
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–direction towards the new large pore zeolite NUD-3†
Fei-Jian
Chen‡
 a,
Zihao Rei
Gao‡
b,
Jian
Li
*c,
Luis
Gómez-Hortigüela
d,
Cong
Lin
e,
Le
Xu
e,
Hong-Bin
Du
*f,
Carlos
Márquez-Álvarez
d,
Junliang
Sun
e and
Miguel A.
Camblor
*b
a,
Zihao Rei
Gao‡
b,
Jian
Li
*c,
Luis
Gómez-Hortigüela
d,
Cong
Lin
e,
Le
Xu
e,
Hong-Bin
Du
*f,
Carlos
Márquez-Álvarez
d,
Junliang
Sun
e and
Miguel A.
Camblor
*b
aDepartment of Chemistry, Bengbu Medical College, Bengbu 233030, China
bInstituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas (ICMM-CSIC) c/Sor Juana Inés de la Cruz 3, Madrid 28049, Spain. E-mail: macamblor@icmm.csic.es
cBerzelii Center EXSELENT on Porous Materials, Department of Materials and Environmental Chemistry, Stockholm University, Stockholm, 10691, Sweden. E-mail: jxpxlijian@pku.edu.cn
dInstituto de Catálisis y Petroleoquímica, Consejo Superior de Investigaciones Científicas (ICP-CSIC), c/Marie Curie 2, Madrid 28049, Spain
eCollege of Chemistry and Molecular Engineering, Peking University, 5 Yiheyuan Road, Beijing, 100871, China
fState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, China. E-mail: hbdu@nju.edu.cn
First published on 24th November 2020
Abstract
The new zeolite NUD-3 possesses a three-dimensional system of large pore channels that is topologically identical to those of ITQ-21 and PKU-14. However, the three zeolites have distinctly different frameworks: a particular single 4-membered ring inside the denser portion of the zeolite is missing in PKU-14, disordered in ITQ-21 and fully ordered in NUD-3. We document these differences and use molecular simulations to unravel the mechanism by which a particular structure directing agent dication, 1,1′-(1,2-phenylenebis(methylene))bis(3-methylimidazolium), is able to orient this inner ring.
Structure directing agents (SDAs) typically play an important role in the synthesis of zeolites.1,2 Over the last several decades there has been huge experimental effort in the design and trial of organic SDAs for the synthesis of zeolites.3 The effort has been rewarded with a large increase in the number of zeolite topologies recognized as unique and viable by the International Zeolite Association (IZA) with a steady rate of over five new topologies accepted per year during the last 40 years.4 However, it is still generally impossible to predict the target zeolite topology before the synthesis, even when the structural relationship among the various zeolite topologies and their “synthesis descriptors” is studied,5 or when the SDA is selected by energy minimization calculations to target a specific topology,6 although some notable cases of success have been reported.7 Occasionally, a very small change in the SDA brings about a large change in the ability to produce one or another zeolite framework.8 One could be tempted to consider structure–direction in pretty simple terms: zeolites that are intrinsically stable may just require a loose pore filling, easily achievable, for instance, by flexible SDAs that can adapt themselves to the zeolite pores (“the cation adapts to the pore”, as in the so called “default zeolites”9), whereas less stable zeolites would require a more specific structure–direction where the pore matches to some extent the geometry of a particular cation (“the zeolite adapts to the cation”). However, the structure–direction by organic agents is far from well understood and it is likely rich in subtleties, although the notion that the size and shape of the organic cation may influence the size and shape of the zeolite pore in which it resides is still popular. We will challenge this notion by presenting the case of three zeolites with distinct structures but topologically identical pores that are synthesized through the use of different cations.
The new zeolite NUD-3 (Nanjing University Du's group zeolite number 3) possesses a hitherto unknown topology that is, however, closely related to those of another two previously reported materials, ITQ-2110 and PKU-14.11 In fact, the three zeolites possess a topologically identical three-dimensional (3D) system of large pores opened through 12-membered ring windows (12MR). The difference between the three materials doesn’t lie in the pores but in the denser portion of the structure, where there may be missing tetrahedral atoms (PKU-14), a disordered array of atoms (ITQ-21) or a fully ordered structure (NUD-3). Since the pores of the three zeolites are topologically identical, it is intriguing that the ability to direct the synthesis towards any of the three materials depends on the host–guest interactions established between the cation and the pore. In this work, we characterize the new zeolite NUD-3 to clearly establish its unique nature and then use molecular simulations to study the mechanism of this special structure–direction effect.
NUD-3 has been discovered using 1,1′-(1,2-phenylenebis(methylene))bis(3-methylimidazolium) dications (SDA1, Fig. S1a, ESI†) and fluoride anions from a gel composition 0.5 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 GeO2:0.25 SDA(OH)2:0.5 HF:1.5 H2O, at 150 °C for 15 days within a static autoclave. It is worth mentioning that one very similar SDA2 (Fig. S1b, ESI†) was used by Boal et.al., but only yielded a layered material plus zeolite Beta.8c Additionally, we have found out that, under the same synthesis conditions, SDA3 (Fig. S1c, ESI†) and SDA4 (Fig. S1d, ESI†), also containing phenyl and imidazolium moieties, are good templates to synthesize ITQ-21, but not NUD-3 (see the ESI,† Table S1).12 An SDA similar to SDA3 but lacking all methyl groups at position 2 of the imidazolium rings directed the synthesis towards ITQ-37 quite specifically.13
:0.5 GeO2:0.25 SDA(OH)2:0.5 HF:1.5 H2O, at 150 °C for 15 days within a static autoclave. It is worth mentioning that one very similar SDA2 (Fig. S1b, ESI†) was used by Boal et.al., but only yielded a layered material plus zeolite Beta.8c Additionally, we have found out that, under the same synthesis conditions, SDA3 (Fig. S1c, ESI†) and SDA4 (Fig. S1d, ESI†), also containing phenyl and imidazolium moieties, are good templates to synthesize ITQ-21, but not NUD-3 (see the ESI,† Table S1).12 An SDA similar to SDA3 but lacking all methyl groups at position 2 of the imidazolium rings directed the synthesis towards ITQ-37 quite specifically.13
The structure of NUD-3 was solved by the state-of-the-art three-dimensional electron diffraction technique, namely continuous rotation electron diffraction (cRED).14 A 4/mmm Laue symmetry with a tetragonal unit cell a = 14.56 Å and c = 13.11 Å can be easily obtained from the cRED dataset (Fig. S2, ESI†). The reflection conditions derived from the reconstructed 3D reciprocal lattice indicated as possible space groups P422 (89#), P4mm (99#), P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m (111#), Pm2 (115#) and P4/mmm (123#). The cRED data were used to solve the structure with space group P4/mmm and the structure, including the SDA location, was refined against synchrotron powder X-Ray diffraction data (Fig. S3, ESI†). All the crystallographic details are provided in the ESI† (Tables S2–S4, ESI†) and the structure was deposited in the CCDC (no. 2045881).
2m (111#), Pm2 (115#) and P4/mmm (123#). The cRED data were used to solve the structure with space group P4/mmm and the structure, including the SDA location, was refined against synchrotron powder X-Ray diffraction data (Fig. S3, ESI†). All the crystallographic details are provided in the ESI† (Tables S2–S4, ESI†) and the structure was deposited in the CCDC (no. 2045881).
The framework of NUD-3 is very similar to those of ITQ-21 and PKU-14. Their structures could be described as 0D [46612] units of 32 T atoms (i.e. units displaying six 4MR and twelve 6MR faces; T atoms: tetrahedral atoms) connected through double 4-membered rings (D4R) along the three dimensions, thus forming a 3D framework with intersecting 12MR channels along the three crystallographic axes (Fig. 1a and b). It should be noted that the only topological difference among NUD-3, ITQ-21 and PKU-14 is the presence, orientation or absence of four additional inner T atoms in that unit, which then may actually contain 32 or 36 T atoms. In ITQ-21, a single 4-membered ring unit (S4R) formed by those four inner atoms is disordered along the a-, b-, and c-axis, so each S4R has an occupancy of 1/3 and the unit contains 36 T atoms (Fig. 1d).10 In PKU-14, the four inner atoms are absent and the unit is effectively a 32 T-atom cage filled with 8 terminal T–OH and 2 extra water molecules (Fig. 1e).11 In contrast, in NUD-3, the four inner atoms form a S4R, which is ordered and oriented perpendicular to the c-axis (Fig. 1c), and the unit is a 36 T-atom unit. Since the structural difference between the three frameworks is restricted to an area inside a dense portion of the structure, the pore system and the microporous surface of the three zeolites are topologically equivalent, which makes intriguing that a simple host–guest filling interaction in the pores might cause a difference in the zeolite that crystallizes.
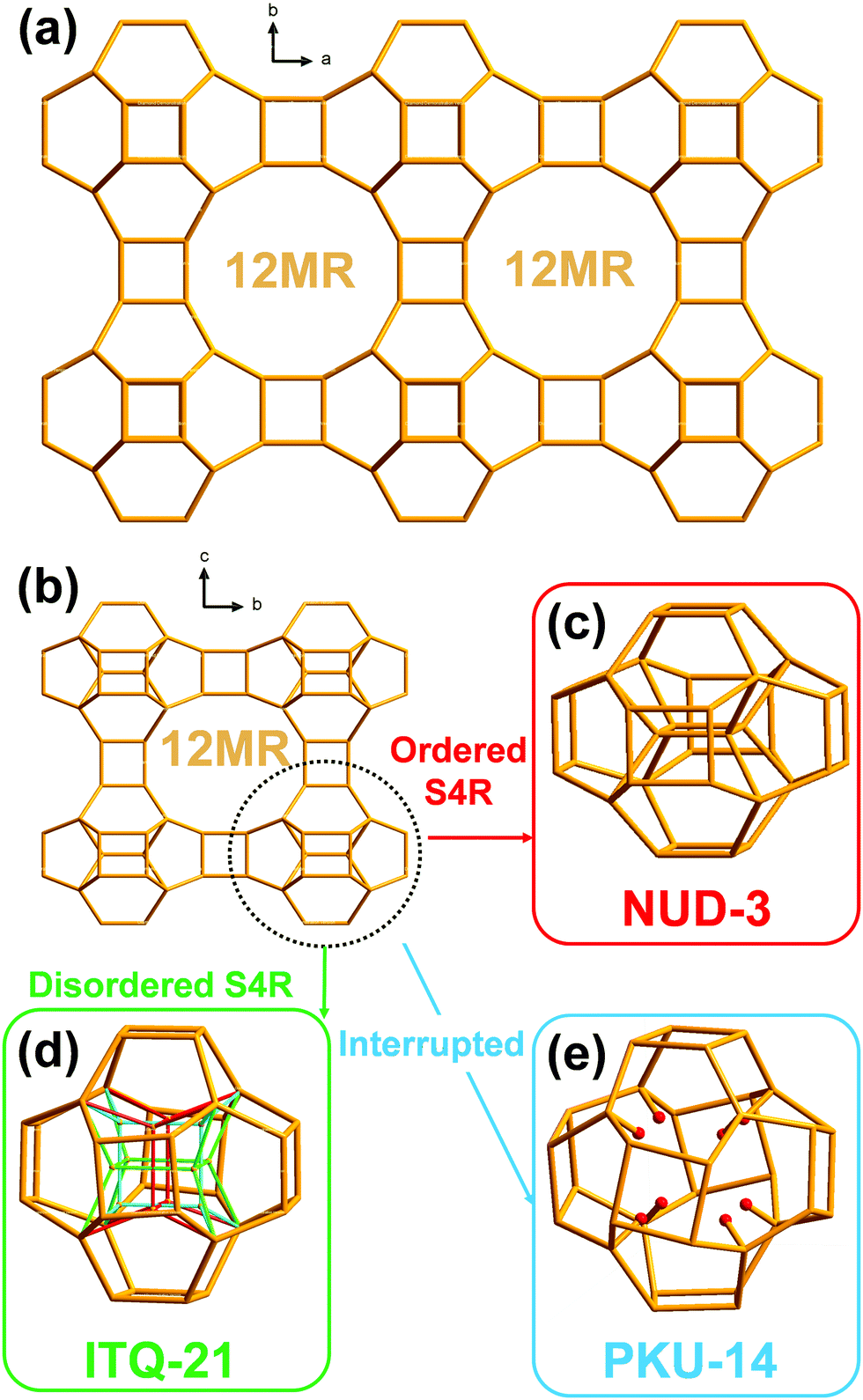 | ||
| Fig. 1 Structural relationship among NUD-3, ITQ-21, and PKU-14: (a) 12MR channel in all three structures, viewed along the c-axis, (b) 12MR channel in NUD-3 with ordered S4R, viewed along the a-axis, where the 36 T unit is encircled, (c) the unit of 36 T atoms in NUD-3, showing the ordered S4R, (d) the unit of 36 T atoms in ITQ-21, with a S4R disordered along each direction and a 1/3 occupancy each (red, green and light blue), and (e) the unit of 32 T atoms in PKU-14, showing the absence of S4R and the presence of 8 terminal T–OH inside the resulting cage. | ||
NUD-3 has a 3D system of intersecting and straight 12MR channels along the a-, b- and c-axis. Along the a- and c-axis (Fig. S4, ESI†), the pore sizes are 6.3 × 8.1 Å and 7.7 × 8.0 Å, respectively. The framework density (FD) of NUD-3 is 13.63 T/1000 Å3, which is very close to that of ITQ-21 (13.55).10b N2 and Ar adsorption data (Fig. S5–S7) are discussed in the ESI.†
The differences between NUD-3, ITQ-21 and PKU-14 are clearly revealed in the PXRD patterns of the three structures and their symmetries (Fig. 2 and Table S5, ESI†). As-made and calcined ITQ-21 are cubic, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, only showing three reflections, (200), (220) and (222), in the region up to 12° (Fig. 2a). As-made PKU-14 is monoclinic, I2/m, which splits the first and second peaks into several partially overlapped peaks (Fig. 2c). However, after heating PKU-14 at 180 °C for 2 hours, the removal of the water molecule inside the [46612] cage results in a cubic symmetry with space group Fmc (Fig. 2b),11 the resulting pattern being much similar to that of ITQ-21 with the same three reflections in the low angle region. This high symmetry reflects the disorder inside the denser portion of both frameworks. By contrast, both as-made and calcined NUD-3 are tetragonal, space group P4/mmm, due to the ordered orientation of S4R, and thus the first two reflections (200) and (220) in the cubic system split into (100), (001) and (110), (101), respectively (Fig. 2d and e). Additionally, when we calcined the NUD-3 samples in air from room temperature to 600 °C, all the patterns kept the same tetragonal symmetry (Fig. S8, ESI†). In conclusion, the three structures can be characterized and distinguished by the differences in their PXRD patterns and crystallographic data.
c, only showing three reflections, (200), (220) and (222), in the region up to 12° (Fig. 2a). As-made PKU-14 is monoclinic, I2/m, which splits the first and second peaks into several partially overlapped peaks (Fig. 2c). However, after heating PKU-14 at 180 °C for 2 hours, the removal of the water molecule inside the [46612] cage results in a cubic symmetry with space group Fmc (Fig. 2b),11 the resulting pattern being much similar to that of ITQ-21 with the same three reflections in the low angle region. This high symmetry reflects the disorder inside the denser portion of both frameworks. By contrast, both as-made and calcined NUD-3 are tetragonal, space group P4/mmm, due to the ordered orientation of S4R, and thus the first two reflections (200) and (220) in the cubic system split into (100), (001) and (110), (101), respectively (Fig. 2d and e). Additionally, when we calcined the NUD-3 samples in air from room temperature to 600 °C, all the patterns kept the same tetragonal symmetry (Fig. S8, ESI†). In conclusion, the three structures can be characterized and distinguished by the differences in their PXRD patterns and crystallographic data.
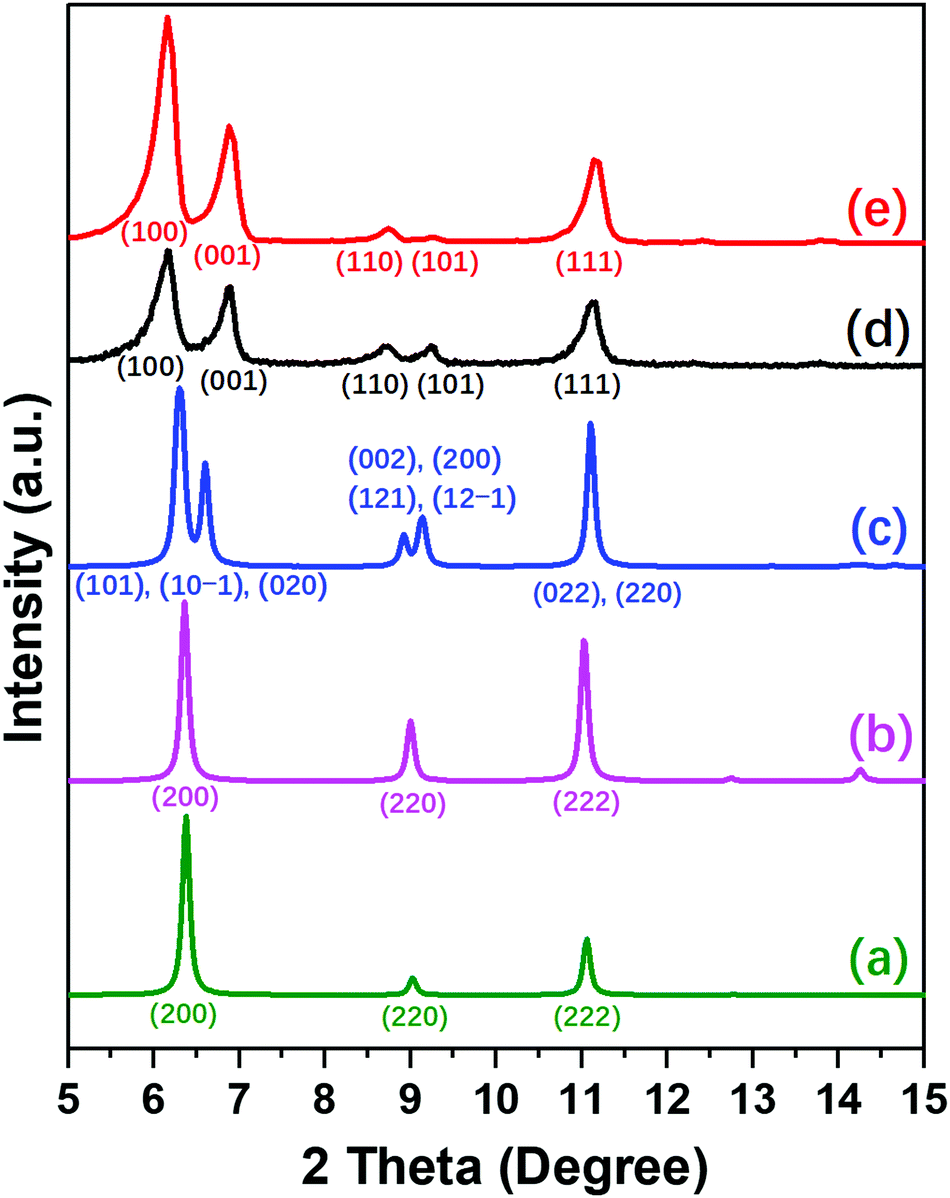 | ||
| Fig. 2 PXRD patterns of simulated (a) as-made ITQ-21, (b) dehydrated PKU-14, (c) as-made PKU-14, and experimental (d) as-made NUD-3 and (e) calcined NUD-3. Each peak ranging from 2θ 5–15° is marked with its Miller index. | ||
The Fourier transform infrared (FT-IR) spectra can additionally prove the difference between NUD-3 and PKU-14. After heating at 180 °C for 2 hours, the FT-IR spectrum of NUD-3 (Fig. S9, ESI†) showed no band in the 3400–3600 cm−1 range while PKU-14 showed a band located at 3528 cm−1 ascribed to the stretching vibrations of the terminal hydroxyl groups.11
The incorporation of SDA1 within the NUD-3 framework was studied by molecular simulations. We started from the location of the organic dication obtained from the Rietveld refinement; in this location, the organic dication sites in the ac (or bc) plane, with one imidazolium ring in the elliptical [100] (or [010]) channels, and the other imidazolium ring aligned along the c-axis (Fig. S10, ESI†). Three different long-range arrangements for packing the organic cations in such a position while avoiding short-contacts were found, which differ in the relative orientation of adjacent SDA1 cations (1, 2 and 3 in Fig. S11, ESI†). In the most stable case (3) (as calculated using Dreiding forcefield), the imidazolium rings along the c axis point in opposite directions, and no π–π stacking between imidazolium rings is developed (Fig. 3, top). From these systems with the NUD-3 framework under the Rietveld refined tetragonal unit cell, we built analogous systems but now with an isometric (pseudocubic) unit cell (with a = 13.8490 Å), and calculated the interaction energy (Table S6, ESI†). In all cases, the host/guest interaction energy developed in the pseudocubic framework is lower in magnitude, showing that SDA1 fits better in the tetragonal framework. In order to confirm this, we built hypothetical NUD-3 frameworks while varying the c parameter from 12.35 to 14.85 Å, and calculated the stability of the resulting structure (Fig. S12, ESI†). Energy results showed a reduced stability when c increased, displaying an energy minimum at the real c value of 12.85 Å, clearly evidencing an optimal geometric relationship of SDA1 and the NUD-3 tetragonal unit cell. Methyl groups in adjacent SDA1 cations of the imidazolium rings aligned with [001] channels become too close when c increases (Fig. 3, bottom), provoking a steric repulsion and providing a potential explanation for the reduced stability of SDA1 confined in the pseudocubic framework.
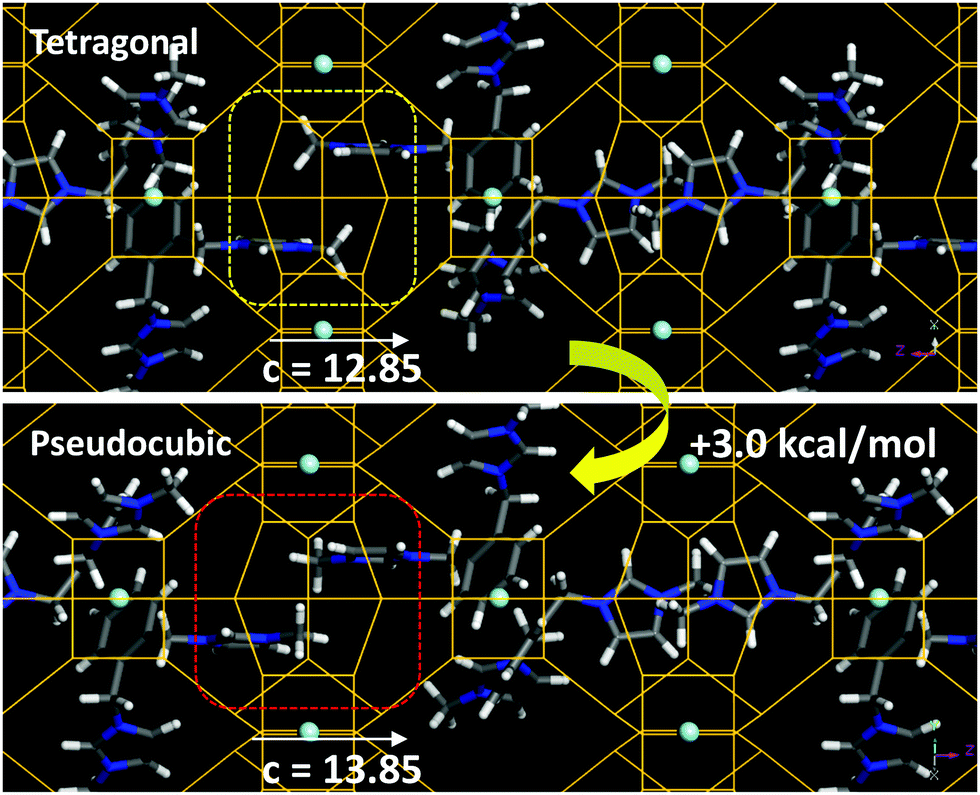 | ||
| Fig. 3 Packing interactions between imidazolium rings along the [001] channels in the tetragonal (top) or pseudocubic (bottom) cell, showing the steric repulsion in the latter case. | ||
As mentioned previously, the reduced c parameter is a direct consequence of the ordered nature of the S4R, and hence of the better fit of the SDA1 packing arrangement within a tetragonal unit cell. Propagation of this favored geometrical host/guest fit along the crystal might be explained because of the particular organic supramolecular arrangement, where the imidazolium rings aligned within the [001] channels develop a well-ordered orientation, with the imidazolium rings of adjacent cations siting parallel to each other to optimize the packing efficiency, spreading the preferred S4R orientation along the c-axis. In addition, adjacent SDA1 cations in the ab plane orient perpendicular to each other to avoid overlapping (Fig. S11, ESI†), thus promoting also propagation of the preferred S4R orientation along the crystal in [100] and [010] directions. We note here that under none of the three potential configurations, π–π stacking interactions between the phenyl rings are established, which is consistent with a major occurrence of phenyl monomers observed by fluorescence spectroscopy (band at 280 nm, Fig. S13, ESI†). Interestingly, this type of long-range organic arrangement achieved by SDA1 is not possible for SDA3 or SDA4 because of their different molecular structure, which could explain the lack of a preferential long-range orientation of the zeolite building units, and hence the crystallization of a cubic framework with disordered S4R instead.
NUD-3 poses an additional intriguing question related to charge balance. In our SDA1-NUD-3 model, there are 2 dications per unit cell, which amounts to 4 positive charges. The organic content in the real system, impossible to model by simulation because of disorder, is even slightly larger. Three of the positive charges would be compensated by three F anions occupying the three D4Rs per unit cell found in the NUD-3 framework, consistent with a 19F MAS NMR resonance at −8.2 ppm (Fig. S14, ESI†) that we assign to fluoride occluded in D4R units with Ge–Ge pairs but with no Ge adjacent to three Ge neighbours (type III resonance).15 The rest of the positive charges could be balanced by T–O− but these are more frequently associated with T–OH that are absent in NUD-3,16 according to the FT-IR spectra commented above. Thus, we propose that additional fluoride anions could be located in the main channels, associated with water clusters or the positively charged imidazolium rings, and could be responsible for an additional broad signal around −100 ppm in the 19F MAS NMR spectrum. A broad signal around a similar chemical shift is present in the spectrum of PKU-12, where fluoride is believed to be present in 8MR pores (in addition to D4R).17
In conclusion, NUD-3 possesses a new topology with a 3D system of large pores. The structure is fully ordered and fully condensed and is closely related to the disordered zeolite ITQ-21 and the interrupted zeolite PKU-14. Its structure has been solved by continuous rotation electron diffraction and refined using synchrotron diffraction data. We propose that the ability of the SDA to direct the crystallization to a fully ordered, fully condensed tetragonal framework relies on better packing of the organic structure directing agent in the tetragonal cell. This can explain the specific structure–direction of SDA1 towards NUD-3 despite the topological equality of the pore system of the three zeolites.
The authors are grateful for financial support from the Spanish Ministry of Science Innovation and Universities (MAT2015-71117-R, PID2019-105479RB-I00 and PID2019-107968RB-I00 projects, AEI, Spain and FEDER, EU), the National Natural Science Foundation of China (No. 22071099, 21673115, 21527803, 21871009, and 21601004), top talent cultivation program of Anhui Educational Committee (gxgwfx2018051), and the Swedish Research Council (VR) and the Knut and Alice Wallenberg Foundation (KAW). Synchrotron experiments were performed at beamline BL04 (MSPD) at ALBA Synchrotron with the collaboration of ALBA staff, and special thanks are given to A. Manjón for support in collecting the data and for helpful comments and suggestions. Z. R. G. specially thanks Yingxia Wang of Peking University for her kind suggestions and help with this paper. Centro Técnico Informático-CSIC is acknowledged for running the calculations, and BIOVIA for providing the computational software. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. F. Lobo, S. I. Zones and M. E. Davis, J. Inclusion Phenom. Macrocyclic Chem., 1995, 21, 47–78 CAS.
- L. Gómez-Hortigüela and M. A. Camblor, Insights Into The Chemistry Of Organic Structure-Directing Agents In The Synthesis Of Zeolitic in Materials, ed. L. Gómez-Hortigüela, Springer, 2018, 175, pp. 1–42 Search PubMed.
- Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, Springer, 2018, p. 175 Search PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, 2020, http://www.iza-structure.org/databases.
- K. Muraoka, Y. Sada, D. Miyazaki, W. Chaikittisilp and T. Okubo, Nat. Commun., 2019, 10, 4459 CrossRef.
- C.-R. Boruntea, G. Sastre, L. F. Lundegaard, A. Corma and P. N. R. Vennestrøm, Chem. Mater., 2019, 31(22), 9268–9276 CrossRef CAS.
- (a) J. E. Schmidt, M. W. Deem and M. E. Davis, Angew. Chem., 2014, 126, 8512–8514 ( Angew. Chem., Int. Ed. , 2014 , 53 , 8372–8374 ) CrossRef; (b) S. K. Brand, J. E. Schmidt, M. W. Deem, F. Daeyaert, Y. Ma, O. Terasaki, M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(20), 5101–5106 CrossRef CAS.
- (a) A. Rojas, L. Gomez-Hortiguela and M. A. Camblor, Dalton Trans., 2013, 42, 2562–2571 RSC; (b) B. W. Boal, M. W. Deem, D. Xie, J. H. Kang, M. E. Davis and S. I. Zones, Chem. Mater., 2016, 28(7), 2158–2164 CrossRef CAS; (c) J. E. Schmidt, C.-Y. Chen, S. K. Brand, S. I. Zones and M. E. Davis, Chem. – Eur. J., 2016, 22, 4022–4029 CrossRef CAS; (d) P. Lu, L. Villaescusa and M. A. Camblor, Chem. Rec., 2018, 18, 713–723 CrossRef CAS; (e) L. Villaescusa, J. Li, Z. Gao, J. Sun and M. A. Camblor, Angew. Chem., 2020, 132, 11379–11382 ( Angew. Chem., Int. Ed. , 2020 , 59 , 11283–11286 ) CrossRef; (f) Z. R. Gao, J. Li, C. Lin, A. Mayoral, J. Sun and M. A. Camblor, Angew. Chem. DOI:10.1002/anie.202011801.
- A. Moini, K. D. Schmitt, E. W. Valyocsik and R. F. Polomski, Zeolites, 1994, 14(7), 504–511 CrossRef CAS.
- (a) A. Corma, M. J. Diaz-Cabanas, J. Martinez-Triguero, F. Rey and J. Rius, Nature, 2002, 418, 514–517 CrossRef CAS; (b) T. Blasco, A. Corma, M. J. Diaz-Cabanas, F. Rey, J. Rius, G. Sastre and J. A. Vidal-Moya, J. Am. Chem. Soc., 2004, 126, 13414–13423 CrossRef CAS.
- J. Liang, J. Su, Y. Wang, Y. Chen, X. Zou, F. Liao, J. Lin and J. Sun, Chem. – Eur. J., 2014, 20, 16097–16101 CrossRef CAS.
- F.-J. Chen, Rational Design and Application of Structure-Directing Agents for Zeolite Synthesis, PhD thesis, Nanjing University, Nanjing, 2014.
- F.-J. Chen, Z.-H. Gao, L.-L. Liang, J. Zhang and H.-B. Du, CrystEngComm, 2016, 18, 2735–2741 RSC.
- (a) J. Li and J. Sun, Acc. Chem. Res., 2017, 50(11), 2737–2745 CrossRef CAS; (b) Z. Huang, E. S. Grape, J. Li, K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427, 213583 CrossRef CAS.
- R. T. Rigo, S. R. G. Balestra, S. Hamad, R. Bueno-Perez, A. R. Ruiz-Salvador, S. Calero and M. A. Camblor, J. Mater. Chem. A, 2018, 6, 15110–15122 RSC.
- H. Koller, R. F. Lobo, S. L. Burkett and M. E. Davis, J. Phys. Chem., 1995, 99, 12588–12596 CrossRef CAS.
- J. Su, Y. Wang, J. Lin, J. Liang, J. Sun and X. Zou, Dalton Trans., 2013, 42, 1360–1363 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, characterization methods, crystallographic details, computational details, and supplementary tables, figures and references. See DOI: 10.1039/d0cc07333d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |