
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous and heterogeneous molecular catalysts for electrochemical reduction of carbon dioxide
Maryam Abdinejad,
M. Nur Hossain and
Heinz-Bernhard Kraatz *
*
Department of Physical and Environmental Sciences, University of Toronto Scarborough, 1265 Military Trail, Toronto, ON M1C 1A4, Canada. E-mail: bernie.kraatz@utoronto.ca
First published on 15th October 2020
Abstract
Carbon dioxide (CO2) is a greenhouse gas whose presence in the atmosphere significantly contributes to climate change. Developing sustainable, cost-effective pathways to convert CO2 into higher value chemicals is essential to curb its atmospheric presence. Electrochemical CO2 reduction to value-added chemicals using molecular catalysis currently attracts a lot of attention, since it provides an efficient and promising way to increase CO2 utilization. Introducing amino groups as substituents to molecular catalysts is a promising approach towards improving capture and reduction of CO2. This review explores recently developed state-of-the-art molecular catalysts with a focus on heterogeneous and homogeneous amine molecular catalysts for electroreduction of CO2. The relationship between the structural properties of the molecular catalysts and CO2 electroreduction will be highlighted in this review. We will also discuss recent advances in the heterogeneous field by examining different immobilization techniques and their relation with molecular structure and conductive effects.
1. Introduction
Carbon dioxide (CO2), as a greenhouse gas, is a significant contributor to climate change. The global average atmospheric CO2 level in 2019 was 409.8 ppm, much higher than the previous highest concentration of 300 ppm, with levels projected to keep increasing unless immediate measures are taken.1,2 These emission levels have raised serious environmental concerns and have translated to noticeable, aberrant meteorological changes.Recent strategies that convert CO2 into value-added materials using photochemically3 or electrochemically4–6 powered reduction reactions have shown promise in recent years. However, this task is challenging due to the high energy required (750 kJ mol−1) to break the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond7,8 and the molecule's stable linear geometry, which makes CO2 reduction reactions (CO2RRs) sluggish and challenging.3,9 Additionally, the electrocatalytic CO2 reduction mechanism is a complex process that involves multiple proton-coupled electron transfer steps and may include several side-reactions and intermediates.10–14
O bond7,8 and the molecule's stable linear geometry, which makes CO2 reduction reactions (CO2RRs) sluggish and challenging.3,9 Additionally, the electrocatalytic CO2 reduction mechanism is a complex process that involves multiple proton-coupled electron transfer steps and may include several side-reactions and intermediates.10–14
The first step of many CO2RRs is the one-electron reduction of CO2 to a CO2˙− radical anion which has a more reactive, bent geometry (Table 1).15,16 Although most CO2RRs describe two-electron reduction to carbon monoxide (CO) and formaldehyde, products of multi-electron transformations such as methane,17 methanol18 and ethanol19 are highly coveted. Table 1 shows the theoretical potentials required to form various multi-electron reductions. Although the theoretical potentials required to form the target products shown in Table 1 appear relatively low, because the products formed are often either thermodynamically similar or more stable than CO2, more negative potentials are required for practical applications to obtain reasonable reaction rates.9 In order to facilitate CO2RR, the use of catalysts is essential and serves several purposes including lowering activation energy barriers, improving reaction rates, and increasing product selectivity.20–23
| CO2 reduction half-reactions | Electrode potentials (V vs. NHE) at pH = 7 |
|---|---|
| CO2 + e− → CO2˙− | −1.90 |
| CO2 + 2H+ + 2e− → HCO2H | −0.61 |
| CO2 + 2H+ + 2e− → CO + H2O | −0.53 |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.48 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
Electroreduction of CO2RR can be completed using either homogeneous or heterogeneous catalysts. Although homogeneous catalysis have shown high selectivity, with near product unity for the production of CO and other reduction products,18,25–29 these systems are dependent on the solubility constraints of the catalysts and are limited by low current densities and instability.30 On the other hand, heterogeneous electrocatalysts minimize the electrode and catalyst distance, allowing for more efficient processes and higher current densities at the expense of product selectivity.31–33 In either case, although a variety of electrocatalysts have been introduced for CO2RR in recent decades,34–36 the performance of these systems has yet to reach a level where they can be successfully implemented industrially.37,38
Recent advancements have found success through incorporating a combination of molecular catalysts and heterogeneous immobilization strategies.39–41 Different molecular approaches such as metal organic frameworks (MOFs), covalent organic frameworks (COFs), and metal-free catalysts have tried to address this issue.31,42–45 It has been shown that applying organic compounds, such as thiols,46,47 polypyrrole,48 N-heterocyclic carbenes (NHCs),49 and N-substituted pyridines,17,50,51 can reduce CO2 to desirable materials such CO, HCOO− and COOH.
The amino functional group in particular has proven to be effective at selectively capturing CO2 from a mixture of gases. This ability is especially pronounced in primary or secondary amines such as monoethanolamine (MEA), diethanolamine (DEA) and decylamine (DCA).52–57 In these reactions, the amino groups initially capture CO2 to form a zwitterionic species that can react with another amino equivalent to form a carbamate salt (Scheme 1).58,59
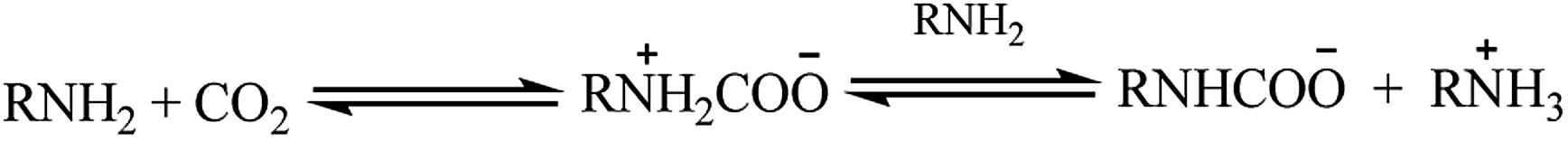 | ||
| Scheme 1 Carbamate formation using primary and secondary amines. | ||
This review will start with a general introduction to the electrochemical reduction of CO2 and the metrics that are used to quantify catalyst efficiency and continue with a summary of the recent developments of amine molecular catalysts in both homogeneous and heterogeneous environments. In general, this report suggests that structural tuning of organic compounds followed by either covalent or non-covalent immobilization onto various conductive surfaces (i.e., graphite, Au, Ag, Pd, and Cu), results in high performing systems.
2. Electroreduction of carbon dioxide
Electrochemical capture and reduction of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60 has received extensive interest in the last decade because of the: (1) controllable nature of the technique (e.g. potential and temperature); (2) flexibility between organic and aqueous media; (3) relative scalability of bench-side reaction setups to industrial application.61
60 has received extensive interest in the last decade because of the: (1) controllable nature of the technique (e.g. potential and temperature); (2) flexibility between organic and aqueous media; (3) relative scalability of bench-side reaction setups to industrial application.61
Typical electrochemical cells consist of a cathode, anode, electrolyte and a membrane (Fig. 1). CO2RR occurs at the cathode, while reciprocal oxidation or oxygen evolution reactions (OERs) occur at the anode. The cathode and the anode are separated by a membrane which maintains charge balance and separates the respective redox products. The electrolyte carries the charge between the electrodes and delivers dissolved CO2 to the catalytically active surface.
 | ||
| Fig. 1 A typical electrochemical CO2 reduction reaction cell. The oxidation and reduction occur at anode and cathode, respectively. The membrane separates the compartments. The electrolyte includes positive and negative species that assist charge and CO2 transport. | ||
3. Quantifying catalytic performance
Several factors are used to quantify catalytic performance. Selectivity is measured by the faradaic efficiency (FE), and the catalyst activity is determined by the current density (j) as a function of the electrode area. The current density can be used to describe either the total current density of all reduced products or the partial current density of one particular product. In the context of CO2RR, current density can be used to describe the rate of reaction. The robustness of the catalyst is calculated with the turnover number (TON) which is determined by dividing the mole of reduced product with the mole of catalyst. The turnover frequency (TOF, s−1) is defined as the mole of reduction product divided by the mole of active catalysts per unit of time.4. Molecular electrocatalysis for CO2RR
Using electrochemical techniques to reduce captured CO2 with small molecules is a promising strategy to produce valuable materials.62 This has been demonstrated previously using amino and pyridine-substituted compounds for electrochemical CO2RR in both homogeneous and heterogeneous media.18,63–69Various catalysts have been developed as both homogeneous molecular catalysts16,18,70–77 and heterogeneous solid-state catalysts,22,40,78 such as metal alloys,79,80 non-metal catalysts81 and single atoms.82 The identity of the metal electrodes have also been shown to play a role in the product distribution.12,83–93 In this section, recent developments in molecular electrocatalysts for CO2 reduction will be discussed with respect to their molecular structure, nano-structuring immobilization and electrode surface modification. The study of the following molecular catalysts highlights the importance of molecular design, electronic factors, and ligand structure in successful experimental design.
4.1. Homogeneous amine molecular catalysts and electrochemical CO2RR
Homogeneous studies of amine-based molecular electrocatalysts have been identified their utility for CO2RR. Using meso-substituted amino groups on metallated porphyrins, we were able to achieve selective reduction of CO2 to CO and methanol (Fig. 2a).18 Comparing the cyclic voltammograms of Co-TPP and Co-TPP–NH2 in the presence of CO2 clearly highlights the importance of the amino group and its role in reducing CO2 (Fig. 2b). The influential presence of the cobalt center in CO2RR, can be seen in Fig. 2d. In this project, H2O was used as an extra proton source to facilitate the C–O bond cleavage (Fig. 2e). To further understand the electroactivity of the amino group, a comparison with nitro porphyrins (TPP–NO2) shows a slightly better performance of the amino group (Fig. 2f).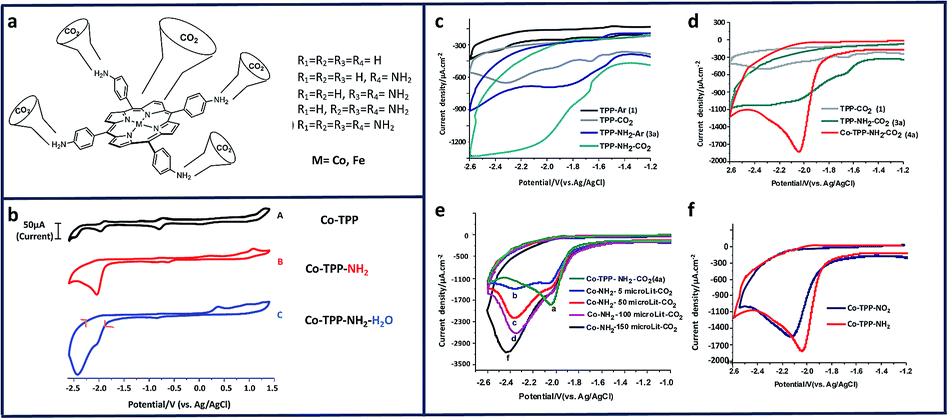 | ||
| Fig. 2 (a) Schematic of metallated amino porphyrins; cyclic voltammograms (CV) of 0.01 mM of (b) Co-TPP, Co-TPP–NH2, and Co-TPP–NH2 with 5% H2O; under CO2; (c) comparison of TPP and TPP–NH2 under Ar and CO2; (d) comparison of TPP, TPP–NH2, and Co-TPP–NH2 under Ar and CO2; (e) Co-TPP–NH2 in 0.1 M NBu4PF6 in DMF solutions at a scan rate of 100 mVs−1 in: (a) (no water), (b) 5 μL (C) 50 μL (d) 100 (e) 150 μL water. (f) Comparison of Co-TPP–NO2 and Co-TPP–NH2 under Ar and CO2 in 0.1 M NBu4PF6 and DMF solutions. Conditions: scan rate, 100 mV s−1; working electrode, glassy carbon; reference electrode, Ag/AgCl; counter electrode, platinum.18 Copyright (2019) American Chemical Society. | ||
Chapovetsky et al.94 also reported a cobalt aminopyridine macrocycle with amine substituents selectively reducing CO2 to CO. From electrochemical experiments, they found that the catalytic activity is strongly dependent on the number of secondary amines (Fig. 3).95 Subsequent studies showed how those amine groups could act as hydrogen bond donors to enhance catalytic performance.
 | ||
| Fig. 3 (a) Experimental catalytic rate constants, kobs (s−1), as a function of the number of pendant secondary amines using 1.5 M TFE under CO2 saturation at a scan rate of 100 mV s−1. (b) Schematic of pendant hydrogen-bond donors in cobalt catalysts independently enhance CO2 electroreduction.95 Copyright (2018) American Chemical Society. | ||
The identity of the electrode used has been found to have a large influence on the catalytic activity of homogeneous amine solutions, with different electrodes such as glassy carbon, copper, and silver each eliciting their own distinctive response.18,46,55,63,96–101 Lue et al.55 reported a systematic study on electrochemical CO2 reduction with 30% (w/w) MEA on, Sn, Pb, Pd, Ag, Cu and Zn metal electrodes. Schmitt et al.100 used in situ surface-enhanced Raman spectroscopy to study 3,5-diamino-1,2,4-triazole (DAT) exposed-Ag electrodes, finding that the amine treated electrode increased FECO due to a weakening of the CO bonding strength.
Many studies of copper (Cu) electrodes have characterized their ability to reduce CO2 to multi-carbon products,14,33,55,87,102–110 whereas when exposed to molecular catalysts it is more common to see CO2 selectively converted to CO,57 formate,111 and formic acid.13,104 We have also investigated the ability of primary amines to selectively reduce CO2 to CO using Cu electrodes (Fig. 4).57 In these studies, ethylenediamine (EDA) proved to be the most effective absorbent for CO2 capture and subsequent reduction to CO among MEA and decylamine (DCA), with a current density of −18 mA cm−2, TON of 252 and a FE of 58% at −0.78 V vs. RHE. Compared to glassy carbon electrodes, the cathodic current was dramatically enhanced when Cu was used as a working electrode (Fig. 4f and g).
 | ||
| Fig. 4 13C NMR spectra of DCA (a), DCA–13CO2 (b) and H2O–13CO2 (c) in CDCl3. Cyclic voltammograms (CV) of: (d) 1–3 under CO2 with GCE; (e) compounds 1–3 under CO2 with GCE stacked; (f) 1–3 under CO2 with Cu electrode; (g) 1–3 under CO2 with Cu electrode stacked; 0.1 mM concentration in 0.1 M NaClO4 solution. Conditions: scan rate, 100 mVs−1; working electrode, glassy carbon/copper; reference electrode, Ag/AgCl; counter electrode, platinum.57 Copyright (2020) American Chemical Society. | ||
Our recent studies on the electrochemical reduction of CO2 in various fractions of MEA solutions at smooth and nanodendrite (ND) Cu, Ag and Au showed that a 0.05 M fraction of MEA exhibited the highest catalytic activity for each surface.112 CO2 electroreduction to HCOO−. The ND electrodes exhibited much higher current efficiencies for CO2 to HCOO− conversion compared to the smooth metal electrodes, revealing the critical role of surface morphology in enhancing catalytic activity.
4.2. Heterogeneous amine molecular catalysts and electrochemical CO2RR
Heterogeneous electrocatalysts have benefits over homogeneous electrocatalysts for CO2RR application due to the catalytically active site being either located directly on the electrode surface or the electrode itself. As a result, catalytic loading concentrations can be lower. Molecular catalysts can be attached to solid, conductive surfaces via covalent/non-covalent immobilization techniques29,113,114 or using polymers and metal–organic frameworks.115–117 This strategy offers higher stability and catalytic efficiency56 with a greater potential of reaching the necessary current densities for industrial implementation.118 Due to its simple preparations, one of the most popular immobilization techniques involves depositing conjugated organic ligands onto carbon surfaces which are stabilized by the non-covalent π–π interactions between the catalyst and solid surface.17,56,119 The molecular catalysts can be also deposited on electrode surfaces through covalent bond.120,121Previous reports on CO2RR selectivity involved either the use of a metal electrode surface, where the electron-transfer efficiency was largely dependent on the material's conductivity, or the incorporation of small inactive molecules39 on the surface of the metal electrode to maximize interaction between the electrode and the molecular catalysts.56,61,122–125 An example of this are electrografting techniques which produce a direct chemical bond between the catalyst and a solid substrate.98,126 The direct connections that arise from these methods are believed to be the primary factor in increasing the reaction rate of CO2RR relative to hydrogen evolution reactions (HERs) and lowering overpotentials.125,127–131 Using this technique, immobilization of terpyridine onto glassy carbon electrodes has been previously reported.66
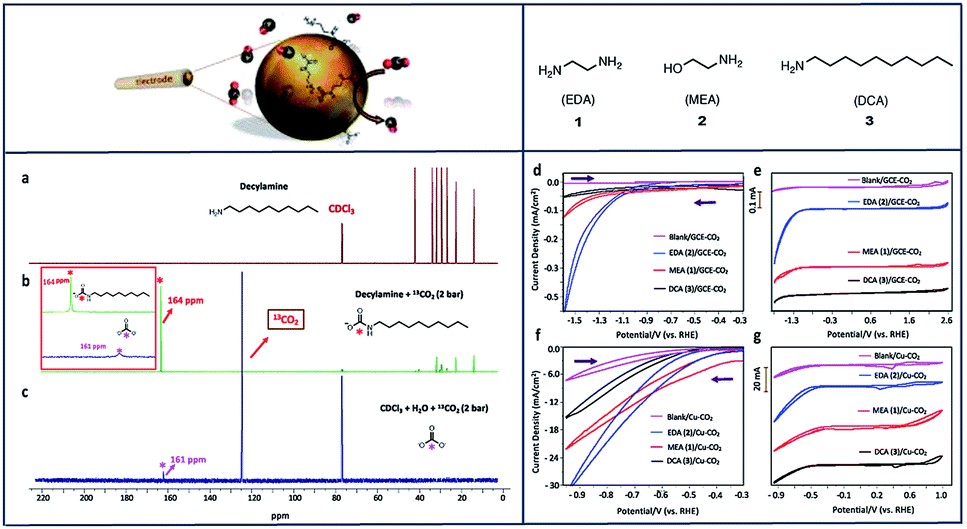
Marianov et al.121 have also successfully electrografted amino porphyrins via electro reduction of diazonium salt onto glassy carbon (Scheme 2). By introducing a conjugated linker between the porphyrin and the electrode, they proved that the CoI/CoII redox couple facilitates the CO2 electroreduction process (Fig. 5a). With the covalently linked catalyst an increase to the current density (4.7 mA cm−2) was seen, compared to the unlinked catalysts (1.4 mA cm−2) (Fig. 5b). In addition to the covalent linkage facilitating electrode-to-catalyst charge transfer, the current density was also observed to be dependent on the catalyst loading concentration and the total active surface area (Fig. 5b and c).
 | ||
| Scheme 2 Preparation of covalently immobilized Co tetraphenylporphyrin (CoTPP-cov).121 Copyright (2019) Elsevier. | ||
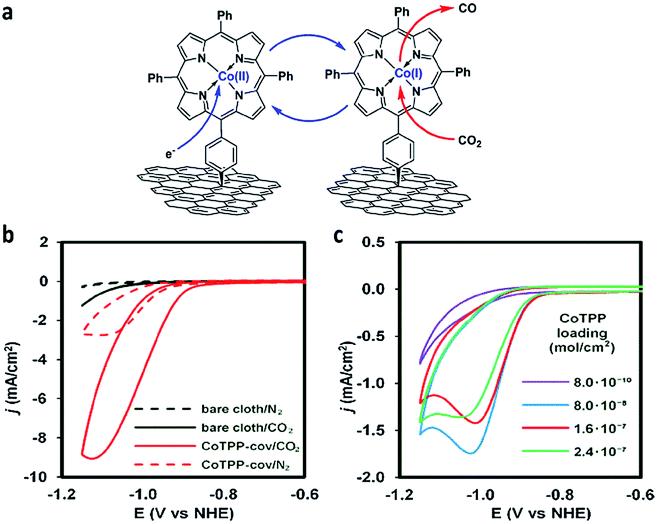 | ||
| Fig. 5 (a) Preparation of covalently immobilised Co tetraphenylporphyrin (CoTPP-cov); (b) CVs of CoTPP-cov in N2- and CO2-purged aqueous electrolyte, CVs of bare carbon cloth are shown for clarity; (c) CV traces of CoTPP-noncov with the variable amount of noncovalently immobilized CoTPP in CO2-saturated solution. Conditions: electrolyte: 0.5 M KHCO3 in all cases, potential scan rate is 100 mV s−1.121 Copyright (2019) Elsevier. | ||
Zouaoui et al.97 investigated the electrocatalytic activity of amine derivatives deposited onto Pb surfaces toward electroreduction of CO2 to formate. Using diazonium chemistry, 4-aminomethylbenzene (4-ABA), 3-aminomethylbenzene (3-ABA), 4-(2-aminoethyl)benzene (4-AEA) and 4-nitrobenzene (4-NB) were grafted onto Pb electrodes (Fig. 6). The Pb-amine modified electrodes showed enhanced activity and selectivity in all cases (Fig. 6a). Fig. 6b shows chronoamperograms of the 4-ABA modified Pb electrode (6.3 × 10−7 mol cm−2) in a 1 M HKCO3 solution saturated with CO2. In this study, 4-ABA reached a maximum current density of −24 mA cm−2 at −1.29 V vs. RHE, with a FE over 80% (Fig. 6c and d).
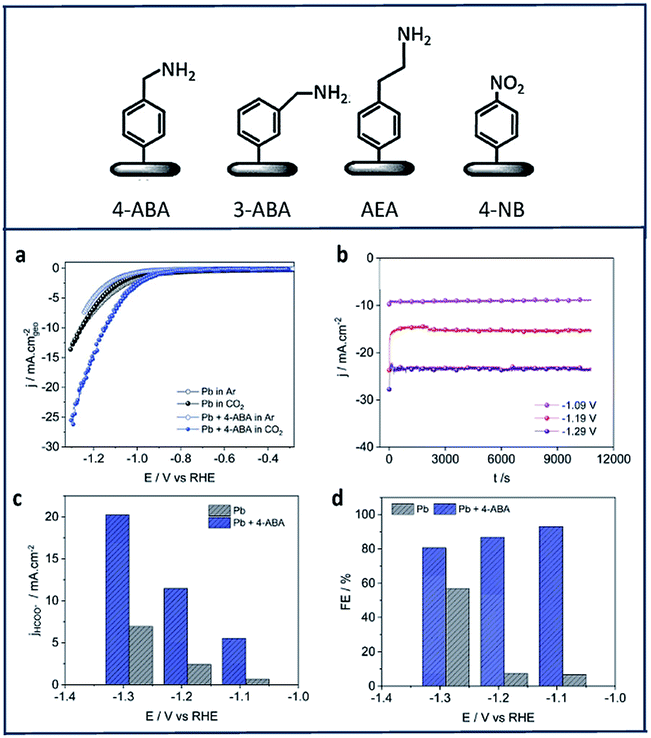 | ||
| Fig. 6 The four different amines used in this work to modify the Pb electrodes: (a) CV of Pb and Pb + 4-ABA; (b) chronoamperograms recorded in CO2-saturated 1 M HKCO3 solution for Pb + 4-ABA electrode (6.3 × 10−7 mol cm−2); comparison of (c) current density and (d) faradic efficiency at different potentials for = Pb + 4-ABA and bare Pb electrodes.97 Copyright (2019) Royal Society of Chemistry. | ||
Gold (Au) has been also found to exhibit catalytic activity towards CO2RR.132–134 Mikoshiba et al.135 showed that imidazolium ions immobilized on Au electrodes suppress H2 generation and accelerate CO2RR. In their study, imidazolium salts with small methylene units (IL-2, Fig. 7A) exhibited greater current densities compared to longer chained units with FEs up to 87% (Fig. 7B).
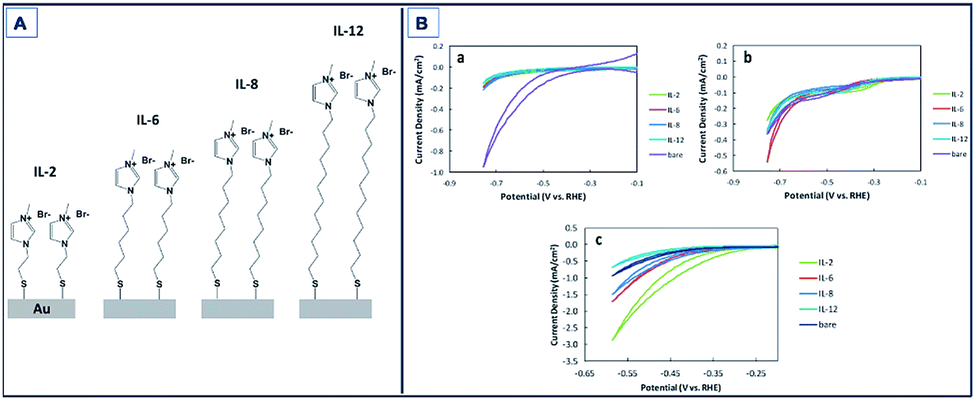 | ||
| Fig. 7 Schematic of Au electrodes with 1-methylimidazolium-terminated SAMs (IL-2, IL-6, IL-8, and IL-12). CV of bare and SAM modified Au electrodes in Na2SO4 aqueous solution purged with (a) N2 and (b) CO2. Scan rate: 100 mV s−1; (c) CV of bare and SAM-modified Au electrodes in NaHCO3 aqueous solution purged with CO2. Scan rate: 100 mV s−1.135 Copyright (2015) Royal Society of Chemistry. | ||
In another study, Au electrodes functionalized with 4-pyridinylethanemercaptan (PEM) thiols showed similar increases in product selectivity and catalytic activity (Fig. 8a).136 The proposed mechanism for formate production shows the pyridine H-atom abstracted by reduction of the aqueous solution and adsorbed onto the Au surface (Fig. 8a). HCO2 is formed through electrophilic attack of CO2 onto the adsorbed proton. The FE of the electroreduction products in this system were observed to be potential dependent. Fig. 8b–g shows the potential-dependent product distribution (formate, CO and H2) of functionalized Au and bare Au surfaces.
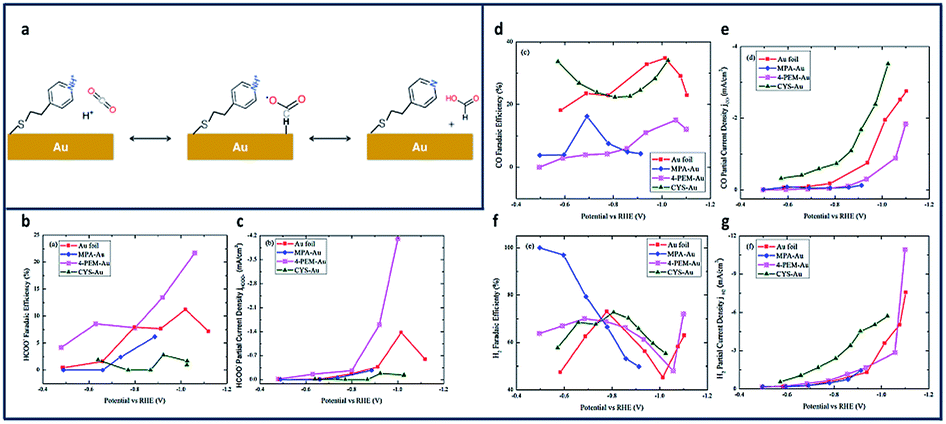 | ||
| Fig. 8 (a) Proposed Mechanism for reduction to formate at PEM-modified Au Electrode; comparison of partial current density and FE for thiolate ligand on polycrystalline Au and pure polycrystalline Au: (b) FE of formate formation (±2.5% at 95% confidence level (CL)), (c) FE of CO formation (±6.2% at 95% CL), (d) FE of H2 formation (±25% at 95% CL); (e) partial current density of formate formation; (f) partial current density of CO formation, and (g) partial current density of H2 formation.136 Copyright (2020) American Chemical Society. | ||
A 2-fold increase in FEformate and a 3-fold increase in current density were achieved and attributed to enhancement of proton and electron transfers using Au foil (Fig. 8b and c).137 This increase in current density is due to the amine's ability to make a complex with CO2 near the Au surface.138 Cystemine modified electrodes saw a 2-fold increase in both CO and H2 production (Fig. 8d–f), while electrodes with 2-mercaptopropionic acid (MPA) ligands reported nearly 100% selectivity for H2 (Fig. 8g).
In another study, it was found that immobilization of Au nanoparticles using N-heterocyclic carbenes facilitated electron transfer from Au to CO2 (Fig. 9a).139 The electrochemical reduction of CO2 to CO catalysed by a Au-1,3-bis(2,4,6 trimethylphenyl)imidazol-2-ylidene nano particle (Au–Cb NP) was found to be greater than that of bare Au nanoparticles (Au NP). Oleylamine-capped Au NPs (Au–Oa NP) were first loaded onto carbon black to make a Au–Oa NP/C mixture.140 The active surface area for Au NP/C and Au-1,3-bis(2,4,6 trimethylphenyl)imidazol-2-ylidene nano particle (Au–Cb NP) electrode were evaluated using Pb underpotential deposition (upd).141–143 The current density increased substantially (Fig. 9c and e) and the FECO increased from 53% to 83% in when the Au nanoparticles were deposited onto CB (Fig. 9d). The kinetics of the CO2 reduction were examined using Tafel analysis (Fig. 9f) which shows a decreasing slope from 138 mV dec−1 to 72 mV dec−1.
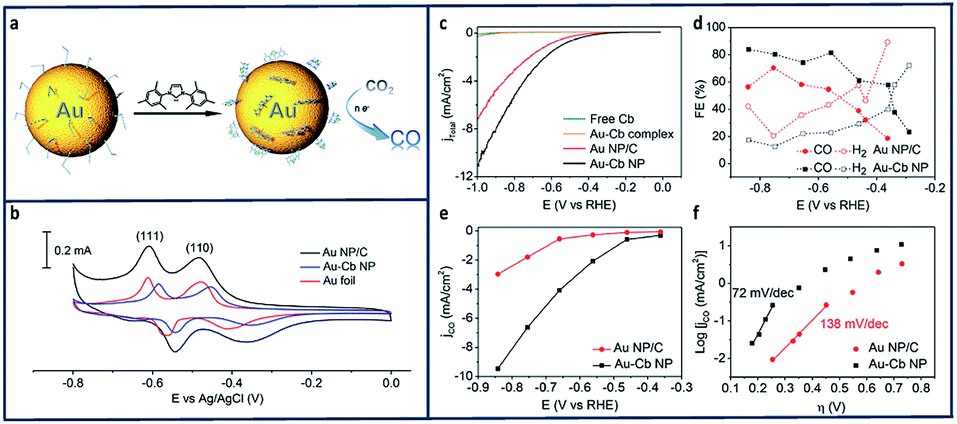 | ||
| Fig. 9 (a) Schematic reduction of CO2 using N-heterocyclic (NHC) carbene-functionalized on a gold electrode. (b) Pb-upd profiles of the Au NP/C, Au–Cb NP, and Au–Cb NP was referenced to the geometric area of the Au foil with scan rate of 50 mV s−1 (c) LSV scans of Au–Cb NP, Au NP/C, free carbene and molecular Au–Cb complex under CO2-saturated 0.1 M KHCO3 at pH 6.8. (d) FEs of reduction product formed from Au–Cb NP and Au NP/C. (e) Specific CO current density (based on electrochemically active surface area) plots for Au–Cb NP and Au NP/C. (f) Tafel plots of Au–Cb NP and Au NP/C.139 Copyright (2016) American Chemical Society. | ||
Other promising active electrocatalytic systems incorporate Ag metal centers or Ag electrodes.47,63,144–147 Compared to Au electrocatalysts, Ag catalyst are cheaper and have comparable activity. Various strategies, such as morphology-nanostructuring have been paired with these electrodes.148,149 Hwang and co-workers100 prepared three different types of Ag nanoparticles with different surface capping agents. These included oleylamine (OLA), having an amine functional group; oleic acid, having a carboxyl functional group; and dodecanethiol (DDT) with a thiol functional group. They discovered that the amine substituent was highly effective in enhancing the electrochemical reduction of CO2 to CO with high selectivity (FE = 94%) at low overpotentials (−0.75 V vs. RHE) due to an exceptional suppression of HER.
Comparing the mass activities of the CO and H2 products in Fig. 10d and e, HER suppression was observed at more negative potentials (lower than −0.9 V vs. RHE). DDT showed the highest CO partial mass activity compared to both OLA and the oleic acid (OA) at −0.4 V to −0.9 V vs. RHE (Fig. 10e). They also compared the immobilization of ethylenediamine (EDA) to cysteamine onto Ag nanoparticles and found that EDA showed a higher selectivity toward CO production due to the presence of the additional amine group.
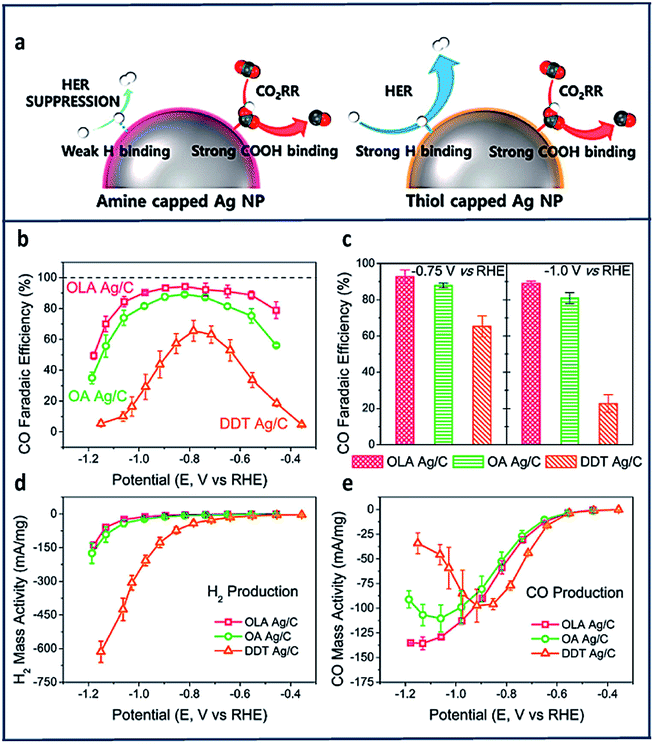 | ||
| Fig. 10 (a) Schematic of CO2RR on amine and thiol-capped Ag nanoparticles. Variation of FECO of OLA, OA and DDT Ag/C with (b) the applied voltage and (c) fixed potential. Mass activity for (d) H2 and (e) CO production of OLA, OA and DDT Ag/C at varied applied potentials.100 Copyright (2017) American Chemical Society. | ||
Carbon-based materials such as CNTs have proven to be a promising conductive solid support for heterogenization of molecular catalysts toward electrochemical CO2RR. This is due to their ability to form a strong noncovalent π–π interactions with aromatic ligands such as pyrene150 and porphyrin.41,151 Hu et al.152 reported reduction of CO2 to CO with an efficiency of over 90% using immobilized cobalt-tetraphenylporphyrins (CoTPPs) onto CNT in aqueous solution. Likewise, previous work by our group demonstrates selective reduction of CO2 to CO with a FE of 90% upon immobilization of iron–porphyrin-dimers onto CNTs.56 This proved to be twice as efficient as when the same catalyst was applied in a homogenous medium.
Similar enhancements to the reduction of CO2 to methane (CH4) and CO with both metalled and non-metallated iron–porphyrin–pyridine (Fe–TPPy) catalysts were seen when non-covalently immobilized onto CNTs.17 Among the synthesized catalysts shown in Fig. 11a, Fe-cis (2b)–pyridine–porphyrin catalysts, exhibited the highest current density (1.32 mA cm−2) and FE (76%) in reducing CO2 to CH4 and CO. Current density and product selectivity were remarkably enhanced to 30 mA cm−2 with the total FE of 92% after immobilization onto CNTs, comparable or higher than that of similarly reported catalysts.
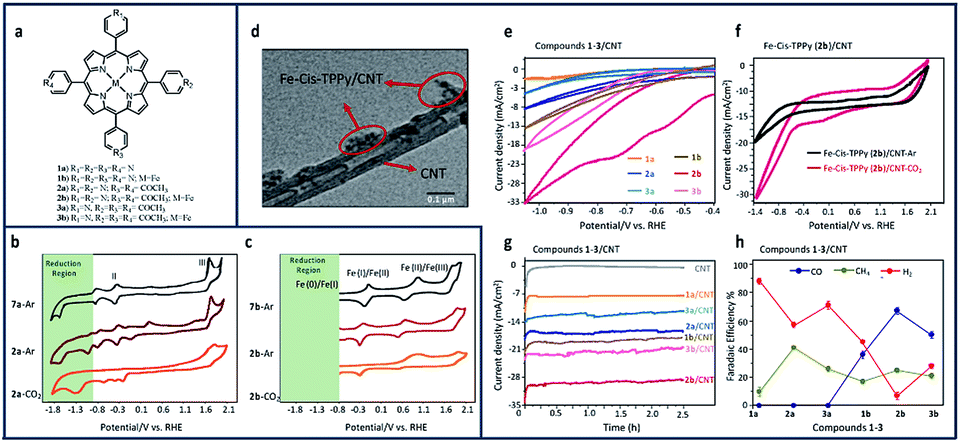 | ||
| Fig. 11 (a) Molecular structure pyridine–porphyrin complexes (b) CV comparison of 0.01 mM of TPP (7a)/GCE under argon, and cis-TPPy (2a)/GCE under argon and CO2; (c) Fe–TPP (7b)/GCE under argon, and Fe–cis–TPPy (2b)/GCE under argon and CO2 in 0.1 M NBu4PF6/DMF solution. Conditions: scan rate: 100 mV s−1; working electrode: glassy carbon; reference electrode: Ag/AgCl; counter electrode: platinum; (d) Transmission electtron microscopy (TEM) image of the porphyrin 2b/CNT with scale bar of 0.1 μm. CV comparison of (e) all compounds 1-3/CNT under CO2; (f) compound 2b/CNT in the presence and absence of CO2; (g) chronoamperometry comparison of 1–3/CNT at −0.6 V vs. RHE; (h) FE comparison of all 1–3/CNT compounds at −0.6 V vs. RHE in 0.1 M aqueous NaHCO3.17 Copyright (2020) American Chemical Society. | ||
Comparing the CV of non-metallated 2a/GCE in Fig. 11b under argon and CO2, an enhancement to the current density can be seen in the CO2 saturated solution stating at ∼−0.8 V vs. RHE. This increase in current density seen after purging 2a/GCE with CO2 demonstrates the important role of pyridine in the capture and electroreduction of CO2 to methane. Metallated isomers increased the number of available capture sites and led to a direct increase in current density for all studied compounds (Fig. 11c). As seen in Fig. 11c, the broad CO2 reduction peak at ∼−1.3 V vs. RHE aligns with the potential range observed for iron-cantered porphyrins.
Another report suggests using polyethylenimine (PEI) (Fig. 12a) will stabilize the electroreduction of CO2 to HCOO− through hydrogen bonding interactions (Fig. 12b).153 As shown in Fig. 12c and d, PEI-NCNT had the highest current density (9 mA cm−2) compared to nitrogen doped carbon nanotubes (NCNT) and bare CNT with a high FE of 87%.
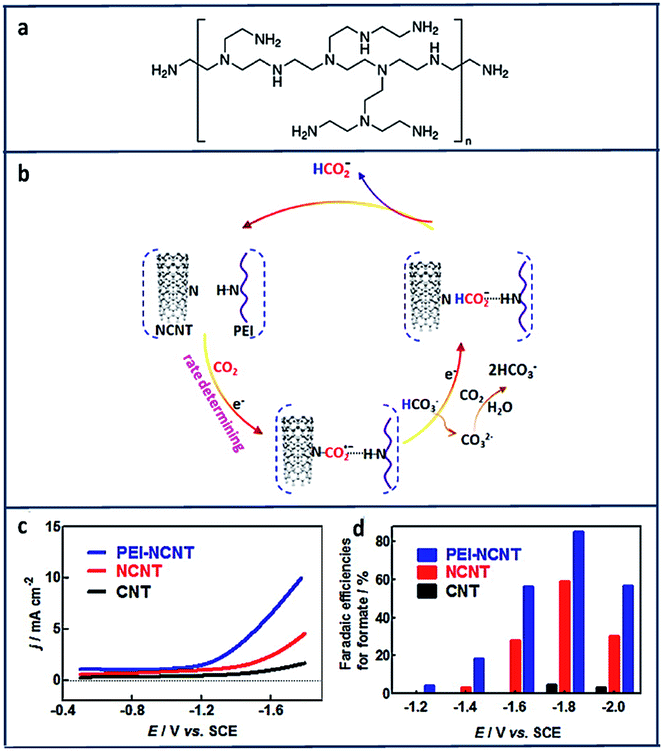 | ||
| Fig. 12 (a) Structure of branched polyethylenimine (PEI). (b) Proposed mechanism for CO2 reduction at PEI Functionalized, nitrogen-doped carbon nanomaterials. (c) cathodic linear sweep voltammetry scans at 50 mV s−1 in a CO2-saturated aqueous 0.1 M KHCO3 solution. (d) Plot of faradaic efficiencies for formate production vs. applied potential at CNT/GC, NCNT/GC, and PEI–NCNT/GC electrodes.153 Copyright (2014) American Chemical Society. | ||
4.3. Enhanced heterogeneous amine molecular catalysts using flow cells
In addition to the aforementioned solid supports and immobilization techniques for heterogeneous molecular catalysis, use of flow cell electrolyzers is another technique that has been proven to enhance overall catalytic performance. This emerging system minimizes the distance between the electrode and the catalytic layer; combining efficient electrode-to-catalyst electron transfers with a continuous, single-pass directional CO2 delivery. These optimizations ultimately result in high energy efficiencies, product selectivities, and a reduction to operational costs.154–160 An additional benefit of flow cell electrolyzers is the translatability of their results to modern industrial practices. Generalized flow cell setups include a gas diffusion layer (GDL) which is directly exposed to the electrolyte solution (Fig. 13).161–163 The catalyst layer is typically deposited directly onto the GDL, allowing for a greater effective catalyst surface area. | ||
| Fig. 13 Schematic of a flow cell.161 Copyright (2020) American Chemical Society. | ||
Recent studies of molecular catalysts operated in flow cells find significant gains to both product selectivity and reaction conversion rate. Cobalt and iron porphyrin and phthalocyanine complexes deposited onto a gas diffusion electrode through non-covalent bonding in a flow cell have been reported to achieve high current densities and selectivities.164,165 An example of immobilized cobalt and iron amino molecular catalysts on carbon paper supports report current densities up to 165 mA cm−2 while maintaining high product selectivity (up to 94%).161,165 These results, confirm the importance of state-of-the-art noble molecular based catalysts for electrochemical CO2RR.
5. Conclusions and future prospects
A wide range of amine-based molecular catalysts has been explored for the electrochemical reduction of CO2 over the years and the contributions of small molecule catalysis to finding insights into the mechanism of electrochemical CO2RR is instrumental to the intelligent design of new catalysts. In particular, the role of amine-based ligands and functional groups were found to play an important role in capturing CO2 itself and being used as covalent linkers for direct immobilization.Although insights into the intricacies of CO2RR have been garnered thanks to thorough studies of immobilization techniques, the influence of metal electrodes, and the role of different metal centers in organometallic compounds, further improvements to catalytic activity and stability are still needed before large-scale application can be realized. As described in this review, noncovalent and covalent immobilization can be achieved through various techniques to positive effect. Expanding on this new approach, many renewed studies on both homogeneous and heterogeneous systems are gaining greater traction with promising bounds being made every year.
Various strategies can be considered to overcome the current limitations in the electrochemical reduction process for CO2 using amine-based molecular catalysts. For homogeneous electrocatalysis; (i) synthesizing small amine molecules that have a high affinity towards CO2 but have a weaker amine–CO2 bond; (ii) developing new nanostructured catalysts with large electrochemically active surface areas to facilitate the reduction process of the amine–CO2 at lower potentials and high catalytic activity and selectivity would be promising next steps. For heterogeneous systems: (i) developing facile synthetic approaches to amine-functionalized MOFs; (ii) preparing high amine content MOFs with improved chemical stability; and (iii) improving immobilization strategies with nanostructured materials instead of the smooth metal surfaces are recommended to achieve higher catalytic performance.
Therefore, further investigations are required to achieve high stability and catalytic activity of the amino electrocatalysts to understand the fundamental kinetics of CO2 reduction, and the effectiveness of the catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Science and Engineering Research Council (NSERC), and the University of Toronto (U of T) for financial support.References
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS.
- M. Ades, R. Adler, R. Allan, R. P. Allan, J. Anderson, A. Argüez, C. Arosio, J. A. Augustine, C. Azorin-Molina, J. Barichivich, J. Barnes, H. E. Beck, A. Becker, N. Bellouin, A. Benedetti, D. I. Berry, S. Blenkinsop, O. Bock, M. G. Bosilovich, O. Boucher, S. A. Buehler, L. Carrea, H. H. Christiansen, F. Chouza, J. R. Christy, E.-S. Chung, M. Coldewey-Egbers, G. P. Compo, O. R. Cooper, C. Covey, A. Crotwell, S. M. Davis, E. de Eyto, R. A. M. de Jeu, B. V. VanderSat, C. L. DeGasperi, D. Degenstein, L. Di Girolamo, M. T. Dokulil, M. G. Donat, W. A. Dorigo, I. Durre, G. S. Dutton, G. Duveiller, J. W. Elkins, V. E. Fioletov, J. Flemming, M. J. Foster, R. A. Frey, S. M. Frith, L. Froidevaux, J. Garforth, S. K. Gupta, L. Haimberger, B. D. Hall, I. Harris, A. K. Heidinger, D. L. Hemming, S. Ho, D. Hubert, D. F. Hurst, I. Hüser, A. Inness, K. Isaksen, V. John, P. D. Jones, J. W. Kaiser, S. Kelly, S. Khaykin, R. Kidd, H. Kim, Z. Kipling, B. M. Kraemer, D. P. Kratz, R. S. La Fuente, X. Lan, K. O. Lantz, T. Leblanc, B. Li, N. G. Loeb, C. S. Long, D. Loyola, W. Marszelewski, B. Martens, L. May, M. Mayer, M. F. McCabe, T. R. McVicar, C. A. Mears, W. P. Menzel, C. J. Merchant, B. R. Miller, D. G. Miralles, S. A. Montzka, C. Morice, J. Mühle, R. Myneni, J. P. Nicolas, J. Noetzli, T. J. Osborn, T. Park, A. Pasik, A. M. Paterson, M. S. Pelto, S. Perkins-Kirkpatrick, G. Pétron, C. Phillips, B. Pinty, S. Po-Chedley, L. Polvani, W. Preimesberger, M. Pulkkanen, W. J. Randel, S. Rémy, L. Ricciardulli, A. D. Richardson, L. Rieger, D. A. Robinson, M. Rodell, K. H. Rosenlof, C. Roth, A. Rozanov, J. A. Rusak, O. Rusanovskaya, T. Rutishäuser, A. Sánchez-Lugo, P. Sawaengphokhai, T. Scanlon, V. Schenzinger, S. G. Schladow, R. W. Schlegel, E. Schmid, Martin, H. B. Selkirk, S. Sharma, L. Shi, S. V. Shimaraeva, E. A. Silow, A. J. Simmons, C. A. Smith, S. L. Smith, B. J. Soden, V. Sofieva, T. H. Sparks, P. W. Stackhouse, W. Steinbrecht, D. A. Streletskiy, G. Taha, H. Telg, S. J. Thackeray, M. A. Timofeyev, K. Tourpali, M. R. Tye, R. J. van der A, V. B. V. van der Schalie, Robin, G. van der Schrier, W. Paul, G. R. van der Werf, P. Verburg, J.-P. Vernier, H. Vömel, R. S. Vose, R. Wang, S. G. Watanabe, M. Weber, G. A. Weyhenmeyer, D. Wiese, A. C. Wilber, J. D. Wild, T. Wong, R. I. Woolway, X. Yin, L. Zhao, G. Zhao, X. Zhou, J. R. Ziemke and M. Ziese, Bull. Am. Meteorol. Soc., 2020, 101, S9–S128 CrossRef.
- P. R. Yaashikaa, P. Senthil Kumar, S. J. Varjani and A. Saravanan, J. CO2 Util., 2019, 33, 131–147 CrossRef CAS.
- C. B. Hiragond, H. Kim, J. Lee, S. Sorcar, C. Erkey and S. In, Catal. Rev., 2020, 10, 1–21 Search PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- M. Papasizza, X. Yang, J. Cheng and A. Cuesta, Curr. Opin. Electrochem., 2020, 23, 80–88 CrossRef.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 RSC.
- Y. Hori, in Modern Aspects of Electrochemistry, Springer New York, New York, NY, 2008, pp. 89–189 Search PubMed.
- A. J. Göttle and M. T. M. Koper, Chem. Sci., 2017, 8, 458–465 RSC.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207–2221 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- M. Abdinejad, C. Dao, B. Deng, F. Dinic, O. Voznyy, X. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 9549–9557 CrossRef CAS.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X. A. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- X. Yang, J. Cheng, B. Fang, X. Xuan, N. Liu, X. Yang and J. Zhou, Nanoscale, 2020, 12, 18437–18445 RSC.
- S. Zhao, S. Li, T. Guo, S. Zhang, J. Wang, Y. Wu and Y. Chen, Nano-Micro Lett., 2019, 11, 62 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- Y. Wu, J. Jiang, Z. Weng, M. Wang, D. L. J. Broere, Y. Zhong, G. W. Brudvig, Z. Feng and H. Wang, ACS Cent. Sci., 2017, 3, 847–852 CrossRef CAS.
- A. A. Al-Omari, Z. H. Yamani and H. L. Nguyen, Molecules, 2018, 23, 2835 CrossRef.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS.
- Y. Hori, Electrochemical CO2 reduction on metal electrodes, Modern aspects of electrochemistry, Springer, 2008, pp. 89–189 Search PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS.
- J. Liu, C. Guo, A. Vasileff and S. Qiao, Small Methods, 2017, 1, 1600006 CrossRef.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Curr. Opin. Electrochem., 2017, 2, 26–31 CrossRef CAS.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- C. L. Yao, J. C. Li, W. Gao and Q. Jiang, Phys. Chem. Chem. Phys., 2017, 19, 15067–15072 RSC.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS.
- Y. Mun, K. Kim, S. Kim, S. Lee, S. Lee, S. Kim, W. Choi, S. Kim, J. W. Han and J. Lee, Appl. Catal., B, 2018, 236, 154–161 CrossRef CAS.
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed. Engl., 2018, 57, 4981–4985 CrossRef CAS.
- D. J. Abdallah and R. G. Weiss, Langmuir, 2000, 16, 352–355 CrossRef CAS.
- A. Thevenon, A. Rosas-Hernández, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 58, 16952–16958 CrossRef CAS.
- Y. Kuwahara, D.-Y. Kang, J. R. Copeland, P. Bollini, C. Sievers, T. Kamegawa, H. Yamashita and C. W. Jones, Chem.–Eur. J., 2012, 18, 16649–16664 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef.
- C. Ferrag, M. Abdinejad and K. Kerman, Can. J. Chem., 2019, 1, 1–8 Search PubMed.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2017, 10, 4109–4118 CrossRef CAS.
- M. Abdinejad, C. Dao, B. Deng, M. E. Sweeney, F. Dielmann, X. Zhang and H. B. Kraatz, ChemistrySelect, 2020, 5, 979–984 CrossRef CAS.
- M. Abdinejad, Z. Mirza, X. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- P. V Danckwerts, Chem. Eng. Sci., 1979, 34, 443–446 CrossRef.
- M. Caplow, J. Am. Chem. Soc., 1968, 90, 6795–6803 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- G. Zhao, X. Huang, X. Wang and X. Wang, J. Mater. Chem. A, 2017, 5, 21625–21649 RSC.
- C. Anex, E. Touzé, L. Curet, F. Gohier and C. Cougnon, ChemElectroChem, 2019, 6, 4963–4969 CrossRef CAS.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS.
- M. Isaacs, F. Armijo, G. Ramírez, E. Trollund, S. R. Biaggio, J. Costamagna and M. J. Aguirre, J. Mol. Catal. A: Chem., 2005, 229, 249–257 CrossRef CAS.
- D. Quezada, J. Honores, M. García, F. Armijo and M. Isaacs, New J. Chem., 2014, 38, 3606–3612 RSC.
- N. Elgrishi, S. Griveau, M. B. Chambers, F. Bedioui and M. Fontecave, Chem. Commun., 2015, 51, 2995–2998 RSC.
- X. Zhou, D. Micheroni, Z. Lin, C. Poon, Z. Li and W. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 4192–4198 CrossRef CAS.
- C. Sun, L. Rotundo, C. Garino, L. Nencini, S. S. Yoon, R. Gobetto and C. Nervi, ChemPhysChem, 2017, 18, 3219–3229 CrossRef CAS.
- L. M. Aeshala and A. Verma, Macromol. Symp., 2015, 357, 79–85 CrossRef CAS.
- J. P. Collin and J. P. Sauvage, Coord. Chem. Rev., 1989, 93, 245–268 CrossRef CAS.
- C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392–1399 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS.
- G. F. Manbeck and E. Fujita, J. Porphyrins Phthalocyanines, 2015, 19, 45–64 CrossRef CAS.
- D. H. Apaydin, S. Schlager, E. Portenkirchner and N. S. Sariciftci, ChemPhysChem, 2017, 3094–3116 CrossRef CAS.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- A. Glüer and S. Schneider, J. Organomet. Chem., 2018, 861, 159–173 CrossRef.
- D. C. Grills, Y. Matsubara, Y. Kuwahara, S. R. Golisz, D. A. Kurtz and B. A. Mello, J. Phys. Chem. Lett., 2014, 5, 2033–2038 CrossRef CAS.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef.
- H. Xie, T. Wang, J. Liang, Q. Li and S. Sun, Nano Today, 2018, 21, 41–54 CrossRef CAS.
- C. Kim, F. Dionigi, V. Beermann, X. Wang, T. Möller and P. Strasser, Adv. Mater., 2019, 31, 1805617 CrossRef.
- D. M. Fernandes, A. F. Peixoto and C. Freire, Dalton Trans., 2019, 48, 13508–13528 RSC.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef.
- A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Chem. Commun., 2018, 54, 11630–11633 RSC.
- H. De Jesús-Cardona, C. del Moral and C. R. Cabrera, J. Electroanal. Chem., 2001, 513, 45–51 CrossRef.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- J. J. Kim, D. P. Summers and K. W. Frese, J. Electroanal. Chem. Interfacial Electrochem., 1988, 245, 223–244 CrossRef CAS.
- H. Sato, Energy Procedia, 2014, 63, 4031–4034 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- T. H. T. Myren, A. M. Lilio, C. G. Huntzinger, J. W. Horstman, T. A. Stinson, T. B. Donadt, C. Moore, B. Lama, H. H. Funke and O. R. Luca, Organometallics, 2019, 38, 1248–1253 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef CAS.
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS.
- N. Zouaoui, B. D. Ossonon, M. Fan, D. Mayilukila, S. Garbarino, G. de Silveira, G. A. Botton, D. Guay and A. C. Tavares, J. Mater. Chem. A, 2019, 7, 11272–11281 RSC.
- A. Grondein and D. Bélanger, Fuel, 2011, 90, 2684–2693 CrossRef CAS.
- L. Ou, W. Long, J. Huang, Y. Chen and J. Jin, RSC Adv., 2017, 7, 11938–11950 RSC.
- K. G. Schmitt and A. A Gewirth, J. Phys. Chem. C, 2014, 118, 17567–17576 CrossRef CAS.
- H. Coskun, A. Aljabour, P. De Luna, D. Farka, T. Greunz, D. Stifter, M. Kus, X. Zheng, M. Liu, A. W. Hassel, W. Schöfberger, E. H. Sargent, N. S. Sariciftci and P. Stadler, Sci. Adv., 2017, 3, e1700686 CrossRef.
- S. Xin, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- J. Li, Y. Kuang, Y. Meng, X. Tian, W.-H. Hung, X. Zhang, A. Li, M. Xu, W. Zhou, C.-S. Ku, C.-Y. Chiang, G. Zhu, J. Guo, X. Sun and H. Dai, J. Am. Chem. Soc., 2020, 142, 7276–7282 CrossRef CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897–898 CrossRef.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef.
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020, 2, 235–247 CrossRef.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS.
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Adv. Mater., 2020, 32, 1908398 CrossRef CAS.
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Energy Fuels, 2006, 20, 409–414 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- X. Lu, Y. Wu, X. Yuan, L. Huang, Z. Wu, J. Xuan, Y. Wang and H. Wang, ACS Energy Lett., 2018, 3, 2527–2532 CrossRef CAS.
- M. N. Hossain, S. Ahmad, I. Santos da Silva and H.-B. Kraatz, Chem.–Eur. J., 2020 DOI:10.1002/chem.202003039.
- C. Sun, R. Gobetto and C. Nervi, New J. Chem., 2016, 40, 5656–5661 RSC.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS.
- X.-M. Hu, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2019, 15, 148–154 CrossRef CAS.
- D. M. Chisholm and J. Scott McIndoe, Dalton Trans., 2008, 3933–3945 RSC.
- B. M. L. Dioos, I. F. J. Vankelecom and P. A. Jacobs, Adv. Synth. Catal., 2006, 348, 1413–1446 CrossRef CAS.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS.
- J. Choi, P. Wagner, R. Jalili, J. Kim, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Adv. Energy Mater., 2018, 8, 1801280 CrossRef.
- R. W. Murray, A. G. Ewing and R. A. Durst, Anal. Chem., 1987, 59, 379A–390A CAS.
- A. N. Marianov and Y. Jiang, Appl. Catal., B, 2019, 244, 881–888 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- C. Yang, S. Li, Z. Zhang, H. Wang, H. Liu, F. Jiao, Z. Guo, X. Zhang and W. Hu, Small, 2020, 16, 2001847 CrossRef CAS.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- D. Bélanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- A. Wang, W. Yu, Z. Huang, F. Zhou, J. Song, Y. Song, L. Long, M. P. Cifuentes, M. G. Humphrey, L. Zhang, J. Shao and C. Zhang, Sci. Rep., 2016, 6, 23325 CrossRef CAS.
- S. Baranton and D. Bélanger, J. Phys. Chem. B, 2005, 109, 24401–24410 CrossRef CAS.
- A. K. D. Dimé, A. Bousfiha and C. H. Devillers, Curr. Opin. Electrochem., 2020, 24, 69–78 CrossRef.
- N. M. Orchanian, L. E. Hong, J. A. Skrainka, J. A. Esterhuizen, D. A. Popov and S. C. Marinescu, ACS Appl. Energy Mater., 2019, 2, 110–123 CrossRef CAS.
- M. Mooste, E. Kibena-Põldsepp, M. Marandi, L. Matisen, V. Sammelselg, F. I. Podvorica and K. Tammeveski, J. Electroanal. Chem., 2018, 817, 89–100 CrossRef CAS.
- J. W. Vickers, D. Alfonso and D. R. Kauffman, Energy Technol., 2017, 5, 775–795 CrossRef CAS.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- J. Tamura, A. Ono, Y. Sugano, C. Huang, H. Nishizawa and S. Mikoshiba, Phys. Chem. Chem. Phys., 2015, 17, 26072–26078 RSC.
- Y. Fang and J. C. Flake, J. Am. Chem. Soc., 2017, 139, 3399–3405 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2017, 139, 9408 CrossRef CAS.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606–4609 CrossRef CAS.
- A. Hamelin, J. Electroanal. Chem. Interfacial Electrochem., 1984, 165, 167–180 CrossRef CAS.
- A. Hamelin and J. Lipkowski, J. Electroanal. Chem. Interfacial Electrochem., 1984, 171, 317–330 CrossRef CAS.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819 RSC.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, X. Ma, M. Kim, S. Adhikari, C. Lu, C. Dun, M. Chi, Y. Qiu and S. M. Geyer, Nat. Commun., 2019, 10, 5724 CrossRef CAS.
- J. Xiao, H. B. Li, W. Q. Zhou, N. Q. Luo, Y. Liang, J. M. Shao, P. Liu, X. Y. Zou, Z. Dai and G. W. Yang, RSC Adv., 2013, 3, 20532–20537 RSC.
- L.-X. Wu, Y.-G. Zhao, Y.-B. Guan, H. Wang, Y.-C. Lan, H. Wang and J.-X. Lu, RSC Adv., 2019, 9, 32628–32633 RSC.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Chem. Commun., 2015, 51, 10226–10228 RSC.
- X. M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS.
- S. Verma, B. Kim, H.-R. “Molly” Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS.
- R. Lin, J. Guo, X. Li, P. Patel and A. Seifitokaldani, Catalysts, 2020, 10, 473 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- M. Abdinejad, C. Dao, X. Zhang and H. Bernhard Kraatz, J. Energy Chem., 2020 DOI:10.1016/j.jechem.2020.09.039.
- B. Endrődi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar and C. Janáky, Prog. Energy Combust. Sci., 2017, 62, 133–154 CrossRef.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS.
- K. Torbensen, C. Han, B. Boudy, N. von Wolff, C. Bertail, W. Braun and M. Robert, Chem.–Eur. J., 2020, 26, 3034–3038 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |