
Controlled ring-opening polymerization of N-(3-tert-butoxy-3-oxopropyl) glycine derived N-carboxyanhydrides towards well-defined peptoid-based polyacids†
Bailee N.
Barrett‡
,
Garrett L.
Sternhagen‡
and
Donghui
Zhang
 *
*
Department of Chemistry and Macromolecular Studies Group, Louisiana State University, Baton Rouge, Louisiana 70803, USA. E-mail: dhzhang@lsu.edu
First published on 5th January 2021
Abstract
Polypeptoids bearing carboxylic acid groups on the N-substituent are useful building blocks for the construction of peptidomimetic supramolecular assemblies with stimuli-responsive properties. Towards this end, N-(3-tert-butoxy-3-oxopropyl) glycine derived N-carboxyanhydride (tBuO2Pr-NCA) has been successfully synthesized and polymerized using primary amine initiators to produce the corresponding poly(N-(3-tert-butoxy-3-oxopropyl) glycine) with molecular weights (Mn) of 5.6–59 kg mol−1 and a narrow molecular weight distribution (PDI = 1.003–1.026). The polymerization was shown to proceed in a controlled manner, evidenced by the good agreement of the experimental molecular weight (Mn) with theoretical values and narrow molecular weight distribution in a wide range of monomer-to-initiator ratios ([M]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]0 = 25:1–200:1), the linear increase of Mn with conversion and the second-order polymerization kinetics. The cloaked carboxyl groups on the poly(N-(3-tert-butoxy-3-oxopropyl) glycine) can be readily unveiled in mild acidic conditions to yield the poly(N-(2-carboxyethyl) glycine), a structural mimic of poly(glutamic acid). The poly(N-(2-carboxyethyl) glycine) polymer is a weak polyelectrolyte whose hydrodynamic size in water can be controlled by the solution pH.
:[I]0 = 25:1–200:1), the linear increase of Mn with conversion and the second-order polymerization kinetics. The cloaked carboxyl groups on the poly(N-(3-tert-butoxy-3-oxopropyl) glycine) can be readily unveiled in mild acidic conditions to yield the poly(N-(2-carboxyethyl) glycine), a structural mimic of poly(glutamic acid). The poly(N-(2-carboxyethyl) glycine) polymer is a weak polyelectrolyte whose hydrodynamic size in water can be controlled by the solution pH.
Introduction
Biomimetic polymers continue to gain popularity and are prized for their biocompatibility, stimuli-responsive characteristics, and similarity to chemical motifs found in nature.1 Significant progress has been made in both the accessibility of biomimetic polymers and the application of these polymers in fields ranging from sensing and tissue culture, to drug delivery.2 As a structural mimic of polypeptides, N-substituted polyglycines, or polypeptoids, have been increasingly investigated as a new class of biopolymers for different applications.3 The small chemical change in the location of the sidechain from the α-carbon, as in peptides, to the nitrogen atom offers significant synthetic and processing advantages.3 This shift in side-chain location along the polymer backbone eliminates stereogenic centers found in polypeptides and significantly limits the hydrogen bonding interaction along and amongst the polymer backbones relative to the polypeptides.While a range of polypeptoids with differing N-substituents have been successfully synthesized by ring-opening polymerization (ROP) of N-substituted glycine derived N-carboxyanhydrides (R-NCAs)1,3 or N-thiocarboxyanhydride (R-NTA),4 the structural diversity of N-substituents has been largely limited to various hydrocarbons (e.g., alkyl,5–10 aromatic,11,12 allyl,13 propargyl)14,15 and thioether groups.16 While propargyl and allyl N-substituents can be further derivatized to install various functional groups on the polypeptoid polymers post-polymerization, the efficiency and extent of derivation vary with the nature of the functional groups and polymer chain length.13,14,17–19 Thus, it is desirable to design and develop controlled ring-opening polymerization of R-NCA monomers bearing functional N-substituents to enable access to well-defined polypeptoids that have quantitative functional sidechain presence and tailorable polymer chain length.
Poly(L-glutamic acid) and their derivatives have been widely investigated for various biomaterials applications (e.g., drug delivery, tissue engineering, theranostic agents, biosensors, etc.) due to their synthetic tunability20 and similarity to the glutamic acid residues found in natural proteins.21 In nature, glutamic acid residues serve functions in protein structure and stability, as well as for ion and other substrate binding in enzyme catalysis.21,22 Poly(L-glutamic acid γ-esters) are typically obtained by ROP of L-glutamic acid γ-ester derived NCA, and hydrolysis of the polymer affords poly(L-glutamic acid).23,24 Several recent studies have also documented the synthesis of poly(L-glutamic acid γ-ester) by ROP of the less reactive but more hydrolytically stable, L-glutamic acid γ-ester derived N-thiocarboxyanhydride (NTA).25,26 By comparison, poly(N-2-carboxyethyl glycine), the polypeptoid analog of poly(L-glutamic acid), has been investigated as a pH-responsive and water-soluble building block for the construction of sequence-defined peptoid oligomers and their hierarchical supramolecular assemblies in water.27–32 In these studies, the N-2-carboxyethyl glycine segment is incorporated into the oligomeric peptoid chains in a stepwise fashion by the sub-monomer method.27 While this method offers precise monomer sequence control in the synthesis of short-chain peptoids, the sequence control and synthetic efficiency are increasingly limited as the targeted chain length increases. Considering the potential uses of poly(N-2-carboxyethyl glycine) as a building block for the construction of stimuli-responsive and hierarchical assemblies, it is important to develop a controlled polymerization method to access the polymer.
In this contribution, we report the design, synthesis and polymerization of an N-substituted glycine derived NCA bearing a cloaked carboxylic acid functionality, namely N-(3-tert-butoxy-3-oxopropyl) glycine derived N-carboxyanhydride (tBuO2Pr-NCA) to produce the poly(N-(3-tert-butoxy-3-oxopropyl) glycine) polymers. The ring-opening polymerization of tBuO2Pr-NCA in toluene was found to proceed rapidly in a controlled manner using primary amine initiators, yielding the corresponding well-defined polypeptoids with tailorable molecular weight and narrow molecular weight distribution. The tert-butyl ester group on the N-substituents can be efficiently deprotected under mild conditions to produce the corresponding pH-responsive peptoid-based polyacids, a structural mimic of poly(L-glutamic acid).
Experimental
Materials
Solvents were all HPLC grade and used as received. Dichloromethane, chloroform, ethyl acetate, ethanol, tetrahydrofuran, toluene, and trifluoroacetic acid were purchased from Fisher Chemical. Hexanes were purchased from Macron Fine Chemicals. Glycine ethyl ester HCl (99%) and triethylamine (99+%) were purchased from Alfa Aesar. tert-Butyl acrylate (>98.0%) was purchased from TCI. Di-tert-butyl pyrocarbonate (99.5%) was purchased from Chem-Impex Int'l Inc. Hydrochloric acid aq. (37%) and potassium carbonate (ACS grade) were purchased from VWR. Phosphorous trichloride (98%) was purchased from Beantown Chemical. Benzylamine (99%) and butylamine (99%) were purchased from Sigma Aldrich. Deuterated NMR solvents CDCl3, DMSO-d6, and Tol-d8 were purchased from Cambridge Isotope Laboratories.Spectroscopic characterization
1H and 13C{1H} NMR spectra were obtained using a Bruker AV-400 Nanobay spectrometer (400 MHz for 1H NMR and 100 MHz for 13C{1H} NMR) and a Bruker AV-500 spectrometer (500 MHz for 1H NMR and 125 MHz for 13C{1H} NMR) at 298 K. Chemical shifts (δ) given in parts per million (ppm) were referenced to protio impurities or the 13C isotopes of deuterated solvents. FTIR spectra were recorded on a Bruker ALPHA II FTIR spectrometer equipped with Platinum ATR. Data were processed using OPUS v7.2 software.Size-exclusion chromatography (SEC)
SEC analysis was performed using a Tosoh Bioscience EcoSEC Elite system (Tosoh Bioscience degasser, isocratic pump, autosampler, and column heater) equipped with two TSKgel Alpha-M 13 μm, 7.8 mm ID × 30 cm columns, a Tosoh Bioscience dual flow RI detector with a 630–670 nm LED light source, and a Tosoh Bioscience LenS3 multiangle light scattering (MALS) detector (30 mW diode laser at λ = 505 nm). HFIP with 3 mg mL−1 CF3CO2K was used as the eluent at a flow rate of 0.450 mL min−1. The pump housing, column oven, and RI detector temperatures were set at 40 °C. All data analysis was performed using SECview software. Polymer molecular weight and molecular weight distribution were obtained by analyzing the RALS-DRI data based on the LS and RI instrument constants that were calibrated with a PMMA standard (Mw(LS) = 32350 g mol−1, PDI = 1.03) in HFIP/CF3CO2K (3 mg mL−1) with known concentration. The refractive index increment (dn/dc) of the polymer was determined to be 0.183 mL g−1 in HFIP/CF3CO2K (3 mg mL−1) at 40 °C.
Matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (MALDI-TOF MS)
MALDI-TOF MS measurements were conducted on a Bruker ultrafleXtreme tandem time-of-flight (TOF) mass spectrometer equipped with a smartbeam-II™ 1000 Hz laser (Bruker Daltonics, Billerica, MA). The instrument was calibrated with Peptide Calibration Standard II (Bruker Daltonics, Billerica, MA). A saturated solution of α-cyano-4-hydroxycinnamic acid (CHCA) in methanol was used as the matrix. The polymer solution samples (10 mg mL−1 in methanol) were mixed with the saturated matrix solutions at 1:1 volume ratio. The mixtures (1 μL) were deposited onto a 384-well ground-steel sample plate and dried in air prior to measurement using positive reflector mode. Data analysis was carried out using FlexAnalysis software.
Electrospray ionization mass spectrometry (ESI MS)
An Agilent 1260 Infinity II quaternary liquid chromatograph coupled to an Agilent 6230 Electrospray Time-of-Flight mass spectrometer was used for detection of analytes. The samples were run in positive mode ionization with a capillary voltage of 4000 V. Drying gas (nitrogen) temperature was 325 °C delivered at 10 L min−1 and the fragmentor voltage was set to 150 V. No LC column was used for sample delivery, only flow through injection was utilized (direct injection from LC to mass spectrometer). Mobile phases used were A: 30% LCMS grade water with 0.1% formic acid and B: 70% LCMS grade acetonitrile with 0.1% formic acid with a flow rate of 0.4 mL min−1.Dynamic light scattering (DLS)
DLS measurements were conducted on a Wyatt Dawn Heleos-II using a laser wavelength of 660 nm at a temperature of 25 °C. The acidic polymer solution (3 mg mL−1) was obtained by direct dissolution of poly(N-(2-carboxyethyl) glycine) (Mn (NMR) = 23 kg mol−1) in pre-boiled ultrapure water at room temperature. The basic polymer solution was obtained by adding one equivalent NaOH aqueous solution (2.5 M). The solution samples were then filtered through polyethersulfone (PES) syringe filters (0.45 μm) into clean scintillation vials before measurement. The correlation functions were fitted using the maximum entropy method (MEM)33 to calculate the diffusion coefficient and hydrodynamic radius (Rh). Maximum Entropy analysis was performed using the Clementine (v1.2) package for Igor Pro (v6.37). The fitting resulted in a distribution of decay times (eqn (1)) which were converted to diffusion coefficients using eqn (2) and hydrodynamic radii using the Stokes–Einstein relationship (eqn (3)). The hydrodynamic size distribution was fitted with a lognormal distribution function and sizes were reported as the distribution mean.| G1(t) = exp−Γt | (1) |
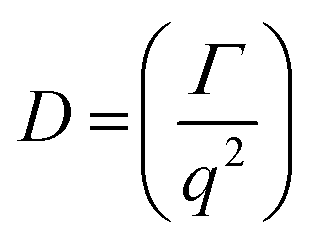 | (2) |
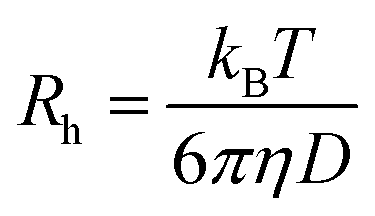 | (3) |
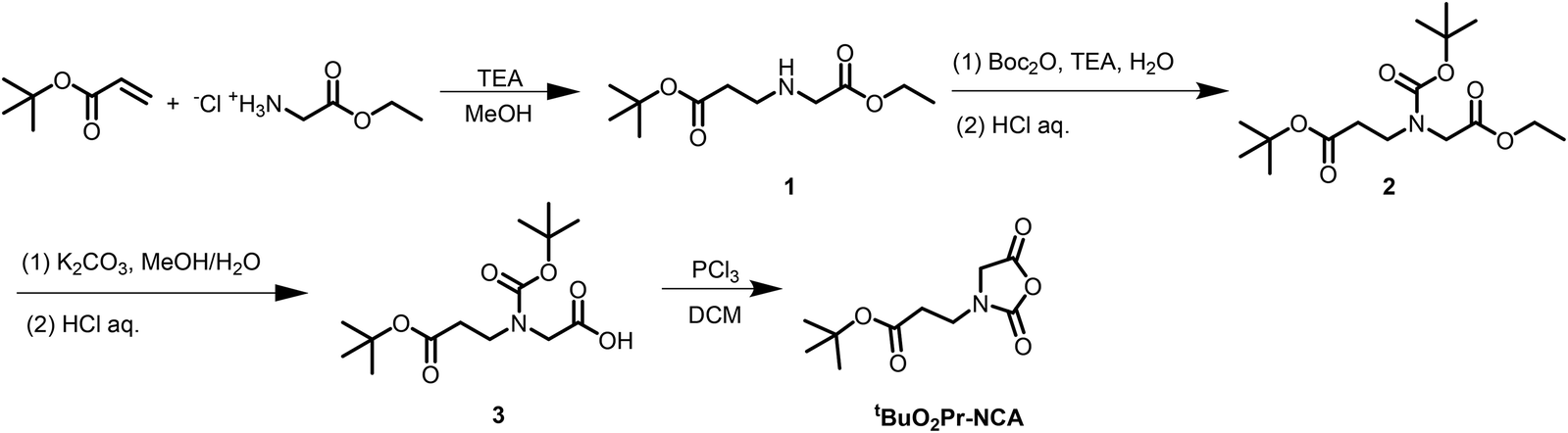
Synthesis of 2-((3-tert-butoxy-3-oxopropyl)amino) acetic acid (1, Scheme 1)
Glycine ethyl ester HCl salt (14.53 g, 62.74 mmol) was dissolved in methanol (0.5 M) with tert-butyl acrylate (9.10 mL, 62.4 mmol) and triethylamine (26 mL, 190 mmol). Reaction was allowed to stir for 12 h and then concentrated under vacuum. The residue was mixed with hexane and filtered. Filtrate was concentrated in vacuum to yield the product as a colorless to pale yellow oil (12.24 g, 52.92 mmol, 85% yield). 1H NMR (δ in CDCl3, ppm): 1.28 (t, –COCH2CH3); 1.45 (s, (CH3)3CCOOCH2CH2–); 1.99 (s, –NH–); 2.43 (t, (CH3)3CCOOCH2CH2–); 2.85 (t, (CH3)3CCOOCH2CH2–); 3.40 (s, –COCH2–); 4.19 (q, –COCH2CH3). 13C{1H} NMR (δ in CDCl3, ppm): 14.36 (–COCH2CH3); 28.24 ((CH3)3CCOOCH2CH2–); 36.11 ((CH3)3CCOOCH2CH2–); 45.11 ((CH3)3CCOOCH2CH2–); 51.04 (–COCH2–); 60.86 (–COCH2CH3); 80.73 ((CH3)3CCOOCH2CH2–); 171.9 ((CH3)3CCOOCH2CH2–); 172.3 (–COCH2–).Synthesis of 2-(N-(tert-butoxycarbonyl)-N-(3-tert-butoxy-3-oxopropyl) amino) acetic acid (3, Scheme 1)
2-((3-tert-Butoxy-3-oxopropyl)amino) acetic acid 1 (8.83 g, 38.7 mmol, 1 M) was stirred with di-tert-butyl dicarbonate (8.30 g, 38.0 mmol) in CH2Cl2 (39 mL) for 12 h. The reaction mixture was concentrated under vacuum and re-dissolved in ethyl acetate. Organic layer was washed with 1 M HCl aq. (3 × 100 mL) and then brine (1 × 100 mL). Organic layer was concentrated to yield the compound 2 (Scheme 1) as a colorless oil. The oil was stirred with K2CO3 (10.50 g, 76.0 mmol) in MeOH:DI H2O (1:1 v/v) for 24 h. Solution was acidified to pH 2 with HCl aq. (4 M) and extracted with ethyl acetate (3 × 150 mL). Organic extracts were combined and concentrated to yield the product as a colorless oil (8.98 g, 29.6 mmol, 78% yield). 1H NMR (δ in CDCl3, ppm): 1.44 (s, (CH3)3CCOOCH2CH2–); 1.48 (s, –NCOOC(CH3)3); 2.54 (t, (CH3)3CCOOCH2CH2–); 3.52 (t, (CH3)3CCOOCH2CH2–); 4.01 and 4.07 (s, –COCH2–); 9.64 (s, –COOH). 13C{1H} NMR (δ in CDCl3, ppm): 28.23 ((CH3)3CCOOCH2CH2– and (CH3)3CCOON–); 35.23 ((CH3)3CCOOCH2CH2–); 44.98 ((CH3)3CCOOCH2CH2–); 50.33 (–COCH2–); 81.04 ((CH3)3CCOOCH2CH2– and (CH3)3CCOON–); 155.56 ((CH3)3CCOON–); 171.9 ((CH3)3CCOOCH2CH2); 175.6 (–COOH).
 | ||
| Scheme 1 | ||
Synthesis of N-(3-tert-butoxy-3-oxopropyl) glycine derived N-carboxyanhydride (tBuO2Pr-NCA) (4, Scheme 1)
PCl3 (3.0 mL, 34 mmol) was added into an anhydrous CH2Cl2 solution of 2-(N-(tert-butoxycarbonyl)-N-(3-tert-butoxy-3-oxopropyl) amino) acetic acid 3 (11.7 g, 38.6 mmol, 0.25 M) under nitrogen flow at room temperature. The solution was stirred for 4 h and then concentrated under vacuum. The residue was filtered through a silica plug with DCM. The filtrate was concentrated to yield the product as a pale yellow oil (2.89 g, 12.6 mmol, 33% yield). Note that the monomer can also be purified by vacuum distillation (70 °C, 40 mTorr). 1H NMR (δ in CDCl3, ppm): 1.46 (s, (CH3)3CCOOCH2CH2–); 2.60 (t, (CH3)3CCOOCH2CH2–); 3.64 (t, (CH3)3CCOOCH2CH2–); 4.24 (s, –COCH2–). 13C{1H} NMR (δ in CDCl3, ppm): 28.19 ((CH3)3CCOOCH2CH2–); 33.94 ((CH3)3CCOOCH2CH2–); 39.84 ((CH3)3CCOOCH2CH2–); 50.34 (–COCH2–); 82.03 ((CH3)3CCOOCH2CH2–); 152.3 (–NCOO–); 165.7 (–COCH2–); 170.7 ((CH3)3CCOOCH2CH2–).Synthesis of poly(N-(3-tert-butoxy-3-oxopropyl) glycine) by ROP of tBuO2Pr-NCA
In a typical polymerization, tBuO2Pr-NCA (4, Scheme 1) (56.1 mg, 0.245 mmol) was dissolved in toluene (0.5 M). A known volume of benzylamine/toluene stock solution (6.2 μL, 1.2 μmol, 0.2 M, [M]0:[I]0 = 200:1) was added to the monomer solution. The reaction mixture was stirred at 25 °C and conversion was monitored by 1H NMR spectroscopy. Upon observation of full monomer conversion, the solution was concentrated under vacuum to yield a sticky residue (41.8 mg, 92% yield). 1H NMR (δ in DMSO-d6, ppm): 1.37 (b, (CH3)3CCOOCH2CH2–); 2.41 (b, (CH3)3CCOOCH2CH2–); 3.41 (b, (CH3)3CCOOCH2CH2–); 3.70–4.90 (b, COCH2–); 13C{1H} NMR tabulated data (δ in DMSO-d6, ppm): 27.67 ((CH3)3CCOOCH2CH2–); 33.08 ((CH3)3CCOOCH2CH2–); 44.06 ((CH3)3CCOOCH2CH2–); 48.93 (–COCH2–); 79.75 ((CH3)3CCOOCH2CH2–); 168.4 (–COCH2–); 170.4 ((CH3)3CCOOCH2CH2–).
Synthesis of poly(N-(2-carboxyethyl) glycine)
A representative procedure is given as follows. Poly(N-(3-tert-butoxy-3-oxopropyl) glycine) (56.1 mg, 0.30 mmol repeating units, Mn (SEC) = 35 kg mol−1, PDI = 1.08) was dissolved in a trifluoroacetic acid (TFA)/chloroform solution (0.565 mL, 1.50 mmol, 20 vol%) and stirred for 24 h at room temperature. Evaporation of the volatiles under a nitrogen flow afforded a sticky solid residue, which was further triturated by stirring in THF at room temperature. The evaporation of THF afforded a white solid (29.7 mg, 76.0% yield). 1H NMR (δ in DMSO-d6, ppm): 2.45 (b, COOHCH2CH2–); 3.43 (b, COOHCH2CH2–); 3.75–4.75 (b, COCH2–); 13C{1H} NMR (δ in DMSO-d6, ppm): 32.18 (COOCH2CH2–); 44.13 (COOCH2CH2-); 48.91 (–COCH2–); 169.0 (–COCH2–); 172.9 (COOHCH2CH2–).Kinetic studies of ROP of tBuO2Pr-NCA
A representative polymerization is given as follows. A known volume of n-butyl amine/tol-d8 stock solution (37.5 μL, 3.75 μmol, 0.1 M) was added to a Tol-d8 solution of tBuO2Pr-NCA (89.4 mg, 0.390 mmol, 0.5 M) at room temperature. The time was recorded as t = 0. The reaction mixture was transferred to an NMR tube, and the reaction was monitored by 1H NMR spectroscopy until full conversion was reached. Reactions were repeated with varying [M]0:[I]0 ratios of 25:1, 50:1, 100:1 and 200:1. Each kinetics measurement was repeated at least 3 times to obtain the mean observed rate constant (kobs) and the standard deviation.
Results and discussion
N-(3-tert-Butoxy-3-oxopropyl) glycine-derived N-carboxyanhydride (tBuO2Pr-NCA) has been synthesized in multi-gram scale in four steps by the Leuch method (Scheme 1). Briefly, 2-((3-tert-butoxy-3-oxopropyl)amino) acetic acid 1 was obtained by Michael addition of tert-butyl acrylate with ethyl glycinate. N-Protection of 1 with di-tert-butyl dicarbonate (Boc2O) afforded 2 which carries two different ester groups, one of which can be selectively hydrolyzed under a basic condition to yield 3. PCl3-mediated cyclization of 3 afforded the desired product tBuO2Pr-NCA in quantitative conversion. Purification of tBuO2Pr-NCA by filtration through a silica plug or vacuum distillation yielded a yellow oil which was used in the subsequent polymerization studies. 1H NMR and 13C{1H} NMR spectroscopic analysis of the yellow oil supports the successful synthesis of tBuO2Pr-NCA (Fig. 1A and S3†).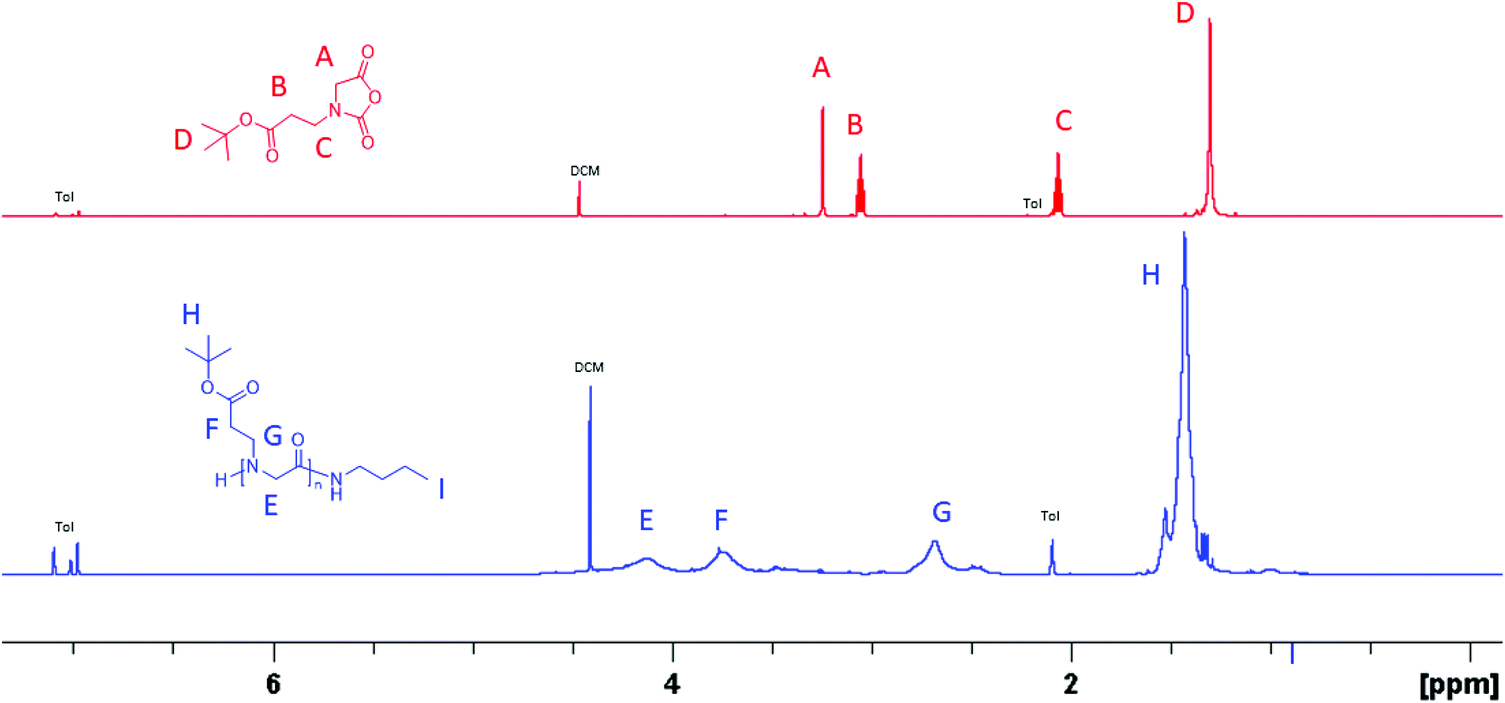 | ||
| Fig. 1 (A) 1H NMR spectra of tBuO2Pr-NCA and (B) poly(N-(3-tert-butoxy-3-oxopropyl) glycine) in toluene-d8 that were obtained by the butylamine-initiated ROPs of tBuO2Pr-NCA. | ||
Polymerizations of tBuO2Pr-NCA were conducted using primary amine initiators (i.e., benzylamine or butylamine) in toluene at room temperature (ca. 22 °C) with a constant initial monomer concentration ([M]0 = 0.5 M) and varying initial monomer-to-initiator ratio ([M]0:[I]0 = 25:1–400:1). All reactions reached quantitative conversion after 24 h. 1H and 13C{1H} NMR analysis of the resulting products supported the formation of the desired polypeptoids, namely poly(N-(3-tert-butoxy-3-oxopropyl) glycine) (Fig. 1B and Fig. S4†). MALDI-TOF MS analysis of a low molecular weight polymer sample revealed the presence of a major envelope of mass ions whose mass corresponds to the summation of n integer of 185.11, 73.09 and 1.01 (H+)/22.99(Na+)/38.96(K+) (Fig. 2). This is consistent with the formation of desired poly(N-(3-tert-butoxy-3-oxopropyl) glycine) polymer bearing a butylamide and a secondary amino chain end, in accord with the expected normal amine mechanism for the ROP of NCAs.1,34,35 In addition, mass ions consistent with poly(N-(3-tert-butoxy-3-oxopropyl) glycine) bearing one or two N-2-carboxyethyl glycine repeating units are also discernable, which is tentatively attributed to the partial hydrolysis of the carboxyl ester group on the N-substituent during MALDI-TOF MS sample preparation or laser induced fragmentation.
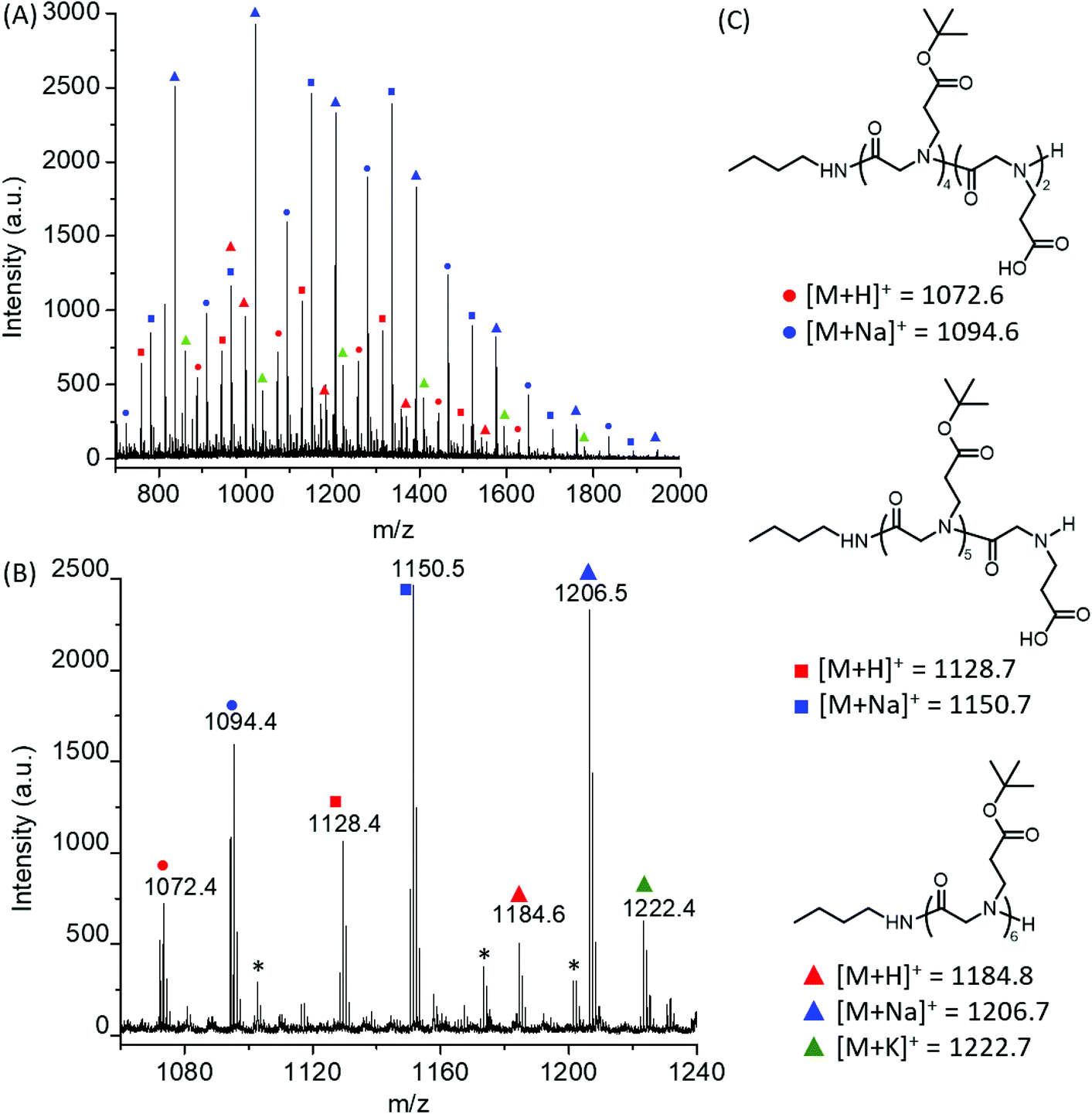 | ||
| Fig. 2 (A) Full and (B) expanded MALDI-TOF MS spectrum of a low molecular weight poly(N-(3-tert-butoxy-3-oxopropyl) glycine) obtained from the butylamine-initiated ROPs of tBuO2Pr-NCA in toluene together with (C) the structural assignment of the molecular ions and corresponding calculated m/z values. * (m/z = 1101.7, 1173.4 and 1201.4) correspond to poly(N-(3-tert-butoxy-3-oxopropyl) glycine) species whose end-group structures are presently unclear. | ||
All poly(N-(3-tert-butoxy-3-oxopropyl) glycine) polymers have been characterized by size-exclusion chromatography (SEC) with tandem differential refractive index (DRI) and multi-angle light scattering (MALS) detectors in HFIP/CF3CO2K (3 mg mL−1) at 40 °C. SEC-MALS-DRI analysis of all polymer samples revealed a monomodal molecular weight distribution (Fig. 3A and S6, S7†), which systematically shifts to lower elution volume as the [M]0:[I]0 ratio is increased, indicating a larger hydrodynamic volume and corresponding higher polymer molecular weight. The polymer molecular weight (Mn) can be systematically adjusted between 5.6–59 kg mol−1 with narrow molecular weight distribution (PDI = 1.003–1.026) by varying the monomer-to-initiator feed ratio ([M]0:[I]0, Table 1). The experimental molecular weights [Mn (SEC)] determined by SEC-MALS-DRI analysis and end-group analysis using 1H NMR spectroscopy [Mn (NMR)] are consistent with one another and agree reasonably well with the theoretical values based on living polymerization of tBuO2Pr-NCA by benzylamine initiator alone up to the [M]0:[I]0 ratio of 200:1 (Fig. 3B). At higher [M]0:[I]0 ratio (i.e., 400:1), the experimental molecular weight was found to be lower than the theoretical value, which is tentatively attributed to presence of unintended nucleophilic impurities that can initiate the polymerization of tBuO2Pr-NCA in additional to the primary amine initiator (entry 5, Table 1). In addition, by monitoring the progression of a butylamine-initiated polymerization of tBuO2Pr-NCA ([M]0:[I]0 = 100:1), the Mn of the forming polymers was found to increase linearly with conversion, and the PDI remains low throughout the course of the polymerization (Fig. 3C, D and Fig. S7†). The Mnversus conversion plot did not pass through (0,0), which suggests an initiation comparable in rate relative to propagation for the butylamine-initiated polymerization of tBuO2Pr-NCA in toluene.
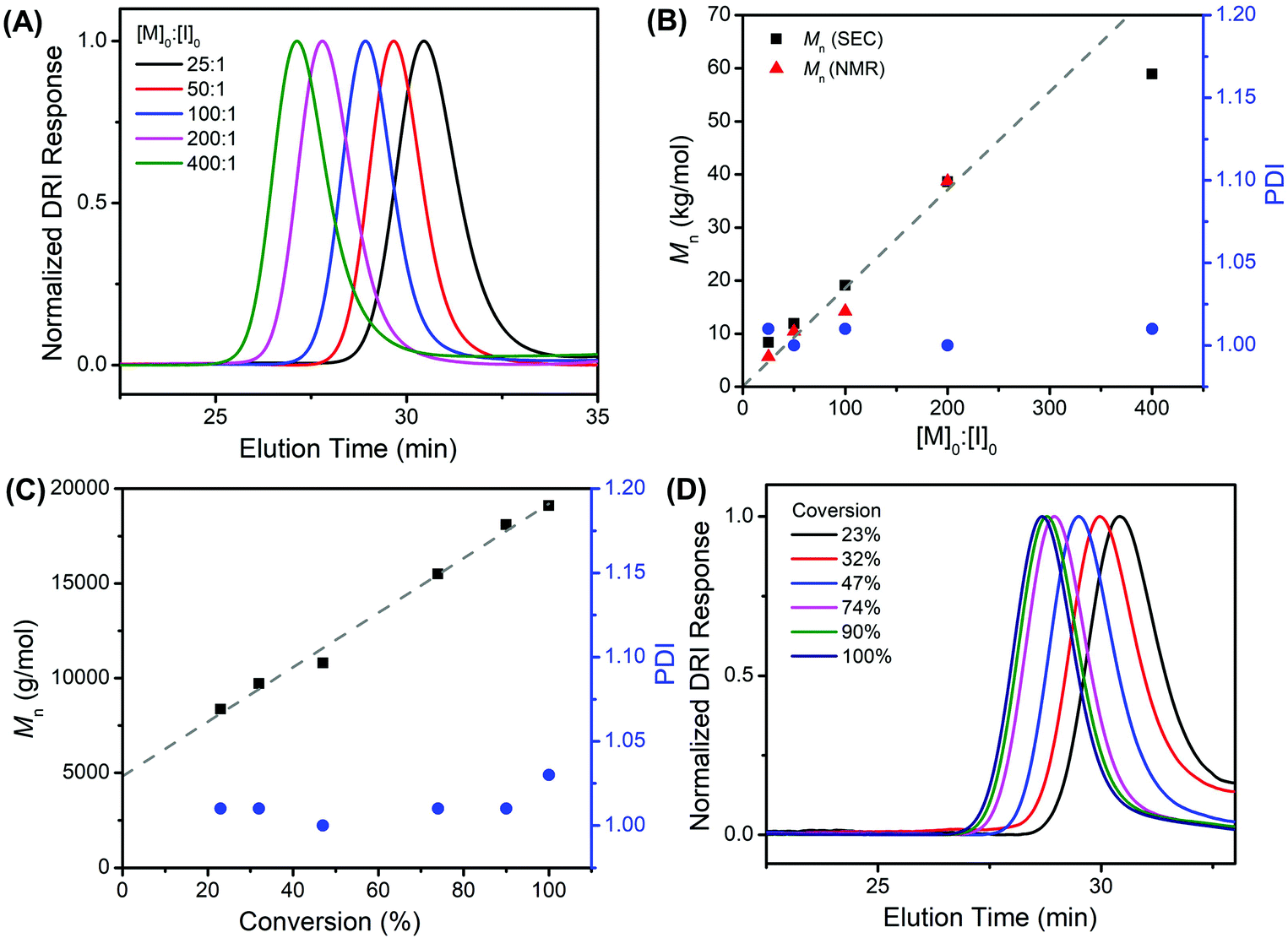 | ||
Fig. 3 (A) Plots of the SEC-DRI chromatograms and (B) the corresponding experimental molecular weights [Mn (SEC) (■), Mn (NMR) ( ) and Mn (theo.) ( ) and Mn (theo.) ( )] and PDI ( )] and PDI ( ) versus the initial monomer-to-initiator ratio (i.e., [M]0:[I]0 = 25:1–400:1), (C) Mn (SEC) (■) and PDI ( ) versus the initial monomer-to-initiator ratio (i.e., [M]0:[I]0 = 25:1–400:1), (C) Mn (SEC) (■) and PDI ( ) versus conversion ([M]0 = 0.5 M, [M]0:[I]0 = 100:1), the linear fit of Mnvs. conversion data ( ) versus conversion ([M]0 = 0.5 M, [M]0:[I]0 = 100:1), the linear fit of Mnvs. conversion data ( ) and (D) the corresponding SEC-DRI chromatograms of polymers formed at different conversion for the primary amine-initiated ROPs of tBuO2Pr-NCA in toluene at room temperature. ) and (D) the corresponding SEC-DRI chromatograms of polymers formed at different conversion for the primary amine-initiated ROPs of tBuO2Pr-NCA in toluene at room temperature. | ||
| Entry # | Initiator | [M]0:[I]0 |
M n (theo.)b (g mol−1) | M n(NMR) c (g mol−1) | M n(SEC)d (g mol−1) | PDId | Conv.e (%) |
|---|---|---|---|---|---|---|---|
|
a All polymerizations proceeded for 24 h in toluene at room temperature with a constant initial monomer concentration ([M]0 = 0.5 M).
b Theoretical Mns are calculated using the initial monomer-to-initiator ratio ([M]0:[I]0) and conversion at 24 h.
c Experimental Mns were determined by end-group analysis using 1H NMR spectroscopy.
d Experimental Mns and PDIs were determined by SEC-MALS-DRI analysis using dn/dc = 0.183 ml g−1 in HFIP/CF3CO2K (3 mg mL−1).
e Polymerization conversions were determined by 1H NMR analysis of reaction aliquot at 24 h.
|
|||||||
| 1 | BnNH2 | 25:1 |
4580 | 5630 | 8360 | 1.015 | >99 |
| 2 | BnNH2 | 50:1 |
9060 | 10400 |
11900 |
1.004 | >99 |
| 3 | BnNH2 | 100:1 |
18900 |
14200 |
19100 |
1.006 | >99 |
| 4 | BnNH2 | 200:1 |
36700 |
38700 |
38600 |
1.003 | >99 |
| 5 | BnNH2 | 400:1 |
68900 |
— | 58900 |
1.009 | >99 |
| 6 | BuNH2 | 25:1 |
4700 | 4300 | 5620 | 1.007 | >99 |
| 7 | BuNH2 | 50:1 |
9330 | 8040 | 11500 |
1.003 | >99 |
| 8 | BuNH2 | 100:1 |
18600 |
18800 |
19100 |
1.026 | >99 |
| 9 | BuNH2 | 200:1 |
37100 |
— | 28100 |
1.012 | >99 |
Kinetic studies revealed that the polymerization of tBuO2Pr-NCA using butylamine initiators exhibited a first-order dependence on the monomer concentration and the initiator concentration respectively with a propagation rate constant kp = 181 ± 8 M−1h−1 (kobs = kp[I]0) (Fig. 4). It is rather surprising that the propagation rate constant of tBuO2Pr-NCA is significantly higher than that of N-2-methoxyethyl glycine derived NCA (MeOEt NCA) and N-2-(2′-methoxyethoxy)ethyl glycine derived NCA (Me(OEt)2 NCA) using primary amine initiators in toluene,36 indicating that both steric and electronic characteristics of the N-substituent influence the relative rate of propagation.6 These combined results strongly support that the primary amine-initiated ROPs of tBuO2Pr-NCA proceeds in a living manner.
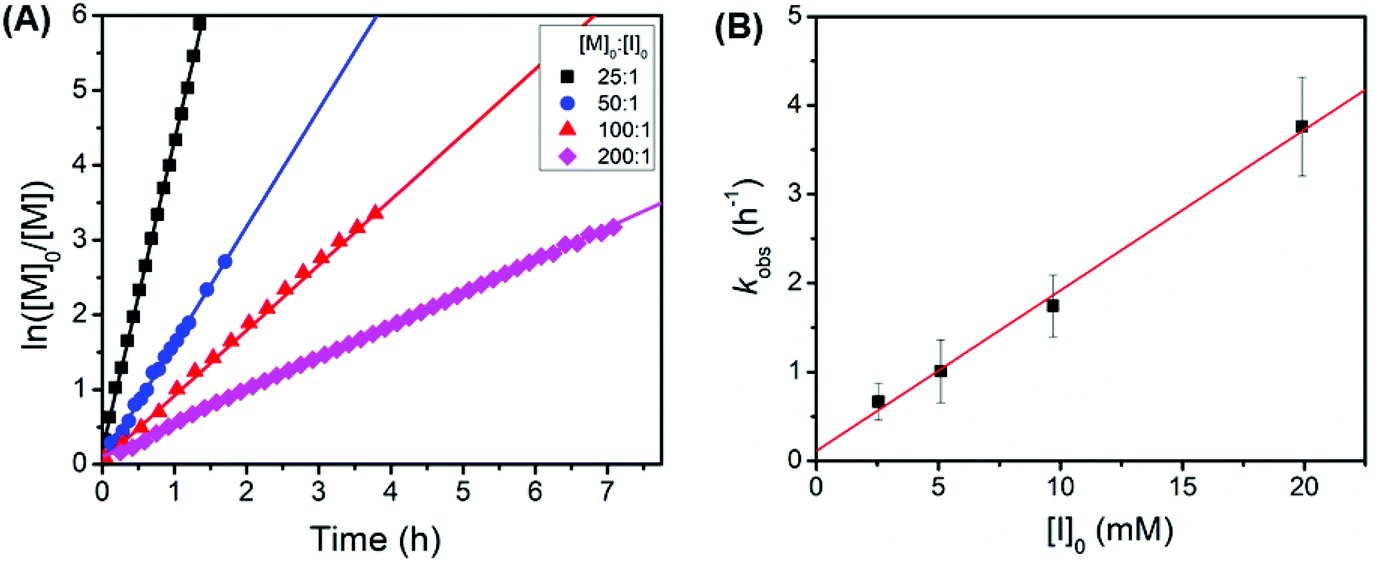 | ||
Fig. 4 (A) Plots of ln([M]0:[M]t) versus time butylamine-initiated ROPs of tBuO2Pr-NCA in toluene at room temperature with a constant initial monomer concentration and varying initiator concentration (i.e., [M]0 = 0.5 M, [M]0:[I]0 = 25:1, 50:1, 100:1, and 200:1), and (B) the pseudo-first order observed polymerization rate constant (kobs, ■) versus the initial initiator concentration ([I]0) and the linear fit ( ) of the data, the slope of which yields the propagation rate constant kp = 181 ± 8 M−1 h−1. ) of the data, the slope of which yields the propagation rate constant kp = 181 ± 8 M−1 h−1. | ||
It should be noted that the alkylene linker length on the N-substituent is important for the controlled polymerization behavior to produce well-defined polypeptoids bearing carboxyl functionality on the sidechain. For example, in contrast to the controlled polymerization behavior observed for tBuO2Pr-NCA, polymerization of EtO2Et-NCA and tBuO2Et-NCA bearing one less methylene unit on the N-substituent relative to tBuO2Pr-NCA using benzyl amine initiators under identical conditions resulted in low conversion (<10%) due to the competitive termination by intramolecular transamidation relative to chain propagation (Scheme 2, Fig. S8†). This mode of termination has also been observed for the polymerization of a N-carboxymethylene glycine derived NTA in THF using a primary amine initiator.37 The suppression of the undesired side reaction in the ROP of tBuO2Pr-NCA has been attributed to the energetically unfavored 7-member rings that would form by the intramolecular transamidation.
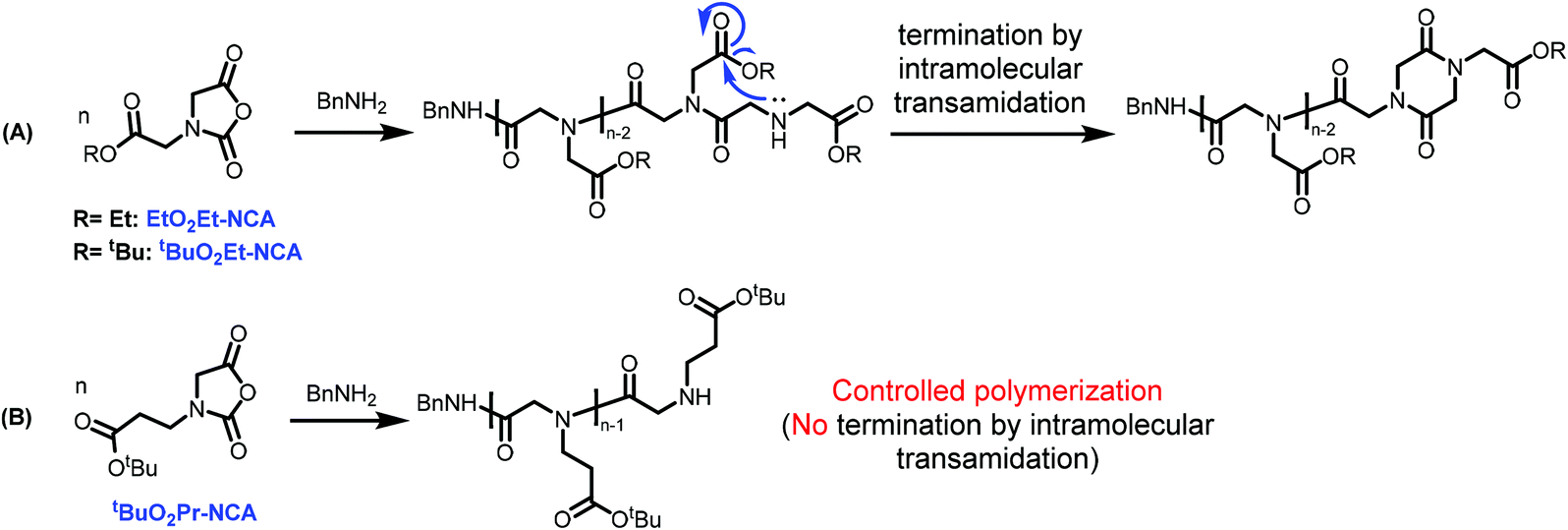 | ||
| Scheme 2 | ||
Poly(N-(3-tert-butoxy-3-oxopropyl) glycine) can be readily deprotected to reveal the carboxyl groups on the sidechain by treatment of trifluoroacetic acid (TFA) in CHCl3 at room temperature for 24 h, evidenced by the disappearance of tert-butyl protons in the 1H NMR spectrum in the final polymer product (Fig. S5 and S9†). The number average degree of polymerization (DPn) of the poly(N-(3-tert-butoxy-3-oxopropyl) glycine) and the poly(N-(2-carboxyethyl) glycine) was determined by integrating the methylene protons of the polymer backbone (A) versus the methyl protons of the butylamide end-group (F, Fig. S9†). The poly(N-(2-carboxyethyl) glycine) was found to have comparable chain length (DPn = 177, Mn = 33 kg mol−1) to that of the poly(N-(3-tert-butoxy-3-oxopropyl) glycine) precursor (DPn = 183, Mn = 34 kg mol−1), indicating the effectiveness of TFA treatment in uncloaking the carboxyl functional groups on the polypeptoid sidechains without causing backbone cleavage.38
Poly(N-(2-carboxyethyl) glycine) is a weak polyelectrolyte whose size in water can be controlled by pH change. For example, DLS analysis of a 3 mg mL−1 dilute aqueous solution of poly(N-(2-carboxyethyl) glycine) (Mn(SEC) = 35 kg mol−1, PDI = 1.08) revealed a hydrodynamic radius (Rh) of 2.5 ± 0.2 nm at low pH (2.4) which increases to 85.6 ± 0.2 nm at high pH (11.8) (Fig. S10†). This has been attributed to the enhanced electrostatic repulsion amongst the N-substituents on the polymers owing to the increased extent of deprotonation of the carboxyl groups on the sidechains. Future work will focus on the investigation of poly(N-(2-carboxyethyl) glycines) as peptidomimetic building blocks in the design and synthesis of stimuli-responsive supramolecular assemblies.
Conclusion
We have designed and synthesized a new class of N-substituted glycine derived NCA bearing a carboxyl ester functionality linked to the nitrogen via an alkyl group. The alkyl linker length of the NCA was found to be important for their polymerization behavior. For N-(3-tert-butoxy-3-oxopropyl) glycine derived N-carboxyanhydride (tBuO2Pr-NCA) bearing ethylene linker, the ring-opening polymerization in the presence of primary initiators proceeds rapidly in a controlled manner, producing well-defined poly(N-(3-tert-butoxy-3-oxopropyl) glycine) with controlled molecular weight and narrow molecular weight distribution. For analogous EtO2Et-NCA and tBuO2Et-NCA monomers with a methylene linker, the polymerization under identical conditions only reaches low conversion due to competitive termination by intramolecular transamindation relative to chain propagation. The cloaked carboxyl functionality on the poly(N-(3-tert-butoxy-3-oxopropyl) glycine) can be readily unveiled under mild acidic conditions without affecting the polymer chain length. The resulting peptoid-based polyacids are attractive peptidomimetic building blocks for stimuli responsive polymeric assemblies that will have appeal to the biomaterials community due to their structural similarity to poly(L-glutamic acid).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
GLS would like to thank David Siefker for fruitful discussions. The work was supported by the National Science Foundation (CHE 1609447 and 2003458).References
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad and R. Luxenhofer, Peptoids and Polypeptoids at the Frontier of Supra- and Macromolecular Engineering, Chem. Rev., 2016, 116(4), 1753–1802 CrossRef CAS.
- B. A. Badeau and C. A. DeForest, Programming Stimuli-Responsive Behavior into Biomaterials, Annu. Rev. Biomed. Eng., 2019, 21(1), 241–265 CrossRef CAS.
- B. A. Chan, S. Xuan, A. Li, J. M. Simpson, G. L. Sternhagen, T. Yu, O. A. Darvish, N. Jiang and D. Zhang, Polypeptoid polymers: Synthesis, characterization, and properties, Biopolymers, 2018, 109(1), e23070 CrossRef.
- X. Tao, M.-H. Li and J. Ling, α-Amino acid N-thiocarboxyanhydrides: A novel synthetic approach toward poly(α-amino acid)s, Eur. Polym. J., 2018, 109, 26–42 CrossRef CAS.
- L. Guo and D. Zhang, Cyclic Poly(α-peptoid)s and Their Block Copolymers from N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-Substituted N-Carboxylanhydrides, J. Am. Chem. Soc., 2009, 131(50), 18072–18074 CrossRef CAS.
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Polypeptoids from N-Substituted Glycine N-Carboxyanhydrides: Hydrophilic, Hydrophobic, and Amphiphilic Polymers with Poisson Distribution, Macromolecules, 2011, 44(17), 6746–6758 CrossRef CAS.
- C.-U. Lee, A. Li, K. Ghale and D. Zhang, Crystallization and Melting Behaviors of Cyclic and Linear Polypeptoids with Alkyl Side Chains, Macromolecules, 2013, 46(20), 8213–8223 CrossRef CAS.
- S. H. Lahasky, X. Hu and D. Zhang, Thermoresponsive Poly(α-peptoid)s: Tuning the Cloud Point Temperatures by Composition and Architecture, ACS Macro Lett., 2012, 1(5), 580–584 CrossRef CAS.
- X. Tao, C. Deng and J. Ling, PEG-Amine-Initiated Polymerization of Sarcosine N-Thiocarboxyanhydrides Toward Novel Double-Hydrophilic PEG-b-Polysarcosine Diblock Copolymers, Macromol. Rapid Commun., 2014, 35(9), 875–881 CrossRef CAS.
- X. Tao, J. Du, Y. Wang and J. Ling, Polypeptoids with tunable cloud point temperatures synthesized from N-substituted glycine N-thiocarboxyanhydrides, Polym. Chem., 2015, 6(16), 3164–3174 RSC.
- L. Guo, J. Li, Z. Brown, K. Ghale and D. Zhang, Synthesis and characterization of cyclic and linear helical poly(α-peptoid)s by N-heterocyclic carbene-mediated ring-opening polymerizations of N-substituted N-carboxyanhydrides, Pept. Sci., 2011, 96(5), 596–603 CrossRef CAS.
- L. Guo and D. Zhang, Synthesis and Characterization of Helix-Coil Block Copoly(α-peptoid)s, in Non-Conventional Functional Block Copolymers, American Chemical Society, 2011, vol. 1066, pp. 71–79 Search PubMed.
- J. W. Robinson and H. Schlaad, A versatile polypeptoid platform based on N-allyl glycine, Chem. Commun., 2012, 48(63), 7835–7837 RSC.
- S. H. Lahasky, W. K. Serem, L. Guo, J. C. Garno and D. Zhang, Synthesis and Characterization of Cyclic Brush-Like Polymers by N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Propargyl N-Carboxyanhydride and the Grafting-to Approach, Macromolecules, 2011, 44(23), 9063–9074 CrossRef CAS.
- J. W. Robinson, C. Secker, S. Weidner and H. Schlaad, Thermoresponsive Poly(N-C3 glycine)s, Macromolecules, 2013, 46(3), 580–587 CrossRef CAS.
- Y. Deng, H. Chen, X. Tao, F. Cao, S. Trépout, J. Ling and M.-H. Li, Oxidation-Sensitive Polymersomes Based on Amphiphilic Diblock Copolypeptoids, Biomacromolecules, 2019, 20(9), 3435–3444 CrossRef CAS.
- C. Secker, S. M. Brosnan, F. R. P. Limberg, U. Braun, M. Trunk, P. Strauch and H. Schlaad, Thermally Induced Crosslinking of Poly(N-Propargyl Glycine), Macromol. Chem. Phys., 2015, 216(21), 2080–2085 CrossRef CAS.
- C. Secker, J. W. Robinson and H. Schlaad, Alkyne-X modification of polypeptoids, Eur. Polym. J., 2015, 62, 394–399 CrossRef CAS.
- X. Fu, J. Tian, Z. Li, J. Sun and Z. Li, Dual-responsive pegylated polypeptoids with tunable cloud point temperatures, Biopolymers, 2019, 110(4), e23243 CrossRef.
- Z. Song, Z. Han, S. Lv, C. Chen, L. Chen, L. Yin and J. Cheng, Synthetic polypeptides: from polymer design to supramolecular assembly and biomedical application, Chem. Soc. Rev., 2017, 46(21), 6570–6599 RSC.
- J. T. Brosnan and M. E. Brosnan, Glutamate: a truly functional amino acid, Amino Acids, 2013, 45(3), 413–418 CrossRef CAS.
- G. J. Bartlett, C. T. Porter, N. Borkakoti and J. M. Thornton, Analysis of Catalytic Residues in Enzyme Active Sites, J. Mol. Biol., 2002, 324(1), 105–121 CrossRef CAS.
- D. G. H. Ballard and C. H. Bamford, The Polymerization of α-Benzyl-L-Glutamate N-Carboxy Anhydride, J. Am. Chem. Soc., 1957, 79(9), 2336–2338 CrossRef CAS.
- P. Doty and R. D. Lundberg, Polypeptides. XA. Additional Comments of the Amine-Initiated Polymerization, J. Am. Chem. Soc., 1957, 79(9), 2338–2339 CrossRef CAS.
- D. Siefker, A. Z. Williams, G. G. Stanley and D. Zhang, Organic Acid Promoted Controlled Ring-Opening Polymerization of α-Amino Acid-Derived N-thiocarboxyanhydrides (NTAs) toward Well-defined Polypeptides, ACS Macro Lett., 2018, 1272–1277 CrossRef CAS.
- J. Cao, D. Siefker, B. A. Chan, T. Yu, L. Lu, M. A. Saputra, F. R. Fronczek, W. Xie and D. Zhang, Interfacial Ring-Opening Polymerization of Amino-Acid-Derived N-Thiocarboxyanhydrides Toward Well-Defined Polypeptides, ACS Macro Lett., 2017, 6(8), 836–840 CrossRef CAS.
- E. J. Robertson, A. Battigelli, C. Proulx, R. V. Mannige, T. K. Haxton, L. Yun, S. Whitelam and R. N. Zuckermann, Design, Synthesis, Assembly, and Engineering of Peptoid Nanosheets, Acc. Chem. Res., 2016, 49(3), 379–389 CrossRef CAS.
- R. V. Mannige, T. K. Haxton, C. Proulx, E. J. Robertson, A. Battigelli, G. L. Butterfoss, R. N. Zuckermann and S. Whitelam, Peptoid nanosheets exhibit a new secondary-structure motif, Nature, 2015, 526(7573), 415–420 CrossRef CAS.
- R. Kudirka, H. Tran, B. Sanii, K. T. Nam, P. H. Choi, N. Venkateswaran, R. Chen, S. Whitelam and R. N. Zuckermann, Folding of a single-chain, information-rich polypeptoid sequence into a highly ordered nanosheet, Pept. Sci., 2011, 96(5), 586–595 CrossRef CAS.
- E. J. Robertson, C. Proulx, J. K. Su, R. L. Garcia, S. Yoo, E. M. Nehls, M. D. Connolly, L. Taravati and R. N. Zuckermann, Molecular Engineering of the Peptoid Nanosheet Hydrophobic Core, Langmuir, 2016, 32(45), 11946–11957 CrossRef CAS.
- E. J. Robertson, E. M. Nehls and R. N. Zuckermann, Structure–Rheology Relationship in Nanosheet-Forming Peptoid Monolayers, Langmuir, 2016, 32(46), 12146–12158 CrossRef.
- B. Sanii, T. K. Haxton, G. K. Olivier, A. Cho, B. Barton, C. Proulx, S. Whitelam and R. N. Zuckermann, Structure-Determining Step in the Hierarchical Assembly of Peptoid Nanosheets, ACS Nano, 2014, 8(11), 11674–11684 CrossRef CAS.
- A. K. Livesey and J. C. Brochon, Analyzing the Distribution of Decay Constants in Pulse-Fluorimetry Using the Maximum Entropy Method, Biophys. J., 1987, 52(5), 693–706 CrossRef CAS.
- D. Zhang, S. H. Lahasky, L. Guo, C.-U. Lee and M. Lavan, Polypeptoid Materials: Current Status and Future Perspectives, Macromolecules, 2012, 45(15), 5833–5841 CrossRef CAS.
- D. Siefker and D. Zhang, Chapter 9 Ring-opening Polymerization of N-carboxyanhydrides Using Organic Initiators or Catalysts, in Organic Catalysis for Polymerisation, The Royal Society of Chemistry, 2019; pp. 367–405 Search PubMed.
- S. Xuan, S. Gupta, X. Li, M. Bleuel, G. J. Schneider and D. Zhang, Synthesis and Characterization of Well-Defined PEGylated Polypeptoids as Protein-Resistant Polymers, Biomacromolecules, 2017, 18(3), 951–964 CrossRef CAS.
- B. Zheng, T. Bai, J. Ling and J. Sun, Direct N-substituted N-thiocarboxyanhydride polymerization towards polypeptoids bearing unprotected carboxyl groups, Commun. Chem., 2020, 3, 144 CrossRef.
- H. Tran, S. L. Gael, M. D. Connolly and R. N. Zuckermann, Solid-phase submonomer synthesis of peptoid polymers and their self-assembly into highly-ordered nanosheets, J. Visualized Exp., 2011, 57, e3373 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C{1H} NMR spectra of the tBuO2Pr-NCA monomers and precursors, poly(N-(3-tert-butoxy-3-oxopropyl) glycine) and poly(N-(2-carboxyethyl) glycine), SEC-DRI-MALS chromatograms, ESI-MS spectrum of oligomers obtained by ROP of EtO2Et-NCA, DLS correlograms and intensity-weighted decay time distribution plot. See DOI: 10.1039/d0py01395a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |