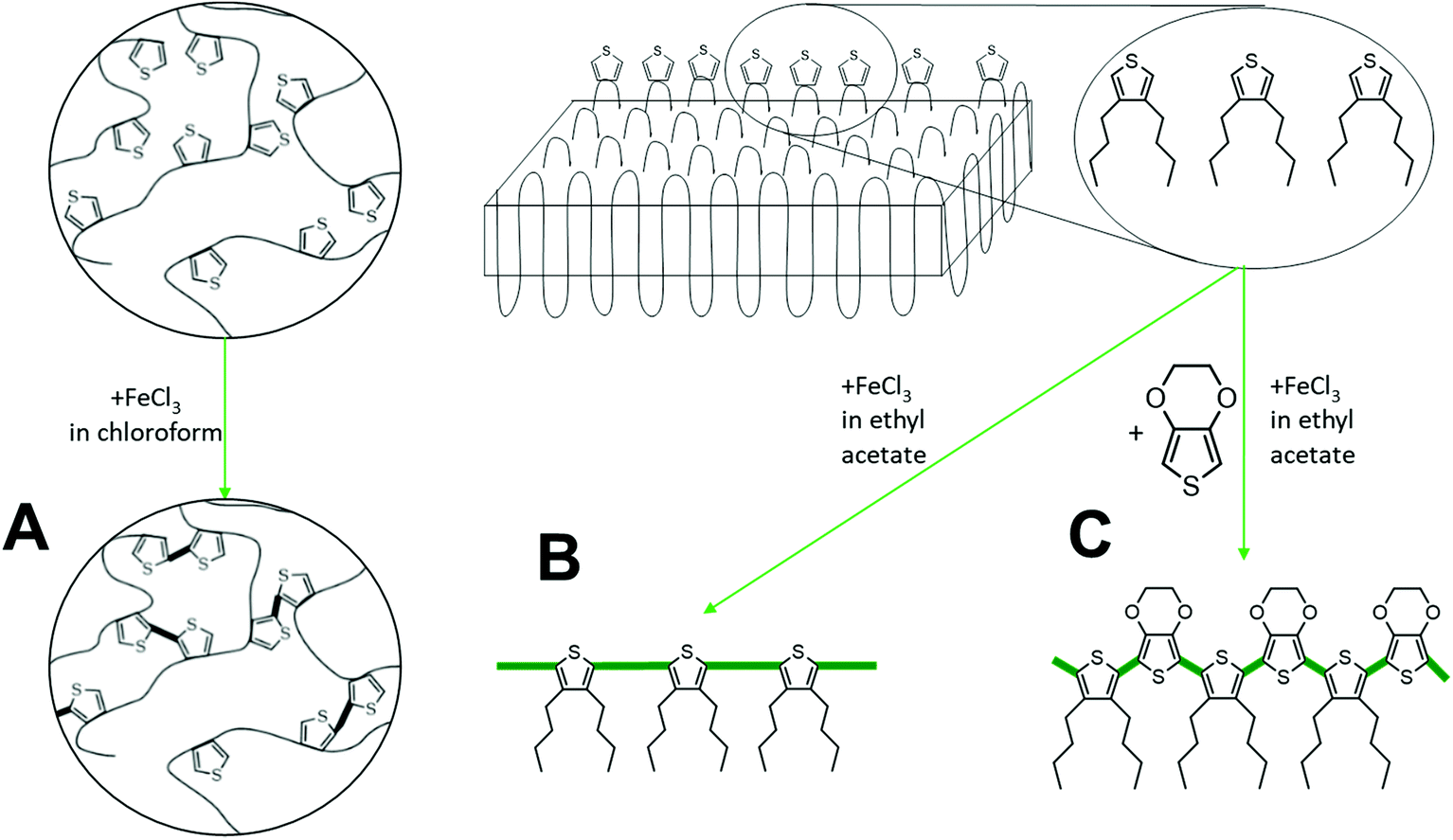 | ||
| Scheme 2 Reaction scheme of the oxidative coupling of TH-38H. The thiophene groups are expelled to the lateral surface of the lamellar polymer crystal. The distance between the thiophene groups is too large to allow charge transport. Accordingly, the thiophene groups need to be polymerized to achieve surface conductivity. The oxidative coupling can be performed in the presence of FeCl3 (A) or with a mixture of FeCl3 and EDOT (B). When the oxidative coupling is performed with dissolved TH-38, insoluble particles precipitate due to uncontrolled crosslinking (C). | ||
Synthesis of Grignard reagent and ADMET diene monomers TH20-m and TH38-m
All reactions have been carried out under an argon atmosphere. Into a flame-dried 25 mL 3-neck round-bottom flask, equipped with reflux and a tube with CaCl2 on the top neck and connected with an argon line on the left neck, magnesium metal pellets were added (0.34 g, 14.2 mmol, 3.45 eq.) and then a magnetic stirrer was used. After 10 min, one iodine crystal and 10 mL of THF were added. The mixture was heated to 40 °C and the first halide of the alkyl bromides, i.e. 11-bromo-1-undecene (1.49 mL, 6.8 mmol, 1.5 eq.) for TH20-m or 18-bromo-1-octadecene (2.25 g, 6.8 mmol, 1.5 eq.) for TH38-m, was added. The reaction mixture slowly changed its color to dark blue, and then the second half of the respective alkyl halide was added. The mixture was allowed to stir until almost the whole magnesium metal pellets disappeared (ca. 30–40 min). After that, a solution of 3,4-dibromothiophene (1) (0.5 mL, 4.5 mmol, 1.0 eq.) in 10 mL THF was added, followed by (1,3-dppp)NiCl2 (0.12 g, 0.2 mmol, 0.05 eq.). The reaction mixture was stirred at room temperature overnight and then poured into a mixture of water and hydrochloric acid. The aqueous layer was extracted with ether twice; combined organic phases were dried over anhydrous MgSO4, filtered and dried under reduced pressure.The product was purified by column chromatography over silica gel eluting with petroleum ether to give 3 as a pale yellow oil. Yield 73% for TH20-m and 75% for TH38-m. 1H NMR TH20-m (Fig. S1 top†) (CDCl3, δ): 6.81 (s, 2H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –S), δ 5.73 (ddt, 2H, H2CC–), 4.94–4.84 (m, 4H, 2CCH–), 2.45–2.40 (t, 4H, C–C2–CH2), 2.00–1.92 (m, 4H, HC2–C2–CH), 1.57–1.52 (m, 4H, C–CH2–C2–CH2), 1.28 (br s, 24H).13C NMR TH20-m (Fig. S2 top†) (CDCl3, ppm): 142.11, 139.25, 119.89, 114.12, 33.82, 29.67–28.82.
–S), δ 5.73 (ddt, 2H, H2CC–), 4.94–4.84 (m, 4H, 2CCH–), 2.45–2.40 (t, 4H, C–C2–CH2), 2.00–1.92 (m, 4H, HC2–C2–CH), 1.57–1.52 (m, 4H, C–CH2–C2–CH2), 1.28 (br s, 24H).13C NMR TH20-m (Fig. S2 top†) (CDCl3, ppm): 142.11, 139.25, 119.89, 114.12, 33.82, 29.67–28.82.
1H NMR TH38-m (Fig. S3 top†) (CDCl3, δ): 6.81 (s, 2H, CC–S), δ 5.73 (ddt, 2H, H2CC–), 4.94–4.84 (m, 4H, 2CCH–), 2.45–2.40 (t, 4H, C–C2–CH2), 1.97–1.88 (m, 4H, HC2–C2–CH), 1.59–1.54 (m, 4H, C–CH2–C2–CH2), 1.28 (br s, 52H). 13C NMR TH-38-m (Fig. S4 top†) (CDCl3, ppm): 142.12, 139.28, 119.87, 114.07, 33.84, 29.71–28.82.
GSMS (EI) for TH20-m (Fig. S5†): calcd for C26H44S: 388.7 (M), found: 388.1 (M). m/z: 789.9 GSMS (EI) for TH38-m (Fig. S8†): calcd for C26H44S: 585.1 (M), found: 585.9 (M). m/z: 663.9, 601.9, 587.9, 586.9, 583.9, 557.9, 335.5
Procedure for ADMET bulk polycondensation of TH20 and TH38
Monomer TH20-m (0.7 g, 1.80 mmol, 1 eq.) or monomer TH38-m (0.6 g, 1.026 mmol, 1 eq.) was charged into a flame-dried 25 mL flask in CH2Cl2 (2 mL) and the solution was stirred for 5 min at room temperature under argon. Then, the 1st generation Grubbs catalyst (0.01 eq.) was added to this solution under an argon atmosphere. After the addition of the catalyst, the reaction mixture was stirred for 1 h at room temperature under argon flow and then evacuated to 5 × 10−2 mbar for 2 h, at 40 °C for 2 h, at 60 °C for 5 h and at 80 °C for 10 h to remove ethylene and remaining solvent. The reaction mixture was cooled to room temperature and the catalyst was terminated by adding ethyl vinyl ether (1 mL in 2 mL of CH2Cl2), followed by 30 min stirring at room temperature. The reaction mixture was precipitated into methanol (100 mL). A brown, highly viscous polymer (4) was obtained after removal of the solvent in vacuo (Yield: 73%). 1H NMR TH20 (Fig. S1 middle†) (CDCl3, δ): 6.81 (s, CC–S), δ 5.33–5.23 (m, CC), 2.44–2.39 (t, C–C2–CH2), 1.96–1.86 (m, HC2–C2–CH), 1.59–1.50 (m, C–CH2–C2–CH2), 1.26 (br s, backbone). 13C NMR TH20 (Fig. S2 middle†) (CDCl3, δ): 142.11, 130.38, 119.89, 33.66, 29.66–28.85. 1H NMR TH38 (Fig. S3 middle†) (CDCl3, δ): 6.81 (s, CC–S), δ 5.32–5.26 (m, CC), 2.45–2.40 (t, C–C2–CH2), 1.97–1.86 (m, HC2–C2–CH), 1.59–1.51 (m, C–CH2–C2–CH2), 1.26 (br s, backbone). 13C NMR TH-38 (Fig. S4 middle†) (CDCl3, ppm): 142.11, 130.36, 119.87, 32.63, 29.72–28.82.
Hydrogenation of polymer TH20
The hydrogenation was performed using a 1st generation Grubbs catalyst modified with ethyl vinyl ether as the catalyst.11 In a glass vessel, TH20 (500 mg) was dissolved in 10 mL THF and argon was bubbled through the solution for 5 min. Upon addition of the catalyst (1% mol), the solution changed its color to orange. The hydrogenation was performed in a 250 mL ROTH autoclave. The system was flushed twice with hydrogen; afterwards the hydrogenation was performed at 40 °C and 70 bar H2 for 2 days. The dark brown solution was concentrated in vacuo before precipitating into cold methanol to yield an off-white solid material (80% yield). 1H NMR (Fig. S1 bottom†) (CDCl3, δ): 6.81 (s, CC–S), 2.45–2.439 (t, C–C2–CH2), 1.59–1.51 (m, C–CH2–C2–CH2), 1.26 (br s, backbone). 13C NMR (Fig. S2 bottom†) (CDCl3, δ): 142.11, 19.86, 29.73–28.82.
Hydrogenation of polymer TH38
The hydrogenation was performed using a 1st generation Grubbs catalyst modified with ethyl vinyl ether as the catalyst. In a glass vessel, TH38 (300 mg) was dissolved in 10 mL toluene and argon was bubbled through the solution for 5 min. Upon addition of the catalyst (1 mol%), the solution changed its color to orange. The hydrogenation was performed in a 250 mL Roth autoclave. The system was flushed twice with hydrogen; afterwards the hydrogenation was performed at 50 °C and 65 bar H2 overnight. The dark brown solution was concentrated in vacuo before precipitating into cold methanol to yield an off-white solid material (80% yield). 1H NMR (Fig. S3 bottom†) (CDCl3, δ): 6.81 (s, CC–S), 2.45–2.40 (t, C–C2–CH2), 1.97–1.86 (m, HC2–C2–CH), 1.59–1.51 (m, C–CH2–C2–CH2), 1.26 (br s, backbone). 13C NMR (Fig. S4 bottom†) (CDCl3, δ): 142.11, 119.87, 29.72–28.82.
Oxidative coupling
All experiments were performed under an argon atmosphere.Results and discussion
Two polyethylene-like polymers carrying precisely spaced thiophene units were prepared by ADMET polymerization. The distance between the thiophene groups was adjusted to either 20 or 38 methylene units, which should influence the thermal properties of the resulting polymers. The monomers TH20-m and TH38-m for ADMET polymerization were prepared according to Scheme 1 in THF with an excess of 11-bromo-1-undecene or 18-bromo-1-octadecene, respectively. After column chromatography, the structure of TH20-m and TH38-m was confirmed by NMR and mass-spectroscopy (Fig. S1 top, S2 top, S3 top, S4 top, Fig. S5 and S8†). Peaks with masses 388.1 and 585.9 are responsible for di-substituted thiophene derivatives. The terminal olefin protons were detected at 4.9 and 5.7 ppm and the terminal olefin carbon signal at 139.3 and 114.1 ppm from 1H and 13C spectra, respectively, indicating that the alkylation reaction was successful.The ADMET polymerization of TH20-m and TH38-m was carried out in the bulk using 1% of the first generation Grubbs catalyst. Instead of terminal double bond multiplets at 4.9 and 5.7 ppm, a new internal double bond multiplet at 5.30 ppm was detected on 1H spectra after ADMET polycondensation (Fig. S1 middle and Fig. S3 middle†). In the 13C NMR spectra, the resonances of the terminal olefins vanished (Fig. S2 middle and Fig. S4 middle†) and a new signal at 130.4 ppm was detected, which was assigned to the internal double bonds of the polymer.
Internal double bonds in TH-20 and TH38 were hydrogenated in the presence of Grubbs catalyst 1st generation modified with ethyl vinyl ether as the catalyst. Structures of successfully hydrogenated polymers TH20-H and TH38-H were confirmed by NMR spectroscopy. Comparison of the polymers before and after hydrogenation reveals the disappearance of the double bond resonances at 5.30 ppm for 1H spectra and at 130.4 ppm for 13C NMR spectra, confirming successful hydrogenation of the polymers (Fig. S1, S2, S3 and S4 bottom†).
Molecular weights of TH20-H and TH38-H were determined by SEC in THF vs. polystyrene standards and were in accordance with the values of the respective unsaturated polymers (Fig. S6 and S9†). The obtained TH20-H and TH38-H revealed an apparent molecular weight Mw of ca. 23400 g mol−1 (by SEC, Mw/Mn = 2.2) for TH20-H and 6300 g mol−1 (by SEC, Mw/Mn = 1.7) for TH38-H. Due to the low solubility of the sample with 38 CH2 units, probably a lower degree of polymerization was achieved.
Solid state characterization
Both the non-hydrogenated and the hydrogenated polymers were able to crystallize, as shown by the DSC thermograms in Fig. 1 and S7.† The hydrogenation increased the melting temperature Tm from −36 to 56 °C for TH20 and from 50 to 77 °C for TH38 as the internal cis-double bonds act as crystallization defects in the unsaturated polymer structures.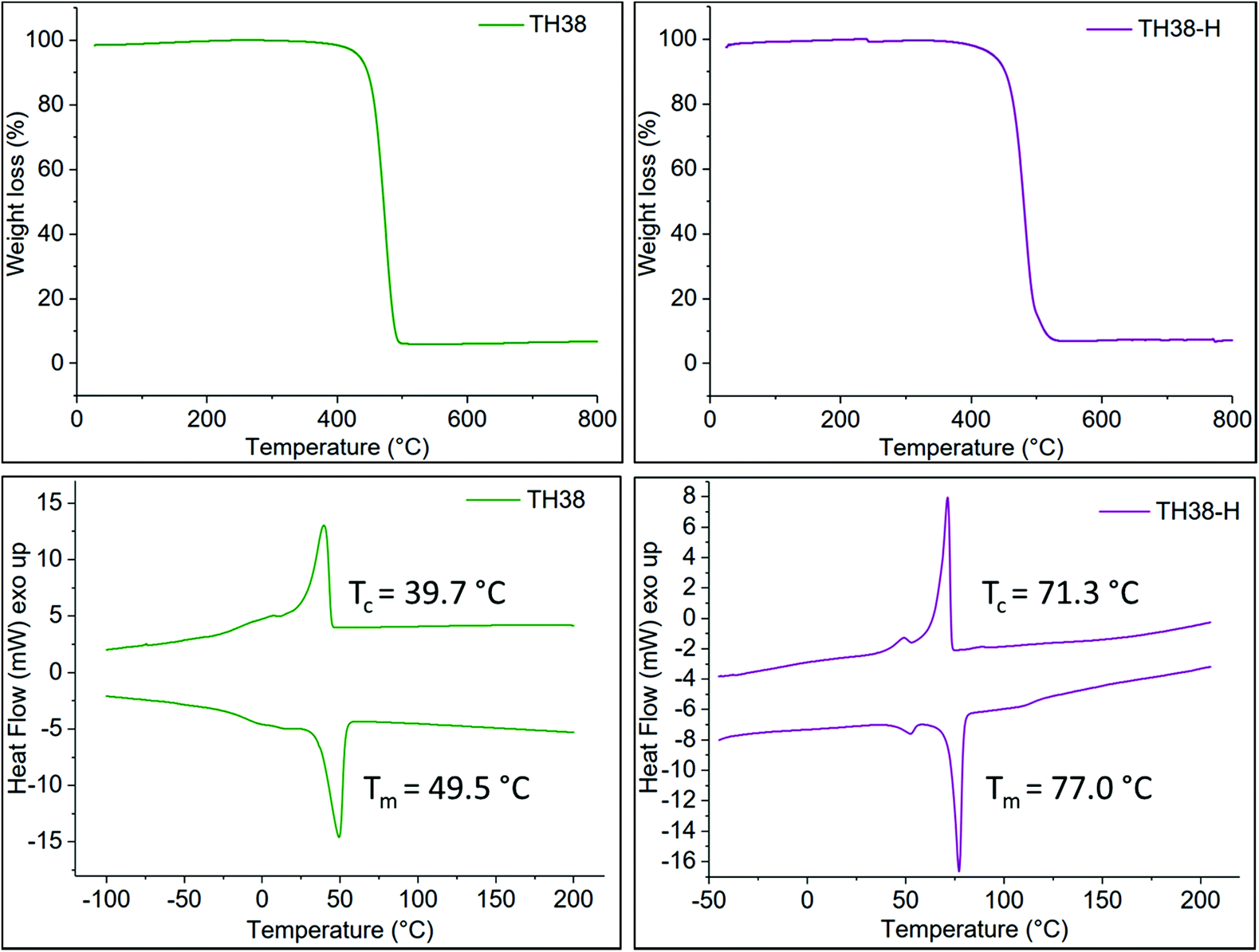 | ||
| Fig. 1 TGA (top) and DSC (bottom) thermograms of unsaturated polymer TH38 (left) and the product after hydrogenation TH38-H (right). The hydrogenation has no significant influence on the decomposition temperature; both materials are thermally stable up to approximately 500 °C. The thermal properties of the crystal, on the other hand, significantly change after the hydrogenation. The melting temperature Tm increases by 27 °C and the crystallization temperature Tc increases by 31 °C. This effect is attributed to the fact that the unsaturated bond in the center of the aliphatic part acts as a defect for the crystallization and hence reduces the melting- and crystallization temperature. | ||
By elongating the aliphatic spacer between the thiophene units, the frequency of crystallization-defects is reduced, which resulted in increased melting temperatures. As the solution crystallization temperature of the TH20-H polymer was relatively low (39 °C), the further oxidative coupling experiments were carried out exclusively with the TH38-H polymer.
The crystal structure of TH38-H lamellar crystals was determined by TEM and WAXS measurements. TEM measurement (Fig. 2A) revealed crystals of truncated lozenge shape. The SAED diffraction pattern proved an orthorhombic structure, with an orthorhombic lattice plane of 4.18 Å for the (110) and 3.75 Å for the (200). This results in the orthorhombic lattice constants of a = 7.5 Å and b = 5.1 Å, which were slightly larger than for conventional polyethylene with a = 7.4 Å and b = 4.9 Å.
 | ||
| Fig. 2 (A) TEM bright-field micrograph and selected area electron diffraction pattern (inset) of a solution grown single crystal of polymer TH38-H from n-octane. The diffraction spots reveal an orthorhombic crystal structure with a lattice distance of 4.18 Å for (110) and 3.75 Å for (200). Assuming an orthorhombic crystal lattice, the lattice constants are a = 7.5 Å and b = 5.1 Å. (B) Wide-angle X-ray scattering of polymer TH38-H corroborates this finding with the two crystal peaks corresponding to a lattice distance of 4.2 Å and 3.8 Å, respectively. | ||
In the WAXS pattern (Fig. 2B) distinct (110) and (200) Bragg reflections at 2θ = 21.2° and 23.3° were observed, which correspond to a lattice distance of 4.2 Å and 3.8 Å, respectively.
Oxidative coupling
To achieve conductive properties, a covalent bond between thiophene groups is necessary to form a conjugated oligomer or polymer.13 For the oxidative coupling of the initial polymer two distinct approaches were chosen – in homogeneous solution or “on-surface” of the 2D nanoplatelets after they had been crystallized from a dilute solution. First, oxidative coupling in solution (Scheme 2A) was performed using anhydrous chloroform as a solvent. This experiment was necessary to check, whether the thiophene groups in the polymer backbone can react with each other despite their relatively low abundance. The polymer was completely soluble in chloroform and during the polymerization agglomeration occurred. After 24 h, a dispersion of black and almost insoluble polymer particles was obtained due to the intra- and intermolecular crosslinking of the thiophene units. Visualization of the dispersion via TEM proved the formation of particles with diameters of ca. 250 nm (Fig. S10B left†). The random crosslinking hinders the formation of continuous conductive layer. Therefore, the polymerized product did not show a significant increase in conductivity and probably only dimers or very short oligomers had been formed during the reaction (Fig. S10C left†). It is well known for thiophene polymers that during polymerization in solution the solubility drastically decreases which practically impedes processing and exploitation of the final product.14 DSC measurement of the randomly cross-linked polymer (Fig. S10D left†) reveals only a minor glass transition temperature peak at 55 °C, which is in good agreement with other polythiophene polymers.15Instead of utilizing the in-chain thiophene groups in solution, we crystallized the polymer prior to the thiophene polymerization from a dilute solution to prepare anisotropic polymer nanoplatelets with expelled thiophene units to their surface as previously proven for structurally similar polyphosphoesters.12 Although the lamellar crystals are supposed to have the thiophene groups at their surface, they were non-conductive (Fig. 3c left column). To achieve conductivity, the thiophene groups need to be covalently bonded, e.g. by polymerization on the crystal surface (Scheme 2).
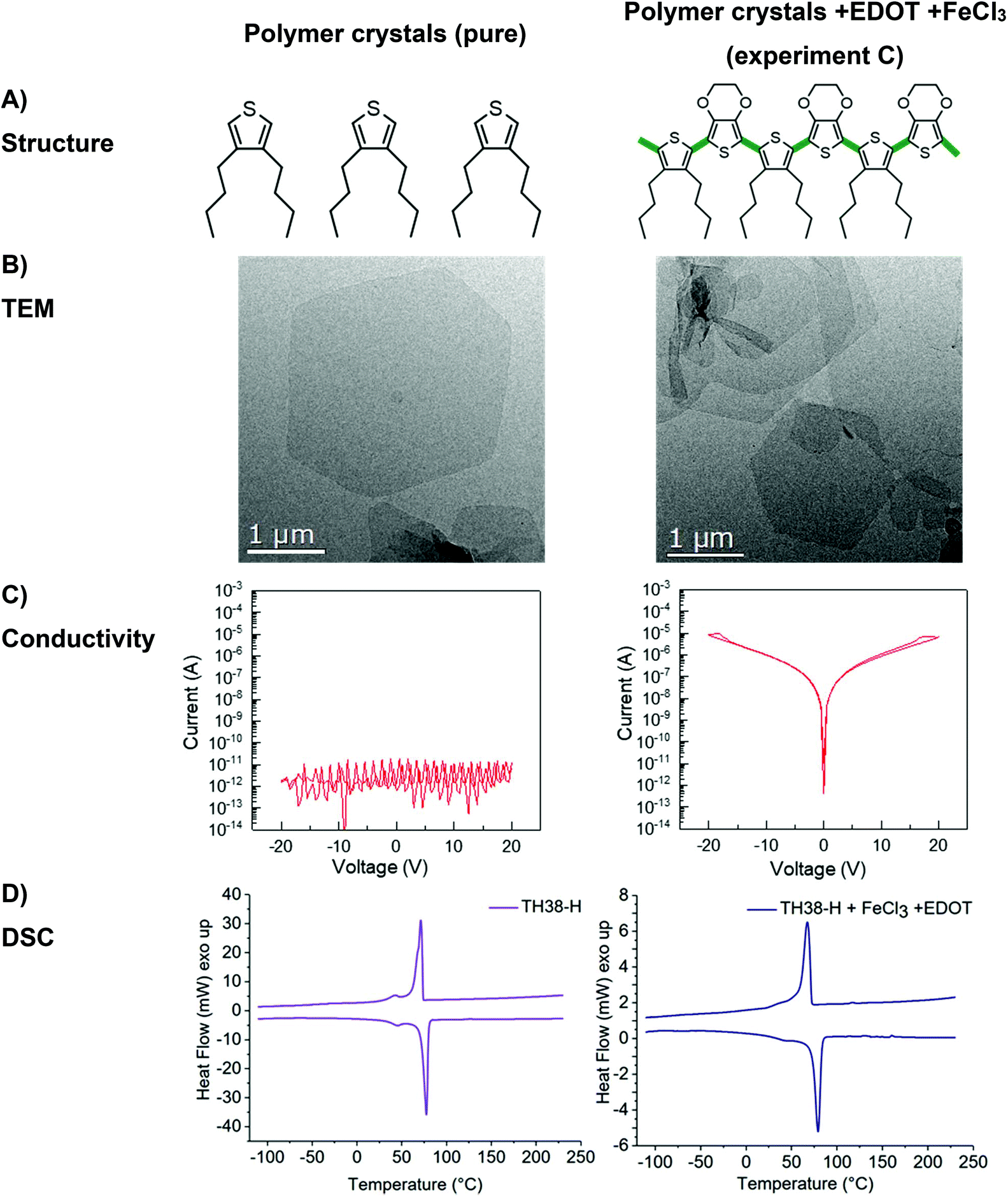 | ||
| Fig. 3 Structure (A), TEM bright-field micrograph (B), conductivity (C) and DSC thermograms (D) of polymer TH38-H before (left) and after oxidative coupling in crystal state with addition of EDOT (right). | ||
For the polymerization of the thiophene groups on the crystal surface, we have to take care that the crystals are not destroyed, e.g. by melting or dissolution during the process. Hence, the oxidative polymerization of the thiophene groups was carried out in cold ethyl acetate, which allowed conserving the polymer crystals, in the presence of FeCl3 as a catalyst (Scheme 2B).16 After 24 h, the color of the dispersion turned light brown, which indicated the partial oxidation of FeCl3. The TEM measurement reveals that the TH-38 crystals stayed intact (Fig. S10B right†), but the electrical conductivity did not show a marked increase as compared to the initial nanoplatelets (Fig. S10C right†). The lack of conductivity after reaction with FeCl3 can be explained by the arrangement of the thiophene groups on the crystal surface. Assuming an orthorhombic crystal structure of the TH-38 (with lattice constants a = 7.5 Å and b = 5.1 Å), the minimal thiophene-to-thiophene distance is 9.1 Å (in (110) direction). Hence, several additional molecules may be necessary to bridge the thiophene units. Thus, it is likely that thiophene groups only react to a low extent in the amorphous parts but could not generate conjugated bonds on the surface of the crystals.
To covalently bond thiophene units on the crystal surface with each other, we added EDOT as a comonomer which should bridge the distance between the thiophene groups on the crystal surface (Scheme 2C). By copolymerization with EDOT as the linker between the thiophenes conductive behavior on the crystal surface should be feasible. Also during the copolymerization special care was taken that the TH-38 nanoplatelets were not destroyed, which was proven by TEM imaging after the polymerization (Fig. 3B right). From the TEM images only nanoplatelets were observed without any additional visible impurities, indicating the successful copolymerization on the surface on the crystals. In addition, the sample proved a substantial increase in conductivity, with a current level of 2.7 × 10−9 A at 1 V and 4.7 × 10−7 A at 20 V. Thus, it is likely that EDOT copolymerized with the thiophene groups “on surface” (Fig. 3C right). As shown in Fig. S11,† the current level was linearly proportional to the applied voltages in the low bias regime, revealing the Ohmic nature of conduction through the poly(EDOT-co-TH-38) surface layer.17 Observation of an Ohmic behavior is indicative of good contact formation between the Au electrodes and the thiophene-EDOT surface of the 2D conductive crystals. Besides the desired copolymerization between the thiophene groups of TH-38 and EDOT, also the homopolymerization of EDOT to form poly(3,4-ethylenedioxythiophene (PEDOT) might occur. After the polymerization, the crystals were purified by centrifugation to remove FeCl3 and PEDOT, which might have formed as a side product. Then, the platelets were visualized by TEM (Fig. 3B); from the TEM imaging, no additional polymer nanoparticles or aggregates could be visualized, which hints that no homopolymer was present after workup. Moreover, no FeCl3 residues were found during TEM examination. These would be visible as small dark spots in the TEM micrographs. In addition, DSC measurements did not show signs of PEDOT (Fig. 3D). In order to prove that the observed conductivity resulted from the formation of bonds between thiophene groups in the polymer chain and EDOT, but not only due to formation of PEDOT, which might even not be detectable by DSC measurements, we performed a control experiment with polymer nanoplatelets carrying no thiophene groups. We prepared similar nanoplatelets using a long-chain polyphosphoester.18 This polyethylene-like polymer carries 20 methylene groups between phosphate units and our previous findings proved that they form very similar nanoplatelets when crystallized from a dilute solution. The nanoplatelets were prepared accordingly and EDOT and FeCl3 were added to initiate the polymerization. As the phosphate units cannot generate a copolymer with EDOT only homopolymerization is possible. After the reaction, we purified the nanoplatelets in the same way and measured their conductivity but, importantly, we did not observe an increased conductivity (Fig. S12†). Together, these experiments prove that we were able to prepare 2D nanoplatelets from polyethylene-like polymers carrying thiophene units on their surface and that conductivity on the crystal surface was achieved by copolymerization with EDOT in dispersion.
Conclusion and outlook
We demonstrated a new method to generate polymer 2D lamellar nanoplatelets with a conductive surface. By crystallizing polyethylene-derivatives from a dilute solution that carry thiophene groups with a precise, equidistant distribution along their backbone, lamellar crystals with a constant thickness and a thiophene surface functionalization were obtained (nanoplatelets). The thiophene groups, expelled from the crystal surface, were copolymerized with EDOT, yielding a conductive surface. This is a new, versatile concept exploiting the crystallization of a polyethylene-derivative with build-in in-chain functional groups to spatially control conductivity on the surface of nanoplatelets. Because the crystallization from solution can be easily controlled, a combination of polymers, doped with different functional groups, can be co-crystallized and will yield a nanostructured polymer crystal with a local control of the chemical groups on the surface. Accordingly, this new method has the potential to serve as a versatile concept to achieve nanostructured surface functionalization, not only for microelectronic applications.Author contributions
OS synthesized the polymers, conducted the investigations and wrote the original draft of the manuscript. BJ and KA performed and analyzed the conductivity measurements. KL supervised the project. FRW supervised the polymer synthesis and contributed to the concept of this work. IL developed the concept of this work and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Max-Planck Society. Our thanks go to Petra Räder and Michael Steiert from the Max Planck Institute for Polymer Research for assistance with the DSC and WAXS measurements. Finally, the authors would like to thank Jasper Michels from the Max Planck Institute for Polymer Research for the stimulating discussion that inspired the idea for this work. Open Access funding provided by the Max Planck Society.References
- K. Yoshino, et al., Electrical and optical properties of conducting polymer-fullerene and conducting polymer-carbon nanotube composites, Fullerene Sci. Technol., 1999, 7, 695–711 CrossRef CAS.
- M. Bajpai, R. Srivastava, R. Dhar and R. S. Tiwari, Review on Optical and Electrical Properties of Conducting Polymers, Indian J. Mater. Sci., 2016, 2016, 1–8 CrossRef.
- M. Kimura, Conductive Polymer Fibers for Sensor Devices, in Handbook of Smart Textiles, 2015, pp. 63–78, DOI:10.1007/978-981-4451-45-1.
- R. D. McCullough, The chemistry of conducting polythiophenes, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- G. Barnes, C. P. Yau and M. Heeney, Poly(thiophene)s, in Encyclopedia of Polymeric Nanomaterials, 2014, pp. 1–15, DOI:10.1007/978-3-642-36199-9_120-1.
- N. Chanunpanich, et al., Grafting polythiophene on polyethylene surfaces, Polym. Int., 2003, 52, 172–178 CrossRef CAS.
- M. D. Mamo, E. S. Shin and Y. Y. Noh, Self-aligned patterning of conductive films on plastic substrates for electrodes of flexible electronics, J. Mater. Chem. C, 2017, 5, 10900–10906 RSC.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Acyclic Diene Metathesis (ADMET) Polymerization, Macromolecules, 1991, 24, 2649–2657 CrossRef CAS.
- N. F. Sauty, L. C. da Silva, M. D. Schulz, C. S. Few and K. B. Wagener, Acyclic diene metathesis polymerization and precision polymers, Appl. Petrochem. Res., 2014, 4, 225–233 CrossRef CAS.
- M. D. Schulz and K. B. Wagener, Precision Polymers through ADMET Polymerization, Macromol. Chem. Phys., 2014, 215, 1936–1945 CrossRef CAS.
- P. Ortmann, F. P. Wimmer and S. Mecking, Long-Spaced Polyketones from ADMET Copolymerizations as Ideal Models for Ethylene/CO Copolymers, ACS Macro Lett., 2015, 4, 704–707 CrossRef CAS.
- T. Haider, et al., Controlling the crystal structure of precisely spaced polyethylene-like polyphosphoesters, Polym. Chem., 2020, 11, 3404–3415 RSC.
- M. Hamzah, E. Saion, A. Kassim, N. Yahya and E. Mahmud, Conjugated Conducting Polymers: A Brief Overview, Springer Ser. Solid-State Sci., 1992, 2, 63–68 Search PubMed.
- A. R. Katritzky, et al., Comprehensive Heterocyclic Chemistry III: A Review of the Literature 1995–2007, Elsevier Science, 2008, DOI:10.1016/C2009-1-28335-3.
- J. Shen, H. Masaoka, K. Tsuchiya and K. Ogino, Synthesis and properties of a novel brush-type copolymers bearing thiophene backbone and 3-(N-carbazolyl)propyl acrylate side chains for light-emitting applications, Polym. J., 2008, 40, 421–427 CrossRef CAS.
- V. M. Niemi, P. Knuuttila, J. E. Österholm and J. Korvola, Polymerization of 3-alkylthiophenes with FeCl3, Polymer, 1992, 33, 1559–1562 CrossRef CAS.
- R. D. Gould, Structure and electrical conduction properties of phthalocyanine thin films, Coord. Chem. Rev., 1996, 156, 237–274 CrossRef CAS.
- Y. R. Zheng, et al., Morphology and Thermal Properties of Precision Polymers: The Crystallization of Butyl Branched Polyethylene and Polyphosphoesters, Macromolecules, 2016, 49, 1321–1330 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00117e |
| This journal is © The Royal Society of Chemistry 2021 |