
Ortho-vanillin derived Al(III) and Co(III) catalyst systems for switchable catalysis using ε-decalactone, phthalic anhydride and cyclohexene oxide†
Wilfred T.
Diment
,
Tim
Stößer
,
Ryan W. F.
Kerr
,
Andreas
Phanopoulos
,
Christopher B.
Durr
and
Charlotte K.
Williams
 *
*
Oxford Chemistry, Chemical Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
First published on 12th January 2021
Abstract
Switchable catalysis is a useful one-pot method to prepare block polyesters utilising a single catalyst exposed to a mixture of monomers. The catalyst is switched between lactone ring-opening polymerization (ROP) and epoxide/anhydride ring-opening copolymerization (ROCOP) by controlling its chain-end chemistry. Here, novel aluminium(III) (1) and cobalt(III) complexes (2), coordinated by ortho-vanillin derived salen ligands, show excellent switchable catalytic activity and selectivity for the preparation of poly(ε-decalactone-block-cyclohexene phthalate-block-ε-decalactone) [PDL-b-PCHPE-b-PDL]. Both complexes have competitive activities with a commercial chromium salen catalyst for epoxide/anhydride ROCOP (TOFCr(III) = 1200 h−1vs. TOFAl(III) = 350 h−1, 1 mol% catalyst loading vs. anhydride, 100 °C) and are significantly more active than the commercial catalyst for lactone ROP (TOFcr(III) = 3 h−1vs. TOFAl(III) = 300 h−1; 0.5 mol% catalyst loading vs. lactone, T = 100 °C). The catalysts are tolerant to low loadings (0.1 mol% vs. anhydride, 0.05% vs. lactone) and produce high molar mass triblock polyesters (Mn = 6–57 kg mol−1). The efficient production of high molar mass polyesters allows for future optimization of the block polyester thermal–mechanical properties and applications.
Introduction
Polyesters are important in packaging, consumer products, clothing and construction applications, as well as in medicine for wound healing, drug delivery and matrices in tissue engineering.1–3 They may be sustainable alternatives to hydrocarbon polymers because the ester linkage can be reversibly cleaved, enabling chemical recycling and, in some cases, biodegradation.4,5 Several commercial aliphatic polyesters are fully bio-based and many monomers could be bio-derived in the future.6–8 Today, most commercial polyesters are prepared by condensation polymerisations, but these uncontrolled methods are problematic for production of block polymers and prevent compositional sequence control. Block polyesters, prepared by controlled lactone ring-opening polymerisations (ROP) or by ring opening copolymerization (ROCOP) of epoxides and anhydrides, are thermoplastic elastomers, plastomers, rigid plastics, adhesives and coatings.9 Such applications benefit from block polyesters comprising a high glass transition temperature (Tg) or semi-crystalline material with a low glass transition temperature block.9–13 Here, catalysts are targeted to deliver block polyesters comprised of poly(ε-decaclactone) (PDL), Tg = −51 °C, and poly(cyclohexylene phthalate) (PCHPE), Tg = 133–146 °C.14–18 Such block polyesters have already shown promising performances, including high elasticity (elongation at break = 1000–1900%), excellent elastic recoverability (98%), wide operating (−51 to +138 °C) and processing (+100 to +200 °C) temperature windows.14 The polymer is fully degradable and the monomers are (ε-decalactone (ε-DL)), or could be (cyclohexene oxide (CHO), phthalic anhydride (PA)), bio-based.14,19,20 Currently, the catalysts used to make these block polyesters through switchable catalysis display low turnover frequencies for at least one of the catalytic cycles. Here, this deficiency is tackled by the developing catalyst systems displaying high activities for both the ROP of ε-DL and ROCOP of PA with CHO.The block polyester synthesis requires combination of two different controlled polymerizations: lactone ROP and ROCOP of epoxides and anhydrides.2 High performance lactone ROP catalysts are well developed and best suited to the production of aliphatic polyesters.9,21–23 Epoxide/anhydride ROCOP catalysts, in contrast, are less well developed but can deliver polyesters showing high rigidity, with aromatic backbones and functional side-chains.2,24 Switchable catalysis allows a single catalyst to access both of the polymerization cycles: it is straightforward to operate in a one-pot procedure (Scheme 1).25 In switchable polymerizations, the catalyst is self-directed between the two different cycles (ROP and ROCOP) by controlling the catalyst-growing polymer chain-end chemistry and this speciation is determined by the presence/absence of specific monomers.25
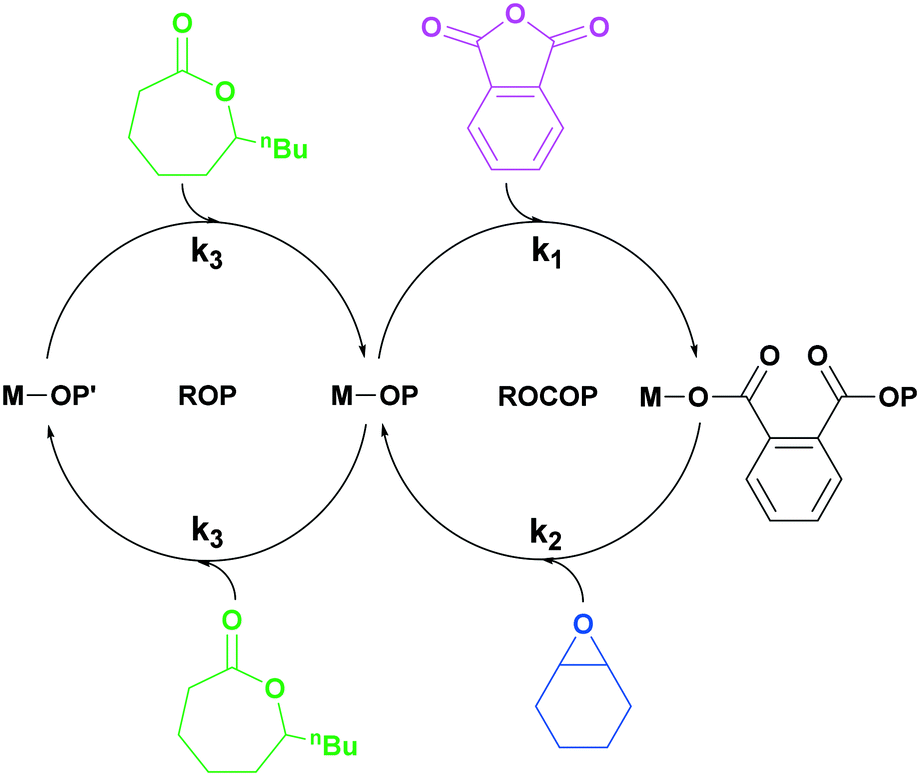 | ||
| Scheme 1 Illustration of switchable polymerization catalytic cycles using a single catalyst. This method allows for the selective formation of block polymers, commonly in these cycles k1 > k3.25 | ||
Although the block copolyesters targeted in this work are accessed through a switch in catalytic mechanism, there are several other methods through which ‘switches’ in catalytic selectivity can be achieved, resulting in changes of monomer selectivity in a range of catalysts.26 In particular, electrochemical switches have been utilised to synthesise a range of poly(lactide-b-ether) copolymers.27,28 Such copolymers have been demonstrated to display favourable mechanical properties when compared to PLA.29
Previously, we reported di-zinc switchable catalysts which selectively enchained block polymers from mixtures of lactones, epoxides, carbon dioxide and/or anhydrides.10,30–33 The two catalytic cycles are bridged by a central metal-alkoxide intermediate common to both pathways. Selectivity for a particular cycle arises due to differences in both the rates and stabilities of the linkages formed after insertion into the metal alkoxide bond. For mixtures of DL, CHO and PA, the dizinc alkoxide reacts selectively and preferentially with the anhydride (PA) compared to the lactone (DL), thus only PA–CHO ROCOP occurs until the complete consumption of PA.32,33 Provided there remains some CHO present, the metal alkoxide intermediate is re-generated and the ε-DL ROP cycle builds up the PDL blocks. Switchable catalysis works with other monomers, e.g. epoxides, anhydrides, carbon dioxide, lactones, lactide or O-carboxyanhydrides.14,20,34 It can be applied to up to three different catalytic cycles, i.e. enabling ABC-type block polymer structures, and, because it is a controlled polymerization, the addition of fresh monomer mixture results in the formation of multi-block polymers.14,20,35–38
The principles of switchable catalysis have now been shown to apply to other homogeneous catalysts, including dizinc, Zn(II)/Mg(II) heterodinuclear complexes, Zn(II) β-diiminates, Cr(III)/Co(III)/Al(III) salen catalyst systems, bicomponent organocatalysts and Lewis bases.35,36,38–45 Metal(III) salen catalyst systems are interesting due to the very broad monomer scope, commercial availability/ease of preparation of the catalyst and precedent for good rates in the homopolymerizations.2,18,46–62 To improve metal salen switchable catalysts two aspects need to be addressed: 1) the catalytic activity and 2) the initiation selectivity. Catalytic activities in switchable catalysis remain quite low, with turn-over-frequency (TOF) values of 1–10 h−1 for the ROP of DL (0. 25 mol% vs. lactone).35 Furthermore, catalyst loadings were generally quite high (1–0.25 mol%) and so should be reduced.35,36,38 To improve activity, metal salen catalysts require an equimolar amount of a co-catalyst, typically bis(triphenyl phosphoranylidene)ammonium chloride (PPNCl), but this complicates the chain end-group chemistry because chains are initiated from catalyst and co-catalyst.63 It is possible to control chain end-group chemistry by the addition of protic compounds, known as chain transfer agents (CTAs).64 By application of an excess of a bifunctional CTA, hydroxyl telechelic chains dominate but with the trade-off of producing low molar mass polymers (Mn < 5 kg mol−1). There is still a need for catalysts that can deliver both high activity and chain end selectivity in order to access high molar mass block polyesters showing block phase separation.
Here, tetradentate Schiff base ligands derived from the bio-based o-vanillin are investigated. There is a rich coordination chemistry for such o-vanillin derived Schiff base ligands, especially in the production of bimetallic complexes and helicates.65 In terms of polymerization catalysis, we previously reported di-Zn(II) o-vanillin catalysts which showed good activity in PA/CHO ROCOP (TOF = 198 h−1, 0.1 mol% catalyst, 100 °C).66 Recently, the same o-vanillin ligands coordinated to Al(III) were reported for lactide ROP catalysis (TOF = 800 h−1, 1 mol%, 120 °C).67 We have also reported bidentate o-vanillin Ti(IV) catalysts, coordinated by two chelating ligands, which showed very high rates in caprolactone ROP (CL ROP, TOF = 1800 h−1, 0.5 mol%, 80 °C).68
Here, we target the effective tetradentate ortho-vanillin ligand for coordination with metal centres known to catalyse both lactone ROP and epoxide/anhydride ROCOP, i.e. Al(III) and Co(III), to overcome the known limitations of metal salen catalysts in switchable catalysis.2,24
Results and discussion
Catalyst synthesis
In order to investigate the tetradentate o-vanillin complexes in switchable catalysis, two complexes were targeted (Scheme 2, see ESI† for experimental details). The proligand, LvanH2, was synthesised through the Schiff base condensation reaction of o-vanillin with 2,2-dimethyl-1,3-propanediamine.66 Complex 1 was synthesised through reaction of LvanH2 with AlEt3 in toluene and was isolated in 68% yield following its precipitation from the reaction solution. Complex 2 was synthesised through reaction of LvanH2 with [Co(OAc)2·4H2O] in THF to give, in situ, the Co(II) adduct, which was subsequently exposed to oxygen, allowing for oxidation to the Co(III) containing complex 2. The complex slowly precipitated from the reaction solution, allowing for its isolation in 70% yield. Both complexes were characterised by solution phase NMR spectroscopy, elemental analysis and single crystal X-ray diffraction (Fig. S1–S11†).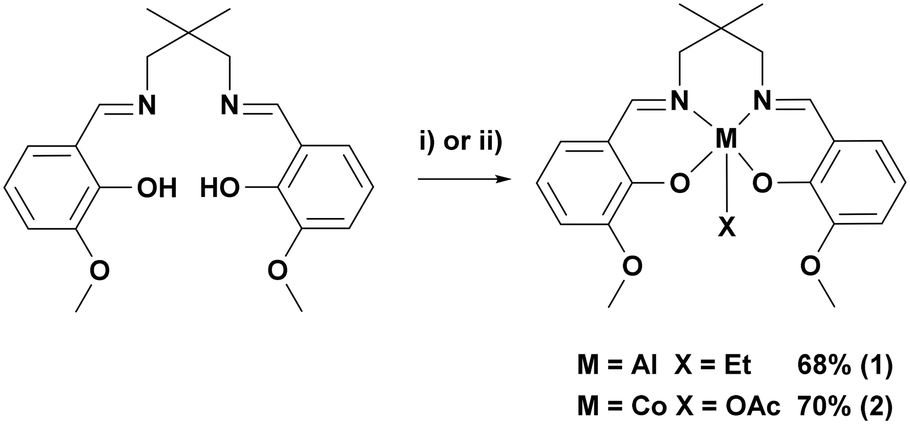 | ||
| Scheme 2 Synthesis of complexes 1 and 2. i) AlEt3, toluene, −30–20 °C, 2 h, 68%. ii) Co(OAc)2·4H2O, THF, 20 °C, 2 h followed by exposure to O2, 20 °C, 48 h, 70%. | ||
Single crystals of complex 1, suitable for X-ray crystallography, were obtained via layering a saturated THF solution of the complex with hexane, whilst single crystals of complex 2 were obtained via layering a saturated DCM solution of the complex with hexane (Fig. 1). In both cases the complexes are monometallic in the solid state, although the coordination geometry of the metal differs. In complex 1, the aluminium possesses a distorted trigonal bipyramidal geometry (N(1)–Al(1)–O(3) ∠ 167.80°, τ5 = 0.79), whilst in complex 2 the cobalt possesses a distorted octahedral geometry, with the vanillin ligand coordinated in a cis-β fashion and a κ2-acetate co-ligand.69,70
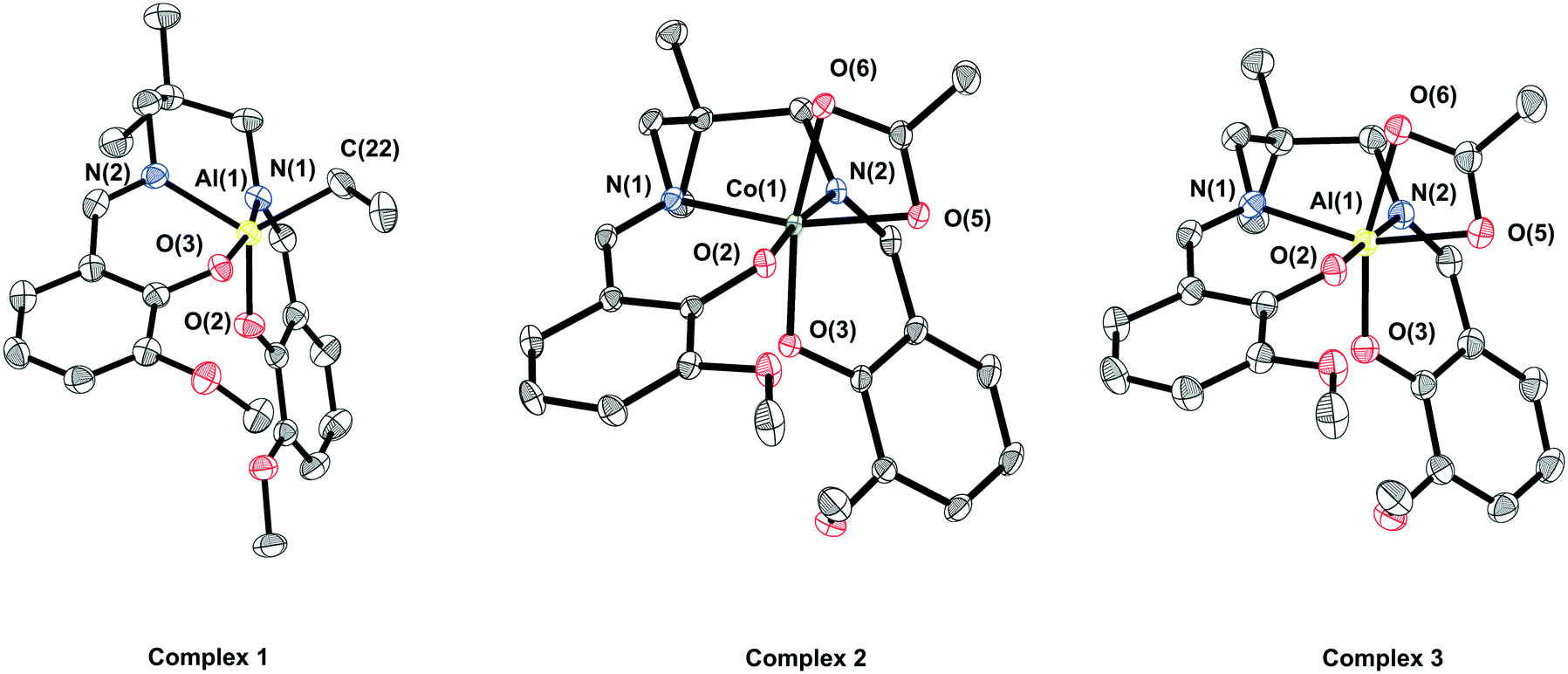 | ||
| Fig. 1 ORTEP representations of structures of complexes 1, 2 and 3 obtained by single crystal X-ray diffraction. H-Atoms have been omitted for clarity and thermal ellipsoids are at 50% probability. Selected bond angles and lengths are presented in Tables S1–S4.† | ||
The 1H NMR spectrum of complex 1 at room temperature is consistent with Cs symmetry in solution. The o-vanillin ligand environments are observed as nine distinct resonances, with both the methyl and methylene protons of the dimethylpropylene backbone being diastereotopic, and with two distinct resonances for the ethyl co-ligand. In comparison, the 1H NMR spectrum of complex 2 is consistent with the C1 symmetry observed in the solid state and sixteen distinct peaks are observed for the o-vanillin ligand environments, together with a single resonance for the methyl group of the acetate co-ligand. This spectroscopic data is consistent with the asymmetric cis-β ligand coordination mode.70 The 13C{1H} NMR spectra for both complexes are also fully consistent with the proposed structures.
To directly compare coordination chemistries, the complex LvanAlOAc (3) was synthesised via the reaction of complex 1 with one equivalent of acetic acid in THF, and was isolated in 79% yield. The complex was characterised by solution phase NMR spectroscopy and X-ray crystallography (Fig. S12–S17†). Compared to complex 2, its 1H NMR is analogous to that of complex 1, i.e. indicative of Cs symmetry. This finding implies either that there is a κ1-acetate co-ligand (and hence a pentacoordinate aluminium centre), or there is rapid interconversion of a κ2-acetate co-ligand with a single averaged coordination isomer being observed by NMR spectroscopy. To further investigate both complexes 1 and 3 were analysed by 27Al{1H} NMR spectroscopy (Fig. S6 and S17,† respectively). Whilst the spectrum of complex 1 features a broad resonance at 5.7 ppm, the spectrum of complex 3 features a sharp peak at 14.8 ppm. Since the Al nucleus is quadrupolar, a sharp resonance will only be observed for a symmetric coordination environment, hence complex 3 should have an octahedrally coordinated aluminium centre. It is therefore proposed that, in solution, complex 3 undergoes rapid interconversion between two octahedrally coordinated isomers.
Single crystals of 3, suitable for X-ray crystallography, were obtained via layering a saturated CHCl3 solution of the complex with hexane (Fig. 1). In the solid state, complex 3 shows a similar structure to complex 2, with the aluminium centre possessing a distorted octahedral geometry, the vanillin ligand showing a cis-β coordination geometry, and a κ2-acetate co-ligand. The solid state data further supports the notion that the symmetric NMR spectra result from the rapid interconversion of coordination isomers, rather than being attributed to a pentacoordinate coordination geometry.
Polymerization catalysis
Prior to investigating catalysts 1 and 2 for switchable catalysis, it was first necessary to understand their performances in the component polymerisations, i.e. PA/CHO ROCOP and ε-DL ROP. A commercial salen catalyst, [SalcyCrCl] previously reported for switchable catalysis, was investigated under equivalent conditions as the benchmark (Table 1).35 The two new catalysts, as well as the commercial [SalcyCrCl] catalyst system, were effective for the ROCOP of PA and CHO, displaying good activity and excellent (>99%) selectivity for polyester linkages (Table 1, entries 1–3). The activity TOF, for complex 1 (350 h−1) was approximately half that of complex 2 (516 h−1), which itself has a TOF approximately half that of commercial [SalcyCrCl] (1160 h−1). Full conversion of PA could be obtained for complexes 1 or 2 by leaving the reactions for 15 minutes (Table S5†). Polymerizations were all conducted with the addition of PPNCl as co-catalyst, since it is known to enhance rates, and with two equivalents of diol, 1,2-cyclohexanediol (CHD), to ensure production of hydroxyl telechelic polyesters.24,50,51 Under these conditions, bimodal molar mass distributions result, with the lower molar mass series being attributed to α-chloride(or acetate)-ω-hydroxy terminated polyester and the higher molar mass series due to α,ω-hydroxy terminated polyester (Fig. S18 and S19†).35,51| Entry | Catalyst | Monomer | [Mon]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Cat] :[Cat] |
Time (min) | Conv. (%) | TONb | TOFc (h−1) | M n,GPC [Đ]d (kg mol−1) | M n,Th (kg mol−1) |
|---|---|---|---|---|---|---|---|---|---|
|
a Catalysis conditions: [Cat]:[PPNCl]:[CHD]:[monomer]:[CHO] = 1:1:2:x:500, where x is given, T = 100 °C.
b TON (turnover number) = number of moles of monomer consumed/number of moles of catalyst.
c TOF (turnover frequency) = TON per unit time.
d Determined by GPC, in THF, using narrow dispersity polystyrene standards.
e Theoretical Mn values. Determined through (TON × Mn(repeat unit))/(number of equivalents of CHD + number of equivalents of PPNCl (+ number of equivalents of catalyst when 2 and [SalcyCrCl] are utilised)).
|
|||||||||
| 1 | 1 | PA | 100 | 5 | 29 | 29 | 350 | 2.2 [1.11] | 2.5 |
| 2 | 2 | PA | 100 | 5 | 43 | 43 | 516 | 3.5 [1.12] | 2.7 |
| 3 | [SalcyCrCl] | PA | 100 | 5 | 97 | 97 | 1160 | 5.1 [1.17] | 6.1 |
| 4 | 1 | DL | 200 | 30 | 82 | 164 | 330 | 15.6 [1.19] | 9.3 |
| 5 | 2 | DL | 200 | 180 | 96 | 192 | 64 | 12.2 [1.27] | 8.1 |
| 6 | [SalcyCrCl] | DL | 200 | 4320 | 94 | 188 | 3 | 7.1 [1.30] | 8 |
The three complexes were also tested for the ROP of ε-DL (Table 1, entries 4–6). All catalysts were highly selective for PDL formation, with no epoxide ROP or ether linkage formation. Both catalysts 1 and 2 show good catalytic activity and, different to PA/CHO ROCOP, complex 1 is the most active (TOF = 330 h−1), while complex 2 is significantly slower (TOF = 64 h−1). Both new catalysts were significantly faster than the commercial [SalcyCrCl] (TOF = 3 h−1). It has previously been demonstrated that the presence of o-methoxy groups can lead to high activity for lactide ROP for Al(III) salen derivative catalysts.68 The presence of both monofunctional (chloride, acetate) and bifunctional (CHD) initiating groups in bimodal PDL molar mass distributions in all cases (Fig. S20†).
As both novel complexes showed good activity in the component polymerisations, next it was necessary to uncover the optimal loading of diol, i.e. chain transfer agent, to deliver a majority of α,ω-hydroxy terminated polyesters. The goal is to enable a high selectivity for hydroxyl telechelic chains, since these are needed to deliver ABA triblock polyester structures, whilst also maximising the overall polyester molar masses to allow for block phase separation. Previous switchable catalysis investigations using [SalcyCrCl] required use of 15 equivalents of diol (vs. catalyst) to deliver monomodal molar mass distributions.35 In switchable catalysis, epoxide–anhydride ROCOP usually occurs first, hence the optimization of molar mass was conducted for PA/CHO ROCOP (Fig. 2).
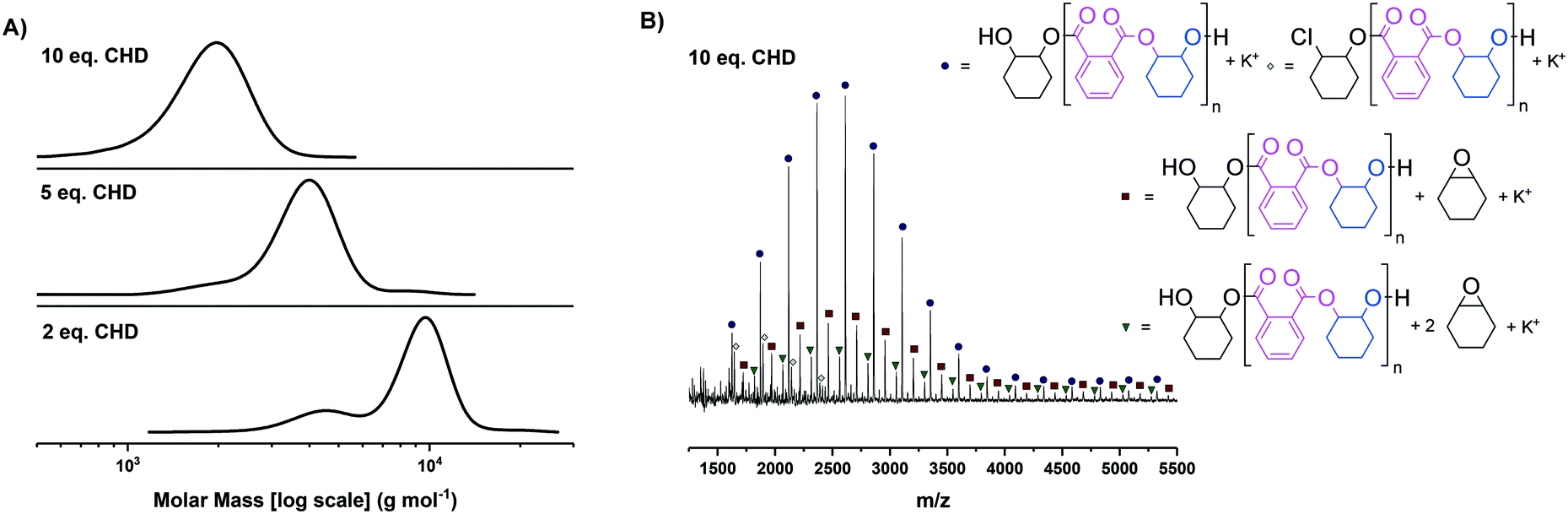 | ||
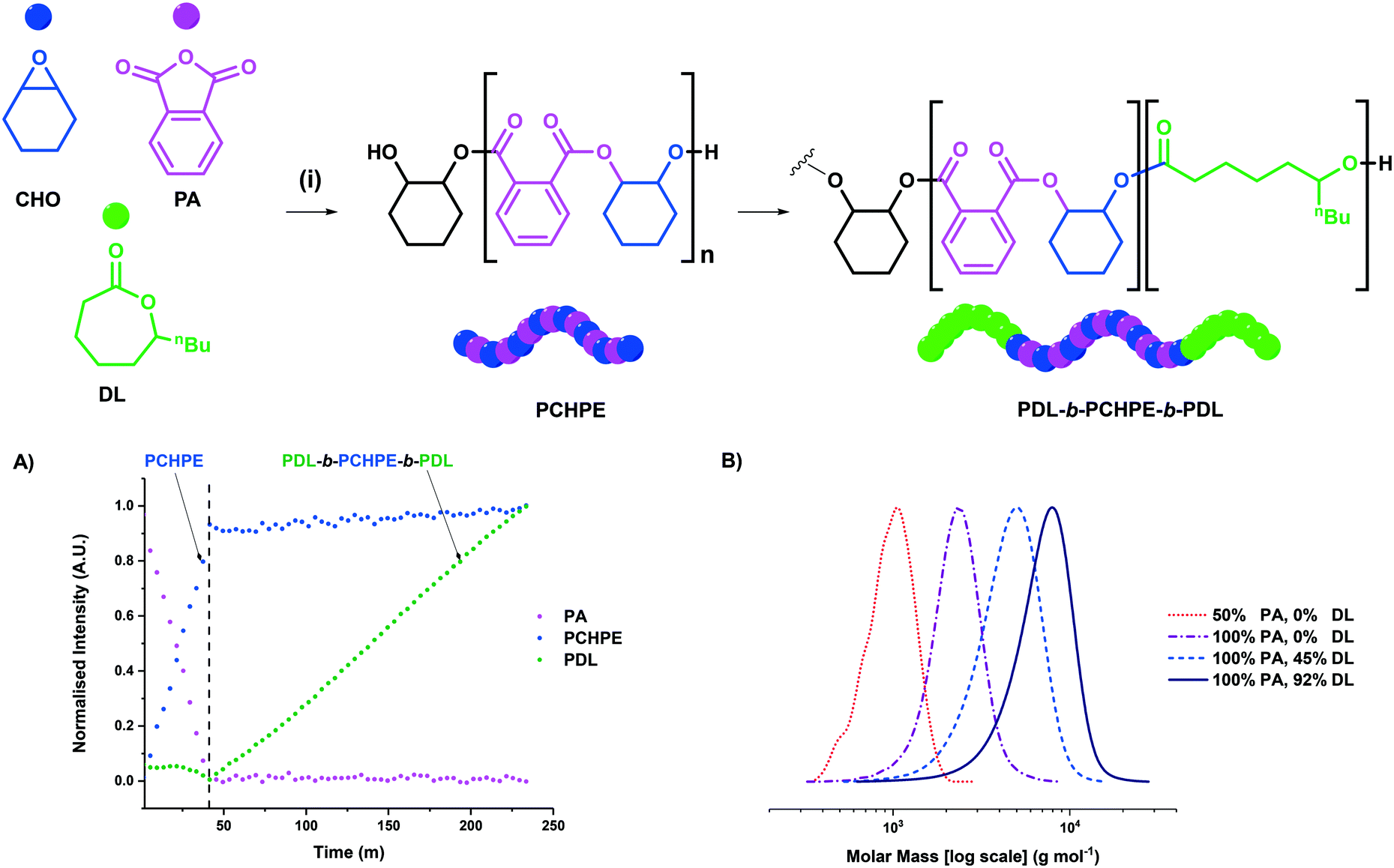
| Fig. 2 A) Molar mass distributions for the PA/CHO ROCOP polyester (poly(cyclohexene-alt-phthalate)) [PCHPE] synthesised with complex 1, showing that 10 equivalents of diol (chain transfer agent) are sufficient to yield monomodal molar mass distributions. B) MALDI-TOF spectrum of PCHPE synthesised with 10 equivalents of 1,2-cyclohexane diol (CHD). | ||
For both complexes 1 and 2, clearly monomodal molar mass distributions were observed only when 10 equivalents of diol (CHD) were applied (Fig. 2A for complex 1, Fig. S21A† for complex 2). The MALDI-TOF analysis of these polyesters showed the majority desired telechelic α,ω-hydroxy polyester with only a minor series attributed to α-chloro-ω-hydroxy polyester (and, for complex 2 α-acetate-ω-hydroxy polyester) (Fig. 2B and S21B†).
Utilising these conditions, complexes 1 and 2 were subsequently applied for switchable catalysis using mixtures of CHO, PA and DL. Polymerizations were monitored using in situ ATR-IR spectroscopy and showed that PA/CHO ROCOP proceeded first with high selectivity. Only after complete PA consumption did DL ROP occur (Fig. 3). By taking regular aliquots from a representative polymerisation reaction, the monomer conversions and polyester molar mass were measured using 1H NMR spectroscopy and GPC. Block polyester formation was observed, as evidenced by a continual increase in polymer molar mass with conversion and the retention of monomodal distributions and narrow dispersities throughout the reaction (Fig. 3). The triblock polyester was isolated, via precipitation from methanol, and chain end-group analysis was conducted using 31P{1H} NMR spectroscopy according to previously published methods (see ESI† for details).25,32,35,36,38 The end-group analysis confirmed triblock polyester formation since only PDL end-groups were detected (Fig. S22†). The 1H DOSY NMR spectroscopy showed that all polyester signals have the same diffusion coefficient, again indicative of block polyester formation (Fig. S23†). Taken together, these data are consistent with successful switchable polymerization catalysis and the formation of triblock polyester PDL-b-PCHPE-b-PDL. It's relevant to note that, following precipitation, the triblock polyester produced using catalyst 1 (Al-catalyst) was colourless.
 | ||
| Fig. 3 Switchable polymerization catalysis using PA, CHO and DL utilising complex 1. (i) [Cat]:[PPNCl]:[CHD]:[PA]:[DL]:[CHO] = 1:1:10:100:200:500, T = 100 °C. A) Data from ATR-IR spectroscopy showing the conversion of the different blocks with time. The green series shows formation of PDL, whilst the pink and blue series indicate PA consumption and PCHPE formation, respectively. B) Evolution of polyester molar mass with conversion, from reaction aliquots. | ||
In switchable polymerizations, catalyst 1 was significantly more active than complex 2 (Table 2, entries 1 and 2). The activity order results from the markedly higher activity of complex 1 for ε-DL ROP (vide supra). Both complexes are significantly more active than the benchmark commercial [SalcyCrCl] catalyst, which required >24 hours to produce the triblock polyester.35 Given the promising performance of catalyst 1, its loading was sequentially reduced from 1 to 0.2 to 0.1 mol%, so as to target progressively higher molar mass polyesters (Table 2, entries 3 and 4). The catalyst remains active under all conditions, and produced triblock polyester with molar masses of up to 57.1 kg mol−1, with low dispersity and monomodal distributions. The polyester molar mass can also be controlled, at the desirable lowest catalyst loading, by the addition of greater quantities of diol (Table 2, entry 5).
| Entry | Catalyst | Loading | CHD | Time (h) | Conv. (PA%, DL%) | M n,GPC [Đ]b (kg mol−1) | M n,Th (kg mol−1) | T g (°C) |
|---|---|---|---|---|---|---|---|---|
|
a Polymerization conditions: [Cat]:[PPNCl]:[CHD]:[PA]:[DL]:[CHO] = 1:1:10:x:y:z, where [PA]:[DL]:[CHO] = 1:2:5 and x is given as loading (i.e. 1% loading: [Cat]:[PA] = 1:100), T = 100 °C.
b Determined by GPC, in THF, using narrow dispersity polystyrene standards.
c Theoretical Mn values. Determined through (TON × Mn(repeat unit))/(number of equivalents of CHD + number of equivalents of PPNCl (+ number of equivalents of catalyst when 2 or [SalcyCrCl])).
d Polymerization conditions: [Cat]:[PPNCl]:[CHD]:[PA]:[DL]:[CHO] = 1:1:15:200:200:250, 2.2 mL toluene, 100 °C.
|
||||||||
| 1 | 1 | 1 | 10 | 2 | 100, 94 | 6.5 [1.16] | 5.3 | −22.3 |
| 2 | 2 | 1 | 10 | 4.5 | 100, 96 | 6.3 [1.16] | 4.8 | −17.7 |
| 3d | [SalcyCrCl] | 0.5 | 15 | >24 | 100, 100 | 6.2 [1.20] | 4.9 | N.d. |
| 4 | 1 | 0.2 | 10 | 7 | 100, 93 | 33.0 [1.14] | 25.7 | −48.2 |
| 5 | 1 | 0.1 | 10 | 18 | 100, 92 | 57.1 [1.07] | 52.0 | −50.5 |
| 6 | 1 | 0.1 | 15 | 18 | 100, 90 | 42.8 [1.07] | 35.9 | −50.5 |
Switchable polymerizations conducted at the lowest 0.1 mol% catalyst loading sometimes show GPC data with a minor intensity low molar mass shoulder (Fig. S24†). This is attributed to a low concentration of AB block polyester formed as a consequence of the residual monofunctional chloride initiators. The speciation was confirmed through MALDI-TOF analysis of aliquots taken during the PA/CHO ROCOP phase where low intensity peaks due to α-chloro-ω-hydroxy polyester are observed (Fig. S25†).
Previously investigated switchable catalysts have often failed to produce high molar mass polymers, owing to both low catalytic activity and the requirement for high loading of CTA to deliver monomodal mass distributions.35,36,38 To achieve block phase separation, using polyesters from DL, PA and CHO, high molar masses are required owing to weak segregation strength of the PDL and PCHPE blocks.14 Achieving block phase separation may be important to deliver the best material properties.14 Complex 1 is notable in showing good activity at loadings as low as [Cat]:[CHO] = 1:5000, enabling access to higher mass polymers. The resulting triblock polyesters were analysed by DSC which showed a single Tg, assigned to the PDL block (Fig. S26†). It is important to note that the upper glass transition temperature, attributed to the PCHPE block, is known to be of low intensity and difficult to observe by DSC.14 Clear evidence for block phase separation in the higher molar mass polyester samples arises from the similarity of the Tg value to pure PDL (Tg(block) = −50 °C, Tg(PDL) = −51 °C). In contrast, lower molar mass triblock polyesters show partial block miscibility.
Discussion
Complexes 1 and 2 show good activity for switchable polymerisations using ε-DL, PA and CHO, with complex 1 capable of producing triblock polyesters with molar mass of 57.1 kg mol−1, within 18 hours. Such activity values are high, particularly when compared against other switchable catalysts (Fig. 4, Table 3). To aid comparison with other switchable catalysts, the measure Mn per hour was used. Catalysts must also display both good chain-end control and high activities in order to achieve high molar mass triblock polymers, therefore, a simple comparison of turn-over-frequency (TOF) values may not be the most suitable. The commercial salen catalyst system, [SalcyCrCl] + PPNCl, produced PDL-b-PCHPE-b-PDL with molar mass values up to 6.2 kg mol−1, but required more than 24 hours to reach this weight, giving a Mn per hour of 0.26 kg mol h−1 (Table 3, entry 2). The low activity largely results from the slow rate for ε-DL ROP (TOF = 3 h−1). The triblock polyester is a fully miscible amorphous polyol, thus chain extension processes are required to yield higher molar mass polymers.35 In 2015, we reported a dizinc catalyst featuring an organometallic co-ligand which is non-initiating, [LZn2Ph2]. This complex was used with a diol (CHD) to selectively produce PDL-b-PCHPE-b-PDL.32 The dizinc catalysts shows lower TOFs for both PA/CHO ROCOP and DL ROP, TOF = 25 h−1 and 160 h−1, respectively, which are ca. 14 times and 2 times lower than those reported for complex 1. Nonetheless, because the di-zinc catalyst does not have competitive initiating groups it can access molar masses up to 34.6 kg mol−1 in 4 hours, a respectable Mn per hour of 6.2 kg mol−1 h−1 (Table 3, entry 3). The resulting triblock polyesters were chain extended to access multi-block polyesters with molar masses up to 56.6 kg mol−1 which were thermoplastic elastomers or shape memory plastics, depending on the block composition.10 Compared with the di-zinc catalyst, complex 1 produced the block polyester of equivalent molar mass without requiring any chain extension process. This performance is feasible because of its better activity and ability to tolerate low catalyst loadings (i.e. 0.1 mol%). The ability of complex 1 to function effectively at 1:1000 [catalyst]:[anhydride] loading is notable since most catalysts in this field are reported at an order of magnitude higher loading (i.e. 1:100 or 1 mol%).
 | ||
| Fig. 4 A selection of literature catalysts reported for switchable catalysis using various monomers, as described in Table 3.10,14,19,35,43 | ||
| Entry | Catalysta | Monomers | Temp. (°C) | Time (h) | M n,GPC, [Đ]b (kg mol−1) | M n,GPC per hourc (kg mol−1 h−1) |
|---|---|---|---|---|---|---|
|
a Representative conditions as reported for each catalyst. [1]:[PPNCl]:[CHD]:[CHO]:[PA]:[DL] = 1:1:10:5000:1000:2000. [SalcyCrCl]: [SalcyCrCl]:[PPNCl]:[CHD]:[CHO]:[PA]:[DL] = 1:1:15:250:200:200, 2.2 mL toluene.35 [LZn2Ph2]:[Cat]:[CHD]:[CHO]:[PA]:[DL] = 1:2:800:100:100.10 [LZnMg(C6F5)2]:[1,4-BDM]:[CHO]:[PA]:[DL] = 1:4:1050:700:1500.14 [tBuP1]:[BDM]:[CHO]:[PA]:[LA] = 1:2:500:100:100.43 [Et3B]:[DBU]:[CTA]:[PO]:[PA]:[LA] = 3:1:5:500:100:100.19
b As reported under conditions described in respective literature.
c Molar mass of block polymer per unit time.
|
||||||
| 1 | 1 | CHO/PA/DL | 100 | 18 | 57.1 [1.07] | 3.2 |
| 2 | SalcyCrCl | CHO/PA/DL | 100 | >24 | 6.2 [1.20] | 0.26 |
| 3 | LZn2Ph2 | CHO/PA/DL | 100 | 4 | 34.6 [1.19] | 6.2 |
| 4 | LZnMg(C6F5)2 | CHO/PA/DL | 80 | 36 | 108 [<1.10] | 3 |
| 5 | t BuP1 | CHO/PA/LA | 100 | 8 | 6.6 [1.18] | 0.83 |
| 6 | Et3B/DBU | PO/PA/LA | 60 | 3 | 6.4 [1.12] | 2.1 |
There have also been a number of reports of effective organocatalysts for switchable catalysis and these warrant comparison against complex 1. Li et al. reported the use of the phosphazine base tBuP1 for switchable catalysis, producing PLA-b-PCHPE-b-PLA with a molar mass of 6.6 kg mol−1, over 8 hours, an Mn per hour of 0.83 kg mol−1 h−1 (Table 3, entry 5).43 One benefit of this system is its ability to function without a co-catalyst, hence it should not require such a large excess of chain transfer agent to yield block polymers. Nonetheless the catalytic activity remains moderate and so accessing higher molar mass polyesters, a common challenge for organocatalysts, was not reported. In the exemplar switchable catalysis using this organocatalyst, the block polyester comprises PLA portions which cannot deliver the desired low Tg for thermoplastic elastomer applications, although it may prove useful in other material contexts. Bicomponent catalysts comprising Et3B/DBU Lewis acid/base pairs, reported by Wang et al., were also capable of switchable catalysis from mixtures of propylene oxide (PO), PA and LA yielding P(PO-alt-PA)-b-PLA.19 By installing a functional chain end-group, RAFT polymerisation of methyl methacrylate formed PMMA-b-P(PO-alt-PA)-b-PLA. In this catalysis, the diblock polyester has a low molar mass (6.4 kg mol−1) but is efficiently produced within 3 hours using 5 equivalents of chain transfer agent and at 60 °C, giving a Mn per hour of 2.1 kg mol−1 h−1 (Table 3, entry 6).
This year, we reported a heterodinuclear catalyst [LZnMg(C6F5)2] which again features an organometallic co-ligand and obviates need for a large excess of chain transfer agent.14 This heterodinuclear catalyst allows for the synthesis of PCHC-b-PDL-b-PCHC or PCHPE-b-PDL-b-PCHPE. In the case of the polyesters, the molar masses reached 100 kg mol−1 (Table 3, entry 4). These materials were accessed through a one-pot, sequential addition catalysis yielding ABA structure triblocks, whilst the switchable catalysis discussed in this work yields the inverse BAB-structure polymers.
Conclusions
In conclusion, two new ortho-vanillin Schiff base Al(III) and Co(III) catalyst systems for switchable catalysis were reported. The under-pinning catalytic activities and selectivities for PA/CHO ROP and ε-DL ROP revealed good performances. Hence when the catalysts were exposed to mixtures of PA, CHO and DL, both were efficiently able to produce triblock polyesters with molar masses up to 57.1 kg mol−1 within 18 hours. These catalysts show objectively fast rates, tolerance to low loadings and an ability to access high molar mass block polyesters compared to other leading catalysts in this field. The resulting triblock polyesters showed evidence of phase separation which highlights the opportunity to use these catalyst systems in future to deliver material property improvements. These Schiff base catalysts are straightforward to synthesise and present a plethora of opportunities for future investigations and structure–activity correlations. For example, it is straightforward to vary the backbone linker (stereo)chemistry, phenolate substituents and to reduce the imine functionalities. Given the promising proof of concept studies in switchable catalysis uncovered in this work, future optimization of catalyst structure and monomer substrate scope are warranted.Conflicts of interest
The authors have no competing interests to declare.Acknowledgements
The EPSRC (EP/S018603/1, EP/R027129/1), Oxford Clarendon Scholarship (W.D.), SCG Chemicals and the Oxford Martin School (Future Plastics Programme), are acknowledged for research funding.Notes and references
- E. Castro-Aguirre, F. Iniguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- R. De Clercq, M. Dusselier and B. F. Sels, Green Chem., 2017, 19, 5012–5040 RSC.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS.
- Y. Zhu, M. R. Radlauer, D. K. Schneiderman, M. S. P. Shaffer, M. A. Hillmyer and C. K. Williams, Macromolecules, 2018, 51, 2466–2475 CrossRef CAS.
- A. Watts, N. Kurokawa and M. A. Hillmyer, Biomacromolecules, 2017, 18, 1845–1854 CrossRef CAS.
- M. T. Martello, D. K. Schneiderman and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2014, 2, 2519–2526 CrossRef CAS.
- D. K. Schneiderman, E. M. Hill, M. T. Martello and M. A. Hillmyer, Polym. Chem., 2015, 6, 3641–3651 RSC.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- M. T. Martello, A. Burns and M. Hillmyer, ACS Macro Lett., 2011, 1, 131–135 CrossRef.
- X. Yu, J. Jia, S. Xu, K. U. Lao, M. J. Sanford, R. K. Ramakrishnan, S. I. Nazarenko, T. R. Hoye, G. W. Coates and R. A. DiStasio, Jr., Nat. Commun., 2018, 9, 2880 CrossRef.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
- G. Si, L. Zhang, B. Han, Z. Duan, B. Li, J. Dong, X. Li and B. Liu, Polym. Chem., 2015, 6, 6372–6377 RSC.
- S. Zhu, Y. Zhao, M. Ni, J. Xu, X. Zhou, Y. Liao, Y. Wang and X. Xie, ACS Macro Lett., 2020, 9, 204–209 CrossRef CAS.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Pena Carrodeguas, G. Trott, A. Santmarti, K. Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazina and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- T. Stößer, T. T. D. Chen, Y. Zhu and C. K. Williams, Philos. Trans. R. Soc., A, 2018, 376, 20170066 CrossRef.
- A. M. Doerr, J. M. Burroughs, S. R. Gitter, X. Yang, A. J. Boydston and B. K. Long, ACS Catal., 2020, 14457–14515, DOI:10.1021/acscatal.0c03802.
- A. Lai, Z. C. Hern and P. L. Diaconescu, ChemCatChem, 2019, 11, 4210–4218 CrossRef CAS.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS.
- C. Diaz, T. Tomković, C. Goonesinghe, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2020, 53, 8819–8828 CrossRef CAS.
- D. C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS.
- S. K. Raman, R. Raja, P. L. Arnold, M. G. Davidson and C. K. Williams, Chem. Commun., 2019, 55, 7315–7318 RSC.
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef.
- T. Stößer, D. Mulryan and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 16893–16897 CrossRef.
- T. T. D. Chen, Y. Zhu and C. K. Williams, Macromolecules, 2018, 51, 5346–5351 CrossRef CAS.
- T. Stößer, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 2668 CrossRef.
- Y. Liu, J.-Z. Guo, H.-W. Lu, H.-B. Wang and X.-B. Lu, Macromolecules, 2018, 51, 771–778 CrossRef CAS.
- S. R. Petersen, J. A. Wilson and M. L. Becker, Macromolecules, 2018, 51, 6202–6208 CrossRef CAS.
- F. Isnard, M. Carratù, M. Lamberti, V. Venditto and M. Mazzeo, Catal. Sci. Technol., 2018, 8, 5034–5043 RSC.
- H. Li, H. Luo, J. Zhao and G. Zhang, ACS Macro Lett., 2018, 7, 1420–1425 CrossRef CAS.
- H. Y. Ji, B. Wang, L. Pan and Y. S. Li, Angew. Chem., Int. Ed., 2018, 57, 16888–16892 CrossRef CAS.
- S. Kernbichl, M. Reiter, J. Mock and B. Rieger, Macromolecules, 2019, 52, 8476–8483 CrossRef CAS.
- Y. Zhou, C. Hu, T. Zhang, X. Xu, R. Duan, Y. Luo, Z. Sun, X. Pang and X. Chen, Macromolecules, 2019, 52, 3462–3470 CrossRef CAS.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2, 586 CrossRef.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS.
- E. Hosseini Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef.
- E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- J. Liu, Y.-Y. Bao, Y. Liu, W.-M. Ren and X.-B. Lu, Polym. Chem., 2013, 4, 1439–1444 RSC.
- J. M. Longo, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 15897–15900 CrossRef CAS.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- J. Y. Jeon, S. C. Eo, J. K. Varghese and B. Y. Lee, Beilstein J. Org. Chem., 2014, 10, 1787–1795 CrossRef.
- U. Biermann, A. Sehlinger, M. A. R. Meier and J. O. Metzger, Eur. J. Lipid Sci. Technol., 2016, 118, 104–110 CrossRef CAS.
- A. M. DiCiccio, J. M. Longo, G. G. Rodriguez-Calero and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 7107–7113 CrossRef CAS.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- N. J. Van Zee, M. J. Sanford and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 2755–2761 CrossRef CAS.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS.
- J. Li, B.-H. Ren, S.-Y. Chen, G.-H. He, Y. Liu, W.-M. Ren, H. Zhou and X.-B. Lu, ACS Catal., 2019, 9, 1915–1922 CrossRef CAS.
- C. Romain, A. Thevenon, P. K. Saini and C. K. Williams, Carbon Dioxide and Organometallics, ed. X. B. Lu, Springer International Publishing, 2016, vol. 53, pp. 101–141 Search PubMed.
- D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- M. Andruh, Dalton Trans., 2015, 44, 16633–16653 RSC.
- A. Thevenon, J. A. Garden, A. J. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS.
- A. Rae, A. J. Gaston, Z. Greindl and J. A. Garden, Eur. Polym. J., 2020, 138, 109917 CrossRef CAS.
- C. B. Durr and C. K. Williams, Inorg. Chem., 2018, 57, 14240–14248 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1359 RSC.
- A. L. Gavrilova and B. Bosnich, Chem. Rev., 2004, 104, 349–383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental protocols, characterization and X-ray crystallographic data. CCDC 2041972–2041974. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy02164d |
| This journal is © The Royal Society of Chemistry 2021 |