
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–activity relationships in well-defined conjugated oligomer photocatalysts for hydrogen production from water†
Catherine M.
Aitchison
 a,
Michael
Sachs
b,
Marc A.
Little
a,
Liam
Wilbraham
c,
Nick J.
Brownbill
ad,
Christopher M.
Kane
a,
Frédéric
Blanc
ad,
Martijn A.
Zwijnenburg
*c,
James R.
Durrant
b,
Reiner Sebastian
Sprick
*ae and
Andrew I.
Cooper
*a
a,
Michael
Sachs
b,
Marc A.
Little
a,
Liam
Wilbraham
c,
Nick J.
Brownbill
ad,
Christopher M.
Kane
a,
Frédéric
Blanc
ad,
Martijn A.
Zwijnenburg
*c,
James R.
Durrant
b,
Reiner Sebastian
Sprick
*ae and
Andrew I.
Cooper
*a
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford St, Liverpool L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk
bDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, 80 Wood Lane, London W12 0BZ, UK
cDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
dStephenson Institute for Renewable Energy, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK
eDepartment of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: sebastian.sprick@strath.ac.uk
First published on 28th July 2020
Abstract
Most organic semiconductor photocatalysts for solar fuels production are linear polymers or polymeric networks with a broad distribution of molecular weights. Here, we study a series of molecular dibenzo[b,d]thiophene sulfone and fluorene oligomers as well-defined model systems to probe the relationship between photocatalytic activity and structural features such as chain length and planarity. The hydrogen evolution rate was found to vary significantly with bridge head atom, chain length, and backbone twisting. A trimer (S3) of only three repeat units has excellent activity for proton reduction with an EQE of 8.8% at 420 nm, approaching the activity of its polymer analogue and demonstrating that high molar masses are not a prerequisite for good activity. The dynamics of long-lived electrons generated under illumination in the S3 oligomer are very similar to the corresponding polymer, both under transient and quasi-continuous irradiation conditions.
Introduction
Solar fuel production has been of interest since the 1970s when Fujishima and Honda showed that TiO2 could photocatalyse the splitting of water into hydrogen and oxygen.1 In recent years, much more photoactive metal oxides, sulfides and oxynitrides have been discovered,2–4 resulting in systems that facilitate overall water splitting in the absence of scavengers.5–7 Organic semiconductors are an alternative class of materials that can act as photocatalysts for hydrogen production and have the potential advantages of molecular tunability, low cost, and earth abundant element compositions. Carbon nitride is by far the most widely studied organic photocatalyst,8–10 but to our knowledge, the first organic material reported for photocatalytic proton reduction was in fact poly(p-phenylene),11 reported as early as 1985. Follow-up studies have focused on a large variety of polymeric materials including conjugated linear polymers,12–17 conjugated microporous polymers,18–22 covalent organic frameworks,23–27 and covalent triazine-based frameworks.28–30Oligomers are far less studied as photocatalysts despite their extensive use in organic photovoltaics31,32 and in other heterogeneous photocatalysis applications.33 Oligomers of phenylene (n = 2–6)12,34 and pyridine35 show low but measurable photocatalytic activity, and it seems that it has been generally assumed, but not proven, that longer chain lengths are required for good photocatalytic activity. This could be based on the assumption that longer conjugation lengths and red-shifted light absorption onsets in higher molecular weight materials are needed. However, this has not been clearly demonstrated: indeed, we do not observe high molecular weights for polymer photocatalysts when studied via matrix-assisted laser desorption/ionisation mass-spectrometry,17,36 although this does not prove definitively that longer polymer chains do not exist in these materials. A study of low molecular weight phenyl–triazine oligomers also showed that these materials outperformed their polymeric analogues.37,38 Besides this, oligomers have the advantage of being well-defined chemical structures with known molecular weights and end-groups.
As molecular compounds, oligomers can also have a well-defined secondary structure. Unlike polymers, their arrangement in the solid-state structures and their molecular conformations can often be studied using X-ray diffraction. Long-range order has been suggested to enhance the photocatalytic activity in carbon nitrides,39 phenyl–triazine oligomers,37,38 and covalent organic frameworks compared to an amorphous analogue,26 although all these materials also show differences in driving-forces or unreacted functional groups from incomplete polymerisation. As a result, it is difficult to decouple the effects of composition and secondary structure on photocatalytic activity. We have shown recently that the packing motif of a photocatalytically active molecular crystal can be hugely important, with crystalline and amorphous versions of the same chemical displaying orders of magnitude differences in the photocatalytic hydrogen production rate.40 Intermolecular interactions were also shown to be a key structural feature in water soluble porphyrin oligomers, whereby aggregation was needed for high photocatalytic activity.41 While it is unclear whether crystallinity is beneficial for photocatalytic activity in all cases, the ability to correlate structural properties with changes in catalytic activity is useful for developing a better understanding of the factors affect activity, thus guiding the design of future photocatalysts. As well as their ability to form crystals, the solubility of oligomers in common organic solvents allows us to compare homogeneous and heterogeneous hydrogen production photocatalysts directly.
In this work, we synthesised three series of oligomers to investigate the effect of various chemical and structural properties, such as chain-length and backbone twisting, on physical properties such as light absorption, excited state lifetime, and hydrogen evolution activity. Dibenzo[b,d]thiophene sulfone has been shown repeatedly to be an active monomer unit in hydrogen producing organic photocatalysts;12,16,26 this is thought to be related to both its hydrophilicity and associated efficient generation of long-lived polarons. We therefore investigated oligomers of dibenzo[b,d]thiophene sulfone (S1–S3, Fig. 1) as well as the more easily processable (that is, more soluble) 9,9-dimethyl-9H-fluorene oligomers MeF1–MeF3 (Fig. 1). To test the impact of crystal packing more directly, we also designed a set of photocatalysts with either phenyl or mesitylene substituents on a 9H-fluorene or dibenzo[b,d]thiophene sulfone ‘core’ (PSP, PFP, MSM and MFM, Fig. 1).
 | ||
| Fig. 1 Structures of the oligomer photocatalysts in this study. | ||
Results
Oligomer synthesis and characterisation
Dibenzo[b,d]thiophene sulfone oligomers (S1–S3, PSP, MSM, Fig. 1), and 9H-fluorene oligomers (MeF1–MeF3, PFP, MFM) were either obtained from commercial sources or prepared by Pd(0)-catalysed Suzuki–Miyaura coupling reaction (see ESI† for full synthetic details). The oligomers were then purified by column chromatography or by recrystallisation, except for S2 and S3 because of their insolubility in common organic solvents. The oligomers were characterised by elemental analysis and solution state 1H and 13C{1H} NMR spectroscopy (Fig. S1–S14†) or, for S2 and S3, by solid state 13C magic angle spinning (MAS) NMR (Fig. S7†). MAS NMR confirmed the structures of S2 and S3 as a dimer and trimer, respectively, and no evidence of multiple solid-state phases was observed, although it is possible this was obscured by the relatively broad line width of the spectra. Very low levels of signals attributed to boronic acid pinacol ester were observed in the 13C MAS NMR spectrum of S3 (Fig. S7†), which are most likely due to residual starting material, or boronic acid pinacol ester end-groups on a product that only underwent one coupling reaction instead of two.Crystal structures of S1 (space group symmetry: C2/c) and MeF1 (I41/a) were determined by single crystal X-ray diffraction using the commercially sourced, as received, chemicals. Crystal structures MeF2 (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), MeF3 (P21/n), PSP (P21/n), MSM (Ia), PFP (P21/n) and MFM (P) were determined by single crystal X-ray diffraction, after growing suitable quality crystals by slowly evaporating the organic solvent mixtures used for purification (see Tables S1–S3† for full details). All of the oligomers were found to crystallise without solvent, and it was these solvent free crystalline phases that were used for the photocatalysis experiments. The poor solubility of S2 and S3 in organic solvents limited crystal growth under similar conditions. Consequently, suitable quality single crystals of S2 (P) and S3 (P) were grown by sublimation at temperatures >400 °C. The crystal structures of MeF1–MeF3 are dominated by edge-to-face and offset π–π interactions between neighboring oligomers (Fig. 2d–f). By comparison, the S1–S3 structures are dominated by face-to-face π–π stacking interactions between the sulfone dimers, which are rotated by 180° in each layer to prevent steric clashes between the sulfone oxygen atoms (Fig. 2j–i).
), MeF3 (P21/n), PSP (P21/n), MSM (Ia), PFP (P21/n) and MFM (P) were determined by single crystal X-ray diffraction, after growing suitable quality crystals by slowly evaporating the organic solvent mixtures used for purification (see Tables S1–S3† for full details). All of the oligomers were found to crystallise without solvent, and it was these solvent free crystalline phases that were used for the photocatalysis experiments. The poor solubility of S2 and S3 in organic solvents limited crystal growth under similar conditions. Consequently, suitable quality single crystals of S2 (P) and S3 (P) were grown by sublimation at temperatures >400 °C. The crystal structures of MeF1–MeF3 are dominated by edge-to-face and offset π–π interactions between neighboring oligomers (Fig. 2d–f). By comparison, the S1–S3 structures are dominated by face-to-face π–π stacking interactions between the sulfone dimers, which are rotated by 180° in each layer to prevent steric clashes between the sulfone oxygen atoms (Fig. 2j–i).
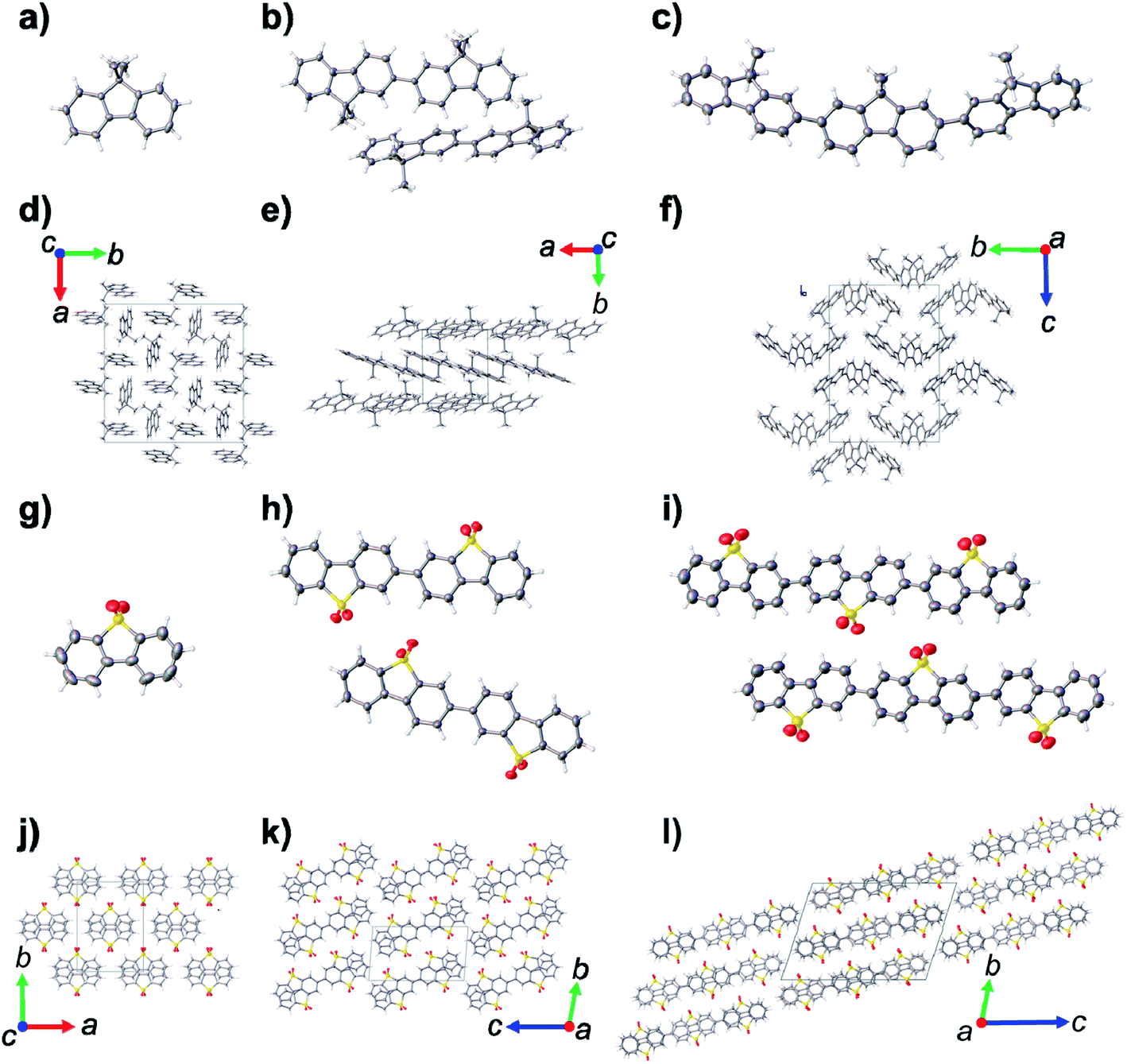 | ||
| Fig. 2 Displacement ellipsoid plots from the single crystal structures of; (a) MeF1, (b) MeF2, (c) MeF3, (g) S1, (h) S2, (i) S3; ellipsoids displayed at 50% probability level. Crystal packing in the single crystal structures of; (d) MeF1, (e) MeF2, (f) MeF3, (j) S1, (k) S2, (l) S3. Grey = carbon, white = hydrogen, yellow = sulfur; red = oxygen. | ||
Powder X-ray diffraction (PXRD) analysis indicated that the bulk prepared crystalline materials of MeF1, MeF2, MeF3, PSP, MSM, and PFP matched the simulated PXRD patterns for the single crystal structures, and these phase pure crystalline materials were used for subsequent photocatalytic experiments. Analysis of the PXRD patterns for S2 and S3 indicated that these materials were semi-crystalline after purification (Fig. S17 and S18†), with SEM analysis showing that S2 was comprised of well-defined cuboidal crystals of circa 100 nm × 500 nm (Fig. S20†), while S3 was made up of globular particulates (Fig. S21†). Although the sublimed materials of S2 and S3 appeared to be more crystalline and of the same phase as the bulk purified semi-crystalline materials (Fig. S17 and S18†), we observed partial decomposition of the oligomers under the sublimation conditions. This prevented us from preparing large enough quantities of the sublimed S2 and S3 materials to allow photocatalytic experiments. Further analyses were therefore conducted on the as-purified semi crystalline materials. The PXRD pattern of MFM indicated that the bulk material was a mixed phase, and we could only determine the structure of the P structure during this study. MFM was therefore used as a mixed phase during the subsequent photocatalytic experiments.
The residual palladium content of the oligomers from their synthesis was measured via inductively coupled plasma mass spectrometry (ICP-MS) after digestion with nitric acid; commercial monomers MeF1 and S1, as well as PSP, showed palladium levels below the detection limit of the instrument (approximately 10 ppm, see ESI† for full details). MSM and PFP also had low Pd contents of 0.0015 and 0.0031 wt% (15 and 31 ppm) while MeF2, MeF3 and MFM had Pd levels between 0.01 and 0.02 wt% (100–200 ppm). The insoluble oligomers S2 and S3 showed the highest Pd levels (0.22 and 0.26 wt% or 2200 and 2600 ppm) probably because, unlike the other materials in this study, they could not be recrystallised from solution.
UV-Visible and photoluminescence spectroscopy was carried out on the oligomers in the solid-state (reflectance) and in chloroform solution (Fig. S23–S32†). The absorption onset of all the oligomers was also predicted using time-dependent density functional theory (TD-DFT) calculations (Table S9†). The families MeF1–MeF3 and S1–S3 both showed the expected redshift in absorption onset with increasing chain length while the absorption onset of the mesityl substituted oligomers MSM and MFM were blue-shifted relative to their phenyl substituted analogues PSP and PFP (ESI, Table S4†). This blue-shift for MSM and MFM results from their less planar structure and the reduced conjugation between the central ring and the two peripheral rings. The experimental and the TD-DFT predicted optical gap were found to be well correlated (Fig. 3b). The extinction coefficients of all soluble oligomers in chloroform were measured at the wavelength of their absorption maxima: the molar extinction coefficient increased from 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1 for MeF1 to 42800 M−1 cm−1 for MeF2 and 71100 M−1 cm−1 for MeF3. The phenyl-substituted oligomers PSP and PFP had extinction coefficients of 35300 and 40300 M−1 cm−1 at their maximum of absorption, while the mesityl substituted analogues MSM and MFM were determined to be 18800 and 29800 M−1 cm−1.
500 M−1 cm−1 for MeF1 to 42800 M−1 cm−1 for MeF2 and 71100 M−1 cm−1 for MeF3. The phenyl-substituted oligomers PSP and PFP had extinction coefficients of 35300 and 40300 M−1 cm−1 at their maximum of absorption, while the mesityl substituted analogues MSM and MFM were determined to be 18800 and 29800 M−1 cm−1.
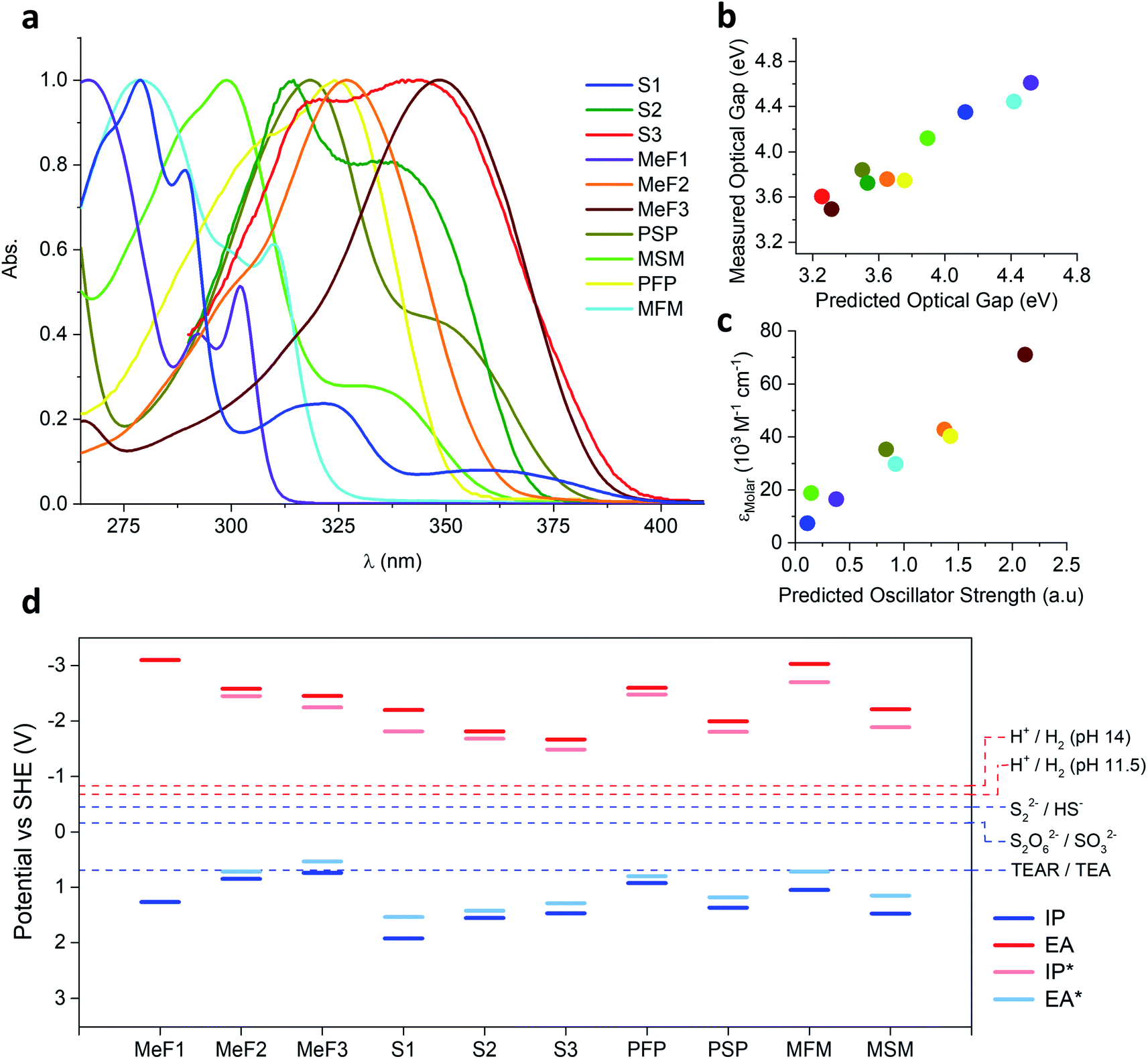 | ||
| Fig. 3 (a) Normalised UV-Vis absorption spectra of the oligomers in chloroform; (b) plot of the TD-DFT predicted optical gap versus the value obtained from absorption maxima in chloroform solution; (c) plot of the oscillator strength predicted by TD-DFT versus the measured molar extinction coefficient (εMolar) in chloroform solution; (d) potentials of the free charge carriers (IP,EA) and excitons (IP*,EA*) in the studied oligomers predicted by DFT and relevant solution reactions (at pH 11.5 for the case of using triethylamine as hole scavenger and pH 14.0 in the case of SO32− and HS−). Potential of the 2-hole oxidation of triethylamine to diethylamine and acetaldehyde not shown as it lies in a similar place as the proton reduction potential for pH 11.5. | ||
Fluorescence quantum yields for all materials were measured in chloroform. We find an increase in photoluminescence quantum yield (PLQY, see ESI for details†) with increasing chain-length for both the dibenzo[b,d]thiophene sulfone oligomers (S1–S3) and 9H-fluorene oligomers (MeF1–MeF3): from 10% for MeF1 to 94% for MeF2 and to unity for MeF3. Likewise, we found an increase in the PLQY from 11% for S1 to 73% for S2 and 77% for S3. The mesityl substituted fluorene unit MFM had a very low PLQY of 9% while the phenyl analogue PFP was highly photoluminescent with a value of 94%. MSM and PSP had more similar PLQYs of 65% and 76%.
The photoluminescence lifetime of the oligomers was measured by time-correlated single photon counting (TCSPC) experiments. All oligomers showed multi-exponential photoluminescence decays and, in general, the oligomers containing dibenzo[b,d]thiophene sulfone showed longer lifetimes than the fluorene based materials. For example, MSM and PSP had weighted-average photoluminescence lifetimes of 2.68 and 2.47 ns whereas MFM and PFP were 0.58 and 1.02 ns (Fig. S36 and Table S5†). This trend was also observed for chloroform solutions; MSM, PSP, PFP and MFM had weighted average lifetimes of 3.86, 2.42, 0.57 and 0.05 ns, respectively (Fig. S38†). In general, longer chain lengths were found to have shorter photoluminescence lifetimes; in the solid state S3 had a weighted-average lifetime of 3.00 ns whilst this increased to 4.85 ns in S2 and 5.71 ns in S1. A sample of S1 was also tested with 0.36 wt% Pd added to give it similar Pd content to S2 and S3 but showed little change from pristine S1 (Fig. S33†) with a weighted average lifetime (5.64 ns), within error. Similarly, the addition of high levels of palladium to S2 and S3 had little effect on the PL lifetimes. S2 loaded with 3 wt% palladium had an identical lifetime to S2 without added palladium, while the photoluminescence lifetime of S3 loaded with additional 3 wt% Pd decreased slightly from 3.00 ns to 2.76 ns (Fig. S33†). The emission spectra of MeF oligomers in the solid-state were more complex with multiple maxima but again showed a decrease in lifetime with chain length; MeF3 had emissions at 428 nm and 446 nm with lifetimes of 0.78 and 1.07 ns, respectively, while MeF2 had lifetimes of 1.43 and 1.98 ns (Fig. S35†). MeF1 was significantly longer lived with weighted lifetimes of 2.87 and 7.15 ns at 326 and 350 nm. In this case, a sample of the MeF1 loaded with Pd to give equivalent levels to the dimer and trimer (0.016 wt%) did show a significant decrease in lifetime with a weighted average lifetime of 1.71 ns at 326 nm and 5.53 ns at 350 nm (Fig. S34†). In chloroform solution, all of the MeF oligomers had very short lifetimes but the general trend was preserved with average lifetimes of 1.15, 1.07 and 0.83 ns for MeF1–MeF3 (Fig. S37 and Table S6†).
Static light scattering experiments were conducted on suspensions of the oligomers in water to determine the particle size of the catalysts (Fig. S39–S42†). The particles of the dibenzo[b,d]thiophene sulfone oligomers were by far the smallest with both S2 and S3 having a significant fraction of material smaller than 1 μm. The Sauter mean diameters (D[3,2])42 were 3.96, 0.87 and 1.79 μm for S1, S2, and S3 respectively. The phenyl and mesityl oligomers were slightly larger with D[3,2] ranging from 5.25 μm for PSP up to 11.6 μm for PFP whilst the MeF oligomers had D[3,2] of 24.2, 13.9 and 33.4 μm for MeF1–MeF3. S1 dissolved in the TEA/MeOH/water mixture, but S2 and S3 suspensions were also measured under these conditions and had D[3,2] values of 0.68 and 0.57 μm.
Hydrogen evolution experiments in suspension
Hydrogen evolution experiments were first conducted on suspensions of the MeF 9H-fluorene oligomers in a mixture of water, methanol and triethylamine (1:1:1). Triethylamine (TEA) acts as the sacrificial hole scavenger,11 and methanol is used to aid TEA miscibility with water.12,16 The photocatalysis experiments were performed using broadband illumination (λ > 295 nm, 300 W Xe light source) and the longest 9H-fluorene oligomer, the trimer MeF3, showed the highest hydrogen production rate of 37 μmol h−1 g−1. The rates were lower for the dimer and monomer with 13 μmol h−1 g−1 for MeF2 and 4.7 μmol h−1 g−1 for MeF1 (Fig. S52†). Under UV light irradiation only using a 275–400 nm filter (see ESI† for the filter transmission characteristics), the results also showed an increase in activity with chain length, with rates of 2.6, 4.3 and 28 μmol h−1 g−1 for MeF1–MeF3 (Fig. S53†). However, it was noted that, while MeF2 and MeF3 are insoluble in the solvent mixture, the monomer MeF1 partially dissolved under these conditions. To probe whether this effects the photocatalytic performance we used an aqueous Na2S/Na2SO3 solution as an alternative sacrificial electron donor, thus removing all organic components that might solubilise the monomer during the photocatalysis experiment. Under these conditions, the MeF oligomers did not dissolve at all. Using broadband illumination (λ > 295 nm), we measured hydrogen production rates of 4.1, 3.4 and 10.1 μmol h−1 g−1 for MeF1–MeF3 (Fig. S46†), respectively, with Na2S/Na2SO3. Under UV light irradiation (using the 275–400 nm filter), the monomer MeF1 displayed a higher rate, producing 6.5 μmol h−1 g−1 hydrogen, which is similar to the trimer (6.6 μmol h−1 g−1) and higher than the dimer (4.4 μmol h−1 g−1) (Fig. S46†). All materials were initially tested as-prepared, without the addition of further noble metals, although it has been shown that residual palladium from the synthesis acts as the co-catalyst for hydrogen production.19,43 We noted that the Pd content of MeF1 was considerably lower than the dimer MeF2 and trimer MeF3. We therefore photodeposited Pd from [Pd(NH4)2Cl4] onto MeF1 increasing the Pd content to 0.016 wt% (which is similar to MeF2 and MeF3), which resulted in increase of the hydrogen evolution rate from 4.1 to 14 μmol h−1 g−1 under broadband illumination (λ > 295 nm) and from 6.5 to 28 μmol h−1 g−1 under UV light irradiation (400 > λ > 255 nm) using the Na2S/Na2SO3 scavenger.
Suspensions of the PSP, MSM, PFP and MFM oligomers in water were also tested for hydrogen evolution using the Na2S/Na2SO3 scavenger (Fig. S48 and S49†). The phenyl substituted dibenzo[b,d]thiophene sulfone material (PSP) had a hydrogen evolution rate of 24.1 μmol h−1 g−1 under broadband illumination (λ > 295 nm filter, 300 W Xe light source), higher than its 9H-fluorene analogue PFP when tested under the same conditions (13.8 μmol h−1 g−1). The mesityl substituted dibenzo[b,d]thiophene sulfone (MSM) and 9H-fluorene (MFM) photocatalysts were significantly less active under broadband illumination with rates of 5.5 and 4.8 μmol h−1 g−1. Under UV light irradiation (275–400 nm filter, 300 W Xe light source) PSP was again the most active material within the series with a HER of 29.6 μmol h−1 g−1, followed by its mesityl analogue MSM (20.4 μmol h−1 g−1). PFP on the other hand was the least active oligomer under these conditions with HER of 8.7 μmol h−1 g−1, slightly less than that of mesityl substituted MFM (10.0 μmol h−1 g−1).
Suspensions of the dibenzo[b,d]thiophene sulfone oligomers were the most active materials in this study: using Na2S/Na2SO3 mixtures under broadband illumination (λ > 295 nm, 300 W Xe light source), the HER increased with oligomer length from of 15 μmol h−1 g−1 for S1 to 81 μmol h−1 g−1 for S2 to 281 μmol h−1 g−1 for S3 (Fig. S50†). Using UV light irradiation (275–400 nm filter), the activity of the monomer S1 increased to 33 μmol h−1 g−1, similar to S2 (50 μmol h−1 g−1) but significantly lower than S3 (162 μmol h−1 g−1) (Fig. S51†). In these experiments, the monomer S1 was loaded with 0.36% Pd by photodeposition to give loadings that were comparable to the dimer and trimer. Without added Pd, S1 had a lower rate of 8 μmol h−1 g−1 at λ > 295 nm. A TEA/MeOH/water (1:1:1) mixture gave rise to significantly higher HERs of 26, 414 and 2073 μmol h−1 g−1 for the monomer (S1), dimer (S2), and trimer (S3), respectively (Fig. S54†). Increased activity with increasing chain length was also shown when only UV light was employed for photolysis, albeit to a lesser extent than when broadband illumination was used; using a 275–400 nm filter S1 had a HER of 20 μmol h−1 g−1, S2 101 μmol h−1 g−1 and S3 526 μmol h−1 g−1 (Fig. S55†). The visible light activity of these materials was also tested using a λ > 420 nm filter. The monomer (S1) produced no detectable amount of hydrogen over 5 hours under these conditions but the dimer (S2) retained some activity with a HER of 26 μmol h−1 g−1 whilst the trimer (S3) was reduced by less than 50% compared to broadband illumination with a HER of 1125 μmol h−1 g−1 (Fig. S55†). Again, it should be noted that the monomer (S1) is partially soluble in the TEA/MeOH/water (1:1:1) mixture, hence, the results cannot be compared directly with the results for S2 and S3, which are completely insoluble during the experiment. The external quantum efficiencies for S2 and S3 were determined using a 420 nm LED as the light source (ESI, General methods†). S2 has a relatively low EQE of 0.4% consistent with its limited light absorption at this wavelength. For S3, on the other hand, a high EQE of 8.8% was determined.
Oligomer stability in heterogeneous photocatalysis experiments
The stability of the photocatalysts that were soluble in chloroform (S1, MeF1–MeF3, PSP, MSM, PFP and MFM) was studied using 1H NMR spectroscopy in CDCl3 and PXRD (Fig S71–S84†) after the photocatalysis experiments. Samples collected after 6 hours of broadband spectrum irradiation (λ > 295 nm, 300 W Xe light source), using Na2S/Na2SO3 as the scavenger, were all found to be chemically stable. There was also no change in crystal packing, as determined by PXRD. In addition, the PXRD patterns and solution NMR spectra of MeF1–MeF3 after 6 hours of photolysis in TEA/MeOH/water (1:1:1) mixture under broadband spectrum irradiation (λ > 295 nm, 300 W Xe light source) also showed no degradation, despite the partial solubility of MeF1 under these conditions.
Dibenzo[b,d]thiophene sulfone (S1) was also soluble in the TEA/MeOH/water mixture but, unlike MeF1, a significant drop of in activity over 5 hours of broadband spectrum irradiation (λ > 295 nm, 300 W Xe light source) was observed. The 1H NMR spectrum of S1 after photocatalysis indicated breakdown of the catalyst. To investigate this further, a sample of S1 was irradiated for 72 hours in the photolysis mixture. Analysis of this material by solution NMR spectroscopy (1H, 13C ATP, 1H COSY and HSQC Fig. S62–S66†) and mass spectrometry indicated the breakdown product was the triethylamine salt of [1,1′-biphenyl]-2-sulfonic acid. This is possibly due to oxidation of S1 by either singlet oxygen or superoxide anions generated from residual oxygen in the system. To further support this interpretation, addition of a singlet oxygen and superoxide anion scavenger (nickel(II) dibutyldithiocarbamate)44,45 caused an increase of the HER under the same conditions to 53 μmol h−1 g−1 and an extended stability for at least 23 hours (Fig. S63†). The material collected after this photolysis experiment was found to be S1 by 1H NMR spectroscopy with no breakdown products present (Fig. S62†).
By contrast, we found that the PXRD patterns of dibenzo[b,d]thiophene sulfone oligomers S2 and S3 showed no change after 5 hours of irradiation (λ > 295 nm, 300 W Xe light source) in TEA/MeOH/water. Longer-term photocatalysis experiments of oligomer S2 irradiated for 50 hours under broadband illumination (λ > 295 nm, 300 W Xe light source) showed good longevity, with a slow drop off in activity and a reduction in the photocatalytic rate of less than 50% after 50 hours (Fig. S57†), which is comparable to many polymer photocatalysts.46 Oligomer S3 showed a more rapid drop off in photocatalytic hydrogen evolution rates from 2080 μmol h−1 g−1 over the first five hours to 576 μmol h−1 g−1 over 50 to 55 hours. However, when the catalyst was collected by filtration, washed and dispersed in new TEA/MeOH/water mixture we observed a recovery of the photocatalytic activity to 2066 μmol h−1 g−1 (Fig. S58†). It is possible that the reduction in activity is caused by accumulation of TEA oxidation products that inhibit photocatalysis. In addition, samples of S2 and S3 irradiated for 72 hours under broadband illumination (λ > 295 nm, 300 W light source) showed very similar PXRD patterns, UV and IR spectra to the as synthesised material (Fig. S67–S70†). It is possible that oxidation by reactive oxygen species does occur in S2 and S3 but at a slower rate than S1. The fact that S1 is stable in the Na2S/Na2SO3 photolysis mixture indicates that insolubility in the dispersant may be required for stability or it is possible that TEA plays a role in catalyst breakdown.
Photocatalytic activity at high palladium loadings
So far we have investigated the oligomers with Pd contents of less than 0.4 wt%; that is, considerably lower than the levels of co-catalysts commonly used in organic photocatalysts, where optimal loadings vary from 1–10 wt%.47–49 In light of our recent studies50 into the role of palladium in photocatalytic hydrogen production with polymers such as P10, which is the polymer analogue of S1–S3, we also tested the effect of the addition of 3 wt% Pd via photodeposition from [Pd(NH4)2Cl4]. The hydrogen evolution rate of S1 (TEA/MeOH/water, λ > 295 nm) was not linear over the first five hours and an initial induction period (due to photodeposition of palladium) was observed. After this induction period the HER reached a maximum of 86 μmol h−1 g−1 before slowing significantly, the reduction in rate was thought to be due to catalyst breakdown as discussed above (Fig. S59†). By contrast, both S2 and S3 displayed stable hydrogen evolution over the 5 hour period (Fig. 4a and b) and in both cases the increased Pd loading led to a significant increase in HER at λ > 295 nm; S2 improved by a factor of 3.3 to 1369 μmol h−1 g−1 while S3 improved to 6550 μmol h−1 g−1, a factor of 3.2. Scanning transmission electron microscopy (STEM) imaging (Fig. 4c–f) of these two materials collected after photocatalysis by filtration showed that Pd nanoparticles of diameters between 5 and 30 nm had formed on the surface of the oligomers. ICP-MS analysis indicated the photodeposition had occurred in reasonable yield; the S2 sample contained 2.1 wt% Pd while the S3 sample contained 2.5 wt% Pd. The large increase in activity for both S2 and S3 upon increasing the Pd content from approximately 2000 ppm to 20000 ppm is similar to the behaviour of P10, where the transfer of long-lived electrons to Pd sites has been identified as a limitation for hydrogen evolution on this polymer.50 When the residual Pd level was reduced for S2 by using sodium N,N-diethyldithiocarbamate as a Pd scavenger51,52 it resulted in a slightly reduced HER of 306 μmol h−1 g−1 indicating some dependence of activity at low concentrations as well. ICP-MS analysis of this sample showed a Pd content of 0.008 wt% (80 ppm).
 | ||
| Fig. 4 Hydrogen evolution of (a) S2 (5 mg) and (b) S3 (5 mg) in TEA/MeOH/water (1:1:1, 5 mL) with and without the addition of 3 wt% Pd by photodeposition of [Pd(NH4)2Cl4], irradiated with a 300 W Xe light source fitted with a λ > 295 nm filter. STEM images of S2 with 3 wt% Pd in (c) bright field mode and (e) high angle dark field mode and S3 with 3 wt% Pd in (d) bright field mode and (f) high angle dark field mode. | ||
Time-resolved spectroscopy
To further elucidate the high activity of S3 in the dibenzo[b,d]thiophene sulfone oligomer series, S2 and S3 were studied using time-resolved spectroscopy in TEA/MeOH/water suspensions under both transient and quasi-continuous excitation conditions. These experiments allow for comparisons of the photophysical behaviour of these molecular materials to their previously studied16 polymer analogue, P10. The exciton transient absorption peak observed upon 355 nm excitation on the ps timescale continuously red-shifts with increasing chain length in the range of 600 nm to 900 nm (Fig. S87a†), which is in good agreement with the concomitant red-shift in the absorption onset and photoluminescence maxima. Interestingly, a pronounced decrease in exciton lifetime with increasing chain length is observed (Fig. S87b†), which is consistent with the time-resolved photoluminescence lifetime data discussed above and demonstrates that the increasing hydrogen evolution activity with longer oligomer length in this dibenzo[b,d]thiophene sulfone series is not due to longer exciton lifetimes.Fig. 5a shows transient absorption decay kinetics of S2 and S3 suspended in TEA/MeOH/water where the probe wavelength was set to 600 nm, corresponding to the transient absorption peak of S3 as shown in the inset. In spectral shape as well as spectral position, this transient absorption feature is in very good agreement with the absorption signature of electron polarons in P10 which we previously observed in the form of a peak at 630 nm,16 and we thus assign the 600 nm peak in the present study to electron polarons in S3. The shown kinetics demonstrate that these electron polarons have long lifetimes up to the millisecond timescale, which is sufficiently long to drive proton reduction.16 In contrast, S2 lacks a comparable polaron absorption peak in the visible range and generally exhibits much lower signal amplitudes over the probed spectral range. The main transient absorption feature above 1000 nm might be assigned to the onset of NIR polaron absorption in this material. In any case, assuming a comparable electron polaron extinction coefficient for S2 and S3, the electron polaron yield for S3 is considerably higher than that of S2, in line with its substantially higher hydrogen evolution activity.
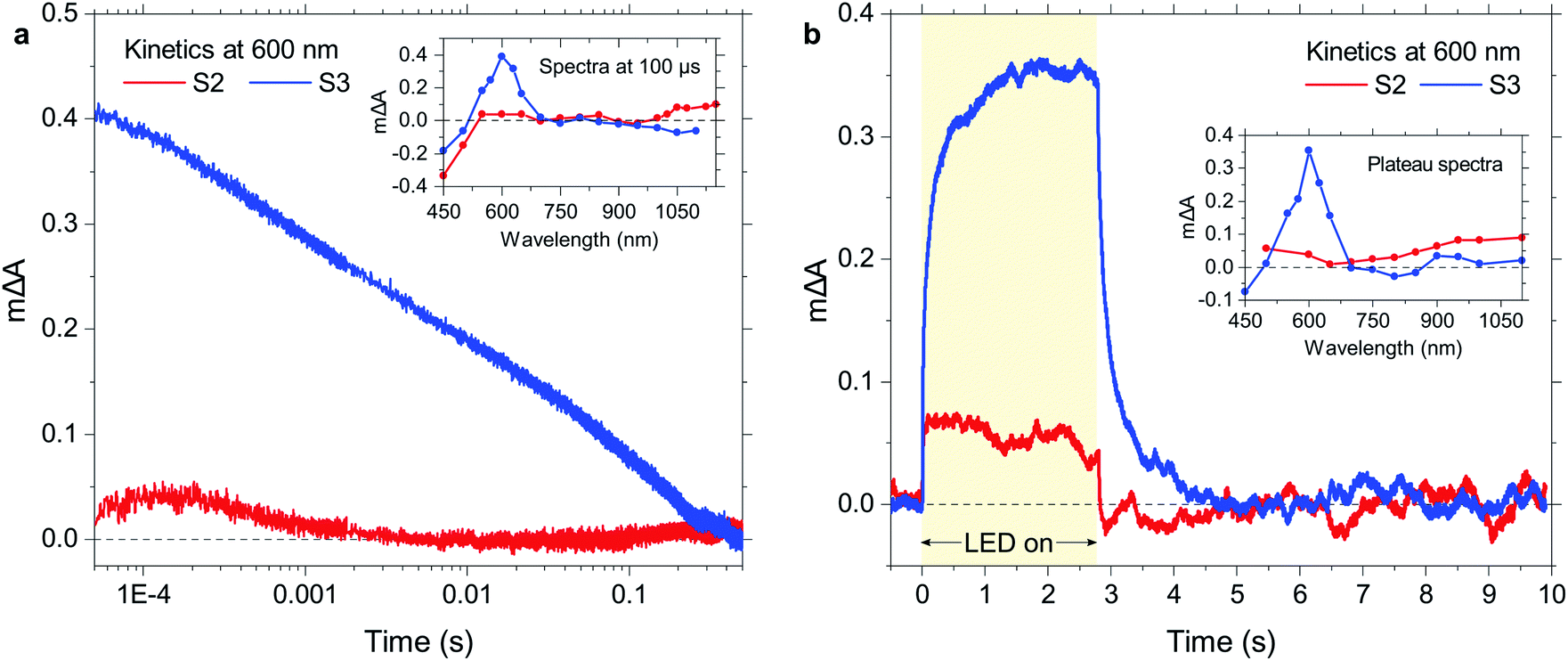 | ||
| Fig. 5 Optical signals from photogenerated charges under transient or quasi-continuous illumination, probed at 600 nm for suspensions of S2 and S3 in TEA/MeOH/water. (a) Transient absorption decay kinetics following laser excitation (pulse duration 4–7 ns), and (b) photoinduced absorption kinetics showing charge accumulation during illumination with a 2.7 s LED pulse and charge decay once the LED is turned off. The insets show the corresponding spectra, probed (a) at 100 μs under transient conditions and (b) at the plateau under quasi-continuous illumination. Excitation conditions: 355 nm, 0.40 mJ cm−2 for laser; 365 nm, 5.5 mW cm−2 for LED. | ||
To probe photogenerated charges under operando photocatalytic reaction conditions, we now turn to quasi-continuous illumination using 365 nm LED pulses with a duration of 2.7 s. As shown in Fig. 5b, we observe the buildup of a population of reaction intermediates when our S3 suspension is illuminated with the LED, and a saturation regime (steady state) is reached once an equilibrium between their generation, recombination, and reaction is established. As shown in the inset, the accumulating reaction intermediates exhibit an essentially identical absorption spectrum to the transiently observed electron polarons in the case of S3, suggesting that this signal is due to the buildup of electron polarons under illumination. This behavior of S3 is again very similar to that of its polymer analogue P10, where electron polarons build up during reaction as their transfer to catalytic Pd clusters is relatively slow.50 Like in the transient experiments (Fig. 5a), S2 exhibits lower signal amplitudes and a much broader spectrum with an absorption towards the NIR. Given the comparable Pd content of S2 and S3, this lower signal amplitude demonstrates that the lower yield of electron polarons in this material can also be observed under reaction conditions.
Overall, S3 behaves very similarly to P10 from a photophysical point of view, which is consistent with the observation that its activity approaches that of its polymer analogue (1125 vs. 2825 μmol h−1 g−1).50 Long-lived oligomer-centred electron polarons are generated upon photoexcitation and accumulate on the oligomer under quasi-continuous illumination, and the differences in polaron yield correlate with the measured hydrogen evolution activity.
Driving force calculations
To estimate the ability of the different oligomers after excitation by light to drive the reduction of protons and oxidation of triethylamine or Na2S, the ionisation potential, IP, and electron affinity, EA, of each, as well as their excited-state analogous IP* and EA*, were predicted. IP and EA determine the available thermodynamic driving force for triethylamine/Na2S oxidation and proton reduction by free electrons and holes respectively, while EA* and IP* determine the same but for the case of excitons (i.e. before dissociation of the exciton in to free electrons and holes, something that is not generally spontaneous for organic systems). These calculations used our standard approach53,54 based around a combination of ΔDFT and TD-DFT (B3LYP55–57/DZP58) in combination with the COSMO59 dielectric screening model (εr 80.1, water) to describe the screening of charges in the oligomers near the oligomer–solution interface. Previously, we showed that this approach yields results comparable to those measured using photoelectron spectroscopy, at least for amorphous polymers.54,60 The results of these calculations (Fig. 3d), suggest for MeF1–MeF3 and S1–S3 that with increasing oligomer length the IP becomes less positive and EA less negative, and hence the driving force for triethylamine/Na2SO3/Na2S oxidation and proton reduction decreases with oligomer length. The calculations also suggest that the sulfone containing oligomers always have a larger driving force for oxidation and a smaller driving force for reduction than the non-sulfone oligomers of a similar size. The same holds for the mesityl-oligomers vs. their non-mesityl counterparts.Discussion
Ideally, a photocatalyst maximises both the thermodynamic driving force for proton reduction and water/sacrificial hole scavenger oxidation, as well as the absorption of sunlight. However, as discussed above, these two material properties are oppositely affected by the chain length of oligomeric/polymeric photocatalysts. Increasing the chain length red-shifts the absorption onset, and hence increases the amount of photons absorbed, but it also reduces the driving force for proton reduction and water oxidation, or here hole scavenger oxidation. As a result, depending on what material property limits the activity of oligomers, the photocatalytic activity might either go up or down with increasing chain length. The trend in activity with chain length is likely also influenced by the molecule (water/hole scavenger) that is oxidised. Triethylamine is easier to oxidise than water, with a predicted17 potential for the two-hole oxidation to diethylamine and acetaldehyde of −0.72 V vs. SHE at pH 11.5, the likely pH of a triethylamine solution. This can be compared with +0.82 V for the oxidation of water to oxygen at pH 7 (and +0.55 V at pH 11.5). In the case of the Na2S/Na2SO3 mixture, both HS−, the anion formed upon the dissolution of Na2S in water, and SO32−, sulfite, can act as hole scavengers. Of the two, sulfite is the easiest to oxidise, even easier than triethylamine, with a reported61 experimental potential of −0.94 V at pH 13.96 (solution Na2S/Na2SO3 pH 13.33) for the 2-hole oxidation to sulfate. However the initial one-hole oxidation of HS− is actually slightly easier (−0.45 V) than that of SO32− (−0.16 V) meaning it is unclear which anion would act as the hole scavenger. The easier the molecule is to oxidize, be it water, triethylamine or sulphite (that is, the more negative the associated standard reduction potential), the less likely it is that an oligomer photocatalyst will be limited by the driving force for oxidation.We can use the analysis above to understand activities trends in Tables 1 and 2. We focus initially on the MeF1–MeF3 and S1–S3 oligomers, and in case of MeF1 and S1 on the activity measured for the samples where additional Pd was added, such that all materials compared have similar Pd loadings. When using triethylamine as sacrificial hole scavenger, the photocatalytic performance of both oligomer series appears to be more limited by light absorption than driving force, with the most active materials being MeF3 and S3, respectively. This does not only follow from the trend in activity with oligomer length but also the fact that when switching from UV to broadband illumination rates uniformly increase. When using the Na2S/Na2SO3 solution as sacrificial hole scavenger, S1–S3 behave as in the presence of triethylamine, though with 5–7× smaller hydrogen evolution rates. By contrast, MeF1–MeF3 appear limited by driving force with MeF1 being the most active material with hydrogen evolution rates, if anything, being higher under UV than broadband illumination. It is perhaps not surprising that MeF1–MeF3 rather than S1–S3 would be limited by driving force as MeF1–MeF3 have a smaller driving force for sacrificial hole scavenger oxidation than S1–S3. As far as we are aware, the first direct evidence of the link between oligomer length, oligomer properties, and oligomer photocatalytic activity, although we have previously observed related structure–property–activity relationships for co-polymers with varying composition17 and 2D networks with different linker lengths.62
| Material | Pd amount by ICP-MS (wt%) | HER λ > 295 nm irradiationa,b (μmol h−1 g−1) | HERa 275 > λ > 400 nm irradiationb (μmol h−1 g−1) |
|---|---|---|---|
| a Photocatalyst (25 mg) suspended in Na2S/Na2SO3 (aq.) (0.2 M/0.35 M, 25 mL), rate calculated as linear regression fit over 5 hours. b See ESI for filter characteristics. c Pd below the detection limit of the instrument, see ESI for full details. | |||
| PSP | > 0.001c | 24.1 ± 0.8 | 29.6 ± 0.4 |
| MSM | 0.002 | 5.5 ± 0.2 | 20.4 ± 0.7 |
| PFP | 0.003 | 13.8 ± 0.8 | 8.7 ± 0.3 |
| MFM | 0.014 | 4.8 ± 0.4 | 10.0 ± 0.5 |
| MeF1 | > 0.001c | 4.1 ± 0.3 | 6.5 ± 0.6 |
| MeF1 + Pd | 0.016 | 14.2 ± 0.3 | 28 ± 1 |
| MeF2 | 0.011 | 3.4 ± 0.2 | 4.4 ± 0.2 |
| MeF3 | 0.014 | 10.1 ± 0.3 | 6.6 ± 0.1 |
| S1 | > 0.001c | 8.1 ± 0.8 | 25.4 ± 0.2 |
| S1 + Pd | 0.36 | 14.7 ± 0.5 | 33 ± 0.7 |
| S2 | 0.22 | 81 ± 2 | 50 ± 1 |
| S3 | 0.26 | 286 ± 4 | 162 ± 6 |
| Material | Pd amounta (wt%) | HER λ > 295 nm irradiationb,c (μmol h−1 g−1) | HER 275 > λ > 400 nm irradiationb,c (μmol h−1 g−1) |
|---|---|---|---|
|
a Determined via ICP-MS after digestion of the sample.
b Photocatalyst (25 mg) suspended in TEA/MeOH/water (1:1:1, 25 mL), rate calculated as linear regression fit over 5 hours.
c See ESI for filter characteristics.
d Catalyst partially soluble in mixture used for photocatalysis experiment.
e Rate determined as linear regression fit over 3 hours or less due to catalyst instability.
f Measurement of the amount of photodeposited Pd not possible due to catalyst solubility.
g Due to lack of material these experiments were conducted with photocatalyst (5 mg) in TEA/MeOH/water (1:1:1, 5 mL), control experiments were also run for S2 and S3 under these conditions and showed little change in rate from the large volume experiments (see Fig. 4).
|
|||
| MeF1 | < 0.001d | 4.7 ± 0.2d | 2.6 ± 0.3d |
| MeF2 | 0.011 | 13 ± 0.4 | 4.3 ± 0.3 |
| MeF3 | 0.017 | 37 ± 1 | 28 ± 1 |
| S1 | < 0.001d | 26 ± 3d,e | 20.1 ± 0.4d,e |
| S2 | 0.22 | 414 ± 9 | 101 ± 1 |
| S3 | 0.26 | 2073 ± 82 | 526 ± 9 |
| S1 + 3 wt% Pd | —f | 43 ± 3d,e | — |
| S2 + 3 wt% Pd | 2.1 | 1369 ± 24g | — |
| S3+ 3 wt% Pd | 2.5 | 6550 ± 150g | — |
Comparing the activities of the MeF1–MeF3 and S1–S3 oligomers, the S1–S3 oligomers showed significantly higher photocatalytic activities compared to the MeF1–MeF3, analogues. This is consistent with our previous work comparing their polymer analogues.12 These oligomers all form close packed structures and are expected to have small internal surface areas. However, external surface area varies significantly between samples, as shown by the factor of 30 variation in D[3,2] values obtained from SLS, and could influence photocatalytic activity. Thus, in addition to the larger driving force and red-shifted light absorption onset discussed above, the much smaller particle sizes of the S1–S3 oligomer compared to the MeF1–MeF3 series (ESI, Fig. S39–S40†) likely to contributes to their much higher hydrogen evolution rates. Aside from primary particle size, the improved dispersibility of the sulfone-containing oligomers is thought to be related to their high hydrophilicity. We also note that the face-to-face stacking observed in the S1–S3 crystal structures, and the accompanying intermolecular orbital overlap, could facilitate improved transfer of excitons or charged species compared to the less-overlapped, edge-to-face packing that dominates in the MeF1–MeF3 structures.
Interestingly, the visible light activity of the trimer S3 (1125 μmol h−1 g−1) is of the same order of magnitude as its polymer analogue, P10, under equivalent conditions (2825 μmol h−1 g−1).50 An external quantum efficiency (EQE) of 8.8% at 420 nm was determined for S3, which is close to the 11.6% reported for P10 and higher than most conjugated polymer photocatalysts.20,63–65 The high performance of S3 is in line with our data showing that its potentials, optical gap, polaron signatures and polaron dynamics under transient and quasi-continuous illumination conditions are similar to P10. S3 challenges the general assumption that long chain lengths are required for significant hydrogen evolution activity, and, as a molecular material, demonstrates that these sulfone-containing photocatalysts can achieve high performance despite primarily relying on intermolecular exciton/charge transfer rather than exciton/charge transport along a polymer backbone.66 Our recent study on 1,3,6,8-tetrakis(p-benzoic acid)pyrene67 showed a highly crystalline, hydrogen-bonded form of the molecule, with idealised pyrene–pyrene stacking, had excellent activity for proton reduction, with an EQE of 4.1%. S3 is a semi-crystalline material, whose secondary structure contains π-stacking interactions over much smaller domain sizes, and yet it shows even higher activity. While it appears that molecular materials can be effective hydrogen production photocatalysts without extended backbones or the long-range stacking found in hydrogen bonded frameworks (HOFs), it seems likely that these materials might achieve even higher performances in the presence of more efficient charge transport, which could enable faster transfer to catalytic sites.50
While the polaron signatures for S3 are very similar to P10, optical signals from reaction intermediates under transient and quasi-continuous illumination are much lower for S2. No sharp polaron peak is observed in the visible range, which is in line with the significantly lower activity of S2 compared to S3 and suggests that S2 likely gives too low polaron yields to be determined reliably via spectroscopy. The performance of S1–S3 improves significantly upon addition of more Pd, which is in line with P10 where transfer of long-lived polarons to catalytic Pd sites limits the hydrogen evolution activity of the material.50 This interpretation is further corroborated by the polaron-like signature of S3, which suggests that long-lived electrons reside on the oligomer rather than on Pd clusters under both transient and charge accumulation conditions. On the other hand, the exciton lifetime in the S1–S3 series, as quantified via fluorescence lifetime measurements, is affected only little by added Pd, suggesting that the performance increase upon addition of more Pd is indeed due to improved catalysis rather than more efficient charge separation at early times.
Switching the focus to the MSM, MFM, PSP and PFP oligomers, it appears that aside from chain length, the effective conjugation length can have a marked effect on the activity of oligomers. Crystal structures show the backbone dihedral angles of MSM (64.0(5)° and 96.6(5)° for conformer 1 in the asymmetric unit; and 86.6(5)° and 115.4(5)° for conformer 2) and MFM (78.0(2)° and 109.4(1)° for conformer 1 in the asymmetric unit; and 91.8(1)° and 108.1(1)° for conformer 2) are significantly larger than the comparable dihedral angles for PSP (19.2(3)° and 33.5(3)°) and PFP (11.80(7)° and 14.09(7)°), which is due to the phenyl groups in PSP and PFP being substituted with the sterically hindered mesityl groups in MSM and MFM (Fig. S16†). This lack of planarity reduces the conjugation length, blue-shifting the absorption onsets in the solid-state by 40 nm when comparing PSP to MSM and by 96 nm for PFP compared to MFM, with similar shifts in solution measurements. Comparing PSP and PFP to MSM and MFM this twisting also appears to affect the intensity of absorption as reflected in the significantly higher extinction coefficient of the phenyl-bearing analogues which is consistent with the higher oscillator strength predicted by DFT. The loss in light absorption for the mesityl substituted oligomers as a result of their reduced conjugation length is reflected in their lower photocatalytic activity under broad band irradiation. However, when using UV light only, the activities of PSP and MSM are similar, as are those of PFP and MFM, where the former have at least twice the activity. The smaller effect of the reduced conjugation length in the UV could be because the UV-only range includes more of the middle-UV (275–300 nm), where the mesitylated oligomers absorb more strongly (Fig. S29†), than the broadband illumination spectrum, reducing the effect of loss of absorption at longer wavelengths. In this case, it is perhaps significant that the dibenzo[b,d]thiophene sulfone-containing PSP and MSM oligomers have a larger driving force for scavenger oxidation than their fluorene-based PFP and MFM analogues. Alternatively, the more hydrophilic nature of dibenzo[b,d]thiophene sulfone containing materials may improve interaction with water at the surface of the crystal. Differences in particle sizes, however, do not seem to play a role here; SLS measurements show that the particle sizes for PSP and MSM are broadly similar to PFP and MFM (Fig. S41†).
Conclusions
In summary, we used a series of oligomers to investigate the effects of chain length and backbone twisting on photocatalytic hydrogen evolution. The higher driving force associated with short conjugation lengths—that is, large fundamental (band) gaps—were only found to be dominant in systems where the overpotentials for the reaction were small. In general, shorter conjugation lengths were found to reduce performance and primarily because of inferior light absorption properties in terms of both onset wavelength and absorption magnitude. Since absorption properties can be easily tuned by the introduction of particular functional groups, this suggests that future investigations into organic photocatalysts for solar fuels production need not be limited to polymers. Chain length was also found to have a considerable impact on excited state lifetime but this did not correlate with hydrogen evolution, particularly in dibenzo[b,d]thiophene sulfone bearing oligomers. Along with the highly Pd-dependent activities of the dibenzo[b,d]thiophene sulfone oligomers, this provides further evidence that performance in these materials is more likely to be limited by the ability of electron polarons to transfer to an active Pd centre. Future studies might therefore focus on measuring charge transport in these materials and developing ways to improve this. In general, long-lived electron polarons in S3 behave very similarly to those in its polymer analogue P10 both under transient and quasi-continuous illumination conditions, as evidenced by transient and photoinduced absorption experiments. This highlights the need to broaden the range of organic semiconductors that is considered for photocatalytic hydrogen production to include molecular materials with new opportunities for substrate modification and where favourable properties, such as high crystallinity,40 are easier to achieve. Oligomers might also be used as a photoactive layer in photoelectrochemical water splitting systems. While the integration of polymeric materials can be achieved using thermal vapour polymerisation67 the greater range of deposition techniques that are possible for solution-processable oligomers could make them ideally suited for the production of high-quality, low-defect crystalline films.Author contributions
C. M. A. synthesised and characterised the materials and performed hydrogen evolution experiments. R. S. S. synthesised PFP and MFM. M. S. performed TAS measurements and together with J. R. D. analysed the data. M. A. L. and C. M. K. performed and analysed SCXRD. L. W. and M. A. Z. performed DFT calculations. N. J. B. and F. B. performed ssNMR experiments and analysis. R. S. S. and A. I. C. supervised the project. The manuscript was written by C. M. A., R. S., M. S., M. A. L., M. A. Z., J. R. D., and A. I. C. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support under Grant EP/N004884/1 and a PhD studentship to N. J. B. M. S. thanks Imperial College for a President's PhD Scholarship and is grateful to EPSRC for a Doctoral Prize Fellowship. The authors acknowledge Diamond Light Source for access to beamlines I19 (CY21726).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- T. Jafari, E. Moharreri, A. Amin, R. Miao, W. Song and S. Suib, Molecules, 2016, 21, 900 CrossRef PubMed.
- S. Shen, X. Chen, F. Ren, C. X. Kronawitter, S. S. Mao and L. Guo, Nanoscale Res. Lett., 2011, 6, 290 CrossRef PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- X. Zong, X. Miao, S. Hua, L. An, X. Gao, W. Jiang, D. Qu, Z. Zhou, X. Liu and Z. Sun, Appl. Catal., B, 2017, 211, 98–105 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- L. Li, W. Y. Lo, Z. Cai, N. Zhang and L. Yu, Macromolecules, 2016, 49, 6903–6909 CrossRef CAS.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 CrossRef CAS PubMed.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- F. Haase, T. Banerjee, G. Savasci, C. Ochsenfeld, B. V. Lotsch, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Faraday Discuss., 2017, 162, 165–169 Search PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- C. B. Meier, R. Clowes, E. Bernardo, K. Jelfs, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Chem. Mater., 2019, 31, 8830–8838 CrossRef CAS PubMed.
- L. Guo, Y. Niu, H. Xu, Q. Li, S. Razzaque, Q. Huang, S. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- Y. Lin, Y. Li, X. Zhan, Y. Qu, Z. Xie, D. Yan, Y. Geng, F. Wang, J.-L. Brédas, M. D. McGehee, A. Sellinger, M. Drees, L. Jiang, C. Shu and C. Wang, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- L. Wang, W. Huang, R. Li, D. Gehrig, P. W. M. Blom, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9783–9787 CrossRef CAS PubMed.
- S. Matsuoka, H. Fujii, T. Yamada, C. Pac, a Ishida, S. Takamuku, M. Kusaba, N. Nakashima, S. Yanagida, K. Hashimoto and T. Sakata, J. Phys. Chem., 1991, 95, 5802–5808 CrossRef CAS.
- S. Yanagida, T. Ogata, Y. Kuwana, Y. Wada, K. Murakoshi, A. Ishida, S. Takamuku, M. Kusaba and N. Nakashima, J. Chem. Soc., Perkin Trans. 2, 1996, 1963–1969 RSC.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Chem. Mater., 2018, 30, 5733–5742 CrossRef CAS.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- V. W. H. Lau, M. B. Mesch, V. Duppel, V. Blum, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2015, 137, 1064–1072 CrossRef CAS PubMed.
- G. Zhang, G. Li, Z.-A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- C. M. Aitchison, C. M. Kane, D. P. McMahon, P. R. Spackman, A. Pulido, X. Wang, L. Wilbraham, L. Chen, R. Clowes, M. A. Zwijnenburg, R. S. Sprick, M. A. Little, G. M. Day and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7158–7170 RSC.
- X. Yang, Z. Hu, Q. Yin, C. Shu, X. Jiang, J. Zhang, X. Wang, J. Jiang, F. Huang and Y. Cao, Adv. Funct. Mater., 2019, 1808156 CrossRef.
- J. Sauter, Die Grössenbestimmung der im Gemischnebel von Verbrennungskraftmaschinen vohrhandenen Brennstoffteilchen, VDI-Verlag, Berlin, 1926 Search PubMed.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- F. Waiblinger, J. Keck, A. P. Fluegge, H. E. A. Kramer, D. Leppard and G. Rytz, J. Photochem. Photobiol., A, 1999, 126, 43–49 CrossRef CAS.
- M. Salvador, N. Gasparini, J. Darío Perea, S. Harish Paleti, A. Distler, L. N. Inasaridze, P. A. Troshin, L. Lüer, H. J. Egelhaaf and C. Brabec, Energy Environ. Sci., 2017, 10, 2005–2016 RSC.
- V. S. Vyas, V. W. H. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CrossRef CAS.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 1–7 CrossRef PubMed.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francàs, S. Corby, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020 DOI:10.1021/jacs.0c06104.
- K. T. Nielsen, K. Bechgaard and F. C. Krebs, Macromolecules, 2005, 38, 658–659 CrossRef CAS.
- R. S. Sprick, M. Hoyos, J. J. Morrison, I. M. Grace, C. Lambert, O. Navarro and M. L. Turner, J. Mater. Chem. C, 2013, 1, 3327–3336 RSC.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, J. Mater. Chem. A, 2014, 2, 11996–12004 RSC.
- P. Guiglion, A. Monti and M. A. Zwijnenburg, J. Phys. Chem. C, 2017, 121, 1498–1506 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, D. S. Ginger, I. Mcculloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J.-S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 125, 1472–1476 CrossRef.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 CrossRef CAS PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef PubMed.
- Y. Fang, Y. Xu, X. Li, Y. Ma and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 9749–9753 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1999747–1999756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02675a |
| This journal is © The Royal Society of Chemistry 2020 |