
Cavity-based applications of metallo-supramolecular coordination cages (MSCCs)
Sarita
Yadav
,
Palanisamy
Kannan
 * and
Guanyinsheng
Qiu
*
* and
Guanyinsheng
Qiu
*
College of Biological, Chemical Science and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, Zhejiang Province, P. R. China. E-mail: ktpkannan@mail.zjxu.edu.cn; qiuguanyinsheng@mail.zjxu.edu.cn
First published on 20th July 2020
Abstract
The microenvironment within self-assembled supramolecular coordination cages (SCCs) inaugurates a new phase of chemistry other than the conventional solid, liquid and gaseous phases. The rational design and ease of synthesis of self-assembled architectures have received tremendous attention mainly in the field of supramolecular chemistry. The structural transition from 2-D metallacycles to 3-D metallacages has led to better encapsulation and enhanced cavity effects, like restricted substrate motion, desolvation of the substrate and covalent binding of the transition state etc. Metal-mediated self-assemblies, principally known as metallo-supramolecular coordination cages (MSCCs), form a separate class of SCCs with unique chemical phenomena inside the cavities. The aesthetic structural diversity of MSCCs obtained from the rational choice of metal knots and organic linkers renders designable and programmable architectures. This review presents cavity-based applications of these MSCCs, such as molecular recognition and separation, stabilization of reactive species by encapsulation, as drug-delivery systems and as “molecular flasks” to foster reactions with unprecedented results. The objective of this review is to inspire and pave the way for researchers working in this field to design more advanced MSCCs and gain deep insight into cavity effects.
 Sarita Yadav | Dr Sarita Yadav obtained her Ph.D. degree from the University of Delhi, India in 2018. During her Ph.D., she worked on the synthesis and applications of transition metal-based (silver, copper and rheniumm) coordination complexes and metallacycles. Currently she is working as a postdoctoral fellow in Prof. Guanyinsheng Qiu's research group, in the College of Biological and Chemical Sciences and Engineering, Jiaxing University, China. Her research interests include supramolecular chemistry and transition metal catalyzed organic synthesis. |
 Palanisamy Kannan | Dr Palanisamy Kannan received his Ph.D. in 2010 from Gandhigram Rural Institute–Deemed University, India. Dr Kannan completed his post-doctoral programs in the Institute of Physical Chemistry, Warsaw, Poland, and Nanyang Technological University, Singapore. He is now Associate Professor in the College of Biological, Chemical Sciences and Engineering, Jiaxing University. China. He has published more than 60 papers in journals including Nano Energy, Journal of Materials Chemistry A, Biosensors and Bioelectronics, ACS Applied Materials and Interfaces, Sensors and Actuators Part B, and Analytical Chemistry. His research interests are mainly the development of inspired organic and inorganic nanomaterials for sensor and energy applications. |
 Guanyinsheng Qiu | Dr Guanyinsheng Qiu was born in Jiangxi (China) in 1984. He received his BS (2008) and MS (2011) from the Department of Chemistry at Jiangxi Normal University. He obtained his Ph.D. degree from Fudan University in 2014. Later, he worked as a Postdoctoral Fellow in l’École Polytechnique Fédérale de Lausanne, Switzerland. He now works in Jiaxing University as a Full Professor. He has published more than 90 papers in journals including Angewandte Chemie, Chemical Society Review, Organic Letters, Chemical Communications, and Advanced Synthesis Catalysis. His current research interests focus mainly on green synthetic methodological development involving transition metal catalysis and photocatalysis. |
1. Introduction
The application of coordination bonds for the design of molecular hosts was a critical development in the area of supramolecular chemistry. Coordination-driven self-assembly has proved to be a fascinating tool for the design and development of discrete supramolecular architectures with diverse geometries and functionalities due to the directional nature of the metal–ligand coordination bond.1,2 Directional metal–ligand dative bonds allow the component units to act as programmed moieties, which self-assemble in solution to give desired architectures.1 Complementarity of size and shape between various components of the SCC is crucial for self-assembly. Hence, supramolecular architectures can be rationally designed in accordance with their application in various fields like catalysis, host–guest chemistry, sensors etc.2–4 Certain reversible metal–ligand bonds thermodynamically control the process of self-assembly, which permits ‘self-error correction’ and fine-tuning of the final architecture. Using self-assembly and incorporating different functional groups, complex supramolecular hosts with diverse shapes, sizes and chemical environments of the cavity could be easily designed from relatively simple components.1,3,5 The metal-directed self-assembly produces stable nanometer-sized hollow complexes as suitable hosts with tunable cavities to encapsulate one large or several smaller molecules. The guests within these isolated cavities are exposed to a completely different microenvironment and interactions with the host alone or with co-encapsulated guests. These reduced interactions alter the properties of bound guests and host complexes, leading to their applications in recognition and separation,6–10 sensitive and reactive species stabilization,11–13 catalysis,2,14–16 biological processes for drug delivery,4,17–19 and as catalyst enhancers (increasing catalytic activity by encapsulation)20 and nanoscale chemical vessels.21 This review systematically describes the use of metal–ligand self-assembled nanocages for cavity-based applications. The selective encapsulation of one or more molecules isolates them from the bulk solution, inducing special conformations, increasing effective concentrations, and stabilizing reactive and sensitive intermediates, resulting in unique features. With the help of this review, the authors aim to draw the attention of researchers working in supramolecular chemistry to a detailed exploration of the microenvironment within cavities.Since the pioneering work of Lehn, Pedersen and Cram, a large number of organic supramolecular coordination complexes, including crown ethers,22,23 cavitands,24 cryptands, cyclodextrins,25,26 calixarenes,27 cucurbiturils,28 dendrimers, and hydrogen-bonded capsules,2 have been developed and reviewed excellently elsewhere,29–32 but the use of the cavity of MSCCs for recognition and separation, the stabilization of reactive species, drug delivery and synthetic transformations within the cavity are discussed herein with specific examples in each section.
The review explains that MSCCs have cavities which can encapsulate specific species, depending on the shape, size and charge of the cages and guests. Since the microenvironment within the cages is completely different from the bulk solution, the guests behave differently in the cage cavity both physically and chemically.21 The selective encapsulation, stabilization of unstable species, unusual chemo- and stereoselectivity and enzyme-like rate acceleration observed in many transformations have made them more exciting. The altered properties encountered by guests are a result of the microenvironment within the cavity of the cage complexes and here we review various cavity effects, like restricted substrate motion, desolvation of the substrate and covalent binding of the transition state.33 The review has been systematically divided into four sections: molecular recognition and separation, stabilization of unstable and sensitive species, drug-delivery systems, and MSCCs as reaction vessels. Each section has been sub-divided into metallo-supramolecular coordination cages that have cavity-based applications in that particular field. The MSCCs have been classified on the basis of their shapes, like octahedral, tetrahedral, trigonal prismatic, cubic, spherical, etc. All cages with similar shapes, studied by different research groups are discussed under the same category.
2. Molecular recognition and separation
In this section a range of metal-based supramolecular coordination (cage) complexes are described with their applications in molecular recognition and separation by encapsulation within their confined cavities.2.1. Octahedral MSCCs (cage I)
In 1995 Fujita and co-workers synthesized M6L4 octahedral cage I (Fig. 1) using a triangular exo-tridentate ligand 2,4,6-tris(4-pyridyl)-1,3,5-triazine (TPT), and cis-protected palladium(II) as the building block.34 This metal complex is now commercially available as it can be self-assembled on large scale by simply mixing the metal salt and ligand in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio in water. The cis end-capped metal unit, Pd(II), occupies the six octahedron vertices, and four exo-tridentate triangular ligands occupy eight alternate triangular faces. Cage I has a roughly spherical cavity with a diameter of ∼2–5 nm and tetrahedral symmetry. Cage I displays some attractive features like the selective recognition of molecules and their encapsulation.35,36
:2 ratio in water. The cis end-capped metal unit, Pd(II), occupies the six octahedron vertices, and four exo-tridentate triangular ligands occupy eight alternate triangular faces. Cage I has a roughly spherical cavity with a diameter of ∼2–5 nm and tetrahedral symmetry. Cage I displays some attractive features like the selective recognition of molecules and their encapsulation.35,36
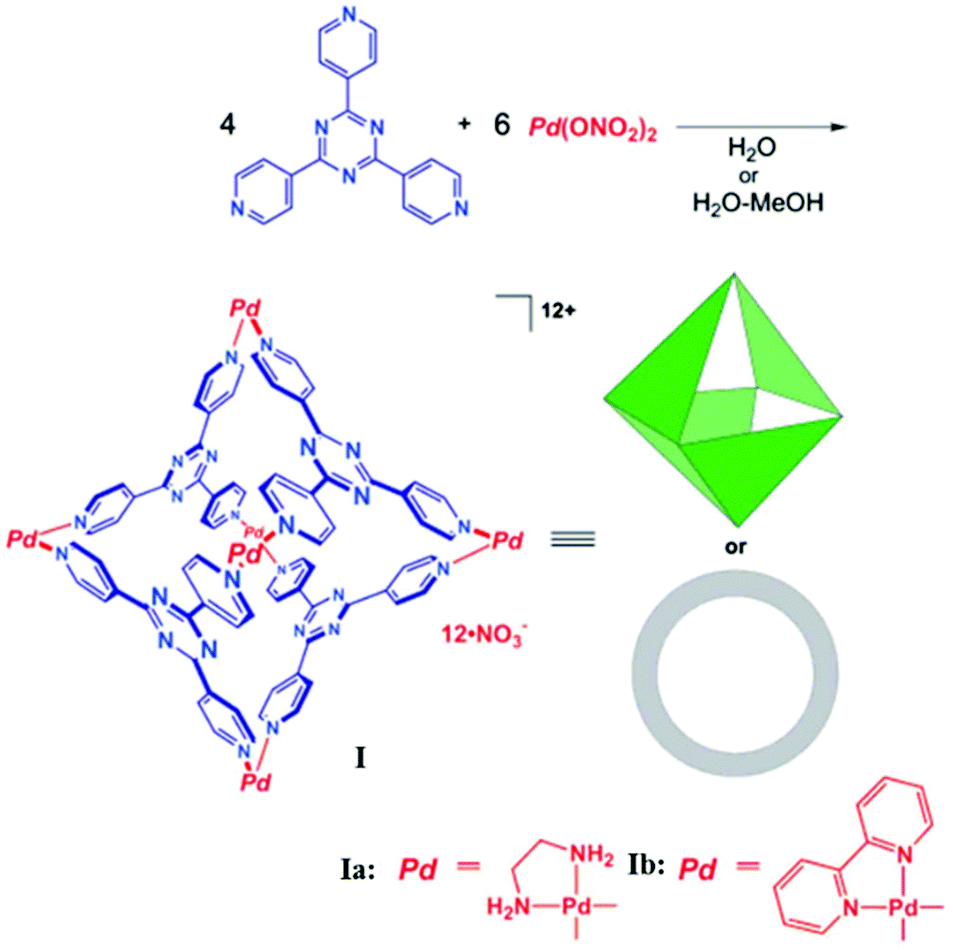 | ||
| Fig. 1 Synthesis and structural representation of octahedral coordination cage I. Reproduced with permission from ref. 35. Copyright 2002, American Chemical Society. | ||
Due to the 12+ charge on the cage and the exposure of cationic Pd(II) units at the periphery, it is highly water soluble and strongly encapsulates both neutral and anionic organic molecules in its hydrophobic cavity.36 For example, the large central void of supramolecular coordination complex I accommodated four adamantyl carboxylate ions, when water-insoluble adamantane (1) gradually dissolved in an aqueous solution of cage I, forming I·(1)4 as an inclusion complex. Fujita and co-workers confirmed the exceptional encapsulation of many large, neutral molecules in the cavity of complex I as I·(G)4 (where G = o-carborane, 1-adamantanol, 2-adamantanol or trimethoxybenzene), I·(G)2 (where G = cis-azobenzene, a stilbene derivative, diphenylmethane or 1,2-bis(4-methoxyphenyl)-1,2-ethanedione) and I·G (where G = tri-tert-butylbenzene or tetrabenzylsilane) (Fig. 2).35
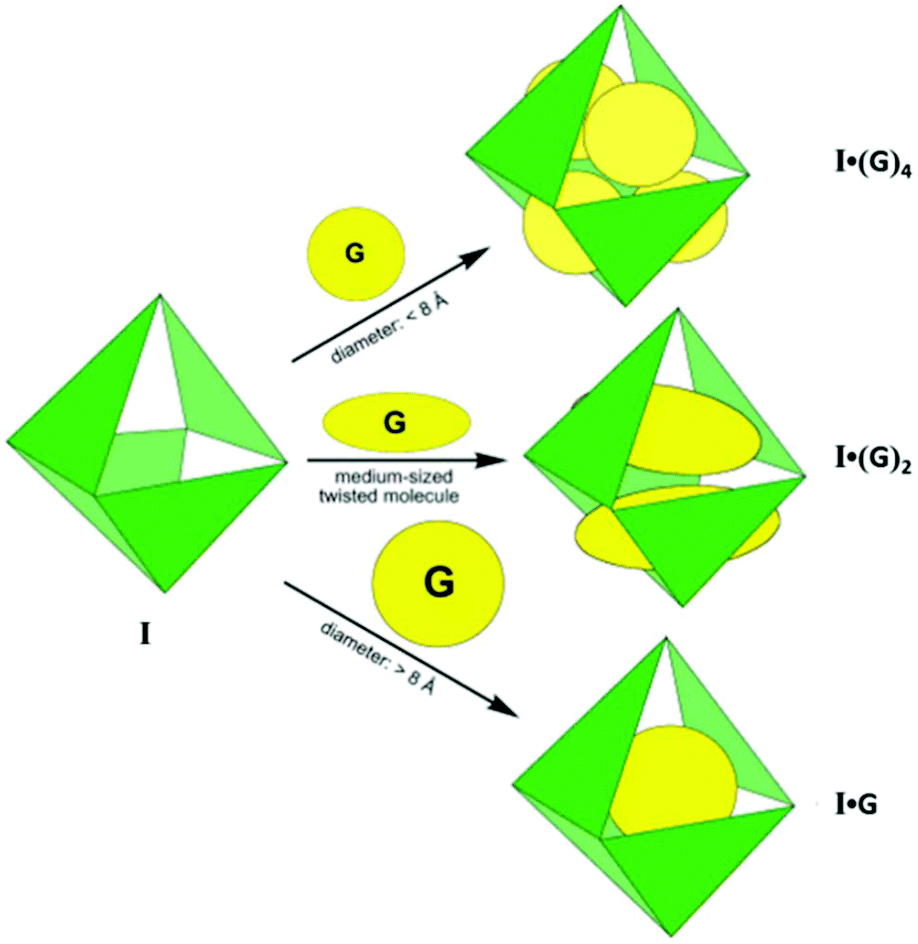 | ||
| Fig. 2 Representation of three types of guest inclusions in cage I, depending upon their shape and size. Reproduced with permission from ref. 35. Copyright 2002, American Chemical Society. | ||
The Fujita and Reek group demonstrated the selective co-encapsulation of a metal complex and a planar aromatic guest in supramolecular coordination cage I. No individual encapsulation of guests was observed, suggesting that steric properties play a vital role in filling the cage cavity by co-enclatheration. The shapes of co-enclatherating guests complement each other, leading to various ternary complexes with different combinations of guests. They demonstrated the co-enclatheration of Ir and Rh–Cp-type metal complexes with planar organic molecules. Moreover, preparation of the most favorable ternary complexes was possible by selective exchange of the guest molecules.37
In a different approach, Fujita and co-workers synthesized cage I following an induced-fit strategy from four 2,4,6-tris(4-pyridyl)-1,3,5-triazine (TPT) ligands and six metal (M = Pt for IPt and Pd for IPd) units with the aid of large template guests like sodium adamantanecarboxylate. Cage IPt maintained its structure even after removal of template guest molecules due to locking of the Pt(II)–py bond, whereas cage IPd collapsed immediately when an acid (HNO3) or a nucleophilic base (NEt3) was added to remove template guests.38 Then in 1999 they described a prototype for a “dynamic receptor library” where individual cage structures were formed upon the addition of appropriate template guest molecules from a thermodynamic mixture of oligomers formed from cage sub-components.39
The same group assembled coordination cage I′ by following same “induced-fit” mechanism, in which a specific guest induces the organization of a host itself. The tridentate ligand 1,3,5-tris(4-pyridy1methyl)benzene on treatment with (en)Pd(NO3)2 resulted in oligomeric products. However, these oligomers disappeared upon addition of sodium 4-methoxyphenylacetate, and supramolecular cage I′ with 4-methoxyphenylacetate as guest was formed in 1:l stoichiometry in aqueous solution. Anionic hydrophobic guests of a size comparable to the inner cage cavity like 1-phenylethyl or adamantyl containing molecules, induced the formation of cages in the highest yields. Monocarboxylates with a phenyl or cyclohexyl group or p-toluenesulfonate also induced the assembly of cage I′ in moderate yields. Less hydrophobic guests like dicarboxylates or acetate and too-large molecules like (1-naphthy1)acetate hardly induced complex formation. Besides anionic molecules, neutral hydrophobic guests like p-xylene also effectively induced cage assembly. Trimethylbenzylammonium bromide, a cationic guest, did not interact with the cage.40 The template guest strategies are helpful in synthesizing MSCCs, as well as in recognizing and separating these template molecules by encapsulation.
2.2. Tetrahedral MSCCs (cage II)
Raymond's group and Nitschke's group have synthesized and comprehensively studied the properties and applications of tetrahedral coordination cages (II).11,41–44 The tetrahedral cages have four metal atoms occupying the vertices, and the edges (M4L6) or faces (M4L4) are occupied by six bis-bidentate or four tridentate ligands, represented as IIeM and IIfM, respectively (Fig. 3). Their research groups have reported metal cages with various metal atoms (M) like Ga3+, Al3+, In3+, and Fe3+. The negatively charged containers have a hydrophobic internal cavity and are soluble in polar solvents as well as water.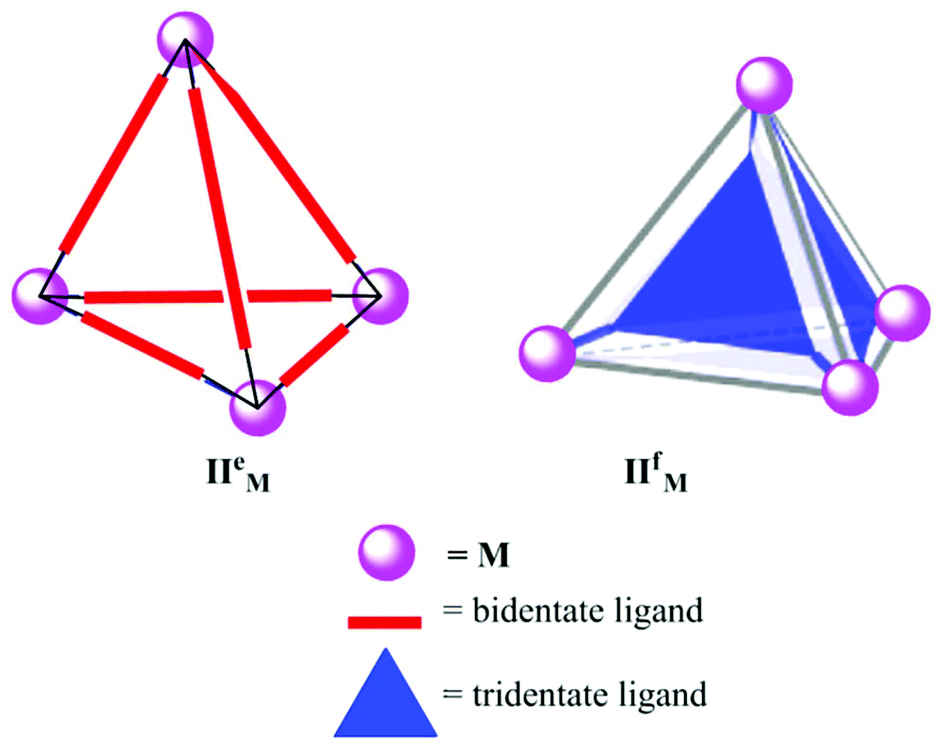 | ||
| Fig. 3 Representation of two types of tetrahedral cages (IIeM and IIfM) synthesized using bidentate (M4L6) and tridentate (M4L4) ligands, respectively. | ||
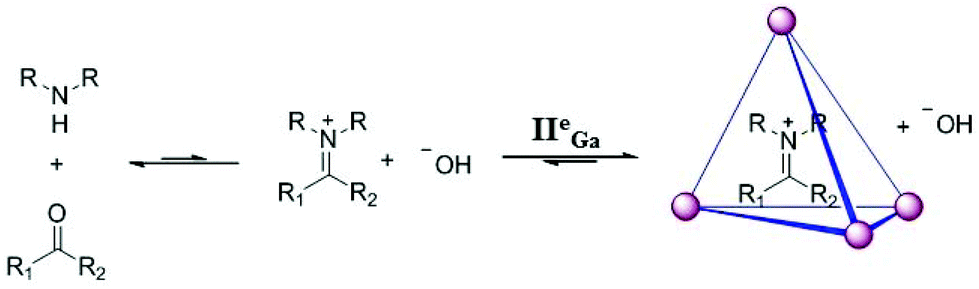
Raymond and co-workers demonstrated the selective encapsulation and stabilization of aqueous cationic guests within [Ga4L6]12− (cage IIeGa).6 They demonstrated the preferable encapsulation of monocationic NR4+ (R = Me, Et, or Pr) guests over neutral and highly charged cationic molecules, owing to the negative charge on the metal cage. Among the monocations, the inclusion of NEt4+ was preferred over the smaller NMe4+ and larger NPr4+, exhibiting the role of the size of the cavity and the hydrophobicity of guests in the encapsulation and specificity of host complexes.45 Later they found that the cage could encapsulate neutral, hydrophobic guests, including saturated hydrocarbons (n-alkanes and cycloalkanes) as well in aqueous solution due to the hydrophobic effect.46
Two positively charged ruthenium complexes, [CpRu(η6-C6H6)]+ (2) and [CpRu(p-cymene)]+ (3) (Cp = C5H5), were also encapsulated in a cage IIeGa assembly by guest exchange with cation NMe4+.47 More importantly, the size of the guests decides their binding affinity with host complexes. Replacement of NMe4+ (lower binding constant) with ruthenium complexes (higher binding constant) was monitored by 1H-NMR spectroscopy. The signals of interior NMe4+ (−0.70 ppm) disappeared while signals for η6-C6H6 at 2.60 ppm and Cp-H 1.89 ppm, corresponding to the encapsulated ruthenium complex, appeared on adding [CpRu(η6-C6H6)]Cl to the K6(NMe4)5[IIGa·NMe4] solution (Fig. 4).
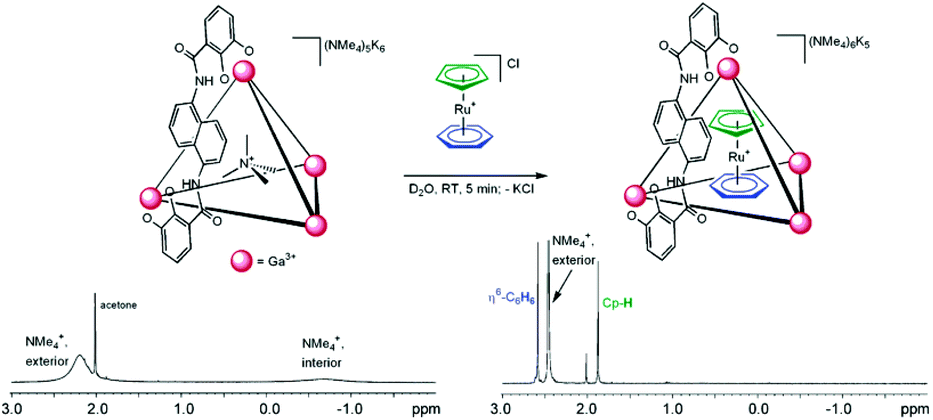 | ||
| Fig. 4 NMe4+ guest exchange with 2, K6(NMe4)5[IIeGa·NMe4] before (left), and after (right) the addition of [CpRu(η6-C6H6)]+. Reproduced with permission from ref. 47. Copyright 2004, American Chemical Society. | ||
Nitschke and co-workers reported the synthesis of tetrahedral cage IIeFe from the reaction of FeII with 4,4′-diaminobiphenyl-2,2′-disulfonic acid and 2-formylpyridine in aqueous medium. Sharp diamagnetic NMR peaks and the dark purple color of the product indicated the presence of exclusively iron(II) in the low-spin state in the cage. The anionic cage encapsulated hydrophobic guest molecules like cyclohexane, cyclopentane, etc. (with no affinity for alcohols like tBuOH or organic cations like [NH4]+ of similar size) in aqueous solution or in the solid state. The hydrophobic effect of the cavity due to the surrounding hydrophobic aryl rings provided remarkable selectivity of the cage for neutral, hydrophobic guests. The high selectivity of the cage for appropriately sized hydrocarbon guests allowed the removal of smaller molecules from the cage while encapsulating larger ones. The encapsulated guests could be removed from the cage cavity by opening the cage by applying chemical signals. Due to the high selectivity for hydrophobic guests, the water-soluble cage could be employed for drug delivery to target areas where “opener” molecules with higher affinity could be placed. Conversely, harmful hydrophobic molecules could be safely sequestered by the cage.48
J. R. Nitschke and co-workers reported a strategy to regulate the rate of exchange of anions encapsulated within another Fe4L6, tetrahedral cage II′eFe, synthesized from a dialdehyde, 18-crown-6 aniline, iron(II) cations and PF6− as an internal template anion. Each FeII vertex of cage II′eFe was bound to three flexible 18-crown-6 receptors to ‘cap’ the vertices of the tetrahedron. The capping was facilitated by strong tritopic interactions through coordination of the crown receptors to tripodal tris(alkylammonium) cations. The mechanism of capping restricted cage flexibility; hence, there was a 20-fold decrease in the rate of anion exchange within the cage cavity. The externally-bound multivalent effector allosterically controlled the guest exchange within the cage.49
The redox-switchable, highly positively (+20) charged FeII4L6 tetrahedral cage II′′eFe was prepared by Nitschke and co-workers using a redox-active dicationic naphthalenediimide (NDI) ligand. Cage II′′eFe had high affinity for anionic guests due to the +20 charge, with no affinity for neutral aromatic molecules. Cage II′′eFe could release anionic molecules following reduction by Cp2Co and re-encapsulate them on subsequent oxidation by AgNTf2. However, reduced cage II′′eFe encapsulated neutral C60, which was subsequently ejected following re-oxidation of the cage (Fig. 5). The anion-mediated transport of cage II′′eFe from organic to aqueous solution facilitated the process of guest release.50
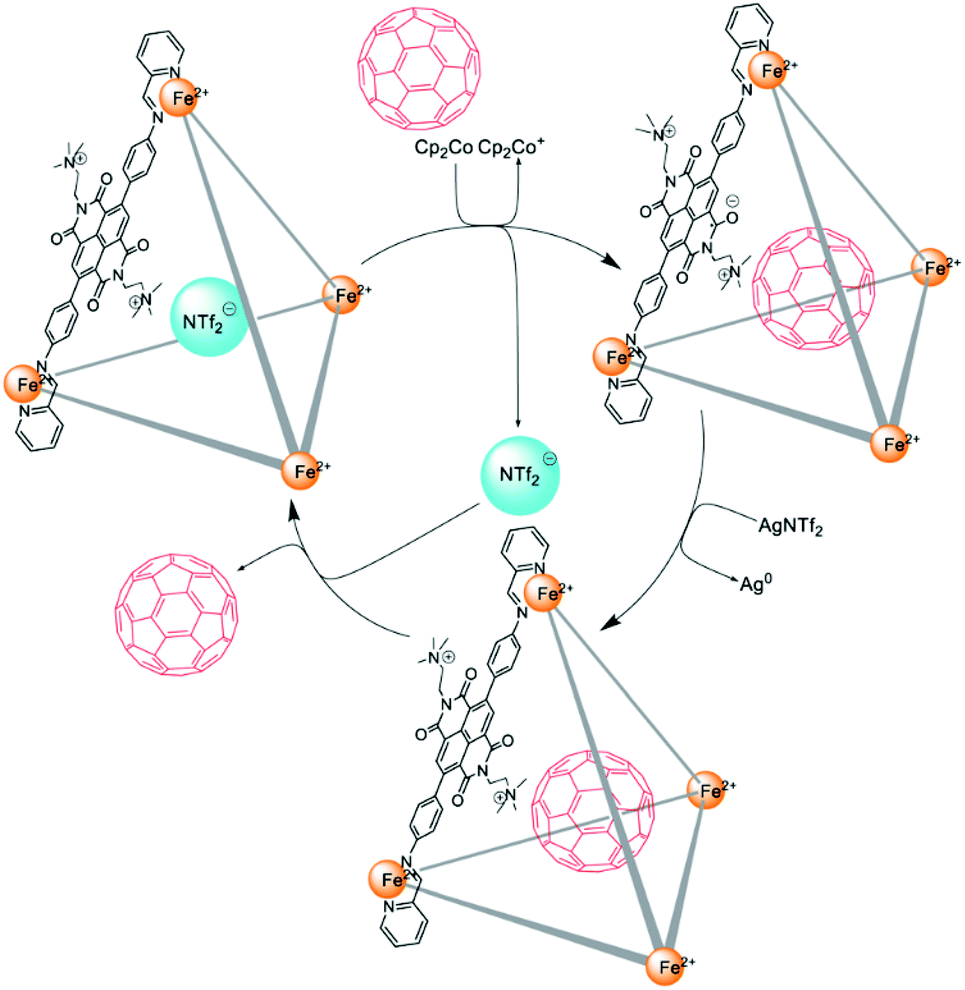 | ||
| Fig. 5 The encapsulation and release of C60 within cage II′′eFe following reduction and subsequent re-oxidation. Reproduced with permission from ref. 50. Copyright 2020, the Royal Society of Chemistry. | ||
The removal of environmental pollutants and the recovery and purification of valuable chemicals by selective anion extraction is a useful tool. In 2018 Nitschke's group reported the use of an FeII4L4, IIfFe cage (Fig. 6a) for the extraction of a high-value anion, ReO4−, from water into nitromethane.51 Importantly, the anion was extracted efficiently even when 10 other common anions were present in water, highlighting the high selectivity of cage IIfFe for ReO4−. Cage IIfFe could be disassembled into an ethyl acetate releasing guest anion in the water and then the cage could be recycled in acetonitrile solvent. The versatile solubility of cage IIfFe facilitated the complete extraction of ReO4− from an organic phase into water. The selectivity of the cage towards the perrhenate anion suggested great potential for the recycling and purification of rhenium compounds.51
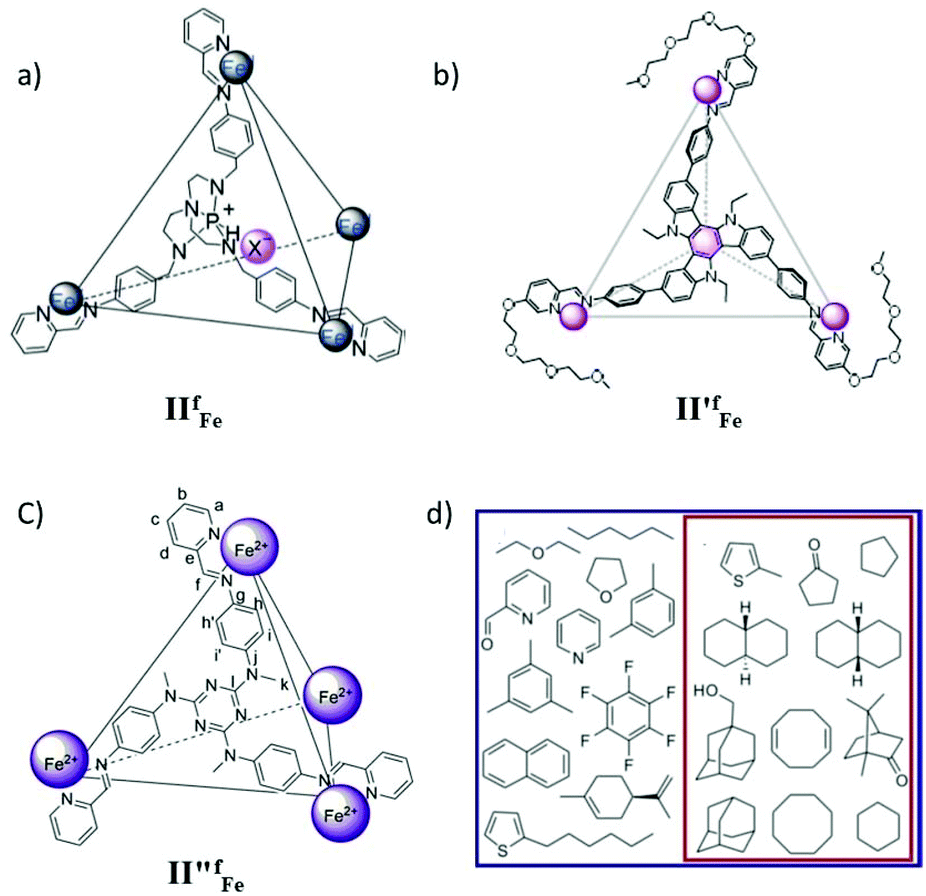 | ||
| Fig. 6 Representation of tetrahedral cages: (a) IIfFe, (b) II′fFe, (c) II′′fFe and (d) guest molecules encapsulated in cage II′′fFe, in D2O all molecules whereas there is a subset in CD3CN. Reproduced with permission from ref. 51–53. Copyright 2018, John Wiley & Sons, Ltd; 2019, and 2014, the American Chemical Society. | ||
Recently, Nitschke's group reported the selective separation of coronene from a mixture of polycyclic aromatic hydrocarbons (PAHs) using supramolecular coordination cage II′fFe (Fig. 6b).52 The tetrahedral MSCC Fe4L4 cage II′fFe was synthesized from four triethyleneglycol-functionalized formylpyridine ligands and FeII metal corners. The cage exhibited a phase transition between aqueous and nitromethane layers by anion metathesis. This property of the cage was employed for selectively encapsulating coronene from a mixture of PAHs (perylene, pyrene, anthracene, chrysene, phenanthrene, triphenylene and corannulene) through phase transfer of the cage.52
In 2014 Nitschke and co-workers synthesized a tetrahedral, face-capped MSCC FeII4L4 cage II′′fFe from a C3-symmetric triamine ligand, 2-formylpyridine and an iron salt by self-assembly and presented its host–guest chemistry (Fig. 6c).53 Remarkably, the cage was soluble either in aqueous or acetonitrile solution depending on the counterion. This allowed solvent-dependent preference of the cage for the encapsulation of certain guests. The ligand flexibility allowed expansion or contraction of the cage upon guest binding, establishing strong binding of the guests. The cage encapsulated a wide variety of aromatic and aliphatic guests in water and a subset of only the aliphatic among them in acetonitrile (Fig. 6d). The confined cage cavity favored the otherwise thermodynamically disfavored conformations of large guest molecules.53
In 2015 Nitschke's group synthesized three tetrahedral MSCCs, ZnII4L4 cages IIfZna, IIfZnb and IIfZnc, by the self-assembly of zinc(II) with tritopic trisiminopyridine ligands formed from (1,3,5-tris(4′-aminophenyl)-benzene and 2-formylpyridine, IIfZna; 1,3,5-tris(2′-formylpyridyl-5′)-benzene and p-toluidine, IIfZnb; 1,3,5-tris(2′-formylpyridyl-5′)-benzene and tris(2-aminoethyl)amine (tren), IIfZnc) (Fig. 7).54 When cages IIfZna and IIfZnb were present in solution together, cage IIfZna selectively encapsulated cyclohexane and cage IIfZnb entrapped chloroform. The addition of tren and perrhenate in a specified order could sequentially release the two guests. Cage IIfZna did not bind anionic guests within its cavity but encapsulated neutral molecule cyclohexane as a guest, while cages IIfZnb and IIfZnc encapsulated anionic guests. A series of anions with different shapes and volumes were probed for encapsulation by cages IIfZnb and IIfZnc. Cages IIfZnb and IIfZnc demonstrated similar anion-binding trends: ReO4− ≫ SbF6− > TfO− > PF6−, except for anions BF4−, NO3−, and ClO4−, which bind only to cage IIfZnb. Cage IIfZnc could be employed for radiopharmaceuticals and nuclear waste treatment, due to its exceptional affinity for ReO4− and stability in aqueous medium.54
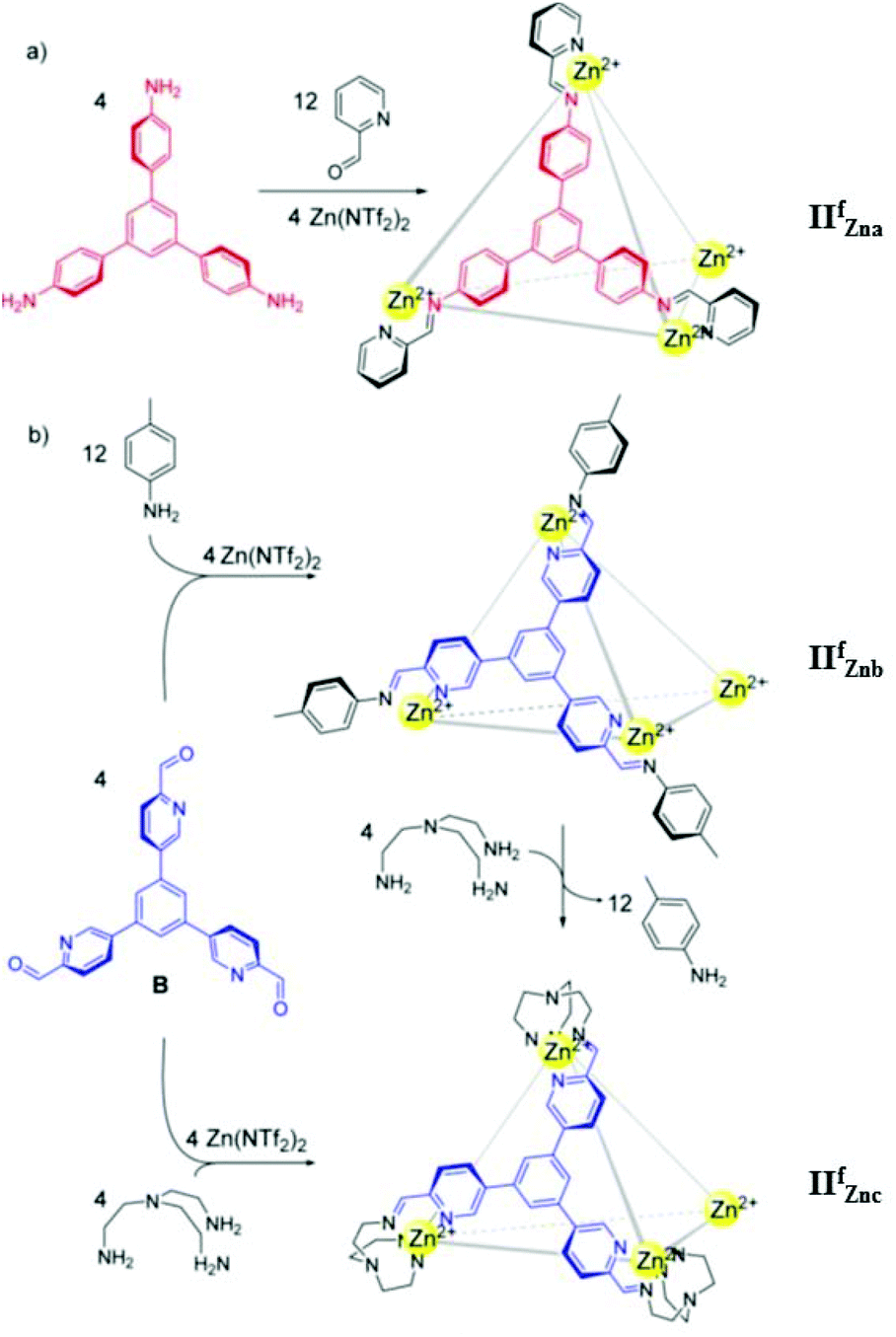 | ||
| Fig. 7 Sub-component self-assembly of cages IIfZna, IIfZnb and IIfZnc. Reproduced with permission from ref. 54. Copyright 2015, the American Chemical Society. | ||
Recently, Boomishankar and co-workers prepared three neutral Pd(II)-based discrete tetrahedral iso-structural porous supramolecular cages, IIPd ([Pd3(NiPr)3PO]4(R-AN)6) with anilate linkers (R-AN, R = H, Cl or Br), for the selective recognition and encapsulation of substituted aromatic hydrocarbons.55 The discrete polyhedral cages offered great selectivity in encapsulating aromatic hydrocarbons within their hydrophobic cavities. The anilate, chloranilate and bromanilate linker tetrahedral cages IIPda, IIPdc and IIPdb selectively encapsulated mesitylene, o-xylene and p-xylene, respectively (Fig. 8).55 They proposed that the cages exhibited selective encapsulation due to interaction between the cages and hydrocarbons and the portal diameter of the cages.
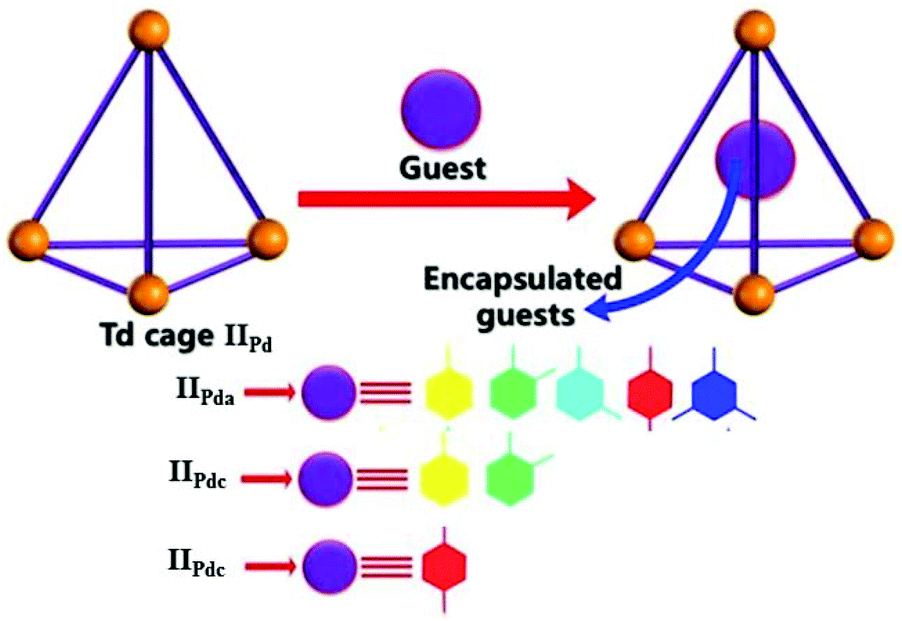 | ||
| Fig. 8 The tetrahedral cages IIPd and selective encapsulation of mesitylene, o-xylene and p-xylene by anilate (IIPda), chloranilate, (IIPdc) and bromanilate (IIPdb) linker cages, respectively. Reproduced with permission from ref. 55. Copyright 2020, John Wiley & Sons, Ltd. | ||
Recently, Gu and co-workers constructed tetrahedral FeII4L6 coordination cages IIFe with cube-like cavities from rigid p-electron aromatic rings (benzene, IIFeb or naphthalene, IIFen) and flexible alkyl units by self-assembly (Fig. 9).56 The rare cube-like cavities of these cages are selective for the encapsulation and separation of fullerene guests. The size and shape of smaller C60 match with the host cavities, rendering it a better guest than C70. According to Julius Rebek's 55% rule, C60 is a good match for cage cavities occupying 56.74% and 61.28% of cage volumes, while C70 occupies about 64.14% and 67.27% of cage volumes, respectively.
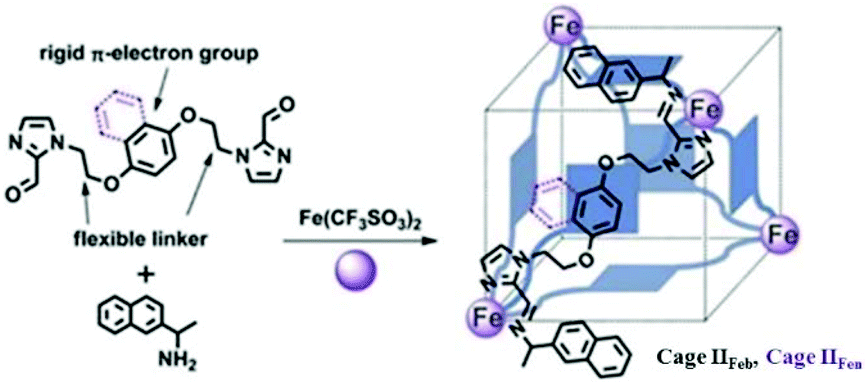 | ||
| Fig. 9 Design and synthesis of tetrahedral cages IIFe (IIFeb and IIFen), with cube-like cavities. Reproduced with permission from ref. 56. Copyright 2018, Royal Society of Chemistry. | ||
2.3. Tetragonal prismatic MSCC (cage III)
Ribas and co-workers designed a tetragonal prismatic supramolecular coordination cage by the metal-assisted self-assembly of four Pd(II) molecular clips and two zinc porphyrin moieties via coordination bonds (Fig. 10).57 Cage III can encapsulate fullerenes of different sizes (from C60 to C84) and can selectively release C60 by solvent washing of the solid host–guest complex of a cage-encapsulated mixture of fullerenes. This finding paved the way to the design and study of similar cages for the encapsulation and purification of higher fullerenes by varying the metalloporphyrin moieties or Pd cations at the molecular clips to finely adjust the selectivity of the cages for endohedral or higher fullerenes.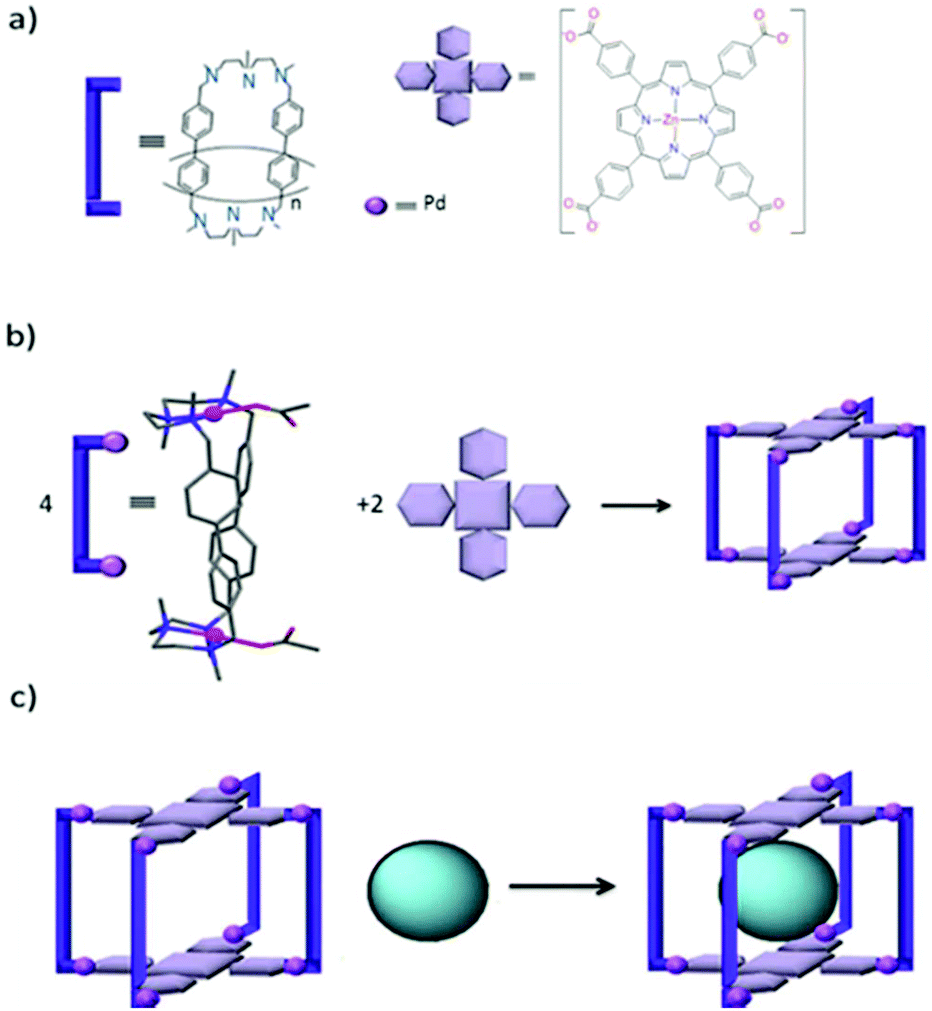 | ||
| Fig. 10 (a) Building blocks of cage III, (b) self-assembly of cage III from its building blocks and (c) encapsulation of Cn-fullerenes into cage III. Reproduced with permission from ref. 57. Copyright 2014, Macmillan Publishers Limited. | ||
Later, Ribas and co-workers also demonstrated the reversible encapsulation and release of an exTTF (tetrapyridyl extended-tetrathiafulvalene)-based ligand (m-Py)exTTF by using the same nanocage III.58 The reversible encapsulation of the (m-Py)exTTF guest was the consequence of ligand oxidation that led to drastic electronic and conformational changes. Addition of (m-Py)exTTF to complex [III·C60] causes ejection of C60 and the formation of [III·(m-Py)exTTF]. The preferentially stronger binding of pyridine moieties of (m-Py)exTTF and Zn-porphyrin units of III triggered C60 ejection. The subsequent redox-triggered release of (mPy)exTTF leaves cage III empty again and available for further C60 encapsulation. Moreover, they also reported the encapsulation of coordination complexes by metallacage III. The coordination of strongly anchored ligands to zinc, copper, and iron metal ions, resulted in metallocomplexes entrapped in the cage.59
2.4. Trigonal prismatic MSCC (cage IV)
Peris and co-workers developed an Ni-based prismatic SCC with an internal cavity of volume 1028 Å3.60 The trigonal prism geometry of the cage IVNi complex was formed via connecting six Ni atoms of bis-nickel-pyrene-di-imidazolylidene complex with two TPT (1,3,5-tripyridyl-triazine) ligands (Fig. 11).60 The large central cavity showed highly selective encapsulation of C70- over C60-fullerene. Hence, cage complex IVNi has shown exceptional potential applications in the separation and purification of mixtures of fullerenes.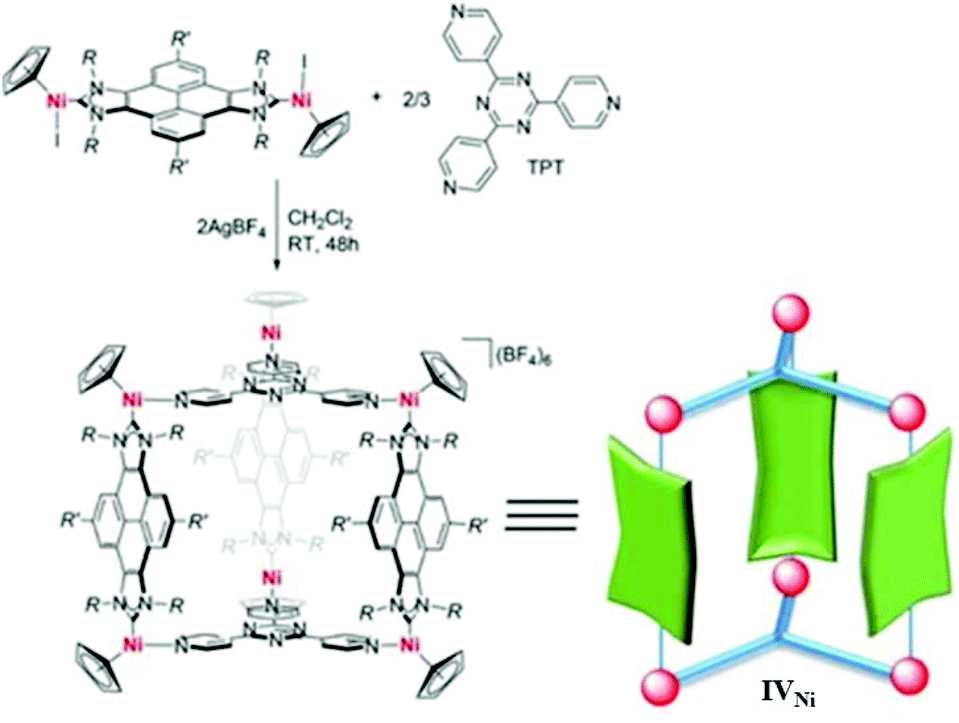 | ||
| Fig. 11 Synthesis of cage IVNi from its components. Reproduced with permission from ref. 60. Copyright 2018, John Wiley & Sons, Ltd. | ||
In 2005 Fujita and co-workers self-assembled an M6L2 L′3 prism-like coordination cage IVPd from six end-capped Pd(II) hinges, three pyridyl-based bidentate rod-like pillars and two large tridentate TPT organic panels (Fig. 12a).61 The large cavity of the metal-hinged, organic-pillared cage accommodated two or more large π-conjugated molecules. The number of guest molecules was determined by the size of the cavity, which was controlled by the length of the pillars. The cage encapsulated two planar π-conjugated guest molecules to form [IVPd·G2]12+ host–guest complexes (G = coronene, pyrene or porphine). Later the self-assembled coordination cage also acted as a molecular press, which flattened the bowl-shaped guest corannulene (C20H10) upon inclusion within the hydrophobic cavity.62 The bidentate pillar ligands were replaced by larger ligands by increasing the value of n in pyridyl-based pillar ligands, to multiply stacking π-systems. The larger cage formed exhibited quadruple and quintuple stacking of large π-conjugated molecules like pyrene/coronene/porphine.61–63 The photochemical and electrochemical properties of such discrete stacked guest molecules could be unique among isolated or infinitely stacked π-systems. They also obtained a prism-like cage IV′Pd by self-assembly of end-capped metal ions, two panel-like ligands and three pyrazine pillar ligands (Fig. 12b).64 A large aromatic compound like hexamethoxytriphenylene was essential for the quantitative formation of cage IV′Pd by the template effect. However, cage IV′Pd remained stable even after the template was removed. The empty cage IV′Pd encapsulated various other organic molecules like pyrene and 1,3-diketones. The confined cage cavity selectively encapsulated the planar enol form of β-diketone, suggesting a stabilization effect within the cage cavity.64
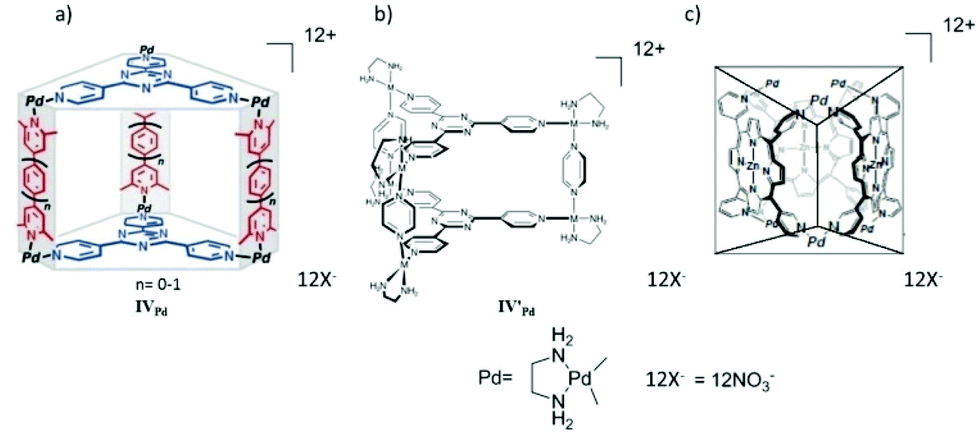 | ||
| Fig. 12 Structural representation of cage (a) IVPd, (b) IV′Pd and IV′′Pd. Reproduced with permission from ref. 61, 64 and 65. Copyright 2005, 2003, and 2001, John Wiley & Sons, Ltd. | ||
Fujita and co-workers demonstrated that guest inclusion triggered structural switching of porphyrin-based prismatic supramolecular cage complex IV′′Pd. The prism-like hollow cage was self-assembled from three porphyrin ligands and six [PdII(en)]2+, (en = ethylenediamine) metal centers (Fig. 12c).65 Neural organic molecules like pyrene and perylene can be encapsulated within the hydrophobic cavity of cage IV′′Pd in an aqueous medium. Structural optimization revealed that the D3h prismatic cage has two possible conformations (one with apical-positioned Pd centers and another with equatorial-positioned Pd centers). The conformation with Pd at the apical position is more stable than the conformation with Pd at the equatorial position. However, detailed NMR spectroscopy analysis elucidated that the D3h symmetry of the host prismatic cage IV′′Pd changed to C2 symmetry upon pyrene encapsulation. They suggested that the binding of large aromatic guests like pyrene, perylene or triphenylene triggered apical-to-equatorial flipping of two Py–Pd–Py diagonal hinges of the host cage which was not possible with encapsulation of small aromatic guests like benzene.65
P. J. Stang and co-workers developed convex trigonal prismatic coordination cage IVPt using Klärner's tetrapyridyl donor ligand clip and 180° diplatinum acceptors. Klärner's ligand clips have benzene and naphthalene units surrounded by convergent aromatic rings. 1H NMR and HRMS analysis demonstrated that these cages have cavities suitable for encapsulating fullerenes via aromatic–aromatic interactions with curved aromatic pyridyl ligands (Fig. 13).66 Fullerene encapsulation by cage IVPt can be useful in their separation and purification.66
 | ||
| Fig. 13 Synthesis of cage IVPt and encapsulation of C60 within its cavity. Reproduced with permission from ref. 66. Copyright 2017, American Chemical Society. | ||
2.5. Lantern-shaped MSCCs (cage V)
Cage V type MSCCs have been mainly explored for their utility in drug-delivery systems.67,68 Recently, Amouri and co-workers explored Pd (VPd) and Pt (VPt) based cage V for the encapsulation of a gold complex.69 Cage VPd failed to trap the Au(III) complex [Au(bdt)2]− (4) (bdt = benzene-1,2-dithiolate) and decomposed in its presence. However, cage VPt successfully captured it inside its cavity (Fig. 14).69 Here cage complex VPt formed a stable host–guest complex, encapsulating the incoming Au complex. However, cage VPd decomposed in the presence of the Au complex which could not be entrapped.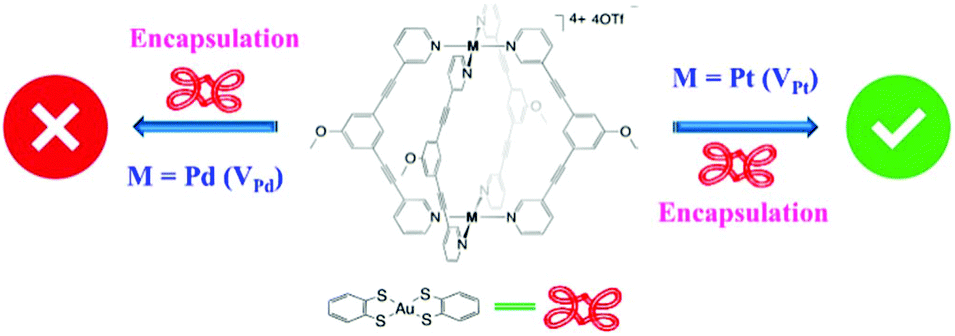 | ||
| Fig. 14 Schematic representation of the encapsulation of Au(III) complex (4) by VPt with no encapsulation noticed for VPd. Reproduced with permission from ref. 69. Copyright 2019, American Chemical Society. | ||
Clever and co-workers in 2012 reported the synthesis of a new lantern-shaped MSCC of type Va composed of two square planar Pd(II) ions coordinated to four dithienylethene (DTE) based bis-monodentate pyridyl photoswitch ligands. The cage was reversibly interconvertible between a conformationally flexible open-ring cage (o-Va) and a rigid closed-ring cage (c-Va) by irradiation with UV light (365 nm) or white light, respectively, due to the photoswitching property of the ligands. The open-ring cage o-Va was smaller in size than its closed-ring isomer c-Va. The cationic cages o-Va and c-Va were studied for the encapsulation of anionic guests due to their ability to reversibly photoswitch between two cages with different sizes. Owing to its spherical shape and size, the dodecafluorododecaborate anion ([B12F12]2−) was employed as a guest molecule (Fig. 15).70 The encapsulation of the guest within the o-Va or c-Va cage resulted in a shifting of the signals corresponding to the inward-pointing protons in 1H-NMR.70
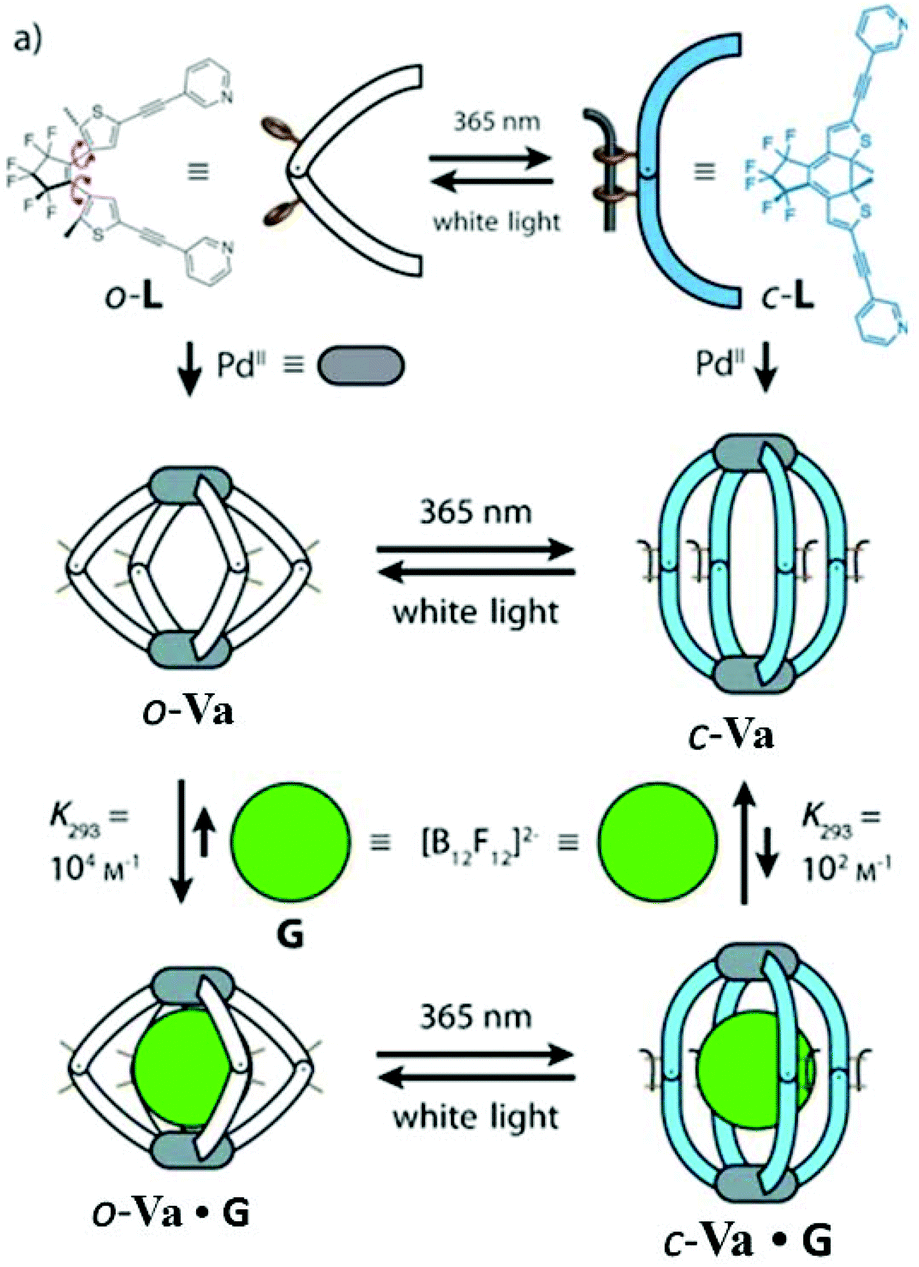 | ||
| Fig. 15 (a) Interconversion of photoswitchable DTE based bis-monodentate pyridyl ligands, (b) synthesis of o-Va and c-Va on addition of Pd(II) ions and (c) encapsulation of ([B12F12]2−) guest by Va cages. Reproduced with permission from ref. 70. Copyright 2013, John Wiley & Sons, Ltd. | ||
Recently, mechanistic studies of guest uptake and release by photochromic cage Va on irradiation by light of different wavelengths were conducted. The study revealed the involvement of four consecutive electrocyclic reactions in the process of converting all the chromophores between their open and closed photo-isomeric forms.71
Clever and co-workers demonstrated the formation of interpenetrated double coordination cages Pd4L8, Vb from the self-assembly of dibenzosuberone-based bis-monodentate pyridyl ligands with PdII cations.72 The BF4− anion within the central cavity ([Pd4L8·BF4]) served as a template for the dimerization of a lantern-shaped Pd2L4 cage, with tremendous affinity of the outer two pockets for allosteric binding of two small chloride anions (Fig. 16a).73 The dimerization of the cage by choice of templating anion and its ability to bind guests in the outer pockets were controlled by functionalization of the ligand backbone with bulky aryl groups. The bulky aryl groups on the ligand formed monomeric cage Pd2L4 due to prevention of large BF4− anion from serving as a template for the formation of interpenetrated dimeric cage Pd4L8. The addition of the small Cl− anionic template induced dimerization, yielding [Pd4L8·Cl], the interpenetrated double cage with Cl− acting as a template anion, whose enlarged outer pockets preferentially encapsulated large anions such as ReO4− (Fig. 16b).73
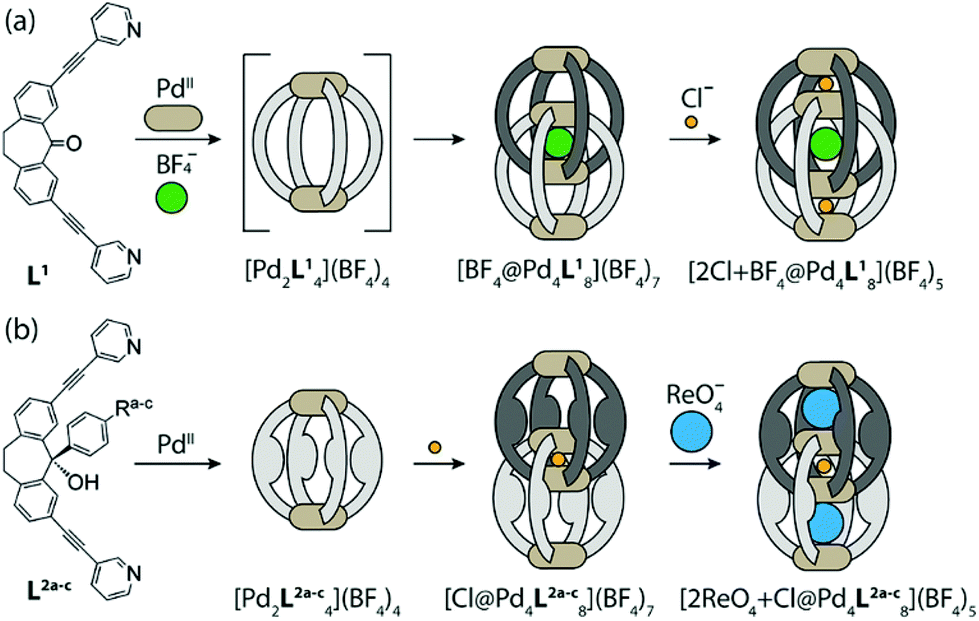 | ||
| Fig. 16 Synthesis of interpenetrated dimer cage: (a) ([Pd4L8·BF4]) with BF4− as a template anion and Cl− anions in the outer pockets, (b) ([Pd4L8·Cl]) with Cl− as a template anion and ReO4− in the outer pockets. Reproduced with permission from ref. 73. Copyright 2013, American Chemical Society. | ||
Later, they designed double cages with three consecutive tetrafluoroborate anion filled pockets, which could be activated for binding neutral guests like benzene, cyclohexane or norbornadiene by the addition of chloride anions. They also conducted a systematic study using more than 50 small neutral guests to reveal the influence of the size, shape and chemical nature of the guests on the binding affinity.72,73
In 2012 Shionoya and Clever's group reported Pd2L4 cage Vc, self-assembled from the coordination of four concave ligands to two square planar PdII ions. The cage quantitatively encapsulated a hexamolybdate dianion [Mo6O19]2− in solution.74 They also proposed that the strength and mode/position of binding (inside vs. outside) of the dianionic guest molecules to the cationic, self-assembled metallosupramolecular coordination cage was controlled by the size and stoichiometry of the guests added.75 Using a ditopic ligand with strong hydrogen-bond donor capacity due to the central, endohedral imide group, another cage Vd was prepared for the encapsulation of octahedral anionic guests.75 The hollow cavity of cage Vd provided a complementary environment for binding anionic octahedral guests through equatorial H-bonding and axial electrostatic interactions. The Oh symmetric octahedral complex [Pt(CN)6]2− was distorted to D4h symmetry upon encapsulation in the cage (Fig. 17).76 Another lantern-shaped Pd2L4 cage was synthesized using curved backbone based bis-pyridyl ligands and used as a selective induced-fit receptor for C60 fullerene.77,78
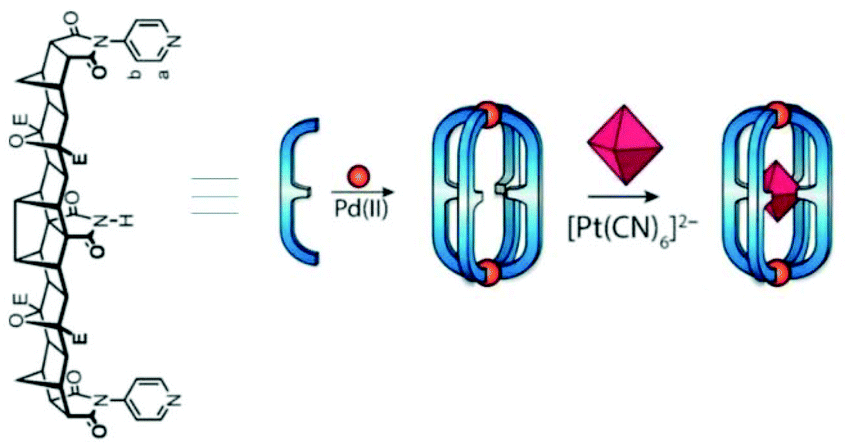 | ||
| Fig. 17 Synthesis of Vd and subsequent encapsulation of octahedral complex [Pt(CN)6]2− within the cage cavity. Reproduced with permission from ref. 76. Copyright 2016, John Wiley & Sons, Ltd. | ||
2.6. Trigonal MSCC (cage VI)
Yu and co-workers prepared hexametallic supramolecular cage VI [(N^N)6Pd6L2]6+ by the self-assembly of fluorescent tritopic tripyrazole ligands, tris(4-(1H-pyrazol-3-yl)phenyl)amine and dimetallic corners, [(N^N)2Pd2(NO3)2](NO3)2 (N^N = 2,2′-bipyridine, 4,4′-dimethylbipyridine or 1,10-phenanthroline) (Fig. 18).79 The positively charged hexametallic cage VI exhibited anion sensitivity toward HSO3− in aqueous solution via fluorescent titration and NMR spectroscopy studies. The cage with fluorescent ligands can act as a cleaner for eliminating pollutant SO2 from water.79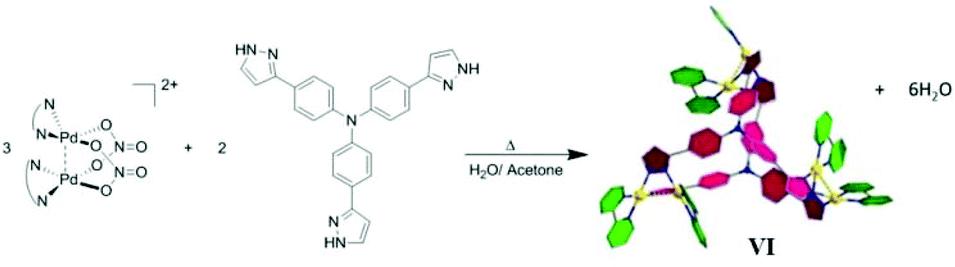 | ||
| Fig. 18 Representation of self-assembly of cage VI, [M6L2] using dimetallic corners. Reproduced with permission from ref. 79. Copyright 2017, Royal Society of Chemistry. | ||
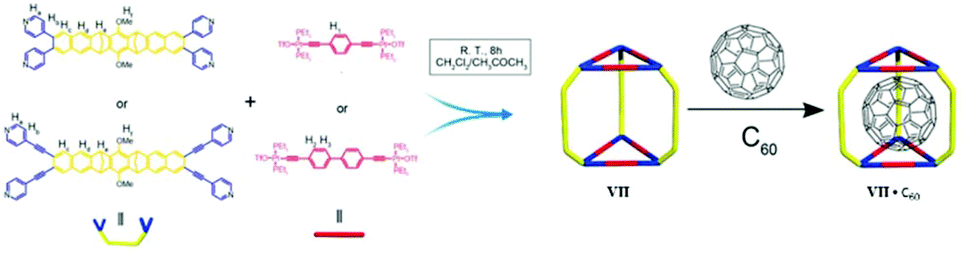
2.7. Cubic MSCC (cage VII)
In 2006 Nitschke and group self-assembled cubic MII8L6 cages VII from porphyrin ligands, forming electron-deficient walls with metal ions (FeII or ZnII) occupying the vertices of the cube. Cages VII selectively encapsulated fullerene bisadducts like C60-indene or C60-anthracene, while monoadducts and unfunctionalized fullerenes were not encapsulated (Fig. 19).80 C60 and anthracene also reacted to yield bisadducts within the cavity of FeII8L6 cage VIIFe, whereas no reaction occurred without a cage under the same conditions. Cages VII could be practically useful in polymer solar cells, where C60 bisadducts are used as electron acceptors but their syntheses and purification are laborious and time consuming.80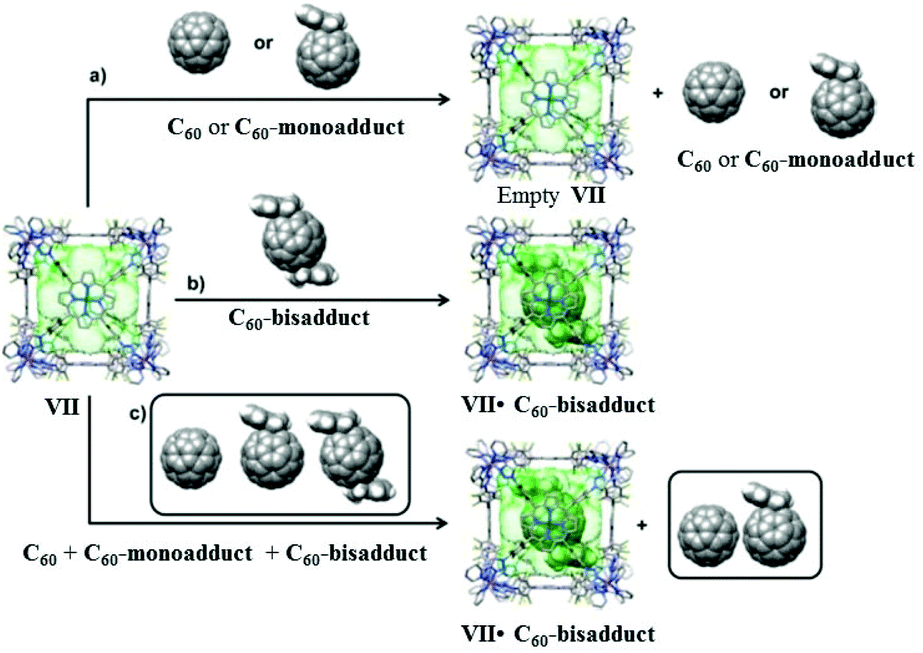 | ||
| Fig. 19 Representation of selective encapsulation of C60-bisadducts by cage VII from a mixture of C60, C60-monoaddcts and C60-bisadducts. Reproduced with permission from ref. 80. Copyright 2017, American Chemical Society. | ||
Another O-symmetric cubic coordination cage VII′, M8L6, was synthesized by J. R. Nitschke and co-workers from the self-assembly of a C4-symmetric tetrakis-bidentate ligand with a C3-symmetric iron(II) tris(pyridylimine) center. The porphyrin-faced hollow cubic cage VII′ had a large central cavity with sufficient volume for guest encapsulation, well isolated from bulk media by small-sized pores and walls made of ligands rich in π-electron density. The cage demonstrated selective encapsulation of higher fullerenes like C70, C76, C78, C82 and C84 and large aromatic guests like coronene.81
2.8. Spherical MSCC (cage VIII)
Spherical M2L4 coordination cages are the simplest and a highly symmetric class of supramolecular architectures. M. Yoshizawa and co-workers synthesized M2L4 type supramolecular coordination cages VIII by self-assembly of M(II) ions (M = Zn, Cu, Pt, Pd, Ni, Co, or Mn) and bent ligands with anthracene fluorophores.82 The spherical bimetallic Pd(II) cage can efficiently encapsulate neutral spherical and planar molecules. Spherical molecules like adamantanes, paracyclophanes and fullerenes (C60) formed host–guest complexes in a 1:1 ratio, while planar molecules like pyrenes and triphenylene formed host–guest complexes in a 1:2 ratio. Competitive encapsulation selectively formed an unusual ternary complex in the case of triphenylene and corannulene (Fig. 20).83 The M2L4 cage selectively encapsulated D-sucrose in water from a mixture of natural disaccharides. The D-sucrose molecules were selectively isolated and entrapped within the cage via shape complementary and CH–π interactions, unlike previous molecular receptors which bind biomolecules through covalent, coordinative or multiple hydrogen bonding. Hence, these cage complexes can be employed for the selective recognition and separation of biomolecules in water.84
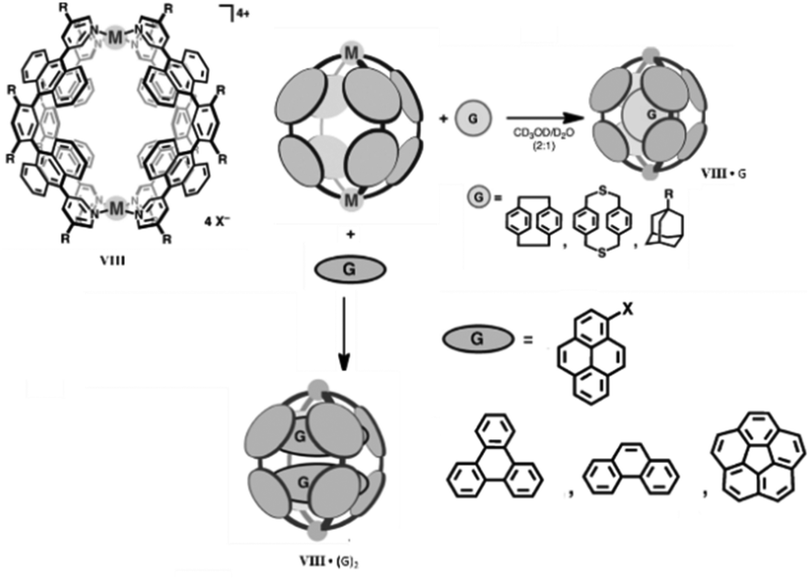 | ||
| Fig. 20 Structural representation of spherical cage VIII and encapsulation of spherical and planar molecules within its cavity. Reproduced with permission from ref. 83. Copyright 2013, John Wiley & Sons, Ltd. | ||
2.9. Square antiprism MSCC (cage IX)
D. Li and co-workers synthesized Cu10L8 bicapped square antiprism supramolecular cage IX with an adaptable cavity from Cu2+ ions and flexible bi-imidazole ligands. The polyhedral metal–imidazolate cages encapsulated anions of different sizes and expanded or compressed their cavities accordingly. The anion-induced flexibility of the cage cavity mimics the ‘induced-fit’ property of enzymes. The volume of the internal cavity of the cage could expand or compress by more than 53% on guest encapsulation: for instance, an expanded ClO4−-bound cage transformed to a highly compressed Cl−-bound one.853. Stabilization of sensitive and unstable species
The ability to encapsulate guest molecules by MSCCs has been exploited for the identification, isolation and stabilization of sensitive and reactive intermediates (radicals, nitrenes, and carbenes, as well as labile products).863.1. M6L4 octahedral MSCCs (cage I)
The encapsulation of guests within the hydrophobic cavity of water-soluble cage I improves their solubility in water. Fujita's group reported the selective encapsulation of C-shaped molecules like cis-azobenzene and -stilbene derivatives.87 The subsequent entering of these derivatives resulted in hydrophobic dimers within the cavity in a “ship-in-a-bottle” manner. In 2006 Fujita and co-workers presented the crystallographic encapsulation of an extremely labile and coordinatively unsaturated Mn-complex within cage I.88 Coordinatively unsaturated transition-metal complexes are reactive intermediates of catalytic organometallic reactions. Mechanistic insights into organic/organometallic reactions are possible through investigation of these labile complexes. Stable Cp′Mn(CO)3, 5 (Cp′ = methylcyclopentadienyl), inside the cage cavity photodissociated to liberate CO and in situ generated 16-electron unsaturated pyramidal manganese complex 5′ (Fig. 21).88 The geometry around the metal center of reactive intermediate species is difficult to assign using conventional methods; however, the crystallographic data showed a clear picture.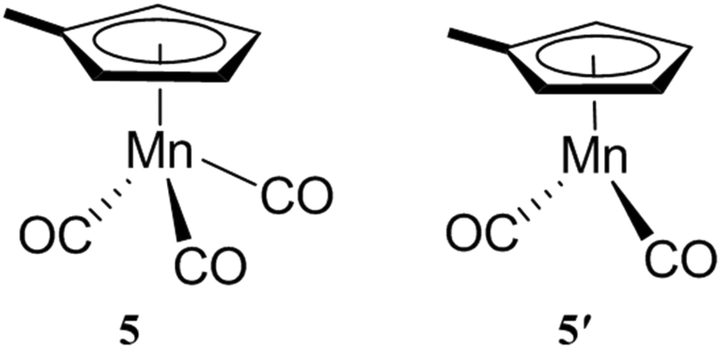 | ||
| Fig. 21 Chemical structure and geometry of complex 5 and pyramidal 16-electron complex 5′. | ||
Cage I was also used as a non-covalent protective group to shield specific reaction sites through encapsulation of the substrate. In the nucleophilic substitution of aryl-substituted allylic halides, cage I non-covalently shielded the internal site of the substrate by encapsulation and directed the incoming nucleophile to the unprotected terminal reaction site (Fig. 22).89 Hence, cage I made regioselectivity control possible without altering the electronic and steric factors or solvent polarity.
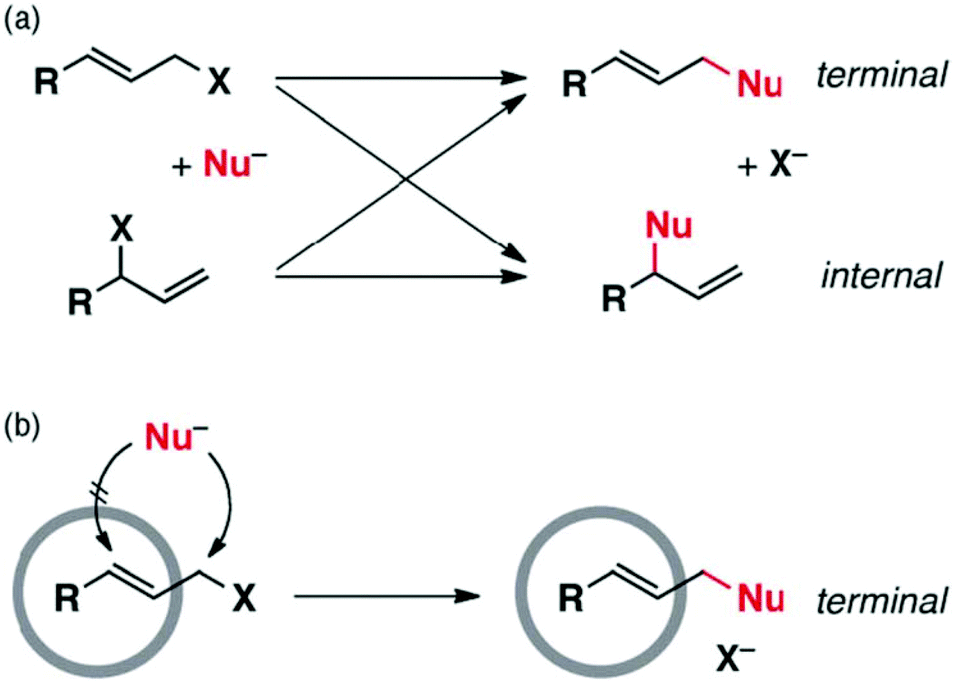 | ||
| Fig. 22 (a) Nucleophilic substitution of allylic halides and their allylic rearrangements shown by crossed arrows. (b) Regioselectivity control in allylic halides due to non-covalent protection by cage I. Reproduced with permission from ref. 89. Copyright 2012, the Royal Society of Chemistry. | ||
Fujita and co-workers showed the encapsulation and stabilization of dinuclear complex [(η5-indenyl)Ru(CO)2]26 with labile metal–metal bonds in the cavity of supramolecular coordination cage I.90 Ruthenium complex 6 is photosensitive and exists in four isomeric forms in rapid equilibrium. In the presence of light the facile Ru–Ru bond cleaves and CO dissociation occurs photochemically. But, cage I froze complex 6 in the CO-bridged cis-configuration within the confined cavity and enhanced its photostability. Recently, Anna and co-workers used ultrafast mid-IR polarization dependent pump–probe spectroscopy to gain an insight into the effects of encapsulation on the dynamics and properties of group VIII metal–carbonyl complexes.91 Fujita and co-workers explained the stabilization of cis-bridged isomeric form of di-ruthenium carbonyl complexes within cage I and their carbonyl photosubstitution without metal–metal bond cleavage.92 Apart from their interesting photochemistry and catalytic properties, the group VIII metal carbonyl complexes exhibit fluxional behavior, where multiple isomers exist in dynamic equilibrium. The encapsulation of guests within confined cage I cavities alters their ultrafast dynamics by restricting diffusional rotational motion and reducing the vibrational lifetime.
Phthalein dyes exist in pink-colored quinone dianion form and colorless lactone form at basic and acidic pH, respectively. Interestingly, the cavity of cage I was exploited to induce chromism in phthalein dye.93 As the shape and size of molecules affect their encapsulation in the host, tetrahedral lactone can ideally fit into the cavity of cage I, and hence is encapsulated and stabilized in the cavity more than quinone. This led to disappearance of the pink color due to shifting of the equilibrium towards lactonization. Therefore, in basic solution phthalein dyes could also stay colorless when encapsulated by cage I.
3.2. Tetrahedral MSCCs (cage II)
The high affinity of Raymond's cage II for positively charged species and its hydrophobic internal cavity led it to being employed for the stabilization of reactive reaction intermediates. Cage IIeGa ([Ga4L6]12−) stabilized the reactive, unstable, cationic species [Me2C(OH)PEt3]+ (7) in aqueous medium (Fig. 23).94 Cationic species 7 had been synthesized and isolated under dry conditions, but it decomposes in aqueous medium. Raymond and co-workers reported that IIeGa contained an acetone molecule (IIeGa·(acetone)) within the cavity. On addition of PEt3 to an aqueous solution of IIeGa·(acetone), protonated phosphine HPEt3+ diffused and formed an unstable cationic species 7 in the presence of protons. As confirmed through 1H-NMR spectroscopy, 7 was stable within the cavity and decomposed when it came out of the hydrophobic tetrahedral cavity.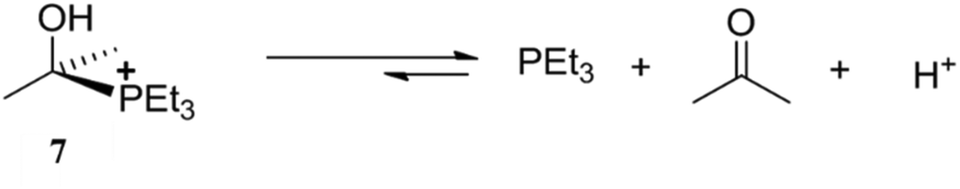 | ||
| Fig. 23 Representation of decomposition equilibrium of [Me2C(OH)PEt3]+ (7) in an aqueous medium. Reproduced with permission from ref. 94. Copyright 2000, John Wiley & Sons, Ltd. | ||
IIeGa also stabilized aromatic diazonium cations and tropylium cations.44 Encapsulation of these cations within IIeGa protected the reactive cations and decelerated their decomposition by solvolysis. Raymond and co-workers used a IIeGa coordination cage for the synthesis and stabilization of iminium ions under unconventional aqueous conditions at basic pH via novel host–guest chemistry.13 The encapsulated iminium ions remained stable at room temperature for several months (Fig. 24). Investigation of various iminium cations generated from a range of ketones and pyrrolidine revealed that the size, shape, charge and hydrophobicity of the guests determined molecular recognition and encapsulation within the cage cavity.
 | ||
| Fig. 24 Synthesis and encapsulation of iminium cation by cage IIGa. Reproduced with permission from ref. 13. Copyright 2006, American Chemical Society. | ||
Nitschke and co-workers found the tetrahedral cage complex IIeFe ([Fe4L6]4−) effective for the encapsulation and stabilization of pyrophoric tetrahedral P4 molecules (Fig. 25).42 Air-sensitive white phosphorus enclatherated within the hydrophobic tetrahedral cavity was stable in air and water soluble. This was because the oxidized species was too large size to fit into the cage cavity. P4 was removed from the cage by simply layering the aqueous layer with benzene via guest exchange. The P4 extracted in benzene was slowly oxidized into phosphoric acid, as confirmed by 31P-NMR spectroscopy.
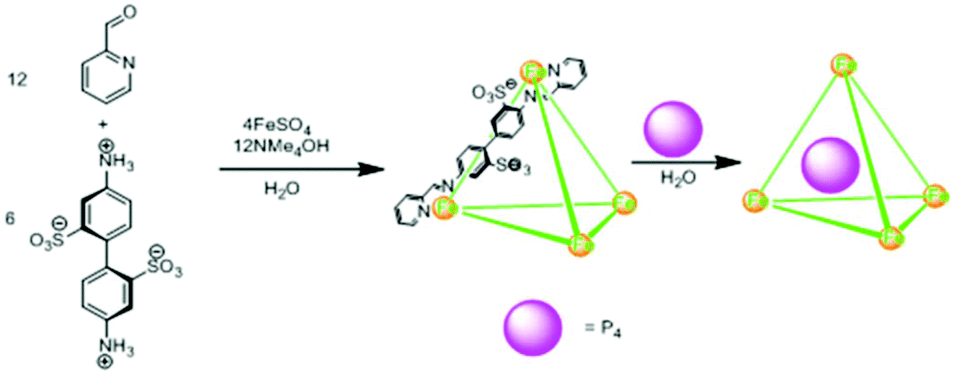 | ||
| Fig. 25 Synthesis of cage IIeFe and trapping of white phosphorus (P4) within tetrahedral cage IIeFe. Reproduced with permission from ref. 42. Copyright 2009, American Association for the Advancement of Science. | ||
Tetrahedral cage IIeFe was also utilized for the reversible and selective capturing of hydrophobic greenhouse gas SF6 (Fig. 25).43 The encapsulated gas can be compressed and stored in the form of an aqueous solution. The cage was found to have no affinity for N2, O2, Ar, Xe, C2H4, N2O or CO2 gases; thus, this coordination cage proved to be a potential candidate for trapping SF6 from gaseous mixtures and storing it at room temperature. The stored SF6 can be easily removed from the cage by increasing the temperature or by adding acid or tris(2-ethylamino) amine. The protective environment of tetrahedral cage IIeFe was further exploited by Nitschke and co-workers to block the Diels–Alder reaction between maleimide and furan by the encapsulation of furan.95 The trapping of furan by the cage blocked it from reacting with the maleimide; however, the competing guest (benzene) released furan which undergoes Diels–Alder cycloaddition (Fig. 26).95
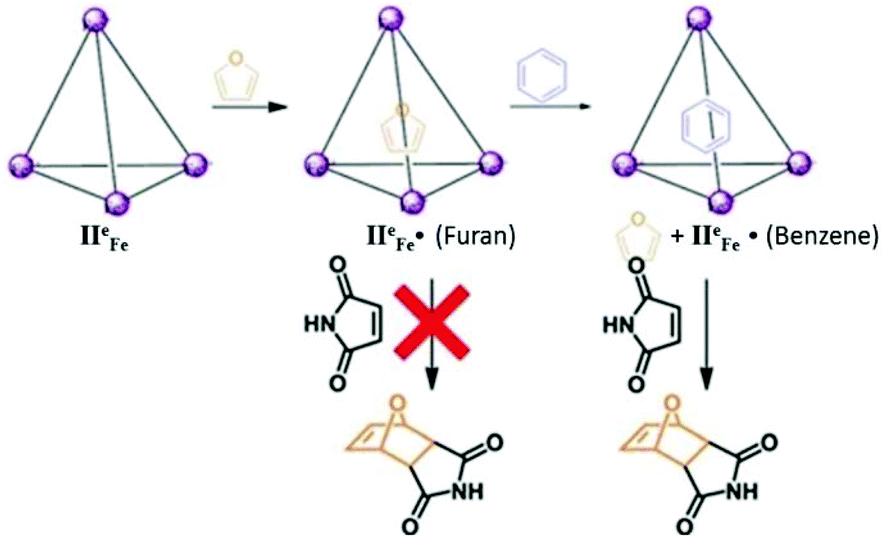 | ||
| Fig. 26 Representation of trapping of furan by cage IIeFe, preventing it from undergoing a Diels–Alder reaction and the release of furan and initiation of a Diels–Alder reaction upon the addition of competing guest benzene. Reproduced with permission from ref. 95. Copyright 2012, Royal Society of Chemistry. | ||
3.3. Cubic MSCCs (cage VII)
J. R. Nitschke and co-workers described the synthesis of a large, cubic host cage VIIFe from the self-assembly of FeII and a zinc-porphyrin-containing ligand. Cage VIIFe selectively encapsulated biomolecular guests (drugs and peptides) with imidazole and thiazole moieties. The interaction between guest functional groups and the zinc porphyrin face of the cubic cage induced selectivity. The encapsulated peptides were protected from trypsin cleavage; however, the physicochemically similar non-encapsulated peptides were rapidly cleaved in the same solution. The large-yet-enclosed cage cavity protected the enclosed guests from the external environment.964. Drug-delivery systems
The crucial parameters for the biological application of metal assemblies are their ability to withstand physiological conditions and water solubility. Cancer chemotherapy is one current field where metal complexes have been practically useful.97,98 However, the selective toxicity of drugs is one of the persistent challenges in chemotherapy in order to reduce their general toxicity. Employing large transporting compounds which can release the drug after penetrating into a cancer cell is one such targeting method, since large molecules selectively aggregate in cancer cells, due to the “enhanced permeability and retention effect”.99 The biological applications of SCCs as drug-delivery systems by the encapsulation of drug molecules within the cavity of a soluble host cage are interesting as they increase the solubility of lipophilic molecules as well as protecting the drug molecules from harsh physiological conditions. The outer surface of host SCCs can be modified with desirable properties like conjugation of targeting moieties or fluorophores without affecting the basic structure of the drug molecule.4.1. Trigonal prismatic MSCCs (cage IV)
Pioneered by Therrien's research group, who first reported them in 2008, the use of self-assembled hexa-ruthenium metallacages (metallaprisms) as drug-delivery systems was demonstrated for lipophilic drugs.100 The hexanuclear cationic metallaprism [(p-cymene)6Ru6(tpt)2(dhbq)3]6+ (tpt = 2,4,6-tris(pyridin-4-yl)-1,3,5-triazine; dhbq = 2,5-dihydroxy-1,4-benzoquinonato) (IVRu) encapsulated the hydrophobic complexes [M(acac)2] (where M = Pd(II) or Pt(II), acac = acetylacetonato) (Fig. 27).100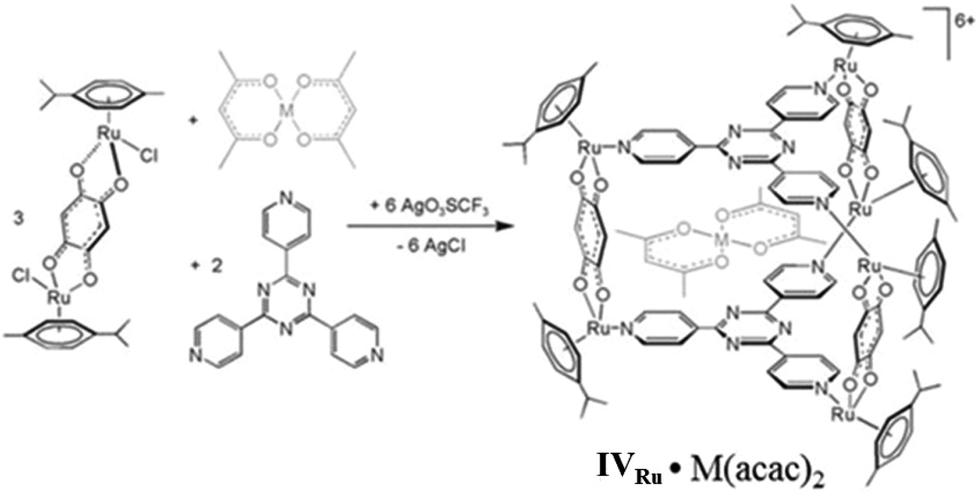 | ||
| Fig. 27 Ruthenium(II)-based drug-delivery system (IVRu)·(M(acac)2). Reproduced with permission from ref. 100. Copyright 2008, John Wiley & Sons, Ltd. | ||
The water-soluble metallaprism IVRu is slightly cytotoxic (IC50 of ca. 23 μM) for human ovarian tumor cells A2780, while [M(acac)2] complexes are virtually insoluble in water with no cytotoxicity. Interestingly, the “complex-in-a-complex” was more active and the cytotoxicity of encapsulated IVRu·Pt(acac)2 was 20-times more toxic (IC50ca. 1 μM) than the empty cage IVRu. This study was the proof-of-concept of Therrien's definition of “the Trojan horse strategy” of screening a cytotoxic agent within the cavity of a coordination cage until internalization inside cancer cells. When it enters the tumor cells, the hexaruthenium cage opens and releases the [M(acac)2] drug complex which performs its cell-killing act. Subsequently, Therrien and co-workers investigated another hexaruthenium metallacage [Ru6(p-iPrC6H4Me)6(tpt)2(C6H2O4)3]6+ (IV′Ru) for the encapsulation and release of a series of fluorescent pyrenes and their in vitro anticancer properties.101,102 Upon encapsulation, the fluorescence of the pyrene derivatives was quenched, so their release could be monitored by fluorescence spectroscopy. At pH 2 or pH 7, there was no release of pyrene derivatives from IV′Ru, while at pH 12 guest molecules were released. IC50 values of IC50 > 20 and 16 ± 2.3 μM for free pyrenes and the vacant-cage complex, respectively, in human ovarian tumor cells A2780 indicated their low cytotoxicity, while the lower IC50 value (6 ± 0.8) μM for encapsulated pyrene in cage IV′Ru showed considerable activity. Using fluorescence microscopy data, they proposed that the uptake of sparingly soluble pyrene derivatives by cancer cells increased after encapsulation in water-soluble IV′Ru, hence increasing their cytotoxicity. Two guest complexes (N-hexadecylpyrene-1-sulfonamide and pyrenyl ethacrynic amide) exhibited antiproliferative effects similar to cisplatin. The pyrenyl derivatives showed differences in cytotoxicities due to their ability to control cell proliferation, which could not be evaluated due to their poor water solubility or the differences in uptake and release of these molecules inside cells. Hence, cage IV′Ru can deliver them inside the cells in vitro successfully and improve their efficacy. Therrien and co-workers thereafter determined the effects of the size of the host complex portal on the encapsulation and retention of guest molecules.103 They synthesized three hexaruthenium cages using 1,4-naphthoquinonato (IV′′Ru), 1,4-anthraquinonato (IV′′′Ru), and 5,12-naphthacenedionato (IV′′′′Ru) analogues in the diruthenium bridging ligands as polycyclic aromatic systems. The portal size of the three cages decreased in sequence as follows: IV′′Ru > IV′′′Ru > IV′′′′Ru, while the internal cavity remained almost the same. Florescence studies described the pyrene derivatives being more efficiently retained by a host complex with a smaller pore size. The highly desirable water solubility has made hexaruthenium metallacages (IVRu) the most investigated future drug-delivery systems. However, a few other SCCs have also been subjected to preliminary encapsulation of drugs and in vitro pharmacological studies to evaluate their drug-delivery potential.
4.2. M2L4 MSCCs (cage V)
Crowley and co-workers designed a cationic quadruply-stranded Pd2L4 cage complex (V′Pd) using the bidentate ligand 2,6-bis(pyridin-3-ylethynyl)pyridine and demonstrated the encapsulation of the anticancer drug cisplatin by XRD studies.67 Cage V′Pd can be reversibly disassembled/reassembled by the subsequent addition and removal of competing ligands. 1H NMR and ESI-MS studies showed that the addition of [4-(dimethylamino)pyridine or Cl−] as competing ligands facilitated the release of cisplatin due to disassembly of the coordination cage (Fig. 28).67 However, these types of Pd2L4 cages are scarcely soluble in aqueous medium.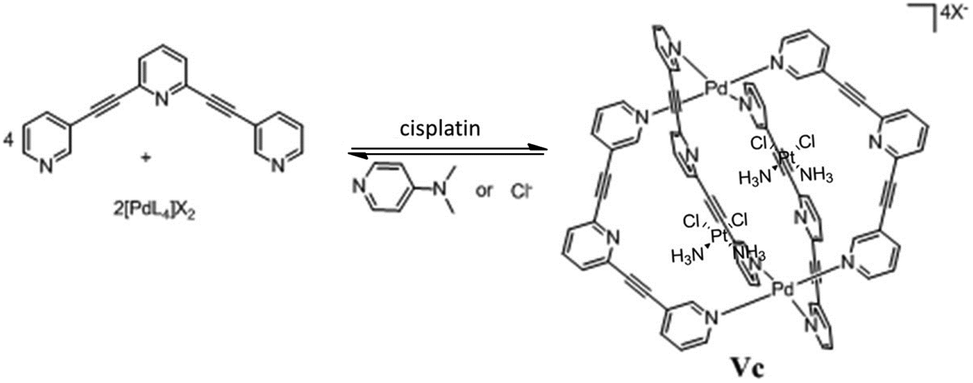 | ||
| Fig. 28 Encapsulation and release of cisplatin by cage V′Pd and subsequent disassembly by addition of competing guests. Reproduced with permission from ref. 67. Copyright 2012, Royal Society of Chemistry. | ||
Following these promising results, various fluorescent exo-functionalized Pd2L4 coordination cages V′′Pd were developed by Casini and co-workers and studied for the enclatheration of cisplatin through NMR spectroscopy,104 which exhibited similar downfield shifts to those observed for the protons in the internal cavity (the result of encapsulation) of Crowley's cage V′Pd. XRD results demonstrated the enclatheration of two cisplatin drug molecules by cage V′′Pd.
Thus, the precursor compounds and palladium cages were tested for their in vitro cytotoxicity against a number of human cancer cells (A549, HepG2 and SKOV-3). The coordination cages were nontoxic in healthy rat liver tissues ex vivo; however, a decreased IC50 value showed that the mixture of cage and cisplatin was more cytotoxic than free cisplatin or a vacant cage. Fluorescence studies conducted to study the uptake of the host–guest complex by cancer cells indicated their accumulation in tumors due to an EPR (enhanced permeability and retention) effect,105 that has been extensively studied for passive drug delivery in cancer therapy.
The exo-functionalization of metallacages by conjugation of cell-specific ligands like tumor-targeting peptides (especially for tumor-surface markers) to their surface could improve the target specificity and efficacy of drug-delivery systems.106 The coating of non-covalent peptide on the surface of self-assembled metallacage M12L24 was explored by Ikemi and co-workers107 and encapsulation of protein within the molecular host Pd12L14 cavity108 initiated the functionalization of MSCCs for biomedical applications. Interestingly in the latter case, the first ligands tethered to the protein while the nanocage self-assembled around the protein on subsequent metal ions and ligand additions. Casini and co-workers reported the bioconjugation of exo-functionalized self-assembled cage V′′Pd by two different approaches (Fig. 29).109
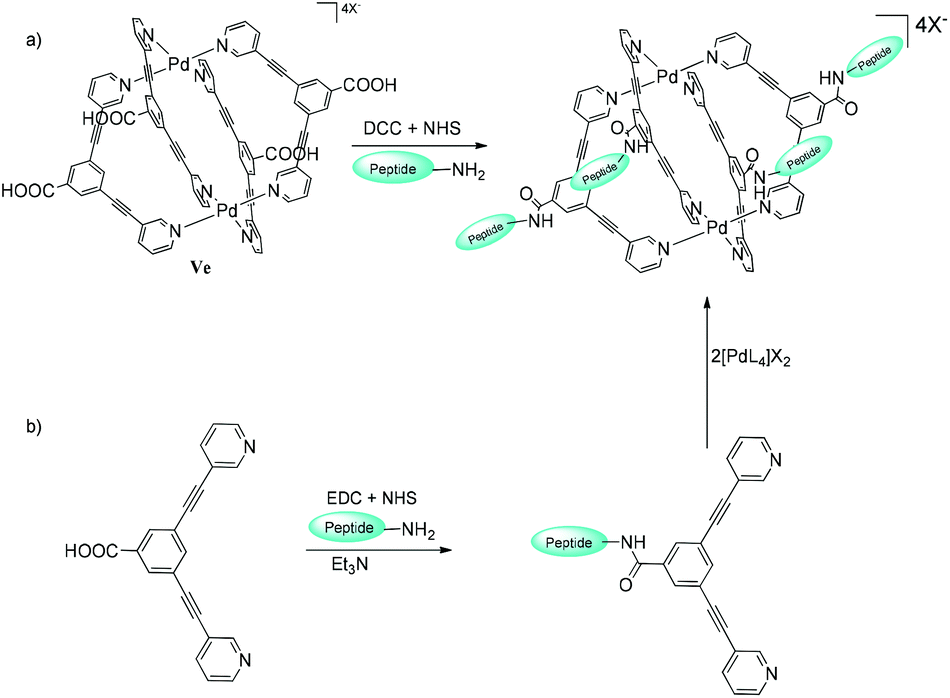 | ||
| Fig. 29 (a) Direct peptide tethering to metallacage V′′Pd and (b) initial ligand anchoring to the peptide, followed by self-assembly of metallacage V′′Pd. Reproduced with permission from ref. 109. Copyright 2017, Royal Society of Chemistry. | ||
Both approaches involved the formation of an amide bond between the amine (or carboxylic acid) functioning as an exo-functionalized ligand/cage and the carboxylic acid (or amine) groups of the side chain of the peptide. Approach II (first the peptide was coupled with the ligand and then the cage was constructed by self-assembly) showed better results, and it opens up the possibility of bioconjugation of metallacages to other target moieties like peptides, affirmers or antibodies. Fluorescence in metallacages due to florescent-exo-functionalized bipyridyl ligands enables one to track their in vitro cellular distribution and cellular accumulation, making them highly desirable components in drug-delivery systems. However, the photoluminescence of the Pd2L4 coordination cage systems is often quenched due to: (i) the “heavy-metal effect” observed on the binding of the fluorescent ligand to a Pd2+ metal-ion and (ii) the destruction of the emissive fluorescent conjugated system.68 So, to study and improve their fluorescent properties, Kühn and co-workers synthesized exo-functionalized cages coupled with fluorescent groups like naphthalenyl or anthracenyl moieties via an amide bond. A carboxy-functionalized system was used to compare the emission properties and anticancer activity of fluorophore-based cage complexes in cancer cells. The carboxy-based ligand exhibited a fluorescent quantum yield higher than that of ligands bonded to fluorescent tags. TD-DFT experiments revealed that a lower HOMO–LUMO transition probability (2%) in fluorophore groups than in carboxyl-functionalized ligands (24%) and an emission in the IR region (2000 nm) could be responsible for the fluorescence quenching. The cages attached to fluorophore groups were also investigated for their in vitro anti-proliferative properties in human ovarian cancer and lung cell lines. The cage complexes displayed higher cytotoxicity than cisplatin in all tested tumor cells, making them appropriate anticancer agents.
Recently, the bispyridyl ligands of the cage were exo-functionalized with boron dipyrromethene (BODIPY) to understand the fate of metallacages after entering the cells using fluorescence microscopy. Encapsulation and release of cisplatin were studied by 1H-NMR and 31P-NMR spectroscopy. The BODIPY-marked cage complexes accumulated in the cytoplasmic vesicles of melanoma cells.110 Kühn and co-workers attempted the conjugation of tris(bipyridine)ruthenium (a highly luminescent fluorophore) to the Pd2L4 metallacage scaffold via an unsaturated amide or cycloazide linker group to avoid the destruction of the emissive fluorescent group.111 The resulting cage exhibited a strikingly high quantum yield of 66%, being categorized among the most emissive metallacages.
4.3. Octahedral MSCCs (cage I)
Lippard and co-workers developed a novel cationic Pt-based drug-delivery system from a cytotoxic adamantylplatinum(IV) prodrug and hexanuclear platinum(II) cage IPt. Structurally, IPt is similar to cage I except that the Pd2+ have been replaced by Pt2+ metal centers (Fig. 30). This system would release cisplatin in the cancerous cells upon reduction.112 The host–guest complex displayed micromolar potency against human cancer cell lines A549, A2780, and A2780CP70, comparable to cisplatin. IC50 values showed that the cytotoxicity profile of the prodrug improved after encapsulation due to the higher cellular uptake of the positively charged host–guest cage nanoconstruct. The platinum(IV) complex prodrug was reduced within the cells by ascorbic acid to release cisplatin, succinic acid and 1-adamantylamine. as indicated by NMR spectroscopy and mass spectrometry data.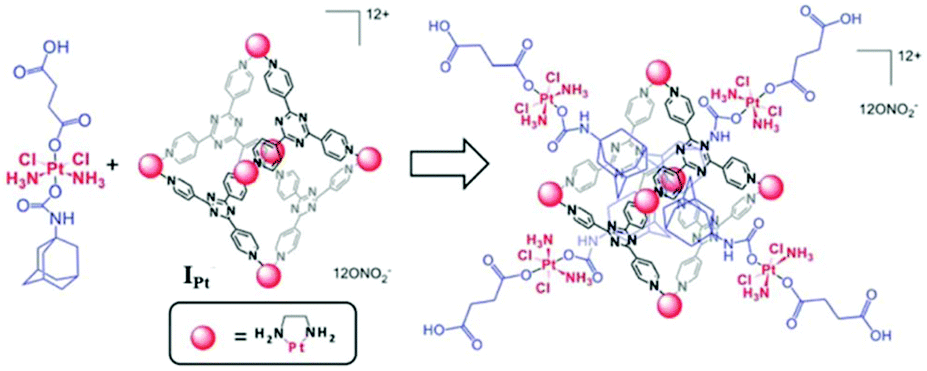 | ||
| Fig. 30 Representation of a Pt drug-delivery system formed from a cytotoxic adamantylplatinum(IV) prodrug and hexanuclear platinum(II) cage IPt. Reproduced with permission from ref. 112. Copyright 2015 Royal Society of Chemistry. | ||
5. MSCCs as reaction vessels
Inspired by enzymes, scientists are persistently working to explore similar highly efficient approaches for synthetic systems.113 This is because the special microenvironment within an enzyme cavity induces many effects, such as substrate pre-organization, protein dynamics, restricted substrate motion, de-solvation of the substrate and covalent binding of the transition state, which play a crucial role in catalysis.15,16,114 The reaction medium greatly affects the chemical reactions; however, inside the enzyme cavity, the substrates and transition/intermediate species are stripped off from solvent molecules and placed in the microenvironment of the cavity. The study of these cavity effects is challenging and has been principally proposed on the grounds of computational studies.115 The nanoreactors (nanosized reaction vessels) enable these cavity effects to be studied experimentally and they can be used as a sustainable future tool for synthesis by harnessing technique used by enzymes in nature. The reactions studied within these nanovessels are highly accelerated and regio- and enantio-selective. A range of MSCCs that have been studied for reaction control within the cavities are described in the following section. The metallacages catalyze the chemical reactions by a unique method without directly participating in the chemical reactions. The confined spaces within the cages provide a microenvironment very different from bulk solution and bring the binding sites of the substrates close together by restricting their movements. So, their catalytic mechanism does not involve active participation of the cages but the alignment of reactants results in unique products and accelerated reaction rates. The encapsulation of substrates to form ternary complexes is described in this review, which are crucial for such unique transformations. However, SCCs with embedded active sites and encapsulated catalysts are not discussed.5.1. Octahedral MSCCs (cage I)
As supramolecular coordination cage I can encapsulate two or more of the same or different molecules as guests in the cavity in restricted positions, the self-assembled coordination nanocage has been explored as a molecular flask by Fujita and co-workers for various chemical reactions.116–119 The range of chemical transformations explored in the cavity of cage I are discussed below.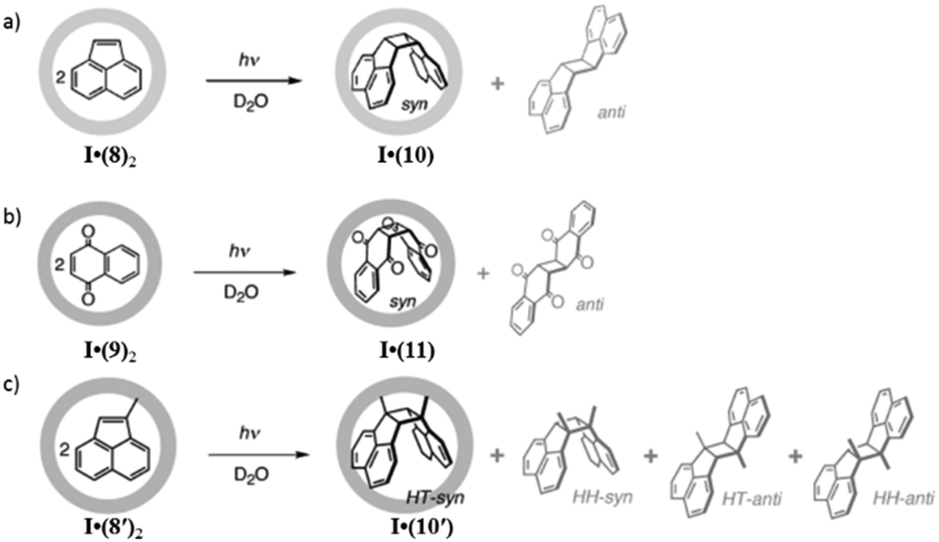 | ||
| Fig. 31 Photodimerization of (a) 8, (b) 9 and (c) 8′ within cage I. Reproduced with permission from ref. 116. Copyright 2002, John Wiley & Sons, Ltd. | ||
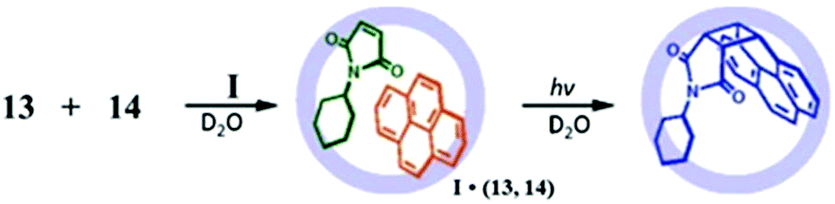 | ||
| Fig. 32 [2 + 2] photo-addition of 13 and 14 within cage I. Reproduced with permission from ref. 118. Copyright 2007, American Chemical Society. | ||
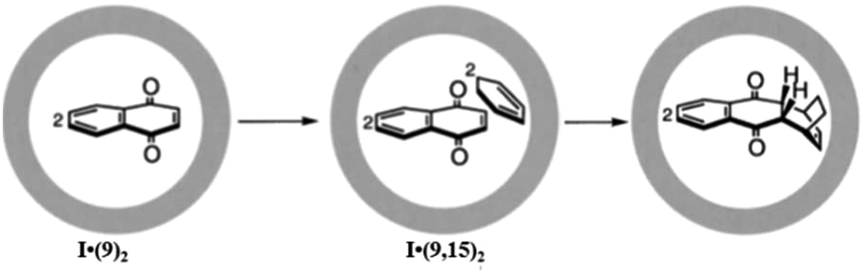 | ||
| Fig. 33 [2 + 4] thermal Diels–Alder reaction of 9 with 15 within cage I. Reproduced with permission from ref. 122. Copyright 2003, Chemical Society of Japan. | ||
Remarkably, the encapsulation of a dienophile with an inert aromatic compound (which shows no reactivity towards a Diels–Alder reaction under typical reaction conditions) exhibited good reactivity inside the cage cavity of I. For example, heating a solution of triphenylene and maleimide (14) in cage I resulted in Diels–Alder cycloaddition where one of the triphenylene benzene rings acted as a diene.118 Triphenylene does not show pericyclic reactions due to its high aromatic stability. However, with maleimide 14 the Diels–Alder reaction proceeded efficiently within the cage I cavity. Similarly, perylene and maleimide (14) enclatherated within cage I in water to produce a ternary complex upon stirring for 24 h that resulted in an exo-adduct. Cage I stabilized the final product from aerial oxidation, which reproduced perylene and acted as a sealed nanosized chamber for unstable products. The same group also investigated the host-mediated [4 + 2] thermal coupling reaction of anthracenes and maleimides. The Diels–Alder reaction of anthracene and maleimide exhibited an unusual regioselectivity in the presence of cage I.123 But the absence of the cage yields a product bridging the (9,10-position) of the center ring of anthracene due to high localization of p-electron density there. This well-established selectivity is altered in the presence of cage I by forming adduct bridging at the electronically unfavourable terminal ring (1,4-position). Molecular modeling determined that the terminal benzene is accessible due to the steric demand of a dienophile (an N-cyclohexyl group) blocking access to the central ring. Previously 1,4-selective adducts of anthracene have been reported only for benzene and 9,10-diarylanthracene addition reactions. The molecular cage I cavity creates neat conditions, where due to local accumulation and pre-organization of the substrate, the reaction profile is switched from bimolecular to pseudo-intramolecular by reducing the high entropic cost of a bimolecular reaction. Unlike previous examples where product inhibition was a serious problem, in this Diels–Alder reaction an exclusion step is involved here, which is similar to enzymatic behavior (the host has an affinity for the reactants and a disaffinity for the product). Pair-selective encapsulation of 9-hydroxymethylanthrancene (16) and N-cyclohexylmaleimide (14) within cage I resulted in the syn-isomer of the 1,4-adduct from the Diels–Alder reaction inside the cavity (Fig. 34).123
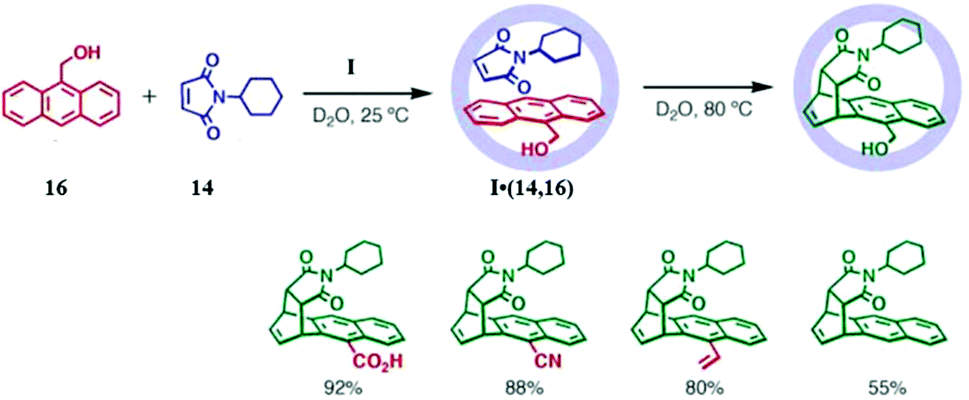 | ||
| Fig. 34 Pair-selective encapsulation followed by Diels–Alder reaction and yields of regioselective syn-1,4-products in cage I. Reproduced with permission from ref. 123. Copyright 2006, American Association for the Advancement of Science. | ||
The intra-cavity Diels–Alder reaction of anthracene with maleimide resulted in product bridging at the usual central ring, when less sterically demanding substituents (like Ph, Et, Me, tBu, or Ph), were used instead of the cyclohexyl group in 14. Hence, the site of reaction on anthracene is determined by the bulkiness of the N-substituted dienophile 14. Naphthalene has extensive utility in synthetic chemistry, but it is notoriously inert towards Diels–Alder reactions with <1% yield even when maleic anhydride is in large excess. Thus, it requires either Lewis acid activation or harsh conditions like high pressures or peri-1,4-substitution to increase its reactivity. Fujita's group studied the reactivity of naphthalene as a diene in cage I due to an increase in effective concentration and pre-organization, but no change in 1H-NMR was observed.124 Then they introduced alkyl groups at positions C2 and C3 of naphthalene to enhance their reactivity.124,125 2,3-Diethylnaphthalene (17) and N-cyclohexylmaleimide (14) formed an I·(14,17) complex in an aqueous solution of cage I, which on heating for 8 h at 100 °C resulted in a Diels–Alder reaction.
The formation of product was confirmed by the disappearance of signals for 14 and 17 from NMR and the appearance of a new set of signals. No evidence of side product was found, but the yield was reduced due to partial sublimation of the reactants. It is noteworthy that the reaction occurred on the non-substituted, less-electron-rich benzene ring of naphthalene, even though the reactivity of the substituted ring is increased by an electron-donating alkyl group giving an unusual regio- and stereo-selective adduct. The unusual selectivity is due to pre-organized substrate encapsulation within cage I and not due to the electronic effects of the alkyl substituents. Analysis of X-ray crystallographic data confirmed the close proximity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 14 and the terminal ring of naphthalene. Reactant packing and pre-organization increased with an increase in steric bulkiness on naphthalene (fused cyclohexyl (17e) < diethyl (17c) < di-n-propyl (17d)), increasing the reaction rate in the same order (Table 1).124
C bond of 14 and the terminal ring of naphthalene. Reactant packing and pre-organization increased with an increase in steric bulkiness on naphthalene (fused cyclohexyl (17e) < diethyl (17c) < di-n-propyl (17d)), increasing the reaction rate in the same order (Table 1).124
| Substrate | Yields | Rate constant (×10−5 s−1) | |
|---|---|---|---|
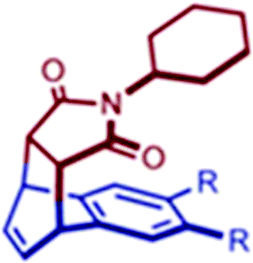
|
17a (R = H) | 0 | — |
| 17b (R = Me) | 8 | — | |
| 17c (R = Et) | 60 | 4.1 | |
| 17d (R = n-Pr) | 64 | 6.2 | |
| 17e (R, R = –CH2)4–) | 62 | 2.8 | |
| 17f (R, R = (CH2)2O–) | 0 | — |
Hence, steric and not electron factors controlled the rate and selectivity of the reaction. The tight packing of the substrates also enabled a [2 + 2] photo-addition reaction. The irradiation of complex I·(17e,14) for 1 h gave a single product with syn-configuration. Fujita's group proposed an alternative method for enhancing the substrate reactivity via fine-tuning of the cage cavity.126 The cavity of cage I was contracted by functionalizing the ancillary ligand 1,10-phenanthroline on Pd(II) with sterically bulky groups. The volume (size) of the cavity of cage I was reduced by 20% on functionalization with a mesityl (Mes) group, resulting in cage I′. The unsubstituted naphthalene (17a) undergoes a Diels–Alder reaction with N-tert-butylmaleimide (14′) in the cavity of contracted cage I′, which was otherwise unreactive in cage I. Thus, the substrate should be closely packed within the cavity of the cage (either by substituting the substrate or tuning the cage volume) for the reaction to take place efficiently. Aceanthrylene (18) and 1H-cyclopenta[l]phenanthrene (19) have stable (inert) aromatic structures due to which they have shown hardly any reactivity for pericyclic reactions. But the confined cavity of cage I unveils the unusual reactivities of these molecules, giving either a [2 + 2] photo- or [2 + 4] thermal adduct with 14 from ternary complexes I·(18,14) and I·(19,14) (Fig. 35).127 The isolation (encapsulation) of guest molecules from bulk in the cavity reduced the activation energy and produced regio- and stereo-selective products with good yield.
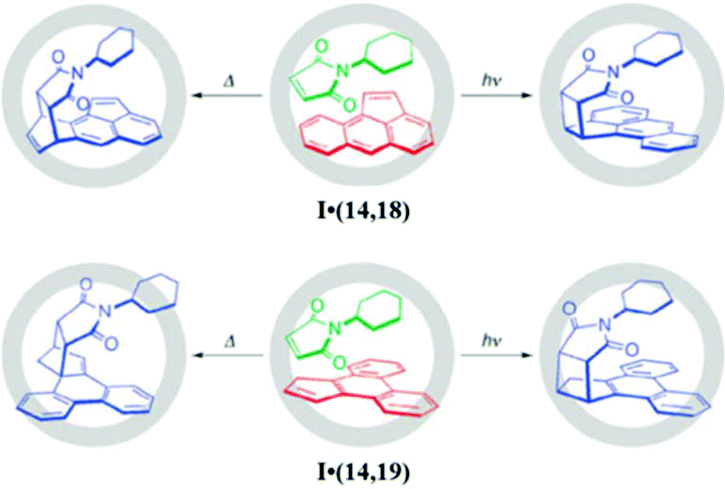 | ||
| Fig. 35 Unconventional pericyclic reactions of (14) with aceanthrylene (18) or 1H-cyclopenta[l]phenanthrene (19) within cage I. Reproduced with permission from ref. 127. Copyright 2010, Royal Society of Chemistry. | ||
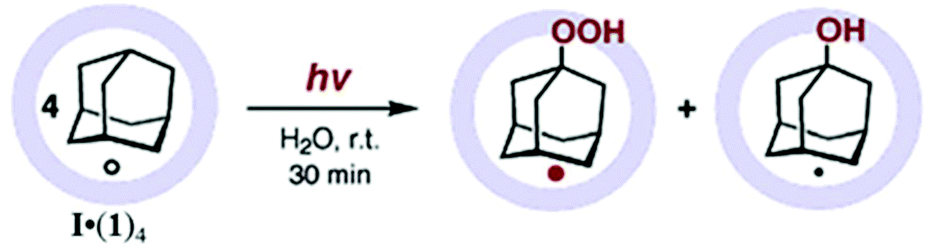 | ||
| Fig. 36 Photo-oxidation of adamantane (1) within cage I. Reproduced with permission from ref. 128. Copyright 2004, American Chemical Society. | ||
Analysis indicated the formation of 24% adamantan-1-ol or 96% assuming oxidation of only one molecule per cavity (mixed with 1-adamantylhydroperoxide). Adamantane (1) was regioselectively oxidized at the tertiary carbon. The proposed pathway included the excitation of a low-lying LUMO triazine ligand followed by the transfer of one electron from the bridgehead CH bond of adamantane to the ligand, which resulted in dissociation of the adamantane radical cation into an adamantyl radical and H+; molecular oxygen or water then trapped the adamantane radical, forming adamantan-1-ol. Electrochemical measurements reveal that cage I can accept only a single electron, hence only one out of four adamantane guests is oxidized (in cage I based 96% yield).
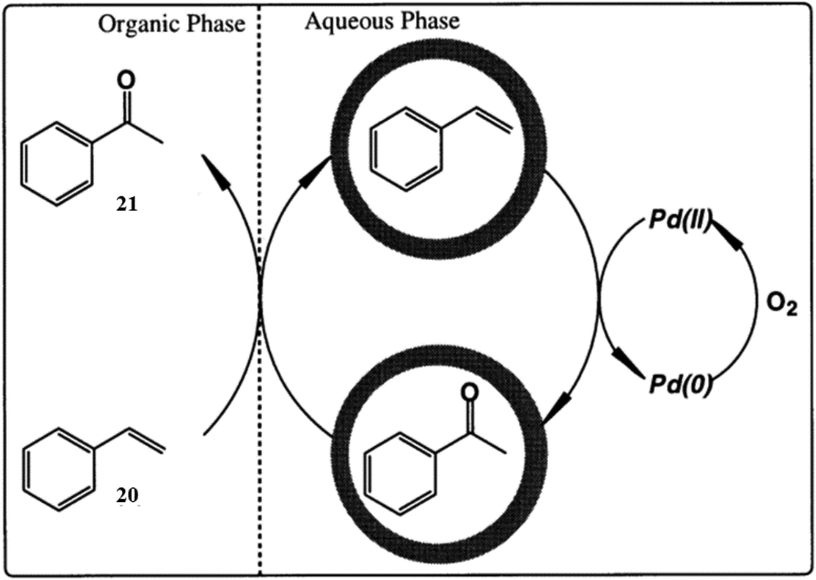 | ||
| Fig. 37 Schematic representation of the Wacker oxidation of (20) by reverse phase-transfer catalysis of I. Reproduced with permission from ref. 119. Copyright 2000, Chemical Society of Japan. | ||
Cage I also promoted the oxidation of linear alkenols in the same manner. For example, heating of 8-nonen-1-ol in cage I for 5 h at 80 °C resulted in Wacker-type oxidation, ensuring a 66% yield of 9-hydroxynonan-2-one.129 The dissolved oxygen in the aqueous solution reoxidized the Pd(0) species produced from Wacker-type oxidation.
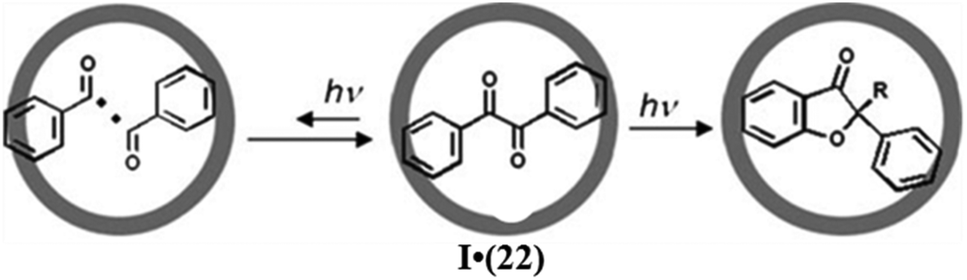 | ||
| Fig. 38 The photoreaction of 22 in the cavity of I. Reproduced with permission from ref. 133. Copyright 2007, John Wiley & Sons, Ltd. | ||
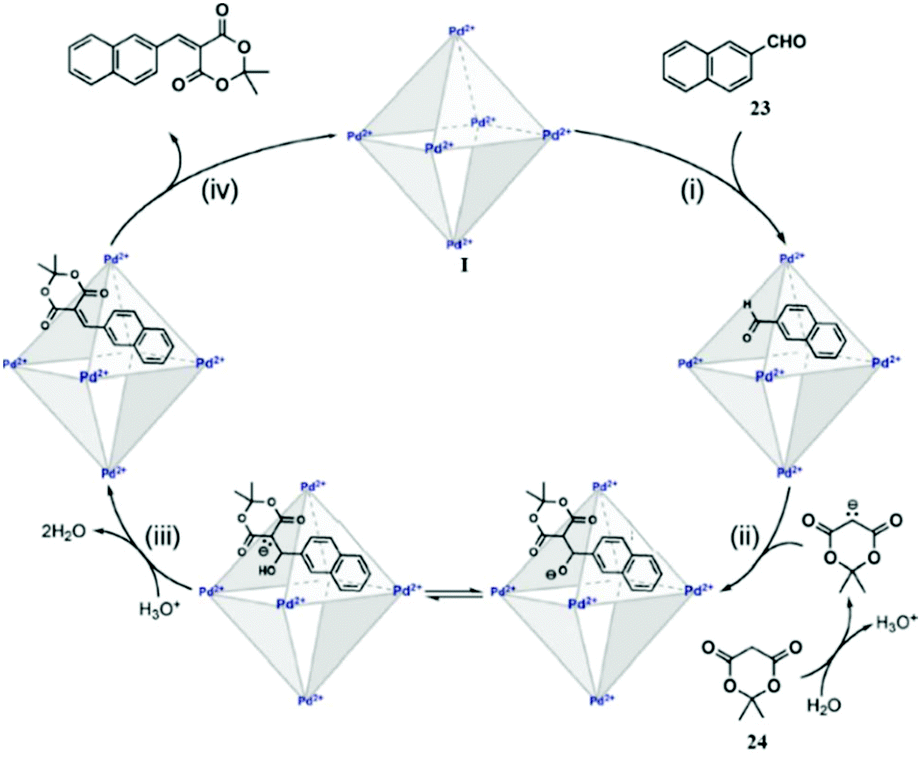 | ||
| Fig. 39 Knoevenagel condensation of aldehyde 23 with 24 in the presence of cage I. Reproduced with permission from ref. 134. Copyright 2012, American Chemical Society. | ||
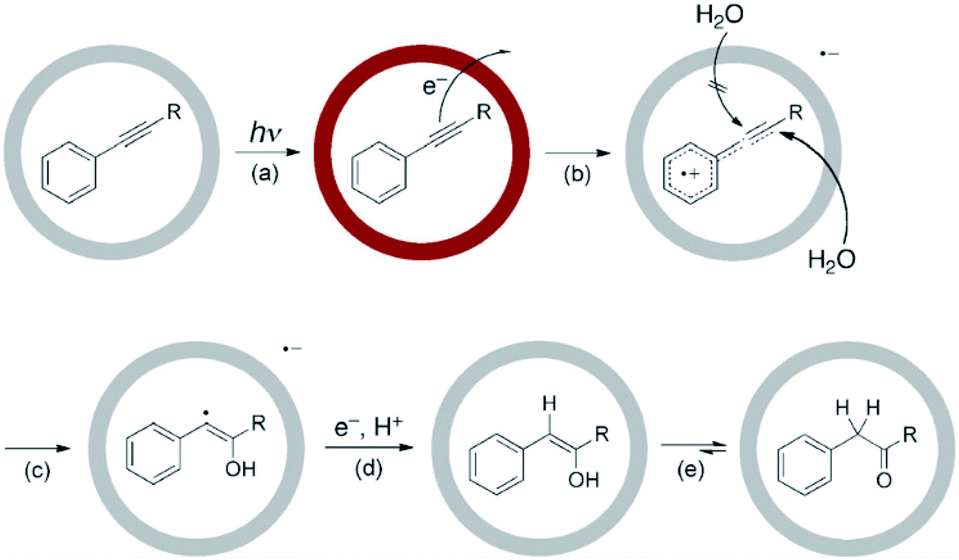 | ||
| Fig. 40 Proposed mechanism for cage I mediated photohydration of arylalkynes. Reproduced with permission from ref. 135. Copyright 2011, Royal Society of Chemistry. | ||
 | ||
| Fig. 41 Photoreaction of quinone 25 with 26 within the cavity of cage I. Reproduced with permission from ref. 136. Copyright 2008, John Wiley & Sons, Ltd. | ||
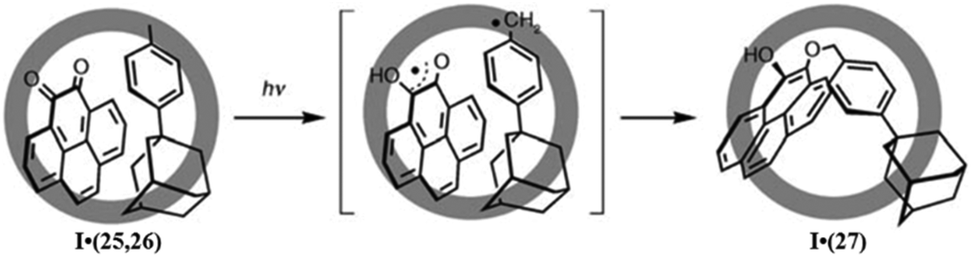
The proposed mechanism involves the photo-excitation of quinone followed by the abstraction of hydrogen from the methyl group of substituted toluene, forming a benzylic radical. 1,4-Addition of a benzylic radical to a semi-quinone radical resulted in product 27. This unusual product is due to the unfavorable arrangement of substrates within the confined cavity of cage I. They also employed phenanthrene-9,10-dione for the selective photomediated addition of a radical within the cage, which gave 80% yield of the 1,4-adduct. A complex mixture with no O-coupled 1,4-adduct was formed in the absence of cage I. Hence, cage I selectively accelerated the O-coupling pathway and suppressed the others.
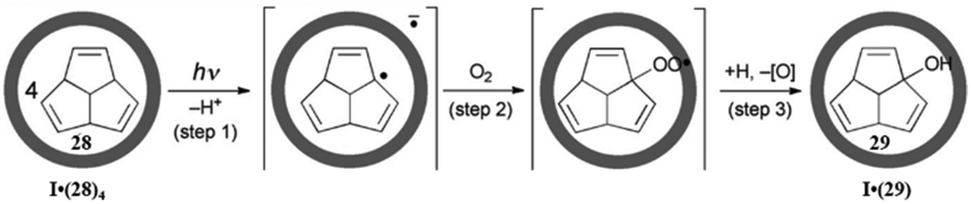 | ||
| Fig. 42 The photo-induced guest-to-host electron transfer for the formation of 29 from 28 within cage I. Reproduced with permission from ref. 137. Copyright 2012, John Wiley & Sons, Ltd. | ||
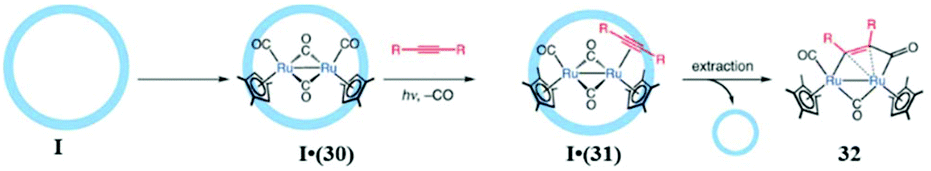 | ||
| Fig. 43 Cage I driven transformation of 30 to 32. Reproduced with permission from ref. 92. Copyright 2012, John Wiley & Sons, Ltd. | ||
C bonds non-covalently and enhanced the site-selective functionalization of the terminal prenyl moiety (Fig. 44a).138
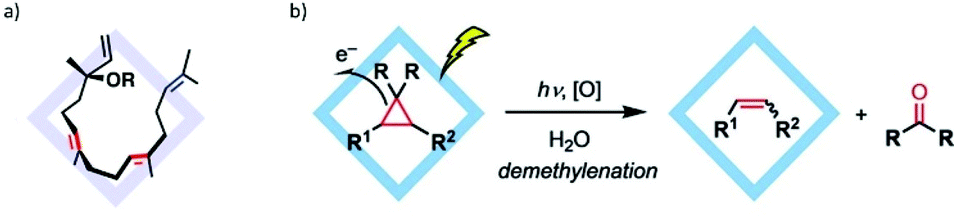 | ||
| Fig. 44 (a) Inclusion of a linear diterpenoid into cage I. (b) Photodemethylenation of cyclopropanes in cage I. Reproduced with permission from ref. 138 and 140. Copyright 2019, the American Chemical Society; and 2019, John Wiley & Sons, Ltd. | ||
5.2. M4L6 tetrahedral MSCCs (cage II)
As we discussed earlier, due to its anionic nature, cage II can encapsulate monocationic guests. These studies opened the way for the application of these tetrahedral coordination cages as reaction containers. Various reactions for which these cages have been investigated, as follows.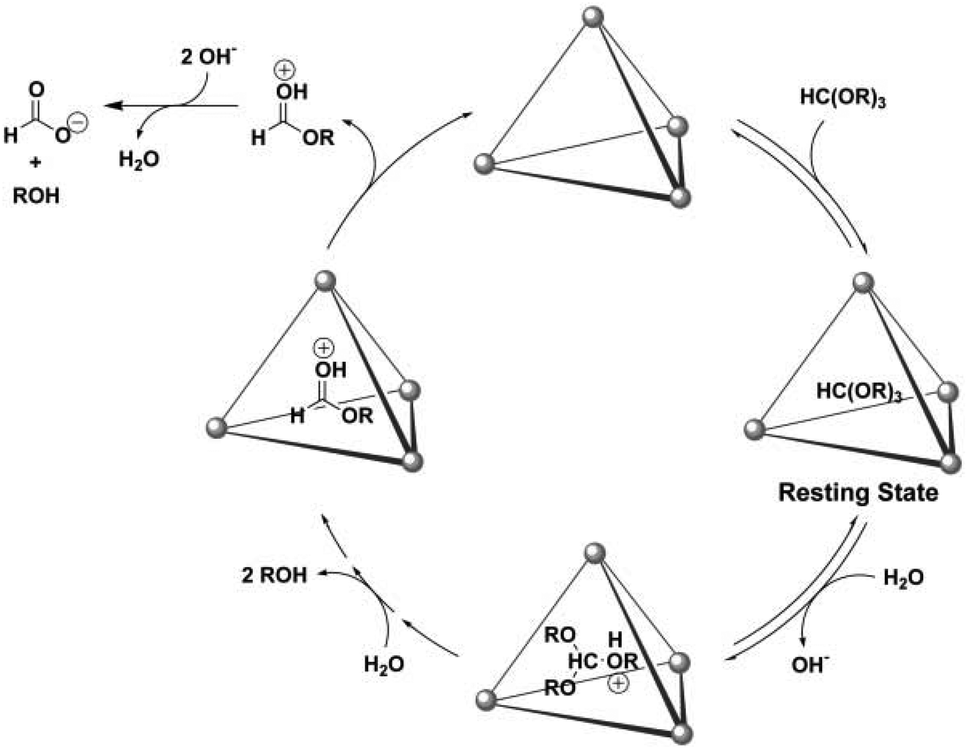 | ||
| Fig. 45 Mechanism for the hydrolysis of orthoformate in cage II. Reproduced with permission from ref. 142. Copyright 2007, American Association for the Advancement of Science. | ||
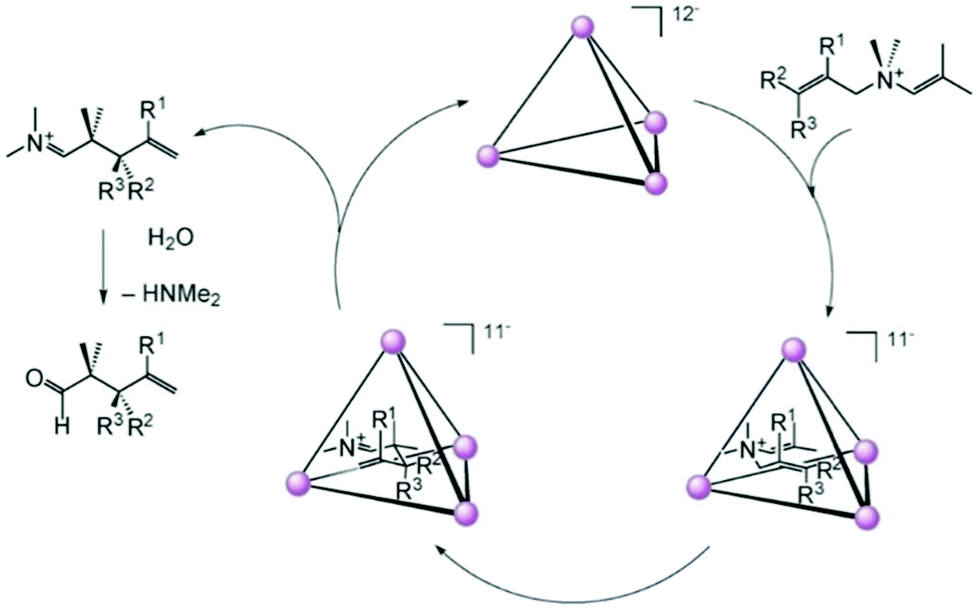 | ||
| Fig. 46 Proposed mechanism for 3-aza–cope rearrangement within cage II. Reproduced with permission from ref. 143. Copyright 2004, John Wiley & Sons, Ltd. | ||
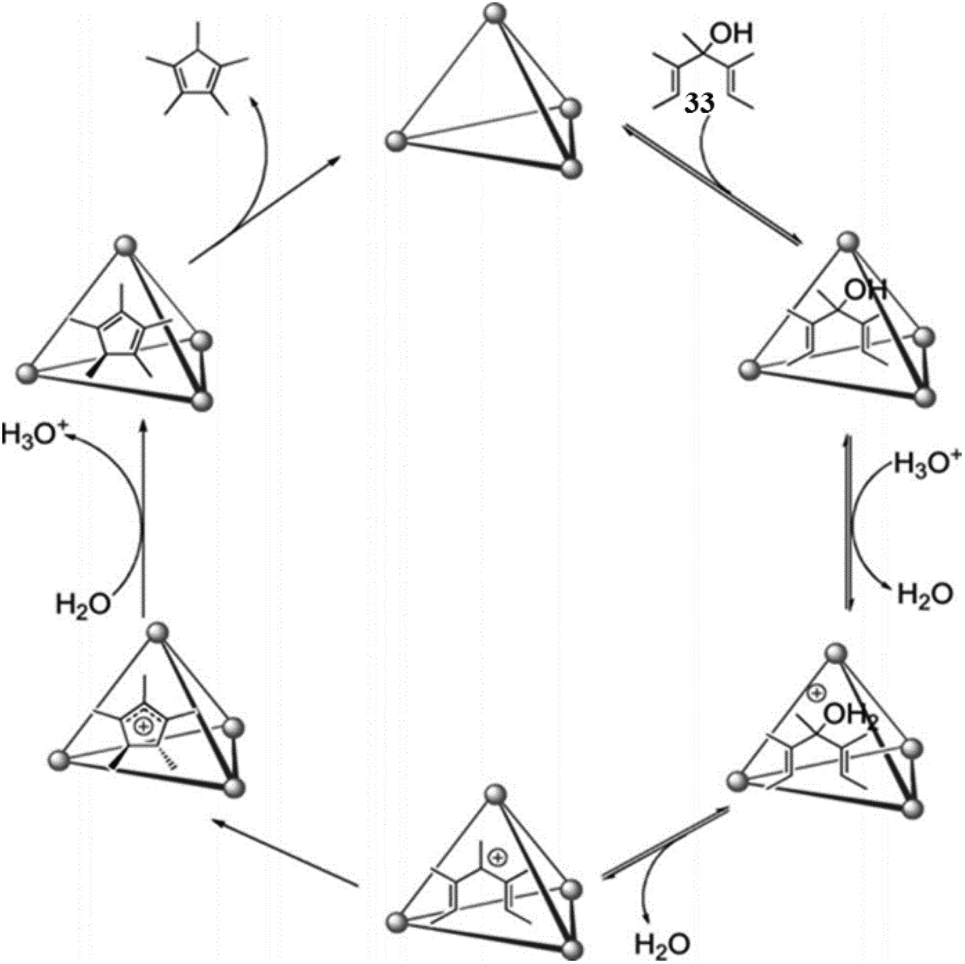 | ||
| Fig. 47 Proposed mechanism for the cage IIeGa-catalyzed Nazarov cyclization of (33). Reproduced with permission from ref. 144. Copyright 2014, John Wiley & Sons, Ltd. | ||
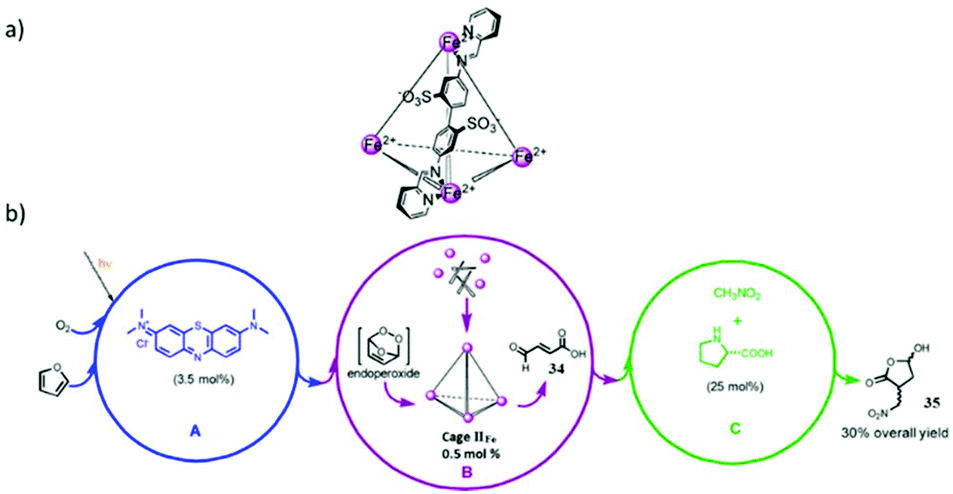 | ||
| Fig. 48 (a) Structure of cage IIFe′. (b) Relay multicatalytic system. Reproduced with permission from ref. 146. Copyright 2013, American Chemical Society. | ||
5.3. Trigonal prismatic MSCC (cage IV)
Mukherjee and co-workers designed a trigonal prismatic Pd6L3 SCC from six Pd(II) and three tetraphenylethylene (TPE) ligands by a self-assembly process.148 They explored the synthesis of xanthenes via a dehydration reaction within a cationic cage in an aqueous medium (Fig. 49).21 Such xanthene synthesis reactions generally require high temperatures and anhydrous conditions and proceed via tetraketone intermediates. The cage can efficiently catalyze the synthesis of tetraketones and corresponding xanthenes within its hydrophobic cavity in water. They could isolate both cyclized xanthenes as well as non-cyclized tetraketones by mere variation of the reaction temperature.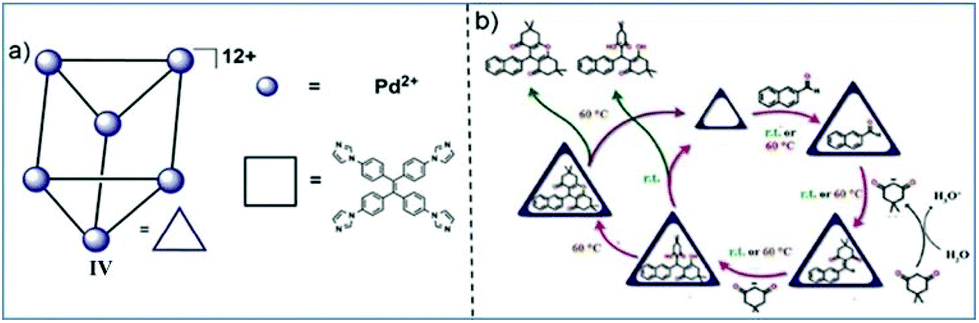 | ||
| Fig. 49 Cage IV catalyzed synthesis of xanthenes via hydrolysis. Reproduced with permission from ref. 21. Copyright 2019, Royal Society of Chemistry. | ||
5.4. M8L12 cubic MSCC (cage VII)
Ward and co-workers synthesized water-soluble cubic octanuclear coordination cages M8L12via self-assembly of Lm-Ph ((1,3-bis-3-methylene-2-phenyl)pyrazolylbenzene) or Lm-Py ((1,3-bis-3-methylene-2-pyridyl)pyrazolylbenzene) with cobalt (VIICo) (Fig. 50a) or zinc (VIIZn) in a 3:2 ratio.149–151 In the cubic cages the vertices are occupied by metal ions, while bridging ligands occupy the edges. The central cavity contained the counter-ions (ClO4− in [Zn8(Lm-Py)12](ClO4)16 or two BF4− in [Zn8(Lm-Ph)12][BF4]16) of metal salts.
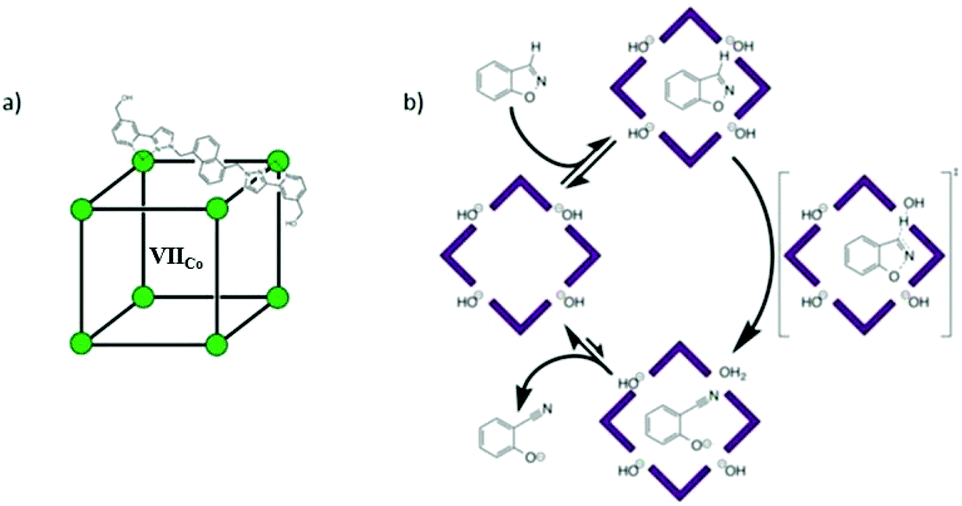 | ||
| Fig. 50 (a) Structure of host cage VIICo ([Co8L12](BF4)16). (b) Catalytic cycle, depending on hydroxide ions pairing to the surface of the cationic cage. Reproduced with permission from ref. 151. Copyright 2018, American Chemical Society. | ||
The water-soluble [Co8L12]16+ cubic coordination cage VIICo was explored for the Kemp elimination reaction of benzisoxazole to produce 2-cyanophenolate.150,151 Benzisoxazole was identified as a possible guest for the cavity from a computational screening experiment. On treatment of cage single crystals with neat liquid benzisoxazole, the guests were found to be taken up into the empty cage cavity. The two key features of the cage catalysis are (a) a very high rate enhancement (kcat/kuncat = 2 × 105 at pD 8.5) because of the accumulation of partially desolvated OH− ions around the trapped guest due to ion pairing with the positively charged cage, making it one of the best supramolecular catalysts; and (b) the catalysis is based on two supramolecular interactions that bring the bimolecular reaction substrates together: (1) hydrophobic interaction of benzisoxazole in the confined cavity and (2) polar interaction of OH− ions with the cage surface. In the catalytic system the product is easily released from the cage due to the formation of an anionic product yielding good catalytic turnover. In the Kemp elimination reaction benzisoxazole transforms into 2-cyano-phenolate through a ring-opening E2 elimination reaction with a hydroxide (base) (Fig. 50b).151 According to the proposed mechanism, ion-pairing effects lead to an accumulation of partially desolvated OH− ions around the positively charged cage, which increases their reactivity. To confirm the cavity effect on the reaction, an excess of a cyclodecanone (a strong affinity competing guest) was added to the reaction mixture. In the presence of the inhibitor, the rate of reaction dropped to that of the uncatalyzed reaction due to prevention of substrate binding inside the cavity of the cage. The catalytic effect of Co2+ ions was also studied by using 1 mM Co2+ ions in the absence of a cage, and there was no effect on the rate of reaction, confirming there no catalytic effect from the metal ions themselves.
5.5. Tetragonal MSCC (cage X)
Mukherjee and co-workers reported the synthesis of a ‘symmetrized’ tetrafacial tube-shaped molecular architecture X with tetragonal geometry by the self-assembly of a tetraimidazole donor ligand and a Pd(II) acceptor unit, [cis-(dppf)Pd(OTf)2] (dppf = diphenylphosphino ferrocene, OTf = CF3SO3−) (Fig. 51a).152 The tube-shaped tetrafacial architecture with four aromatic walls and a large cavity was used as a molecular vessel for catalyzing an intramolecular hetero [4 + 2] cycloaddition reaction and domino-type reactions. The cavity-confined intermolecular [4 + 2] cycloaddition reactions are generally stoichiometric and the large-sized products formed are trapped within the cage cavity if the cage aperture is smaller than the product size. But, the large windows in X easily release the products, making them a suitable choice for cycloaddition reactions in a catalytic fashion.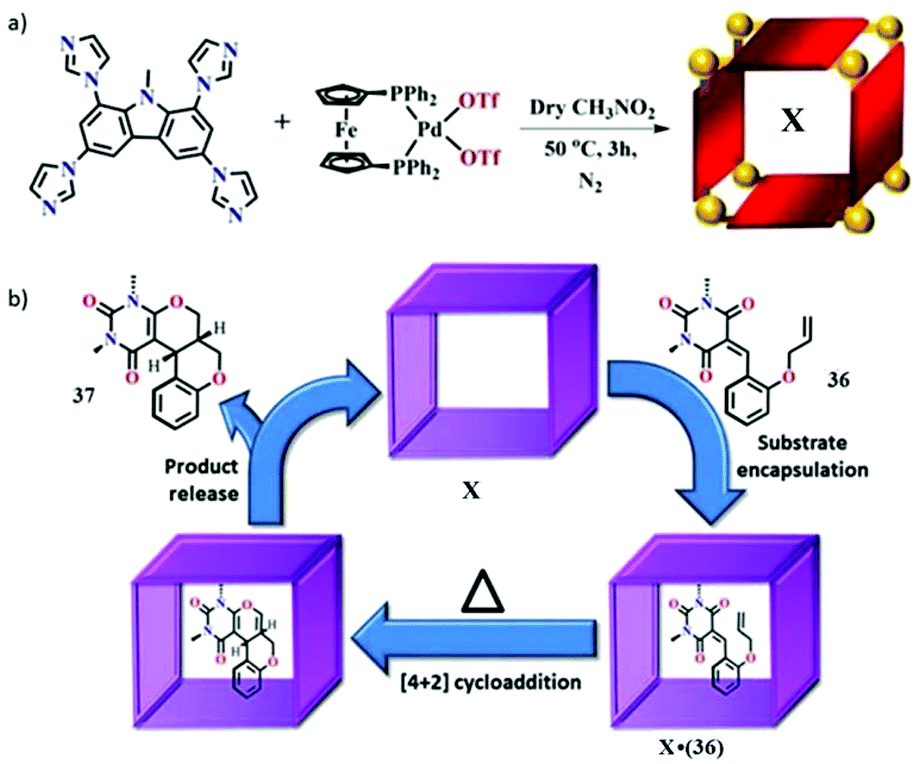 | ||
| Fig. 51 (a) Synthesis of the ‘symmetrized’ molecular tube-shaped cage X. (b) Plausible mechanism of intramolecular hetero Diels–Alder reaction within cage X. Reproduced with permission from ref. 152. Copyright 2017, John Wiley & Sons, Ltd. | ||
The intramolecular hetero Diels–Alder reaction (IMHDA) of O-allylatedbenzylidinebarbituric acid derivatives (36) was catalyzed using the confined space of cage X. The corresponding tetra-/penta-cyclouracil derivatives were obtained with good yield as products (37) and displayed both regio- and stereo-selectivity under mild reaction conditions in the presence of cage X (Fig. 51b).152 The restricted conformational flexibility of the substrates within the confined cavity increased the selectivity of the product. According to literature reports, the synthesis of similar chromenopyranpyrimidinedione derivatives needs reflux at a high temperature or a solid-state melt reaction and yields mostly isomeric mixtures, which makes this protocol more potent.153 Molecular tube X was also found to be an efficient catalyst for the domino synthesis of tetracyclo-uracil (39) directly from O-allylated salicylaldehyde (38).152 Whereas, no detectable quantity of products was formed in the absence of a cage, which demonstrates the catalytic value of X in a domino Knoevenagel hetero Diels–Alder reaction.
5.6. Triangular orthobicupolar MSCC (cage XI)
Recently Mukherjee and co-workers synthesized another water-soluble M12L6 coordination cage XI from a symmetrical tetrapyridyl donor ligand and an acceptor unit cis-(tmeda)Pd(NO3)2 [tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine] by the process of self-assembly (Fig. 52a).154 This is an unusual example of a triangular orthobicupolar-like geometry. The barrel-like cage was used for the intramolecular cycloaddition reaction of O-propargylated benzylidinebarbituric acids in a concerted manner. Through a hetero Diels–Alder reaction fused poly-heterocyclic compounds can be synthesized efficiently by the concerted formation of two or more rings. The low reactivity of alkynes compared to alkenes makes them less efficient dienophiles. The [4 + 2] cycloaddition of unactivated alkynes requires high temperatures and high catalyst loading with a still longer reaction time, resulting in poor or moderate product yields. Coordination cage XI was used as a catalyst for the hetero Diels–Alder reaction of reactive alkynes as dienophiles. Heating of naphthylidenebarbituric acid derivative (40) in cage XI in aqueous solution for 24 h resulted in only 22% pentacyclic chromenopyranpyrimidine dione framework (41) and 78% hydrolysis product hydroxy naphthaldehyde. On heating 40 with 5 mol% cage XI at 70–80 °C for 24 h in nitromethane, a pentacyclic framework (41) was formed in 98% yield as the Diels–Alder product, while the same reaction in the absence of cage XI resulted in 25% product, indicating the supremacy of the coordination cage for the intramolecular Diels–Alder reaction. A range of propargyl-containing benzylidenebarbituric acid derivatives were used as substrates, which contained a heterodiene (1-oxa-1,3-butadiene) and a dienophile (a terminal alkyne moiety) to show the efficiency of cage XI as a catalyst. The large windows in XI make product release easy, enabling it to be used as a catalyst (Fig. 52b).154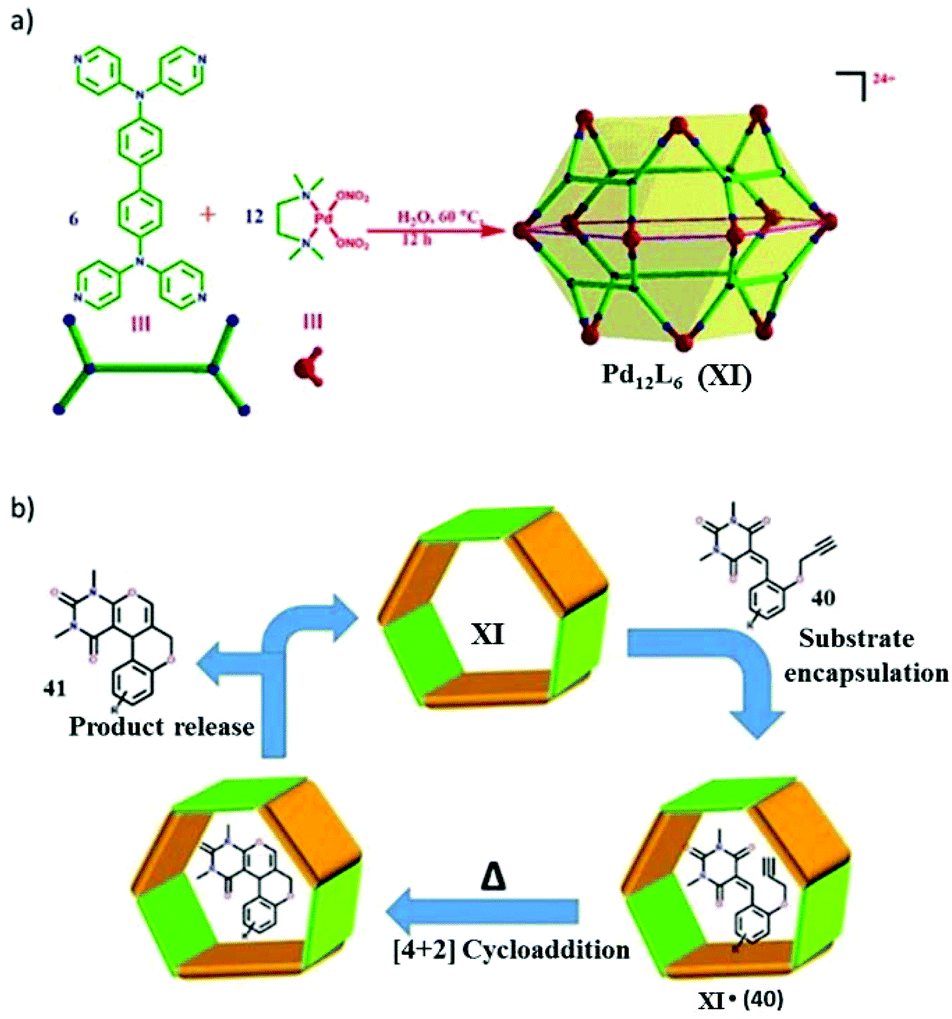 | ||
| Fig. 52 (a) Synthesis of orthobicupolar Pd12 cage XI. (b) Proposed pathways for the intramolecular hetero Diels–Alder reaction of 40 within cage XI. Reproduced with permission from ref. 154. Copyright 2018, John Wiley & Sons, Ltd. | ||
5.7. Hexahedral MSCC (cage XII)
Recently, Cui and co-workers demonstrated the synthesis and application of hexahedral cages (XII) Zn8L6, [(Zn8L6)(OTf)16] by the self-assembly of six bulky and electron-rich aromatic tetraphenylethylene-derived tetrakis-bidentate linkers with eight zinc(II)tris(pyridylimine) centers to induce cascade reactions (Fig. 53).155 | ||
| Fig. 53 Self-assembly of cage XII′ (above) and XII′′ (below) from sub-components. Reproduced with permission from ref. 155. Copyright 2018, Springer Nature. | ||
The flexible and hydrophobic cage cavities encapsulated aromatic substrates anthranilamides (42) and aromatic aldehydes (43) via hydrophobic and CH⋯π interactions and catalytically accelerated a cascade of condensation and cyclization steps to form bent-shaped 2,3-dihyroquinazolinones (44) (Fig. 54).155 The reaction was highly efficient with a 38000-fold maximum rate enhancement. The host–guest geometric discrepancy arising from the unfavorable nonplanar configurations of the products promoted expulsion of the product from the cage cavity, leading to multiple catalytic turnovers. Through molecular simulations and control experiments, they suggested that the cavity-confined microenvironment triggered the inherent power of the binding affinity of two substrates via hydrophobic and π⋯π interactions.155
 | ||
| Fig. 54 Condensation of anthranilamide with aldehyde and subsequent cyclization within the cage XII. Reproduced with permission from ref. 155. Copyright 2018, Springer Nature. | ||
5.8. Extended-octahedral MSCC (cage XIII)
Sun and co-workers self-assembled a water-soluble molecular flask Pd4L2 cage XIII from two pyridinium-functionalized bis-bidentated ligands and four cis-blocked palladium corners units (Fig. 55a).156 Metal cage XIII (Pd4L2) is similar to cage I (Pd6L4) reported by the Fujita group.34 But this cage has a more expanded internal cavity than cage I due to the insertion of two p-xylene spacers. Moreover, with the presence of strongly electron-deficient pyridinium rings on the TPT (2,4,6-tri-4-pyridyl-1,3,5-triazine) panels induced by two p-xylene spacers, cage XIII can bind several guests compared to cage I. The cage cavity of XIII was recently employed to obtain functionalized coumarins (47) from a range of salicylaldehydes (45) and cyanoacetates/malononitrile (46) via Knovenagel condensation (Fig. 55b).156 In the absence of cage XIII, guest 45 with 46 gave rise to 47 in a much lower yield (37.5%). Other control experiments employing the cage components and the same reaction conditions did not progress the reaction. Reactions of a range of salicylaldehyde derivatives with cyanoacetates/malononitrile were carried out with the cage to elucidate the applicability of the method, and all the cases resulted in an increased product yield within the cage compared to background experiments. Michaelis–Menten kinetic studies revealed that the reaction rate had been increased over 23-fold. Also, the fine-tuning of the cage cavity in terms of size and symmetry are critical for the supramolecular transformation because the catalysis of the same reaction by a smaller Pd6L4 cage gave a mixture of products in very low yield.156 | ||
| Fig. 55 (a) Structure of cage XIII with cartoon representation. (b) Formation of inclusion complex XIII·(45)4 and Knoevenagel condensation/intramolecular cyclization within cage XIII. Reproduced with permission from ref. 156. Copyright 2019, Science China Press and Springer-Verlag GmbH Germany. | ||
5.9. Cuboctahedral MSCC (cage XIV)
The cuboctahedral coordination cage XIV synthesized by Nitschke and co-workers directed the self-assembly of otherwise unstable complexes within its cavity. The 24+ charged cage XIV simultaneously encapsulated three distinct sub-components and self-assembled them into complexes that cannot be prepared outside within the cage cavity. The inner cavity provided unique chemical environments for self-assembly.1576. Conclusions and outlook
The modular design and combinatorial divergence of supramolecular coordination complexes have provided a richly diverse array of architectures, ranging from metallacycles to metallacages. The exploration of cavity-based applications of self-assembled coordination cages (SCCs) is fruitful and has the potential for remarkable growth in the fields not only of chemistry but also of medical and material sciences. The unique behaviour of hosts and guests encapsulated within their cavity have found application in various fields of chemistry and biochemistry. The nanocages discussed above are no longer obscure and of limited utility and represent next-generation nanoscale laboratory equipment. The vast structural and synthetic studies of supramolecular coordination cages have made possible the commercial availability of some of them. The coordination cages discussed here represent the beginning of a new field for the design and application of cost-efficient multifunctional MSCCs. The thriving interest in biological system inspired multifunctionalities and hierarchical structures have laid the foundations for a branch of synthetic chemistry with applications in all the fields of science (chemistry, biology, physics).158 The ease of synthesis of these cage complexes has already provided us with a rich library of architectures varying in shape, size and geometry. The functionalization of these architectures to afford multifunctional assemblies is the challenge ahead. We await that along with chemists; physicists, material scientists and biologists are also discovering their application as nanocontainers, enzyme mimetics, chemosensors and delivery systems.We hope this review will inspire new researchers to explore the microenvironment of supramolecular cages and their cavity-based applications. One of the main challenges in the field of designing metal-mediated supramolecular cages is the fine-tuning of cage cavities in terms of their dimensions as well as functional groups, according to their application. The incorporation of various functional groups in the interior of a cavity, like hydrogen-bond donors or acceptors, or chromophoric groups to encapsulate specific guests, could be the next step towards the rational design of MSCCs. Such functionalized architectures would be easy to characterize and track using spectroscopic and analytical techniques. Also, functionalization would also make it possible to study the microenvironment and chemical effects within cavities, which have not yet been studied but have been proposed using computational methods like DFT (Density Functional Theory). A detailed study of the internal cavities of these architectures would lead to an exploration of their advanced applications. Eventually, better structural characterization will be most important. Various spectroscopic techniques like NMR, fluorescence, and UV-Vis are certainly informative for discussing the structural impacts of molecular architectures; however, X-ray crystallography delivers the clearest structural evidence of molecular interactions. A detailed study of the internal cavity environment and chemical transformations within cages by using direct and powerful techniques, such as in situ X-ray diffraction or single-crystal transformation, could be the future for understanding a new phase within MSCCs.
Developing more systematic and precise characterization techniques to investigate microenvironment inside the cavity158 and interaction of encapsulated guests within the cage cavity have great potential for growth in the future. This would help in the design and synthesis of multifunctional cage complexes. The coming era can be imagined with smart, multiple guest-specific sites engineered within a single MSCC host, serving as multifunctional systems. Overall, the design and synthesis of more sophisticated coordination cages along with powerful techniques will ultimately lead to advanced sensing systems, energy storage systems, drug-carrier systems and multifunctional reaction vessels.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PK thanks Jiaxing University for providing financial support (Grant No.: CD70518066).References
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular coordination: Self-assembly of finite two- and three-dimensional ensembles, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- J. Rebek, Molecular behavior in Small Spaces, Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef CAS PubMed.
- S. R. Seidel and P. J. Stang, High-symmetry coordination cages via self-assembly, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed.
- F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, Organometallic cages as vehicles for intracellular release of photosensitizers, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed.
- D. L. Caulder and K. N. Raymond, Supermolecules by design, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- T. N. Parac, D. L. Caulder and K. N. Raymond, Selective encapsulation of aqueous cationic guests into a supramolecular tetrahedral [M4L6]12− anionic host, J. Am. Chem. Soc., 1998, 120, 8003–8004 CrossRef CAS.
- M. D. Pluth and K. N. Raymond, Reversible guest exchange mechanisms in supramolecular host–guest assemblies, Chem. Soc. Rev., 2007, 36, 161–171 RSC.
- P. D. Beer and P. A. Gale, Anion recognition and sensing: The state of the art and future perspectives, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS PubMed.
- H.-N. Zhang, Y. Lu, W.-X. Gao, Y.-J. Lin and G.-X. Jin, Selective encapsulation and separation of dihalobenzene isomers with discrete heterometallic macrocages, Chem. – Eur. J., 2018, 24, 18913–18921 CrossRef CAS PubMed.
- X. Han, Y. X. Xu, J. Yang, X. Xu, C. P. Li and J. F. Ma, Metal-assembled, resorcin[4]arene-based molecular trimer for efficient removal of toxic dichromate pollutants and knoevenagel condensation reaction, ACS Appl. Mater. Interfaces, 2019, 11, 15591–15597 CrossRef CAS PubMed.
- B. Breiner, J. K. Clegg and J. R. Nitschke, Reactivity modulation in container molecules, Chem. Sci., 2011, 2, 51–56 RSC.
- R. Warmuth, Inner-phase stabilization of reactive intermediates, Eur. J. Org. Chem., 2001, 423–437 CrossRef CAS.
- V. M. Dong, D. Fiedler, B. Carl, R. G. Bergman and K. N. Raymond, Molecular recognition and stabilization of iminium ions in water, J. Am. Chem. Soc., 2006, 128, 14464–14465 CrossRef CAS PubMed.
- D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Self-assembled nanoreactors, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed.
- M. J. Dewar and D. M. Storch, Alternative view of enzyme reactions, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 2225 CrossRef CAS PubMed.
- J. Gao, S. Ma, D. T. Major, K. Nam, J. Pu and D. G. Truhlar, Mechanisms and free energies of enzymatic reactions, Chem. Rev., 2006, 106, 3188–3209 CrossRef CAS PubMed.
- N. Ahmad, H. A. Younus, A. H. Chughtai and F. Verpoort, Metal–organic molecular cages: Applications of biochemical implications, Chem. Soc. Rev., 2015, 44, 9–25 RSC.
- Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Encapsulation of Pt(IV) prodrugs within a Pt(II) cage for drug delivery, Chem. Sci., 2015, 6, 1189–1193 RSC.
- A. Ahmedova, R. Mihaylova, D. Momekova, P. Shestakova, S. Stoykova, J. Zaharieva, M. Yamashina, G. Momekov, M. Akita and M. Yoshizawa, M2L4 coordination capsules with tunable anticancer activity upon guest encapsulation, Dalton Trans., 2016, 45, 13214–13221 RSC.
- Y. Fang, Z. Xiao, A. Kirchon, J. Li, F. Jin, T. Togo, L. Zhang, C. Zhu and H.-C. Zhou, Bimolecular proximity of a ruthenium complex and methylene blue within an anionic porous coordination cage for enhancing photocatalytic activity, Chem. Sci., 2019, 10, 3529–3534 RSC.
- Y. Fang, J. A. Powell, E. Li, Q. Wang and H. C. Zhou, Catalytic reactions within the cavity of coordination cages, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- D. J. Cram, Molecular container compounds, Nature, 1992, 356, 29–36 CrossRef CAS.
- C. J. Pedersen, Cyclic polyethers and their complexes with metal salts, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- D. J. Cram, Cavitands: Organic hosts with enforced cavities, Science, 1983, 219, 1177–1183 CrossRef CAS PubMed.
- C. O. Mellet, J. M. G. Fernández and J. M. Benito, Cyclodextrin-based gene delivery systems, Chem. Soc. Rev., 2011, 40, 1586–1608 RSC.
- G. Crini, Review: A history of cyclodextrins, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- V. Böhmer, Calixarenes, macrocycles with (almost) unlimited possibilities, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef.
- J. M. Lehn, Perspectives in chemistry–aspects of adaptive chemistry and materials, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- J. Shi, Y. Chen, Q. Wang and Y. Liu, Construction and efficient radical cation stabilization of cyclodextrin/aniline polypseudorotaxane and its conjugate with carbon nanotubes, Adv. Mater., 2010, 22, 2575–2578 CrossRef CAS PubMed.
- R. Eelkema, K. Maeda, B. Odell and H. L. Anderson, Radical cation stabilization in a cucurbituril oligoaniline rotaxane, J. Am. Chem. Soc., 2007, 129, 12384–12385 CrossRef CAS PubMed.
- L. S. Kaanumalle, J. Nithyanandhan, M. Pattabiraman, N. Jayaraman and V. Ramamurthy, Water-soluble dendrimers as photochemical reaction media: Chemical behavior of singlet and triplet radical pairs inside dendritic reaction cavities, J. Am. Chem. Soc., 2004, 126, 8999–9006 CrossRef CAS PubMed.
- W. Wei, W. Li, Z. Li, W. Su and M. Hong, Stabilization and controlled release of reactive molecules by solid-state van der Waals capsules, Chem. – Eur. J., 2013, 19, 469–473 CrossRef CAS PubMed.
- F. D. Toste, Beyond the Molecule, Acc. Chem. Res., 2018, 51, 2980–2981 CrossRef CAS PubMed.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Self-assembly of ten molecules into nanometre-sized organic host frameworks, Nature, 1995, 378, 469–471 CrossRef CAS.
- T. Kusukawa and M. Fujita, Self-assembled M6L4-type coordination nanocage with 2,2′-bipyridine ancillary ligands: Facile crystallization and X-ray analysis of shape-selective enclathration of neutral guests in the cage, J. Am. Chem. Soc., 2002, 124, 13576–13582 CrossRef CAS PubMed.
- T. Kusukawa and M. Fujita, Encapsulation of large, neutral molecules in a self-assembled nanocage incorporating six palladium(II) ions, Angew. Chem., Int. Ed., 1998, 37, 3142–3144 CrossRef CAS PubMed.
- S. H. A. M. Leenders, R. Becker, T. Kumpulainen, B. de Bruin, T. Sawada, T. Kato, M. Fujita and J. N. H. Reek, Selective co-encapsulation inside an M6L4 cage, Chem. – Eur. J., 2016, 22, 15468–15474 CrossRef CAS PubMed.
- F. Ibukuro, T. Kusukawa and M. Fujita, A thermally switchable molecular lock. guest-templated synthesis of a kinetically stable nanosized cage, J. Am. Chem. Soc., 1998, 120, 8561–8562 CrossRef CAS.
- S. Hiraoka and M. Fujita, Guest-selected formation of Pd(II)-linked cages from a prototypical dynamic library, J. Am. Chem. Soc., 1999, 121, 10239–10240 CrossRef CAS.
- M. Fujita, S. Nagao and K. Ogura, Guest-Induced organization of a three-dimensional palladium(II) cage-like omplex: A prototype for “Induced-Fit” molecular recognition, J. Am. Chem. Soc., 1995, 117, 1649–1650 CrossRef CAS PubMed.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Selective molecular recognition, C–H bond activation, and catalysis in nanoscale reaction vessels, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. Nitschke, White phosphorus is air-stable within a self-assembled tetrahedral capsule, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Encapsulation, storage and controlled release of sulfur hexafluoride from a metal–organic capsule, Chem. Commun., 2011, 47, 457–459 RSC.
- J. L. Brumaghim, M. Michels, D. Pagliero and K. N. Raymond, Encapsulation and stabilization of reactive aromatic diazonium ions and the tropylium ion within a supramolecular host, Eur. J. Org. Chem., 2004, 5115–5118 CrossRef CAS.
- D. L. Caulder, R. E. Powers, T. N. Parac and K. N. Raymond, The self-assembly of a predesigned tetrahedral M4L6 supramolecular cluster, Angew. Chem., Int. Ed., 1998, 37, 1840–1843 CrossRef CAS.
- S. M. Biros, R. G. Bergman and K. N. Raymond, The hydrophobic effect drives the recognition of hydrocarbons by an anionic metal–ligand cluster, J. Am. Chem. Soc., 2007, 129, 12094–12095 CrossRef CAS PubMed.
- D. Fiedler, D. Pagliero, J. L. Brumaghim, R. G. Bergman and K. N. Raymond, Encapsulation of cationic ruthenium complexes into a chiral self-assembled cage, Inorg. Chem., 2004, 43, 846–848 CrossRef CAS PubMed.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, An unlockable–relockable iron cage by subcomponent self-assembly, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed.
- L. K. S. von Krbek, D. A. Roberts, B. S. Pilgrim, C. A. Schalley and J. R. Nitschke, Multivalent crown ether receptors enable allosteric regulation of anion exchange in an Fe4L6 tetrahedron, Angew. Chem., Int. Ed., 2018, 57, 14121–14124 CrossRef CAS PubMed.
- Z. Lu, T. K. Ronson and J. R. Nitschke, Reversible reduction drives anion ejection and C60 binding within an FeII4L6 cage, Chem. Sci., 2020, 11, 1097–1101 RSC.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Selective anion extraction and recovery using a FeII4L4 cage, Angew. Chem., Int. Ed., 2018, 57, 3717–3721 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, R. Lavendomme and J. R. Nitschke, Selective separation of polyaromatic hydrocarbons by phase transfer of coordination cages, J. Am. Chem. Soc., 2019, 141, 18949–18953 CrossRef CAS PubMed.
- J. L. Bolliger, T. K. Ronson, M. Ogawa and J. R. Nitschke, Solvent effects upon guest binding and dynamics of a FeII4L4 cage, J. Am. Chem. Soc., 2014, 136, 14545–14553 CrossRef CAS PubMed.
- A. M. Castilla, T. K. Ronson and J. R. Nitschke, Sequence-dependent guest release triggered by orthogonal chemical signals, J. Am. Chem. Soc., 2016, 138, 2342–2351 CrossRef CAS PubMed.
- A. Yadav, M. Sarkar, S. Subrahmanyam, A. Chaudhary, E. Hey-Hawkins and R. Boomishankar, Anilate tethered neutral tetrahedral Pd(II) cages exhibiting selective encapsulation of xylenes and mesitylene, Chem. – Eur. J., 2020, 26, 4209–4213 CrossRef CAS PubMed.
- W.-K. Han, H.-X. Zhang, Y. Wang, W. Liu, X. Yan, T. Li and Z.-G. Gu, Tetrahedral metal-organic cages with cube-like cavities for selective encapsulation of fullerene guests and their spin-crossover properties, Chem. Commun., 2018, 54, 12646–12649 RSC.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Sponge-like molecular cage for purification of fullerenes, Nat. Commun., 2014, 5, 5557 CrossRef PubMed.
- C. Colomban, G. Szalóki, M. Allain, L. Gómez, S. Goeb, M. Sallé, M. Costas and X. Ribas, Reversible C60 ejection from a metallocage through the redox-dependent binding of a competitive guest, Chem. – Eur. J., 2017, 23, 3016–3022 CrossRef CAS PubMed.
- C. Colomban, V. Martin-Diaconescu, T. Parella, S. Goeb, C. García-Simón, J. Lloret-Fillol, M. Costas and X. Ribas, Design of Zn-, Cu-, and Fe-coordination complexes confined in a self-assembled nanocage, Inorg. Chem., 2018, 57, 3529–3539 CrossRef CAS PubMed.
- V. Martínez-Agramunt, D. G. Gusev and E. Peris, A shape-adaptable organometallic supramolecular coordination cage for the encapsulation of fullerenes, Chem. – Eur. J., 2018, 24, 14802–14807 CrossRef PubMed.
- K. Ono, M. Yoshizawa, T. Kato, K. Watanabe and M. Fujita, Porphine dimeric assemblies in organic-pillared coordination cages, Angew. Chem., Int. Ed., 2007, 46, 1803–1806 CrossRef CAS PubMed.
- B. M. Schmidt, T. Osuga, T. Sawada, M. Hoshino and M. Fujita, Compressed corannulene in a molecular cage, Angew. Chem., Int. Ed., 2016, 55, 1561–1564 CrossRef CAS PubMed.
- M. Yoshizawa, J. Nakagawa, K. Kumazawa, M. Nagao, M. Kawano, T. Ozeki and M. Fujita, Discrete stacking of large aromatic molecules within organic-pillared coordination cages, Angew. Chem., Int. Ed., 2005, 44, 1810–1813 CrossRef CAS PubMed.
- K. Kumazawa, K. Biradha, T. Kusukawa, T. Okano and M. Fujita, Multicomponent assembly of a pyrazine-pillared coordination cage that selectively binds planar guests by intercalation, Angew. Chem., Int. Ed., 2003, 42, 3909–3913 CrossRef CAS PubMed.
- N. Fujita, K. Biradha, M. Fujita, S. Sakamoto and K. Yamaguchi, A porphyrin prism: Structural switching triggered by guest inclusion, Angew. Chem., Int. Ed., 2001, 40, 1718–1721 CrossRef CAS PubMed.
- M. Zhang, H. Xu, M. Wang, M. L. Saha, Z. Zhou, X. Yan, H. Wang, X. Li, F. Huang, N. She and P. J. Stang, Pt(II)-based convex trigonal prismatic cages via coordination-driven self-assembly and C60 encapsulation, Inorg. Chem., 2017, 56, 12498–12504 CrossRef CAS PubMed.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Stimuli-responsive Pd2L4 metallosupramolecular cages: Towards targeted cisplatin drug delivery, Chem. Sci., 2012, 3, 778–784 RSC.
- A. Schmidt, M. Hollering, M. Drees, A. Casini and F. E. Kühn, Supramolecular exo-functionalized palladium cages: Fluorescent properties and biological activity, Dalton Trans., 2016, 45, 8556–8565 RSC.
- E. Puig, C. Desmarets, G. Gontard, M. N. Rager, A. L. Cooksy and H. Amouri, Capturing a square planar gold(III) complex inside a platinum nanocage: A combined experimental and theoretical study, Inorg. Chem., 2019, 58, 3189–3195 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Light-triggered guest uptake and release by a photochromic coordination cage, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed.
- R.-J. Li, J. J. Holstein, W. G. Hiller, J. Andréasson and G. H. Clever, Mechanistic interplay between light switching and guest binding in photochromic [Pd2Dithienylethene4] coordination cages, J. Am. Chem. Soc., 2019, 141, 2097–2103 CrossRef CAS PubMed.
- S. Löffler, A. Wuttke, B. Zhang, J. J. Holstein, R. A. Mata and G. H. Clever, Influence of size, shape, heteroatom content and dispersive contributions on guest binding in a coordination cage, Chem. Commun., 2017, 53, 11933–11936 RSC.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, Template control over dimerization and guest selectivity of interpenetrated coordination cages, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed.
- M. Han, J. Hey, W. Kawamura, D. Stalke, M. Shionoya and G. H. Clever, An inclusion complex of hexamolybdate inside a supramolecular cage and its structural conversion, Inorg. Chem., 2012, 51, 9574–9576 CrossRef CAS PubMed.
- G. H. Clever, W. Kawamura and M. Shionoya, Encapsulation versus aggregation of metal-organic cages controlled by guest size variation, Inorg. Chem., 2011, 50, 4689–4691 CrossRef CAS PubMed.
- M. D. Johnstone, E. K. Schwarze, J. Ahrens, D. Schwarzer, J. J. Holstein, B. Dittrich, F. M. Pfeffer and G. H. Clever, Desymmetrization of an octahedral coordination complex inside a self-assembled exoskeleton, Chem. – Eur. J., 2016, 22, 10791–10795 CrossRef CAS PubMed.
- B. Chen, J. J. Holstein, S. Horiuchi, W. G. Hiller and G. H. Clever, Pd(II) Coordination sphere engineering: Pyridine cages, quinoline bowls, and heteroleptic pills binding one or two fullerenes, J. Am. Chem. Soc., 2019, 141, 8907–8913 CrossRef CAS PubMed.
- B. Chen, S. Horiuchi, J. J. Holstein, J. Tessarolo and G. H. Clever, Tunable fullerene affinity of cages, bowls and rings assembled by PdII coordination sphere engineering, Chem. – Eur. J., 2019, 25, 14921–14927 CrossRef CAS PubMed.
- Y. Cui, Z.-M. Chen, X.-F. Jiang, J. Tong and S.-Y. Yu, Self-assembly and anion sensing of metal–organic [M6L2] cages from fluorescent triphenylamine tri-pyrazoles with dipalladium(II,II) corner, Dalton Trans., 2017, 46, 5801–5805 RSC.
- W. Brenner, T. K. Ronson and J. R. Nitschke, Separation and selective formation of fullerene adducts within an MII8L6 cage, J. Am. Chem. Soc., 2017, 139, 75–78 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, A self-assembled M8L6 cubic cage that selectively encapsulates large aromatic guests, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- Z. Li, N. Kishi, K. Yoza, M. Akita and M. Yoshizawa, Isostructural M2L4 molecular capsules with anthracene shells: Synthesis, crystal structures, and fluorescent properties, Chem. – Eur. J., 2012, 18, 8358–8365 CrossRef CAS PubMed.
- N. Kishi, Z. Li, Y. Sei, M. Akita, K. Yoza, J. S. Siegel and M. Yoshizawa, Wide-ranging host capability of a PdII-linked M2L4 molecular capsule with an anthracene shell, Chem. – Eur. J., 2013, 19, 6313–6320 CrossRef CAS PubMed.
- M. Yamashina, M. Akita, T. Hasegawa, S. Hayashi and M. Yoshizawa, A polyaromatic nanocapsule as a sucrose receptor in water, Sci. Adv., 2017, 3, e1701126 CrossRef PubMed.
- X.-Z. Wang, M.-Y. Sun, J. Zheng, D. Luo, L. Qi, X.-P. Zhou and D. Li, Coordination-driven self-assembly of M10L8 metal–organic bi-capped square antiprisms with adaptable cavities, Dalton Trans., 2019, 48, 17713–17717 RSC.
- A. Galan and P. Ballester, Stabilization of reactive species by supramolecular encapsulation, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- T. Kusukawa and M. Fujita, “Ship-in-a-Bottle” Formation of stable hydrophobic dimers of cis-azobenzene and -stilbene derivatives in a self-assembled coordination nanocage, J. Am. Chem. Soc., 1999, 121, 1397–1398 CrossRef CAS.
- M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, Direct crystallographic observation of a coordinatively unsaturated transition-metal complex in situ generated within a self-assembled cage, J. Am. Chem. Soc., 2006, 128, 6558–6559 CrossRef CAS PubMed.
- Y. Kohyama, T. Murase and M. Fujita, A self-assembled cage as a non-covalent protective group: Regioselectivity control in the nucleophilic substitution of aryl-substituted allylic chlorides, Chem. Commun., 2012, 48, 7811–7813 RSC.
- S. Horiuchi, T. Murase and M. Fujita, Noncovalent trapping and stabilization of dinuclear ruthenium complexes within a coordination cage, J. Am. Chem. Soc., 2011, 133, 12445–12447 CrossRef CAS PubMed.
- R. Gera, S. L. Meloni and J. M. Anna, Unraveling confined dynamics of guests trapped in self-assembled Pd6L4 nanocages by ultrafast mid-IR polarization-dependent spectroscopy, J. Phys. Chem. Lett., 2019, 10, 413–418 CrossRef CAS PubMed.
- S. Horiuchi, T. Murase and M. Fujita, A Remarkable organometallic transformation on a cage-incarcerated dinuclear ruthenium complex, Angew. Chem., Int. Ed., 2012, 51, 12029–12031 CrossRef CAS PubMed.
- H. Takezawa, S. Akiba, T. Murase and M. Fujita, Cavity-directed chromism of phthalein dyes, J. Am. Chem. Soc., 2015, 137, 7043–7046 CrossRef CAS PubMed.
- M. Ziegler, J. L. Brumaghim and K. N. Raymond, Stabilization of a reactive cationic species by supramolecular encapsulation, Angew. Chem., Int. Ed., 2000, 39, 4119–4121 CrossRef CAS PubMed.
- M. M. J. Smulders and J. R. Nitschke, Supramolecular control over Diels–Alder reactivity by encapsulation and competitive displacement, Chem. Sci., 2012, 3, 785–788 RSC.
- J. Mosquera, B. Szyszko, S. K. Y. Ho and J. R. Nitschke, Sequence-selective encapsulation and protection of long peptides by a self-assembled FeII8L6 cubic cage, Nat. Commun., 2017, 8, 14882 CrossRef CAS PubMed.
- B. Stordal, N. Pavlakis and R. Davey, Oxaliplatin for the treatment of cisplatin-resistant cancer: A systematic review, Cancer Treat. Rev., 2007, 33, 347–357 CrossRef CAS PubMed.
- M. J. Cleare, Transition metal complexes in cancer chemotherapy, Coord. Chem. Rev., 1974, 12, 349–405 CrossRef CAS.
- D. F. Baban and L. W. Seymour, Control of tumour vascular permeability, Adv. Drug Delivery Rev., 1998, 34, 109–119 CrossRef CAS PubMed.
- B. Therrien, G. Suss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, The “complex-in-a-complex” cations [(acac)2M subset Ru6(p-iPrC6H4Me)6(tpt)2(dhbq)3]6+: A trojan horse for cancer cells, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed.
- O. Zava, J. Mattsson, B. Therrien and P. J. Dyson, Evidence for drug release from a metalla-cage delivery vector following cellular internalisation, Chemistry, 2010, 16, 1428–1431 CrossRef CAS PubMed.
- J. Mattsson, O. Zava, A. K. Renfrew, Y. Sei, K. Yamaguchi, P. J. Dyson and B. Therrien, Drug delivery of lipophilic pyrenyl derivatives by encapsulation in a water soluble metalla-cage, Dalton Trans., 2010, 39, 8248–8255 RSC.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Excellent correlation between drug release and portalsize in metalla-cage drug-delivery systems, Chem. – Eur. J., 2011, 17, 9669–9677 CrossRef CAS PubMed.
- A. Schmidt, V. Molano, M. Hollering, A. Pöthig, A. Casini and F. Kühn, Evaluation of new palladium cages as potential delivery systems for the anticancer drug cisplatin, Chemistry, 2016, 22, 2253–2256 CrossRef CAS PubMed.
- B. Therrien, Drug delivery by water-soluble organometallic cages, Top. Curr. Chem., 2012, 319, 35–55 CrossRef CAS PubMed.
- X. Wang, X. Wang and Z. Guo, Functionalization of platinum complexes for biomedical applications, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS PubMed.
- M. Ikemi, T. Kikuchi, S. Matsumura, K. Shiba, S. Sato and M. Fujita, Peptide-coated, self-assembled M12L24 coordination spheres and their immobilization onto an inorganic surface, Chem. Sci., 2010, 1, 68–71 RSC.
- D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Protein encapsulation within synthetic molecular hosts, Nat. Commun., 2012, 3, 1093 CrossRef PubMed.
- J. Han, A. Schmidt, T. Zhang, H. Permentier, G. M. M. Groothuis, R. Bischoff, F. E. Kühn, P. Horvatovich and A. Casini, Bioconjugation strategies to couple supramolecular exo-functionalized palladium cages to peptides for biomedical applications, Chem. Commun., 2017, 53, 1405–1408 RSC.
- B. Woods, D. Döllerer, B. Aikman, M. N. Wenzel, E. J. Sayers, F. E. Kühn, A. T. Jones and A. Casini, Highly luminescent metallacages featuring bispyridyl ligands functionalised with BODIPY for imaging in cancer cells, J. Inorg. Biochem., 2019, 199, 110781 CrossRef CAS PubMed.
- A. Schmidt, M. Hollering, J. Han, A. Casini and F. E. Kühn, Self-assembly of highly luminescent heteronuclear coordination cages, Dalton Trans., 2016, 45, 12297–12300 RSC.
- Y. R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Encapsulation of Pt(IV) prodrugs within a Pt(II) cage for drug delivery, Chem. Sci., 2015, 6, 1189–1193 RSC.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Enzyme mimics based upon supramolecular coordination chemistry, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- X. Zhang and K. N. Houk, Why enzymes are proficient catalysts: Beyond the pauling paradigm, Acc. Chem. Res., 2005, 38, 379–385 CrossRef CAS PubMed.
- A. N. Petelski, S. C. Pamies, A. G. Sejas, N. M. Peruchena and G. L. Sosa, Impact of confinement in multimolecular inclusion compounds of melamine and cyanuric acid, Phys. Chem. Chem. Phys., 2019, 21, 8205–8214 RSC.
- M. Yoshizawa, Y. Takeyama, T. Kusukawa and M. Fujita, Cavity-directed, highly stereoselective [2 + 2] photodimerization of olefins within self-assembled coordination cages, Angew. Chem., Int. Ed., 2002, 41, 1347–1349 CrossRef CAS PubMed.
- K. Takaoka, M. Kawano, T. Ozeki and M. Fujita, Crystallographic observation of an olefin photodimerization reaction that takes place via thermal molecular tumbling within a self-assembled host, Chem. Commun., 2006, 1625–1627 RSC.
- Y. Nishioka, T. Yamaguchi, M. Yoshizawa and M. Fujita, Unusual [2 + 4] and [2 + 2] cycloadditions of arenes in the confined cavity of self-assembled cages, J. Am. Chem. Soc., 2007, 129, 7000–7001 CrossRef CAS PubMed.
- H. Ito, T. Kusukawa and M. Fujita, Wacker oxidation in an aqueous phase through the reverse phase-transfer catalysis of a self-assembled nanocage, Chem. Lett., 2000, 29, 598–599 CrossRef.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Photodimerization of anthracenes in fluid solution: Structural aspects, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- M. Yoshizawa and M. Fujita, Self-assembled coordination cage as a molecular flask, Pure Appl. Chem., 2005, 1107 CAS.
- T. Kusukawa, T. Nakai, T. Okano and M. Fujita, Remarkable acceleration of Diels–Alder reactions in a self-assembled coordination cage, Chem. Lett., 2003, 32, 284–285 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Diels-Alder in aqueous molecular hosts: Unusual regioselectivity and efficient catalysis, Science, 2006, 312, 251 CrossRef CAS PubMed.
- T. Murase, S. Horiuchi and M. Fujita, Naphthalene Diels–Alder in a self-assembled molecular flask, J. Am. Chem. Soc., 2010, 132, 2866–2867 CrossRef CAS PubMed.
- S. Horiuchi, T. Murase and M. Fujita, Diels–Alder reactions of inert aromatic compounds within a self-assembled coordination cage, Chem. – Asian J., 2011, 6, 1839–1847 CrossRef CAS PubMed.
- Y. Fang, T. Murase and M. Fujita, Cavity-promoted Diels–Alder reactions of unsubstituted naphthalene: Fine reactivity tuning by cavity shrinkage, Chem. Lett., 2015, 44, 1095–1097 CrossRef CAS.
- S. Horiuchi, Y. Nishioka, T. Murase and M. Fujita, Both [2 + 2] and [2 + 4] additions of inert aromatics via identical ternary host–guest complexes, Chem. Commun., 2010, 46, 3460–3462 RSC.
- M. Yoshizawa, S. Miyagi, M. Kawano, K. Ishiguro and M. Fujita, Alkane oxidation via photochemical excitation of a self-assembled molecular cage, J. Am. Chem. Soc., 2004, 126, 9172–9173 CrossRef CAS PubMed.
- M. Yoshizawa, N. Sato and M. Fujita, Selective enclathration of linear alkanols by a self-assembled coordination cage: Application to the catalytic wacker oxidation of ω-alkenols, Chem. Lett., 2005, 34, 1392–1393 CrossRef CAS.
- R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Roesky, Hetero- and metallasiloxanes derived from silanediols, disilanols, silanetriols, and trisilanols, Chem. Rev., 1996, 96, 2205–2236 CrossRef CAS PubMed.
- M. Yoshizawa, T. Kusukawa, M. Fujita and K. Yamaguchi, Ship-in-a-bottle synthesis of otherwise labile cyclic trimers of siloxanes in a self-assembled coordination cage, J. Am. Chem. Soc., 2000, 122, 6311–6312 CrossRef CAS.
- M. Yoshizawa, T. Kusukawa, M. Fujita, S. Sakamoto and K. Yamaguchi, Cavity-directed synthesis of labile silanol oligomers within self-assembled coordination cages, J. Am. Chem. Soc., 2001, 123, 10454–10459 CrossRef CAS PubMed.
- T. Furusawa, M. Kawano and M. Fujita, The confined cavity of a coordination cage suppresses the photocleavage of α-diketones to give cyclization products through kinetically unfavorable pathways, Angew. Chem., Int. Ed., 2007, 46, 5717–5719 CrossRef CAS PubMed.
- T. Murase, Y. Nishijima and M. Fujita, Cage-catalyzed Knoevenagel condensation under neutral conditions in water, J. Am. Chem. Soc., 2012, 134, 162–164 CrossRef CAS PubMed.
- T. Murase, H. Takezawa and M. Fujita, Photo-driven anti-Markovnikov alkyne hydration in self-assembled hollow complexes, Chem. Commun., 2011, 47, 10960–10962 RSC.
- T. Yamaguchi and M. Fujita, Highly selective photomediated 1,4-radical addition to o-quinones controlled by a self-assembled cage, Angew. Chem., Int. Ed., 2008, 47, 2067–2069 CrossRef CAS PubMed.
- T. Murase, Y. Nishijima and M. Fujita, Unusual photoreaction of triquinacene within self-assembled hosts, Chem. – Asian J., 2012, 7, 826–829 CrossRef CAS PubMed.
- H. Takezawa, T. Kanda, H. Nanjo and M. Fujita, Site-Selective functionalization of linear diterpenoids through U-shaped folding in a confined artificial cavity, J. Am. Chem. Soc., 2019, 141, 5112–5115 CrossRef CAS PubMed.
- H. Takezawa, K. Shitozawa and M. Fujita, Enhanced reactivity of twisted amides inside a molecular cage, Nat. Chem., 2020, 12, 574–578 CrossRef CAS PubMed.
- W. Cullen, H. Takezawa and M. Fujita, Demethylenation of cyclopropanes via photoinduced guest-to-host electron transfer in an M6L4 cage, Angew. Chem., 2019, 131, 9269–9271 CrossRef.
- C. A. Bunton and R. H. D. Wolfe, The hydrolysis of carboxylic ortho Eeters, J. Org. Chem., 1965, 30, 1371–1375 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Acid catalysis in basic solution: A supramolecular host promotes orthoformate hydrolysis, Science, 2007, 316, 85 CrossRef CAS PubMed.
- D. Fiedler, R. G. Bergman and K. N. Raymond, Supramolecular catalysis of a unimolecular transformation: Aza-cope rearrangement within a self-assembled host, Angew. Chem., Int. Ed., 2004, 43, 6748–6751 CrossRef CAS PubMed.
- C. J. Hastings, R. G. Bergman and K. N. Raymond, Origins of large rate enhancements in the Nazarov cyclization catalyzed by supramolecular encapsulation, Chem. – Eur. J., 2014, 20, 3966–3973 CrossRef CAS PubMed.
- W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, Selective monoterpene-like cyclization reactions achieved by water exclusion from reactive intermediates in a supramolecular catalyst, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS PubMed.
- A. G. Salles, S. Zarra, R. M. Turner and J. R. Nitschke, A self-organizing chemical assembly line, J. Am. Chem. Soc., 2013, 135, 19143–19146 CrossRef CAS PubMed.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Molecular containers in complex chemical systems, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- P. Das, A. Kumar, P. Howlader and P. S. Mukherjee, A self-assembled trigonal prismatic molecular vessel for catalytic dehydration reactions in water, Chem. – Eur. J., 2017, 23, 12565–12574 CrossRef CAS PubMed.
- M. D. Ward, Polynuclear coordination cages, Chem. Commun., 2009, 4487–4499 RSC.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Highly efficient catalysis of the Kemp elimination in the cavity of a cubic coordination cage, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, Catalysis in a cationic coordination cage using a cavity-bound guest and surface-bound anions: Inhibition, activation, and autocatalysis, J. Am. Chem. Soc., 2018, 140, 2821–2828 CrossRef CAS PubMed.
- B. Roy, A. Devaraj, R. Saha, S. Jharimune, K.-W. Chi and P. S. Mukherjee, Catalytic intramolecular cycloaddition reactions by using a discrete molecular architecture, Chem. – Eur. J., 2017, 23, 15704–15712 CrossRef CAS PubMed.
- M. V. S. N. Maddipatla, L. S. Kaanumalle, A. Natarajan, M. Pattabiraman and V. Ramamurthy, Preorientation of olefins toward a single photodimer: Cucurbituril-mediated photodimerization of protonated azastilbenes in water, Langmuir, 2007, 23, 7545–7554 CrossRef CAS PubMed.
- I. A. Bhat, A. Devaraj, E. Zangrando and P. S. Mukherjee, A discrete self-assembled Pd12 triangular orthobicupola cage and its use for intramolecular cycloaddition, Chem. – Eur. J., 2018, 24, 13938–13946 CrossRef CAS.
- J. Jiao, Z. Li, Z. Qiao, X. Li, Y. Liu, J. Dong, J. Jiang and Y. Cui, Design and self-assembly of hexahedral coordination cages for cascade reactions, Nat. Commun., 2018, 9, 4423 CrossRef PubMed.
- S.-C. Li, L.-X. Cai, L.-P. Zhou, F. Guo and Q.-F. Sun, Supramolecular synthesis of coumarin derivatives catalyzed by a coordination-assembled cage in aqueous solution, Sci. China: Chem., 2019, 62, 713–718 CrossRef CAS.
- F. J. Rizzuto, W. J. Ramsay and J. R. Nitschke, Otherwise unstable structures self-assemble in the cavities of cuboctahedral coordination cages, J. Am. Chem. Soc., 2018, 140, 11502–11509 CrossRef CAS PubMed.
- L. Feng, K.-Y. Wang, G. S. Day and H.-C. Zhou, The chemistry of multi-component and hierarchical framework compounds, Chem. Soc. Rev., 2019, 48, 4823–4853 RSC.
| This journal is © the Partner Organisations 2020 |