DOI:
10.1039/D0QO00685H
(Review Article)
Org. Chem. Front., 2020,
7, 2576-2597
Advances in the catalytic asymmetric synthesis of quaternary carbon containing cyclobutanes
Received
8th June 2020
, Accepted 20th July 2020
First published on 20th July 2020
Abstract
Chiral cyclobutanes with quaternary stereogenic centers are motifs frequently found in various natural products and bioactive compounds. In addition, they are also useful intermediates for chemical synthesis, as they could undergo ring-expansion or ring-cleavage reactions to deliver various cyclic and acyclic chiral molecules. Therefore, the development of efficient catalytic methods for the highly enantioselective construction of chiral quaternary carbon containing cyclobutanes is of great synthetic value and has broad application prospects. The current review provides a summarization on the advances in catalytic asymmetric synthesis of quaternary carbon containing cyclobutanes.
1. Introduction
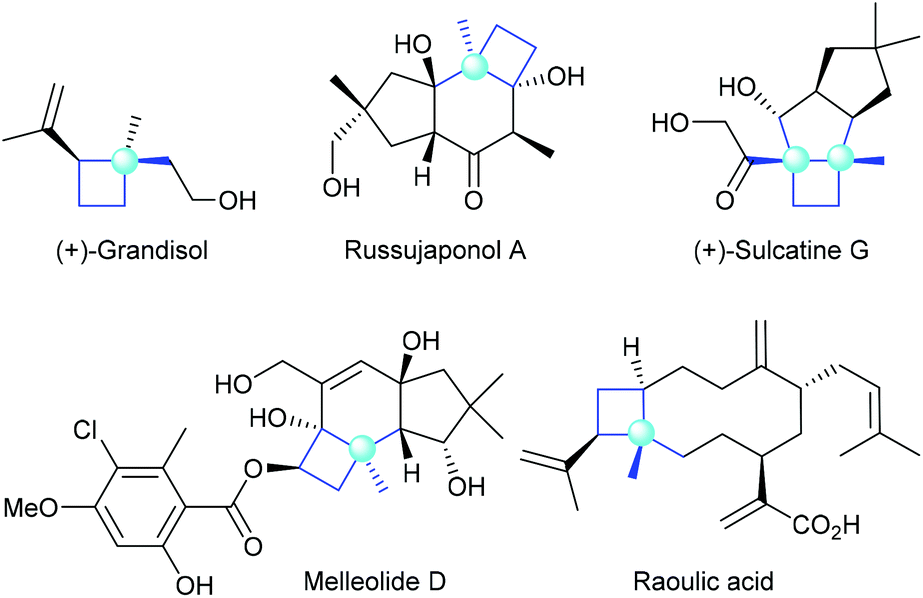
Chiral cyclobutanes with quaternary stereogenic carbon centers are important structural motifs frequently found in various bioactive natural products (Fig. 1).1 Due to their structural rigidity and defined spatial arrangement of their substituents, a feature that is often favorable in drug discovery, cyclobutanes bearing quaternary centers are identified as privileged structural motifs in structure-based drug design.2 In addition, quaternary carbon containing cyclobutanes are useful intermediates for chemical synthesis, as they could undergo ring-expansion or ring-cleavage reactions to deliver a variety of cyclic or acyclic chiral molecules bearing quaternary carbon centers.3 Therefore, the development of catalytic methods for the highly enantioselective construction of chiral quaternary carbon containing cyclobutanes is of great synthetic value and has broad application prospects.
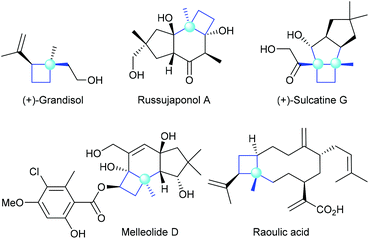 |
| | Fig. 1 Selected natural products bearing quaternary carbon containing cyclobutanes. | |
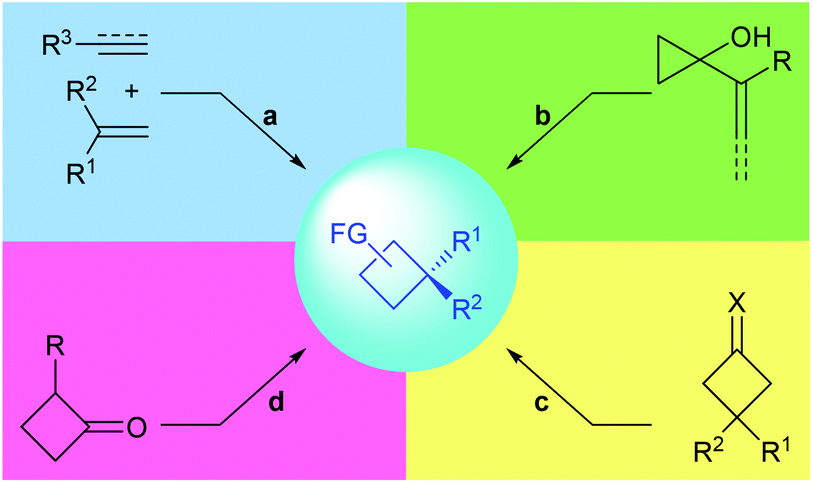
In this context, the last decades have witnessed rapid progress in this area and many novel synthetic methods have been reported. These methods could be classified into four major categories (Fig. 2): (a) asymmetric [2 + 2] cycloaddition reaction of 1,1-disubstituted alkenes with another multiply carbon–carbon bond; (b) enantioselective ring-expansion rearrangement of prochiral cyclopropanol derivatives; (c) catalytic desymmetrization of prochiral substrates, such as cyclobutanes and cyclobutanones; and (d) enantioselective alkylation of cyclobutenolate.
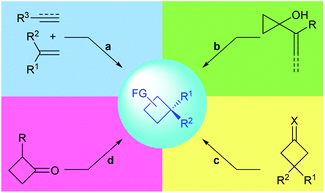 |
| | Fig. 2 Synthetic strategies to quaternary carbon containing cyclobutanes. | |
To date, the enantioselective construction of chiral 4-membered carbon ring has been the topic of many nice reviews.4 Early in 2003, Lee-Ruf et al. introduced the synthesis of enantiomerically pure cyclobutane derivatives and their use in organic synthesis.3a The groups of Butenschön, Brown and Bach summarized the application of [2 + 2] cycloaddition reactions in the synthesis of cyclobutanes in 2008, 2015 and 2016, respectively.4a–c Secci and co-workers reviewed the stereocontrolled synthesis and functionalization of cyclobutanes and cyclobutanones.4d Lu and Ding reviewed the recent advances in the total synthesis of cyclobutane-containing natural products in 2018 and 2020, respectively.1 Very interestingly however, and to the best of our knowledge, no survey focusing on the construction of quaternary carbon containing cyclobutanes has been documented. In view of this, the current review aims to give a summary of the advances in the catalytic asymmetric synthesis of quaternary carbon containing cyclobutanes and is organized according to the above-mentioned four major strategies (Fig. 2).
2. Asymmetric [2 + 2] cycloaddition
Catalytic asymmetric [2 + 2] cycloadditions are the most extensively studied and widely used strategies in the synthesis of cyclobutanes and their derivatives, as they could deliver cyclobutane scaffolds from simple and readily available alkene and alkyne starting materials and are completely atom-economical. As for the construction of quaternary carbon containing cyclobutanes, the reaction requires the use of 1,1-disubstituted alkenes. Since both of the substituents on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 1,1-disubstituted alkenes are non-hydrogen substituents, the steric difference between these two substituents is relatively small, thus making stereoselectivity control more challenging. Though challenging, the last decade still witnessed the development of a series of highly enantioselective methods to construct quaternary carbon containing cyclobutanes.
C bond of 1,1-disubstituted alkenes are non-hydrogen substituents, the steric difference between these two substituents is relatively small, thus making stereoselectivity control more challenging. Though challenging, the last decade still witnessed the development of a series of highly enantioselective methods to construct quaternary carbon containing cyclobutanes.
2.1. Asymmetric Lewis acid catalysis
2.1.1. Sigma Lewis acid catalysis.
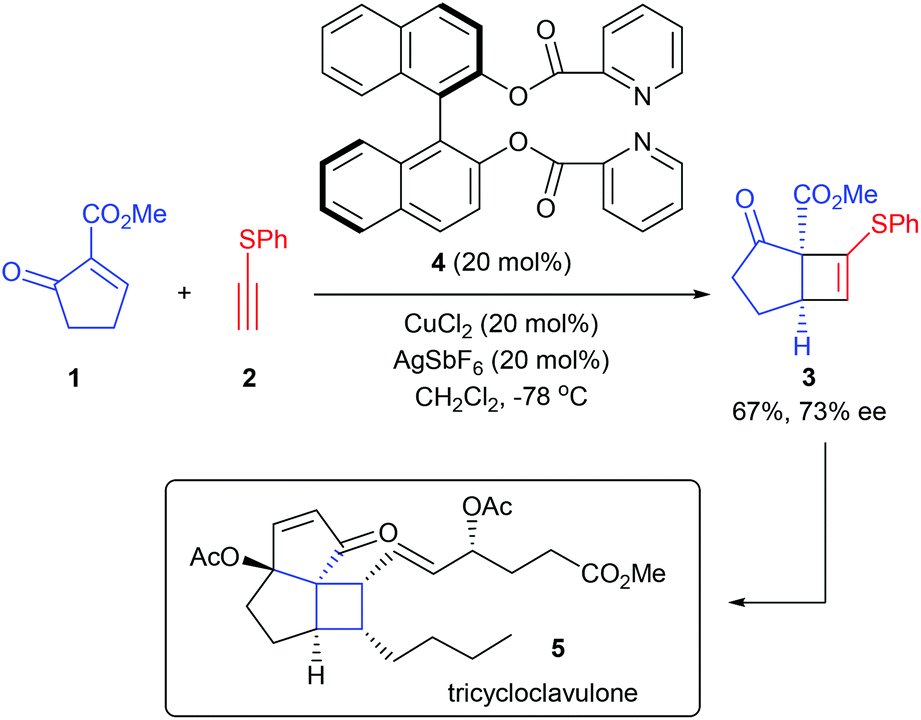
Early in 2004, in their efforts to achieve the total synthesis of (+)-tricycloclavulone (5), Iguchi and co-workers disclosed a catalytic enantioselective [2 + 2]-cycloaddition reaction of 2-methoxycarbonyl-2-cyclopenten-1-one 1 with thioacetylene 2 for the construction of chiral bicyclic systems containing chiral quaternary carbon centers (Scheme 1).5a,b In the presence of 20 mol% Cu(II) complex derived from ligand 4, which was easily synthesized from BINOL and picolinic acid, the desired cycloaddition product 3 could be obtained in up to 67% yield and with 73% ee value at −78 °C. It should be mentioned that this newly developed catalyst is more effective than the known chiral catalysts, such as Ti-TADDOL and Cu-BOX. In a following report from the same group, this reaction was further utilized for the enantioselective formal synthesis of (+)-precapnelladiene.5c
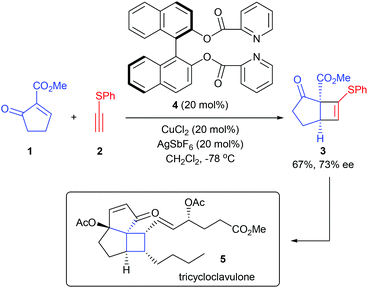 |
| | Scheme 1 [2 + 2]-Cycloaddition reaction of cyclopenten-1-one with thioacetylene. | |
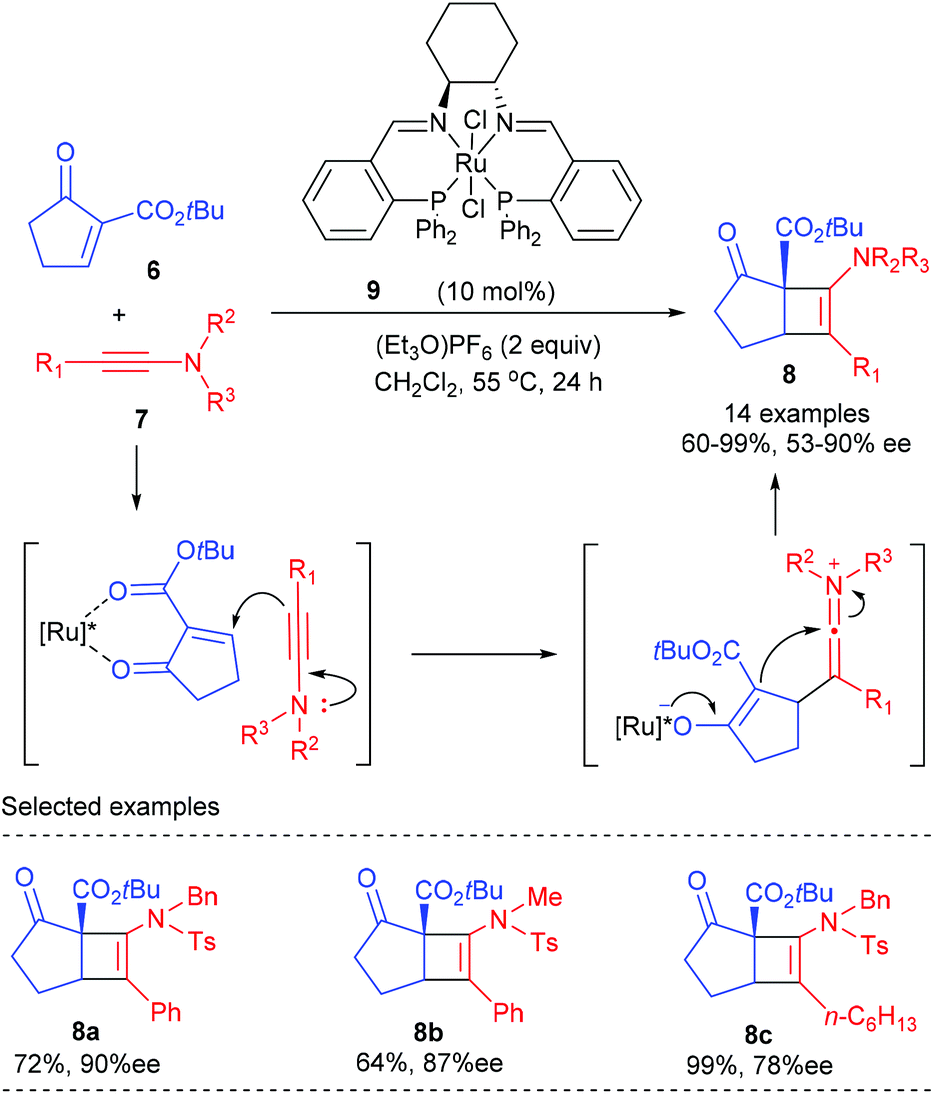
In 2011, the Mezzetti group reported the first enantioselective Ficini reaction to construct amidocyclobutenes bearing quaternary chiral centers (Scheme 2).6 In the presence of 10 mol% dicationic ruthenium/PNNP complex 9 as the catalyst, together with 2.0 equivalents of (Et3O)PF6 as the activator, the [2 + 2] reaction of α-methylene β-keto ester 6 with a number of dienes 7 proceeded smoothly to deliver the desired amidocyclobutenes in 64–99% yield and with 53–90% ee values. The reaction showed a good functional group tolerance. By and large, bulky substituents at the α position of ynamides are required for high enantioselectivity, and good electron donating substituents afforded higher yields (8c). Although the reaction is formally a [2 + 2] cycloaddition process, it is believed to occur stepwise by nucleophilic attack of the β-carbon atom of the ynamide onto the electrophilic position of the enone, which was originally suggested by Ficini for enamines.
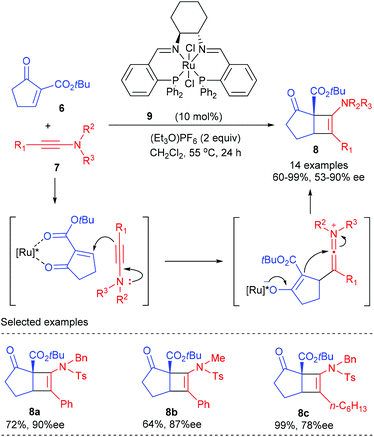 |
| | Scheme 2 Ruthenium catalyzed [2 + 2] cycloaddition of ynamides with cyclic enones. | |
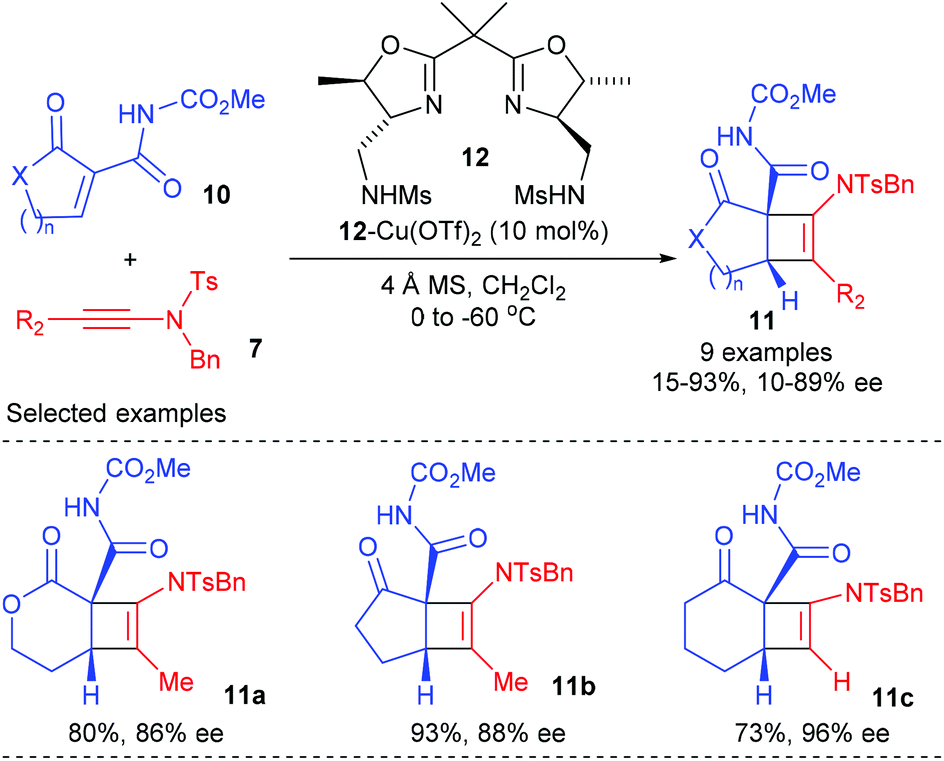
Later in 2015, the Nakada group extended this ynamide participated catalytic enantioselective [2 + 2] cycloadditions to cyclic α-alkylidene-β-oxo imides (Scheme 3).7 Under the catalysis of 10 mol% bisoxazoline ligand 12 derived Cu(II) complex, the reaction of terminal ynamide and α-methyl ynamide with various cyclic α-alkylidene β-oxo imides 10 could give the desired bicyclic products 11a–c with good to high yields and ee values at low temperature, whereas the reaction of α-phenyl ynamide resulted in significantly decreased yield and ee values, likely because of the steric effect of the substituents at the alkyne terminal. The current catalytic asymmetric [2 + 2] cycloaddition is highly useful for natural product synthesis, as the imide group in the resulting products could be successfully transformed into amide, nitrile, and ester groups.
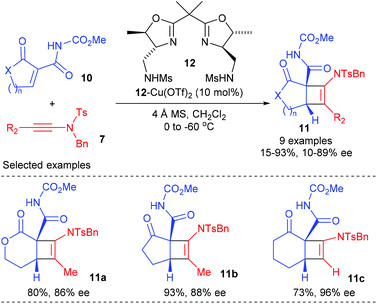 |
| | Scheme 3 [2 + 2]-Cycloadditions of cyclic α-alkylidene β-oxo imides with ynamides. | |
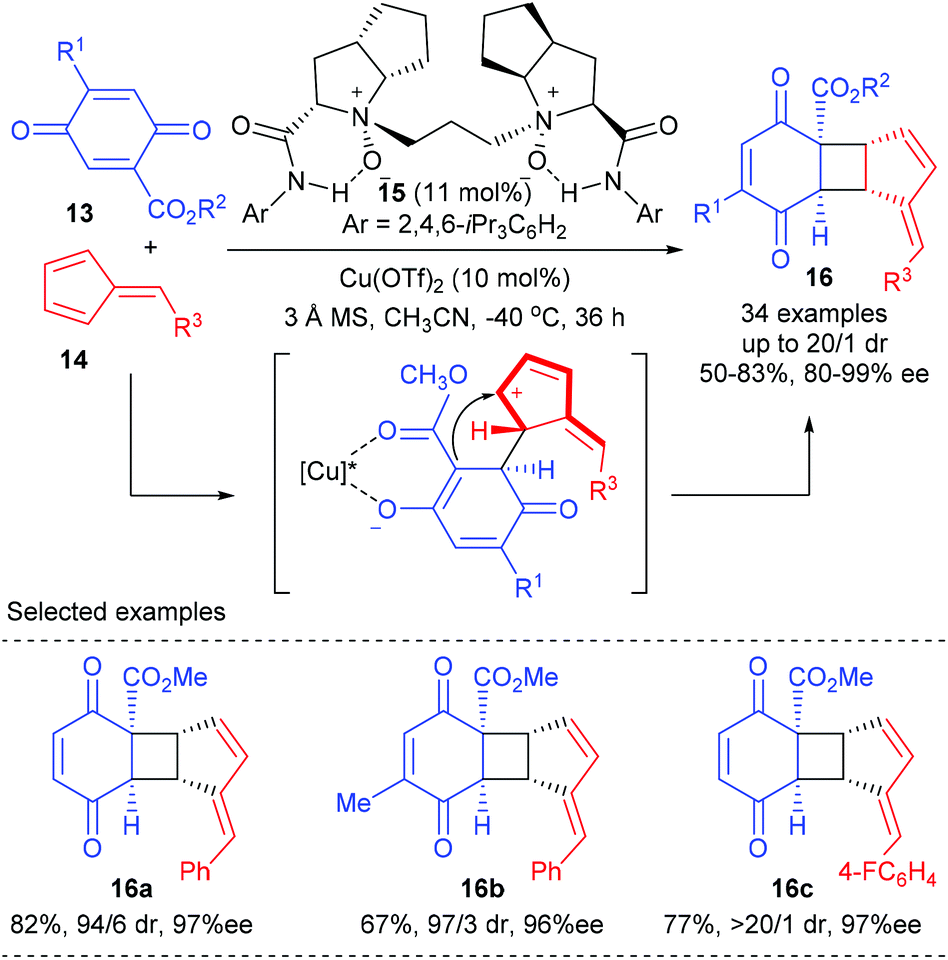
Feng and co-workers achieved the first catalytic asymmetric [2 + 2] cycloaddition between quinones and fulvenes using their chiral N,N′-dioxide ligand derived Cu(II) complex in 2017 (Scheme 4).8 After efforts toward optimization of the chiral catalysts, ligand 15 was identified as the optimal choice, with which the targeted [6,4,5]-tricyclic cyclobutane derivatives 16 could be obtained with good yields and excellent regio- and stereoselectivities, whereas the use of a BOX ligand led to substantial polycondensation of quinine and trace adducts were obtained. In addition, the thus obtained cyclobutane 16 can be easily converted into cyclopenta[b]benzofuran structures diastereo- and enantioselectively under the catalysis of In(OTf)3.
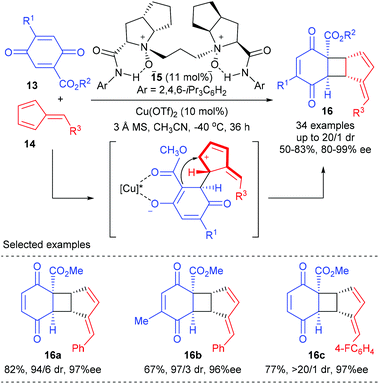 |
| | Scheme 4 Asymmetric [2 + 2] cycloaddition between quinones and fulvenes. | |
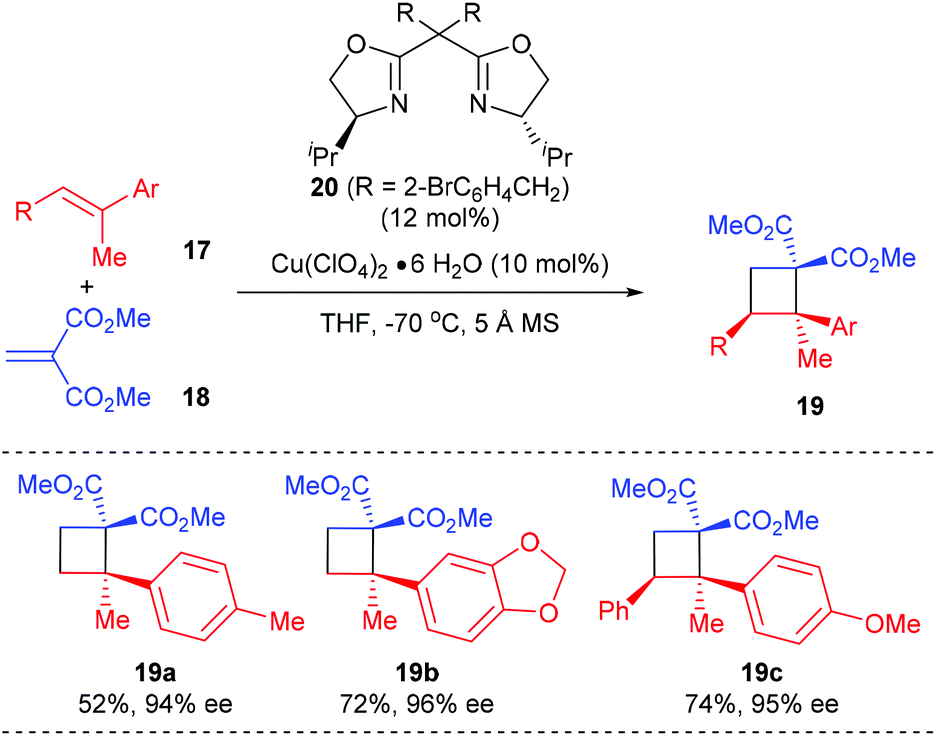
Besides using electron deficient cyclic alkenes as the π-components, the Tang group reported the first asymmetric [2 + 2] cycloaddition of dimethyl methylidenemalonate with polysubstituted olefins for the synthesis of chiral quaternary carbon containing cyclobutanes, using their SaBOX (side-arm-modified BOX) 20 derived Cu(II) complex as the catalyst (Scheme 5).9 Under optimized conditions, the reaction of acyclic methylidenemalonate 18 with 1,1-disubstituted alkenes 17a andb afforded quaternary carbon containing cyclobutanes 19a andb in 52–72% yields and with 94–95% ee values. Moreover, trisubstituted alkene 17c was also a compatible substrate for this reaction, giving product 19c in 74% yield and with excellent enantioselectivity.
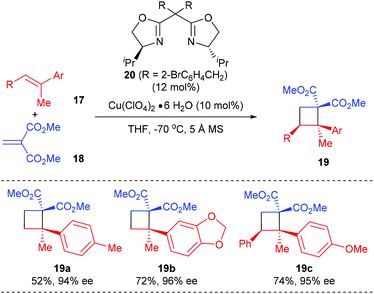 |
| | Scheme 5 Cu(II)/SaBOX-catalyzed [2 + 2] reaction of alkenes with methylidenemalonate. | |
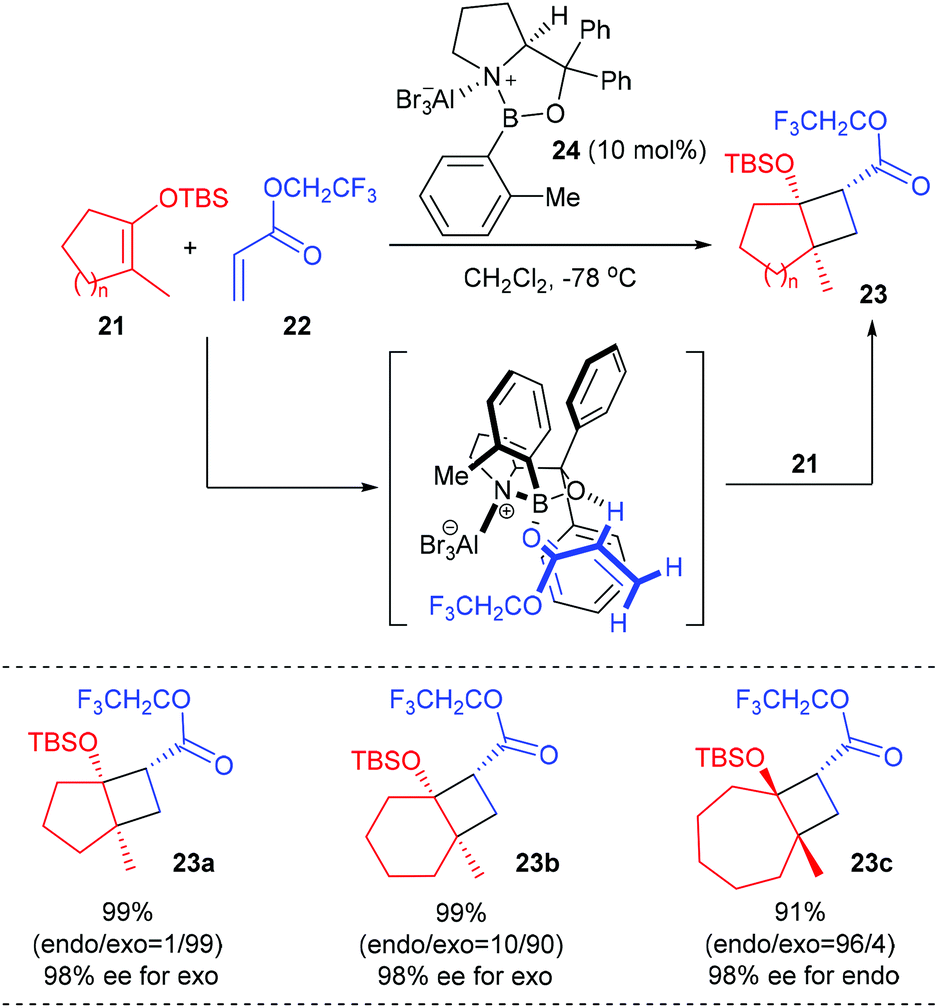
Based on their finding that the oxazaborolidine–AlBr3 complex 24 is an exceedingly effective chiral catalyst for cycloaddition reactions of achiral components, Corey and co-workers developed a new and useful methodology for the synthesis of chiral cyclobutanes bearing a quaternary carbon stereocenter starting from vinyloxysilanes and α,β-unsaturated esters (Scheme 6).10 In the presence of the chiral aluminum bromide complex 24 (10 mol%) as the Lewis acid catalyst, a series of cyclic TBS enolate silyl ethers 21 could react with trifluoroethyl acrylate 22 to give [2 + 2]-cycloaddition products 23a–c, which are very useful chiral intermediates for further synthetic elaboration, with up to 99% yields and 98% ee values at −78 °C. It is hypothesized that the [2 + 2] cycloaddition occurs by an asynchronous process involving the α-C–H hydrogen-bonded complex of catalyst 24 and trifluoroethyl acrylate.
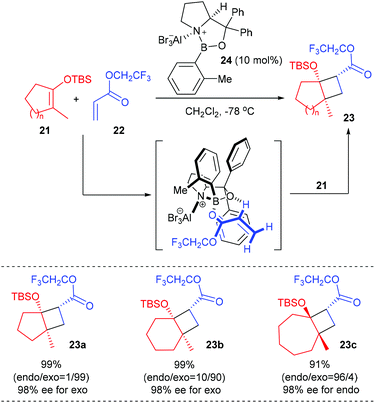 |
| | Scheme 6 Chiral AlBr3 complex catalyzed enantioselective [2 + 2]-cycloaddition reactions. | |
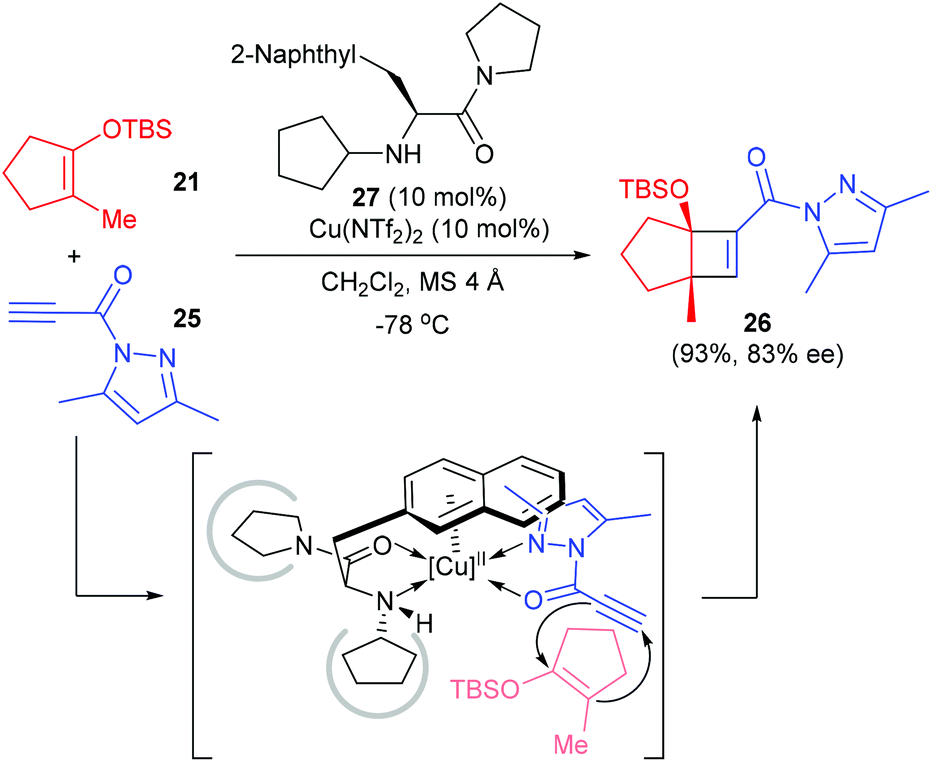
Besides electron deficient alkenes, the electron deficient alkynes are also viable counterparts for enantioselective [2 + 2] cycloaddition reactions with cyclic enolate silyl ethers. Early in 2008, Ishihara and co-workers reported a catalytic and highly enantioselective [2 + 2] cycloaddition reaction of cyclic enolate silyl ethers with electron-deficient propiolamide derivatives 25 (Scheme 7).11 Under the catalysis of the 3-(2-naphthyl)-L-alanine amide 27 derived Cu(II) complex (10 mol%), 25 could react with 2-methyl 1-silyloxy-1-cyclopentenes 21 to give quaternary cyclobutenecarboxamide 26 in 93% yield and with 83% ee value, but the scope of this reaction is yet to be explored.
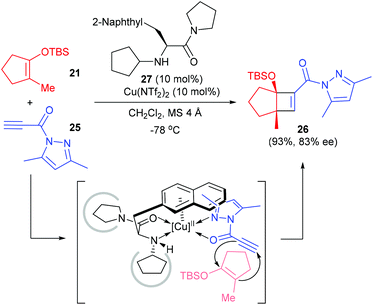 |
| | Scheme 7 [2 + 2] Cycloaddition of cyclic enolate silyl ether with propiolamide derivatives. | |
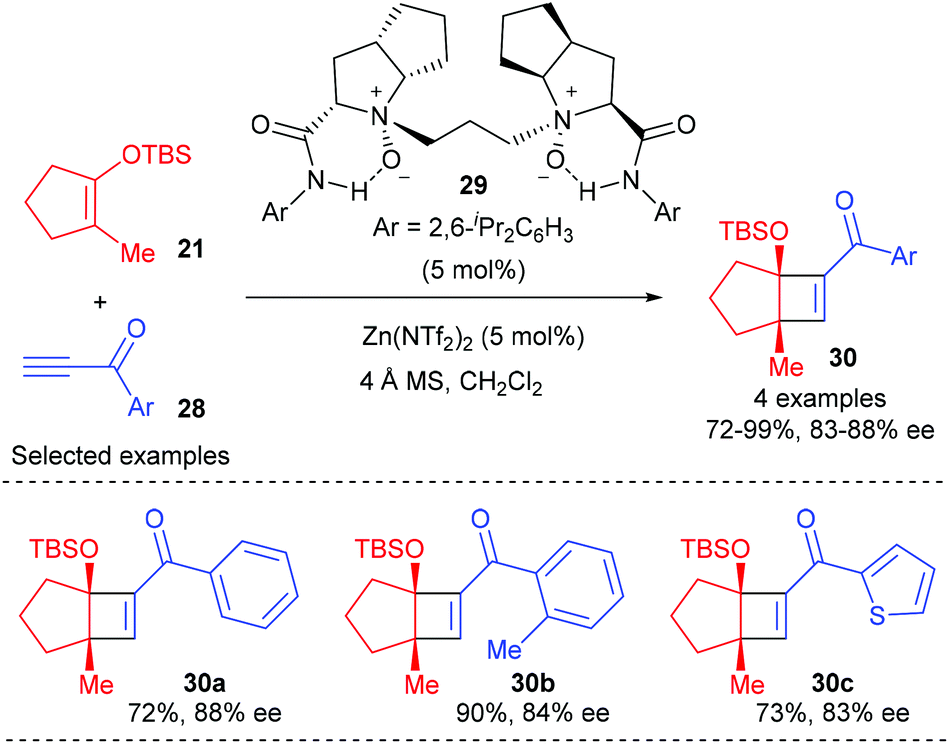
More recently, Feng and co-workers have achieved a highly efficient enantioselective [2 + 2] cycloaddition between alkynones and cyclic enol silyl ethers using a chiral N,N′-dioxide–zinc(II) complex as the catalyst.12 A variety of terminal alkynes and internal alkynes were well tolerated to react with cyclic enol silyl ethers to give good to excellent enantioselectivity. Although the authors focused their attention mainly on 1,2-disubstituted cyclic enol silyl ethers, the reaction of β-methyl substituted enol silyl ether 21 with a range of terminal alkynones (28) with different substituted aryl groups was also explored, affording cyclobutenes (30) with two contiguous stereocenters in 72–99% yields and with 83–88% ee values (Scheme 8).
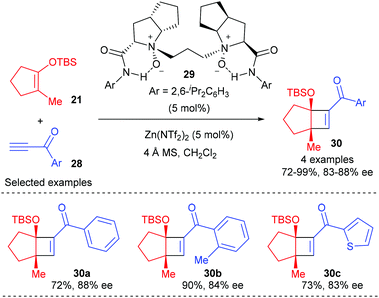 |
| | Scheme 8 Enantioselective [2 + 2] cycloaddition between alkynones and cyclic enol silyl ethers. | |
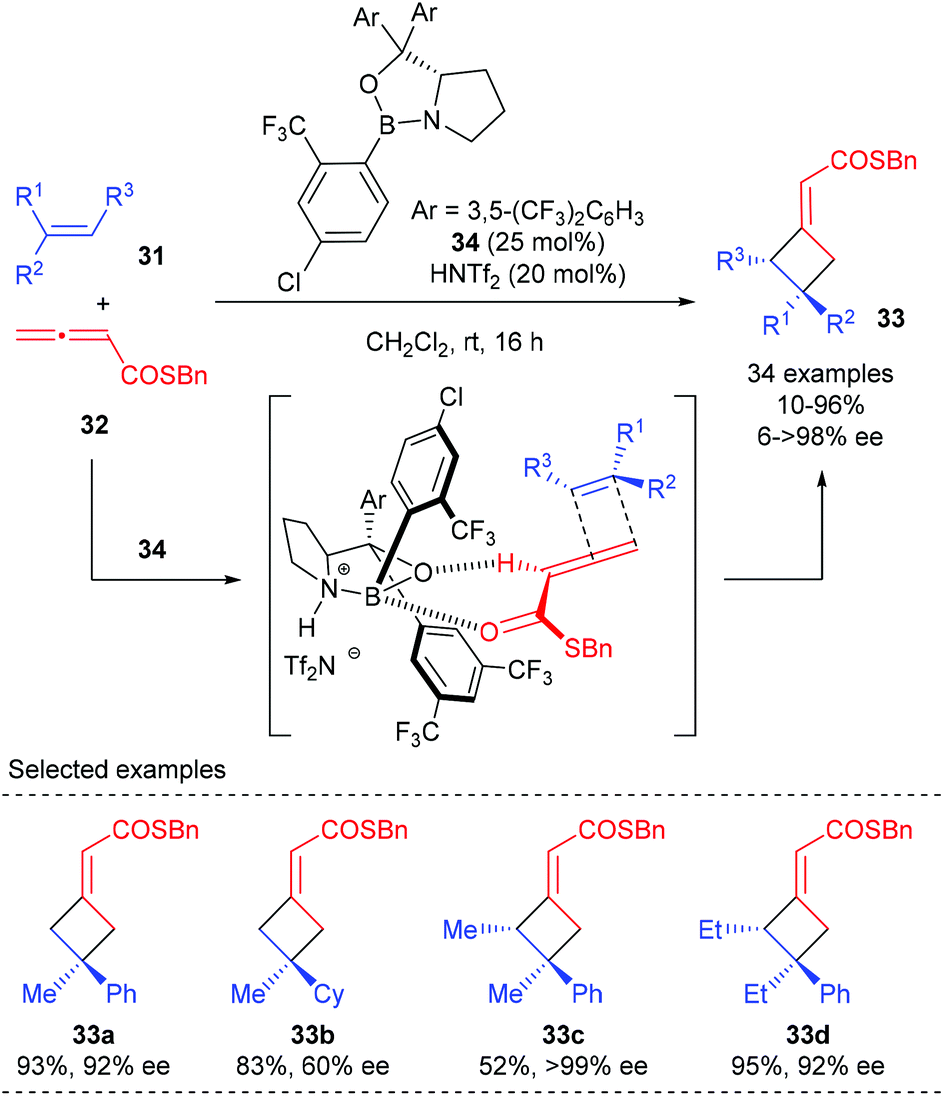
All the reactions discussed above involve the Lewis acid activation of unsaturated carbonyl compounds and cycloaddition via a stepwise process. The Brown group has recently disclosed a catalytic enantioselective [2 + 2] cycloaddition of allenoates and alkenes that is proposed to proceed via a concerted mechanism (Scheme 9).13 Using Brønsted acid HNTf2 as the activator for chiral oxazaborolidine 34, the [2 + 2] cycloaddition reaction of α-alkyl styrenes 31 with allenoates 32 could give the corresponding products (33a) with high enantioselectivity. Replacement of the aryl group with a cyclohexyl group resulted in a substantial decrease in the ee value (33b), demonstrating that the aryl group is necessary to obtain high ee value. Acyclic trisubstituted alkenes are also compatible substrates to give cyclobutanes 33c andd bearing adjacent stereocenters in a highly stereospecific and enantioselective fashion.
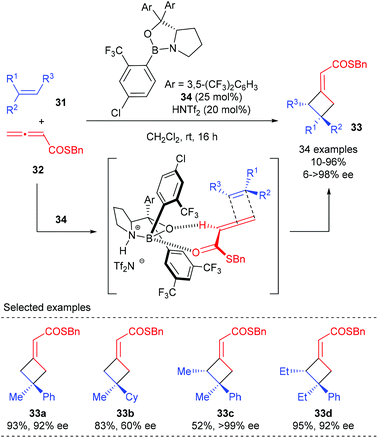 |
| | Scheme 9 Enantioselective [2 + 2] cycloaddition of alkene with allenoates. | |
2.1.2. π-Philic Lewis acid catalysis.
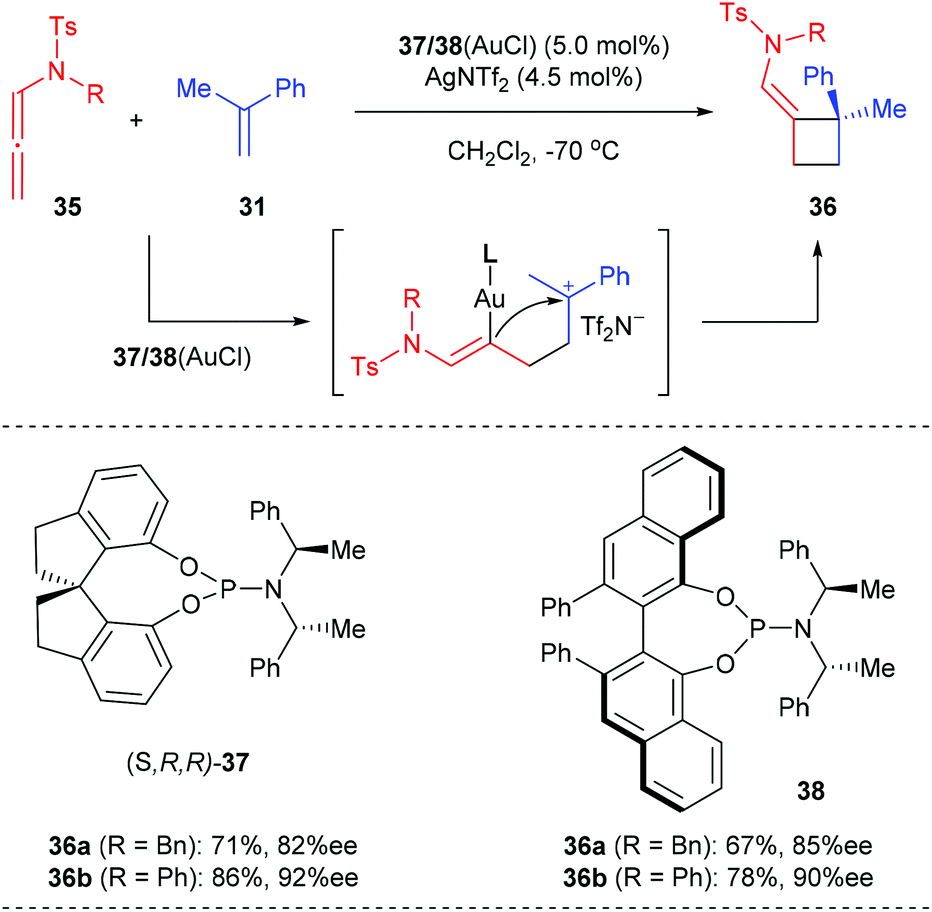
González et al. reported the first asymmetric gold-catalyzed intermolecular [2 + 2] cycloaddition of readily available sulfonylallenamides and vinylarenes to provide a straightforward entry into optically active cyclobutanes in 2012 (Scheme 10).14 After several trials, they found that when ligands 37/38 derived from (R,R)-bis(1-phenylethylamine) and (S)-1,1′-spirobiindane-7,7′-diol or (R)-VANOL were used, the reaction of N-benzyl sulfonylallenamides 35a with α-methylstyrene 31 could afford the desired chiral cyclobutane 36a with 82–85% ee values, whereas the reaction of N-phenyl sulfonylallenamides 35b with 31 generally afforded cycloadduct 36b with higher ee values (90–92%). This reaction is believed to proceed via a stepwise pathway including the activation of the allene by the gold(I) catalyst to facilitate a subsequent attack on the olefin followed by the evolution of the cationic vinyl gold intermediate to give the cyclobutane product. The current reaction also represents one of the first uses of an allene in an intermolecular gold catalyzed process involving enantioselective carbon–carbon bond formation.
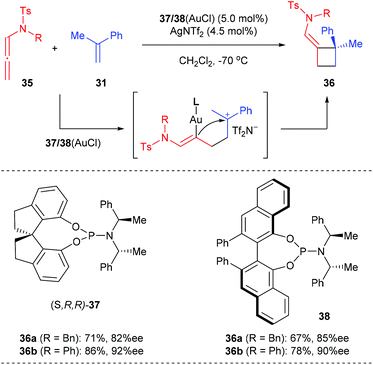 |
| | Scheme 10 Au(I)-Catalyzed intermolecular [2 + 2] cycloaddition. | |
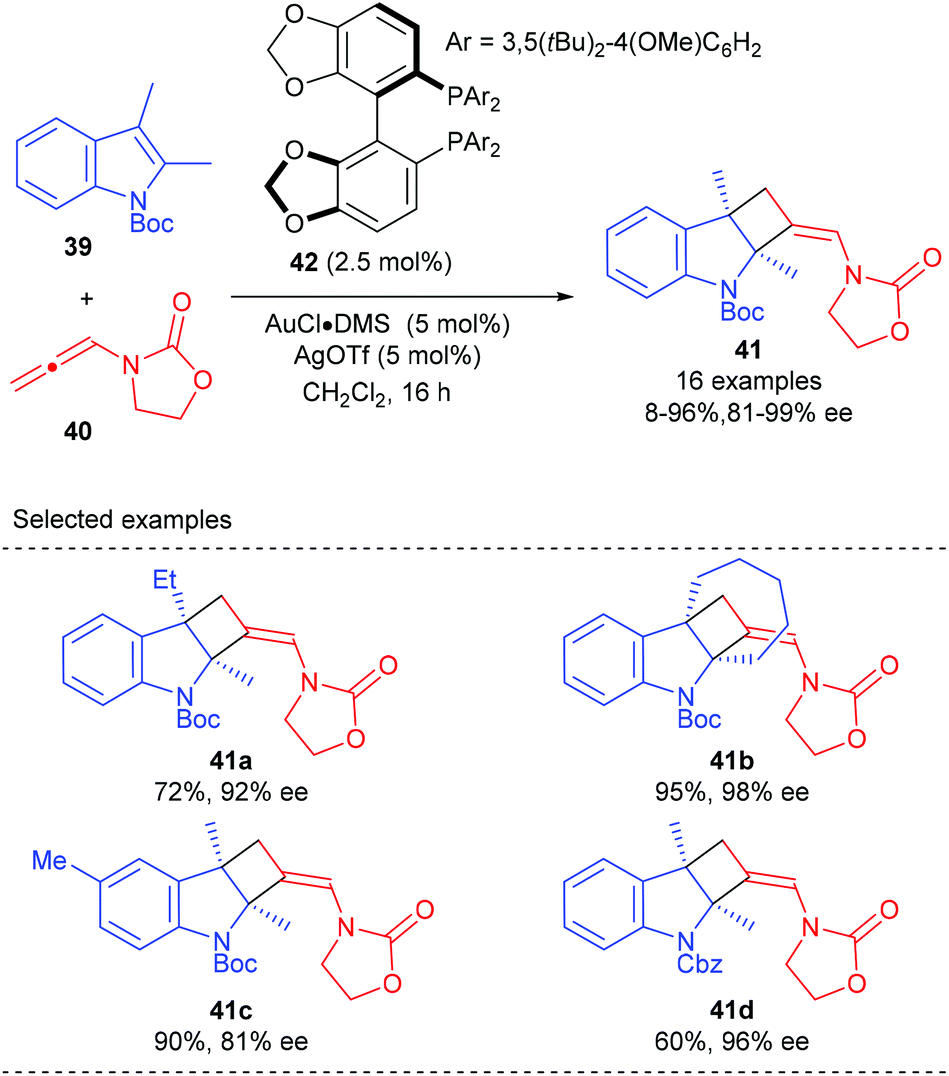
As a continuation of their research interest on the [Au(I)]-mediated functionalization of indoles, the Bandini group reported the first enantioselective gold catalyzed dearomative [2 + 2] cycloaddition between indoles and allenamide for the construction of densely functionalized 2,3-indoline-cyclobutanes (Scheme 11).15 The merger of commercially available (R)-DTBM-segphos (42) and AuCl·DMS was identified as the best catalyst, in the presence of which a series of methylenecyclobutane-fused indolines, featuring two consecutive quaternary carbon centers, could be accessed with excellent stereochemical control (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and up to 99% ee).
:1 dr and up to 99% ee).
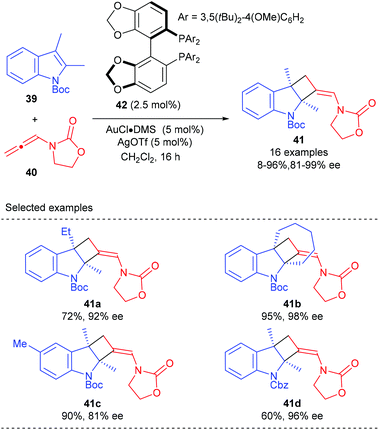 |
| | Scheme 11 Enantioselective [2 + 2] cycloaddition reaction between indoles and allenamides. | |
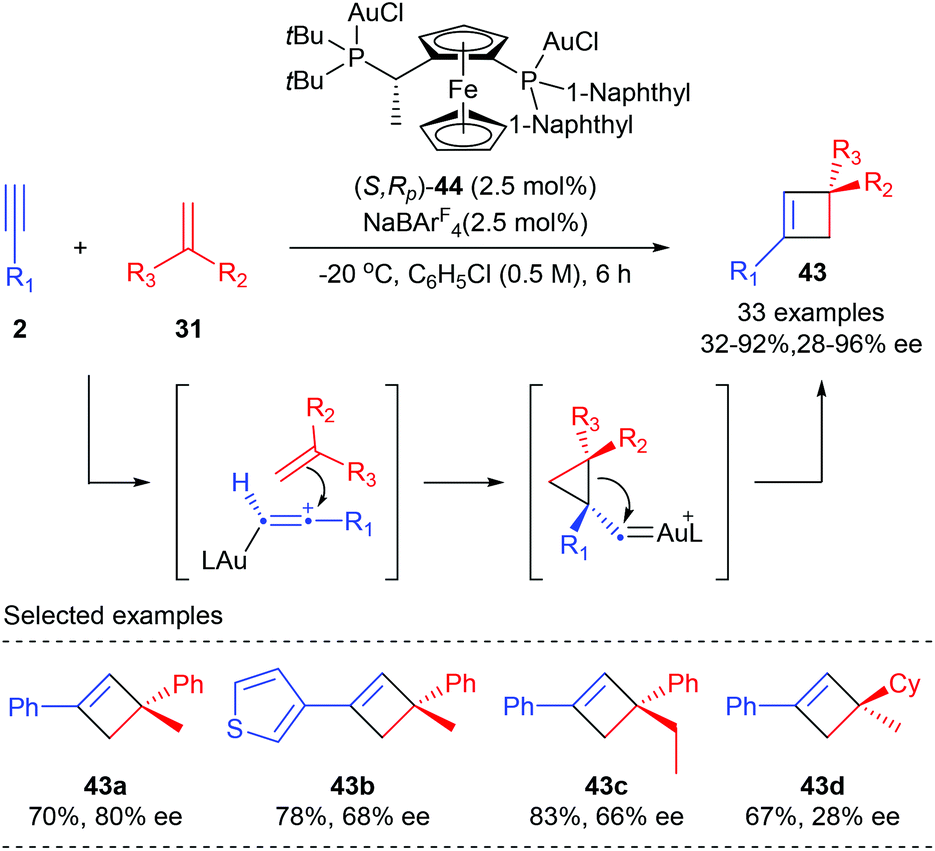
Unlike the above examples using activated alkenes and alkynes, Echavarren et al. have recently reported the first general enantioselective intermolecular [2 + 2] cycloaddition of terminal alkynes with alkenes for the synthesis of cyclobutanes (Scheme 12).16 After evaluation of about 90 chiral ligands, the chiral non-C2 symmetrical Josiphos derived digold(I) complex (S,RP)-44 turned out to be the optimal choice. In the presence of only 2.5 mol% 44, together with 2.5 mol% NaBArF4 as the chloride scavenger, the reaction of terminal aromatic alkynes with various 1,1-disubstituted alkenes could afford quaternary carbon containing cyclobutanes 43 with moderate to high yields and enantioselectivities. Generally, satisfactory results could be expected from aryl alkynes bearing electron rich substituents. Good ee values were obtained with α-alkyl styrenes. However, 1,1-dialkyl substituted alkenes or simple styrenes resulted in a significant loss of enantioselectivity (43d).
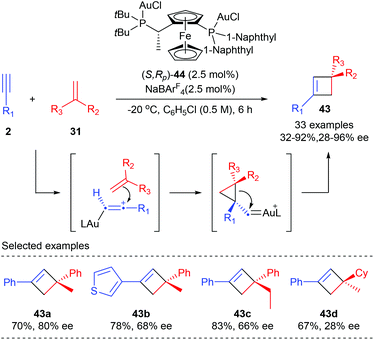 |
| | Scheme 12 Gold-(I)-catalyzed [2 + 2] cycloaddition of terminal alkynes with alkenes. | |
2.2. Asymmetric organocatalysis
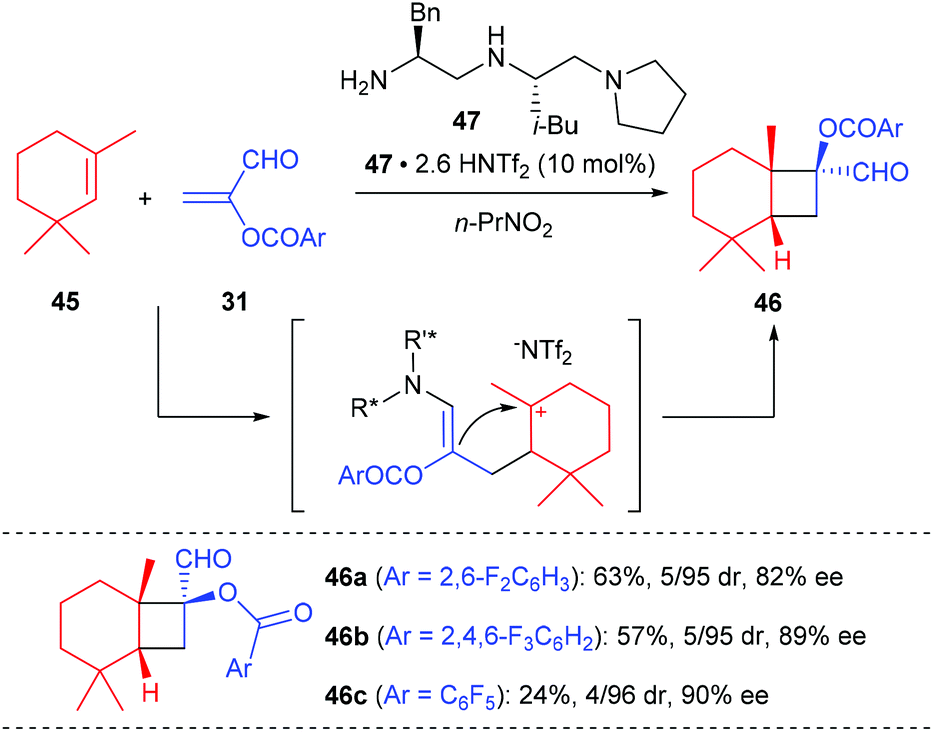
The first organocatalytic enantioselective [2 + 2] cycloaddition reaction of unactivated alkenes with α-acyloxyacroleins was reported by Ishihara and Nakano in 2007 (Scheme 13).17 This reaction was proposed to proceed via a stepwise enantioselective Michael addition of alkenes to a (Z)-iminium intermediate and intramolecular cyclization of a carbocation intermediate. Under optimized conditions, using the organosalt of a chiral secondary amine 47 (10 mol%) as the catalyst, the reaction of α-(fluorobenzoyloxy)acroleins 31 with 1,3,3-trimethylhexene 45 gave bicyclic cyclobutanecarbaldehydes 46 in moderate to good yields and with high ee values. In addition, acyclic alkenes, such as α-methylstyrene, are also compatible substrates for this reaction, giving the desired products with slightly lower enantioselectivities.
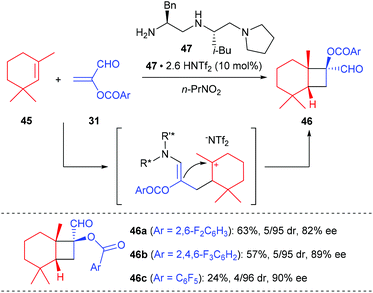 |
| | Scheme 13 First organocatalytic enantioselective [2 + 2] cycloaddition reaction. | |
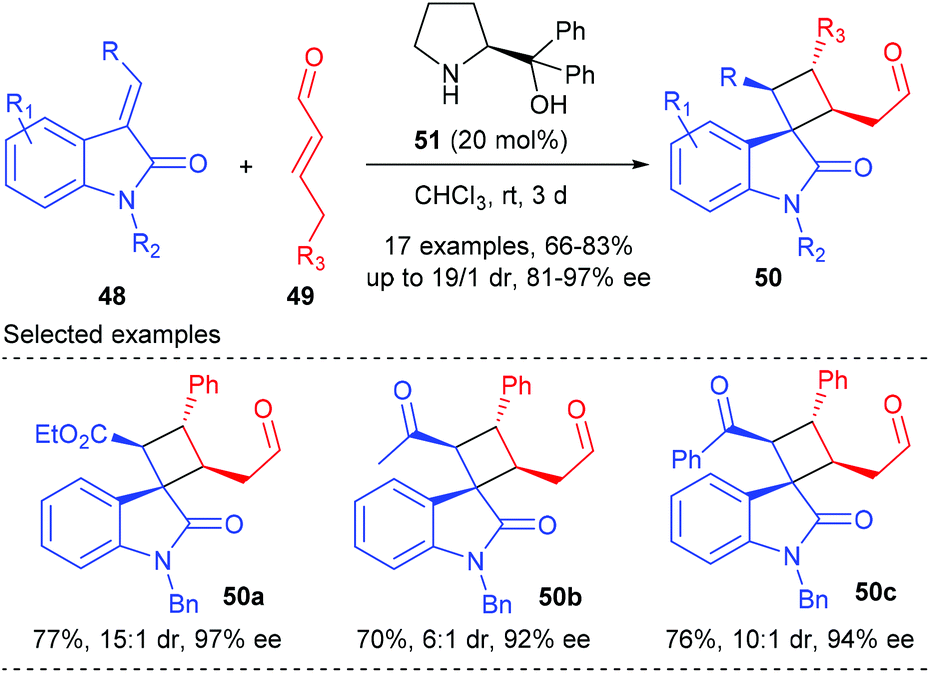
After Ishihara's pioneering work, the groups of Jørgensen, Vicario and Xu also reported their efforts in organocatalytic [2 + 2] cycloaddition reactions, but not for the construction of chiral quaternary carbon containing cyclobutanes.18 Later in 2014, Wang and co-workers achieved the first organocatalytic formal [2 + 2] cycloaddition reaction of ethyleneindolinones with α,β-unsaturated aldehydes via the H-bond-directing dienamine activation strategy (Scheme 14).19 In the presence of α,α-diphenylprolinol 51 (20 mol%) as the catalyst, the reaction could afford the structurally complex spirocyclobutyloxindole products 50 in good yields and with up to >19:1 dr and 97% ee values at room temperature. A variety of substituents on both the oxindole benzene ring and the nitrogen atom were well tolerated. Furthermore, when the ester group was replaced by an acetyl or a benzoyl group, the reactions also proceeded smoothly with excellent stereocontrol to give products 50b andc.
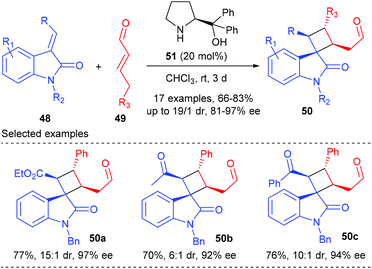 |
| | Scheme 14 Organocatalytic [2 + 2] cycloaddition for the synthesis of spirocyclobutyloxindole. | |
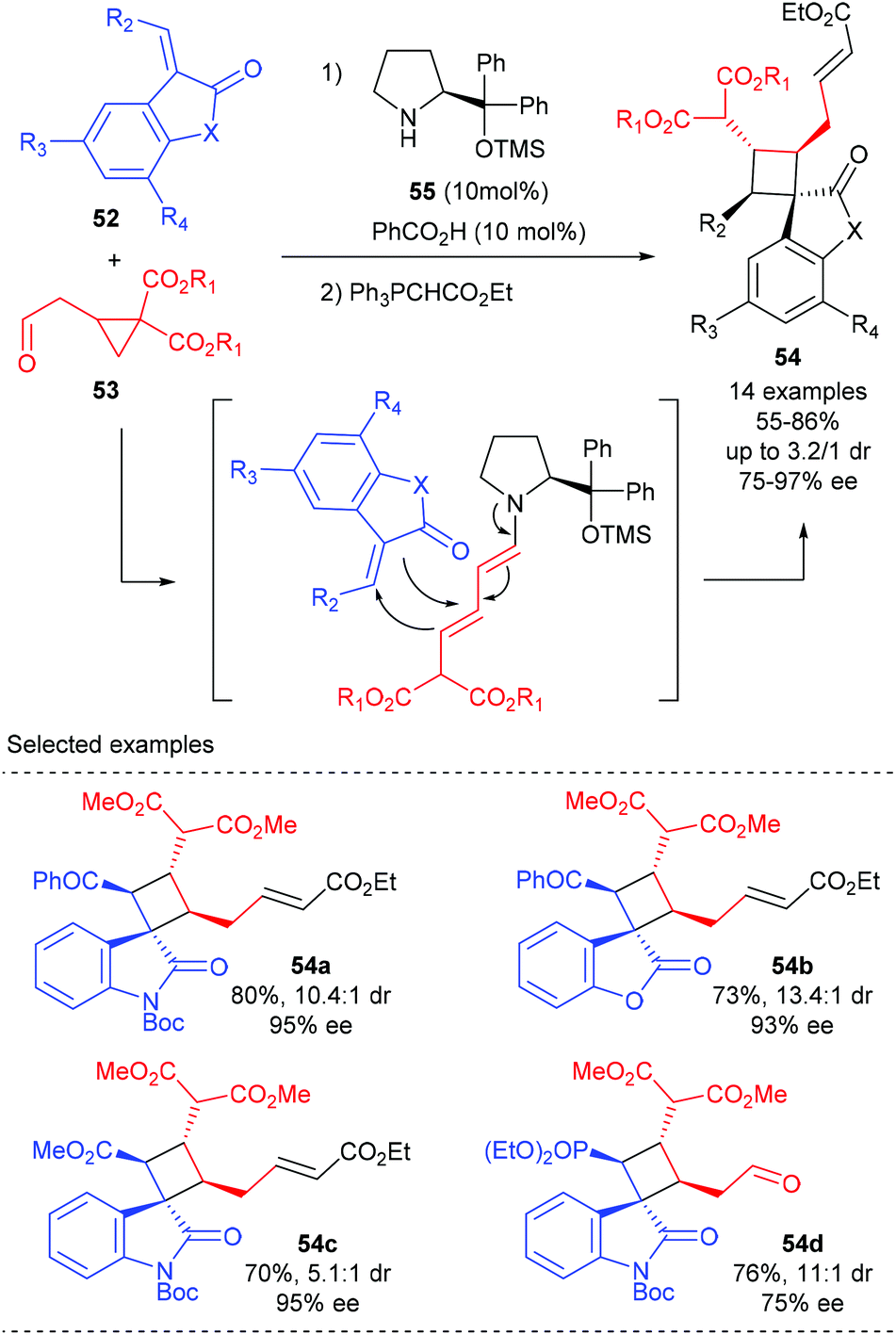
Jørgensen and co-workers described a novel organocatalytic activation mode of cyclopropane and its application in the synthesis of spirocyclobutane derivatives (Scheme 15).20 It is proposed that the treatment of cyclopropylacetaldehyde 53 with aminocatalyst 55 could lead to the ring-opening of cyclopropyl functionality and form a dienamine intermediate, which could then react in a formal [2 + 2]-cycloaddition with benzoyl 3-olefinic oxindoles and benzofuranone to form the desired products 54a andb in good yields and with high dr and ee values. Oxindoles carrying an ester or a phosphonate on the olefinic moiety are also well tolerated, albeit gave diminished diastereoselectivity (54c) or enantioselectivity (54d), respectively.
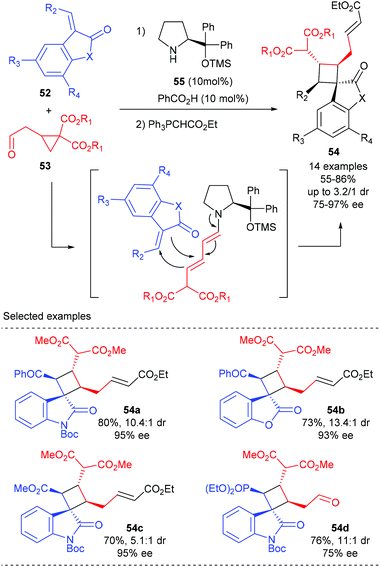 |
| | Scheme 15 Enamine-activation of cyclopropanes for highly stereoselective synthesis of cyclobutanes. | |
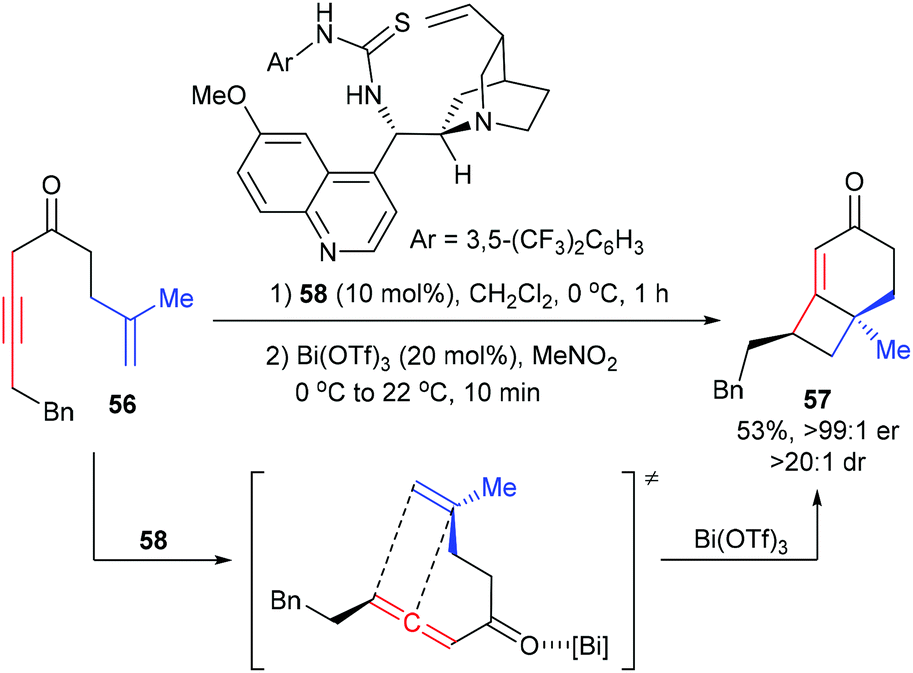
The Brown group reported a chirality transfer [2 + 2] cycloaddition of allenic ketone with tethered alkene for the enantioselective synthesis of [4.2.0]-fused bicycles (Scheme 16).21 Under the catalysis of 10 mol% thiourea catalyst 58, the achiral β,γ-alkynyl ketone 56 was first enantioselectively isomerized to provide a chiral allene intermediate. After adding Bi(OTf)3 (10 mol%), the chiral allene moiety could directly undergo chirality transfer [2 + 2] cycloaddition with the tethered 1,1-disubstituted alkene to form product 57, which contains a quaternary carbon center, with good yield and enantioselectivity.
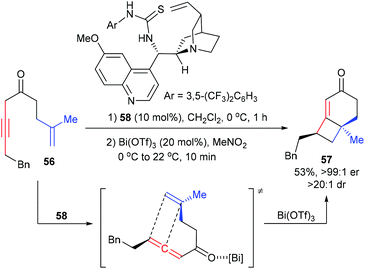 |
| | Scheme 16 Chirality transfer [2 + 2] cycloadditions of allenic ketones with alkenes. | |
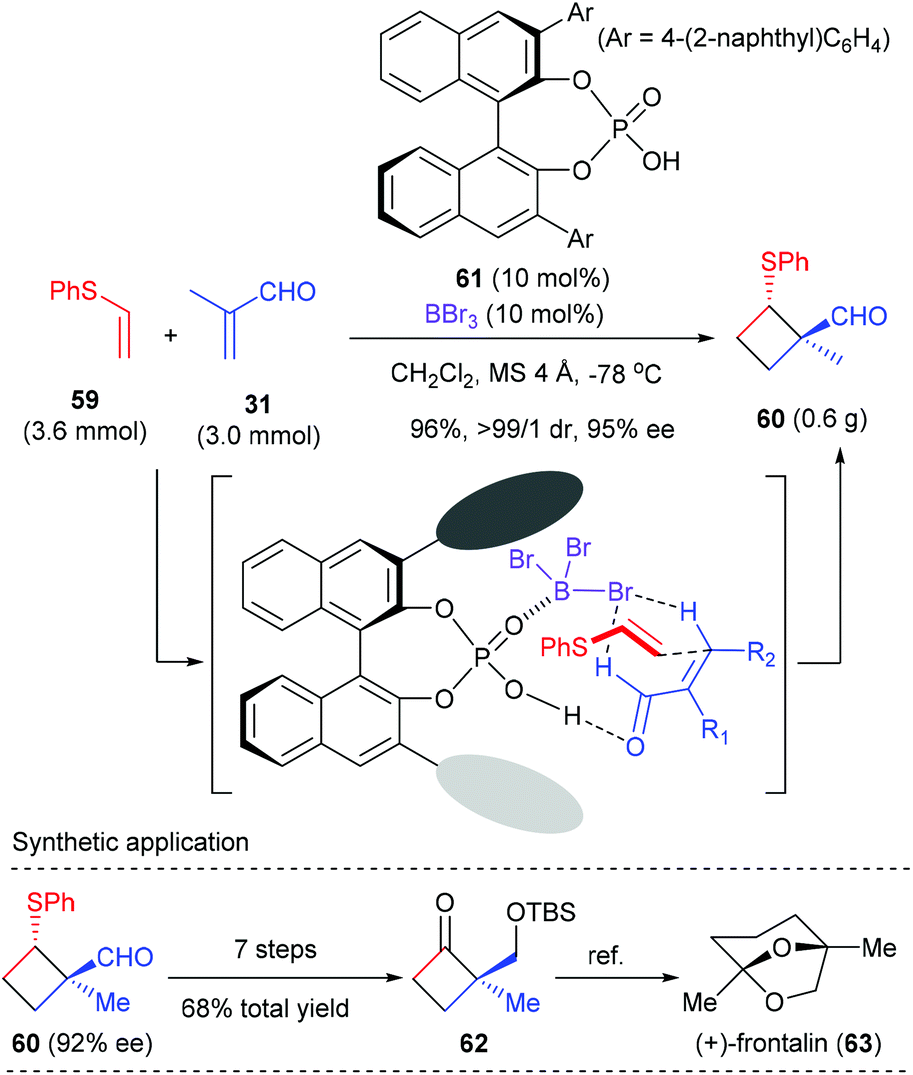
As a continuation of their interest in Lewis acid-activated chiral Brønsted acid (LBA) catalysis,22 Ishihara et al. developed BBr3-assisted chiral phosphoric acid catalysts for enantioselective [2 + 2] cycloaddition. Using phosphoric acid 61 as the catalyst, together with equal amount of BBr3 as the Lewis acid activator, the reaction of phenyl vinyl sulfide 59 with α-methyl acrolein 31 proceeded smoothly to deliver [2 + 2] cycloadduct 60 with high ee value (Scheme 17).23 The thus obtained 60 was a synthetically useful chiral cyclobutane, as is exemplified by the transformation to a key intermediate 62 for the total synthesis of (+)-frontalin (63), a pheromone in Asian elephants. It should be mentioned that this reaction represents the first chiral Brønsted acid catalyzed enantioselective [2 + 2] cycloaddition reaction.
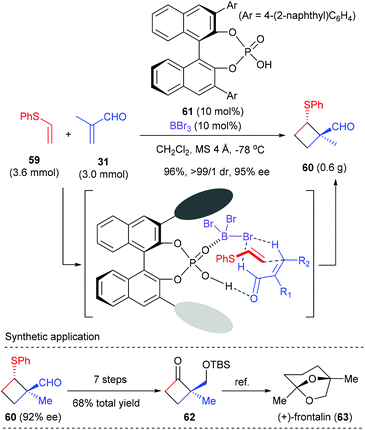 |
| | Scheme 17 BBr3-assisted chiral phosphoric acid catalysts for asymmetric [2 + 2] cycloaddition. | |
2.3. Transition metal catalysis
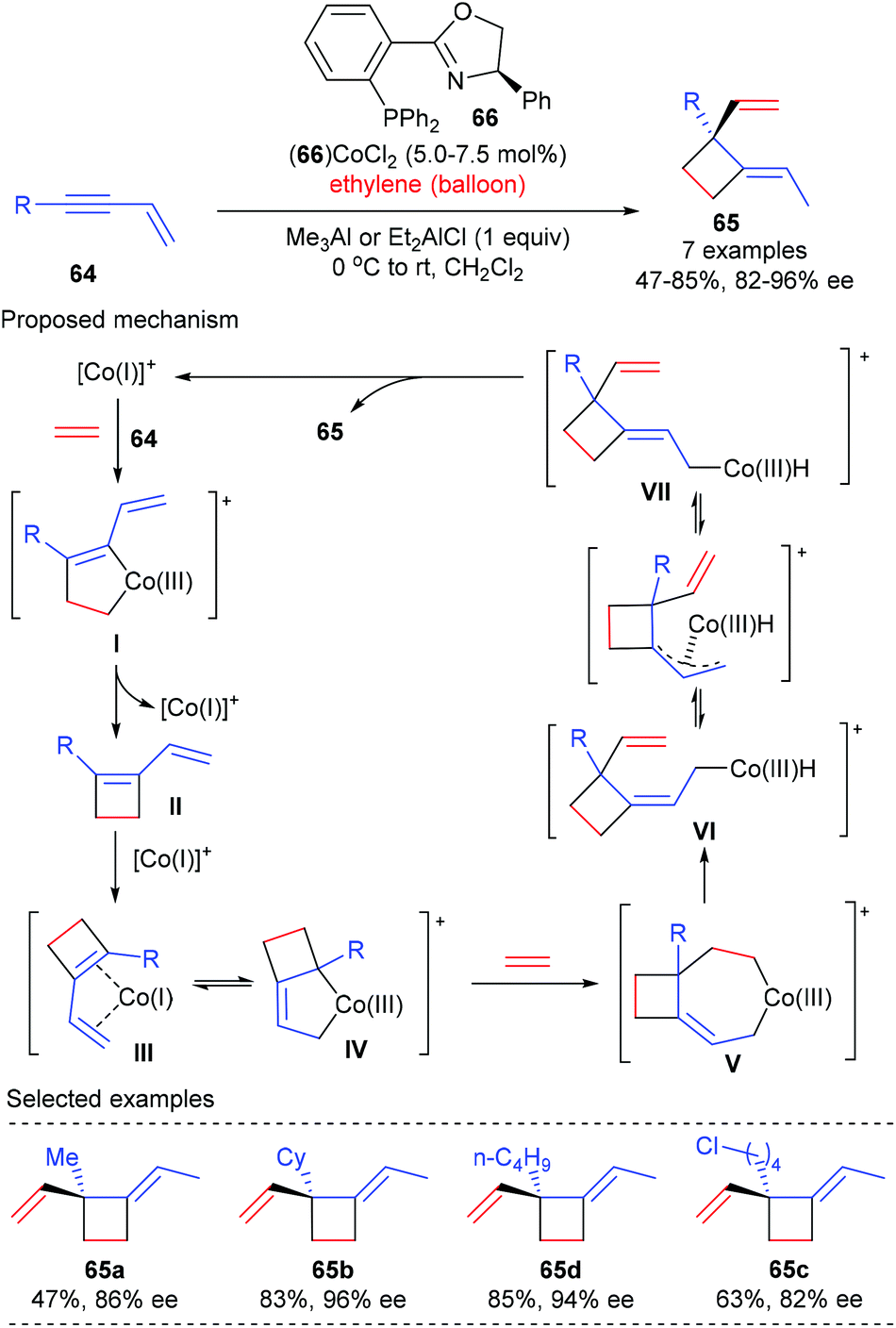
Based on their interest on transition metal catalyzed enantioselective coupling reaction of alkenes, RajanBabu and Pagar achieved a catalytic asymmetric tandem coupling of ethylene and enynes to functionalized quaternary carbon containing cyclobutanes (Scheme 18).24 Starting from 1,3-enynes and ethylene, using the phosphinooxazoline ligand 66 derived Co(II) complex as the chiral catalyst, an initial oxidative dimerization of ethylene and enyne 64 delivered a metallacyclopentene I, which could undergo reductive elimination to afford a cyclobutene intermediate II. Another oxidative dimerization between II and ethylene gave a metallacycloheptene V, which then underwent β-hydrogen elimination to form a Co(III)–hydride intermediate VI. The sterically encumbered VI could undergo (Z)/(E)-isomerization to produce the (E)-4-isomer VII as the major product. A further reductive elimination of VII could deliver cyclobutanes 65, bearing an all-carbon quaternary center, as the (E)-isomer in generally very good yield and with excellent enantioselectivity. This process is highly efficient and desirable, as three highly selective carbon–carbon bonds were formed in one pot using a single chiral cobalt catalyst from simple precursors.
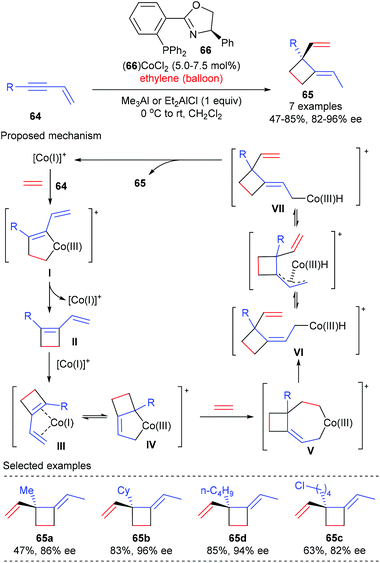 |
| | Scheme 18 Asymmetric tandem coupling of ethylene and enynes to quaternary carbon containing cyclobutanes. | |
2.4. Photochemical [2 + 2] cycloadditions
2.4.1. Lewis acid catalysis.
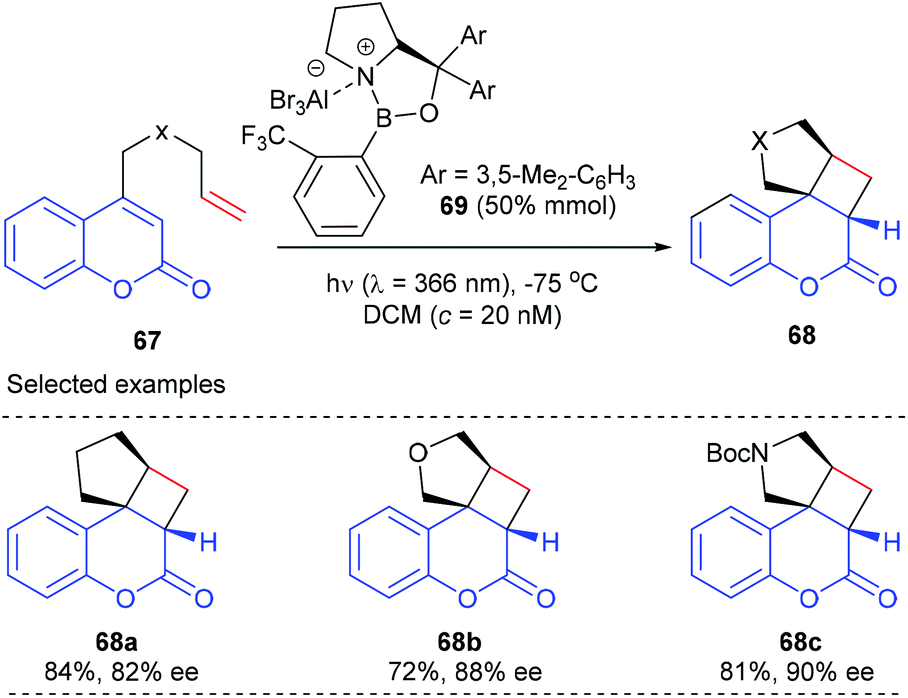
Early in 2010, the Bach group reported that the oxazaborolidine–AlBr3 complex could also serve as a chiral catalyst for a photocycloaddition reaction. In order to compete with an achiral background reaction, a catalyst loading of 50 mol% was required to achieve a good level of enantioselectivity. When the cationic oxazaborolidine–AlBr3 complex 69 was used, the intramolecular [2 + 2] photocycloaddition reaction of 4-alkenyl-substituted coumarins 67a could proceed smoothly to give product 68a in 84% yield and with 82% ee value (Scheme 19).25a
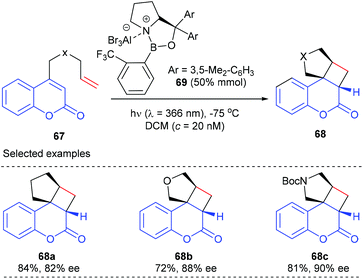 |
| | Scheme 19 Chiral AlBr3 complex catalyzed enantioselective [2 + 2]-cycloaddition reactions. | |
In a following report, they further demonstrated that the above-mentioned catalytic system is also applicable to the [2 + 2] photocycloaddition of heteroatom analogues to give products 68b and c with good to high yields and ee values.25b A mechanism study using UV/Vis spectra indicated that the coordination of the oxazaborolidine–AlBr3 complex 69 to coumarins 67 leads to a bathochromic absorption shift, which in turn facilitates their excitation at λ = 366 nm.
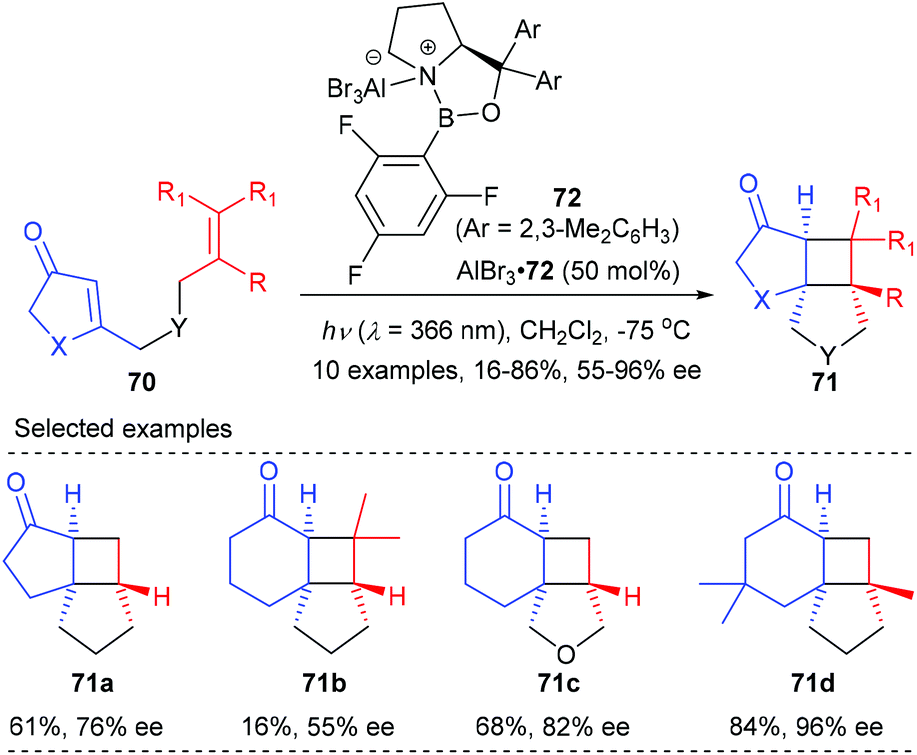
In a recent report, they have further achieved an AlBr3-activated oxazaborolidine catalyzed intramolecular [2 + 2] photocycloaddition of 3-alkenyl-2-cycloalkenones (Scheme 20).25c After extensive screening, oxazaborolidine 72 that bears two 2,3-dimethylphenyl groups on the prolinol and a 2,4,6-trifluorophenyl group on the boron atom was identified as the optimal choice. Upon irradiation at λ = 366 nm, the first asymmetric intramolecular [2 + 2] cycloaddition of both 3-alkenyl 2-cyclohexenones and 2-cyclopentenones proceeded well to give the desired products 71 in 16–86% yields and with up to 96% ee values in the presence of 50 mol% AlBr3-activated 72.
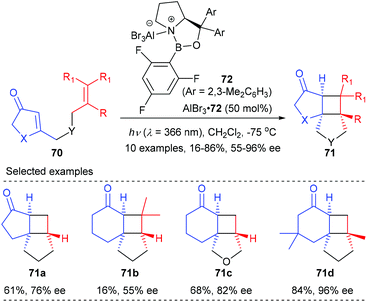 |
| | Scheme 20 Chiral AlBr3 complex catalyzed enantioselective [2 + 2]-cycloaddition reaction of 3-alkenyl-2-cycloalkenones. | |
Intermolecular [2 + 2] photocycloaddition reactions were also achievable using the AlBr3-activated oxazaborolidine complex as the Lewis acid catalyst.26 As shown in Scheme 21, the intermolecular [2 + 2] photocycloaddition of typical cyclic α,β-unsaturated enones 73 with 1,1′-disubstituted olefins 31 could deliver products 74 in moderate to good yields and with high enantioselectivity under the catalysis of 50 mol% 72. The synthetic potential of this methodology was illustrated by the enantioselective total synthesis of monoterpene (−)-grandisol in 6 steps with an overall yield of 13%.
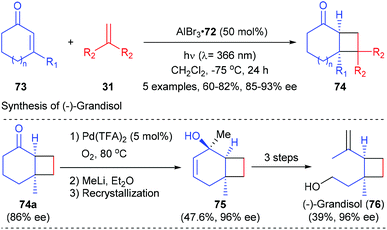 |
| | Scheme 21 Asymmetric intermolecular [2 + 2] photocycloaddition reaction of cyclic enones. | |
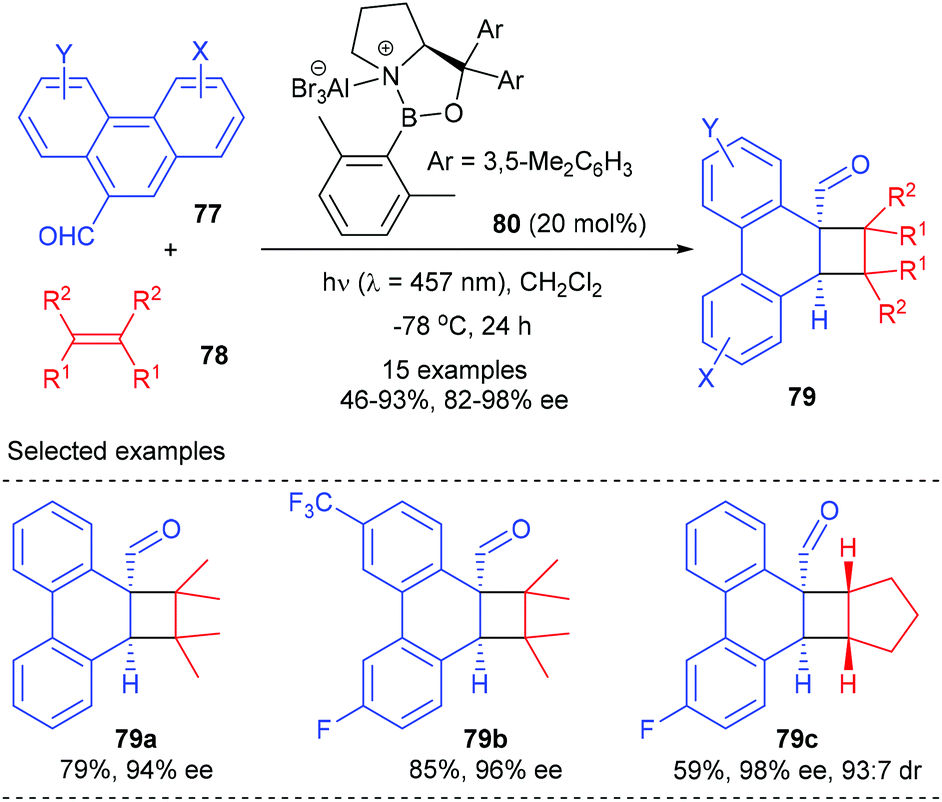
In addition to cyclic α,β-unsaturated enone substrates, Bach et al. further showed that the chiral AlBr3 complex could also activate phenanthrene-9-carboxaldehydes 77 to enable their enantioselective ortho photocycloaddition with olefins under visible-light irradiation conditions (Scheme 22).27 Upon coordination to a Lewis acid, 77 would undergo an extensive bathochromic shift to stretch beyond the absorption bands of uncoordinated phenanthrene-9-carboxaldehydes, thus enabling a selective excitation of the Lewis acid complex and alleviating the achiral background reaction. Therefore, the loading of the chiral oxazaborolidine–AlBr3 complex 80 could be lowered to 20 mol%. A series of substituted phenanthrene-9-carboxaldehydes reacted smoothly with 2,3-dimethyl-2-butane and cyclopentene to give products 79 with 46–93% yields and 82–98% ee values.
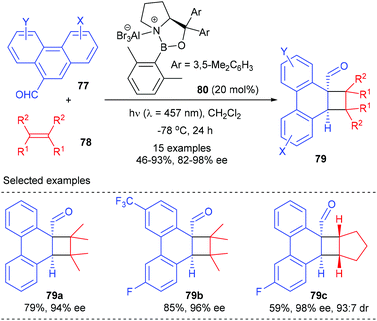 |
| | Scheme 22 Enantioselective photocycloaddition of olefins with phenanthrene-9-carboxaldehydes. | |
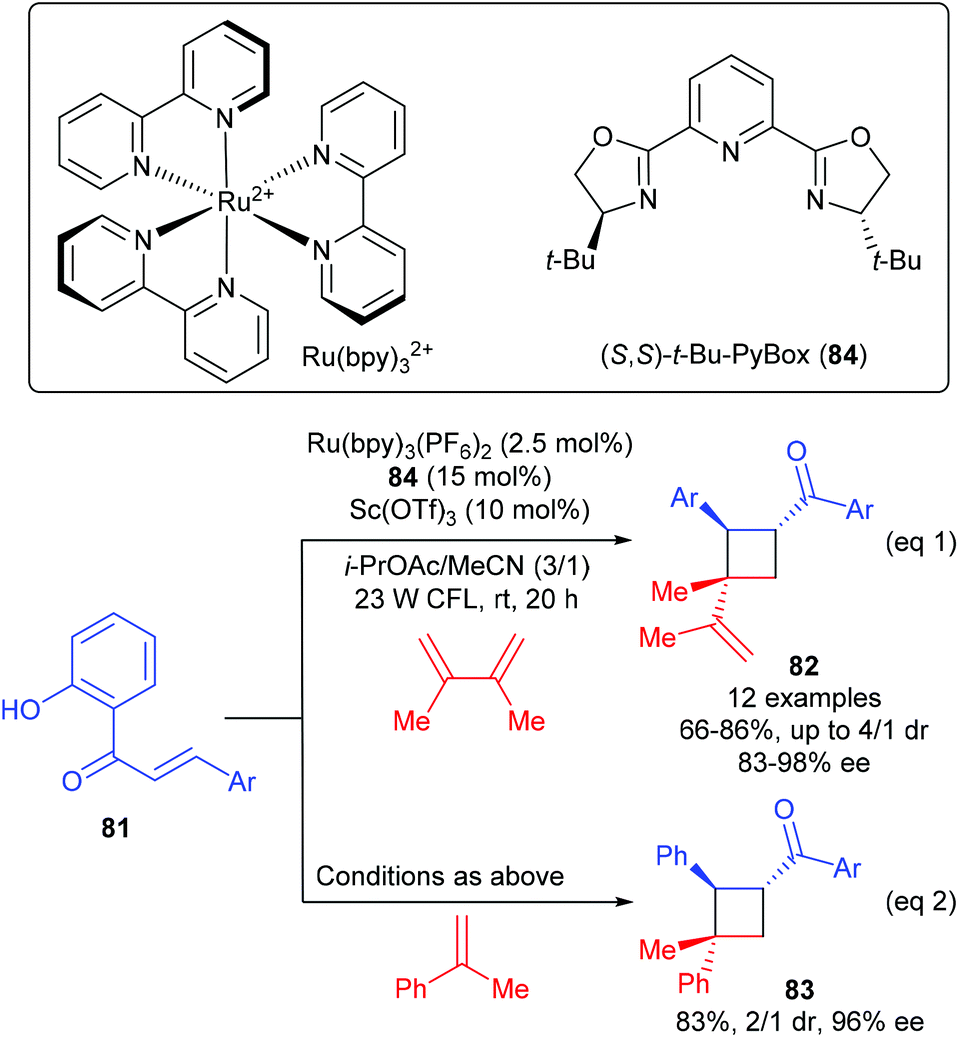
Yoon and workers discovered that chiral Lewis acids could also work cooperatively with a photosensitizer to facilitate the first asymmetric [2 + 2] photocycloadditions of acyclic excited-state 2′-hydroxychalcones with alkenes (Scheme 23).28 In the presence of the PyBox (84) derived Sc(III) complex (15 mol%) as the Lewis acid catalyst, together with 2.5 mol% Ru(bpy)3(PF6)2 as the photosensitizer, a series of 2′-hydroxychalcones 81 reacted with 2,3-dimethylbuta-1,3-diene to afford vinyl cyclobutanes 82 in 66–86% yields and with 83–98% ee values (eqn (1)), whereas the reaction of 81 with α-methyl styrene could give product 83 in 83% yield and with 96% ee (eqn (2)). After a variety of mechanistic studies, they ruled out substrate activation by means of photo-induced electron transfer and proposed an unprecedented mechanism, i.e. Lewis acid coordination could lower the triplet energy of the chalcone substrate and the photosensitizer is responsible for absorbing visible light and the subsequent energy transfer to the substrate–Lewis acid complex.
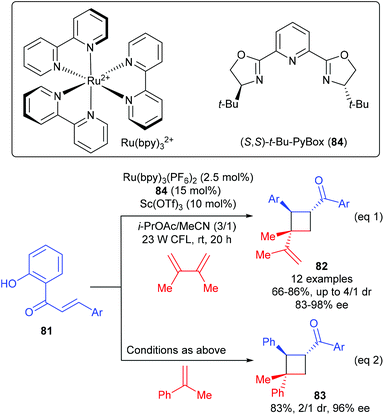 |
| | Scheme 23 Asymmetric [2 + 2] photocycloaddition through Lewis acid–catalyzed triplet energy transfer. | |
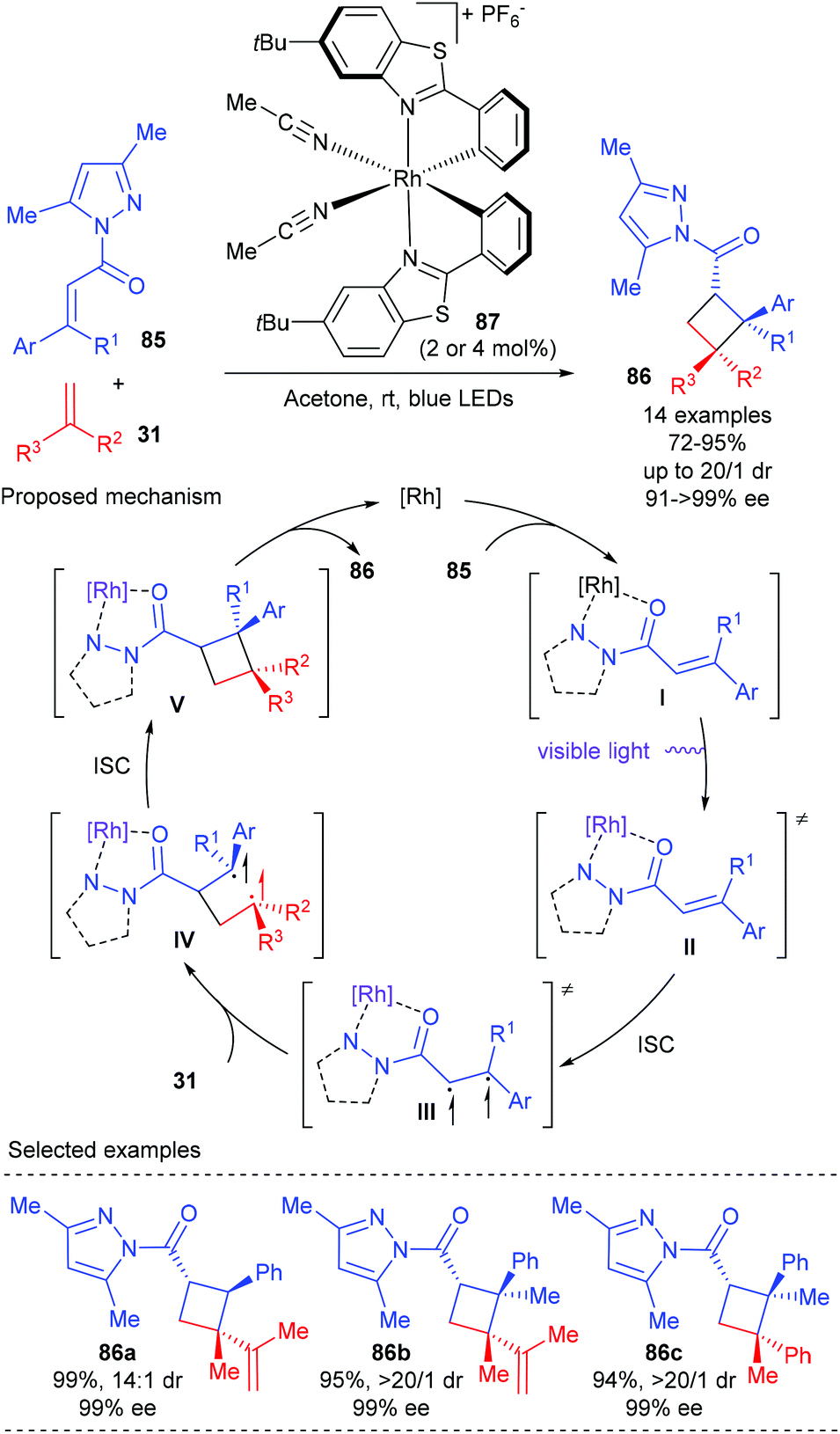
In 2017, Meggers et al. reported that their bis-cyclometalated chiral-at-metal complexes could serve as bifunctional catalysts, combining visible-light-induced electronic activation and reaction stereocontrol, for asymmetric [2 + 2] photocycloadditions. Using 2–4 mol% Δ-RhS (87) as the single catalyst, a variety of α,β-unsaturated imidazoles 85 could react smoothly with 1,1-disubstituted alkene 31 to deliver [2 + 2] addition products 86 in good to excellent yields and with up to 16:1 dr and up to >99% ee values (Scheme 24).29 It is worth noting that cyclobutanes (86b–c) with three contiguous stereogenic centers and vicinal all-carbon quaternary stereocenters can be constructed in a single step using this new methodology in high yields and with excellent dr and ee values. The authors proposed that the complex of 85 and the catalyst were directly excited by visible light to give its lowest singlet state (II). A subsequent intersystem crossing (ISC) of II could give an excited triplet state (III), which then reacted with alkene 31 under control of the stereochemistry by the chiral catalyst to generate a rhodium-bound 1,4-diradical intermediate (IV). The desired [2 + 2] cycloaddition product 86 was obtained after an ISC followed by cyclization and chiral catalyst dissociation.
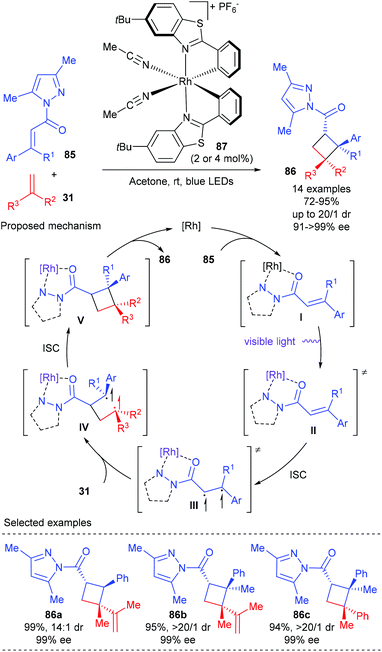 |
| | Scheme 24 Chiral-at-metal complex catalyzed intermolecular [2 + 2] photocycloaddition. | |
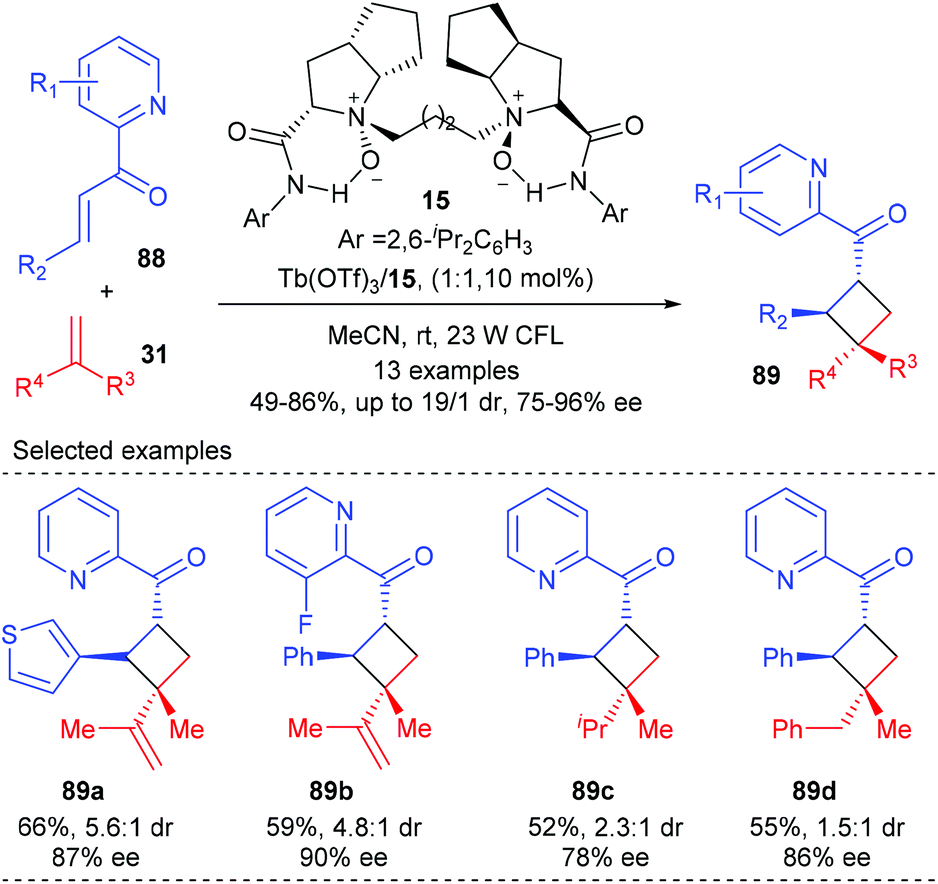
Feng and Liu showed that their chiral N,N′-dioxide derived Tb(III) complexes alone could catalyze the direct visible-light-excited asymmetric [2 + 2] cycloaddition of 2-alkenoylpyridines with various alkenes (Scheme 25).30 In the presence of the chiral N,N′-dioxide 15 coordinated Tb(III) complex (10 mol%), together with 23 W CFL as the irradiator, 1,1-disubstituted alkenes and diene could react smoothly with 2-alkenoylpyridines 88 to deliver products 89 in 52–66% yields and with 78–90% ee and moderate dr values. It was believed that the 2-alkenoylpyridine bounded N,N′-dioxide–Tb(III) complex itself could absorb visible light to reach its excited state and provide facial selectivity of the bound enone, thus enabling the generation of chiral cyclobutane derivatives in the absence of an additional photosensitizer. Additionally, 2′-hydroxychalcones are also compatible substrates for this reaction using a chiral N,N′-dioxide–Sc(III) complex.
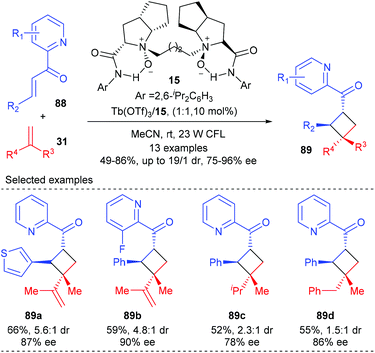 |
| | Scheme 25 Asymmetric [2 + 2] photocycloaddition reactions of enones with olefins. | |
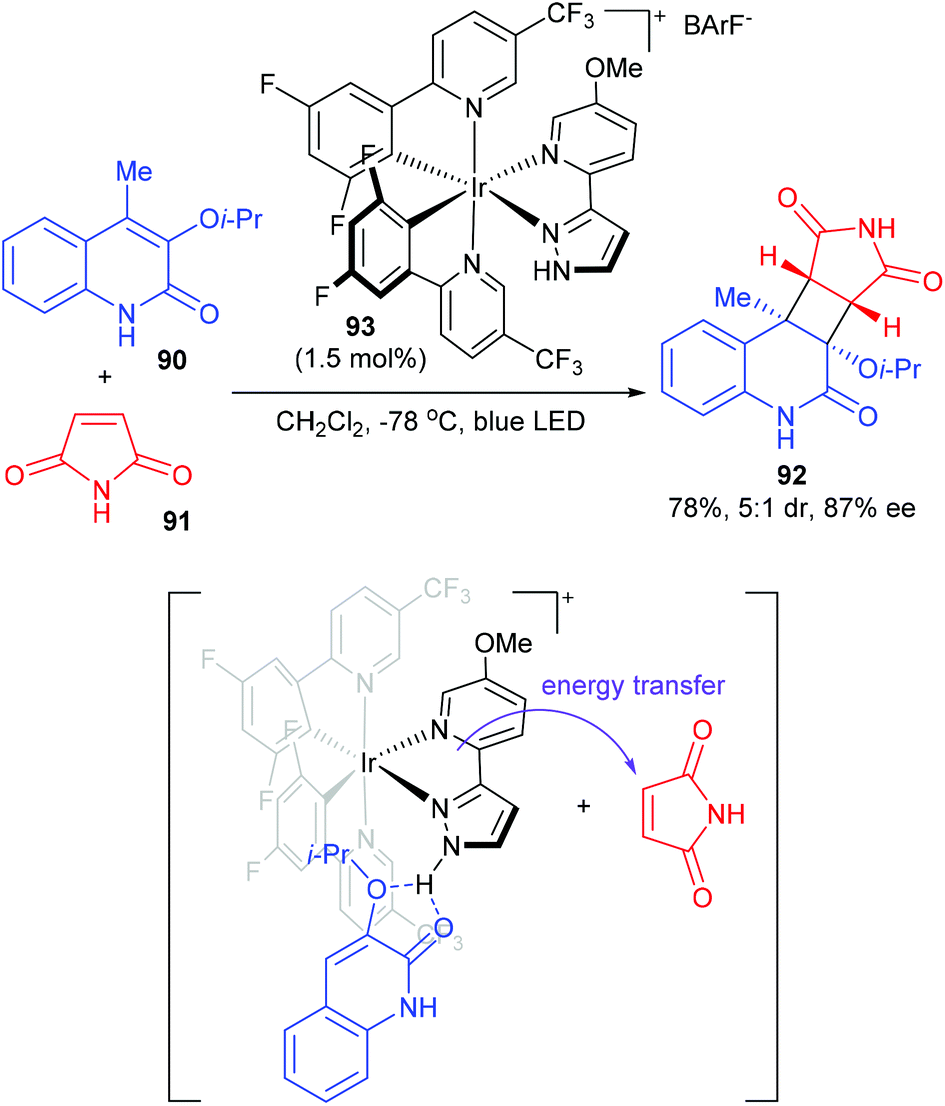
The Yoon group achieved a chiral hydrogen-bonding iridium photosensitizer catalyzed intermolecular [2 + 2] cycloaddition of 3-alkoxyquinolones (Scheme 26).31 In the presence of only 1.5 mol% chiral Ir complex 93, 4-methyl-3-isopropoxy-quinolone 90 reacted with maleimide to afford product 92 in 78% yield and with 5/1 dr and 87% ee values. After a series of mechanistic studies, the authors proposed that photocatalyst 93 and quinolone could form a hydrogen-bonded complex to realize stereocontrol. Upon photoexcitation of this complex, energy transfer sensitization of maleimide rather than quinolone is preferred. Then, the sensitized maleimide reacted with the quinolone–photocatalyst complex to afford the enantioenriched cycloadduct.
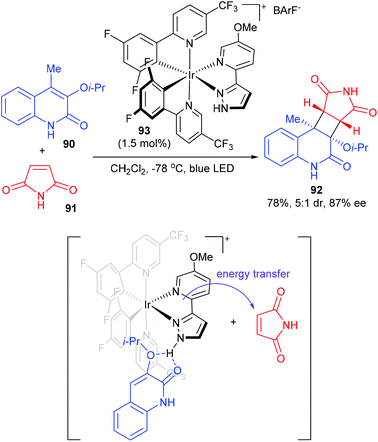 |
| | Scheme 26 Intermolecular [2 + 2] cycloaddition of maleimide with 3-alkoxyquinolones. | |
2.4.2. Organocatalytic [2 + 2]-photocycloaddition.
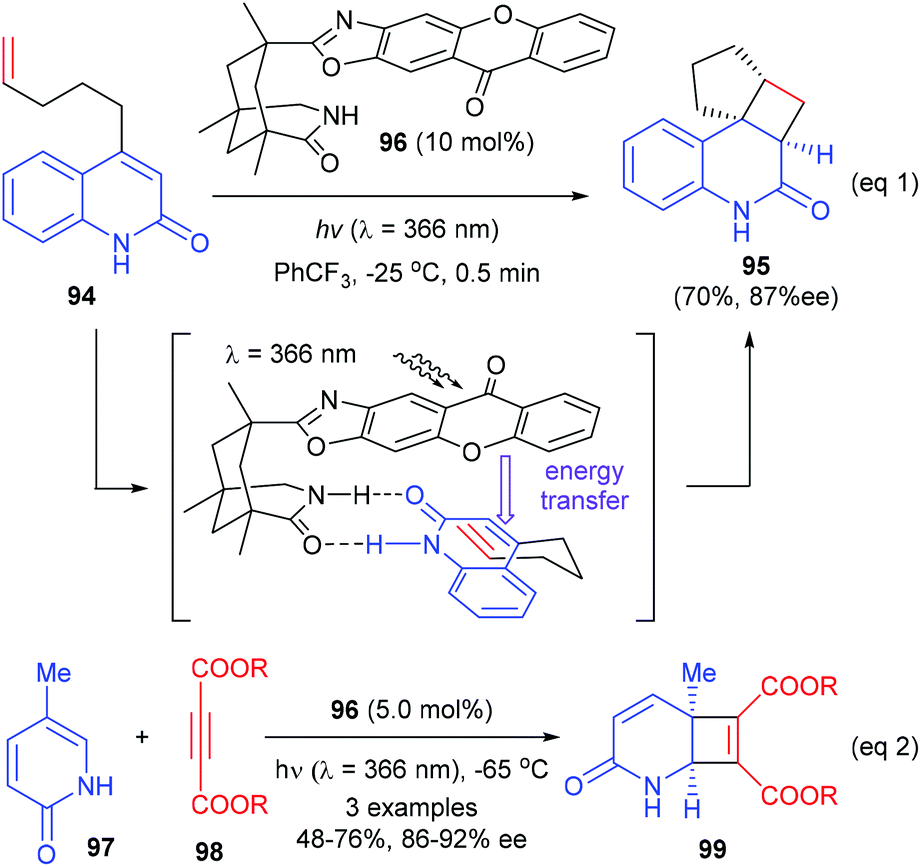
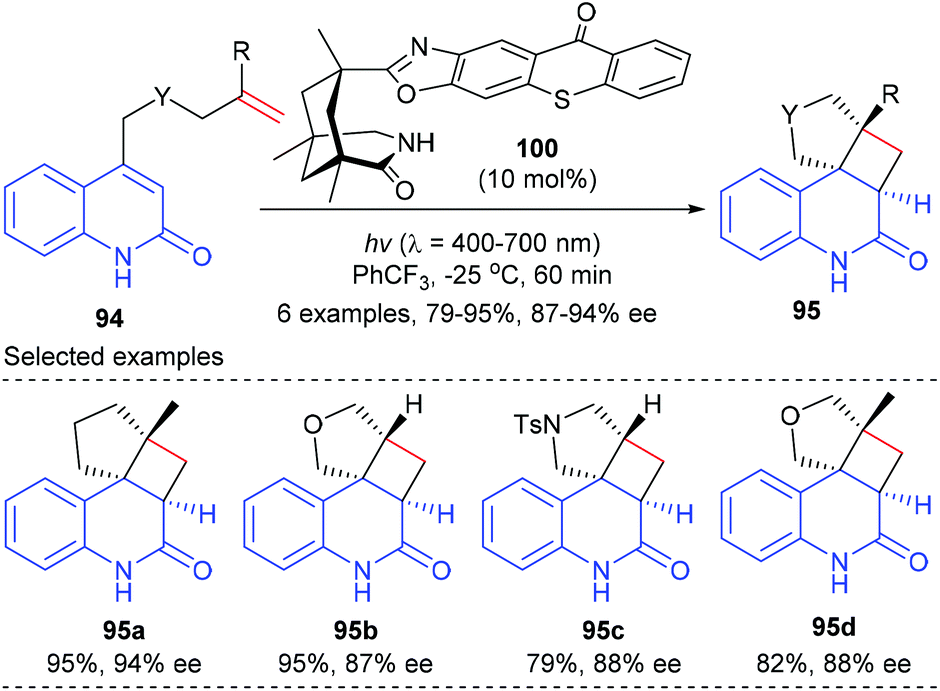
Besides using Lewis acid catalysts, the [2 + 2]-photocycloaddition could also be achieved using organocatalysts as the chiral sources. Early in 2011, Bach and co-workers reported the synthesis of a series of organic chiral sensitizers and their application in asymmetric intramolecular [2 + 2] photocycloaddition reactions (Scheme 27).32 The authors proposed that when quinolone substrate 94 associated with the chiral catalyst, the xanthone moiety could in one hand act as the light-harvesting antenna to transmit energy of the absorbed photon to quinolone and shield the top face of the substrate, so that the tethered alkene moiety could attack the quinolone double bond enantioselectively from the bottom face. On irradiation at λ = 366 nm, 96 could catalyze the intramolecular [2 + 2] reaction of pent-4-enyl quinolone 94 to give the desired product 95 in 70% yield and with 87% ee value (eqn (1)). This catalyst is also effective in catalyzing an intermolecular [2 + 2] photocycloaddition to form enantioenriched quaternary carbon containing cyclobutanes, as is demonstrated by the reaction between 2-pyridones 97 and acetylenedicarboxylates 98, giving products 99 in 48–76% yields and with 86–92% ee values (eqn (2)).
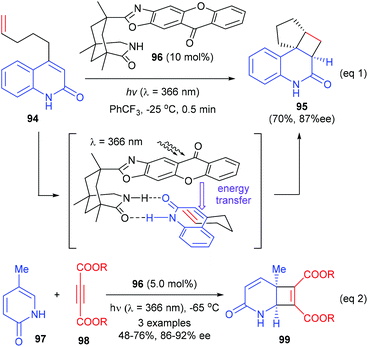 |
| | Scheme 27 [2 + 2]-Photocycloaddition reactions catalyzed by xanthone-based organocatalyst. | |
Based on the above success, the Bach group further synthesized a chiral thioxanthone that is capable of processing visible light for a highly enantioselective photochemical reaction (Scheme 28).33 With a visible source, thioxanthone 100 (10 mol%) facilitated the intramolecular [2 + 2] reaction of 4-(pent-4-enyl) quinolone 94a to deliver tetracyclic product 95a in 95% yield and with 94% ee at −25 °C. The enantioselectivities obtained for the heteroatom analogues 95b, 95c and 95d were slightly lower (87–88%).
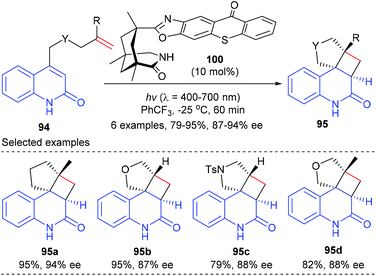 |
| | Scheme 28 Chiral thioxanthone catalyzed [2 + 2] photocycloaddition reactions of 4-tethered quinolones. | |
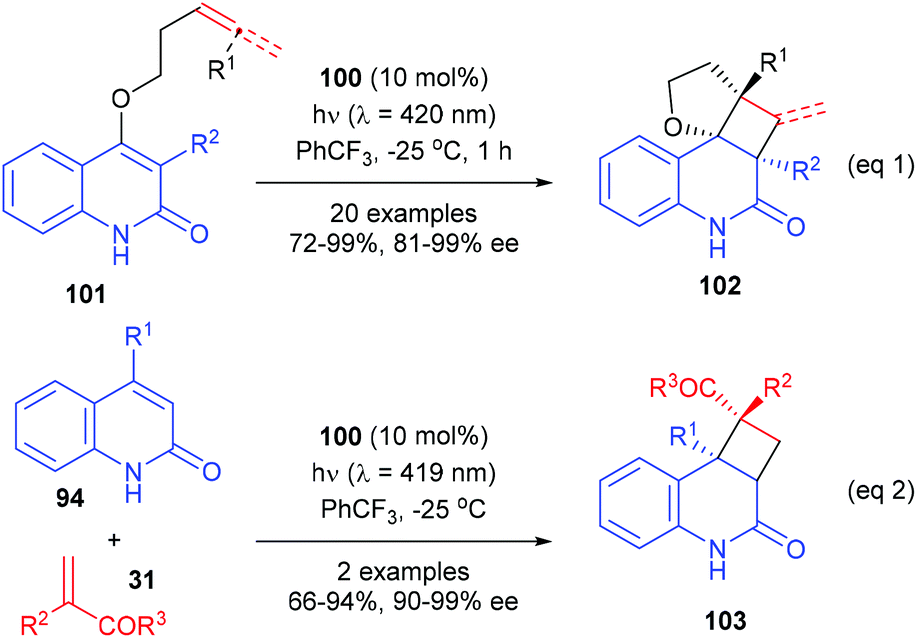
In a most recent report, Bach et al. have further used the chiral thioxanthone catalyst for intramolecular [2 + 2] photocycloaddition reactions of 3-alkylquinolones with 4-O-tethered alkenes and allenes (eqn (1), Scheme 29).34 These reactions could form up to four defined stereogenic centers, including two quaternary carbon centers, in a single step. The alkyl group in the 3-position of 101 could significantly decrease the triplet energy, and it is therefore crucial for the success of the reaction. The intermolecular [2 + 2] reaction of 2(1H)-quinolones 94 with electron-deficient olefins 31 were also possible using 100 as the catalyst, but only two examples of the synthesis of quaternary chiral cyclobutanes were explored (eqn (2)).
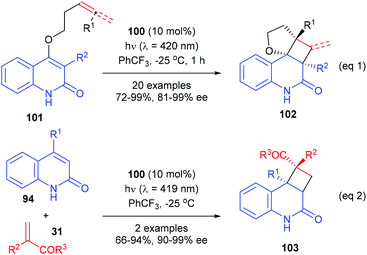 |
| | Scheme 29 Chiral thioxanthone catalyzed intra- and intermolecular [2 + 2] photocycloaddition reactions. | |
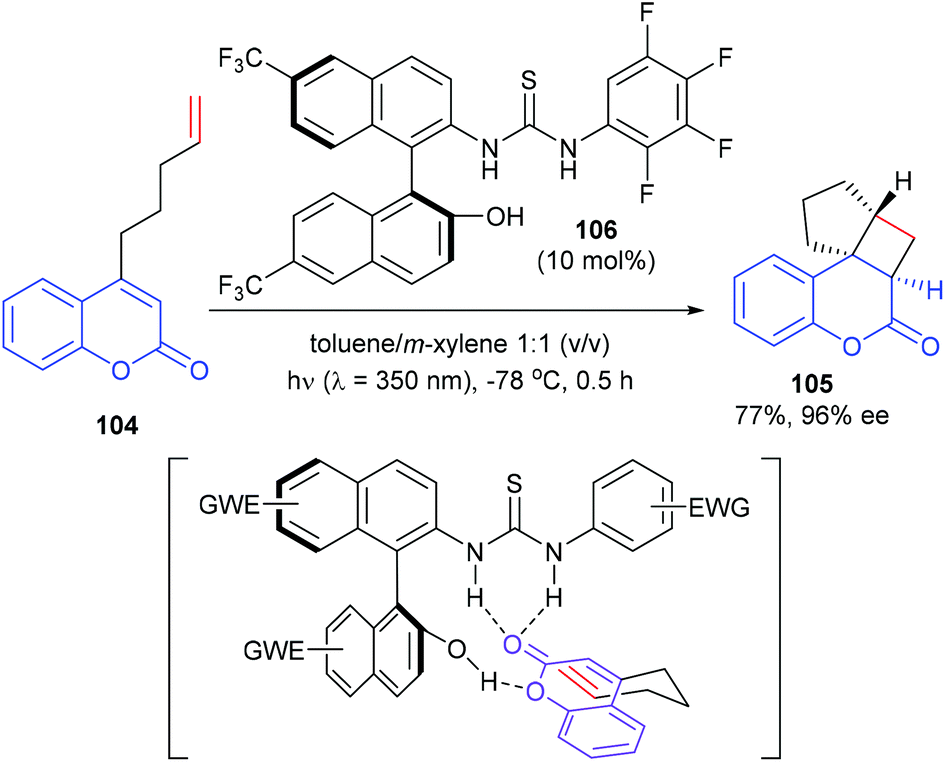
Sivaguru and co-workers reported that a series of chiral binaphthyl-based thiourea derivatives were also efficient organocatalysts for a [2 + 2] photocycloaddition (Scheme 30).35 Among these newly designed catalysts, 106 was found to be the most promising in terms of both yield and enantioselectivity. In the presence of 106 (10 mol%), the intramolecular [2 + 2] reaction of 4-alkenyl-substituted coumarins 104 could afford product 105 with 77% yield and high enantioselectivity. With the aid of hydrogen bonding, the catalyst and the substrate could form a complex, which could be excited efficiently at λ = 350 nm and react to form the desired photoproduct.
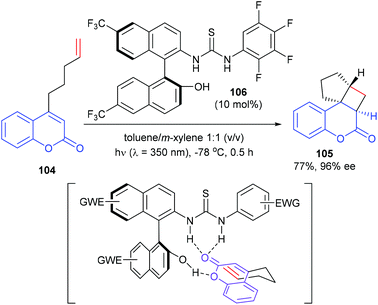 |
| | Scheme 30 Atropisomeric thiourea derivative catalyzed asymmetric [2 + 2] photocycloaddition. | |
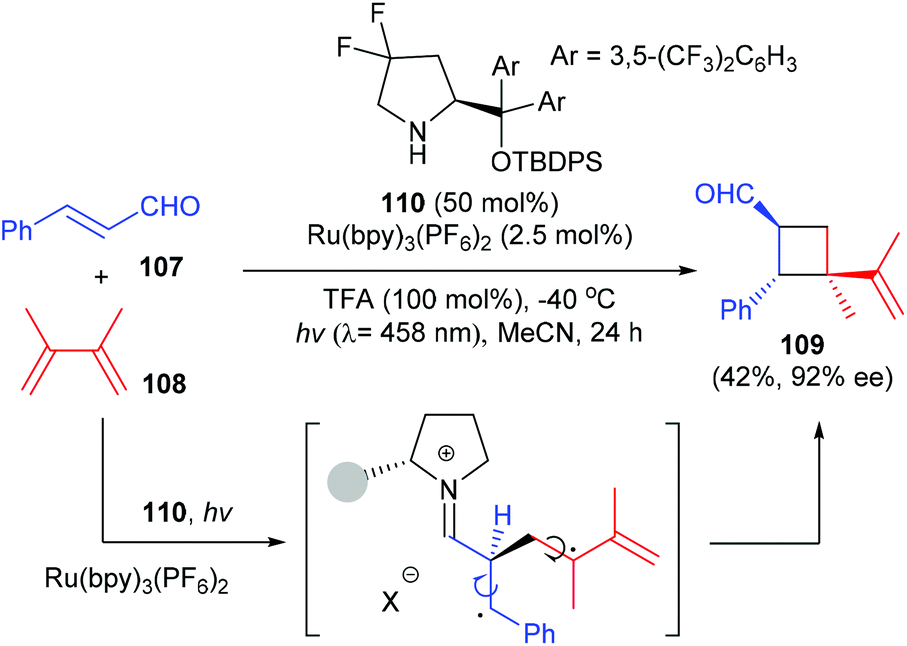
Besides hydrogen bond recognition, the organocatalysts could also mediate a [2 + 2] photocycloaddition by covalent activation. In a very recent work, Bach et al. have found that a chiral Proline derived secondary amine catalyst could activate α,β-unsaturated aldehydes to undergo an intermolecular [2 + 2] photocycloaddition with olefins to afford quaternary carbon containing cyclobutane derivative 109 in 42% yield and with 92% ee, upon irradiation with visible light using a ruthenium (2.5 mol%) complex as the photocatalyst (Scheme 31).36
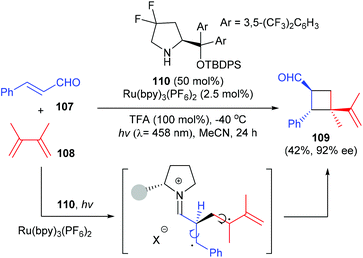 |
| | Scheme 31 Intermolecular [2 + 2] photocycloaddition of olefins with α,β-unsaturated aldehydes. | |
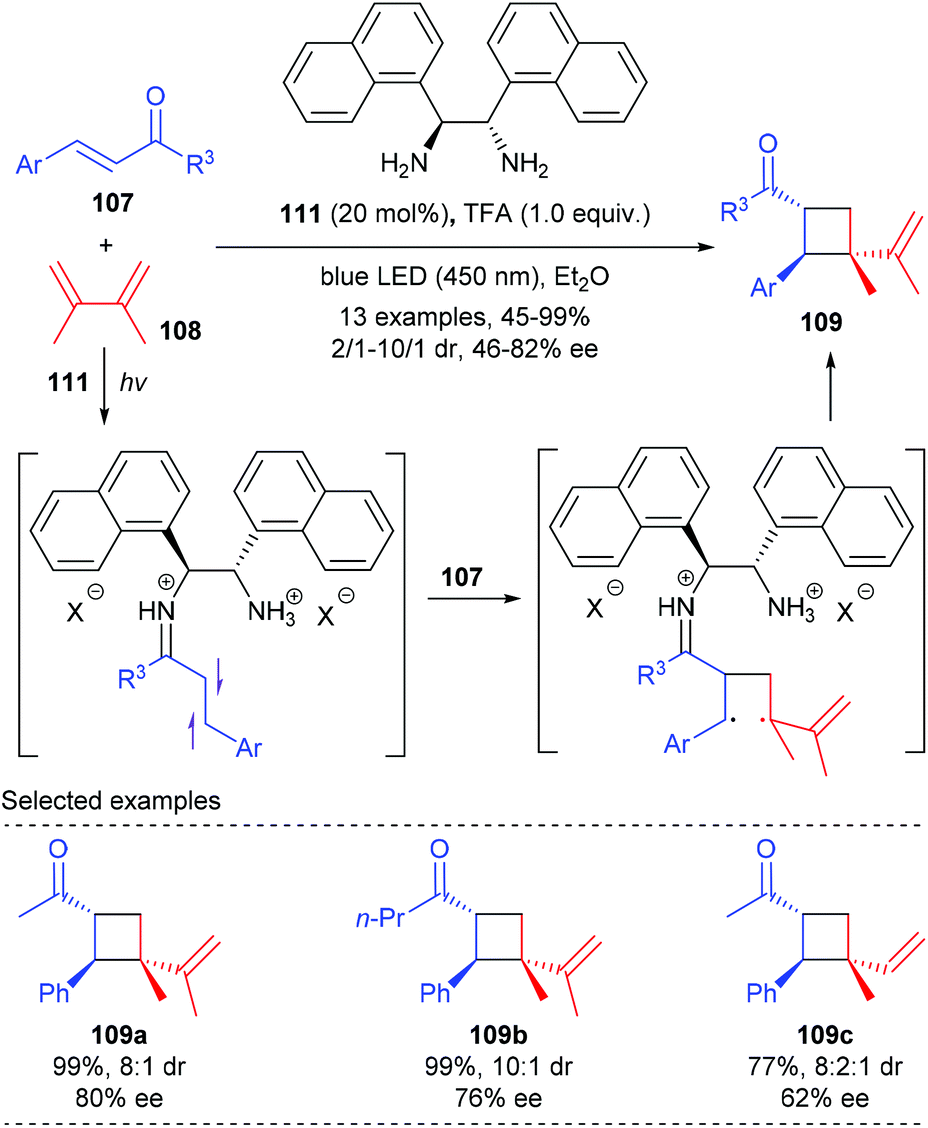
Soon after that, Alemán reported a chiral diamine catalyzed enantioselective [2 + 2] photocycloaddition of α,β-unsaturated ketones (Scheme 32).37 Unlike the above catalytic system, the current reaction does not require the use of any external photocatalyst, and the loading of diamine catalyst 111 could be lowered to 20 mol%. Under blue LED irradiation, together with naphthyl substituted chiral ethane-1,2-diamine 111 as the single catalyst and TFA as an acid promoter, the reaction of enones 107 with 2,3-dimethylbuta-1,3-diene 108 could deliver chiral quaternary carbon containing cyclobutane 109 with good enantiomeric and diastereoisomeric ratios and high yields. The authors proposed that upon condensation with the enone substrate, the diamine catalyst could form an iminium ion intermediate that absorbs in the visible light region. The direct excitation of such an intermediate leads to a charge transfer excited state that unlocks the desired asymmetric [2 + 2] photocycloaddition.
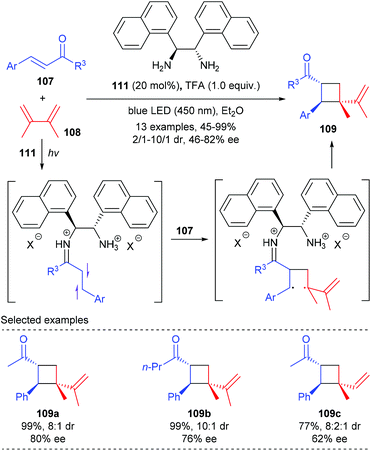 |
| | Scheme 32 Enantioselective aminocatalytic [2 + 2] cycloaddition through visible light excitation. | |
3. Ring-expansion rearrangement
The high ring strain energy associated with the three-membered ring allows cyclopropane derivatives to undergo a variety of synthetically useful ring-expansion reactions to deliver cyclic products that are less strained. For example, the cyclopropylcarbinyl-cyclobutyl rearrangement of cyclopropanols offered efficient methods for the construction of chiral quaternary cyclobutanones.38
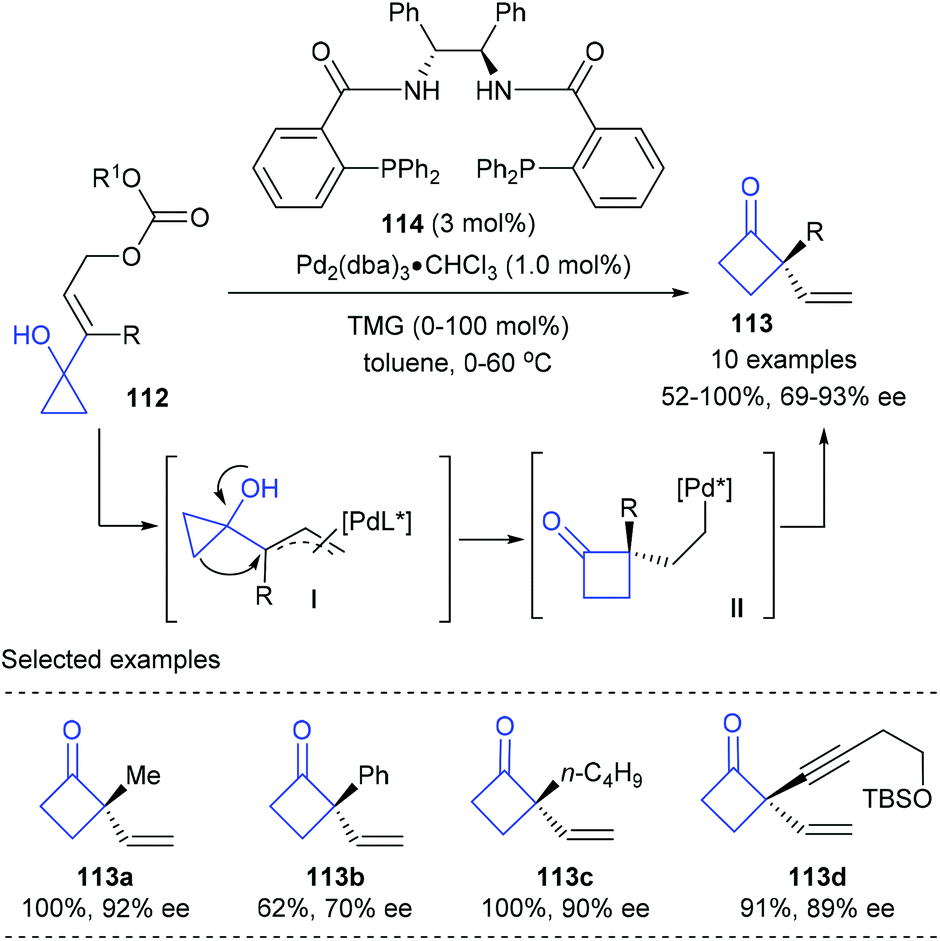
Early in 2001, Trost and co-workers reported the first catalytic enantioselective Wagner–Meerwein rearrangement of prochiral vinylcyclopropanols for the synthesis of cyclobutanones bearing quaternary carbon centers (Scheme 33).39 In the presence of only 1 mol% chiral Pd(0) complex derived from Trost ligand 114 as the catalyst, the targeted quaternary vinylcyclobutanones 113 could be obtained in 52–100% yields and with 69–93% ee values. The current reaction was believed to be initiated by the formation of an π–allylpalladium intermediate (I) via the reaction of the chiral Pd(0) complex with allyl carbonate 112, which is proposed to be the enantio-discriminating step; then a subsequent 1,2-migration and reductive elimination resulted in the final product.
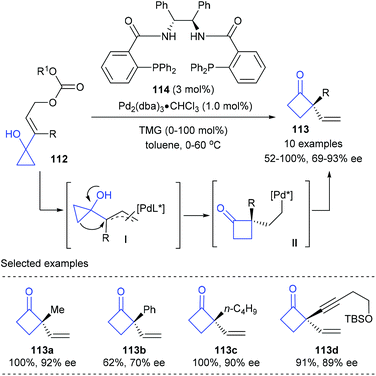 |
| | Scheme 33 First catalytic asymmetric Wagner–Meerwein rearrangement. | |
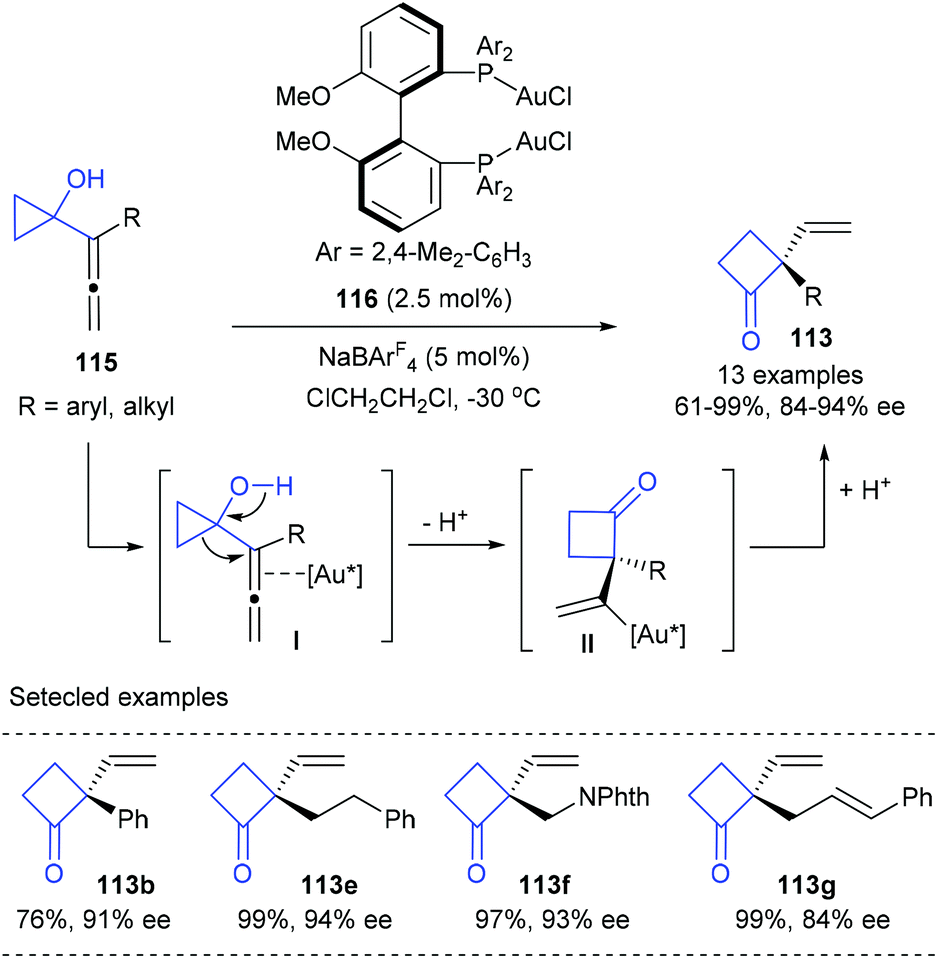
Besides Pd(0) catalysis, cationic Au(I) catalysis has also shown its potential in the desymmetric ring-expansion of prochiral cyclopropanols. In 2009, a enantioselective Wagner–Meerwein rearrangement of 1-allenylcyclopropanols 115 to form cyclobutanones 113 possessing a vinyl-substituted quaternary stereogenic centre in 61–99% yields and with 84–94% ee values was developed by Toste and Kleinbeck, using 2.5 mol% MeO-DM-BIPHEP derived Au(I) complex 116 and 5 mol% NaBArF4 as the catalyst (Scheme 34).40 The cationic gold(I) catalyst was proposed to coordinate with the internal double bond of the allene moiety (I) and trigger ring-expansion by a Wagner–Meerwein rearrangement to generate a vinylgold intermediate (II), which could then undergo a protodemetalation to deliver the final product 113. It should be noted that this work was also the first report of enantioselective gold-catalyzed 1,2-alkyl migration reactions.
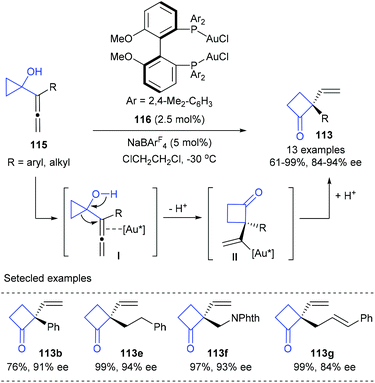 |
| | Scheme 34 Asymmetric ring expansion of allenylcyclopropanols. | |
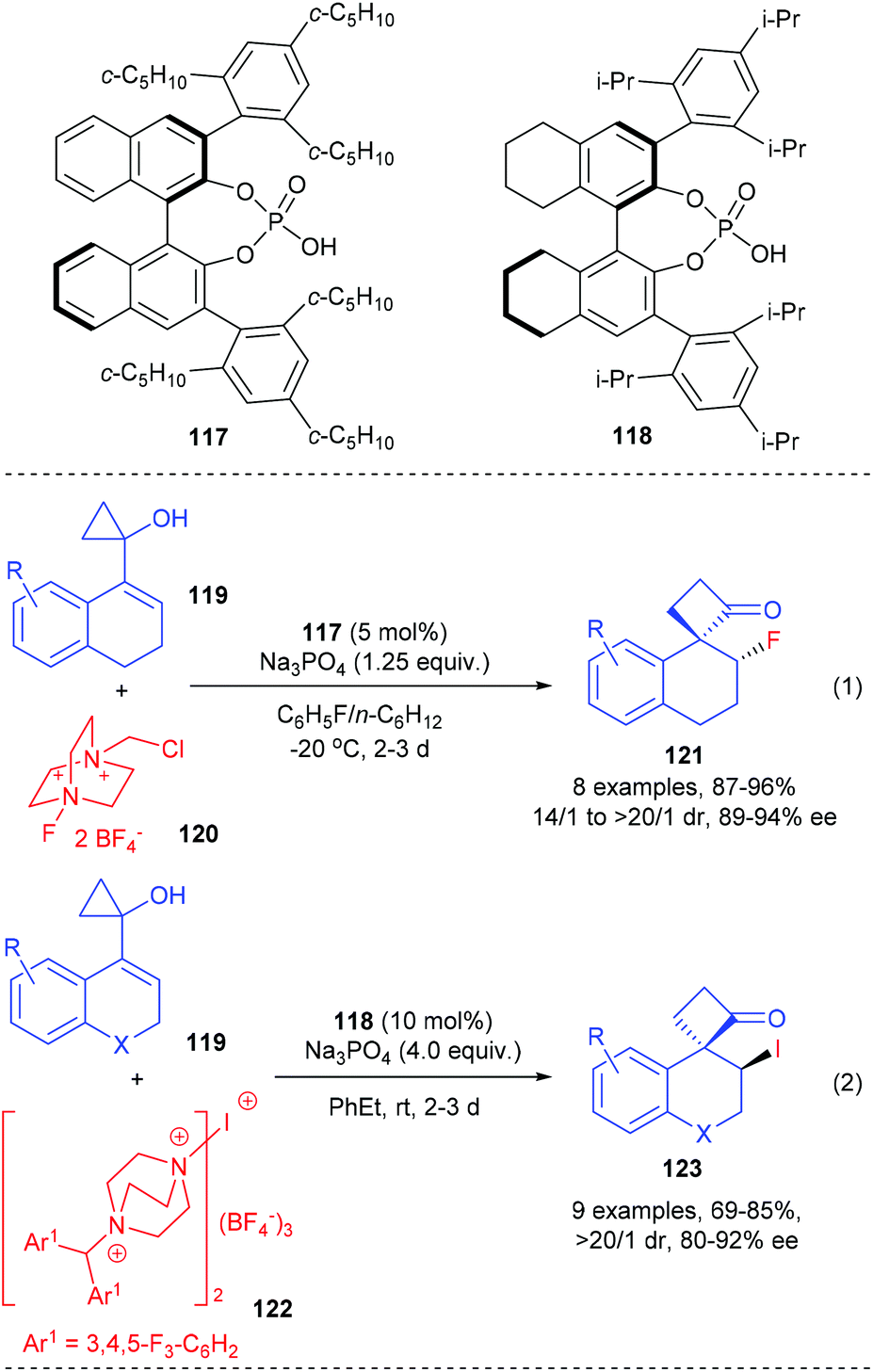
In addition, such ring-expansion reactions could also be initiated by enantioselective halogenations. The Alexakis group described that the fluorination and iodination initiated ring-expansion reaction of allylic cyclopropanols could be achieved using the chiral-counterion catalysis strategy (Scheme 35).41 In the presence of 5 mol% chiral phosphoric acid 117 as the chiral source, Na3PO4 as base and Selectfluor as the fluorinating reagent, the fluorination/ring-expansion reaction of tetralone based allylic cyclopropanols 119 could give the desired β-fluoro spirocyclobutanones 121 with up to 96% yields, >20/1 dr and 94% ee values (eqn (1)). The chiral phosphoric acid counterion is believed to control the facial selectivity and activate the hydroxyl moiety of the substrate via deprotonation.
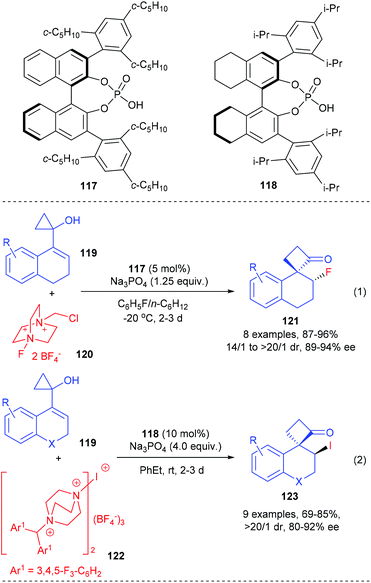 |
| | Scheme 35 Halogenation initiated ring-expansion reaction. | |
A similar phosphoric acid catalyst 118 was found to be an efficient catalyst for the electrophilic iodination initiated ring-expansion rearrangement of strained allylic cyclopropanols 119 to give β-iodo spirocyclobutanones 123 (eqn (2)). After screening a variety of iodinating reagents, a bulky and insoluble cationic iodinating reagent 122 was identified as the optimal choice, with which the desired product 123 could be obtained with up to 92% ee values and 69–85% yields.
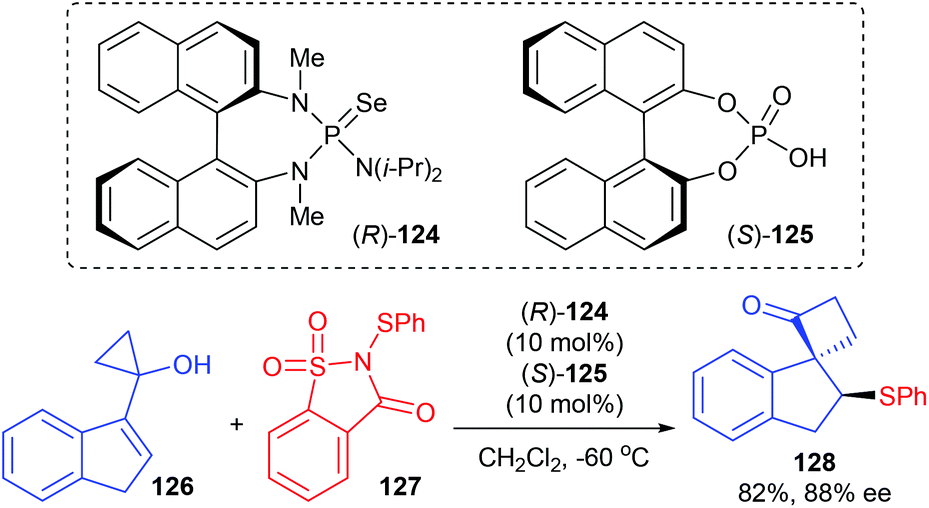
As a continuation of efforts in developing an asymmetric semipinacol rearrangement reaction for synthetic application, Tu and co-workers presented a chiral Lewis base 124 and chiral Brønsted acid 125 co-catalyzed asymmetric sulfenylation/semipinacol rearrangement reaction of 1,1-disubstituted and trisubstituted allylic alcohols for the synthesis of β-arylthio ketones. Although they focused their attention mainly on allylic cyclobutanol substrates, a cyclopropanol substrate 126 was also tried and 82% yield and 88% ee were obtained for product 128 (Scheme 36).42
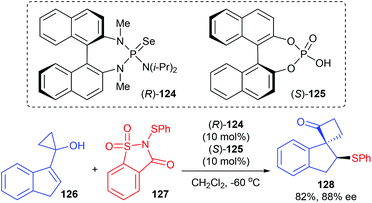 |
| | Scheme 36 Asymmetric sulfenylation/semipinacol rearrangement. | |
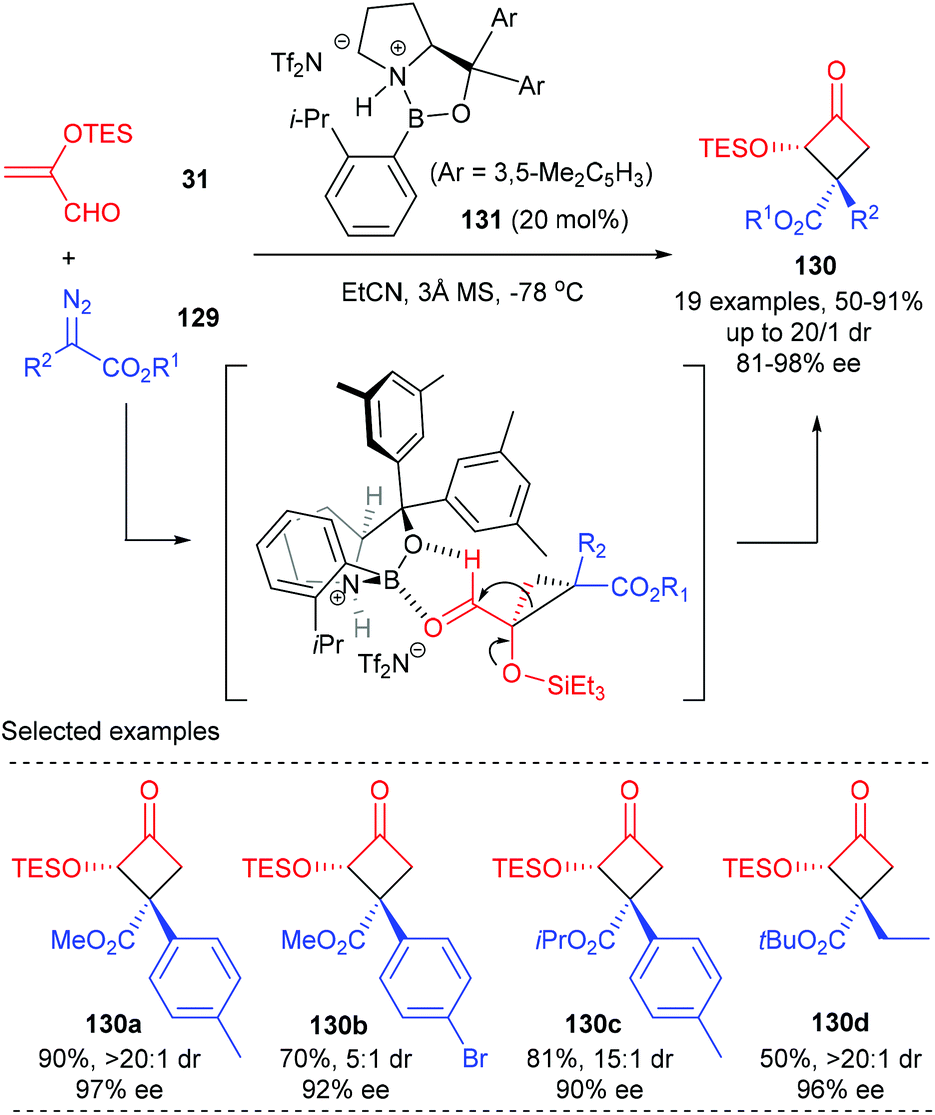
Ryu et al. achieved the first Lewis acid catalyzed asymmetric cyclopropanation/semipinacol rearrangement tandem process to produce quaternary cyclobutanones (Scheme 37).43 Under optimized conditions, using the chiral oxazaborolidinium ion 131 as the catalyst, various α-silyloxycyclobutanones 130 possessing a chiral β-quaternary centre were obtained with up to 91% yields and excellent enantio- and diastereoselectivities (up to 98% ee and >20:1 dr values). Such a process was proposed to start from asymmetric cyclopropanation between α-silyloxyacrolein 31 and diazoester 129 to form a donor–acceptor cyclopropane intermediate, which was further activated by a Lewis acid catalyst to undergo a concerted 1,2-alkyl shift of C1 with an electron-withdrawing group, leading to the formation of α-silyloxycyclobutanones 130 after the subsequent silyl group migration.
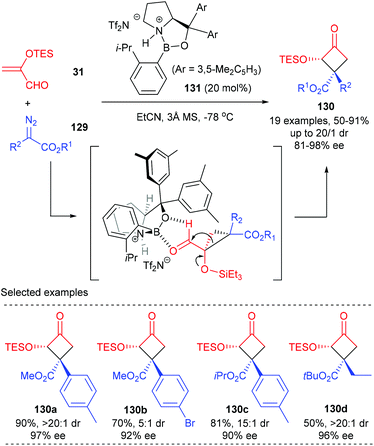 |
| | Scheme 37 Tandem cyclopropanation/semipinacol rearrangement. | |
4. Desymmetrization
The desymmetrization strategy allows the chiral determining step to proceed at the tethered functionality rather than at the prochiral carbon centre, which could alleviate the unfavourable steric repulsion to some extent during the formation of quaternary carbon centers. On the other hand, the reaction site is at least one covalent bond away from the existing quaternary carbon, thus making it more difficult to control the stereoselectivity. Therefore, catalytic desymmetrization reactions are highly potential, but also challenging, for the construction of chiral quaternary carbon containing cyclobutanes.44
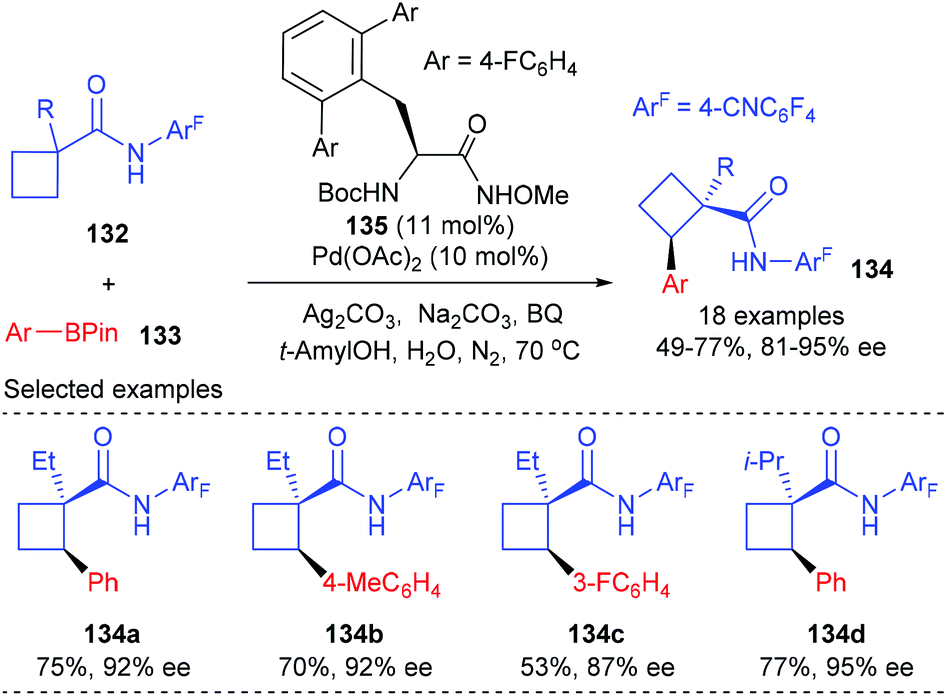
In 2014, Yu and co-workers reported that a Pd(II)-catalyzed desymmetric methylene C(sp3)–H bond reaction of prochiral cyclobutanecarboxylic acid derivatives with arylboron reagents can provide an alternative approach for the enantioselective synthesis of cyclobutane carboxylates containing α-chiral quaternary stereocenters (Scheme 38).45 Under the catalysis of 10 mol% Pd(II) complex derived from newly developed chiral mono-N-protected α-amino-O-methylhydroxamic acid 135, the intermolecular cross-coupling reaction could give the desired optical active cyclobutanecarboxylates 134 in 49–77% yields and with 81–95% ee values.
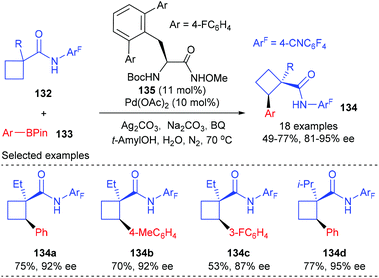 |
| | Scheme 38 Enantioselective C(sp3)–H activation of prochiral cyclobutanecarboxylic acid derivatives. | |
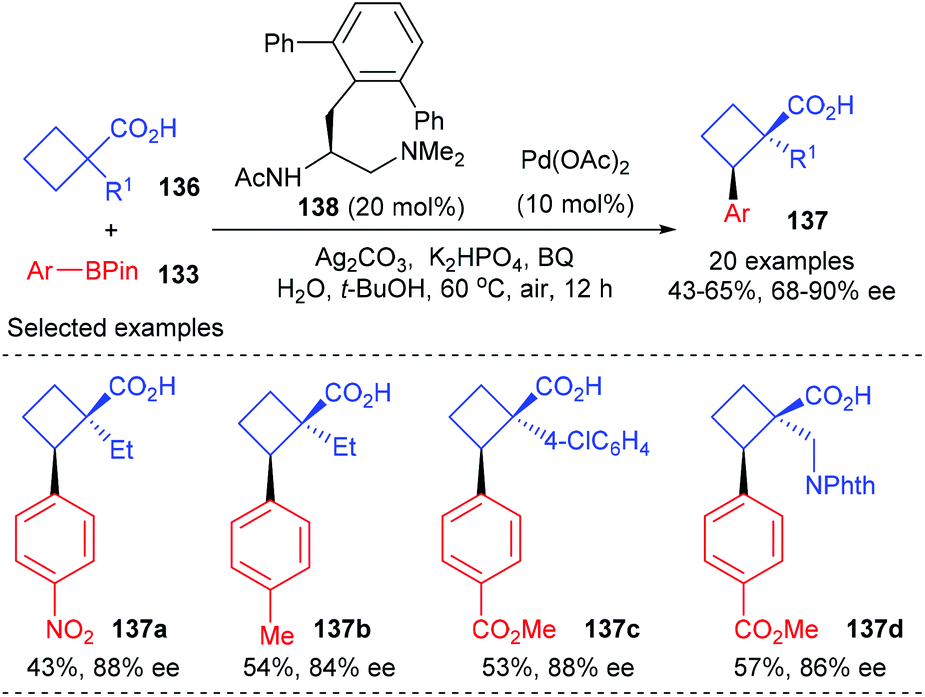
In a more recent work of the same group, they have described that the above-mentioned desymmetric Pd(II)-catalyzed enantioselective C(sp3)–H cross-coupling reaction could be extended to simpler free cyclobutanecarboxylic acids (Scheme 39).46 In the presence of 10 mol% Pd(OAc)2 as the metal catalyst and 20 mol% mono-protected amino ethyl amine 138 as the chiral ligand, prochiral cyclobutanecarboxylic acid could be desymmetrized to afford cyclobutanecarboxylic acids 137 containing quaternary stereocenters in up to 65% yields and with 90% ee values.
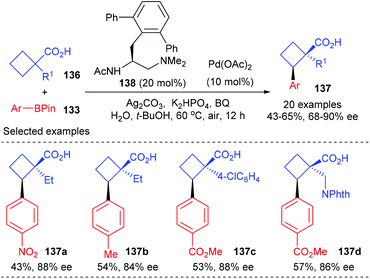 |
| | Scheme 39 Enantioselective C(sp3)–H activation of free prochiral cyclobutanecarboxylic acids. | |
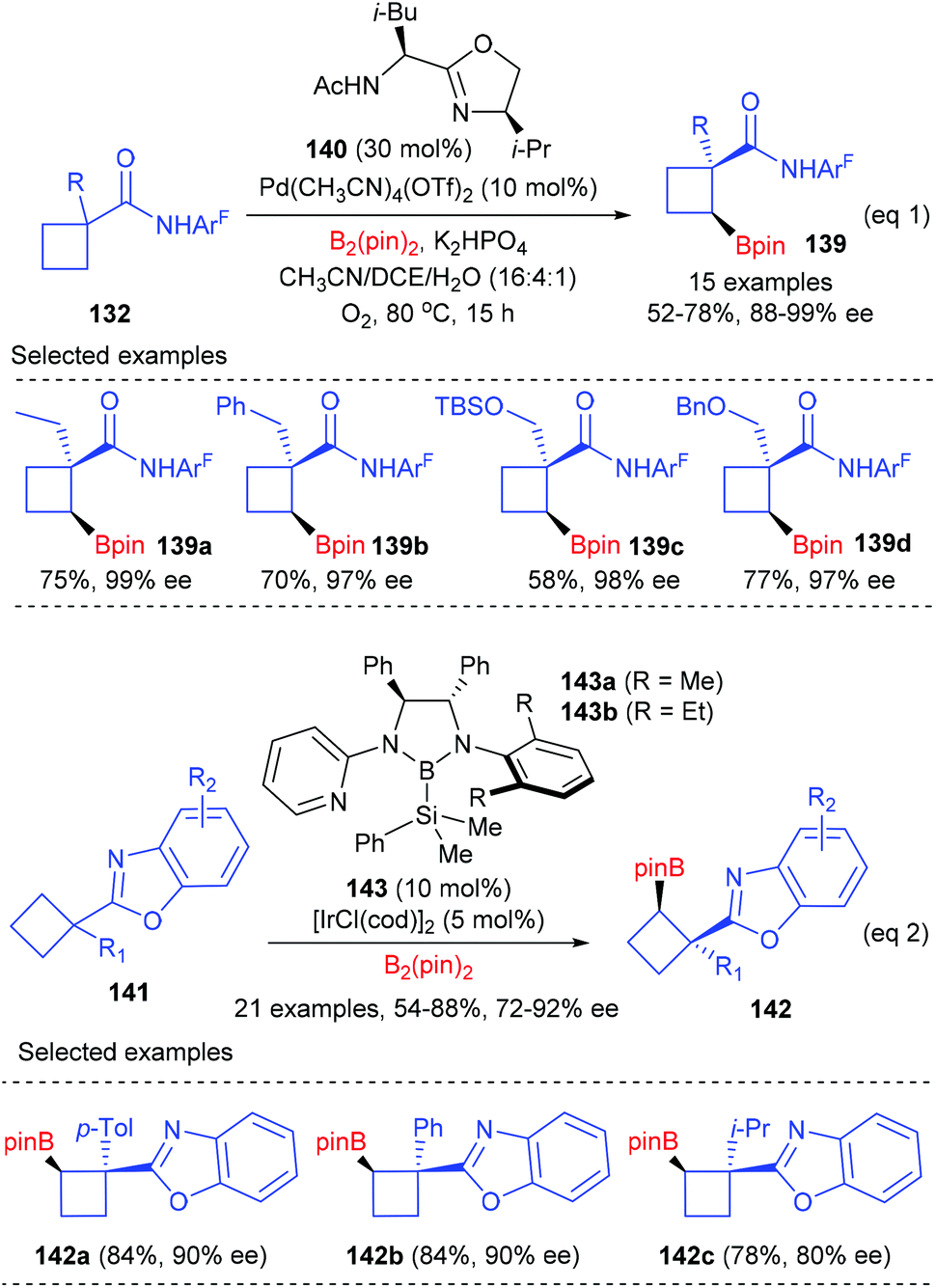
Besides enantioselective C(sp3)–H bond arylation, the desymmetrization of cyclobutanecarboxylic acid derivatives could also be achieved via enantioselective C(sp3)–H bond borylation reactions (Scheme 40).47a After evaluation of a diverse group of newly designed bidentate oxazoline ligands, Yu et al. found that the merger of (S,R)-140 (30 mol%) and Pd(CH3CN)4(OTf)2 (10 mol%) was the best catalyst, which could facilitate the cross-coupling reactions of cyclobutanecarboxylic amides 132 and B2(pin)2 to give chiral cyclobutanecarboxylate products 139 in up to 78% yields and with 88–99% enantioselectivities (eqn (1)). Most recently, Shen and Xu have reported the first benzoxazoline-directed C(sp3)–H borylation of prochiral cyclobutane to give cyclobutylboronates (eqn (2)).47b In the presence of the chiral bidentate boryl ligand 143 coordinated Iridium complex (10 mol%), a variety of functionalized prochiral cyclobutanes could be enantioselectively desymmetrized to afford products 142 with good to excellent ee values, which are synthetically useful intermediates to provide optically active trisubstituted cyclobutane derivatives.
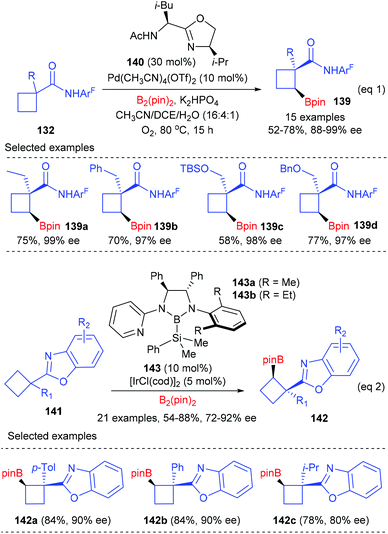 |
| | Scheme 40 Desymmetrization of cyclobutanes via enantioselective C(sp3)–H borylation. | |
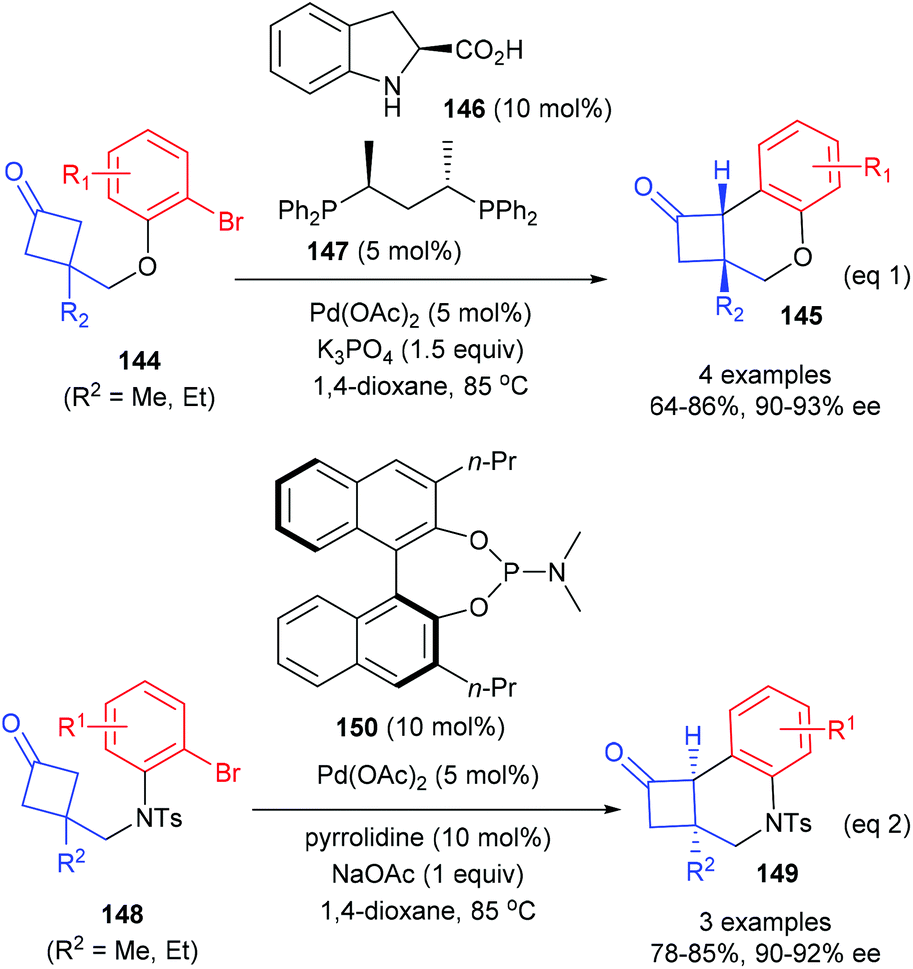
In 2018, Lu and co-workers described that the catalytic enantioselective desymmetrization of cyclobutanones is also an effective route for the synthesis of quaternary carbon containing cyclobutanes (Scheme 41).48 Two different enantioselective control strategies were developed according to the types of substrates. As for the reaction of cyclobutanone substrates bearing O-tethered aryl bromides 144, the merger of (S)-indoline-2-carboxylic acid 146 (10 mol%) as the enamine catalyst and (S,S)-BDPP 147 derived Pd(II) complex (5 mol%) could enable the intramolecular arylation reaction to afford chiral cyclobutanones 145 in 64–86% yields and with 90–93% ee values (eqn (1)), whereas the reaction of substrates bearing N-tethered aryl bromides 144 only requires a chiral phosphoramidite ligand 150 as the chiral source and the targeted products 149 could be obtained with 78–85% yields and up to 92% ee values (eqn (2)).
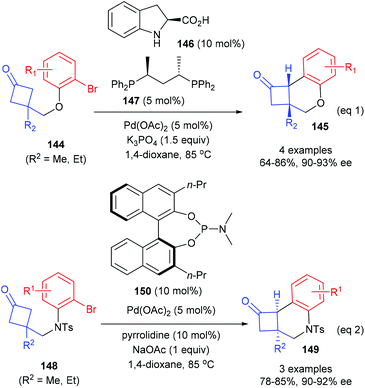 |
| | Scheme 41 Enantioselective desymmetrization of cyclobutanones bearing a tethered aryl bromide. | |
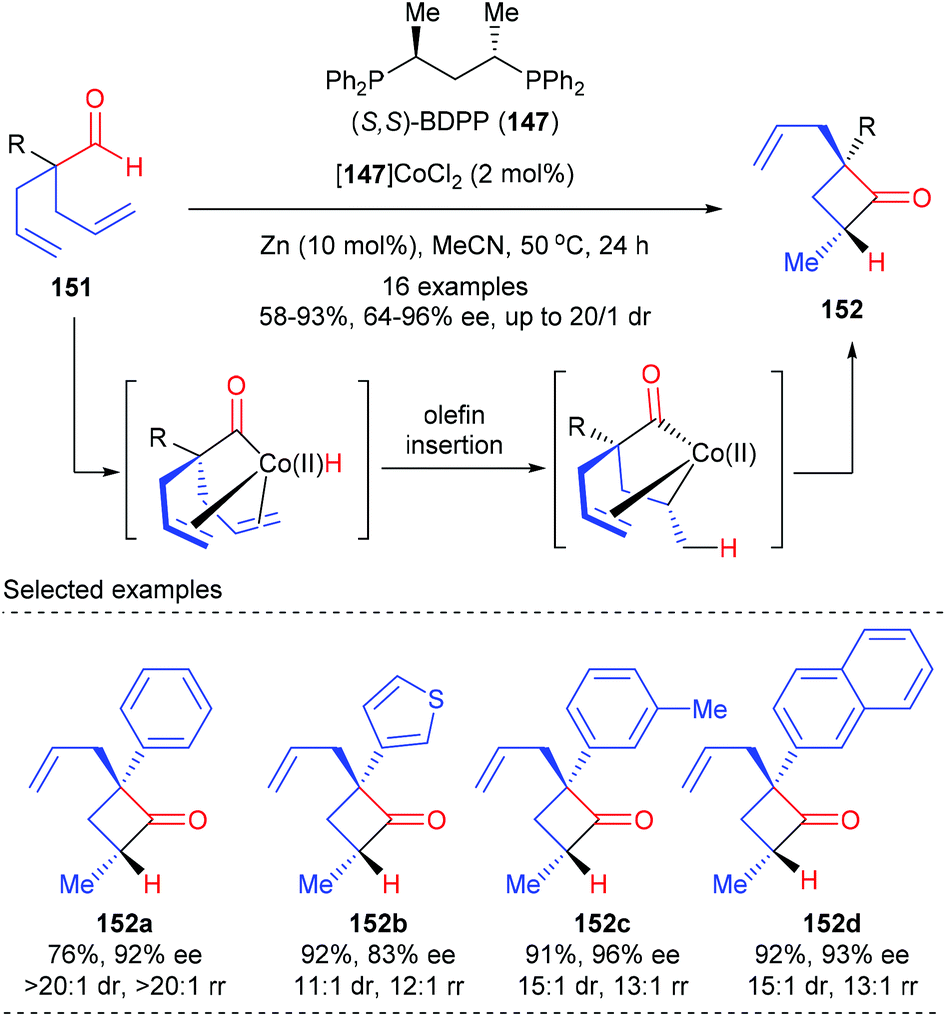
Besides prochiral 4-membered ring substrates, Dong and co-workers have recently reported an alternative cobalt catalyzed intramolecular hydroacylation of acyclic α-aryl dienyl aldehydes to prepare chiral cyclobutanones (Scheme 42).49 In the presence of the (S,S)-BDPP 147 derived Co(II) complex as the optimal catalyst, together with 10 mol% zinc as the reducing agent, a variety of α-aryl dienyl aldehydes 151 underwent intramolecular hydroacylation to afford cyclobutanones 152 as the major product with excellent regio-, diastereo-, and enantiocontrol. The authors proposed that this cyclization was initiated by oxidative addition of Co(0) to the aldehyde C–H bond to form an acyl-Co-hydride intermediate, which underwent subsequent olefin insertion and reductive elimination to construct the strained ring.
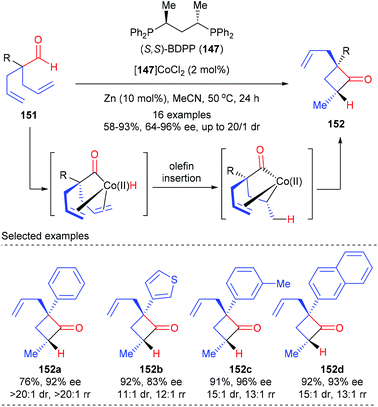 |
| | Scheme 42 Cobalt catalyzed intramolecular hydroacylation of prochiral α-aryl dienyl aldehydes. | |
5. Asymmetric alkylation and miscellaneous
Asymmetric alkylation of in situ generated enolates is one of the most important synthetic routes to α-quaternary carbonyl compounds and has become a fundamental transformation in organic synthesis. As expected, this strategy has also found its application in the synthesis of enantiomerically enriched cyclobutanes bearing quaternary carbon centers.
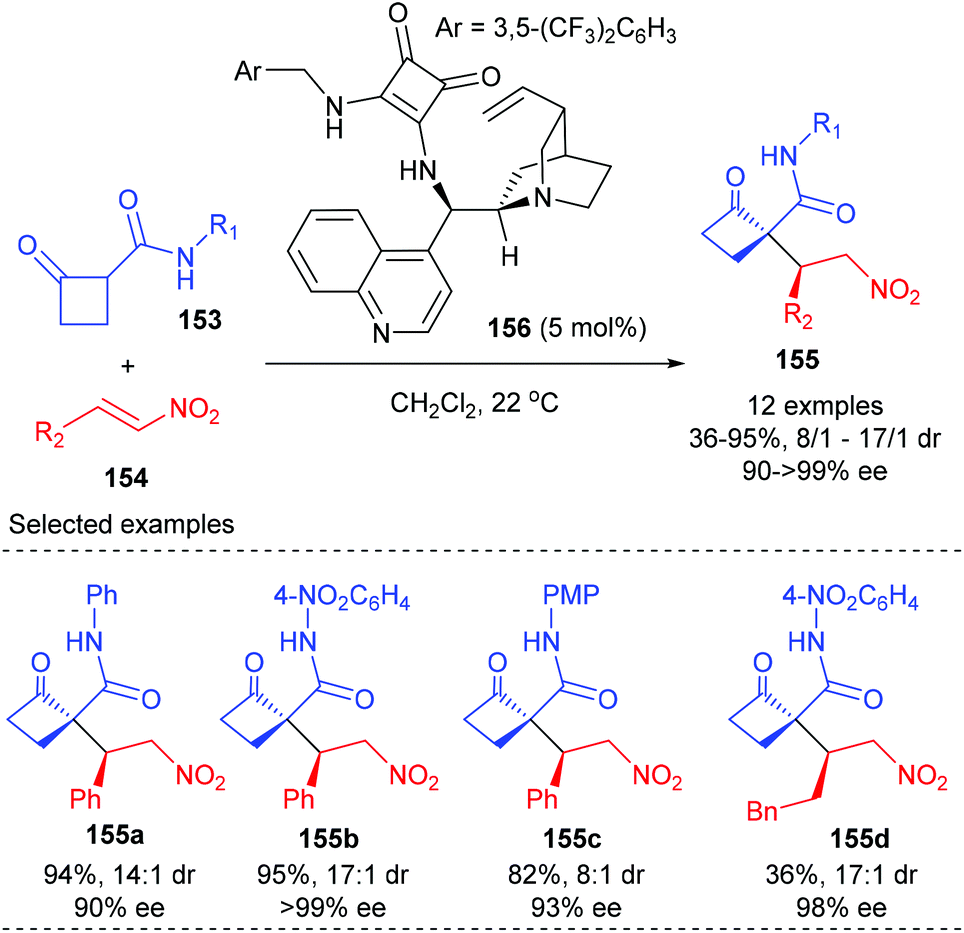
In 2012, Rodriguez and co-workers reported efficient and stereoselective Michael addition reactions of 2-substituted cyclobutanones with nitroalkenes for the construction of densely functionalized cyclobutanones, which are versatile building blocks for further elaboration (Scheme 43).50 Under the catalysis of 5 mol% squaramide-containing organocatalyst 156, the addition of various cyclobutanones 153 to 2-phenyl nitroalkenes 154 could deliver products 155 in good to excellent yields and with >90% ee values. The reaction of 2-alkyl-substituted nitroolefin also gave product 155d with excellent ee value, but with poor yield. It should be mentioned that this work represents the first enantioselective route to cyclobutanones displaying a chiral quaternary carbon center adjacent to an additional controlled stereocenter.
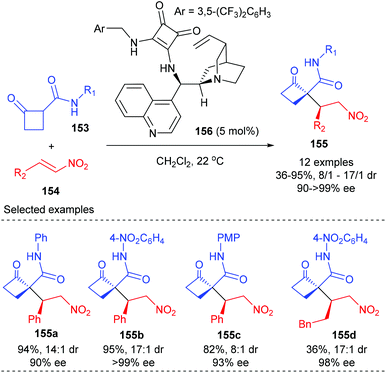 |
| | Scheme 43 Enantioselective Michael addition of cyclobutanones to nitroalkenes. | |
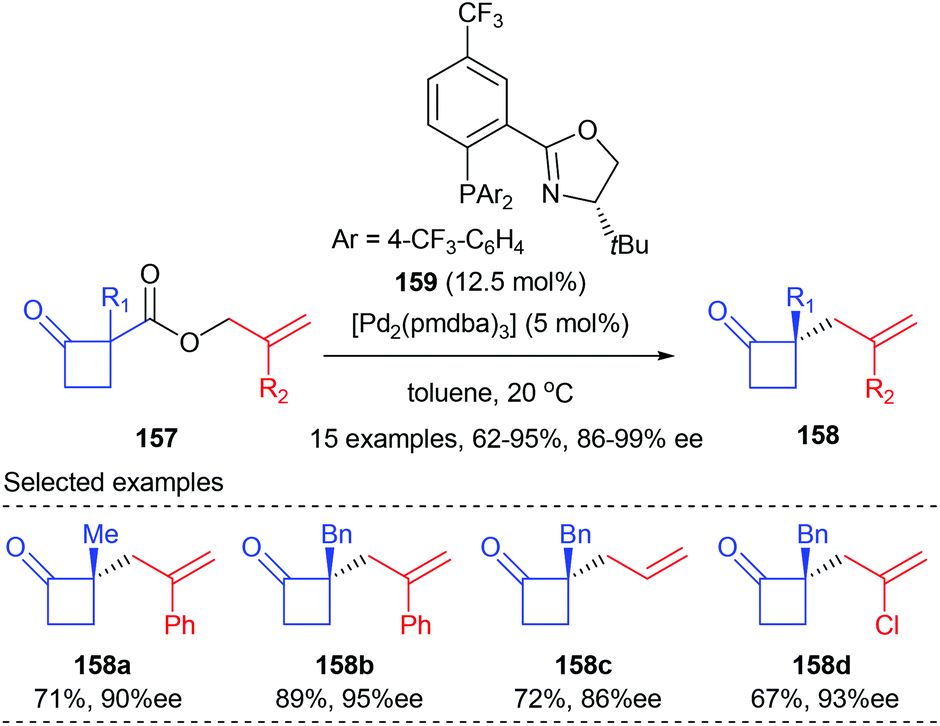
Later in 2013, the Stoltz group demonstrated that the asymmetric alkylation of cyclobutenolates could also be achieved by transition metal catalysis. In the presence of 12.5 mol% PHOX ligand 159 and 5 mol% [Pd2(pmdba)3], allyl cyclobutanecarboxylate 157 could undergo asymmetric decarboxylative allylic alkylation reactions to give the corresponding chiral cyclobutanone products 158 with up to 92% yields and 86–99% ee values (Scheme 44).51 A wide range of substituents are tolerated at both the α-keto and 2-allyl positions. The synthetic utility of the resultant chiral cyclobutanones was demonstrated by transforming into a variety of enantioenriched derivative compounds, such as dialkyl γ-lactams, dialkyl γ-lactones, and α-quaternary cyclopentanones.
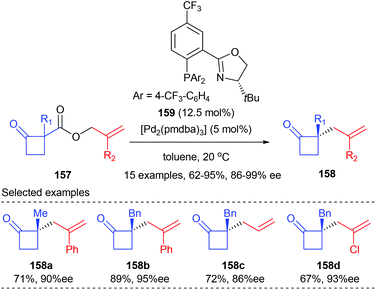 |
| | Scheme 44 Catalytic asymmetric allylic alkylation of cyclobutanones. | |
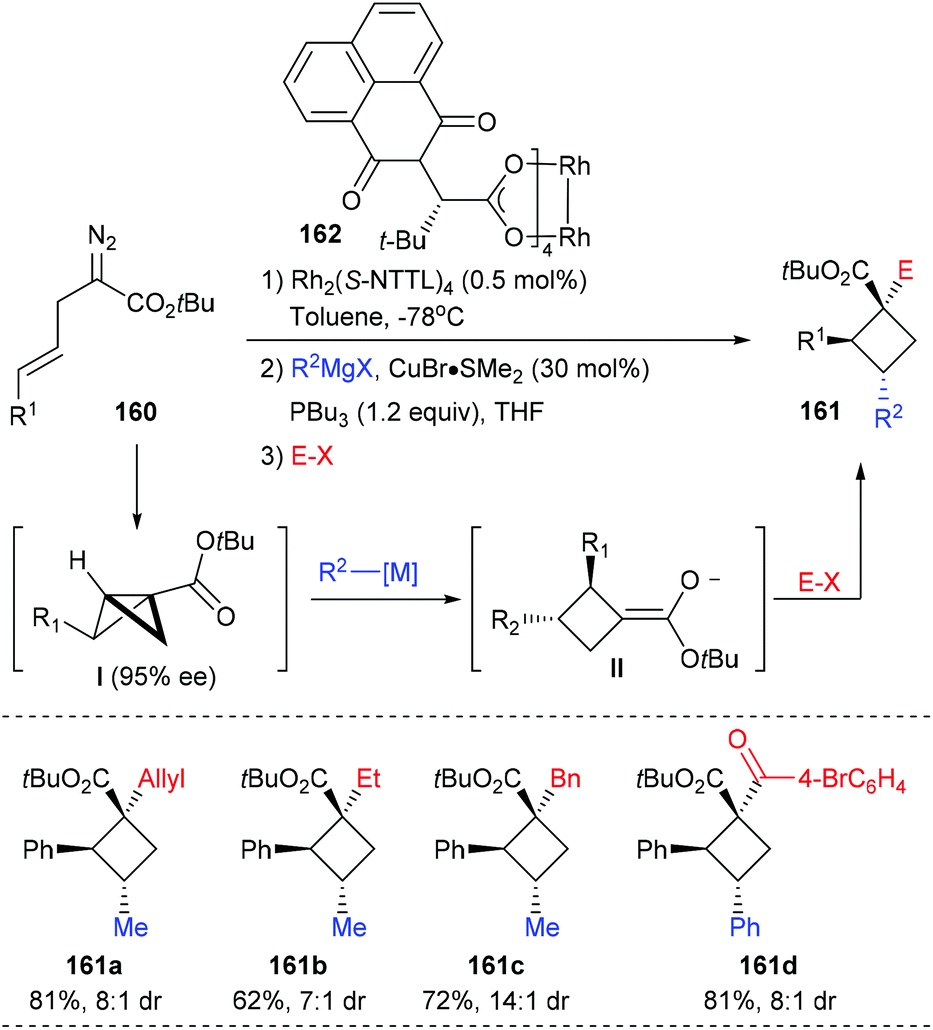
In the same year, Fox et al. reported the construction of chiral quaternary carbon containing cyclobutanes via a 3-component, 2-catalyst, single-flask process (Scheme 45).52 Using 0.5 mol% Rh2(S-NTTL)4 as the catalyst, (E)-2-diazo-5-arylpent-4-enoates 160 first underwent an intramolecular cyclopropanation to afford an enantiomerically enriched bicyclobutane intermediate I, which could then undergo a Cu-catalyzed homo conjugate addition to give exocyclic enolate II. The desired cyclobutanes 161 could be obtained in up to 81% yields after the addition of electrophiles. This process has high potential as many versatile chiral cyclobutanes could be obtained by altering any of the reaction components.
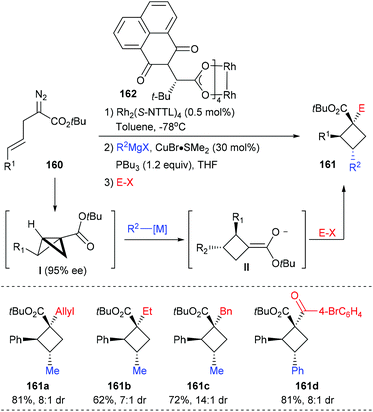 |
| | Scheme 45 Synthesis of enantiomerically enriched cyclobutanes by a three-component process. | |
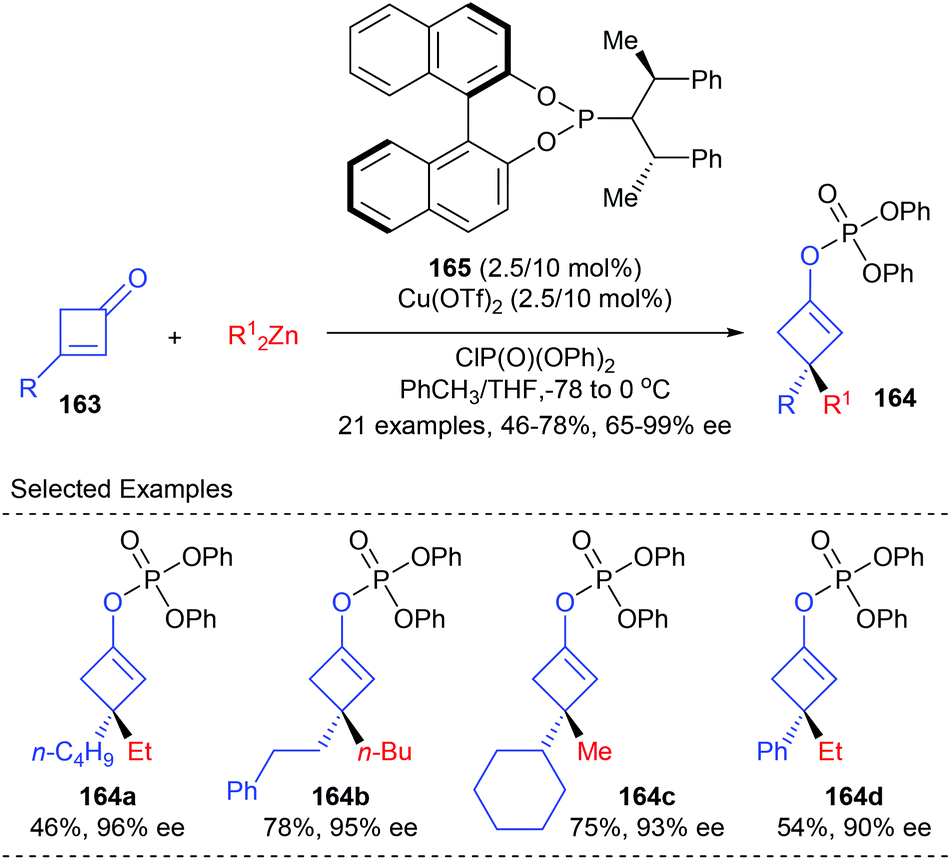
As a part of their ongoing effort to construct enantioenriched cyclobutanes, Lu and co-workers envisioned that the 1,4-addition/trapping reaction of 3-substituted cyclobutenones may offer a direct route to 3,3-disubstituted cyclobutene derivatives (Scheme 46).53 It was found that using the phosphoramidite ligand 165 coordinated Cu(II) complex as the catalyst, dialkyl zinc could react with 3-substituted cyclobutenones 163 to give a chiral metal enol intermediate, which was then in situ trapped by diphenyl chlorophosphate to furnish chiral 3,3-disubstituted cyclobutene derivatives 164 in 46–78% yields and with 65–99% ee values.
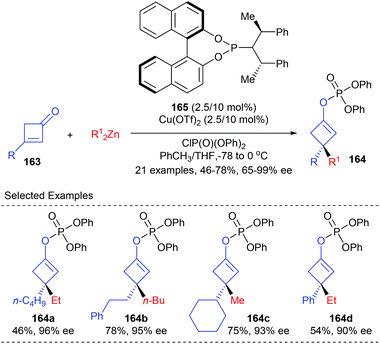 |
| | Scheme 46 Enantioselective 1,4-addition/trapping reaction of 3-substituted cyclobutenones. | |
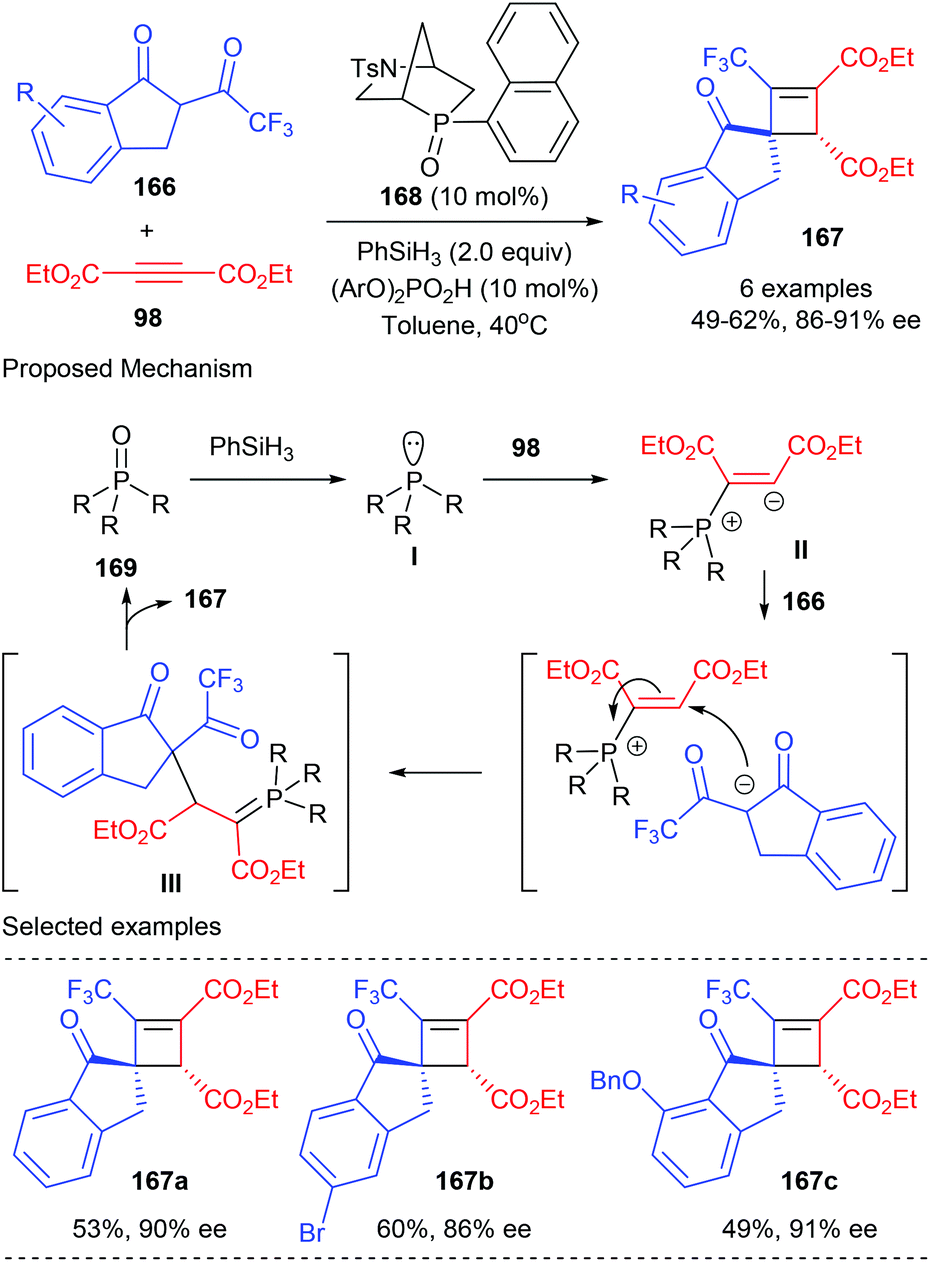
In 2019, the Voituriez group developed the first catalytic asymmetric Michael addition/Wittig reaction to synthesize highly functionalized CF3-substituted spirocyclobutene 167 bearing two contiguous stereogenic centers (Scheme 47).54 This process started from the reduction of the phosphine oxide catalyst 168 to afford chiral phosphine I, which could then add to dialkyl acetylene dicarboxylate 98 to form the zwitterionic species II. Intermolecular proton transfer that occurred between II and CF3-dihydro-1H-indenone 166 and the subsequent addition reaction could form in situ ylide intermediate III. After an intramolecular Wittig olefination reaction, the desired chiral cyclobutene product 167 could be obtained with the regeneration of the catalyst. In the presence of bicyclic chiral phosphine oxides HypPhos 168 (20 mol%), developed by Ohyun Kwon,55 together with phenyl silane as the reducing agent, good isolated yields and up to 91% ee values could be obtained for products 167. Different substituents in different positions of the indenone backbone were well tolerated.
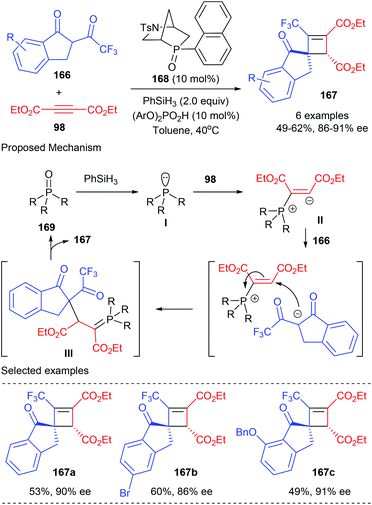 |
| | Scheme 47 Michael addition/Wittig olefination reaction of CF3-substituted spirocyclobutene. | |
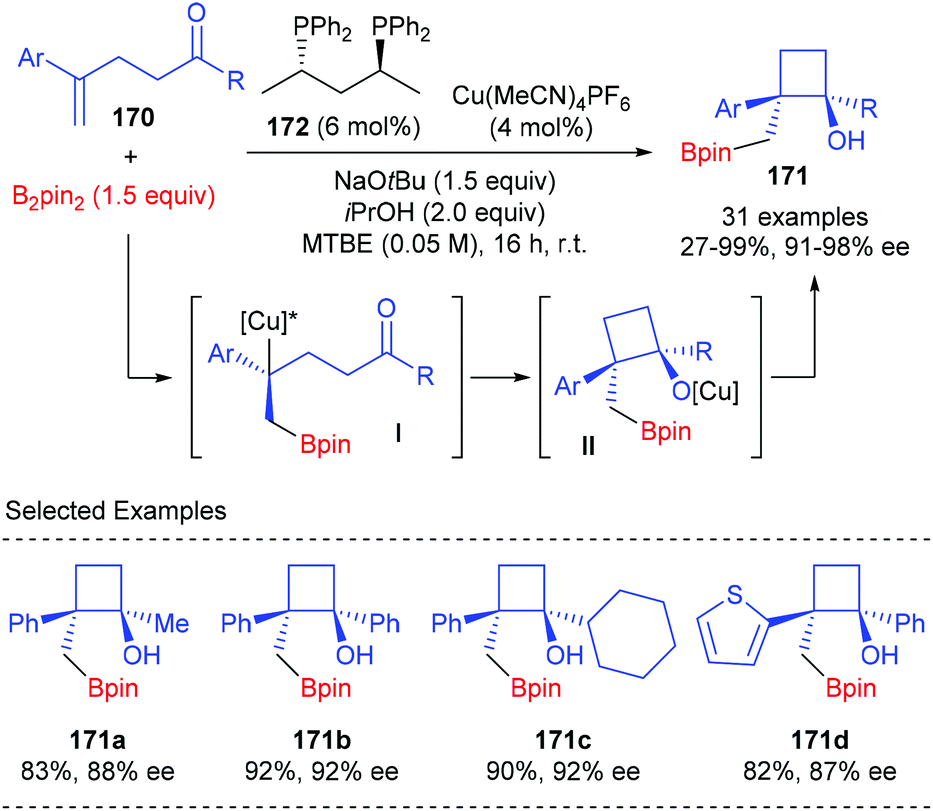
Lautens et al. described a chiral Cu(I) complex catalyzed asymmetric approach to construct boryl-functionalized monocyclic cyclobutanols using 1,1-disubstituted styrenes tethered with a ketone moiety (Scheme 48).56 The reaction was believed to start with the enantioselective borylcupration of 1,1-disubstituted styrenes 170 to generate a chiral benzylic copper intermediate I, which underwent an intramolecular addition with the tethered ketone to form quaternary monocyclic cyclobutanols. In the presence of the (S,S)-BDPP derived Cu(I) complex (4 mol%) as the transition metal catalyst, sodium tert-butoxide as the Lewis base promoter, and isopropanol as an additive, this sequence could afford products 171 in 27–99% yields and with 91–98% ee values.
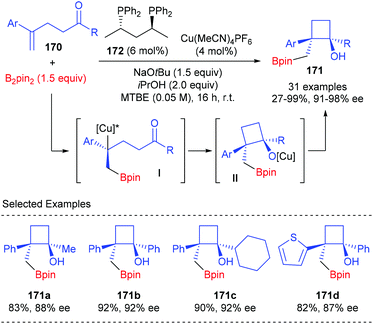 |
| | Scheme 48 Asymmetric synthesis of functionalized cyclobutanols via borylcupration. | |
6. Conclusions and perspectives
Due to the wide existence of enantiomerically active quaternary carbon containing cyclobutane moieties in natural products and bioactive compounds, substantial efforts have been made to develop highly efficient and enantioselective catalytic asymmetric strategies to construct these useful structures. Up to now, four major synthetic routes, including [2 + 2] cycloaddition, ring-expansion, desymmetrization and asymmetric alkylation, have been developed and the asymmetric [2 + 2] cycloaddition reaction is the most widely utilized route. Despite the impressive progress, a number of problems remain to be overcome.
Firstly, catalytic systems that could facilitate a highly enantioselective [2 + 2] cycloaddition between two acyclic alkenes to construct monocyclic cyclobutanes are still rare and the scope of the reported methodologies are relatively limited. Secondly, desymmetrization and ring expansion have emerged as highly potential strategies to obtain chiral quaternary carbon containing cyclobutanes, but the types of substrates used in these reactions are still very limited. In addition, the number of applications of these reactions remains limited compared with the numerous catalytic systems that have already been developed. Therefore, the development of new synthetic strategies to enable the highly enantioselective construction of structurally-unique quaternary carbon containing cyclobutanes and identification of new types of substrates of known strategies are required and these would undoubtedly benefit the total synthesis of related natural products and the development of new drugs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are thankful for the financial support from the National Natural Science Foundation of China (21702080), the Open Project Program of Key Laboratory of Functional Small Organic Molecule, Ministry of Education, Jiangxi Normal University (KLFS-KF-201603) and the foundation of Jiangxi Educational Committee (170223).
References
- For two recent reviews:
(a) M. Wang and P. Lu, Catalytic Approaches to Assemble Cyclobutane Motifs in Natural Product Synthesis, Org. Chem. Front., 2018, 5, 254–259 RSC;
(b) J. Li, K. Gao, M. Bian and H. Ding, Recent Advances in the Total Synthesis of Cyclobutane-Containing Natural Products, Org. Chem. Front., 2020, 7, 136–154 RSC.
-
(a) E. M. Carreira and T. C. Fessard, Four-Membered Ring-Containing Spirocycles: Synthetic Strategies and Opportunities, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed;
(b) C. M. Marson, New and Unusual Scaffolds in Medicinal Chemistry, Chem. Soc. Rev., 2011, 40, 5514 RSC;
(c) P.-W. Xu, J.-S. Yu, C. Chen, Z.-Y. Cao, F. Zhou and J. Zhou, Catalytic Enantioselective Construction of Spiro Quaternary Carbon Stereocenters, ACS Catal., 2019, 9, 1820–1882 CrossRef CAS.
- For selected reviews:
(a) E. Lee-Ruff and G. Mladenova, Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis, Chem. Rev., 2003, 103, 1449–1483 CrossRef CAS PubMed;
(b) D. J. Mack and J. T. Njardarson, Recent Advances in the Metal-Catalyzed Ring Expansions of Three- and Four-Membered Rings, ACS Catal., 2013, 3, 272–286 CrossRef CAS;
(c) E. M. Carreira and T. C. Fessard, Four-Membered Ring-Containing Spirocycles: Synthetic Strategies and Opportunities, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed;
(d) A. Misale, S. Niyomchon and N. Maulide, Cyclobutenes: At a Crossroad between Diastereoselective Syntheses of Dienes and Unique Palladium-Catalyzed Asymmetric Allylic Substitutions, Acc. Chem. Res., 2016, 49, 2444–2458 CrossRef CAS PubMed;
(e) G. Fumagalli, S. Stanton and J. F. Bower, Recent Methodologies That Exploit C–C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed.
-
(a) H. Butenschön, Oxazaborolidines as Catalysts for Enantioselective Cycloadditions: Now [2 + 2]!, Angew. Chem., Int. Ed., 2008, 47, 3492–3495 CrossRef PubMed;
(b) Y. Xu, M. L. Conner and M. K. Brown, Cyclobutane and Cyclobutene Synthesis: Catalytic Enantioselective [2 + 2] Cycloadditions, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS PubMed;
(c) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed;
(d) F. Secci, A. Frongia and P. P. Piras, Stereocontrolled Synthesis and Functionalization of Cyclobutanes and Cyclobutanones, Molecules, 2013, 18, 15541–15572 CrossRef PubMed.
-
(a) H. Ito, M. Hasegawa, Y. Takenaka, T. Kobayashi and K. Iguchi, Enantioselective Total Synthesis of (+)-Tricycloclavulone, J. Am. Chem. Soc., 2004, 126, 4520–4521 CrossRef CAS PubMed;
(b) Y. Takenaka, H. Ito, M. Hasegawa and K. Iguchi, Catalytic Enantioselective [2 + 2]-Cycloaddition Reaction of 2-Methoxycarbonyl-2-Cyclopenten-1-One by Chiral Copper Catalyst, Tetrahedron, 2006, 62, 3380–3388 CrossRef CAS;
(c) Y. Takenaka, H. Ito and K. Iguchi, Enantioselective Formal Synthesis of (+)-Precapnelladiene by Chiral Copper-Catalyzed Asymmetric [2 + 2]-Cycloaddition Reaction, Tetrahedron, 2007, 63, 510–513 CrossRef CAS.
- C. Schotes and A. Mezzetti, Enantioselective Ficini Reaction: Ruthenium/PNNP-Catalyzed [2 + 2] Cycloaddition of Ynamides with Cyclic Enones, Angew. Chem., Int. Ed., 2011, 50, 3072–3074 CrossRef CAS PubMed.
-
(a) K. Enomoto, H. Oyama and M. Nakada, Highly Enantioselective Catalytic Asymmetric [2 + 2] Cycloadditions of Cyclic α-Alkylidene β-Oxo Imides with Ynamides, Chem. – Eur. J., 2015, 21, 2798–2802 CrossRef CAS PubMed; For pioneering work on the development and application of ligand 12:
(b) A. Sakakura, R. Kondo, Y. Matsumura, M. Akakura and K. Ishihara, Rational Design of Highly Effective Asymmetric Diels-Alder Catalysts Bearing 4,4′-Sulfonamidomethyl Groups, J. Am. Chem. Soc., 2009, 131, 17762–17764 CrossRef CAS PubMed;
(c) Y. Matsumura, T. Suzuki, A. Sakakura and K. Ishihara, Catalytic Enantioselective Inverse Electron Demand Hetero-Diels-Alder Reaction with Allylsilanes, Angew. Chem., Int. Ed., 2014, 53, 6131–6134 CrossRef CAS PubMed.
- H.-F. Zheng, C.-R. Xu, Y. Wang, T.-F. Kang, X.-H. Liu, L.-L. Lin and X.-M. Feng, Catalytic Asymmetric [2 + 2] Cycloaddition Between Quinones and Fulvenes and a Subsequent Stereoselective Isomerization to 2,3-Dihydrobenzofurans, Chem. Commun., 2017, 53, 6585–6588 RSC.
- J.-L. Hu, L.-W. Feng, L. Wang, Z. Xie, Y. Tang and X. Li, Enantioselective Construction of Cyclobutanes: A New and Concise Approach to the Total Synthesis of (+)-Piperarborenine B, J. Am. Chem. Soc., 2016, 138, 13151–13154 CrossRef CAS PubMed.
- E. Canales and E. J. Corey, Highly Enantioselective [2 + 2]-Cycloaddition Reactions Catalyzed by a Chiral Aluminum Bromide Complex, J. Am. Chem. Soc., 2007, 129, 12686–12687 CrossRef CAS PubMed.
- K. Ishihara and M. Fushimi, Catalytic Enantioselective [2 + 4] and [2 + 2] Cycloaddition Reactions with Propiolamides, J. Am. Chem. Soc., 2008, 130, 7532–7533 CrossRef CAS PubMed.
- T.-F. Kang, S.-L. Ge, L.-L. Lin, Y. Lu, X.-H. Liu and X.-M. Feng, A Chiral N,N′-Dioxide-ZnII Complex Catalyzes the Enantioselective [2 + 2] Cycloaddition of Alkynones with Cyclic Enol Silyl Ethers, Angew. Chem., Int. Ed., 2016, 55, 5541–5544 CrossRef CAS PubMed.
- J. M. Wiest, M. L. Conner and M. K. Brown, Allenoates in Enantioselective [2 + 2] Cycloadditions: From a Mechanistic Curiosity to a Stereospecific Transformation, J. Am. Chem. Soc., 2018, 140, 15943–15949 CrossRef CAS PubMed.
- S. Suárez-Pantiga, C. Hernndez-Díaz, E. Rubio and J. M. González, Intermolecular [2 + 2] Reaction of N-Allenylsulfonamides with Vinylarenes: Enantioselective Gold(I)-Catalyzed Synthesis of Cyclobutane Derivatives, Angew. Chem., Int. Ed., 2012, 51, 11552–11555 CrossRef PubMed.
- M. Jia, M. Monari, Q.-Q. Yang and M. Bandini, Enantioselective Gold Catalyzed Dearomative [2 + 2]-Cycloaddition between Indoles and Allenamides, Chem. Commun., 2015, 51, 2320–2323 RSC.
- C. García-Morales, B. Ranieri, I. Escofet, L. López-Suarez, C. Obradors, A. I. Konovalov and A. M. Echavarren, Enantioselective Synthesis of Cyclobutenes by Intermolecular [2 + 2] Cycloaddition with Non-C2 Symmetric Digold Catalysts, J. Am. Chem. Soc., 2017, 139, 13628–13631 CrossRef PubMed.
- K. Ishihara and K. Nakano, Enantioselective [2 + 2] Cycloaddition of Unactivated Alkenes with α-Acyloxyacroleins Catalyzed by Chiral Organoammonium Salts, J. Am. Chem. Soc., 2007, 129, 8930–8931 CrossRef CAS.
-
(a) L. Albrecht, G. Dickmeiss, F. Cruz Acosta, C. Rodriguez-Escrich, R. L. Davis and K. A. Jorgensen, Asymmetric Organocatalytic Formal [2 + 2]-Cycloadditions via Bifunctional H-Bond Directing Dienamine Catalysis, J. Am. Chem. Soc., 2012, 134, 2543–2546 CrossRef CAS;
(b) G. Talavera, E. Reyes, J. L. Vicario and L. Carrillo, Cooperative Dienamine/Hydrogen-Bonding Catalysis: Enantioselective Formal [2 + 2] Cycloaddition of Enals with Nitroalkenes, Angew. Chem., Int. Ed., 2012, 51, 4104–4107 CrossRef CAS PubMed;
(c) G.-J. Duan, J.-B. Ling, W.-P. Wang, Y.-C. Luo and P.-F. Xu, Organocatalytic Formal [2 + 2] Cycloaddition Initiated by Vinylogous Friedel-Crafts Alkylation: Enantioselective Synthesis of Substituted Cyclobutane Derivatives, Chem. Commun., 2013, 49, 4625–4627 RSC.
- L.-W. Qi, Y. Yang, Y.-Y. Gui, Y. Zhang, F. Chen, F. Tian, L. Peng and L.-X. Wang, Asymmetric Synthesis of 3,3′-Spirooxindoles Fused with Cyclobutanes through Organocatalytic Formal [2 + 2] Cycloadditions under H-Bond-Directing Dienamine Activation, Org. Lett., 2014, 16, 6436–6439 CrossRef CAS PubMed.
- K. S. Halskov, F. Kniep, V. H. Lauridsen, E. H. Iversen, B. S. Donslund and K. A. Jørgensen, Organocatalytic Enamine-Activation of Cyclopropanes for Highly Stereoselective Formation of Cyclobutanes, J. Am. Chem. Soc., 2015, 137, 1685–1691 CrossRef CAS PubMed.
- N. J. Line, B. P. Witherspoon, E. N. Hancock and M. K. Brown, Synthesis of ent,-[3]-Ladderanol: Development and Application of Intramolecular Chirality Transfer [2 + 2] Cycloadditions of Allenic Ketones and Alkenes, J. Am. Chem. Soc., 2017, 139, 14392–14395 CrossRef CAS PubMed.
-
(a) H. Yamamoto and K. Futatsugi, “Designer Acids”: Combined Acid Catalysis for Asymmetric Synthesis, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef CAS PubMed;
(b)
J. Zhou, Multicatalyst System in Asymmetric Catalysis, John Wiley & Sons, New York, 2014, ch. 6 Search PubMed.
- T. Sakamoto, T. Mochizuki, Y. Goto, M. Hatano and K. Ishihara, Boron Tribromide-Assisted Chiral Phosphoric Acid Catalysts for Enantioselective [2 + 2] Cycloaddition, Chem. – Asian J., 2018, 13, 2373–2377 CrossRef CAS PubMed.
- V. V. Pagar and T. V. RajanBabu, Tandem Catalysis for Asymmetric Coupling of Ethylene and Enynes to Functionalized Cyclobutanes, Science, 2018, 361, 68–72 CrossRef CAS.
-
(a) H. Guo, E. Herdtweck and T. Bach, Enantioselective Lewis Acid Catalysis in Intramolecular [2 + 2] Photocycloaddition Reactions of Coumarins, Angew. Chem., Int. Ed., 2010, 49, 7782–7785 CrossRef CAS PubMed;
(b) R. Brimioulle, H. Guo and T. Bach, Enantioselective Intramolecular [2 + 2] Photocycloaddition Reactions of 4-Substituted Coumarins Catalyzed by a Chiral Lewis Acid, Chem. – Eur. J., 2012, 18, 7552–7560 CrossRef CAS PubMed;
(c) S. Poplata, A. Bauer, G. Storch and T. Bach, Intramolecular [2 + 2] Photocycloaddition of Cyclic Enones: Selectivity Control by Lewis Acids and Mechanistic Implications, Chem. – Eur. J., 2019, 25, 8135–8148 CrossRef CAS PubMed.
- S. Poplata and T. Bach, Enantioselective Intermolecular [2 + 2] Photocycloaddition Reaction of Cyclic Enones and Its Application in a Synthesis of (-)-Grandisol, J. Am. Chem. Soc., 2018, 140, 3228–3231 CrossRef CAS PubMed.
- S. Stegbauer, C. Jandl and T. Bach, Enantioselective Lewis Acid Catalyzed ortho Photocycloaddition of Olefins to Phenanthrene-9-carboxaldehydes, Angew. Chem., Int. Ed., 2018, 57, 14593–14596 CrossRef CAS PubMed.
-
(a) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Enantioselective Photochemistry through Lewis Acid-Catalyzed Triplet Energy Transfer, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed;
(b) Z. D. Miller, B. J. Lee and T. P. Yoon, Enantioselective Crossed Photocycloadditions of Styrenic Olefins by Lewis Acid Catalyzed Triplet Sensitization, Angew. Chem., Int. Ed., 2017, 56, 11891–11895 CrossRef CAS.
- X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, Direct Visible-Light-Excited Asymmetric Lewis Acid Catalysis of Intermolecular [2 + 2] Photocycloadditions, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS PubMed.
- H. Yu, S. Dong, Q. Yao, L. Chen, D. Zhang, X. Liu and X. Feng, Enantioselective [2 + 2] Photocycloaddition Reactions of Enones and Olefins with Visible Light Mediated by N,N′-Dioxide-Metal Complexes, Chem. – Eur. J., 2018, 24, 19361–19367 CrossRef CAS PubMed.
- J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M.-H. Baik and T. P. Yoon, Enantioselective Intermolecular Excited-State Photoreactions Using a Chiral Ir Triplet Sensitizer: Separating Association from Energy Transfer in Asymmetric Photocatalysis, J. Am. Chem. Soc., 2019, 141, 13625–13634 CrossRef CAS PubMed.
-
(a) C. Müller, A. Bauer, M. M. Maturi, M. C. Cuquerella, M. A. Miranda and T. Bach, Enantioselective Intramolecular [2 + 2]-Photocycloaddition Reactions of 4-Substituted Quinolones Catalyzed by a Chiral Sensitizer with a Hydrogen-Bonding Motif, J. Am. Chem. Soc., 2011, 133, 16689–16697 CrossRef PubMed;
(b) M. M. Maturi and T. Bach, Enantioselective Catalysis of the Intermolecular [2 + 2] Photocycloaddition between 2-Pyridones and Acetylenedicarboxylates, Angew. Chem., Int. Ed., 2014, 53, 7661–7664 CrossRef CAS PubMed.
-
(a) R. Alonso and T. Bach, A Chiral Thioxanthone as
an Organocatalyst for Enantioselective [2 + 2] Photocycloaddition Reactions Induced by Visible Light, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS PubMed.
-
(a) A. Tröster, R. Alonso, A. Bauer and T. Bach, Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions of 2(1H)-Quinolones Induced by Visible Light Irradiation, J. Am. Chem. Soc., 2016, 138, 7808–7811 CrossRef PubMed;
(b) X. Li, C. Jandl and T. Bach, Visible-Light-Mediated Enantioselective Photoreactions of 3-Alkylquinolones with 4-O-Tethered Alkenes and Allenes, Org. Lett., 2020, 22, 3618–3622 CrossRef CAS PubMed.
- N. Vallavoju, S. Selvakumar, S. Jockusch, M. P. Sibi and J. Sivaguru, Enantioselective Organo-Photocatalysis Mediated by Atropisomeric Thiourea Derivatives, Angew. Chem., Int. Ed., 2014, 53, 5604–5608 CrossRef CAS.
- F. M. Hörmann, C. Kerzig, T. S. Chung, A. Bauer, O. S. Wenger and T. Bach, Triplet Energy Transfer from Ruthenium Complexes to Chiral Eniminium Ions: Enantioselective Synthesis of Cyclobutanecarbaldehydes by [2 + 2] Photocycloaddition, Angew. Chem., Int. Ed., 2020, 59, 9659–9668 CrossRef PubMed.
- T. Rigotti, R. Mas-Ballesté and J. Alemán, Enantioselective Aminocatalytic [2 + 2] Cycloaddition through Visible Light Excitation, ACS Catal., 2020, 10, 5335–5346 CrossRef CAS.
- O. G. Kulinkovich, The Chemistry of Cyclopropanols, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS PubMed.
- B. M. Trost and T. Yasukata, A Catalytic Asymmetric Wagner-Meerwein Shift, J. Am. Chem. Soc., 2001, 123, 7162–7163 CrossRef CAS PubMed.
- F. Kleinbeck and F. D. Toste, Gold(I)-Catalyzed Enantioselective Ring Expansion of Allenylcyclopropanols, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS PubMed.
-
(a) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Enantioselective Organocatalytic Fluorination-Induced Wagner-Meerwein Rearrangement, Angew. Chem., Int. Ed., 2013, 52, 9266–9270 CrossRef CAS PubMed;
(b) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Enantioselective Organocatalytic Iodination-Initiated Wagner-Meerwein Rearrangement, Org. Lett., 2013, 15, 5890–5893 CrossRef CAS PubMed;
(c) F. Romanov-Michailidis, M. Pupier, L. Guénée and A. Alexakis, Enantioselective Halogenative Semi-Pinacol Rearrangement: A Stereodivergent Reaction on a Racemic Mixture, Chem. Commun., 2014, 50, 13461–13464 RSC;
(d) F. Romanov-Michailidis, M. Romanova-Michaelides, M. Pupier and A. Alexakis, Enantioselective Halogenative Semi-Pinacol Rearrangement: Extension of Substrate Scope and Mechanistic Investigations, Chem. – Eur. J., 2015, 21, 5561–5583 CrossRef CAS PubMed.
- Y.-Y. Xie, Z.-M. Chen, H.-Y. Luo, H. Shao, Y.-Q. Tu, X. Bao, R. F. Cao, S.-Y. Zhang and J.-M. Tian, Lewis Base/Brønsted Acid Co-Catalyzed Enantioselective Sulfenylation/Semipinacol Rearrangement of Di- and Trisubstituted Allylic Alcohols, Angew. Chem., Int. Ed., 2019, 58, 12491–12496 CrossRef CAS PubMed.
- S. Y. Shim, Y. Choi and D. H. Ryu, Asymmetric Synthesis of Cyclobutanone via Lewis Acid Catalyzed Tandem Cyclopropanation/Semipinacol Rearrangement, J. Am. Chem. Soc., 2018, 140, 11184–11188 CrossRef CAS PubMed.
- X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Catalytic Enantioselective Desymmetrization Reactions to All-Carbon Quaternary Stereocenters, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS PubMed.
- K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, Palladium(II)-Catalyzed Enantioselective C(sp3)-H Activation Using a Chiral Hydroxamic Acid Ligand, J. Am. Chem. Soc., 2014, 136, 8138–8142 CrossRef CAS PubMed.
- L. Hu, P.-X. Shen, Q. Shao, K. Hong, J. X. Qiao and J.-Q. Yu, PdII-Catalyzed Enantioselective C(sp3)-H Activation/Cross-Coupling Reactions of Free Carboxylic Acids, Angew. Chem., Int. Ed., 2019, 58, 2134–2138 CrossRef CAS PubMed.
-
(a) J. He, Q. Shao, Q. Wu and J.-Q. Yu, Pd(II)-Catalyzed Enantioselective C(sp3)-H Borylation, J. Am. Chem. Soc., 2017, 139, 3344–3347 CrossRef CAS PubMed;
(b) X. Chen, L.-L. Chen, H.-L. Zhao, Q. Gao, Z.-L. Shen and S.-M. Xu, Iridium-Catalyzed Enantioselective C(sp3)-H Borylation of Cyclobutanes, Chin. J. Chem., 2020, 38 DOI:10.1002/cjoc.202000240.
- M. Wang, J. Chen, Z. Chen, C. Zhong and P. Lu, Enantioselective Desymmetrization of Cyclobutanones Enabled by Synergistic Palladium/Enamine Catalysis, Angew. Chem., Int. Ed., 2018, 57, 2707–2711 CrossRef CAS PubMed.
- D. K. Kim, J. Riedel, R. S. Kim and V. M. Dong, Cobalt Catalysis for Enantioselective Cyclobutanone Construction, J. Am. Chem. Soc., 2017, 139, 10208–10211 CrossRef CAS PubMed.
-
(a) D. Mailhol, M. d. M. S. Duque, W. Raimondi, D. Bonne, T. Constantieux, Y. Coquerel and J. Rodriguez, Enantioselective Organocatalytic Michael Addition of Cyclobutanones to Nitroalkenes, Adv. Synth. Catal., 2012, 354, 3523–3532 CrossRef CAS;
(b) Y. Zhou, Y.-L. Wei, J. Rodriguez and Y. Coquerel, Enantioselective Organocatalytic Four-Atom Ring Expansion of Cyclobutanones: Synthesis of Benzazocinones, Angew. Chem., Int. Ed., 2019, 58, 456–460 CrossRef CAS PubMed;
(c) Y.-L. Wei, Y. Ren, D. Mailhol, M. Rajzmann, J. Rodriguez and Y. Coquerela, An Organocatalytic Two-atom Ring Expansion Approach to Optically Active Glutarimides, Adv. Synth. Catal., 2019, 361, 2992–3001 CrossRef CAS.
- C. M. Reeves, C. Eidamshaus, J. Kim and B. M. Stoltz, Enantioselective Construction of α-Quaternary Cyclobutanones by Catalytic Asymmetric Allylic Alkylation, Angew. Chem., Int. Ed., 2013, 52, 6718–6721 CrossRef CAS PubMed.
- R. Panish, S. R. Chintala, D. T. Boruta, Y. Fang, M. T. Taylor and J. M. Fox, Enantioselective Synthesis of Cyclobutanes via Sequential Rh-catalyzed Bicyclobutanation/Cu-catalyzed Homoconjugate Addition, J. Am. Chem. Soc., 2013, 135, 9283–9286 CrossRef CAS PubMed.
- C. Zhong, Y. Huang, H. Zhang, Q. Zhou, Y. Liu and P. Lu, Enantioselective Synthesis of 3-Substituted Cyclobutenes by Catalytic Conjugate Addition/Trapping Strategies, Angew. Chem., Int. Ed., 2020, 59, 2750–2754 CrossRef CAS PubMed.
- C. Lorton, T. Castanheiro and A. Voituriez, Catalytic and Asymmetric Process via PIII/PV=O Redox Cycling: Access to (Trifluoromethyl)cyclobutenes via a Michael Addition/Wittig Olefination Reaction, J. Am. Chem. Soc., 2019, 141, 10142–10147 CrossRef CAS.
-
(a) C. E. Henry, Q. H. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, Hydroxyproline-Derived Pseudoenantiomeric [2.2.1] Bicyclic Phosphines: Asymmetric Synthesis of (+)- and (−)-Pyrrolines, J. Am. Chem. Soc., 2014, 136, 11890–11893 CrossRef CAS PubMed;
(b) Q. Xu, N. J. Dupper, A. J. Smaligo, Y. C. Fan, L. Cai, Z. Wang, A. D. Langenbacher, J.-N. Chen and O. Kwon, Catalytic Enantioselective Synthesis of Guvacine Derivatives through [4 + 2] Annulations of Imines with α-Methylallenoates, Org. Lett., 2018, 20, 6089–6093 CrossRef CAS PubMed.
- A. Whyte, B. Mirabi, A. Torelli, L. Prieto, J. Bajohr and M. Lautens, Asymmetric Synthesis of Boryl-Functionalized Cyclobutanols, ACS Catal., 2019, 9, 9253–9258 CrossRef CAS.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*