
Tailoring radicals by asymmetric electrochemical catalysis
Qifeng
Lin
ab and
Sanzhong
Luo
 *abc
*abc
aKey Laboratory of Molecular Recognition and Function Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
bUniversity of Chinese Academy of Sciences, Beijing, China
cCenter of Basic Molecular Science, Department of Chemistry, Tsinghua University, Beijing, China. E-mail: luosz@tsinghua.edu.cn; Web: http://www.luoszgroup.com:8080
First published on 20th August 2020
Abstract
Asymmetric catalysis with radicals is challenging due to the fleeting and neutral nature of radical species. We highlight herein asymmetric electrochemical catalysis as a powerful yet exquisite tool in facilitating chiral radical transformations by the works of Lin, Meggers and Guo. The mild and readily tunable features of electrochemical redox processes not only enable mild generations of chiral radical intermediates but also provide exquisite control of both chemo- and stereoselectivity.
 Qifeng Lin | Qifeng Lin received his B.S. degree in bio-tech from Beijing University of Technology, China, in 2017. He is currently a Ph.D. student in organic chemistry at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), under the supervision of Prof. Sanzhong Luo. His research focuses on asymmetric electrochemical catalysis. |
 Sanzhong Luo | Sanzhong Luo received his Ph.D. from the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), in 2005 under the supervision of Prof. Jin-Pei Cheng. Then, he started his in-dependent career at ICCAS, where he became a Full Professor in 2011. In 2009, he was a Visiting Scholar at Stanford University (with B. M. Trost). He moved to Tsinghua University in 2018. The is now Guoqiang Professor and the director of Center of Basic Molecular Science (CBMS), Tsinghua University. His laboratory focuses on asymmetric catalysis, bio-inspired catalysis and the application of machine learning in molecular catalysis and synthesis. |
Radicals are neutral and highly fleeting intermediates and their capture in a temporally and spatially controlled manner has remained a great challenge in asymmetric catalysis. There recently appeared break-throughs in achieving effective asymmetric radical transformations via inner-sphere stereocontrol with chiral radical intermediates (Scheme 1).1a–c In this inner-sphere strategy, the expected radical reaction is initialized with the catalyst-bound radical species (Scheme 1, I and II) or when the in situ generated radical was trapped with chiral metal catalysts to form a chiral intermediate (Scheme 1, III). By doing so, non-controlled radical pathways, normally encountered in out-sphere asymmetric radical transformations, can be largely suppressed, and fine tune and control of stereoselectivity can then be achieved by focusing mainly on the selections of the radical generating or reacting catalytic motif. In addition, the development of new and mild methods in the generation of radicals also underlies this advance. In this regard, both chemical and photochemical means have been demonstrated to be enabling tools and the advent of new approach has been and continues to be the driving force in the evolution of synthetically useful enantioselective radical transformations.1 We highlight herein the recent breakthroughs in the use of novel electrochemical approaches for catalytic asymmetric radical reactions.
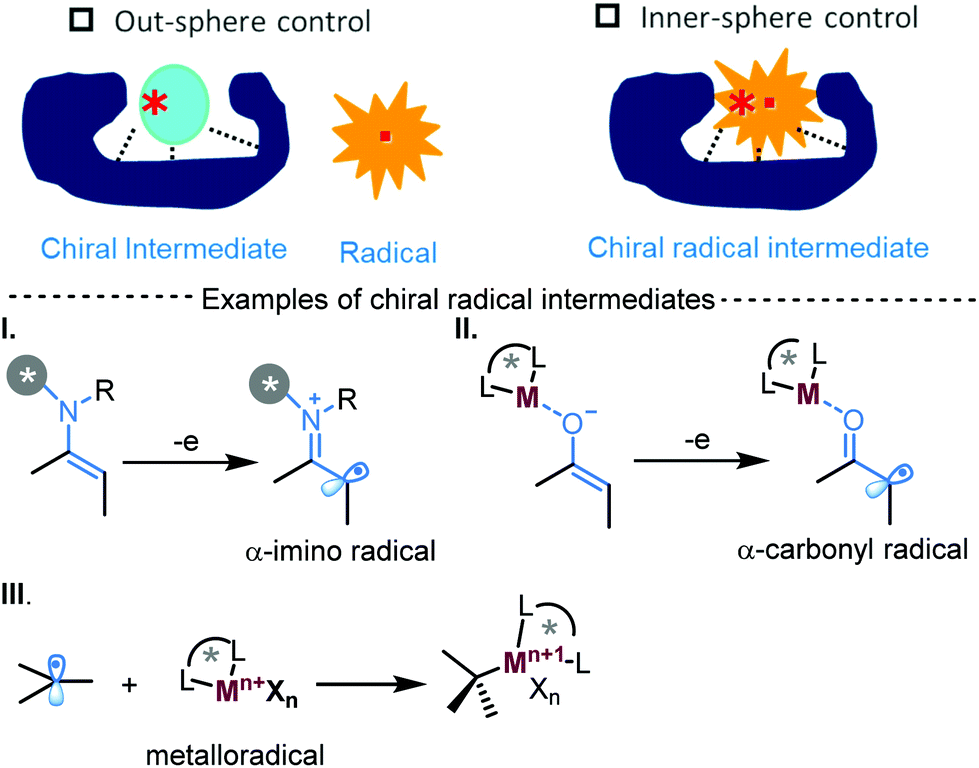 | ||
| Scheme 1 Inner-sphere control in asymmetric radical transformations. | ||
With electron transfer as its core, electrochemical process is intrinsically radical in nature and the early electrode radical reaction can be traced back to 1840s when electrolysis of carboxylic acid, known as Kolbe reaction, was developed.2 The recent reinvigoration of organic electrosynthesis further witness tremendous potential and versatility of electrochemical radical reactions and metal catalysis in organic synthesis.3 However, asymmetric catalysis with electrochemical radicals remains largely underdeveloped despite of the long history of asymmetric electrochemical catalysis.4 An early example to investigate electrochemical SOMO catalysis (with enamine radical cation intermediate) (Scheme 1, I) led to serious reduction in both reactivity and stereoselectivity with the identical chiral aminocatalysts utilized under chemical oxidation conditions, suggesting stereocontrol in an electrochemical setting is substantially different from its chemical counterpart even with the same catalytic system. In addition, there is strong tendency of radical-polar crossover amid the interfacial e-transfer with electrode, adding further challenge in the pursuit of chiral radical transformation. In fact, asymmetric electrochemical catalysis was dominated with 2e-process wherein the initial generated radical species readily undergo further electron-transfer to furnish the ending oxidized/reduced products. Recently, two electrochemical strategies based on α-carbonyl radicals and metalloradicals, have been developed to facilitate enantioselective radical coupling reactions, providing a new paradigm in pursuing catalytic asymmetric radical transformations (see below).
In 2019, Meggers5 and Guo6 independently reported asymmetric electrochemical catalysis with anodic chiral α-carbonyl radicals by chiral Lewis acid complexes (Scheme 1, II and 2). In both works, anodic oxidation was used to generate the key chiral intermediate 2 and 2-acylimidazoles were employed as carbonyl substrates for enantioselective α-alkylation, a difficult yet fundamental transformation in carbonyl chemistry. A bidentate coordination between Lewis acid and acyl imidazole would facilitate the formation of a bidentate coordination between Lewis acid and acyl imidazole would facilitate the formation of a chiral metal enolate 1 in the presence of base, and this enolate intermediate is also more easily oxidized with much lower oxidation potential comparing with its parent carbonyl compounds to form the key chiral radical intermediate 2 (Scheme 2). Meggers used a chiral-at-metal rhodium catalyst (cat-1) to drive the coupling of radical 2 and silyl enol ether of acetophenone with high enantioselectivity. The catalysis could also be applied to the construction of all carbon quaternary stereogenic centers with high enantioselectivity. In Guo's work, a Ni-chiral diamine complex (cat-2) was identified to function similarly and enable the coupling with p-cresols in excellent enantioselectivity via anodic oxidation, but the reaction was limited to those 2,6-disubstituted cresols. In both cases, the metal center performs solely as a Lewis acid promoter to form the oxidation-susceptible metal enolate, not participating directly in electron-transfer with electrode.
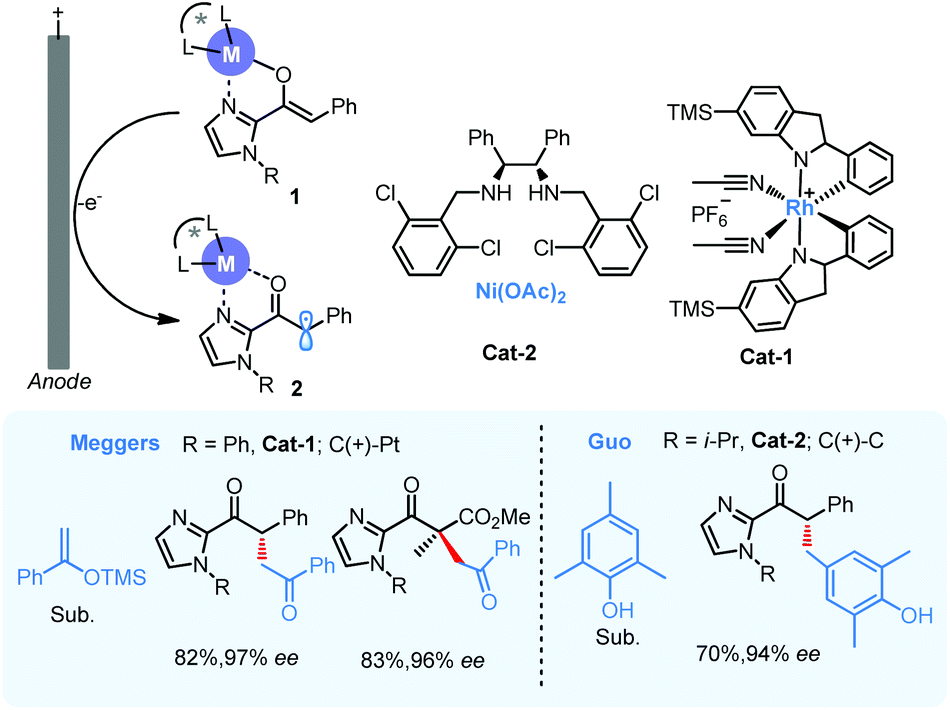 | ||
| Scheme 2 Asymmetric electrochemical catalysis with α-carbonyl radicals 2. | ||
Recently, the group of Lin developed a distinctive electrocatalytic approach with redox metals in asymmetric radical functionalization of alkenes.7 Two concurrent single-electron-transfer cycles between anode and metal were meticulously combined to enable a cascade of in situ formation of a radical intermediate and its eventual trapping with metal for the desired productive pathway (Scheme 3). The success of this cascade anodic radical sequence is built on the authors’ previous development of metal-mediated electrochemical radical reactions8 as well as Liu's pioneering work on copper catalyzed asymmetric radical cyanation reactions.9 Though redox metals as electrocatalysts have been extensively explored in energy chemistry and recently also in metal mediated C–H functionalizations,10 their application in electrochemical radical reactions remains a great challenge particularly in the context of asymmetric catalysis. First of all, redox metals, especially high valent metal complexes, are susceptible to cathodic reduction, causing catalyst deactivation. In addition, the behaviors of radical in the electrode interphase are still poorly understood and the intermolecular trapping of transient radical species could be very difficult as they have strong tendency to undergo radical-polar crossover.
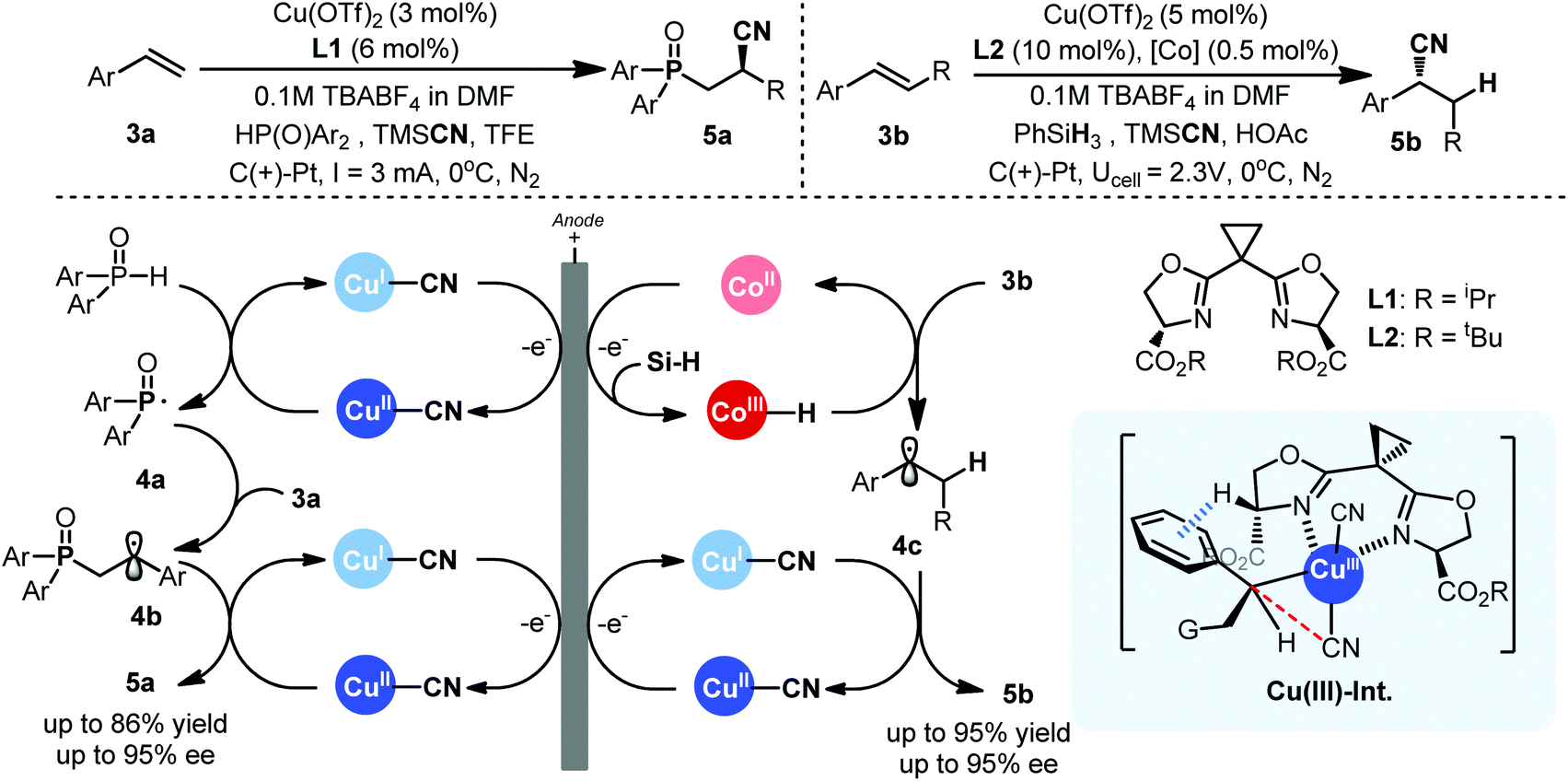 | ||
| Scheme 3 Asymmetric electrochemical functionalization of alkenes catalyzed by metals. | ||
In Lin's radical cyanofunctionalization process, critical to success is the identification of a copper complex as dual catalyst for both radical generation and the following functionalization step (Scheme 3, left). The Cu(II)–cyanide complex, instantaneously formed in situ, not only helped in preventing copper plating on cathode, but also served as a rather mild redox mediator for radical generation. In comparison, the similar chemical process would require t-BuOOH/TMSCN and copper catalyst for the generation of the same phosphinoyl radical.11 Another key factor in Lin's work is the development of new ligands sBOX (L1) that worked more favorably in an electrochemical setting. It should be noted that the chemical process for the same reaction employed a related but structurally different BOX ligand for optimal stereocontrol, further validating that the electrochemical stereocontrol can be very different comparing with its chemical counterpart even for the same process. The Cu(II) complex is also believed to trap the key cascade radical species 4b to give the final cyanophosphoinoyl adduct upon reductive elimination, presumably via a Cu(III)–Int (Scheme 3), with good yields and high enantioselectivity.
Very recently, Lin further advanced their chiral electrochemical catalysis to asymmetric hydrocyanation of alkenes. Enantioselective hydrocyanation of alkenes is a fundamental, yet challenging transformation in asymmetric catalysis with respective to both scope and stereoselectivity.12 Previous examples have mainly utilized Ni–H processes,13 and an enantioselective radical approach was unknown. Lin and coworkers elegantly combined metal–hydride hydrogen-atom transfer catalysis with Co(III)–salen and the copper-radical catalysis with similar sBOX (L2) to achieve asymmetric radical hydrocyanation in an electrochemical setting (Scheme 3, right). Both of the two redox metal cycles are driven by anodic oxidation to diver H˙ and CN˙ radical equivalents, respectively, for addition to double bond and the required electrode potential is as low as 0.2 V. Such a mild condition enables unprecedented scopes to tolerate functional groups that are normally unstable under oxidative stress such as nitrogen heterocycles, electron-rich arenes, sulfide and aldehydes beyond the reach by chemical oxidation approach as demonstrated. The choice of solvent, proton source, and ligand is critical to prevent electroreduction of metal catalysts as well as radical-polar crossover. Again, the use of carboxylate-appended sBOX ligand L2 was found to be essential. The origin of the uniqueness of sBOX ligand remains obscure, and a likely explanation was provided by considering the enhanced acidity of α-carboxlate proton that is favorable for C–H⋯π interaction as revealed by DFT calculations (Scheme 3). From synthetic point of view, Lin's chemistry is still limited to those conjugated alkenes and expanding to include non-conjugated olefins is warranted due to their synthetic versatility.
Conclusions
As illustrated by these studies, electrochemical approach has become a powerful yet exquisite tool in generating catalytic chiral intermediate by anodic oxidation either directly or via a redox mediator and facilitating chiral radical transformations in a highly controlled manner. Beyond their green credentials as sustainable synthetic strategies in terms of redox economy, asymmetric electrochemical catalysis also provides a new twist in achieving the most challenging transformations as shown in Lin's work and the potentials in uncovering new chemistry along this line are enormous. In-depth mechanistic studies in elucidating interphase electron transfer and stereocontrol will certainly aid in its rational development and evolution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science & Technology Fundamental Resource Investigation Program of China (2018FY201200), Tsinghua University Initiative Scientific Research Program (2019Z07L01005) and the Natural Science Foundation of China (21672217, 21572232 and 21933008) for financial support. S. L. is supported by the National Program of Top-notch Young Professionals.References
-
(a) F. Wang, P. Chen and G. Liu, Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed
; (b) Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Copper(I)-Catalyzed Asymmetric Reactions Involving Radicals, Acc. Chem. Res., 2020, 53, 170–181 CrossRef CAS PubMed
-
(a) H. Kolbe, Beobachtungen über die oxydirende Wirkung des Sauerstoffs, wenn derselbe mit Hülfe einer elektrischen Säule entwickelt wird, J. Prakt. Chem., 1847, 41, 137–139 CrossRef
-
(a) Y. Yuan and A. Lei, Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed
-
(a) Q. Lin, L. Li and S. Luo, Asymmetric Electrochemical Catalysis, Chem. – Eur. J., 2019, 25, 10033–10044 CrossRef CAS PubMed
- X. Huang, Q. Zhang, J. Lin, K. Harms and E. Meggers, Electricity-driven asymmetric Lewis acid catalysis, Nat. Catal., 2019, 2, 34–40 CrossRef CAS
- Q. Zhang, X. Chang, L. Peng and C. Guo, Asymmetric Lewis Acid Catalyzed Electrochemical Alkylation, Angew. Chem., Int. Ed., 2019, 58, 6999–7003 CrossRef CAS PubMed
- N. Fu, L. Song, J. Liu, Y. Shen, J. C. Siu and S. Lin, New Bisoxazoline Ligands Enable Enantioselective Electrocatalytic Cyanofunctionalization of Vinylarenes, J. Am. Chem. Soc., 2019, 141, 14480–14485 CrossRef CAS PubMed
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Metal-catalyzed electrochemical diazidation of alkenes, Science, 2017, 357, 575 CrossRef CAS PubMed
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed
-
(a) L. Ackermann, Metalla-electrocatalyzed C–H Activation by Earth-Abundant 3d Metals and Beyond, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed
- G. Zhang, L. Fu, P. Chen, J. Zou and G. Liu, Proton-Coupled Electron Transfer Enables Tandem Radical Relay for Asymmetric Copper-Catalyzed Phosphinoylcyanation of Styrenes, Org. Lett., 2019, 21, 5015–5020 CrossRef CAS PubMed
- L. Song, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. DiStasio and S. Lin, Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes, Nat. Chem., 2020, 12, 747–754 CrossRef PubMed
- H. Zhang, X. Su and K. Dong, Recent progress in transition-metal-catalyzed hydrocyanation of nonpolar alkenes and alkynes, Org. Biomol. Chem., 2020, 18, 391–399 RSC
| This journal is © the Partner Organisations 2020 |