
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanical switching of orientation-related photoluminescence in deep-blue 2D layered perovskite ensembles†
Balaji
Dhanabalan
 ab,
Andrea
Castelli‡
a,
Luca
Ceseracciu
a,
Davide
Spirito§
a,
Francesco
Di Stasio
a,
Liberato
Manna
a,
Roman
Krahne
a and
Milena P.
Arciniegas
*a
ab,
Andrea
Castelli‡
a,
Luca
Ceseracciu
a,
Davide
Spirito§
a,
Francesco
Di Stasio
a,
Liberato
Manna
a,
Roman
Krahne
a and
Milena P.
Arciniegas
*a
aIstituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy. E-mail: Milena.Arciniegas@iit.it
bDipartimento di Chimica e Chimica Industriale, Università degli Studi di Genova, Via Dodecaneso, 31, 16146, Genova, Italy
First published on 15th February 2021
Abstract
The synergy between the organic component of two-dimensional (2D) metal halide layered perovskites and flexible polymers offers an unexplored window to tune their optical properties at low mechanical stress. Thus, there is a significant interest in exploiting their PL anisotropy by controlling their orientation and elucidating their interactions. Here, we apply this principle to platelet structures of micrometre lateral size that are synthesized in situ into free-standing polymer films. We study the photoluminescence of the resulting films under cyclic mechanical stress and observe an enhancement in the emission intensity up to ∼2.5 times along with a switch in the emission profile when stretching the films from 0% to 70% elongation. All the films recovered their initial emission intensity when releasing the stress throughout ca. 15 mechanical cycles. We hypothesize a combined contribution from reduced reabsorption, changes on in-plane and out-of-plane dipole moments that stem from different orientation of the platelets inside the film, and relative sliding of platelets within oriented stacks while stretching the films. Our results reveal how low-mechanical stress affects 2D layered perovskite aggregation and orientation, an open pathway toward the design of strain-controlled emission.
 Milena P. Arciniegas | Milena P. Arciniegas is a very active female scientist with a strong commitment and a solid expertise in material science and technology. She graduated in Mechanical Engineering (Colombia) and obtained her PhD from the Polytechnic University of Catalonia (Spain) in 2008. In 2009 she joined the Advanced Manufacturing Research Center with Boeing (England) and in 2012 she started as postdoctoral fellow in the group of Prof. Liberato Manna at IIT (Italy), working on the mechanics of nanocomposites. Since 2018 she is leading a highly motivated and young research team at IIT that seeks to understand and discover fascinating properties from emitting layered structures for non-conventional applications. She is also currently leading the tasks for data management and the use of Machine Learning for the design of novel layered materials within the Nanochemistry research line at IIT. |
Metal halide layered perovskites (MHLP) are emerging as promising semiconductors for the next generation of optoelectronic devices, such as light emitting diodes, solar cells, lasers, and photodetectors. In addition to their structural and chemical stability, which is reduced in the 3D structures, they are enriched with a variety of fascinating optoelectronic and excitonic properties. For example, they show quantum-well confinement, large exciton binding energy, fast radiative recombination rates, and both narrow and broadband photoluminescence.1–3 Structurally, these properties can be tuned by simple changes on (i) the chemical composition of the inorganic cage; (ii) variations on the type of large organic cations used in their synthesis; and (iii) the number of octahedra layers. Most efforts have been focused on controlling the nature of organic moieties, allocated between the inorganic layers, to modify the inter-connection and orientation of the metal–halide octahedral slabs, as it occurs in the case of Ruddlesden-Popper structures.4–7 In this way, for instance, highly distorted lattices of bromide-based LP can be fabricated, which induce the formation of self-trapped excitons, resulting in white light emission from intra-gap states.8–11 Mechanically, MHLP are relatively flexible hybrids compared to their 3D family, due in part to the soft character of the organic cations, which create a stiffness mismatch in their organic/inorganic layered architecture. The compliance of the organic layer is a key feature for accessing to bright emitting MHLP. In the case of blue-emitting MHLP, it was shown that structures made with molecules containing an aromatic ring (phenethylamine) have higher structural rigidity than those fabricated with a shorter aliphatic amine (butylamine), resulting in enhanced exciton–phonon coupling.1,12 Moreover, similar to other layered nanomaterials, the weak bonding between adjacent organic layers in MHLP allows for mechanical exfoliation and has provided a route for multicolour lasing13 and to increase the photoluminescence (PL) quantum yield.12 Therefore, it comes as no surprise that mechanical forces can be a tool to tune the electronic band structure of these materials,14–17 with strong benefits from their overall mechanical flexibility. Because this is only possible by creating significant changes on the bond-lengths and angles, and related octahedral rotations in the inorganic lattice, pressures in the order of few GPa are typically used. However, in contrast to their 3D counterparts, MHLP can withstand higher pressures (above 3 GPa)18–20 without PL quenching and the reduction of the interlayer space offers an additional mean for strain accommodation at extremely high pressure of around 45 GPa.15,21,22 These conditions can induce strong blue- or red-shifts in PL, longer PL lifetimes, bandgap narrowing, enhanced conductivity and power conversion efficiency that are not observed at ambient pressure,23–28 but can also cause lattice damage, permanent deformations, and an overall reduction of the mechanical integrity of the structures. Alternatively, engineering of the orientation of MHLP, with their characteristic plate-like shape, can grant access to emission tunability through lower mechanical stress/strain, due to their associated PL anisotropy.24 This has been proved effective for other 2D nanomaterials such as Cu2−xSe nanosheets and, more recently, graphene, offering the additional key feature of reversibility in a confined space.29,30 In this work, we transfer this concept to MHLP and report the efficient orientation-related PL switching of benzylammonium (BzA)-lead bromide platelets ((BzA)2PbBr4) as a prototype MHLP material, embedded in an elastomeric matrix. Strain transfer requires a strong contact interface between MHLP platelets and the polymer, which is difficult to achieve by their direct deposition on an elastomeric substrate or by mixing them with an elastomer in a viscous solution to produce a film. Here, we nucleated the MHLP platelets in situ in an elastomeric solution and fabricated macroscopic free-standing thin films. We tested the films through multiple stretch/release cycles and recorded in situ their PL for each cycle at the maximum applied strain, ε, and when the samples were fully released from the tensile stress. Increasing the strain up to 70% results in an enhanced PL intensity by a factor of ∼2.5 in all the cycles, with contributions from differently orientated platelets. The intensity returns to the original value once the strain is removed, evidencing the reversible nature of such mechanism without mechanical damage of the composites. The modulation in reabsorption and re-emission events within the film during stretching is also responsible for the changes observed in emission profile. To better understand the dynamics of the process, we analysed the films through a step-by-step stretching to achieve strain up to ε = 70%, and observed that even a rather low ε of ca. 20% (<1 MPa) induces a change of the PL profile, displaying a maximum intensity at 416 nm that is redshifted by ca. 10 nm (∼70 meV) with respect to the emission peak of the unstrained film centred at 406 nm. This spectral change is associated to the directional emission of the platelets,24 induced by a change of the orientation of the platelets inside the films. Such a PL modulation is significantly larger than those reported from other emitting nanosystems under mechanical stress31–33 and those typically observed from 3D perovskite structures, which are obtained at much higher stress.19 These findings provide promising directions on the use of emitting layered perovskites in stretchable polymer films as mechanically switchable 2D nanomaterials. Our results extend the functionalities of such materials to the monitoring of deformations at very low impact forces or cyclic stress in the order of few MPa, without permanent damage, which is ideal in applications such as biomechanics, detection of barely visible surface defects, mechanical damage monitoring, and optoelectronic skins.
Results and discussion
Synthesis and film fabrication
In our experiments, we prepared a mixture of perovskite precursors (PbBr2 powder dissolved in HBr and acetone) with polydimethylsiloxane (PDMS) in toluene. Then, benzylamine was added to the resulting mixture, triggering immediately the formation of layered perovskite crystals in solution. The milky dispersions were mould cast on custom-made polytetrafluoroethylene stamps to produce MHLP–PDMS free-standing films of 20 × 4 × 0.3 mm3 (see Experimental section for details). Polymer films with different MHLP loading (2 to 6% in vol. of precursors in 1 ml of PDMS in toluene) were fabricated by controlling the amount of amine added to the PDMS solutions, as reported in Table S1.† We also prepared the MHLP platelets following the same protocol described above, but without polymer (only toluene was added) as reference samples. The resulting MHLP–PDMS films show a deep-blue emission upon UV light exposure, as displayed in Fig. 1a (that includes a sketch of the MHLP platelets inside the film). A complete set of photographs of the samples is displayed in Fig. S1.† The observed changes on the film transparency are due to the gradual increment of the crystal content when increasing the amount of MHLP precursors in the solutions. We discuss here the results obtained from the MHLP–PDMS films prepared with the lowest perovskite concentration in solution (ca. 2% in vol); see Fig. S13 and S14† for details on the experiments conducted with higher MHLP concentrations.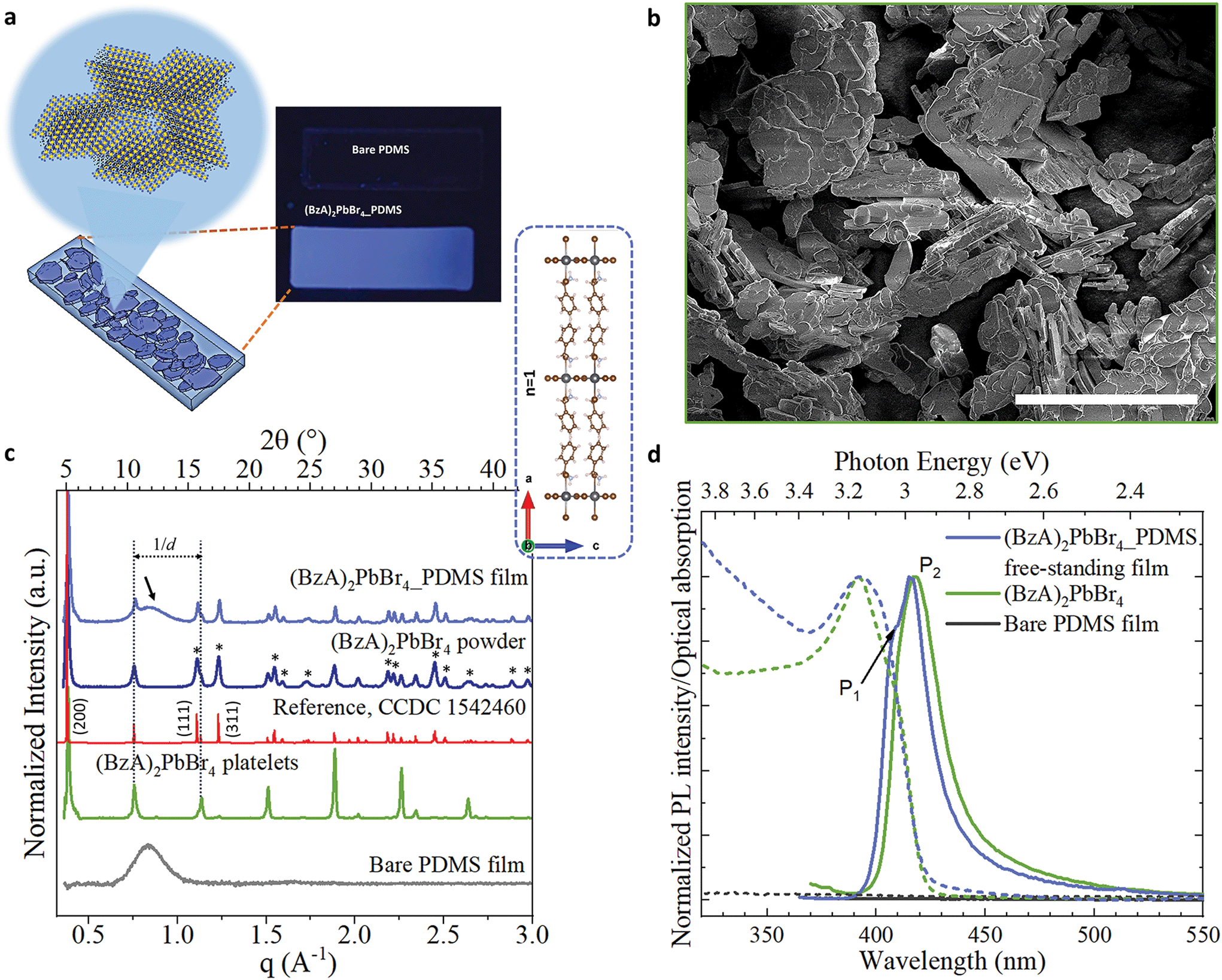 | ||
| Fig. 1 (a) Scheme showing the atomic layered structure of the (BzA)2PbBr4 platelets nucleated in situ in the PDMS by injecting the amine in the mixture of PDMS, PbBr2, HBr and acetone. The photograph displays the resulting blue-emitting film under UV lamp irradiation. (b) SEM images of the platelets without PDMS and deposited on a Si substrate. Scale bar: 50 μm. (c and d) XRD patterns (c) and emission spectra and optical absorption (d) collected from bare PDMS film (grey), ensembles of the perovskite platelets (green), powder sample of platelets, and free-standing film of 2D layered perovskite–PDMS (blue). In red, the reference from ref. 34. In (c): the inset displays the b-view of the orthorhombic unit cell of (BzA)2PbBr4 crystals from ref. 34; the dotted lines indicate the highly intense periodic diffraction peaks of the platelet ensembles from their preferential crystal orientation parallel to the substrate, which is partially retained in the films; the black arrow highlights the broad peak from the PDMS in the composite films and the asterisks indicate the most intense Bragg reflections associated to different diffraction planes. | ||
Structural and optical properties of the as-fabricated MHLP–PDMS free-standing films
Fig. 1b shows a SEM image of the crystals nucleated without polymer, following the protocol described above. The structures are platelets that reach up to 50 μm of lateral sizes. It is reasonable to expect that such morphology will be preserved inside the MHLP–PDMS films. X-ray diffraction (XRD) patterns collected from the platelets and films are displayed in Fig. 1c. We collected XRD patterns from both platelets directly deposited on a substrate (in green) and from their powders (in blue). The pattern from the platelets shows equally spaced diffraction peaks that are indicated in the figure with parallel dotted lines, which stem from the (0 0 2l) Bragg reflections. Note that in the powders, as well as in the MHLP–PDMS film, various diffraction peaks of lower intensity (highlighted with asterisks), arising from other diffraction planes, accompany the set of periodic diffraction peaks, which correlate well with the reported orthorhombic phase of (BzA)2PbBr4 crystals (see reference in red in Fig. 1c).34 These results are in agreement with the nucleation of MHLP crystals in different orientations inside the films. Also, there are no additional reflections in the collected XRD patterns that could be attributed to spurious phases or unreacted precursors (see closer view of the XRD patterns in Fig. S2†), which confirms the phase purity of the in situ formed crystals. We could also observe a trace signal of the amorphous nature of the PDMS in the XRD pattern of the hybrid films – a broad peak around 2θ values of 10–15° indicated by an arrow in Fig. 1c.The lattice periodicity inferred from the equally spaced peaks translates to a d-spacing of 16.75 ± 0.05 Å. This corresponds to the sum of the thickness of a single layer of corner sharing [PbBr4]2− octahedra extending in (001) planes (6 Å) plus the thickness of a double layer of passivating BzA cations (see inset in Fig. 1c). Moreover, elemental analysis performed on the films via energy-dispersive X-ray spectroscopy (EDS) in scanning electron microscopy (SEM) mode revealed a Pb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br atomic ratio of 1:4 (Fig. S3†), evidencing the in situ formation of (BzA)2PbBr4 platelets in the films.
:Br atomic ratio of 1:4 (Fig. S3†), evidencing the in situ formation of (BzA)2PbBr4 platelets in the films.
The absorption and PL spectra collected from the bare PDMS and MHLP–PDMS films, as well as from polymer-free deposits of platelets, are shown in Fig. 1d. The bare PDMS film (in black) had no emission in the investigated portion of the spectrum whereas the MHLP–PDMS films (in blue) exhibits two main peaks at 406 nm (P1) and 416 nm (P2) with a long tail that spans into the red region of the visible spectrum (see PL profile analysis in Fig. S4†). These features are characteristics of the (BzA)2PbBr4 platelets, with the tail typically attributed to self-trapped excitons, i.e. due to the lattice deformation in the presence of excitons within the crystal layers.6,12,35 The absorption spectra show a relatively broad peak centred at 395 nm, which is the sum of the contributions from confined excitons with in-plane and out-of-plane dipole moments. The peak is broader and more symmetric in the composite samples, highlighting a higher contribution of in-plane dipole moment in the low energy side of the peak,35 further confirming the random orientation of the MHLP crystals inside the film. The collected optical characteristics of the composite films and bare platelets are reported in Table S2.†
In situ opto-mechanical study on the MHLP–PDMS films
The cyclic stress–strain behaviour of the composite films was recorded by using an Instron Universal Testing dynamometer with a stretching ramp set in displacement control, with ε varying from 0% to 70% and applying up to 15 stretch/release cycles, as detailed in Experimental section. Fig. S5† shows an outline of our customized set-up. Fig. 2a displays a sketch of the performed stretching/release cyclic experiments. The PL of the films was recorded in situ under stretch (ε = 70%) and release (ε = 0%) at the maximum stretched region. After the first cycle, the strain was released to a minimum of ca. 5% in the following mechanical cycles; see the cyclic stress (calculated as force over section area) vs. strain curves in Fig. 2b. A maximum stress of 1.6 MPa was applied to reach 70% of strain. We tested 5 different composite films produced each from different batches, and a minimum of 5 mechanical cycles were applied to each sample (Fig. S6†) for a total number of 55 measurements recorded in situ (Fig. S7†). The average emission enhancement of the first 5 cycles resulted in 2.21 ± 0.15 (Table S3†). Note that our measurements are conservative as can be inferred from consideration regarding the film elastic strain state: since the in situ PL measurements were recorded by keeping the optics fixed, the spot size did not change; therefore, with increased stretching the volume contributing to the emission is slightly reduced from the striction of the sample thickness only. This is estimated from our experiments of around 20% for a large deformation of 70%. For platelets uniformly distributed in the polymer film this means a reduction on the number of platelets, and thus the PL enhancement per number of flakes may be larger by a factor of 1.25 (see Experimental details). Fig. 2c shows the optical response collected from the composite films for each mechanical cycle from a representative sample. The overall emission of the platelets in the film was increased at least of 2.5 times the initial intensity when the films reached maximum strain on each loading cycle and reversed back to its initial value upon releasing. Over cycling, we observed a slight reduction in the emission intensity recorded at 70% (Fig. 2d), which could be attributed to the degradation over time of the MHLP under continuous laser illumination. We also observed a stronger increase of the intensity of P2 emission as compared to P1 when stretching the films up to 70% in all the cycles (Fig. S8†), while there was no significant change observed in the peak positions (Fig. S9†): the P1 position was maintained at around 408.5–409 nm and the P2 position around 415–416.5 nm upon applying the tensile strain for all the mechanical cycles.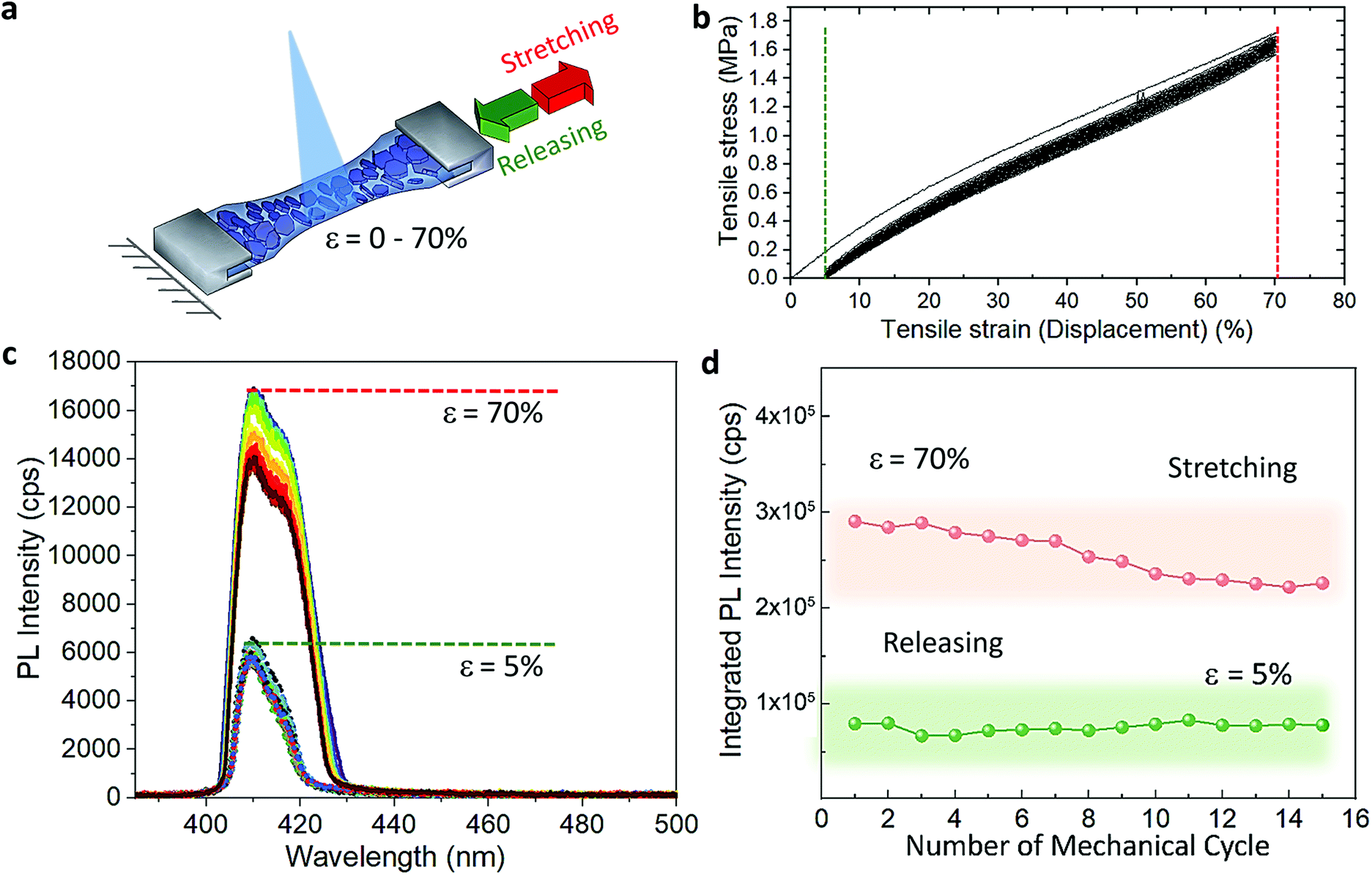 | ||
| Fig. 2 (a) Sketch illustrating the in situ PL experiment of the layered platelets in the polymer film under stretching/releasing cycles. (b) Applied tensile stress as a function of the strain for all the mechanical cycles. (c) Representative emission spectra acquired from the 2% (BzA)2PbBr4–PDMS free-standing film under 15 continuous stretch/release mechanical cycles. The dashed lines indicate the stretching up to a strain of 70% (in red) and the releasing up to 5% (in green). (d) Integrated area under the PL profile collected at the maximum stretch and after releasing the film as a function of the number of cycles. | ||
To understand the photocarrier dynamics in the MHLP–PDMS films, we performed time-resolved PL on the samples and compared the decay traces with those obtained from the (BzA)2PbBr4 platelets without polymer. The results are displayed in Fig. 3a and b (see Experimental section for details). From the fitting parameters in Table S4† we see that τ1 does not change upon embedding the platelets in the polymer, but τ2 becomes relatively faster. Since typically the fast component (with life time around one to few nanoseconds) is attributed to non-radiative decay, we tentatively assign this small but overall faster decay to an enhanced radiative rate. This is in line with the increased radiative recombination rates that we observed from exfoliated flakes in our previous work and in agreement with reduced reabsorption.35
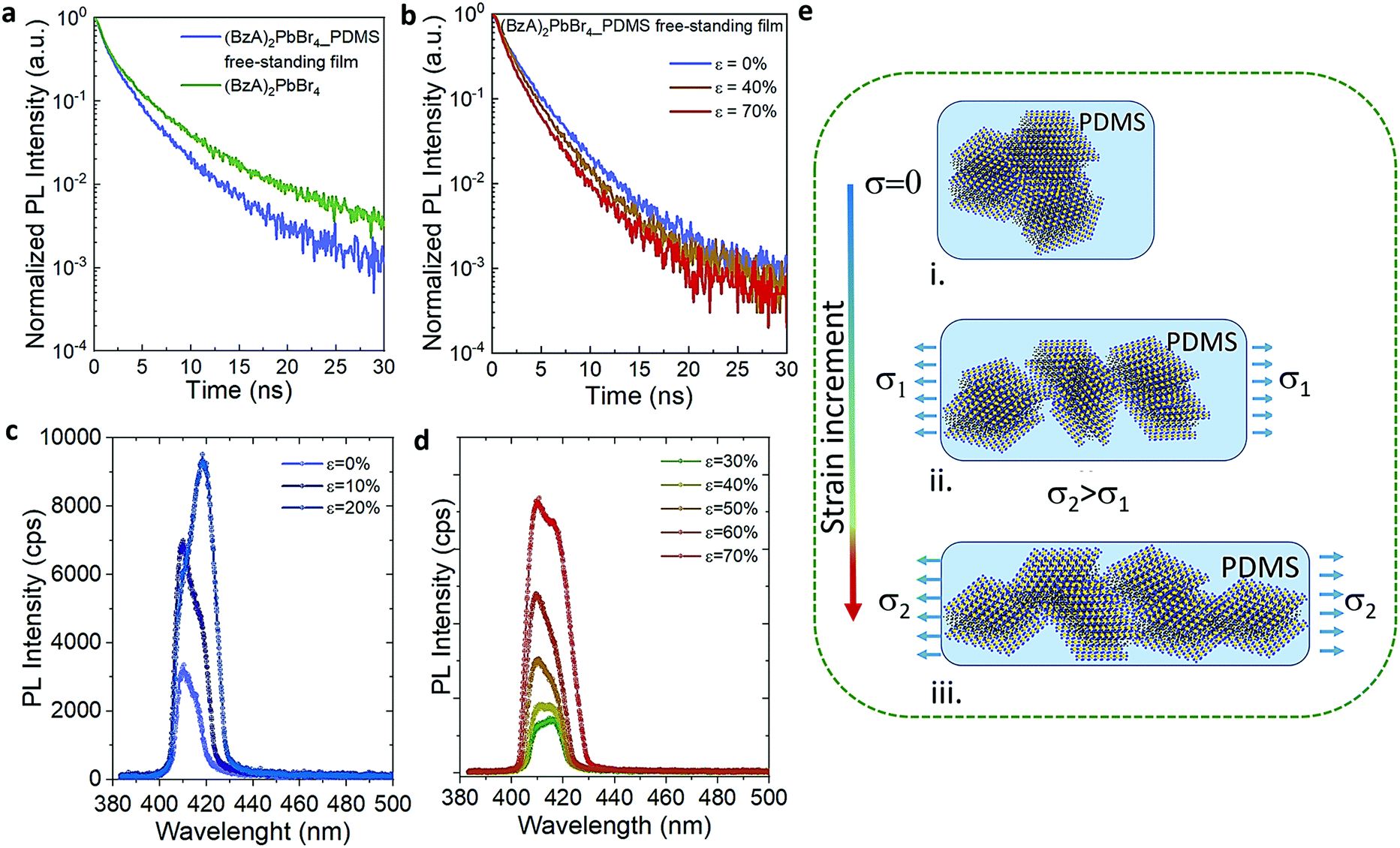 | ||
| Fig. 3 (a and b) PL decay profiles collected from a deposit of the (BzA)2PbBr4 platelets (green), and the (BzA)2PbBr4–PDMS free-standing film (blue) without stretching in (a) and under stretching up to 70% in (b). (c and d) Emission spectra collected from the films by applying step-by-step stretching. (e) Cartoon explaining the orientation switching of the platelets in the polymer film under continuous tensile strain: (i) without stretching, the platelets are stacked one on top of the other and randomly oriented in the film; (ii) under an initial stress (σ1), the ensemble of platelets start to separate from each other and adopt a preferential orientation parallel to the applied stress; (iii) by increasing the stress (σ2), preferentially oriented ensembles become more separated and a sliding of platelets within the ensembles takes place, providing thinner structures in the film, from which an enhanced PL intensity is observed. | ||
To elucidate the evolution of the PL profile under stretching and gain insight about the underlying mechanism, we collected the PL from the films through step-by-step stretching (up to ε of 70%), as shown in Fig. 3c and d. At ε = 10%, the intensity of both P1 and P2 emission peaks increases while at 20% we observe an enhanced intensity of P2 peak and only a tenuous increase in the intensity of P1 peak. Further increment in the tensile strain shows the increment in P1 alongside P2 emission (ε = 30–40%) and both the peaks showed equal contributions at 40% strain. At higher strain (>50%), the relative contribution of the P2 emission peak decreases and the resulting PL profile matches that observed on the multiple cycle experiments (Fig. 3d). Fig. S10† displays the intensity of P1 and P2 as a function of the applied stress in one mechanical cycle. We rationalize these observations as follows (Fig. 3e): (i) the in situ growth of MHLP inside the polymer matrix generates ensembles of platelets packed one on the top of the other with random relative orientations, as it was observed from the samples prepared without polymer (Fig. S11†). (ii) At relatively low ε, different ensembles start to separate from each other. This effect reduces reabsorption and, hence, increases the intensity of both emission peaks from platelets. By increasing the strain further, the ensembles, dragged by the polymer matrix, adopt a preferential orientation that is parallel to the elongation direction and perpendicular to the excitation beam. This results in a preferential in-plane dipole emission and leads to a marked increment of P2 emission intensity.35 (iii) At high strain, we hypothesize that the induced shear stress between platelets within the aligned ensembles leads to a mechanically induced sliding of platelets via polymer-mediated interactions, as predicted by theoretical analysis and observed in other experimental works.36,37 These mechanisms can explain the overall enhancement of the emission intensity favoured by a reduction of the thickness of the platelet ensembles in the film, which alters the scattering path length of emitted light. To evidence this point, we obtained the absorbance spectra of both stretched and unstretched films via diffuse reflectance measurements using an integrating sphere. The results in Fig. S12† show a strong reduction of the absorbance intensity from the stretched films. Note that the in situ growth of the platelets in the polymer might favour the intercalation of polymer molecules among platelets, facilitating their relative displacement at higher elongation. The emission intensity returns to the initial values when releasing the stress, indicating a reversible aggregation process. In line with our explanation, we found that the films prepared with higher content of (BzA)2PbBr4 platelets (3 and 6% in vol.) do not show significant change in their emission intensity (Fig. S14†) over cyclic stretching, with only a 1.2–1.4 fold increase after cycle 1. This indicates that the applied strain can induce a relatively low displacement of the ensembles in the film in this strongly packed environment, which hinders platelet mobility and therefore full changes in their orientation and sliding process.
Finally, to showcase the structural flexibility and tunable optoelectronic properties of the MHLP in the composite films, we fabricated in situ Mn-doped (BzA)2PbBr4 platelets and investigated the potential changes induced by mechanical stretching on their emission. Mn doping of MHLP triggers interesting properties such as magneto-optic and magneto-transport properties,38–40 large Stokes shift between absorbance edge and emission, which is good for down conversion, for example in solar concentrators,41 along with significant improvement in LED device performance by suppressing the electron–phonon interactions.42 In particular, Mn doping in perovskites provides a route to dual emission from a single material, which has been mostly exploited in 3D lead halide structures.43–45 In our fabrication strategy we used a ratio of Mn:Pb precursors of 1:2 and followed the protocol for the preparation of the polymer films. XRD patterns collected from the doped films displayed a slight shift in the peaks towards higher angles (Fig. S15†), which confirms the successful substitution of Pb2+ with smaller Mn2+ in the 2D layered perovskite lattice. This is reflected in the emission spectra from the doped samples, as shown in Fig. 4a. The spectrum displays two emission peaks: the band-edge emission that shared with the undoped samples centred at ca. 414 nm with a FWHM of 22 nm, and a broader (FWHM of 85 nm) emission peak centred at 607 nm that stems from the Mn2+ d–d transitions.46,47 The presence of this Mn-related peak drastically changes the perceived colour of the composites emission, as reflected in their Commission Internationale de l'Eclairage (CIE) coordinates shown in Fig. 4b. We noticed a relatively high contribution of 80% of the red emission with respect to the amplitude of blue band edge peak from the Mn-doped structures in the polymer. Both emission peaks are preserved under stretching. The blue emission peak shows an enhanced intensity at 70% strain that persists over 10 stretch/release cycles. The red peak maintains similar intensity through the first 3 cycles (Fig. S16a†) and then it slightly increases under release in the following, with ca. 2 fold increase after fully releasing the stress with respect to its initial intensity (Fig. S16b†).
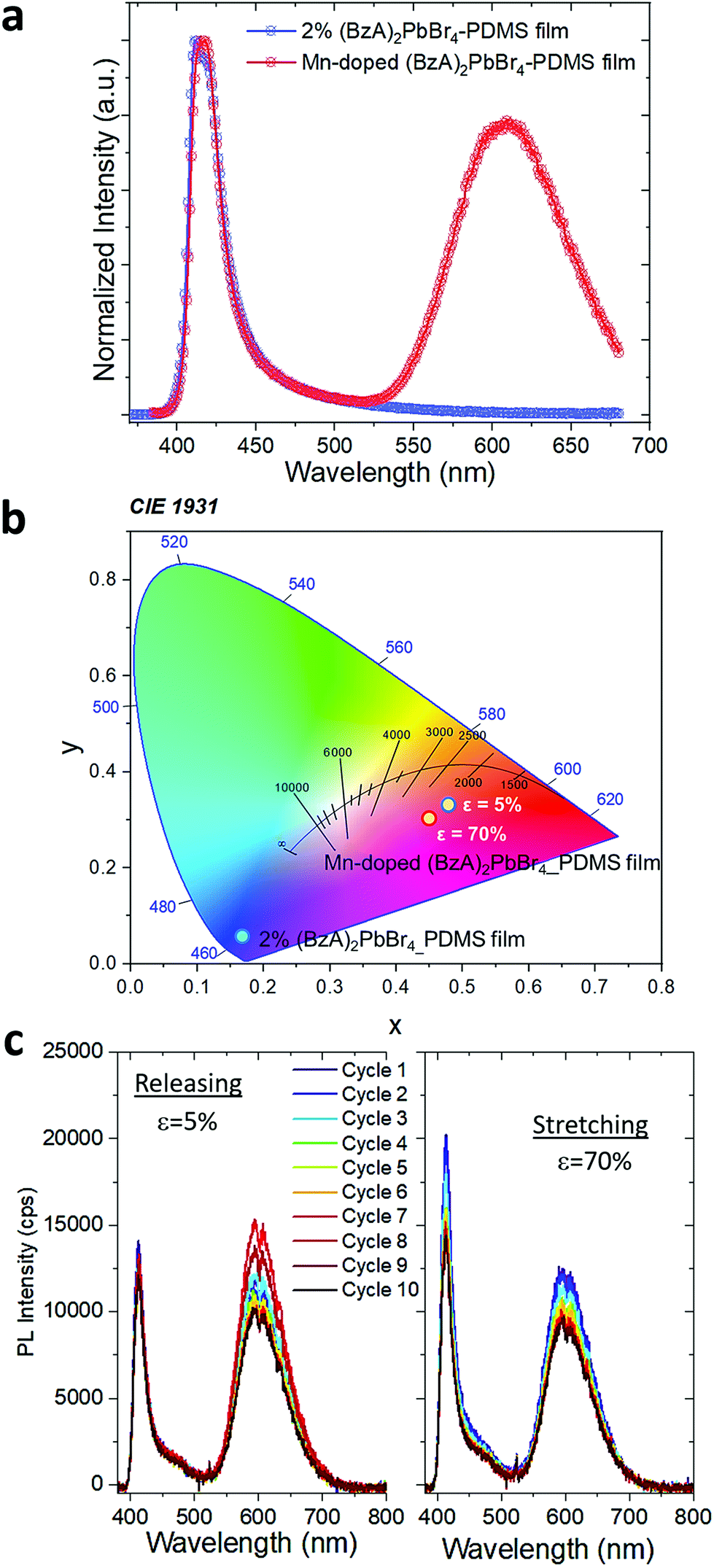 | ||
| Fig. 4 (a) Emission spectrum of the Mn-doped (BzA)2PbBr4–PDMS free-standing film showing a dual emission profile. (b) CIE chromaticity coordinates of the composite films. (c) Opto-mechanical response of Mn-doped (BzA)2PbBr4–PDMS free-standing film under continuous stretch/release mechanical cycles. | ||
The observed strain dependence of the blue emission intensity from the Mn-doped films is in agreement with our results from the undoped composite films and it appears as the dominant factor on the overall PL changes with cyclic stretching. Further assessment is needed to reveal the detailed impact of both platelet orientation and doping distribution over ensembles on their broad red emission while stretching the films, which probably requires contribution from different mechanisms.
Conclusions
In summary, we fabricated 2D layered perovskites embedded in an elastomeric free-standing film in situ, which allowed us to avoid any prior surface modification of the 2D nanomaterials. The resulting composites demonstrated up to 2.5 times enhanced emission under mechanical stretching. Their PL intensity can be modulated by the applied strain with reversibility for the entire duration of the test (up to 15 cycles). Furthermore, we observed a notable change in the emission profile of the platelets due to significant platelet re-orientation under low mechanical tensile stress of less than 2 MPa, without compromising the overall mechanical integrity of the composites. These observations are ascribed to a combined mechanism of reduced reabsorption, platelet orientation, and relative sliding of the platelets within the ensembles. We also demonstrated a simple yet convenient way to obtain dual emission from composite polymer films by using a Mn-doping strategy. The facile integration of stable layered structures in flexible films with tunable properties is a highly desired feature. This work offers new avenues to engineer composite films by controlling the strain-dependent aggregation and orientation of the embedded 2D layered perovskites through low mechanical stress.Experimental
Chemicals
Benzylamine (99%), PbBr2 (98%), hydrobromic acid (HBr, 48% m m−1 in water), SYLGARD®184 based PDMS, toluene and acetone were purchased from Sigma-Aldrich and employed without further purification.MHLP–PDMS film fabrication
All the films reported here were prepared by following this general protocol. The concentration of MHLP was controlled by using different amounts of BzA in the starting solutions. A perovskite stock solution of PbBr2 (0.41 mmol, 150 mg) dissolved in HBr (0.82 mmol, 95 μl) and acetone (640 μl) was prepared in a vial. The vial was shaken to produce a transparent solution. The SYLGARD® 184 based PDMS base and curing agents (SYLGARD® 184 part A and part B) in a weight ratio of 10:1 (5 g/0.5 g) were mixed and 500 μl of toluene were added to reduce the viscosity of the solution. Selected aliquots of the perovskite stock solution – 0 μl, 20 μl, 30 μl, and 50 μl – were each homogeneously dispersed in 1 ml of the polymer solution by vigorous shaking. Then, specific amounts of benzylamine, slightly above stoichiometry, were added to each vials, as reported in Table S1.† 500 μl of the resulting MHLP–PDMS solutions were carefully injected into a PTFE mould of 30 × 10 × 3 mm3. All the solutions were degassed for 1 h to remove air bubbles formed during the preparation. Finally, the moulds were closed with a glass slide and cured at 80° overnight in an oven. In parallel, the same protocol was followed to prepare the MHLP without polymer solution and used as a control sample, together with the films prepared without MHLP. The resulting crystals nucleated without polymer were collected from the vials, deposited on filter paper, and dried at room temperature. For the preparation of Mn-doped films, 0.2 mmol of MnBr2 were dissolved in a perovskite stock solution along with PbBr2 (0.41 mmol) and HBr (0.82 mmol) in acetone (640 μl) under shaking to produce a transparent solution. The Mn-doped film was prepared by following the same protocol reported for the preparation of undoped films. The cured MHLP–PDMS films were peeled-off from the moulds and cut in burr-free prismatic shapes of 20 × 4 mm2.
Structural and optical characterization
XRD patterns were collected from the MHLP synthesized without/with polymer at the reported concentrations by using a PANalytical Empyrean X-ray diffractometer operated at 45 kV and 40 mA and equipped with a 1.8 kW CuKα ceramic X-ray tube. The MHLP crystals were deposited directly on zero diffraction Si substrate for the analysis. In the case of the polymer films, the samples were carefully laid down flat on the substrates. SEM images of the MHLP crystals were collected on a FEI Nova 600 NanoLab instrument. Energy dispersive spectroscopy (EDS) analysis from the films was performed on a JEOL JSM-7500AS in scanning electron microscope mode. The initial assessment of photoluminescence (in the range from 370 to 680 nm) was performed on an Edinburgh Instruments fluorescence spectrometer (FLS920) equipped with a xenon lamp, exciting at 350 nm. The time-resolved photoluminescence analysis of the samples was performed after stretching the MHLP–PDMS composite films manually and placing the stretched film by using home-made clamps outside the Edinburgh Instruments fluorescence spectrometer (FLS920) with a time-correlated single-photon counting (TCSPC) unit coupled to a pulsed diode laser. The samples were excited at 375 nm using an optical fibre with 1 μs pulses at a repetition rate of 1 MHz and a spectral collection window of 10 nm. The absorbance spectra of the samples without polymer and the films loaded with 20 μl of perovskite stock solution were recorded on a Varian Cary 5000 UV-vis-NIR spectrophotometer equipped with an external diffuse reflectance accessory, and operating in absorption geometry. In the case of the absorbance measurements reported in Fig. S12,† the instrument was operated in diffuse reflectance employing the integrating sphere accessory and by using a solid holder supporting the stretched films.In situ PL recording under stretching
The MHLP–PDMS films were loaded into the Instron tensile dynamometer by pneumatic clamps as shown in Fig. S5.† A bifurcated optical fibre (with transmission in the range 300–1100 nm) was connected to a 349 nm ns-laser with a repetition of 1000 Hz and a Horiba i-hr320 spectrophotometer (grating: 600 grooves per mm), equipped with a CCD detector (Synapse). The common end of the fibre was used both to excite the sample and to collect the spectra from the same illuminated spot of around 0.5 mm localized on the region of maximum deformation. The samples were stretched at a rate of 20 mm min−1 until strain reached 70%, which corresponded to a tensile stress of ∼1.6 MPa, and released in a likewise manner, until 5% strain to avoid negative stress due to viscous hysteresis of the film. The PL response was recorded at the maximum and minimum of each cycle with a laser power of 100 μW. The maximum strain the film could withstand without breaking was about 80% and for the safe cyclic operation the in situ opto-mechanical response were collected below the maximum value. The effect of film deformation on the PL measurement was evaluated as follows: since the material is an elastomer, its Poisson ratio, ν, is close to 0.5. We assume ν = 0.45 to use a conservative value, taking into account the presence of the flakes. For large deformations, ΔL/L, the variations of width, W, and thickness, T, i.e. the strain along the respective directions, can be calculated with the classic equation: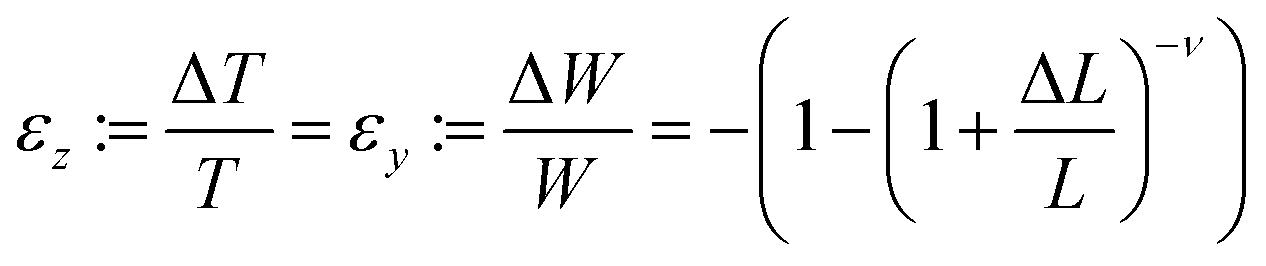
In our experiments only variations in the film thickness are considered since the spot size did not change.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MA and BD thank R. Pothuraju for the technical support on the SEM imaging of the bare platelets and S. Lauciello from the Electron Microscopy Facility at the Istituto Italiano di Tecnologia for the technical support on the EDS analysis of the platelets inside the polymer films. We thank the Materials Characterization Facility at the Istituto Italiano di Tecnologia for the technical support on the XRD and mechanical characterization. The research leading to these results has received funding from the European Union under the Marie Skłodowska-Curie RISE project COMPASS No. 691185. FDS acknowledge financial support from the European Research Council through the ERC Starting Grant “NANOLED” (851794).References
- F. Thouin, D. A. Valverde-Chávez, C. Quarti, D. Cortecchia, I. Bargigia, D. Beljonne, A. Petrozza, C. Silva and A. R. Srimath Kandada, Nat. Mater., 2019, 18, 349–356 CrossRef CAS.
- Y. Fu, H. Zhu, J. Chen, M. P. Hautzinger, X. Y. Zhu and S. Jin, Nat. Rev. Mater., 2019, 4, 169–188 CrossRef CAS.
- G. Grancini and M. K. Nazeeruddin, Nat. Rev. Mater., 2019, 4, 4–22 CrossRef CAS.
- S. Zeng, S. Shi, S. Wang and Y. Xiao, J. Mater. Chem. C, 2020, 8, 1319–1325 RSC.
- A. Fakharuddin, U. Shabbir, W. Qiu, T. Iqbal, M. Sultan, P. Heremans and L. Schmidt-Mende, Adv. Mater., 2019, 31, 1807095 CrossRef CAS.
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS.
- C. M. Mauck and W. A. Tisdale, Trends Chem., 2019, 1, 380–393 CrossRef CAS.
- D. Cortecchia, J. Yin, A. Petrozza and C. Soci, J. Mater. Chem. C, 2019, 7, 4956–4969 RSC.
- M. D. Smith and H. I. Karunadasa, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS.
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS.
- M. D. Smith, B. L. Watson, R. H. Dauskardt and H. I. Karunadasa, Chem. Mater., 2017, 29, 7083–7087 CrossRef CAS.
- X. Gong, O. Voznyy, A. Jain, W. Liu, R. Sabatini, Z. Piontkowski, G. Walters, G. Bappi, S. Nokhrin, O. Bushuyev, M. Yuan, R. Comin, D. McCamant, S. O. Kelley and E. H. Sargent, Nat. Mater., 2018, 17, 550–556 CrossRef CAS.
- Y. Liang, Q. Shang, Q. Wei, L. Zhao, Z. Liu, J. Shi, Y. Zhong, J. Chen, Y. Gao, M. Li, X. Liu, G. Xing and Q. Zhang, Adv. Mater., 2019, 31, 1903030 CrossRef.
- H. Li, Y. Qin, B. Shan, Y. Shen, F. Ersan, E. Soignard, C. Ataca and S. Tongay, Adv. Mater., 2020, 32, 1907364 CrossRef CAS.
- S. Liu, S. Sun, C. K. Gan, A. G. Del Águila, Y. Fang, J. Xing, T. T. H. Do, T. J. White, H. Li and W. Huang, Sci. Adv., 2019, 5, eaav9445 CrossRef CAS.
- Q. Tu, I. Spanopoulos, S. Hao, C. Wolverton, M. G. Kanatzidis, G. S. Shekhawat and V. P. Dravid, ACS Energy Lett., 2019, 4, 796–802 CrossRef CAS.
- S. Wang, Z. Gong, G. Li, Z. Du, J. Ma, H. Shen, J. Wang, W. Li, J. Ren, X. Wen and D. Li, Nanoscale, 2020, 12, 6644–6650 RSC.
- G. Xiao, Y. Cao, G. Qi, L. Wang, C. Liu, Z. Ma, X. Yang, Y. Sui, W. Zheng and B. Zou, J. Am. Chem. Soc., 2017, 139, 10087–10094 CrossRef CAS.
- D. Ghosh, A. Aziz, J. A. Dawson, A. B. Walker and M. S. Islam, Chem. Mater., 2019, 31, 4063–4071 CrossRef CAS.
- Y. Cao, G. Qi, L. Sui, Y. Shi, T. Geng, D. Zhao, K. Wang, K. Yuan, G. Wu, G. Xiao, S. Lu and B. Zou, ACS Mater. Lett., 2020, 2, 381–388 CrossRef CAS.
- Y. Yuan, X.-F. Liu, X. Ma, X. Wang, X. Li, J. Xiao, X. Li, H.-L. Zhang and L. Wang, Adv. Sci., 2019, 6, 1900240 CrossRef.
- A. Weingarten, Nat. Rev. Chem., 2019, 3, 513–513 CrossRef.
- A. Jaffe, S. A. Mack, Y. Lin, W. L. Mao, J. B. Neaton and H. I. Karunadasa, Angew. Chem., Int. Ed., 2020, 59, 4017 CrossRef CAS.
- A. Castelli, G. Biffi, L. Ceseracciu, D. Spirito, M. Prato, D. Altamura, C. Giannini, S. Artyukhin, R. Krahne, L. Manna and M. P. Arciniegas, Adv. Mater., 2019, 31, 1805608 CrossRef.
- Y. Shi, Z. Ma, D. Zhao, Y. Chen, Y. Cao, K. Wang, G. Xiao and B. Zou, J. Am. Chem. Soc., 2019, 141, 6504–6508 CrossRef CAS.
- C. Lv, X. Yang, Z. Shi, L. Wang, L. Sui, Q. Li, J. Qin, K. Liu, Z. Zhang, X. Li, Q. Lou, D. Yang, J. Zang, R. Liu, B. Liu and C.-X. Shan, J. Phys. Chem. C, 2020, 124, 1732–1738 CrossRef CAS.
- S. Mirershadi, F. Sattari, S. Golghasemi Sorkhabi and A. M. Shokri, J. Phys. Chem. C, 2019, 123, 12423–12428 CrossRef CAS.
- Y. Fang, L. Zhang, L. Wu, J. Yan, Y. Lin, K. Wang, W. L. Mao and B. Zou, Angew. Chem., 2019, 131, 15393–15397 CrossRef.
- S. Vikulov, F. Di Stasio, L. Ceseracciu, P. L. Saldanha, A. Scarpellini, Z. Dang, R. Krahne, L. Manna and V. Lesnyak, Adv. Funct. Mater., 2016, 26, 3670–3677 CrossRef CAS.
- D. Rhee, J. T. Paci, S. Deng, W.-K. Lee, G. C. Schatz and T. W. Odom, ACS Nano, 2020, 14, 166–174 CrossRef CAS.
- S. N. Raja, A. C. K. Olson, K. Thorkelsson, A. J. Luong, L. Hsueh, G. Chang, B. Gludovatz, L. Lin, T. Xu, R. O. Ritchie and A. P. Alivisatos, Nano Lett., 2013, 13, 3915–3922 CrossRef CAS.
- M. A. Koc, S. N. Raja, L. A. Hanson, S. C. Nguyen, N. J. Borys, A. S. Powers, S. Wu, K. Takano, J. K. Swabeck, J. H. Olshansky, L. Lin, R. O. Ritchie and A. P. Alivisatos, ACS Nano, 2017, 11, 2075–2084 CrossRef CAS.
- M. Xia, J.-H. Yuan, J. Luo, W. Pan, H. Wu, Q. Chen, K.-H. Xue, X. Miao, G. Niu and J. Tang, J. Mater. Chem. C, 2020, 8, 3814–3820 RSC.
- K.-Z. Du, Q. Tu, X. Zhang, Q. Han, J. Liu, S. Zauscher and D. B. Mitzi, Inorg. Chem., 2017, 56, 9291–9302 CrossRef CAS.
- B. Dhanabalan, A. Castelli, M. Palei, D. Spirito, L. Manna, R. Krahne and M. Arciniegas, Nanoscale, 2019, 11, 8334–8342 RSC.
- R. B. Pandey and B. L. Farmer, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2696–2710 CrossRef CAS.
- C. H. Swanson, T. Stimpfling, A.-L. Troutier-Thulliez, H. Hintze-Bruening and F. Leroux, J. Appl. Polym. Sci., 2013, 128, 2954–2960 CrossRef CAS.
- Q. Ba, A. Jana, L. Wang and K. S. Kim, Adv. Funct. Mater., 2019, 29, 1904768 CrossRef CAS.
- W. D. Rice, W. Liu, T. A. Baker, N. A. Sinitsyn, V. I. Klimov and S. A. Crooker, Nat. Nanotechnol., 2016, 11, 137–142 CrossRef CAS.
- R. Fainblat, C. J. Barrows, E. Hopmann, S. Siebeneicher, V. A. Vlaskin, D. R. Gamelin and G. Bacher, Nano Lett., 2016, 16, 6371–6377 CrossRef CAS.
- F. Meinardi, Q. A. Akkerman, F. Bruni, S. Park, M. Mauri, Z. Dang, L. Manna and S. Brovelli, ACS Energy Lett., 2017, 2, 2368–2377 CrossRef CAS.
- L. Zhang, T. Jiang, C. Yi, J. Wu, X.-K. Liu, Y. He, Y. Miao, Y. Zhang, H. Zhang, X. Xie, P. Wang, R. Li, F. Gao, W. Huang and J. Wang, J. Phys. Chem. Lett., 2019, 10, 3171–3175 CrossRef CAS.
- S. Das Adhikari, R. K. Behera, S. Bera and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 1530–1536 CrossRef CAS.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS.
- T. Sheikh and A. Nag, J. Phys. Chem. C, 2019, 123, 9420–9427 CrossRef CAS.
- H. Shao, X. Bai, H. Cui, G. Pan, P. Jing, S. Qu, J. Zhu, Y. Zhai, B. Dong and H. Song, Nanoscale, 2018, 10, 1023–1029 RSC.
- A. K. Guria, S. K. Dutta, S. D. Adhikari and N. Pradhan, ACS Energy Lett., 2017, 2, 1014–1021 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08043h |
| ‡ Current address: School of Engineering, Cardiff University, Queen's Buildings, The Parade, Cardiff CF24 3AA, Wales UK. |
| § Current address: IHP - Leibniz-Institut für Innovative Mikroelektronik, Im Technologiepark 25, D-15236 Frankfurt (Oder), Germany. |
| This journal is © The Royal Society of Chemistry 2021 |