
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradation induced lattice anchoring self-passivation in CsPbI3−xBrx†
Jingwei
Xiu
ab,
Bo
Dong
 a,
Elizabeth
Driscoll
a,
Xiyuan
Feng
b,
Abubakar
Muhammad
a,
Shaoqing
Chen
b,
Zheng
Du
ab,
Yudong
Zhu
b,
Zheng
Zhang
b,
Zhaoheng
Tang
b,
Zhubing
He
*b and
Peter Raymond
Slater
*a
a,
Elizabeth
Driscoll
a,
Xiyuan
Feng
b,
Abubakar
Muhammad
a,
Shaoqing
Chen
b,
Zheng
Du
ab,
Yudong
Zhu
b,
Zheng
Zhang
b,
Zhaoheng
Tang
b,
Zhubing
He
*b and
Peter Raymond
Slater
*a
aSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: p.r.slater@bham.ac.uk
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen, 518055, P. R. China
First published on 30th April 2020
Abstract
The all-inorganic halide perovskite (CsPbI3) holds promise for photovoltaic applications but suffers from a detrimental phase transformation to a non-perovskite phase δ-CsPbI3 at low-temperature. Of the different perovskite polymorphs, there has been a wide range of studies on γ-CsPbI3 due to its kinetic stability at near room-temperature. However, synthesis routes to this and other all-inorganic halide perovskites are still not ideal, requiring uneconomical elimination of humidity as well as quenching from elevated temperature. Water/moisture is commonly meticulously avoided due the fact that it can accelerate the detrimental degradation of the perovskite. In our synthesis, we used an alternative approach of engineering an in situ degradation process to form a dual-functional PbI(OH) protective covering and succeeded in performing the first room-temperature synthesis of γ-CsPbI3 under ambient humidity. The vastly improved stability benefits from both lattice anchoring and physical coverage of γ-CsPbI3 by an ultra-thin PbI(OH) layer. The resultant γ-CsPbI3 is stable for more than 2 months under ambient conditions (25 °C, RH = 30–60%) and more than 12 hours at 175 °C in air without any degradation. Furthermore, we show that this novel facile method can be successfully applied to mixed halide perovskites such as CsPbI2Br, and this has allowed the first experimental synthesis of the γ-polymorph of CsPbI2Br. Thus, our work provides an efficient degradation-induced lattice-anchoring self-stabilization strategy and a new avenue to the economical synthesis of all-inorganic perovskite materials at room-temperature under ambient conditions.
Introduction
Organic–inorganic halide perovskites have attracted tremendous attention in the past decade because of their good photovoltaic performance.1–4 However, the thermal instability resulting from the volatile nature of the organic cations (e.g., CH3NH3+) impedes their development toward commercialization.5,6 All-inorganic perovskites, realized by replacing the unstable organic cations with inorganic Cs+, are promising candidates for more stable perovskite solar cells.7,8 Unfortunately, α-CsPbI3 is only stable above 325 °C because of the structural instability of the perovskite resulting from the smaller size of Cs+ cations.9 It is first converted into the low-temperature β/γ perovskite phase and then finally to non-perovskite δ-CsPbI3 at room-temperature as shown in Fig. 1a.9–11 This phase instability of CsPbI3 has attracted considerable interest in the development of strategies to achieve the formation of these low-temperature photoactive β/γ perovskite phases, particularly the γ phase.12,13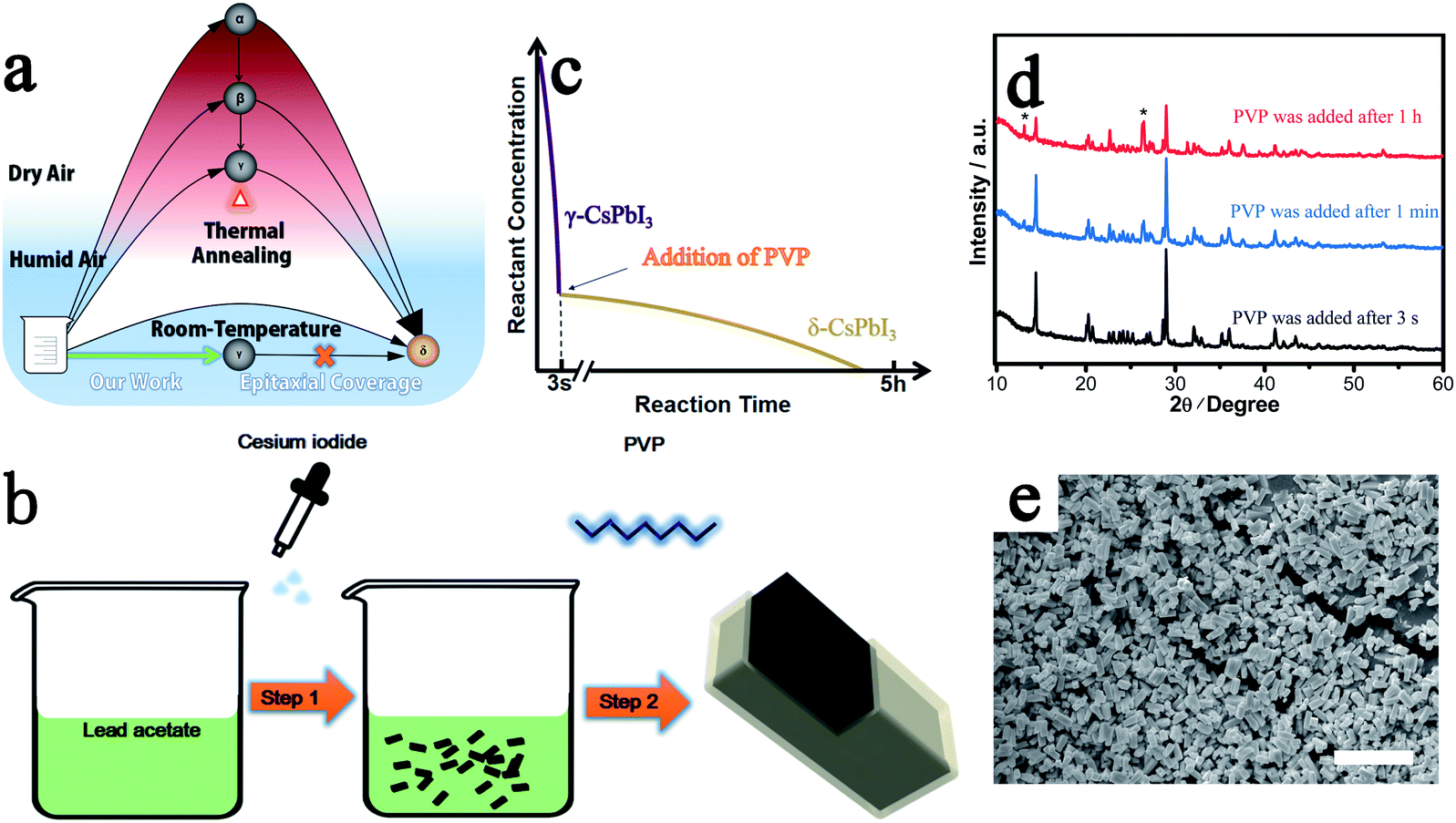 | ||
| Fig. 1 Preparation of all-inorganic perovskite. (a) Synthesis processes and phase relations of CsPbI3 in dry/humid air at low/high temperature. (b) Schematic diagram for the preparation of CsPbI3@PbI(OH). (c) The precipitation of γ-CsPbI3 and δ-CsPbI3vs. time. The addition of PVP is suggested closely following the formation of γ-CsPbI3. (d) PXRD patterns of CsPbI3@PbI(OH) prepared with PVP added after different time intervals. Peaks from δ-CsPbI3 are marked with *. (e) SEM image of CsPbI3@PbI(OH) on conductive carbon adhesive. Scale bar: 10 μm. | ||
Single crystals of γ-CsPbI3 have been grown via a solid-state method through quenching with strict control over humidity.14 Thin-films of γ-CsPbI3 have also been widely studied in recent years.13,15–17 In order to reduce the synthesis temperature, HI has been introduced for the preparation of the γ-CsPbI3 film so as to decrease the annealing temperature for γ-CsPbI3 to 100 °C.13 More recently, co-evaporation of CsI and PbI2 onto the substrate has allowed a lower temperature annealing of 50 °C.17 However, the preparation of γ-CsPbI3 at room-temperature, especially in the ambient environment (with its associated humidity challenges), has not been reported previously. To this end, we have been examining the possibility of synthesizing this phase via a solution route. Such solution-processed perovskites have various merits such as the potential commercial benefits of low-cost room-temperature preparation and scale-up. Nevertheless, this approach is not without its challenges, especially for the preparation of γ-CsPbI3 as a result of the greater inherent thermodynamic stability of the undesirable non perovskite δ-CsPbI3 product. In particular, the presence of moisture or heat is known to accelerate the phase transformation into δ-CsPbI3.17,18 Various strategies, such as reducing the grain size and composition engineering, have been examined to improve the stability.16,19,20 However, these routes entail their own issues, such as the introduction of potentially undesirable defects and the consequent influence on the band gap of the perovskite. Other potential strategies include surface passivation strategies, such as via ligand passivation, which show some promise, although the uniformity and compactness of any resultant passivation layer remains a challenge to be addressed.21,22
In view of these challenges, our aim was to find a feasible solution-processing approach to prepare this all-inorganic perovskite at room-temperature in air in order to kick-start future advanced applications of this material (Fig. 1a). In particular we aimed to address the issue of relying on uneconomical dry synthesis environments, by designing a surface degradation process that would lead to uniform growth of a surface passivation layer, which would be expected to provide a high-quality surface protective covering while preventing uncontrolled degradation throughout the particles. In our thoughts behind this strategy, we were drawn to the degradation reaction of γ-CsPbI3 in water, which is a universal problem for perovskite and has led researchers to naturally avoid its presence for the preparation of these perovskite phases. Our approach was to try to utilize this process to provide a surface degradation layer to passivate against further degradation. The challenge is to achieve a uniform passivation layer, whose lattice matches that of the perovskite in order to prevent delamination and so creating a fresh surface. As a common example, iron readily rusts, due to the fact that the corrosion product delaminates, creating a fresh surface, while Al is kinetically stabilized by a surface layer of the oxide which protects the metal from further corrosion. The aim of stabilizing γ-CsPbI3 therefore is to create a lattice matched surface decomposition product to provide a similar protective layer. In particular, it is known that water can degrade these perovskite systems, and so we explored solution routes containing water to examine whether we could induce and then cap this degradation process to form a stable protective layer.
Here we report this new solution processing preparation route to stable γ-CsPbI3 at room-temperature in an ambient environment. By controlling the degradation process of perovskite with the presence of water, an ultra-thin single-crystal PbI(OH) epitaxial shell is grown on the surface of γ-CsPbI3 micro-crystals assisted by the presence of acetate and PVP. This in situ produced PbI(OH) passivation shell provides a dense protective covering for the perovskite, resulting for the first time in both excellent thermal stability and moisture stability of γ-CsPbI3 in air. This method has also been successfully applied to CsPbI2Br and has allowed the preparation of the low-temperature γ-CsPbI2Br phase for the first time.
Experimental
Materials
Anhydrous isopropanol (99.8%) was purchased from Acros Organics. Methanol (99.8%) and ethanol (99.8%) were purchased from Alfa Aesar. Lead acetate trihydrate (99.995%) and cesium iodide (CsI, 99.995%) were purchased from Alfa Aesar. PVP 4000 was purchased from Sigma Aldrich. All materials were used as received.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). 0.5 mmol lead acetate trihydrate was dissolved in 10 ml methanol. For the preparation of CsPbI3@PbI(OH), 400 μl Pb2+ precursor solution was added into a vial with 4 ml ethanol. The ethanol was used as an anti-solvent, in order to accelerate the precipitation of the perovskite by changing the polarity of the solvent system. The absence of ethanol leads to rapid degradation of the black phase in a few seconds owing to the strong polarity of water and methanol. γ-CsPbI3 was obtained when 300 μl Cs+ precursor solution was injected. Stable CsPbI3@PbI(OH) was obtained by adding 200 mg PVP 4000 closely following the formation of γ-CsPbI3. The CsPbI3@PbI(OH) was washed with ultra-dry isopropanol (IPA) and dried in air naturally. Note: the ultra-dry solvent is not required during the synthesis and is only used for washing after synthesis.
:1). 0.5 mmol lead acetate trihydrate was dissolved in 10 ml methanol. For the preparation of CsPbI3@PbI(OH), 400 μl Pb2+ precursor solution was added into a vial with 4 ml ethanol. The ethanol was used as an anti-solvent, in order to accelerate the precipitation of the perovskite by changing the polarity of the solvent system. The absence of ethanol leads to rapid degradation of the black phase in a few seconds owing to the strong polarity of water and methanol. γ-CsPbI3 was obtained when 300 μl Cs+ precursor solution was injected. Stable CsPbI3@PbI(OH) was obtained by adding 200 mg PVP 4000 closely following the formation of γ-CsPbI3. The CsPbI3@PbI(OH) was washed with ultra-dry isopropanol (IPA) and dried in air naturally. Note: the ultra-dry solvent is not required during the synthesis and is only used for washing after synthesis.
Results and discussion
Characterization of γ-CsPbI3
Through our new approach, γ-CsPbI3 is directly prepared via a solution route at room-temperature in an ambient environment even in the presence of high humidity, bypassing the previous need to quench high-temperature α-CsPbI3 to obtain this phase. The perovskite is then further self-stabilized by surface degradation to prevent conversion into the undesired non-perovskite phase δ-CsPbI3 (Fig. 1a). When the Cs+ and Pb2+ precursor solutions are added into ethanol, a black colloid appears immediately through a simple one-step reaction as shown in Fig. 1b and eqn (1).| 3CsI + Pb(CH3COO)2 → γ-CsPbI3 + 2Cs(CH3COO) | (1) |
As in the prior method of rapid quenching from high temperature to “freeze in” the desired γ-CsPbI3,14 in our method, a similar rule is obeyed in that a high reactant concentration and a rapid precipitation process, which is completed in seconds, are required to produce high quality γ-CsPbI3 (Fig. 1c) while a low concentration and slow reaction speed results in significant amounts of the undesired δ-CsPbI3 phase. This is further illustrated by the slow growth of yellow phase δ-CsPbI3 from the residual reactants over a few hours after the rapid formation of the black phase γ-CsPbI3 perovskite in our experiment (needle-like δ-CsPbI3 can be seen to appear over time as shown in Fig. S1†). The novelty of our approach is that the reaction is performed both at room temperature and in the presence of water, with this route leading to the growth of a protective PbI(OH) lattice anchored shell coating the CsPbI3 particles. The challenge, however, remains to block the concomitant growth of undesired δ-CsPbI3, due to continued degradation, and so PVP was added to the solution after the formation of γ-CsPbI3 to cap the degradation process as shown in step 2 in Fig. 1b. We investigated addition of PVP during different times in the synthesis. The results showed that when PVP was added 1 h after the initial formation of black phase γ-CsPbI3, strong peaks from undesired δ-CsPbI3 appeared in the powder X-ray diffraction (PXRD) patterns (Fig. 1d); however, when PVP was added within 1 min, the intensity of the peaks from δ-CsPbI3 was diminished. Furthermore, when PVP is added immediately (within 3 s), peaks belonging to δ-CsPbI3 can be avoided. The results therefore show that the addition of PVP is efficient in eliminating the gradual growth of δ-CsPbI3. Structural refinement using PXRD data at room temperature confirms the formation of the orthorhombic Pbnm (no. 62) structure of γ-CsPbI3 with lattice parameters a = 8.579(1) Å, b = 12.472(1) Å, and c = 8.867(1) Å as shown in Fig. S2,† which are in agreement with previous reports.13 SEM studies show that the γ-CsPbI3 particles are crystallized into regular micro-rods as shown in Fig. 1e.
The key to the success of this route is the control of the degradation process to ensure both the formation and epitaxial growth of the PbI(OH) lattice anchoring shell. Here, we discuss why PbI(OH) is formed rather than other reported degradation products, such as PbI2.5,23 In particular, we believe that the acetate ligand from the precursor is important to ensure the one-step surface degradation of perovskite into pure PbI(OH). To prove this postulate, black colloids of γ-CsPbI3 synthesized without PVP were isolated from the mother liquor and re-dispersed in 2 ml ethanol with/without cesium acetate in order to study the role of the acetate ligand during the degradation process. To initiate degradation, water was then added to both, which resulted in complete decomposition of the black colloids within 5 minutes. Interestingly, a pale-yellow product was formed with the presence of cesium acetate, while a yellow compound was obtained without cesium acetate, as shown in Fig. 2a. The pale yellow and yellow products were confirmed to be pure PbI(OH) and a mixture of PbI(OH) and PbI2, respectively. Thus, with the presence of the acetate ligand, pure PbI(OH) is formed in one step as shown in eqn (2).
| γ-CsPbI3 + Cs(CH3COO) + H2O → PbI(OH) + 2CsI + CH3COOH | (2) |
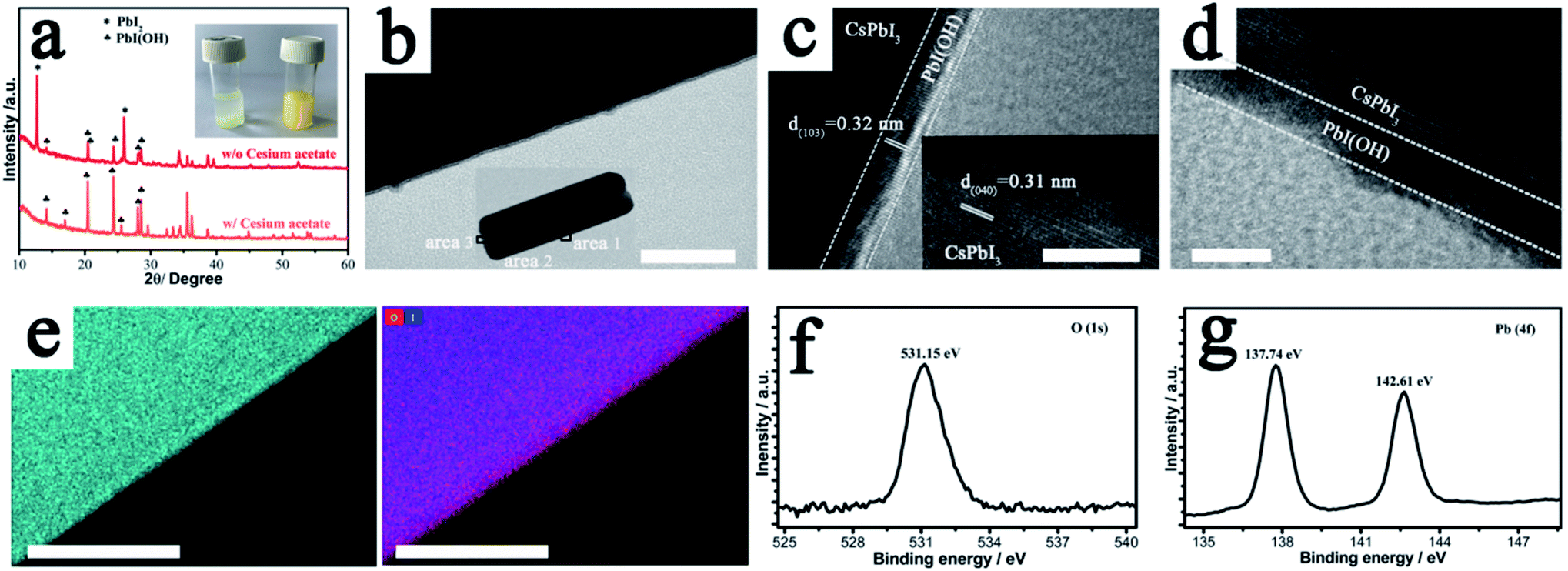 | ||
| Fig. 2 Characterization of epitaxial PbI(OH) on CsPbI3. (a) PXRD pattern of CsPbI3 degraded by water with/without the presence of cesium acetate. Inset: digital photograph of the corresponding degraded products. (b) TEM image of the PbI(OH) layer (scale bar: 100 nm) and selected areas 1–3 for the High-resolution TEM image (HrTEM). (c and d) HrTEM images of PbI(OH) (area 1 and area 3) and CsPbI3 (area 2). Scale bar: 10 nm. (e) STEM-EDX map of CsPbI3@PbI(OH). Left: map of Pb2+. Right: stacked map of O2− and I− (red: O2−, blue: I−, cyan: Pb2+). Scale bar: 100 nm. (f and g) X-ray photoelectron spectrum of CsPbI3@PbI(OH). | ||
In comparison, both PbI2 and PbI(OH) appear from the degradation of the perovskite with the absence of the acetate ligand. We believe that the rapid formation of pure PbI(OH) is essential for the formation of a dense epitaxial shell and that the acetate ligand works though influencing the pH of the solution, as it has been reported in the literature that PbI(OH) can be obtained from lead acetate when the pH is above 7, while PbI2 is produced when the pH is reduced to 7 or below.24 The subsequent addition of PVP is also crucial to cap the degradation process and ensure the formation of a uniform, dense high-quality PbI(OH) protective shell. This is illustrated by the fact that performing the synthesis of CsPbI3, without adding PVP, leads to a product that turns yellow (conversion to δ-CsPbI3) when dried in air. Nevertheless, when formed in the presence of PVP and separated from solution, the CsPbI3@PbI(OH) remains stable under ambient conditions even after removal of the PVP by washing with isopropanol (see later). It has been reported that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in PVP interacts with the CsPbI3 surface.21 Therefore, there is expected to be an initial chemical interaction between the fresh CsPbI3 surface and PVP as it is added into the reaction. As the perovskite surface degrades due to interaction with water into PbI(OH), this initial interaction will be broken. Instead, the PVP will be absorbed again on the PbI(OH) layer coordinated with the OH group, which might be expected to be a stronger interaction and so it will be hard to totally remove by the washing process. The role of this surface absorbed PVP is mostly likely to be limiting the speed of the degradation process and helping to contribute to the uniformity of PbI(OH) shell.
O group in PVP interacts with the CsPbI3 surface.21 Therefore, there is expected to be an initial chemical interaction between the fresh CsPbI3 surface and PVP as it is added into the reaction. As the perovskite surface degrades due to interaction with water into PbI(OH), this initial interaction will be broken. Instead, the PVP will be absorbed again on the PbI(OH) layer coordinated with the OH group, which might be expected to be a stronger interaction and so it will be hard to totally remove by the washing process. The role of this surface absorbed PVP is mostly likely to be limiting the speed of the degradation process and helping to contribute to the uniformity of PbI(OH) shell.
In order to confirm the formation of a protective shell of PbI(OH), we carried out high-resolution transmission electron microscope (HRTEM) measurements to characterize this shell. The results showed that the shell is uniform and forms a continuous surface as shown in Fig. 2b. HRTEM images of bulk perovskite shows the separation of the (040) planes with a spacing of 3.1 Å (Fig. 2c). In the PbIOH shell, the HRTEM image shows the separation of the similarly aligned (103) planes with a lattice spacing of 3.2 Å, leading to a small size mismatch of about 2.9% with the perovskite phase, and thus suggesting epitaxial growth of the degradation layer.25 Representations of these lattice planes are shown in Fig. S3† illustrating the good match between the lattices. While there is good crystallinity of the PbIOH shell on the long axes of the CsPbI3 microrods, the PbI(OH) layer on the end surfaces of these microrods does not show good crystallinity, which may indicate less effective lattice matching here (Fig. 2d). The growth of the PbI(OH) layer was further investigated by energy dispersive X-ray (EDX) spectroscopy using a scanning transmission electron microscope (STEM). The PbI(OH) layer can be distinguished by STEM-EDX mapping of the Pb element because of the different Pb densities in PbI(OH) compared to CsPbI3 as shown in Fig. 2e. The stacked mapping of the elements O and I reveals an O-rich area at the edge of the crystal, corresponding to the PbI(OH) shell. The CsPbI3@PbI(OH) particles were further characterized by X-ray photo-electron spectroscopy (XPS) measurements. The XPS signal at a bonding energy of 531.15 eV, corresponding to the featured peak of O in a metal hydroxide, is assigned to PbI(OH). The Pb 4f signal in this PbI(OH) surface layer is observed at a bonding energy of 137.74 eV, while the absence of a Pb 4f signature peak belonging to CsPbI3 at 137.30 eV indicates that the surface covering is compact so that there is no perovskite phase exposed as shown in Fig. 2g.16 Thus, these XPS results further provide strong evidence for the uniform coverage of the perovskite by the PbI(OH) shell. Elemental analysis through XPS shows an approximately 1:1 ratio of O atoms to I atoms (I:O = 1.08:1), which supports the presence of a PbI(OH) shell.
Stability of γ-CsPbI3
In order to probe the stability in air of these CsPbI3@PbI(OH) particles and confirm the protective nature of the PbI(OH) shell, the particles were washed with ultra-dry isopropanol three times in order to remove any absorbed PVP. To confirm PVP removal, Fourier Transform Infrared Spectra (FTIR) were recorded, which shows that the PVP is mostly removed after washing 3 times (see Fig. S4†). Significantly, even after removal of the PVP, the PXRD pattern shows that CsPbI3@PbI(OH) doesn't show any degradation after keeping it in ambient air (RH = 30–60%) for more than 2 months as shown in Fig. 3a. Furthermore, it shows no degradation after heating at 175 °C for 12 h in air with an environmental relative humidity of 60% as shown in Fig. 3b. At higher temperatures (200 °C), it does transform into δ-CsPbI3, which can be attributed to the dehydration and hence decomposition of the PbI(OH) surface layer.24 To further prove its excellent stability, we recorded the digital photos, SEM images and UV-vis transmittance spectra of the sample before and after heating in air for 12 h. These showed that no obvious changes triggered by degradation can be observed as shown in Fig. S5 and S6.† This heating experiment, along with the previous results for samples where the PVP has been washed away, support the fact that the origin of the improved stability is due to the PbI(OH) shell, rather than other factors, such as PVP coverage. Rather the PVP is present to cap the initial degradation process and ensure that a uniform surface covering is obtained. Thus, our results show that this new synthesis method is efficient in the stabilization of micron-sized particles of γ-CsPbI3 towards both moisture and heating.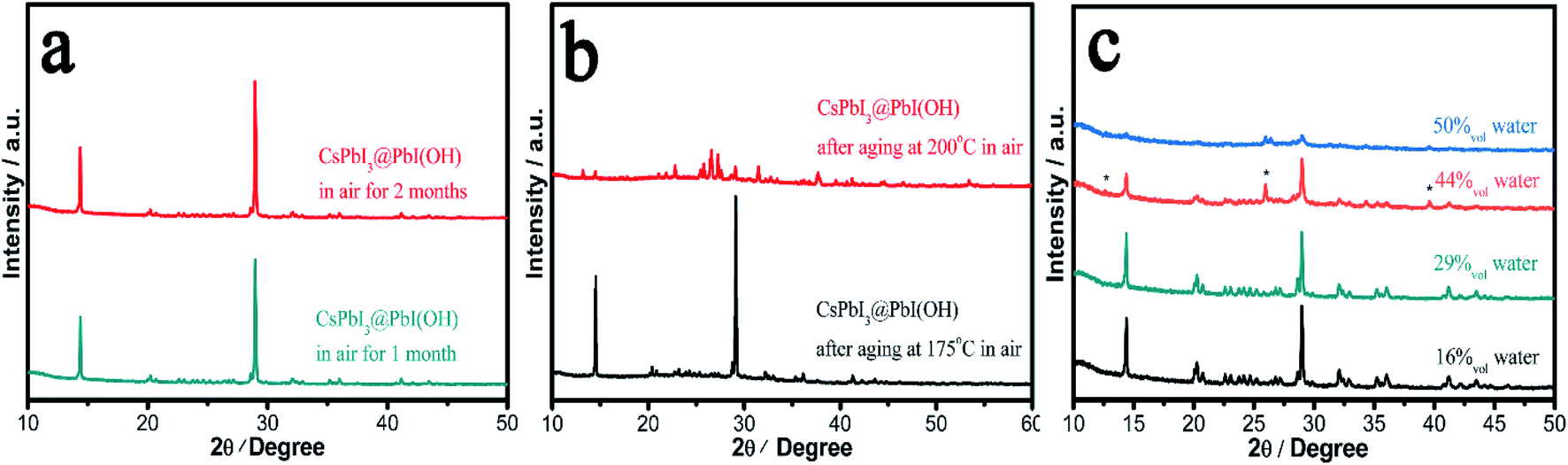 | ||
| Fig. 3 Stability of CsPbI3@PbI(OH) in air and solution. (a) PXRD measurement of CsPbI3@PbI(OH) stored in air. (b) PXRD patterns of CsPbI3@PbI(OH) aged at different temperatures in air showing stability up to 175 °C. (c) PXRD patterns of CsPbI3@PbI(OH) soaked in ethanol solution with different amounts of water, showing stability in up to 29% water. Peaks from PbI2 are marked with *. Note that XRD data show significant preferred orientation due to the rod-shaped crystals. | ||
We have further studied the tolerance of CsPbI3@PbI(OH) to the amount of water in solution and remarkably found that the perovskite is stable with a water concentration as high as 29%. In these experiments, 400 μl, 800 μl, 1.6 ml, and 2 ml water were added to γ-CsPbI3 in 2 ml mother liquor (containing PVP and cesium acetate). The volume concentrations of water in the above solutions are calculated to be 16%, 29%, 44%, and 50%. Fig. 3c shows the PXRD patterns of samples after soaking in the above solutions for 12 h at room temperature. Remarkably, the perovskite is stable under these conditions in a water concentration up to 29%. At higher water concentrations, peaks belonging to PbI2 begin to appear, indicating the decomposition of the PbI(OH) shell in such high water concentrations, with γ-CsPbI3 completely decomposed in the 50% water solution. This high tolerance of CsPbI3@PbI(OH) to water explains why our synthesis can be readily conducted in its presence.
In order to demonstrate the promise of this new method for scalable preparation of stable perovskite semiconductors in solution, we successfully scaled up to 100 ml volume as shown in Fig. S5.† We then considered whether the method could be successfully applied for the synthesis of other all-inorganic perovskites, such as CsPbI2Br.
Synthesis and characterization of γ-CsPbI2Br
CsPbI2Br has attracted considerable interest worldwide because it has been reported to show improved stability by partial substitution of I− with the smaller Br− anion. Prior studies have shown the synthesis of photoactive α-CsPbI2Br through heating followed by quenching to room temperature to kinetically stabilise this phase. Nevertheless, the low-temperature β/γ-CsPbI2Br polymorphs are also expected to show similar properties to α-CsPbI2Br, but their synthesis has proved elusive.26 Our method has the merit of preparation at room-temperature and so omits the need to prepare the high-temperature phase. Thus, through our room temperature solution method, we are able to prepare the low temperature γ-CsPbI2Br perovskite polymorph for the first time. Fig. 4a shows photographs of the CsPbI3@PbI(OH) and CsPbI2Br@PbI(OH) colloidal solutions. The reddish colour of CsPbI2Br@PbI(OH) confirms the introduction of Br− into the lattice. XRD data for this phase were collected and structural refinement confirmed this phase to be γ-CsPbI2Br with lattice parameters a = 8.598(1) Å, b = 12.224(2) Å, and c = 8.496(1) Å (Fig. S6†). Interestingly these structural studies showed the presence of some I/Br ordering, with the Br located on the I2 site (see ESI Table 1†). Furthermore, TEM images confirm a similar core–shell structure for CsPbI2Br@PbI(OH) as shown in Fig. 4b. As observed for CsPbI3, CsPbI2Br was degraded into the yellow non-perovskite phase rapidly in samples when there was no PbI(OH) protective layer, as shown in Fig. 4c.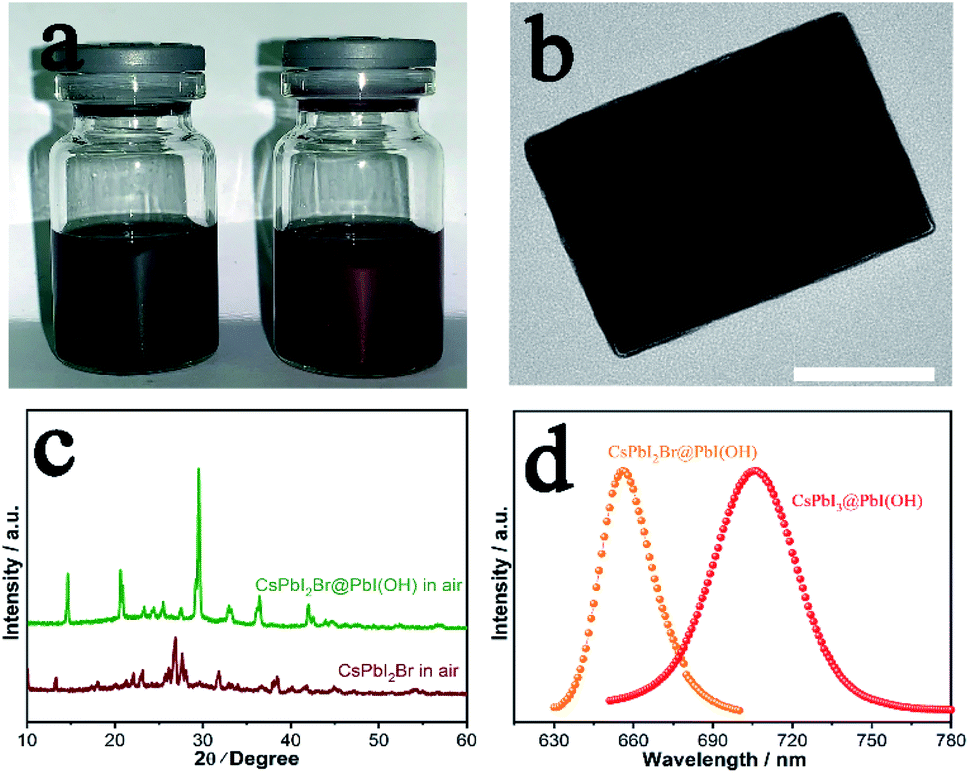 | ||
| Fig. 4 CsPbI2Br prepared by our solution method. (a) Photographs of CsPbI3@PbI(OH) and CsPbI2Br@PbI(OH) prepared in solution. (b) TEM image of CsPbI2Br@PbI(OH). Scale bar: 150 nm. (c) PXRD patterns of CsPbI2Br@PbI(OH) and γ-CsPbI2Br in air, showing degradation for the latter, which does not have a PbI(OH) protective shell. (d) Photo-luminescence spectra of CsPbI3@PbI(OH) and CsPbI2Br@PbI(OH). | ||
Photo-luminescence (PL) spectra have been recorded for CsPbI3@PbI(OH) and CsPbI2Br@PbI(OH) as shown in Fig. 4d. CsPbI3@PbI(OH) exhibits an emission peak at 705 nm, which is consistent with previous results at 709 nm.13 CsPbI2Br@PbI(OH) exhibits a band edge emission at 655 nm, similar to that reported for the α polymorph of CsPbI2Br.4,27 These results show that the ultra-thin PbI(OH) shell does not have a significant effect on the photo-electronic properties of the perovskite.
Conclusions
In summary, we have shown a facile solution approach to prepare γ-CsPbI3 micro-crystals in an ambient environment for the first time. The rapid precipitation process of γ-CsPbI3 ensures the production of the desired perovskite phase at room-temperature, which is then stabilized by the spontaneous degradation of the perovskite in water, which leads to the epitaxial growth of an ultra-thin and single-crystal PbI(OH) shell on the perovskite surface. The acetate anion is believed to play a key role in controlling the degradation of perovskite to ensure formation of PbI(OH), while PVP contributes to capping the process and ensuring the PbIOH shell compactness and uniformity. These coated γ-CsPbI3 microcrystals exhibit vastly improved stability both towards moisture and heating in air, which is attributed to the lattice anchoring and uniform physical coverage of the PbI(OH) shell. Our method has also been successfully extended to other inorganic perovskites with the synthesis of CsPbI2Br. Benefiting from our room-temperature synthesis, CsPbI2Br forms the low-temperature γ perovskite phase, which represents the first experimental report of γ-CsPbI2Br. Uniquely, our study provides a new avenue for the synthesis of stable solar perovskite materials, making use of degradation processes to form a protective stabilization layer on the perovskite. It shows that the bane of many a solar perovskite synthesis (water) can also be a boon, providing that, as in the case here, the degradation products can be tailored to induce surface stability.Author contributions
J. X., Z. H., and P. R. S. conceived the idea and wrote the manuscript. J. X., B. D., E. D., and A. M. collected the XRD data and performed the structural refinements. J. X. synthesised all the materials and performed the initial characterisation measurements and stability studies. X. F., Z. Z., and S. C. contributed to the TEM and EDX measurements. Z. D, Y. Z., and Z. T. contributed to the data analysis. Z. H. and P. R. S. supervised this project. All the authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the technical support from the SUSTech Core Research Facilities (SUSTech CRF). We acknowledge the help from Zenglong Guo (Department of Materials Science and Engineering, Southern University of Science and Technology) for TEM measurements and Zehua Li (School of Chemistry, University of Birmingham) for Fourier Transform Infrared Spectroscopy measurements. Abubakar Muhammad acknowledges the scholarship funding from the Petroleum Technology Development Fund (PTDF), Nigeria. Elizabeth Driscoll acknowledges scholarship support from the University of Birmingham.References
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. J. N. Grätzel, Nature, 2013, 499, 316 CrossRef CAS PubMed.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. G. De Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang and Y. J. S. Zhao, Science, 2017, 355, 722–726 CrossRef CAS PubMed.
- W. S. Yang, B. W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim and J. H. J. S. Noh, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- W. Chen, H. Chen, G. Xu, R. Xue, S. Wang, Y. Li and Y. J. J. Li, Joule, 2019, 3, 191–204 CrossRef CAS.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca and E. Mosconi, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- Y. Wang, T. Zhang, M. Kan and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 12345–12348 CrossRef CAS PubMed.
- J. Liang, C. Wang, Y. Wang, Z. Xu, Z. Lu, Y. Ma, H. Zhu, Y. Hu, C. Xiao and X. Yi, J. Am. Chem. Soc., 2016, 138, 15829–15832 CrossRef CAS PubMed.
- R. J. Sutton, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, F. Giustino and H. J. Snaith, ACS Energy Lett., 2018, 3, 1787–1794 CrossRef CAS.
- B. Wang, N. Novendra and A. Navrotsky, J. Am. Chem. Soc., 2019, 141, 14501–14504 CrossRef CAS PubMed.
- J. A. Steele, H. Jin, I. Dovgaliuk, R. F. Berger, T. Braeckevelt, H. Yuan, C. Martin, E. Solano, K. Lejaeghere and S. M. Rogge, Science, 2019, 365, 679–684 CrossRef CAS PubMed.
- Y. Wang, M. I. Dar, L. K. Ono, T. Zhang, M. Kan, Y. Li, L. Zhang, X. Wang, Y. Yang and X. Gao, Science, 2019, 365, 591–595 CrossRef CAS PubMed.
- B. Zhao, S. F. Jin, S. Huang, N. Liu, J. Y. Ma, D. J. Xue, Q. Han, J. Ding, Q. Q. Ge, Y. Feng and J. S. Hu, J. Am. Chem. Soc., 2018, 140, 11716–11725 CrossRef CAS PubMed.
- D. B. Straus, S. Guo and R. J. Cava, J. Am. Chem. Soc., 2019, 141, 11435–11439 CrossRef CAS PubMed.
- J. Zhang, D. Bai, Z. Jin, H. Bian, K. Wang, J. Sun, Q. Wang and S. Liu, Adv. Energy Mater., 2018, 8, 1703246 CrossRef.
- H. Zhao, J. Xu, S. Zhou, Z. Li, B. Zhang, X. Xia, X. Liu, S. Dai and J. Yao, Adv. Funct. Mater., 2019, 29, 1808986 CrossRef.
- P. Becker, J. A. Márquez, J. Just, A. Al-Ashouri, C. Hages, H. Hempel, M. Jošt, S. Albrecht, R. Frahm and T. Unold, Adv. Energy Mater., 2019, 9, 1900555 CrossRef.
- A. Marronnier, G. Roma, S. Boyer-Richard, L. Pedesseau, J. M. Jancu, Y. Bonnassieux, C. Katan, C. C. Stoumpos, M. G. Kanatzidis and J. Even, ACS Nano, 2018, 12, 3477–3486 CrossRef CAS PubMed.
- A. K. Jena, A. Kulkarni, Y. Sanehira, M. Ikegami and T. Miyasaka, Chem. Mater., 2018, 30, 6668–6674 CrossRef CAS.
- E. M. Sanehira, A. R. Marshall, J. A. Christians, S. P. Harvey, P. N. Ciesielski, L. M. Wheeler, P. Schulz, L. Y. Lin, M. C. Beard and J. M. Luther, Sci. Adv., 2017, 3, eaao4204 CrossRef PubMed.
- B. Li, Y. Zhang, L. Fu, T. Yu, S. Zhou, L. Zhang and L. Yin, Nat. Commun., 2018, 9, 1076 CrossRef PubMed.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili and A. H. Emwas, J. Am. Chem. Soc., 2017, 140, 562–565 CrossRef PubMed.
- G. Yuan, C. Ritchie, M. Ritter, S. Murphy, D. E. Gómez and P. Mulvaney, J. Phys. Chem. C, 2017, 122, 13407–13415 CrossRef.
- J. Dennis, H. Henisch and P. Cherin, J. Electrochem. Soc., 1965, 112, 1240–1241 CrossRef CAS.
- Y. Chen, Y. Lei, Y. Li, Y. Yu, J. Cai, M. H. Chiu, R. Rao, Y. Gu, C. Wang and W. Choi, Nature, 2020, 577, 209–215 CrossRef CAS PubMed.
- Y. Chen, T. Shi, P. Liu, W. Xie, K. Chen, X. Xu, L. Shui, C. Shang, Z. Chen, H. L. Yip, G. Zhou and X. Wang, J. Mater. Chem. A, 2019, 7, 20201–20207 RSC.
- G. Yin, H. Zhao, H. Jiang, S. Yuan, T. Niu, K. Zhao, Z. Liu and S. Liu, Adv. Funct. Mater., 2018, 28, 1803269 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta02210a |
| This journal is © The Royal Society of Chemistry 2020 |