
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous catalytic C(sp3)–H functionalization of gaseous alkanes
Antonio
Pulcinella†
,
Daniele
Mazzarella†
and
Timothy
Noël
 *
*
Flow Chemistry Group, Van’t Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam Science Park 904, 1098 XH, Amsterdam, The Netherlands. E-mail: t.noel@uva.nl; Web: http://www.NoelResearchGroup.com
First published on 1st September 2021
Abstract
The conversion of light alkanes into bulk chemicals is becoming an important challenge as it effectively avoids the use of prefunctionalized alkylating reagents. The implementation of such processes is, however, hampered by their gaseous nature and low solubility, as well as the low reactivity of the C–H bonds. Efforts have been made to enable both polar and radical processes to activate these inert compounds. In addition, these methodologies also benefit significantly from the development of a suitable reactor technology that intensifies gas–liquid mass transfer. In this review, we critically highlight these developments, both from a conceptual and a practical point of view. The recent expansion of these mechanistically-different methods have enabled the use of various gaseous alkanes for the development of different bond-forming reactions, including C–C, C–B, C–N, C–Si and C–S bonds.
 Antonio Pulcinella | Antonio Pulcinella received his BSc in Chemistry in 2018 at the University of Roma Tor Vergata and carried out his thesis at IRBM Science Park Spa. In 2020, he completed a Master in Chemistry in the context of the Double Degree Programme between the University of Giessen and the University of Padova, working in the group of Prof. Dr Peter Schreiner on the synthesis of 2-deoxy-C-glycoside amino acids. Currently, he is a Marie S. Curie PhD candidate at the University of Amsterdam under the supervision of Prof. Dr Noël, working on the continuous-flow C–H activation of aliphatic scaffolds promoted by photochemistry and/or electrochemistry. |
 Daniele Mazzarella | Daniele Mazzarella studied chemistry at the universities of Rome and Bologna (Italy). After a brief experience in Japan working for Nippon Kayaku Co. Ltd, in 2016, Daniele joined Prof. Paolo Melchiorre's group at the ICIQ (Tarragona, Spain) to carry on doctoral studies in the fields of organocatalysis and synthetic photochemistry. After having obtained his PhD degree cum laude in 2020, Daniele joined the group of Prof. Timothy Noël at the University of Amsterdam (The Netherlands), where he is currently pursuing the development of novel electrochemical and photochemical transformations in a microfluidic environment. |
 Timothy Noël | Timothy Noël currently holds a position as Full Professor at the University of Amsterdam, where he is the chair of Flow Chemistry. His research interest ranges from organic chemistry to chemical engineering and encompass more specifically flow chemistry, organic synthesis and synthetic catalytic methodology development. His work received several awards, including the DECHEMA prize (2017), the Hoogewerff Jongerenprijs (2019), the IUPAC-ThalesNano prize in flow chemistry and microfluidics (2020) and the KNCV Gold Medal (2021). He also serves as editor-in-chief of Journal of Flow Chemistry. |
Introduction
Gaseous alkanes are among the most abundant and cheapest carbon-based feedstocks, which are generally used as a source of energy for heating, propulsion or electricity generation.1 In conjunction with these uses, methane, ethane, and heavier analogues could also be employed as building blocks for chemical synthesis.2 This is an increasingly fascinating area of research as the conversion of greenhouse gases into chemical feedstocks and/or bulk chemicals would be of extreme value for the chemical industry as well as for the environment. Indeed, the direct use of alkanes would also bypass the need for functionalized reagents, e.g. haloalkanes and organometallic reagents.3 However, there are several issues that need to be addressed in order to realize this goal (Fig. 1), such as the general inertness of the C–H bonds and the poor solubility of the gases in common organic solvents. Moreover, the desired products are often easier to activate than the gaseous alkanes, leading to issues related to over-reaction.4 | ||
| Fig. 1 General representation of the issues related to the C–H functionalization of gaseous alkanes. BDE: bond dissociation energy; FG: functional group. | ||
While huge attention has been directed to the use of methane to produce methanol,5 less focus has been devoted to its valorization for the production of different methyl-containing derivatives. Additionally, the vast majority of these processes are carried out under heterogeneous conditions,5a,c which are well known to pose an additional problem in terms of selectivity. Notably, reports that exploit the use of a homogeneous catalyst, outside of the field of methanol formation,5b are rare and remain largely underdeveloped.
The scope of this article is to cover the state-of-the-art in the functionalization of gaseous alkanes under homogeneous catalytic conditions, with the aim of showcasing the main achievements, as well as the different activation mechanisms and the future challenges within the field. We have divided the review based on the nature of the bond formed after the functionalization of the gaseous alkane, namely C–B, C–C, C–N, C–Si and C–S. Specifically for the C–C bond formation, processes able to induce chirality have also been highlighted. Regarding the formation of C–O bonds, possibly the most studied reaction for the functionalization of methane and other gaseous alkanes, this topic has been recently reviewed by Periana and co-workers5b and will not be discussed within this article.
C–B bond formation
Protocols capable of converting feedstock chemicals such as light alkanes in added-value building blocks are highly sought after. In the field of catalytic C–H functionalization, particular interest has been directed towards the borylation of inert aliphatic substrates.6 This is motivated by the high synthetic versatility of the resulting organoboron compound, which can be easily converted in a wide range of other functional groups such as amines, alkenes and alcohols or can be engaged in C–C bond forming reactions.7 Earlier works demonstrated that Re, Rh, Ru and Ir are all competent catalysts for the homogeneous borylation of liquid alkanes.6f,h,j Specifically, Rh-based catalysts are capable of forming metal bis(boryl) complexes after oxidative addition of a diboron precursor (B2pin2, Fig. 2).6a,b These species have been reported to react with alkanes under neat conditions, leading to the formation of alkyl-metal boryl complex I. This intermediate undergoes reductive elimination to produce one molecule of the borylated product and metal hydride II. The latter reacts with the borylating reagent affording intermediate III that, upon extrusion of HBpin,6h is able to activate C(sp3)–H bonds to restore active species I. Another plausible and concomitantly operating reaction pathway concerns the oxidative addition of the formed HBpin, leading in this case to the formation of the borylated alkyl species along with molecular hydrogen.6a However, from a thermodynamic viewpoint, diboron species determine a greater enthalpic driving force due to the weaker B–B bond compared to the B–H bond in boranes.8 | ||
| Fig. 2 General representation of the Rhodium-catalyzed borylation of C–H bonds. Pin: pinacolato; Cp*: pentamethylyclopentadienyl. | ||
Despite these literature precedents, translating these thermal catalytic protocols to the activation of C–H bonds within light alkanes faced some major challenges. The first problem to address is the choice of an appropriate solvent that should not outcompete the gaseous alkane during the C–H activation step. Specifically, the higher BDE of methane (bond dissociation energy, BDE = 105 kcal mol−1 for methane) compared to other liquid alkanes poses a unique challenge.5b In addition, the over-functionalization of the product is another likely scenario given the presence of more labile C–H bonds compared to the ones present in the gaseous alkane.9–11
The borylation of gaseous hydrocarbons was pioneered by the groups of Sanford9 and Mindiola,10 who simultaneously reported two transition metal-catalyzed methodologies capable of borylating methane and ethane under thermal conditions.
In their report, Sandford and coworkers9 identified optimal conditions for the borylation of methane using a high-pressure batch reactor employing Rh-based catalyst [Cp*Rh(η4-C6Me6)], 35 bar of methane and B2pin21 as the limiting reagent in cyclohexane at 150 °C (Fig. 3). The choice of the catalyst was made based on earlier density functional theory (DFT) studies reported by Hall et al., proving the theoretical feasibility of the process.12 Crucial for the success of the desired transformation was the choice of the solvent: cyclohexane granted good solubility of the catalyst without compromising the selectivity. Indeed, due to the more strained and hindered nature of the C–H bonds in cyclohexane, only traces of the borylated solvent byproduct 3 were observed.13 Under the optimized reaction conditions, high selectivity and yield were obtained for the mono-borylated methane 2. Additionally, by testing other known catalysts previously employed in the borylation of heavy alkanes, the authors demonstrated that the fine-tuning of the catalyst structure had significant impact on the selectivity of the process.6a The relationship between steric hindrance and selectivity was further corroborated by a competition experiment using known molar quantities of methane and ethane: all the tested catalysts preferentially functionalized the less sterically-encumbered methane.
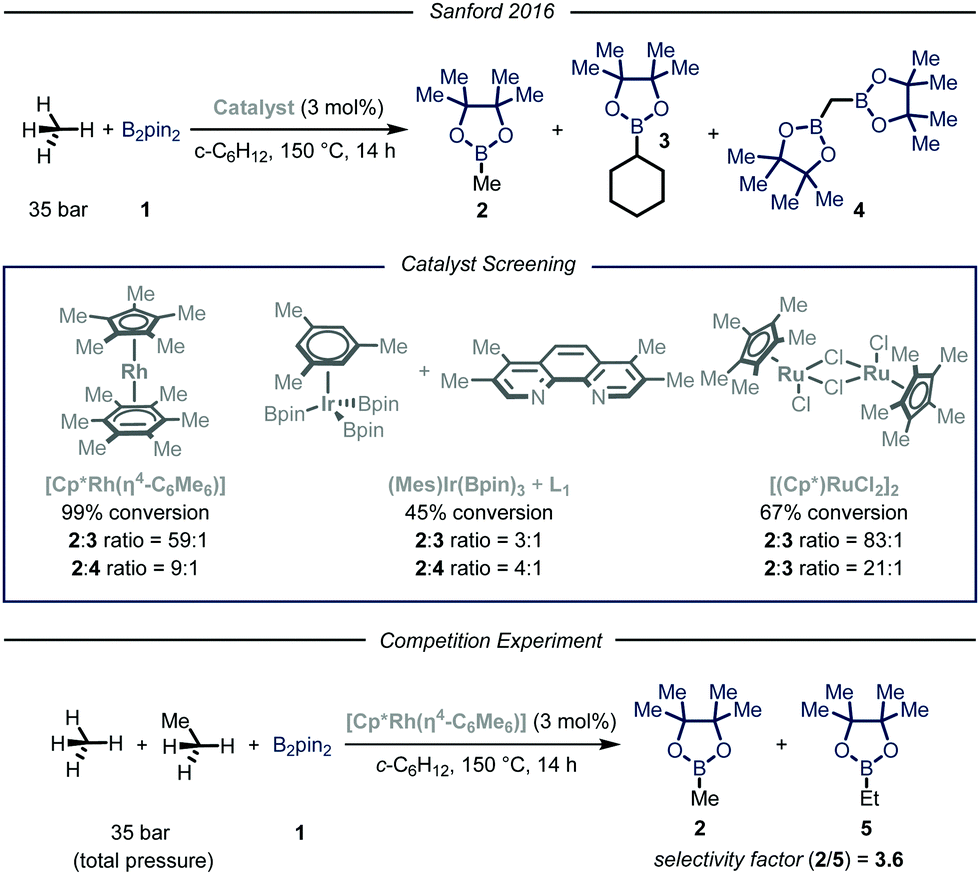 | ||
| Fig. 3 Borylation of methane developed by Sanford and co-workers. Pin: pinacolato; Cp*: pentamethylyclopentadienyl. | ||
Simultaneously, the group of Mindiola10 reported an elegant iridium-catalyzed borylation of methane (Fig. 4). Given the well-known efficacy of iridium-based catalytic systems for the borylation of inert C–H bonds,6f,14 the authors conducted an extensive screening of commercially available iridium catalysts and nitrogen-based ligands. Preliminary results revealed that the highest conversion and selectivity for the monoborylated product 2 were obtained in the presence of an Ir(I) precatalyst and L1 (3,4,7,8-tetramethyl-1,10-phenanthroline) as ligand using tetrahydrofuran as a solvent under 35 bar of methane at 120 °C. Under these conditions, the desired product was obtained in 4.1% yield (determined via gas-chromatography (GC) analysis). The mechanism, as predicted by DFT calculations, begins with the substitution of the ligand to afford Ir(III) complex V, which is the putative resting state of the catalyst. After methane coordination at one of the vacant sites of the complex, the rate-determining oxidative addition occurs via transition state A yielding intermediate VII. Ligand reorganization, followed by reductive elimination, affords the desired product 2 and complex IX, which further regenerates the catalyst resting state by reacting with B2pin2. Since the C–H bond cleavage implies the oxidation of Ir(III) to Ir(IV) within intermediate VI (transition state A), the authors postulated that, in light of their reduced Lewis basicity, phosphine ligands might enhance the polarizability of the metal center, thus facilitating oxidative addition. Indeed, employing L2 as ligand under the optimized conditions, the desired product was obtained in 52% yield with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chemoselectivity over the monofunctionalized compound. Isotopic labeling studies using 13CH4 confirmed that the light alkane is the methylating reagent under the described reaction conditions.
:1 chemoselectivity over the monofunctionalized compound. Isotopic labeling studies using 13CH4 confirmed that the light alkane is the methylating reagent under the described reaction conditions.
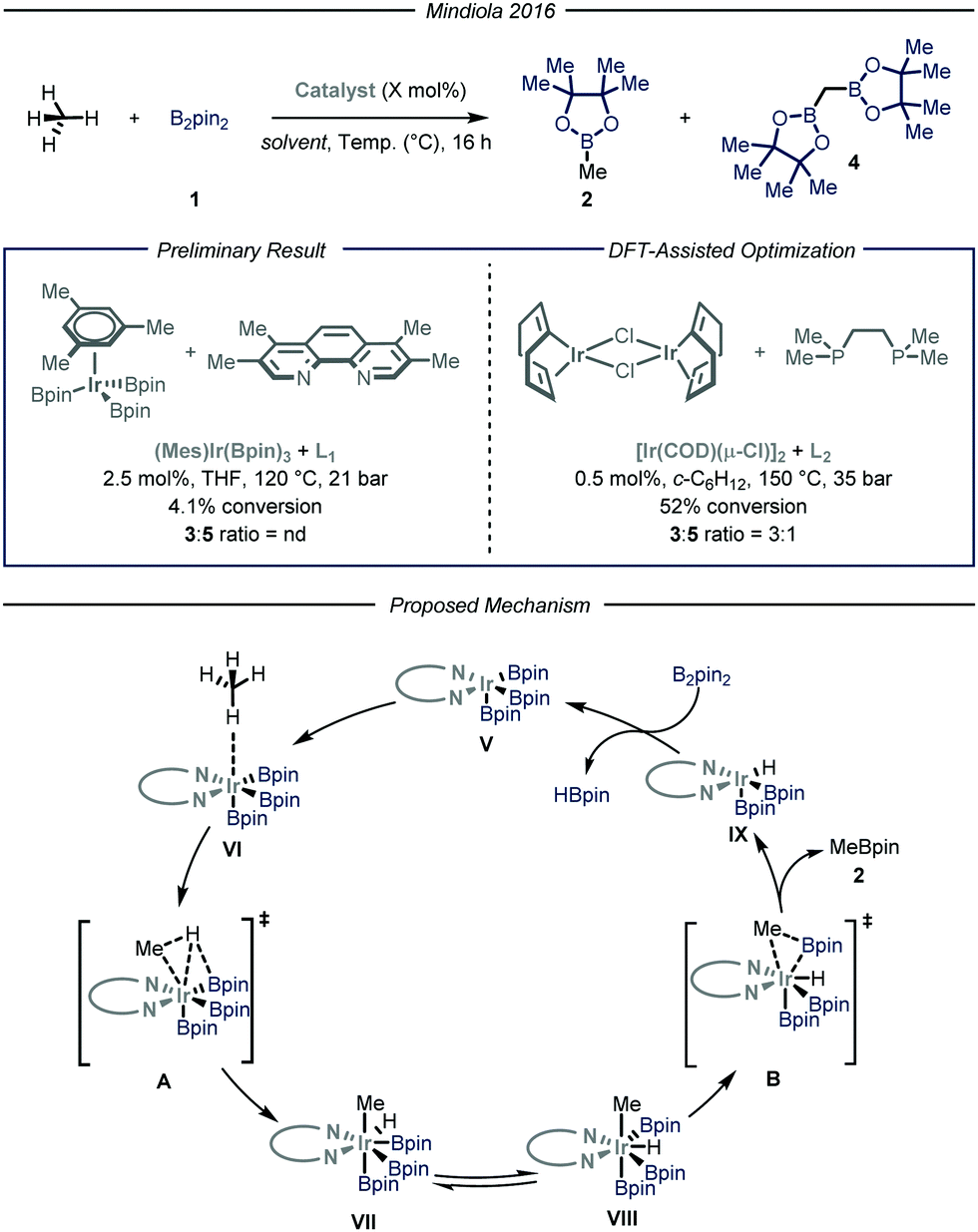 | ||
| Fig. 4 Iridium-catalyzed borylation of methane developed by Mindiola and co-workers. Pin: pinacolato; THF: tetrahydrofuran. | ||
C–C bond formation
Carbon–carbon bond-forming reactions are essential tools for the synthetic community as these bonds constitute the framework of every organic molecule. Within this field, the installment of light alkyl fragments, especially the methyl one, can strongly alter the binding affinities of the parent molecule, boosting its potency as a drug, and has therefore deep implications in medicinal chemistry.15 However, these reactions are generally performed using reactive prefunctionalized alkyl derivatives, either halides or organometallic ones. As such, the direct use of gaseous alkanes would avoid unnecessary functionalization steps and would generally provide a more sustainable process.Several types of C–H functionalization strategies have been exploited to engage volatile alkanes directly in carbon–carbon bond-forming reactions. The first one, reported by Fujiwara and co-workers,16 is based on a transition–metal-catalyzed C–H activation mechanism.17 In these seminal contributions, the authors proved how light alkanes could be converted into carboxylic acids 6 through the action of palladium and/or copper catalysis, using CO as the carboxylating reagent and K2S2O8 as the oxidant in an autoclave (Fig. 5). Specifically for the palladium catalysis, the authors suggest an electrophilic C–H activation of the gaseous alkane by the catalyst to afford intermediate XI. The latter can undergo CO insertion to yield carbonyl-palladium species XII that ultimately is converted into the target carboxylic acid 6 while the palladium catalyst X is regenerated by the oxidant (1–41% yield). Notably, the same group also expanded this mode of action to the aminomethylation of gaseous alkanes by employing in situ generated iminium ions in place of carbon monoxide as electrophilic partners.18
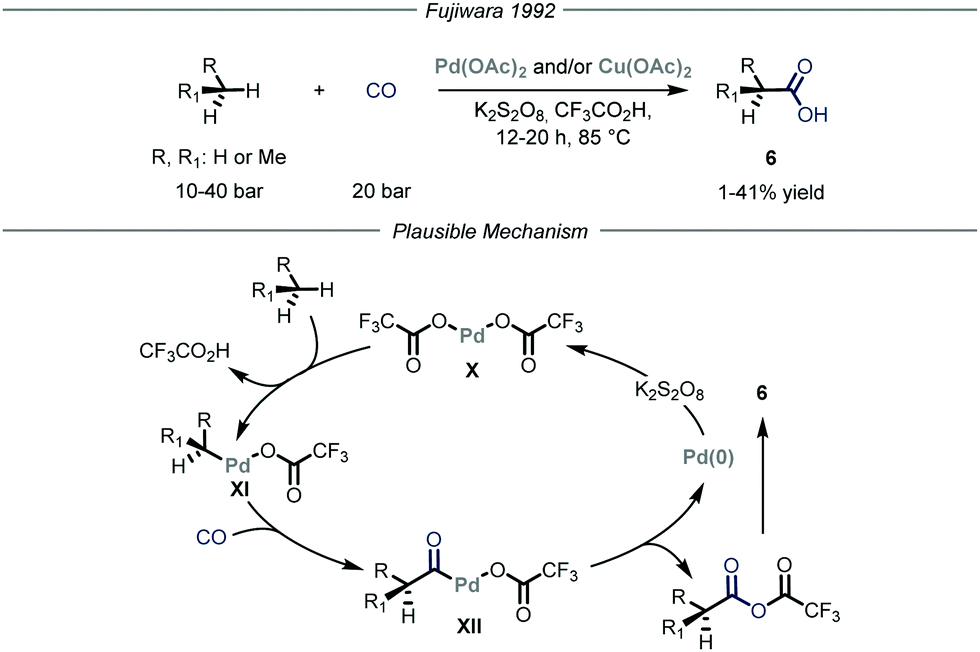 | ||
| Fig. 5 Palladium-catalyzed carboxylation of methane by Fujiwara and co-workers. | ||
Later on, Lin and Sen19 described a rhodium-based catalytic system able to promote the same carboxylation reaction of methane under much milder conditions, employing water instead of trifluoroacetic acid as solvent and oxygen as the oxidant.
In 2020, Chang and co-workers reported a copper-catalyzed process to promote the cross-dehydrogenative-coupling (CDC)20 between gaseous alkanes and polyfluorinated arenes 7 (Fig. 6).21 After an extensive optimization screening, the authors identified β-diketimine L3 as the optimal ligand for the copper catalyst, t-butyl peroxide as the oxidant, t-BuONa as the base in benzene at 80 °C. Apart from the efficient functionalization of liquid and solid alkanes, the authors demonstrated how, in a high-pressure reactor, also n-butane and propane could be productively converted into the corresponding arylated products 8 (39–51% yield).
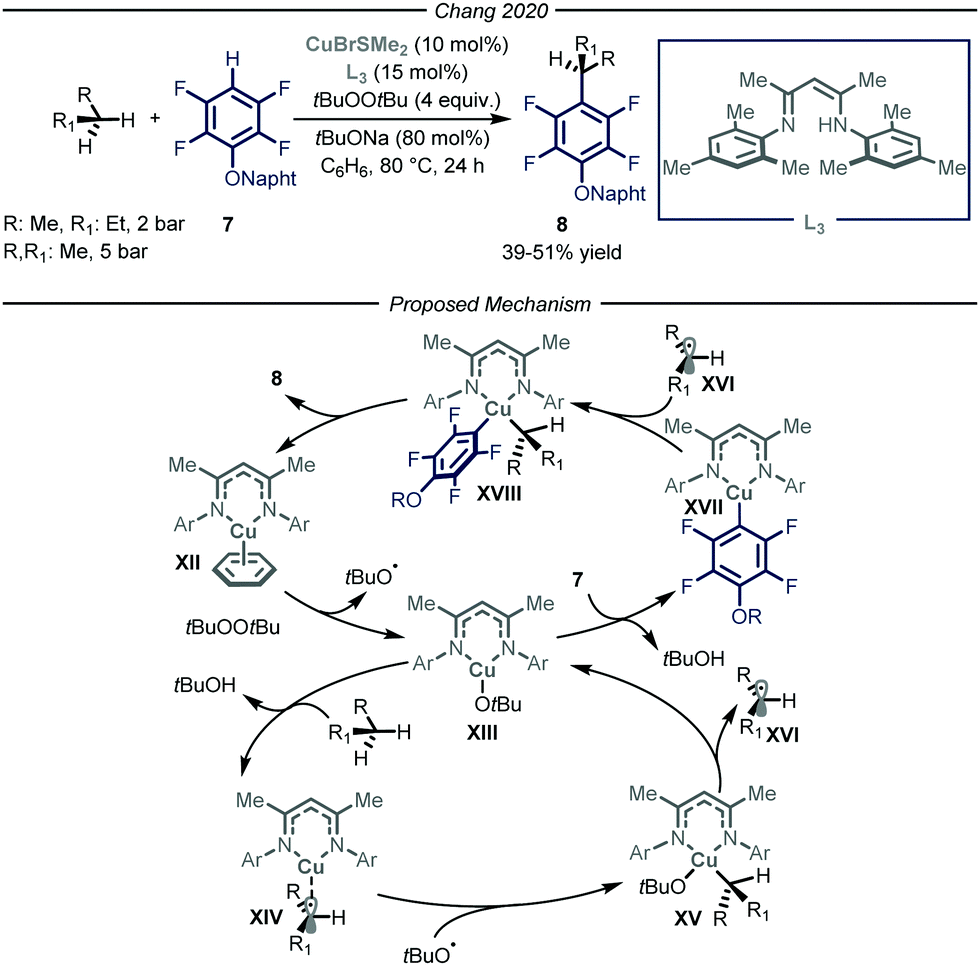 | ||
| Fig. 6 Copper-catalyzed cross-dehydrogenative coupling of alkanes and polyfluoroarenes. | ||
The authors performed a thorough mechanistic investigation, encompassing both experimental and computational work. Overall, the process relies on a dual activation mode, where the copper catalyst is activating the C–H bonds of both polyfluorinated arene 7 and the alkane. Specifically, active copper complex XII reacts with t-butyl peroxide to afford tBuO˙ and Cu(II) intermediate XIII. The latter is responsible for both the activation of the alkane through a radical pathway and of the fluoroarene via a polar mechanism. The ensuing Cu(III) species XVIII undergoes reductive elimination to afford product 8 and close the catalytic cycle.
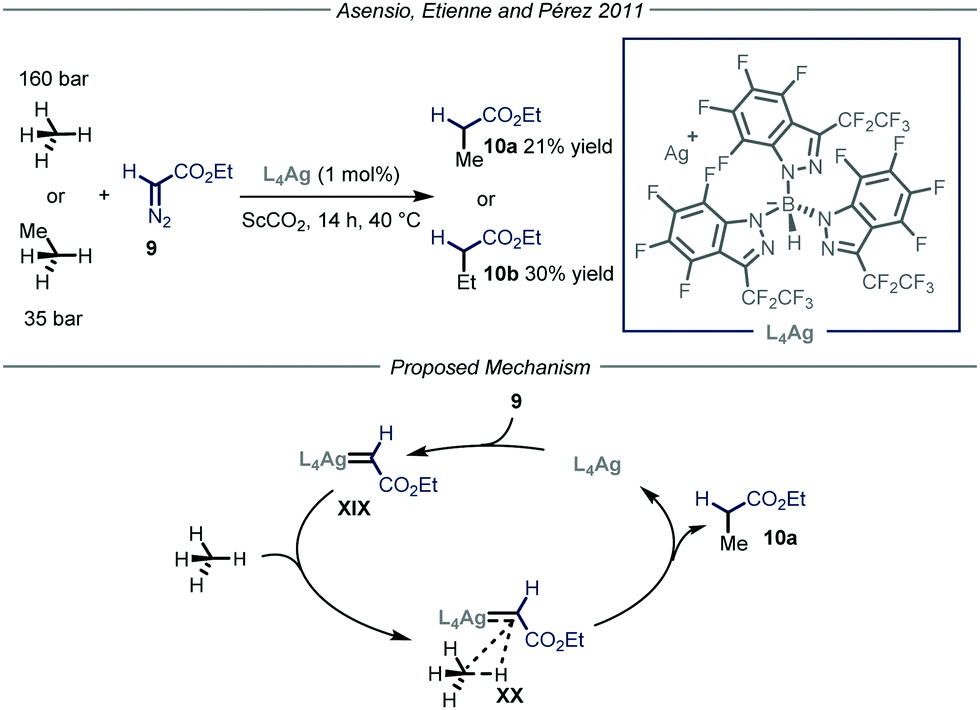
Apart from direct C–H activation methods, the groups of Asensio, Etienne and Pérez22 reported in 2011 a method that takes advantage of the formation of a metal carbenoid species23 between a silver-based catalyst L4Ag and ethyl diazoacetate 9 (Fig. 7). This intermediate is capable of inserting into the C–H bond of methane and ethane, producing ethyl acetate and propionate respectively. Two parameters were crucial for the good outcome of the whole process. First of all, because of the pronounced reactivity of metal carbenoids and of the strength of the C–H bonds within methane and ethane, as well as the low solubility of the gaseous reagents, the choice of the reaction solvent becomes extremely important. Indeed, the use of a solvent possessing weaker C–H bonds with respect to methane would lead to the functionalization of the former. Consequently, the authors used supercritical CO2 (ScCO2) in a high-pressure reactor thermostated at 40 °C; ScCO2 provides the required inertness and displays complete miscibility with methane. The second crucial parameter is the choice of the ligand to tune the reactivity of the metal catalyst. In order to activate a strong chemical bond such as the C–H bonds in methane and ethane, a highly electrophilic metal carbenoid is required. The authors, based on their experience in the development of coinage metal complexes to catalyze C–H insertion reactions,24 selected catalyst L4Ag due to the enhanced electrophilic properties granted by the ligand. Under the optimized conditions, methane and ethane were converted into ethyl acetate and propionate respectively with good efficiency (21–30% yield) taking into account the difficulty of the transformation. As for the mechanism, in analogy with other metal carbenoid-promoted C–H functionalizations, the authors proposed a metal-catalyzed N2 extrusion from compound 9 to form silver carbenoid XIX that subsequently undergoes carbene transfer onto methane or ethane to give the corresponding products 10.
 | ||
| Fig. 7 Use of silver carbenoid for the functionalization of methane and ethane. ScCO2: supercritical CO2. | ||
A third strategy used to drive the C–H functionalization of gaseous alkanes relies on the formation of radical intermediates. The advent of photoredox catalysis25 has enabled the generation of open-shell species under extremely mild conditions, namely light illumination in the presence of substoichiometric quantities of a photocatalyst at room temperature.26 Upon photoexcitation, the catalyst has enhanced redox properties and can either promote single electron transfer (SET) events or directly cleave C(sp3)–H bonds through a hydrogen atom transfer (HAT) pathway.27 Notably, both these mechanisms of activation have been used with the aim of functionalizing gaseous alkanes.
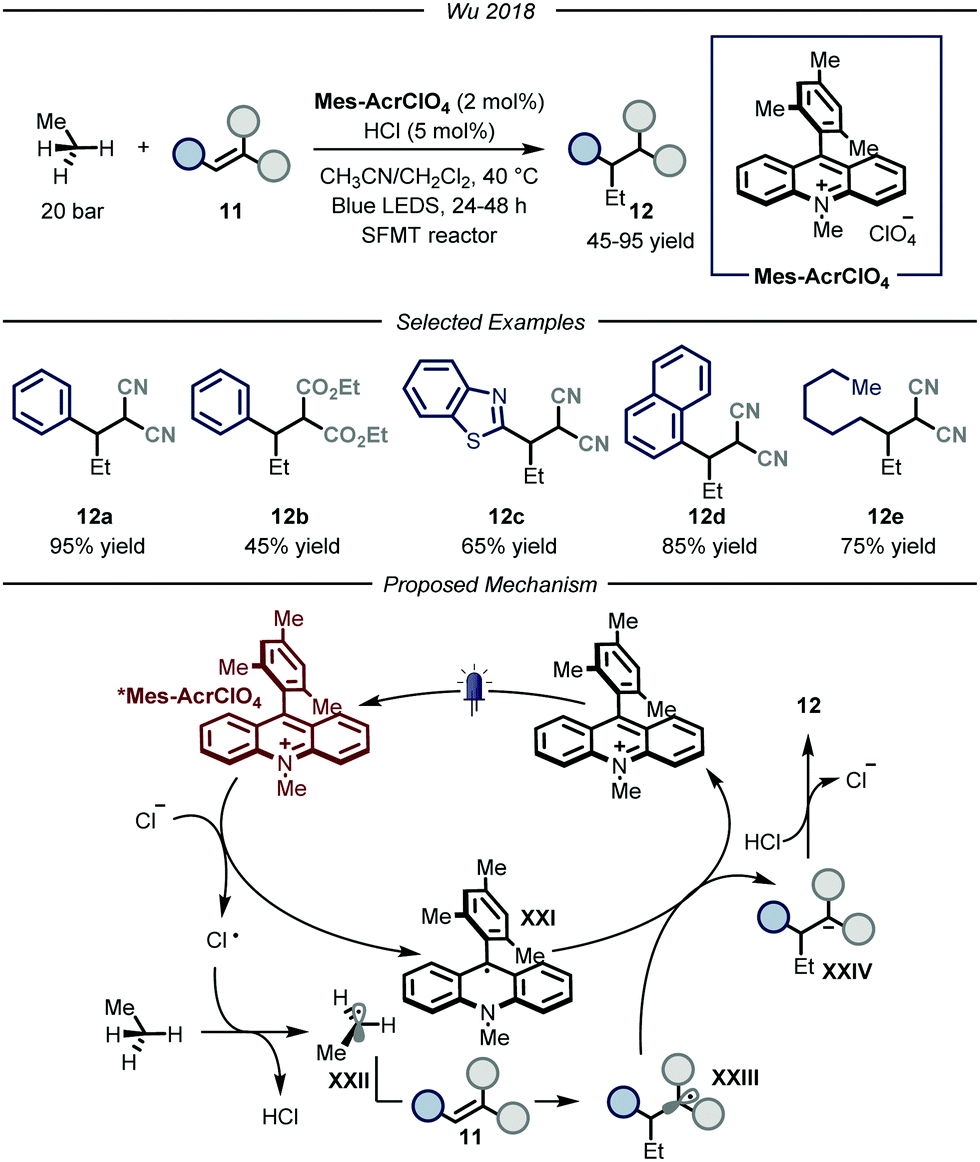
In 2018, the group of Wu reported a photocatalytic process that could promote the functionalization of ethane by using hydrochloric acid as an efficient HAT catalyst (Fig. 8).28 The authors, based on previous findings on the photocatalytic value of chlorine radicals as HAT reagents,29 envisioned that, upon single electron oxidation, HCl itself could be used to generate Cl˙ under catalytic conditions. Nonetheless, the development of such a process poses several challenges. First of all, because of the gaseous nature of both ethane and hydrochloric acid, the use of classical batch reactors was not a viable option. Moreover, because of the necessity to establish uniform irradiation to efficiently promote the photochemical steps, the use of high pressure reactors was also discarded. Within the field of photocatalytic transformations, microreactor technologies have provided a platform based on short length scales which ensure homogeneous irradiation of the reaction mixture while simultaneously guaranteeing efficient gas–liquid mass transfer to saturate the gaseous reagents in the reaction mixture.30 Taking this into consideration, the group of Wu decided to develop the aforementioned process under micro-scale conditions using narrow capillaries. Specifically, the authors employed a so-called “stop-flow” microtubing (SFMT) reactor,31 consisting of a capillary equipped with two shut-off valves at each end of the reactor (PFA (perfluoroalkoxy alkanes), I.D. = 762 μm, V = 3.0 mL). Under the optimized conditions, several electron-poor olefins 11 could be functionalized with ethane, affording adducts 12 in good yields (45–95% yield). Mechanistically, the reaction kicks off with the photoexcitation of the organic photocatalyst Mes-AcrClO4 to afford species *Mes-AcrClO4 that features enhanced oxidizing properties (ERED = 2.05 V vs. SCE)32 and is therefore able to take one electron from the chloride anion (EOX = 2.03 V vs. SCE),33 affording Cl˙. The latter can perform HAT on ethane to produce hydrochloric acid alongside the nucleophilic ethyl radical XXII that is intercepted by electron-poor olefin 11 to afford radical XXIII. This intermediate is prone to single electron reduction by the reduced form of the photocatalyst (XXI) to yield, after protonation by hydrochloric acid, product 12 while regenerating both the catalysts. As a general limitation, only strongly electron-poor olefins could be employed due to the fleeting nature of the ethyl radical, which requires a fast trapping by the acceptor, and due to the limited oxidizing properties of the radical adducts to turn-over the photocatalytic cycle. Moreover, under these conditions, the functionalization of methane proved to be unsuccessful, presumably due to the too high BDE of its C–H bonds.
 | ||
| Fig. 8 Use of chlorine radical as HAT catalyst for the activation of ethane. LED: light emitting diode; SFMT: stopped flow microreactor. | ||
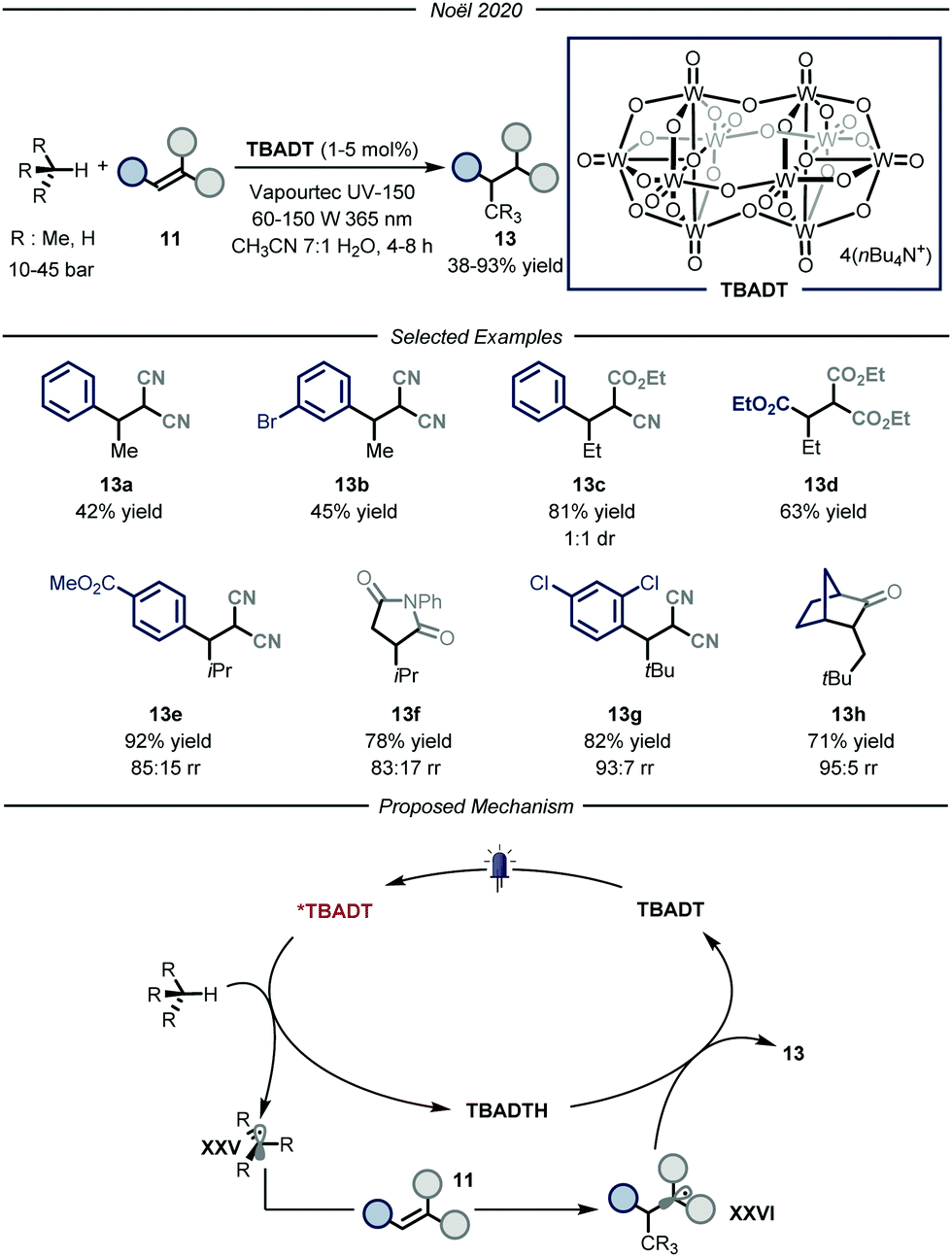
The group of Noël developed a direct photocatalytic HAT method, catalyzed by tetrabutylammonium decatungstate (TBADT, Fig. 9).34 Addressing the same problems faced by the group of Wu, namely the need for a uniform irradiation and an efficient gas–liquid mass transfer, the authors developed a continuous-flow procedure, using the commercially available Vapourtec UV-150 (PFA, for i-butane and propane: I.D. = 1300 μm, V = 10.0 mL; for ethane and methane: I.D. = 762 μm, V = 10.0 mL). Thanks to the good photocatalytic properties of TBADT,35 as well as the use of back-pressure regulators to enable higher pressures within the microflow reactor that completely solubilizes the gaseous alkane into the liquid phase, these processes enabled the functionalization of several substrates. The scope of the reaction is quite broad for what concerns the gaseous alkanes: i-butane, propane, ethane and even methane could all be employed to afford the corresponding products 13 in good to excellent yields (38–93% yield). Mechanistically, the process is initiated by the absorption of a near-UV light irradiation by TBADT to reach an electronically excited state. Upon photoexcitation, the ensuing TBADT* is able to split C(sp3)–H bonds within the alkane substrate, affording carbon-centered nucleophilic radical XXV which is trapped by the electron-poor olefin 11 to generate the second radical intermediate XXV. The latter, having now an opposite polarity, is able to take back the hydrogen atom from the reduced form of the photocatalyst, TBADTH, to afford product 13 and turn over the catalyst.
 | ||
| Fig. 9 Use of TBADT as HAT photocatalyst for the activation of i-butane, propane, ethane and methane. | ||
Enantioselective C–C bond formation
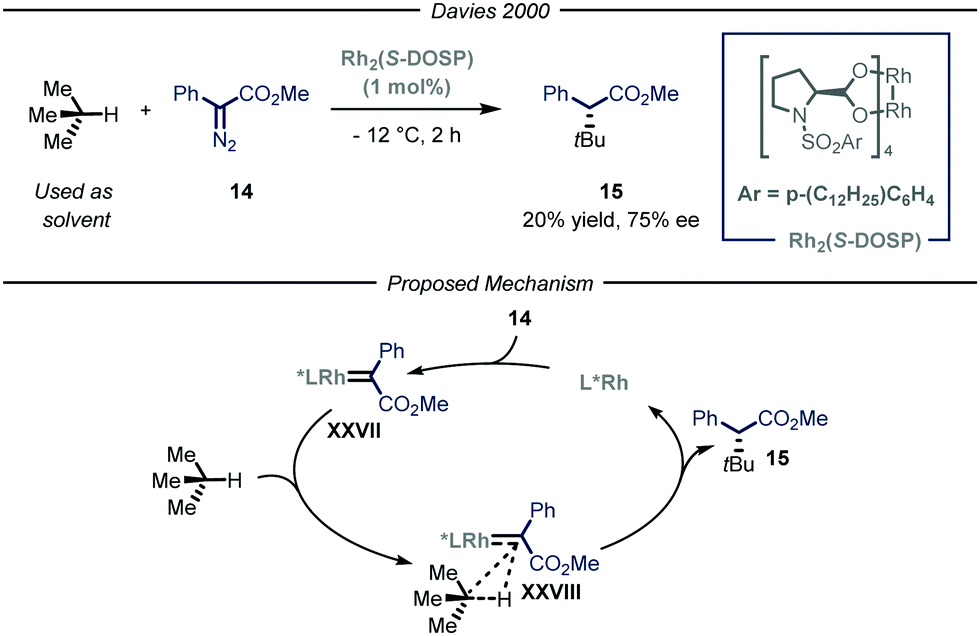
The previously described catalytic processes have shown the potential of both transition metal and photocatalysis to enable the conversion of feedstock chemicals such as gaseous alkanes into added-value compounds. Despite these undeniable contributions, the development of asymmetric variants would be highly relevant as it could serve as a platform for the asymmetric installment of light alkyl fragments. Enantioselective catalysis is one of the most efficient chemical approaches to target chiral compounds with high optical purity and in an economical and environmental benign way. As such, the possibility to combine the aforementioned processes for the activation of gaseous alkanes and enantioselective catalysis would greatly enrich the chemical toolbox of the synthetic community.The group of Davies36 expanded the reactivity of catalytically generated metal carbenoid species23 towards the asymmetric C(sp3)–H functionalization of alkane derivatives.37 Within this seminal contribution, the authors also demonstrated the viability of i-butane as starting alkyl substrate (Fig. 10). As the reaction was performed in batch, for this specific compound, the process was performed at −12 °C to condense the gaseous reagent and use it directly as solvent. Moreover, in order to exert stereocontrol over the process, the authors used a chiral dirhodium-based catalyst, Rh2(S-DOSP), responsible for the formation of chiral carbenoid intermediate XXVII that can insert into the C–H bond of i-butane and ultimately yield the enantioenriched product 15 (20% yield, 75% ee).
 | ||
| Fig. 10 Use of rhodium carbenoids for the asymmetric functionalization of i-butane. | ||
Similarly to the organometallic approach, also the photocatalytic HAT approach has been combined with enantioselective catalysis. The development of asymmetric photocatalytic methods has been one of the major area of research in the past two decades,38 however the enantioselective photochemical functionalization of gaseous reagents has remained underdeveloped so far. In 2019, Gong and co-workers39 reported a procedure that merged photo-HAT activation and chiral Lewis acid catalysis to enable the stereocontrolled functionalization of imine derivatives 16 with, amongst others, propane and i-butane (Fig. 11). Specifically, this cooperative system is composed by a chiral bisoxazoline ligand chelated to a copper metal center as the Lewis catalyst and pentacenetetrone (PT) as the photocatalyst under blue light irradiation for 48 hours. The mechanism of the reaction relies on the visible-light excitation of the photocatalyst to afford a biradical species that is able to perform HAT on the alkyl derivatives, affording radical intermediates XXIX and XXX. In parallel, the copper-based Lewis acid is chelated by imine substrate 16 affording complex XXXI which is reduced by XXX to generate the second open-shell intermediate XXXII and turnover the photocatalytic cycle. At this point, the authors propose a stereocontrolled intermolecular radical–radical coupling, driven by the chiral ligand of the Lewis acid catalyst, to afford the desired product 17 (35–79% yield, 39–69% ee).
 | ||
| Fig. 11 Merger of photocatalyzed HAT and Lewis acid catalysis for the enantioselective functionalization of i-butane and propane. LED: light emitting diode. | ||
Despite the fact that both these methodologies are affected by either low chemical yields and/or limited enantiomeric excess, they serve as a valuable proof of concept for the development of asymmetric procedures for the functionalization of gaseous alkanes.
C–N bond formation
Given the widespread presence of carbon–nitrogen bonds in biologically active molecules and pharmaceuticals, new methods capable to rapidly establish these bonds are of high interest for the scientific community.40 Despite remarkable achievements in this field,41 the development of new catalytic methodologies addressing the direct conversion of earth-abundant inert alkanes into valuable fine chemicals featuring a C–N bond is highly attractive from an industrial perspective. Moreover, installing nitrogen-containing functional groups in gaseous hydrocarbons by C–N bond forming processes has proved to be highly challenging.The first example of such a transformation was reported by Crabtree and co-workers describing the formation of imines from methane and ammonia employing Hg photosensitization (Fig. 12).42 The protocol takes advantage of the formation of an exciplex [Hg–NH3]*, where ammonia is strongly bound to the mercury atom.43 Indeed, the so-formed three electrons bond ensures proximity and facilitates an energy transfer process from mercury to ammonia, thus promoting N–H bond cleavage.44 The authors envisioned that an electrophilic N-centered radical (N–H BDE = 107 kcal mol−1) might promote the C–H abstraction of inert gaseous alkanes.45 In order to prevent further over-functionalization of the product, the authors devised a reactor where a peristaltic pump continuously introduces the gas mixture in a photolysis chamber and a cold trap (−20 °C), which ensures condensation of the products. When an equimolar mixture of alkane/ammonia, containing a trace of mercury vapor was irradiated with UV light (254 nm), the corresponding imine 18 was obtained. Mechanistically, after C–H bond cleavage by the HAT reagent, the alkyl radical undergoes radical–radical coupling with the N-centered radical. The ensuing amine can undergo two consecutive HAT events to yield the final imine.
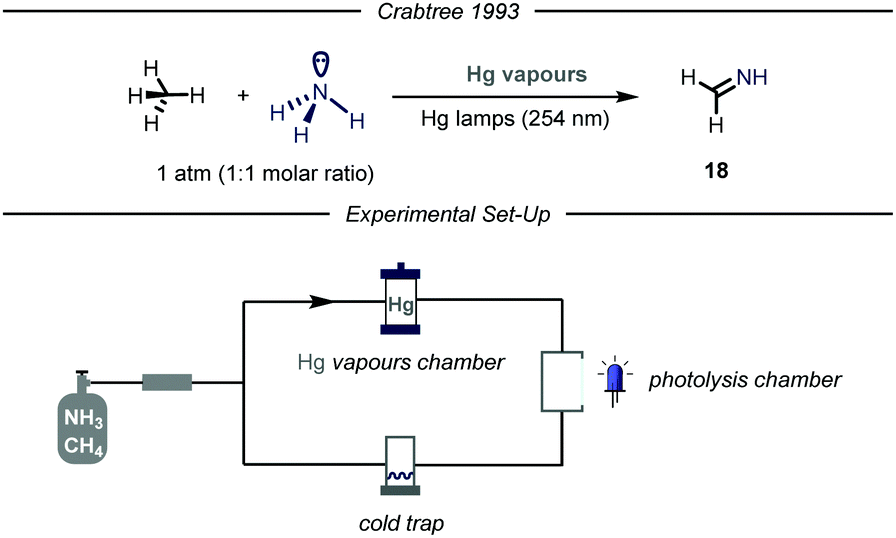 | ||
| Fig. 12 Functionalization of methane using ammonia and Hg sensitization. | ||
More recently, the group of Zuo reported a photocatalytic C–N bond formation protocol capable of converting methane, ethane and higher alkanes into the corresponding protected N-alkylated hydrazines 20 by using a homogeneous cerium-based photocatalyst CeCl3 and an alcohol co-catalyst subjected to 400 nm light irradiation (Fig. 13).46 Under the optimized conditions, using a standard pressure batch reactor, methane, ethane, propane and n-butane were all converted to the desired products in good to excellent yields (15–97% yield) using a variety of diazodicarboxylates 19 as radical traps. Additionally, the authors proved the scalability of the methodology by using a continuous-flow reactor.47 For ethane, an excellent yield of 90% was obtained after 6 minutes of residence time and a high productivity of 2.0 mmol h−1 was accomplished at 15 bar pressure and ambient temperature. The same transformation using methane as the alkylating agent led to a maximum of 15% yield due to the low pressure limit of the reactor employed, which hampered adequate solubilization of the gas. Furthermore, the authors demonstrated the possibility to establish new C–C bonds by intercepting the nucleophilic radicals with both electron poor olefins and electron deficient heteroaromatic scaffolds.48
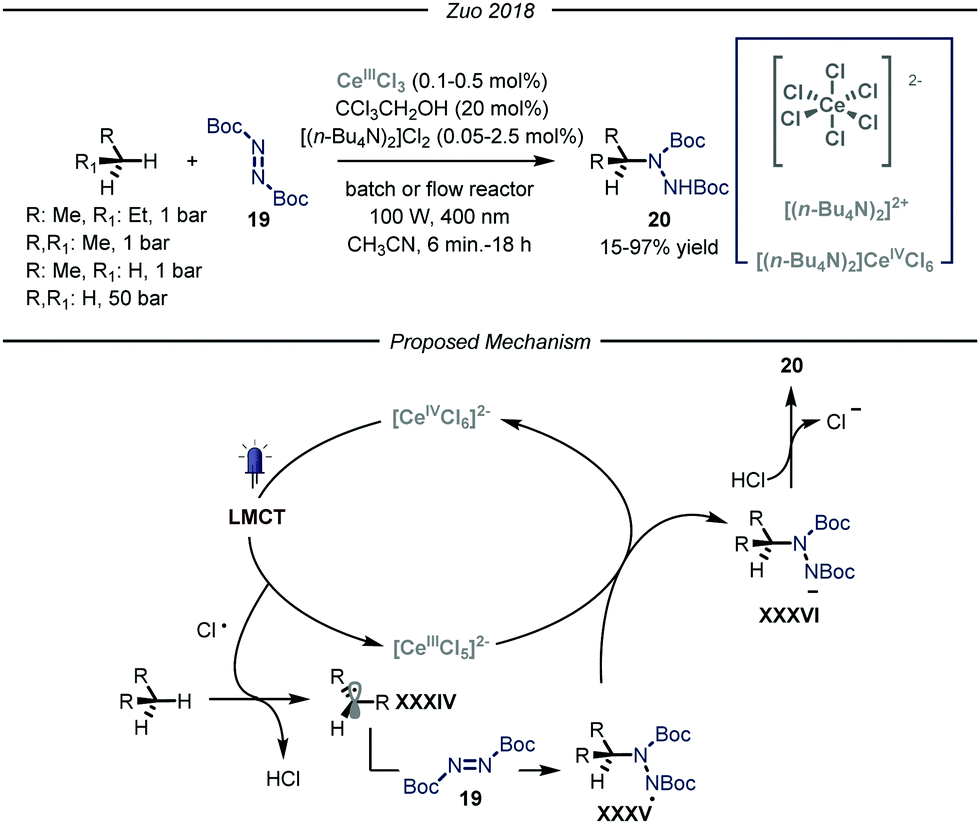 | ||
| Fig. 13 Photocatalyzed C–N bond formation using light alkanes by Zuo and co-workers. LMCT: ligand to metal charge transfer. | ||
Concerning the reaction mechanism, the authors initially postulated that a photoinduced ligand-to-metal charge transfer (LMCT) of a transient Ce(IV)-alkoxide species ([CeCl5(OCH2CCl3)]2−) could lead to the formation of an oxygen-centered radical, responsible for the HAT step. However, recent studies by Schelter, Walsh and co-authors convincingly showed that oxygen-centered radicals are not involved in the catalytic cycle.49 Indeed, titration of cerium pre-catalyst ([NEt4]2[CeCl6]) with increasing amounts of CCl3CH2OH did not determine any change in its UV-visible absorption spectrum. Preparation of the Ce(IV)-alkoxide complex ([CeCl5(OCH2CCl3)]2−) was pursued, and UV-visible spectrum of this species displayed lower absorptivity and a blue shift, thus demonstrating the absence of this intermediate. Additionally, preliminary studies demonstrated that visible light irradiation of ([NEt4]2[CeCl6]), formed in situ from CeCl3 and [NnBu4]Cl, promotes the formation of Cl˙ upon LMCT.50 Kinetic studies showed that the initial rate of photolysis of ([NEt4]2[CeCl6]) has no dependence on the concentration of HOCH2CCl3. In addition, control experiments confirmed the presence of chlorine-radical trapping products. Based on these observations, an alternative mechanistic scenario was suggested in which a photoinduced LMCT of [CeIVCl6]2− generates a chlorine radical atom which is capable of performing HAT on the alkane substrate, thus generating radical XXXIV that is intercepted by the electrophilic nitrogen source 19. The formed N-centered radical intermediate XXXV is then reduced by [CeIIICl5]2− through a single electron transfer and protonated by HCl affording the targeted N-alkylated protected hydrazine 20.
Recently, the groups of Hartwig and Pérez, reported a copper-catalyzed C–H amidation protocol using gaseous hydrocarbons as alkylating agents (Fig. 14).51 Under the optimized conditions, using t-butyl peroxide as both the oxidant and HAT reagent and CuI–phenantroline complex as the catalyst, ethane, propane, n-butane and i-butane were smoothly converted to the corresponding N-alkyl amides 22 (6–94% yield). Regarding the selectivity of the process, substrates containing two different C–H bonds were functionalized at both sites yielding mixtures of regioisomers. For all the tested gaseous alkanes, N-methyl-benzamide 23 was detected as a side-product and accounted for the remaining mass balance. The recurring presence of 23 implies the involvement of a t-butoxy radical in the catalytic cycle, which is prone to undergo β-Me scission,52 thus generating methyl radical XL which ultimately leads to the formation of the methylated side-product. Indeed, removal of the alkane substrate led to the selective formation of 23. In order to distinguish between the two pathways when using methane as a substrate, the amidation of the latter was carried out employing the perdeuterated analogue of t-butyl peroxide. Under these conditions, 3% yield of the product derived from methane incorporation was observed along with 50% yield of the side-product bearing a deuterated methyl unit. Mechanistically, in accordance with a previous report,52a the Cu(I) complex XXXVII promotes the reductive fragmentation of the peroxide, thus leading to Cu(II) species XXXVIII and an oxygen centered radical. While the former reacts with one equivalent of amide 21, the latter can either abstract a hydrogen atom from the gaseous substrate to generate XXXIX or undergo fragmentation affording methyl radical XL. Radical combination with Cu(II) intermediate XXXVIII, followed by reductive elimination, affords the N-alkylated product 22 and regenerates the Cu(I) catalyst.
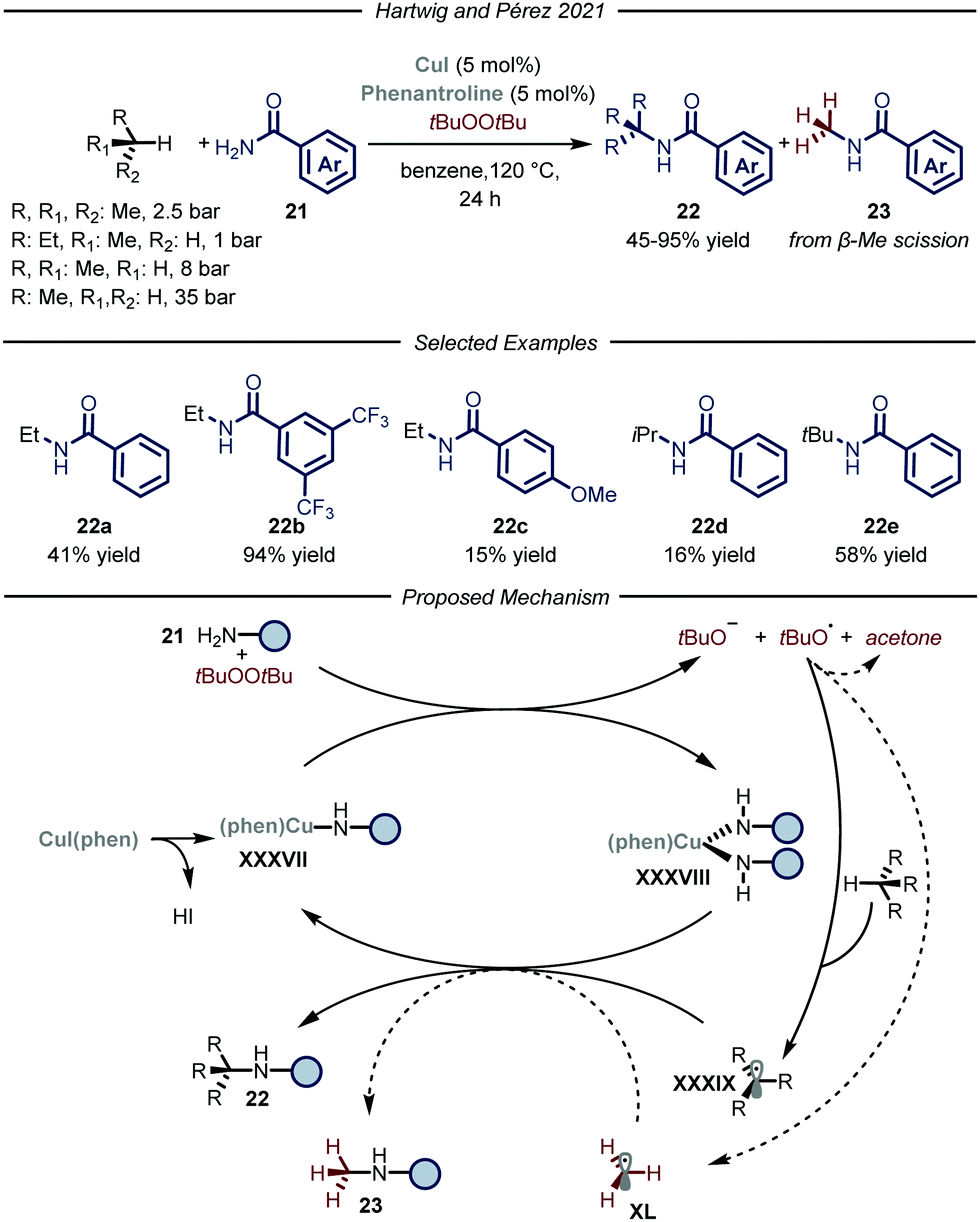 | ||
| Fig. 14 Copper-catalyzed amidation of light alkanes by Hartwig, Pérez co-workers. | ||
In the same year, Chang and co-workers reported a highly selective C–N bond formation methodology capable of converting a wide range of inactivated alkanes into the corresponding N-alkyl carbamic acid ester derivatives 25 (Fig. 15).53 Among the tested compounds, gaseous substrates, such as n-butane, propane and ethane, afforded the desired products 25 in moderate to good yields (34–73% yield). The protocol takes advantage of the formation of an electrophilic cobalt-nitrenoid species XLI that is responsible for the putative hydrogen atom abstraction. The design of a novel Cp*Co(III)(L7) catalyst, featuring enhanced electrophilicity and adequate steric hindrance, proved to be of key-importance to obtain the desired reactivity and site-selectivity. Concerning the first aspect, the authors envisioned that installing an electron-withdrawing group within the bidentate ligand scaffold, might boost the overall electrophilicity of the cobalt–nitrenoid intermediate XLI, enabling the intermolecular activation of unbiased alkanes. This constitutes a transformation which is still elusive via similar catalytic manifolds.54 Another advantage of the disclosed method is the high site-selectivity: indeed, for n-butane and propane, high preference for the functionalization of the methylene unit over the methyl one was observed. Interestingly, neither undesired over-functionalization nor solvent activation occurred. Based on a set of mechanistic studies, including electron paramagnetic resonance (EPR), DFT computations, radical clock experiments and kinetic isotope effect (KIE) studies, the authors proposed a cobalt(III)-nitrene radical activation mode in agreement with previous studies.55 Coordination of Cp*Co(III)(L7) to substrate 24 followed by a rate-determining nitrogen extrusion affords key metal-nitrenoid intermediate XLI. This species is then capable of abstracting a hydrogen atom from the aliphatic substrate, which in turn rapidly undergoes radical rebound affording the desired N-alkylated carbamic acid ester 25.
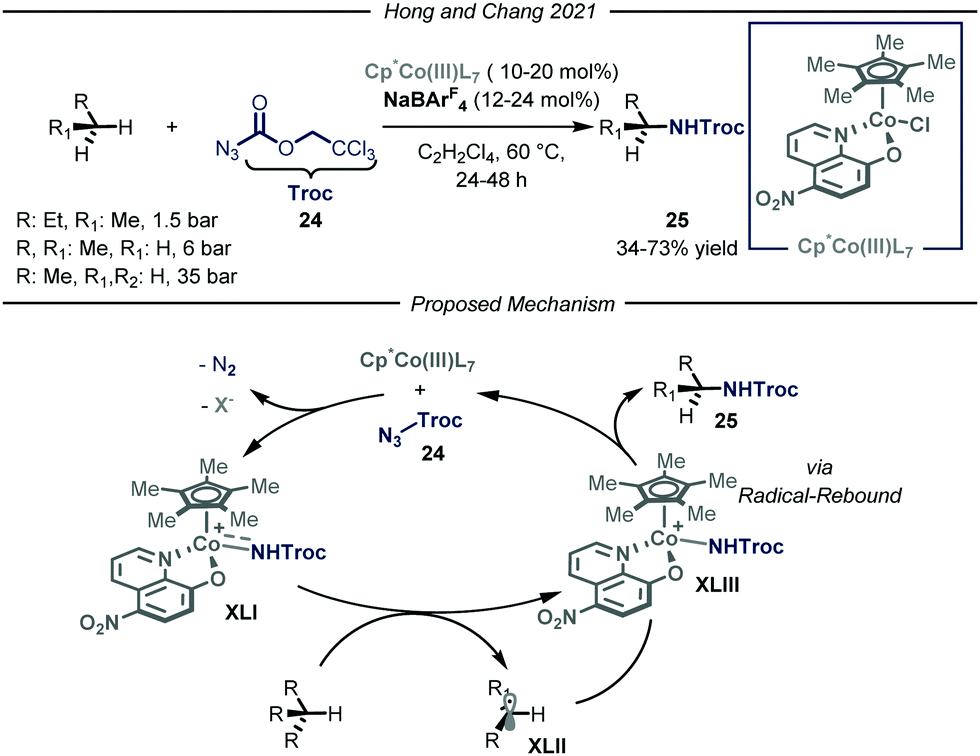 | ||
| Fig. 15 Cp*Co(III)LX-catalyzed site-selective C–N bond formation of light alkanes. Cp*: pentamethylyclopentadienyl; Troc: trichloroethyl chloroformate. | ||
C–Si bond formation
The synthesis of organosilicon compounds is an increasingly important field because of its ramifications in both medicinal56 and material chemistry.57 Moreover, the ensuing compound is a useful synthon due to its feature of acting as a carbon-based nucleophile in cross-coupling reactions.58 Taking this into account, several methods to perform the direct silylation of C(sp3)–H bonds have been reported.59 Within this field, efforts for the installment of the silicon functionality on gaseous alkanes have been made. Specifically, the group of Tilley,60 based on previous findings on the activation of methane by Cp*2ScR2 complexes,61 used this type of catalysts to install the silicon-based group on both methane and cyclopropane in a Parr high-pressure vessel (Fig. 16). After a thorough mechanistic study under both stoichiometric and catalytic conditions, the reaction has been proposed to initiate with an intra-complex C–H activation of one of the methyl substituents on the cyclopentadienyl ring and subsequent release of molecular hydrogen to afford complex XLIV. This intermediate is responsible for the activation of the alkane substrate, thus generating XLV which reacts with the silylating reagent 26, to yield product 27a and regenerate Cp*2ScH. Notwithstanding that the overall reactions are slow, this is an intriguing and significant achievement as it is one of the few processes catalyzed by an early metal that proceed under a non-radical mechanism.62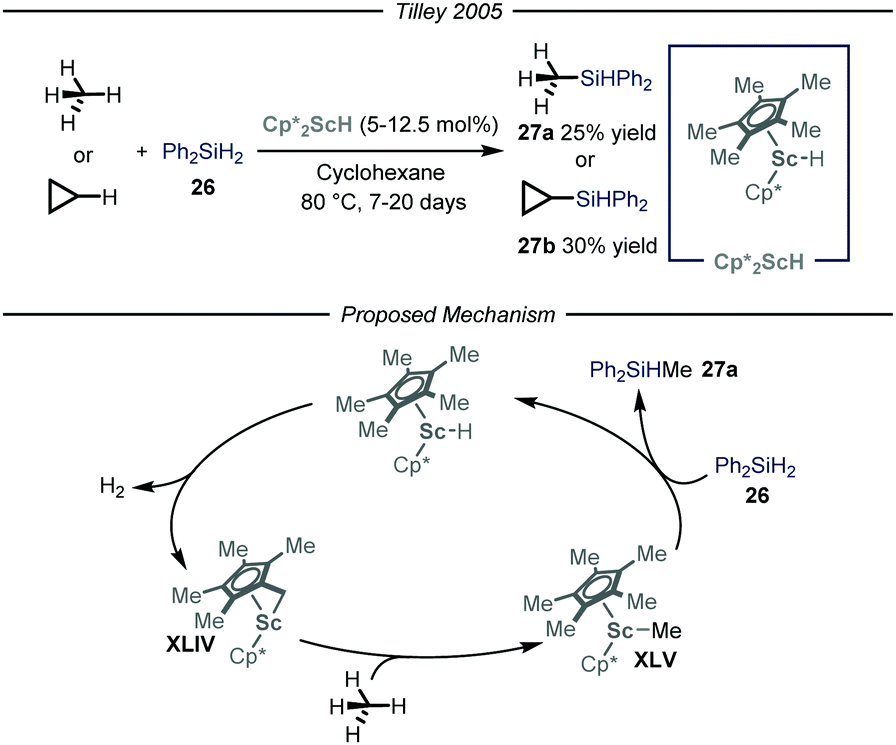 | ||
| Fig. 16 Scandium-catalyzed silylation of light alkanes through C–H activation. Cp*: pentamethylyclopentadienyl. | ||
C–S Bond formation
Owing to their involvement in many processes such as protein folding and electron transfer events, sulfur containing molecules occupy a privileged role in biochemistry.63 Additionally, this class of compounds is routinely selected as novel candidates in the field of agricultural and pharmaceutical chemistry.64 In the context of feedstock valorization, particular emphasis has been given on identifying chemoselective synthetic routes capable of converting methane into methane sulfonic acid (MSA). Indeed, MSA has recently emerged as an ideal electrolyte for many electrochemical industrial processes such as the electrodeposition of metals on electronic devices.65Early examples of methane conversion into MSA took advantage of either K2SO4 or metal peroxides as radical initiators and SO3 as the sulfonating agent, with sulfuric acid as solvent medium.66 In the same period, inspired by an elegant vanadium-catalyzed sulfoxidation of inactivated alkanes,67 Bell and coworkers published the first synthesis of MSA using methane as the alkylating agent and SO2 as the sulfur source.68 The reaction was carried out in triflic acid, with potassium persulfate and calcium salts used as, respectively, the oxidant and the promoter. Shortly after, the same group reported the first homogeneous catalytic sulfonation of methane using SO2.69 The highest conversion of sulfur dioxide into MSA was achieved using a dual catalytic system of Pd(II) and Cu(II) salts, with O2 acting as an effective oxidant in a Parr autoclave reactor under 83 bar of methane (Fig. 17, 20% yield). As for the solvent, only strong acids proved suitable for the desired transformation. Despite the mechanism was not fully elucidated, similarly to Fujiwara and co-workers,16 the authors postulate that the Pd species might promote an electrophilic activation of methane, followed by sulfur dioxide insertion and oxidation. Cu(II) and oxygen are proposed to be involved in the reoxidation of Pd(0) to Pd(II). Indeed, omission of Cu(II) resulted in formation of Pd-black with consequent inhibition of the reactivity.
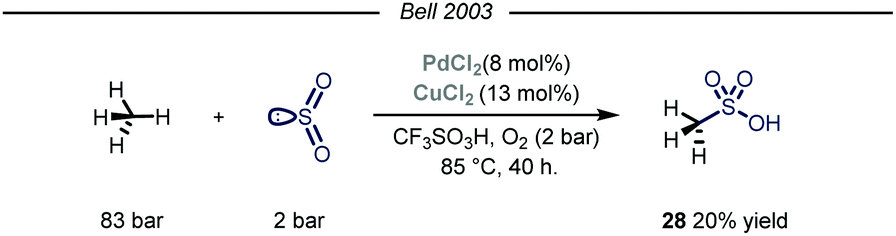 | ||
| Fig. 17 Pd(II)–Cu(II) dual catalytic manifold for the conversion of methane into methanesulfonic acid. | ||
Recently, the chemical division of Grillo-Werke AG reported a highly selective thermal process (99% selectivity and yield) for the conversion of methane and sulfur trioxide into MSA (Fig. 18).70 The protocol employs a highly electrophilic sulfonyl peroxide initiator, formed in situ from MSA, H2O2 and Oleum (SO3 36% weight in H2SO4). The initial optimization, carried out in a high pressure Parr autoclave, revealed that the highest conversion was obtained at 50 °C using 100 bar of methane. Concerning the reaction mechanism, the authors carried out a series of experiments aimed at demonstrating the non-radical nature of the presented transformation. Given the known tendency of the sulfonylperoxides to undergo homolytic cleavage of the O–O bond,71 the authors conducted the same transformation at ambient temperature under exposure to UV-light (190 nm). After eight hours of irradiation no change in the methane pressure was observed, thus excluding the involvement of a radical pathway. Furthermore, inclusion of molecular oxygen in the reaction mixture did not inhibit the desired reactivity.72 Despite these evidences, the reaction mechanism of the catalytic transformation remains unclear and computational studies demonstrated the unfeasibility of a polar cationic pathway.73
 | ||
| Fig. 18 Highly selective conversion of methane and sulfur trioxide into methanesulfonic acid. | ||
In light of the industrial relevance, the authors realized a pilot plant to scale-up the presented transformation with a projected capacity of 20 metric tons year−1. As shown in Fig. 18, the facility features three stainless-steel reactors connected in series. In the first tank, methane and SO3 are introduced and immediately reacted with the pre-formed electrophilic initiator. When the reaction mixture reaches the last reactor, the excess of SO3 is quenched while methane is recycled. The crude mixture obtained after quenching is transferred to a distillation apparatus (D, Fig. 18) which delivers pure MSA. The exhausted solution containing H2SO4 and MSA is further recirculated to the first tank in order to form back the initial solution.
Conclusions
Over the past years, several groups have contributed to the homogeneous functionalization of gaseous alkanes. In this review, we have outlined the different catalytic tools used to perform this transformation, including transition metal-catalyzed C–H activation, carbenoid C–H functionalization and photocatalytic HAT, encompassing both polar and radical reactivity modes. Up to now, these methods have enabled the forging of C–B, C–C, C–N, C–O, C–Si and C–S bonds. Specifically for the C–C bond formation, enantioselective variants have been developed. Moreover, we have also detailed the importance of suitable setups, such as high-pressure Parr vessels or continuous-flow reactors.Future efforts should be directed to construct new chemical bonds, such as C–P or C–Se, with these gaseous coupling partners. In addition, the functional group tolerance of these transformations needs to be further increased to enable the use of volatile alkanes as alkylating reagents within the synthetic routes towards more complex molecules. Other catalytic tools and activation modes should be identified and exploited to promote the functionalization of these recalcitrant substrates. At the same time, as technology clearly plays a crucial role to support these transformations, future endeavours should be also directed towards the development of suitable reactors, which can handle these chemicals efficiently and safely, and can ensure the scale up of the developed transformations. Finally, to bridge the gap with asymmetric catalysis, more efforts are required to ensure high enantioselectivities across various bond-forming reactions.
Author contributions
The concept of this article was conceived and agreed upon after discussions with all authors. D. M. and A. P. carried out the literature study and wrote the article (equal contributions). The entire manuscript was corrected and edited by T. N.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. and T. N. have received funding from the European Union H2020 research and innovation program under the Marie S. Curie Grant Agreement no. 860762 (MSCA ITN: CHAIR).Notes and references
- (a) R. A. Kerr, Science, 2010, 328, 1624 CrossRef CAS PubMed; (b) J. Cooper, L. Stamford and A. Azapagic, Energy Technol., 2016, 4, 772 CrossRef.
- A. Caballero and P. J. Pérez, Chem. Soc. Rev., 2013, 42, 8809 RSC.
- For selected reviews on the topic of the C(sp3)–H functionalization of liquid and solid alkanes see: (a) W. Ali, G. Prakash and D. Maiti, Chem. Sci., 2021, 12, 2735 RSC; (b) J. Das, S. Guin and D. Maiti, Chem. Sci., 2020, 11, 10887 RSC; (c) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS PubMed.
- (a) R. H. Crabtree, Chem. Rev., 1995, 95, 987 CrossRef CAS; (b) H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096 CrossRef CAS PubMed.
- (a) M. Ravi, M. Ranocchiari and A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464 CrossRef CAS PubMed; (b) N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521 CrossRef CAS PubMed; (c) P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580 RSC; (d) V. N. Cavaliere, B. F. Wicker and D. J. Mindiola, Adv. Organomet. Chem., 2012, 60, 1 CrossRef CAS.
- (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (b) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS PubMed; (c) S. Kawamorita, R. Murakami, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2013, 135, 2947 CrossRef CAS PubMed; (d) S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8157 CrossRef CAS PubMed; (e) C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714 CrossRef CAS PubMed; (f) C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 12422 CrossRef CAS PubMed; (g) R. Oeschger, B. Su, I. Yu, C. Ehinger, E. Romero, S. He and J. F. Hartwig, Science, 2020, 368, 736 CrossRef CAS PubMed; (h) H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS PubMed; (i) M. R. Jones, C. D. Fast and N. D. Schley, J. Am. Chem. Soc., 2020, 142, 6488 CrossRef CAS PubMed; (j) H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391 CrossRef CAS.
- (a) Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (c) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31 CrossRef CAS; (d) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed.
- P. R. Rablen and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 4648 CrossRef CAS.
- A. K. Cook, S. D. Schimler, A. J. Matzger and M. S. Sanford, Science, 2016, 351, 1421 CrossRef CAS PubMed.
- K. T. Smith, S. Berritt, M. Gonzalez-Moreiras, S. Ahn, M. R. Smith III, M. H. Baik and D. J. Mindiola, Science, 2016, 351, 1424 CrossRef CAS PubMed.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551 CrossRef CAS PubMed.
- J. F. Hartwig, K. S. Cook, M. Hapke, C. D. Incarvito, Y. Fan, C. E. Webster and M. B. Hall, J. Am. Chem. Soc., 2005, 127, 2538 CrossRef CAS PubMed.
- For a pertinent review on the topic: (a) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC for selected examples: ; (b) J. D. Lawrence, M. Takahashi, C. Bae and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 15334 CrossRef CAS PubMed; (c) C. S. Wei, C. A. Jiménez-Hoyos, M. F. Videa and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3078 CrossRef CAS PubMed.
- M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633 CrossRef CAS PubMed.
- (a) H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256 CrossRef PubMed; (b) C. S. Leung, S. S. F. Leung, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2012, 55, 4489 CrossRef CAS PubMed.
- (a) Y. Fujiwara, K. Takaki and Y. Taniguchi, Synlett, 1996, 591 CrossRef CAS; (b) T. Nishiguchi, K. Nakata, K. Takaki and Y. Fujiwara, Chem. Lett., 1992, 1141 CrossRef CAS; (c) K. Nakata, T. Miyata, T. Jintoku, A. Kitani, Y. Taniguchi, K. Takaki and Y. Fujiwara, Bull. Chem. Soc. Jpn., 1993, 66, 3755 CrossRef CAS; (d) K. Nakata, Y. Yamaoka, T. Miyata, Y. Taniguchi, K. Takaki and Y. Fujiwara, J. Organomet. Chem., 1994, 473, 329 CrossRef CAS.
- R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481 CrossRef CAS PubMed and related thematic issue.
- Y. Taniguchi, S. Horie, K. Takaki and Y. Fujiwara, J. Organomet. Chem., 1995, 504, 137 CrossRef CAS.
- M. Lin and A. Sen, Nature, 1994, 368, 613 CrossRef CAS.
- For reviews on the topic, see: (a) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2013, 53, 74 CrossRef PubMed; (b) C. J. Scheuermann, Chem. – Asian J., 2010, 5, 436 CrossRef CAS PubMed.
- W. Xie, J. Heo, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 7487 CrossRef CAS PubMed.
- A. Caballero, E. Despagnet-Ayoub, M. M. Díaz-Requejo, A. Díaz-Rodríguez, M. E. González-Núñez, R. Mello, B. K. Muñoz, W.-S. Ojo, G. Asensio, M. Etienne and P. J. Pérez, Science, 2011, 332, 835 CrossRef CAS PubMed.
- (a) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (b) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (c) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed; (d) M. M. Díaz-Requejo, T. R. Belderraín, M. C. Nicasio and P. J. Pérez, Dalton Trans., 2006, 5559 RSC.
- (a) J. Urbano, T. R. Belderraín, M. C. Nicasio, S. Trofimenko, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2005, 24, 1528 CrossRef CAS; (b) E. Despagnet-Ayoub, K. Jacob, L. Vendier, M. Etienne, E. Álvarez, A. Caballero, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2008, 27, 4779 CrossRef CAS.
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111 CrossRef CAS.
- (a) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (c) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed; (d) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (f) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (g) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- (a) L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376 RSC; (b) L. Guillemard and J. Wencel-Delord, Beilstein J. Org. Chem., 2020, 16, 1754 CrossRef CAS PubMed; (c) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056 CrossRef CAS PubMed.
- H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661 CrossRef CAS PubMed.
- (a) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719 CrossRef CAS PubMed; (b) M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191 CrossRef CAS PubMed; (c) H. Deng, X. Fan, Z. Chen, Q. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579 CrossRef CAS PubMed.
- For reviews on the benefits of flow technology for C–H functionalization chemistry, see (a) C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92 CrossRef CAS; (b) S. Govaerts, A. Nyuchev and T. Noël, J. Flow Chem., 2020, 10, 13 CrossRef CAS; (c) S. Santoro, F. Ferlin, L. Ackermann and L. Vaccaro, Chem. Soc. Rev., 2019, 48, 2767 RSC; (d) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef PubMed.
- F. Xue, H. Deng, C. Xue, D. K. B. Mohamed, K. Y. Tang and J. Wu, Chem. Sci., 2017, 8, 3623 RSC.
- D. J. Wilger, J.-M. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720 CrossRef CAS PubMed.
- A. A. Isse, C. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92 CrossRef CAS PubMed.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701 CrossRef CAS.
- H. M. L. Davies, T. Hansen and M. R. Churchill, J. Am. Chem. Soc., 2000, 122, 3063 CrossRef CAS.
- For a review on the topic, see: H. M. L. Davies and K. Liao, Nat. Rev. Chem., 2019, 3, 347 CrossRef CAS PubMed.
- For reviews on the topic see: (a) A. Lipp, S. O. Badir and G. A. Molander,, Angew. Chem., Int. Ed., 2021, 60, 1714 CrossRef CAS PubMed; (b) C. Prentice, J. Morrisson, A. D. Smith and E. Zysman-Colman, Beilstein J. Org. Chem., 2020, 16, 2363 CrossRef PubMed; (c) H.-H. Zhang, H. Chen, C. Zhu and S. Yu, Sci. China: Chem., 2020, 63, 637 CrossRef CAS; (d) D. Saha, Chem. – Asian J., 2020, 15, 2129 CrossRef CAS PubMed; (e) C. Jiang, W. Chen, W.-H. Zheng and H. Lu, Org. Biomol. Chem., 2019, 17, 8673 RSC; (f) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed.
- Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016 CrossRef CAS.
- (a) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS PubMed; (b) S. R. Chemler, Science, 2013, 341, 624 CrossRef CAS PubMed; (c) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed; (d) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed.
- D. Michos, C. A. Sassano, P. Krajnik and R. H. Crabtree, Angew. Chem., Int. Ed. Engl., 1993, 32, 1491 CrossRef.
- M. C. Duval, B. Soep and W. H. Breckenridge, J. Phys. Chem., 1991, 95, 7145 CrossRef CAS.
- M. Z. Hoffmann, M. Goldwasser and P. L. Damour, J. Chem. Phys., 1967, 47, 2195 CrossRef.
- (a) A. W. Hofmann, Chem. Ber., 1883, 16, 558 CrossRef; (b) G. Kumar, S. Pradhan and I. Chatterdjee, Chem. – Asian J., 2020, 15, 651 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668 CrossRef CAS PubMed.
- Z. Dong, Z. Wen, F. Zhao, S. Kuhn and T. Noël, Chem. Eng. Sci.: X, 2021, 10, 100097 CAS.
- The group of Duan reported a similar functionalization of light alkanes for forging C–C bonds mediated by an iron catalyst and visible light: Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021 10.1039/D1GC01563J.
- Q. Yang, Y.-H. Wang, Y. Qiao, M. G. P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847 CrossRef CAS PubMed.
- (a) H. Yin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266 CrossRef CAS PubMed; (b) L. L. Costanzo, S. Pistara and G. Condorelli, J. Photochem., 1983, 21, 45 CrossRef CAS.
- M. A. Fuentes, R. Gava, N. I. Saper, E. A. Romero, A. Caballero, J. F. Hartwig and P. J. Pérez, Angew. Chem., Int. Ed., 2021, 60, 18467 CrossRef CAS PubMed.
- (a) B. L. Tran, B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 2555 CrossRef CAS PubMed; (b) A. Vasilopoulos, S. W. Krska and S. S. Stahl, Science, 2021, 372, 398 CrossRef CAS PubMed.
- J. Lee, S. Jin, D. Kim, S. H. Hong and S. Chang, J. Am. Chem. Soc., 2021, 143, 5191 CrossRef CAS PubMed.
- (a) J. Lee, J. Lee, H. Jung, D. Kim, J. Park and S. Chang, J. Am. Chem. Soc., 2020, 142, 12324 CrossRef CAS PubMed; (b) Y. Baek, A. Das, S. L. Zheng, J. H. Reibenspies, D. C. Powers and T. A. Betley, J. Am. Chem. Soc., 2020, 142, 11232 CrossRef CAS PubMed; (c) H. Lu, V. Subbarayan, J. Tao and X. P. Zhang, Organometallics, 2010, 29(2), 389 CrossRef CAS; (d) F. Ragaini, A. Penoni, E. Gallo, S. Tollari, C. L. Gotti, M. Lapadula, E. Mangioni and S. Cenini, Chem. – Eur. J., 2003, 9, 249 CrossRef CAS PubMed.
- V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264 CrossRef CAS PubMed.
- (a) G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551 CrossRef CAS PubMed; (b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed.
- S. A. Ponomarenko and S. Kirchmeyer, Adv. Polym. Sci., 2011, 235, 33 CrossRef.
- T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631 CrossRef CAS.
- B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2021, 50, 5062 RSC.
- (a) A. D. Sadow and T. D. Tilley, J. Am. Chem. Soc., 2005, 127, 643 CrossRef CAS PubMed; (b) A. D. Sadow and T. D. Tilley, Angew. Chem., Int. Ed., 2003, 42, 803 CrossRef CAS PubMed.
- M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203 CrossRef CAS.
- The group of Tilley exploited this mode of activation to also drive C–C bond formation: A. D. Sadow and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 7971 CrossRef CAS PubMed.
- (a) M. Fontecave, S. Ollagnier-de-Choudens and E. Mulliez, Chem. Rev., 2003, 103, 2149 CrossRef CAS PubMed; (b) R. Zabel and G. Weber, Anal. Bioanal. Chem., 2016, 408, 1237 CrossRef CAS PubMed; (c) M. Kazemi, S. Sajjadifar, A. Aydi and M. M. Heydari, J. Med. Chem. Sci., 2018, 1, 1 CAS.
- (a) P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed; (b) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC.
- M. D. Gernon, M. Wu, T. Buszta and P. Janney, Green Chem., 1999, 1, 127 RSC.
- (a) L. J. Lobree and A. T. Bell, Ind. Eng. Chem. Res., 2001, 40, 736 CrossRef CAS; (b) S. Mukhopadhyay and A. T. Bell, Ind. Eng. Chem. Res., 2002, 41, 5901 CrossRef CAS; (c) S. Mukhopadhyay and A. T. Bell, Angew. Chem., Int. Ed., 2003, 42, 1019 CrossRef CAS PubMed; (d) S. Mukhopadhyay and A. T. Bell, Org. Proc. Res. Dev., 2003, 7, 161 CrossRef CAS.
- Y. Ishii, K. Matsunaka and S. Sakaguchi, J. Am. Chem. Soc., 2000, 122, 7390 CrossRef CAS.
- S. Mukhopadhyay and A. T. Bell, J. Am. Chem. Soc., 2003, 125, 4406 CrossRef CAS PubMed.
- S. Mukhopadhyay and A. T. Bell, Chem. Commun., 2003, 1590 RSC.
- C. Díaz-Urrutia and T. Ott, Science, 2019, 363, 1326 CrossRef PubMed.
- F. Minisci, A. Citterio and C. Giordano, Acc. Chem. Res., 1983, 16, 27 CrossRef CAS.
- N. Basickes, T. E. Hogan and A. Sen, J. Am. Chem. Soc., 1996, 118, 13111 CrossRef CAS.
- V. A. Roytman and D. A. Singleton, Science, 2019, 364, eaax7083 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |