DOI:
10.1039/D0QO00563K
(Review Article)
Org. Chem. Front., 2020,
7, 2873-2898
Recent advances in nitro-involved radical reactions
Received
10th May 2020
, Accepted 17th July 2020
First published on 17th July 2020
Abstract
Nitro-containing compounds are important structural moieties in drugs, natural products, and small molecule therapeutics, and are widely used in a variety of organic transformations. During the past decades, these compounds have received considerable attention from the synthetic chemistry community, since they have been successfully applied in the preparation of amines, oximes, alkenes, nitrones and alcohols through the radical-initiated pathway. Additionally, significant progress in the chemistry of nitro radicals has been witnessed, providing efficient and rapid access to nitro-containing compounds as well as isoxazoline derivatives. Recent advances in the radical reactions involving the nitro group and various transformations have been summarized in this review.
1. Introduction
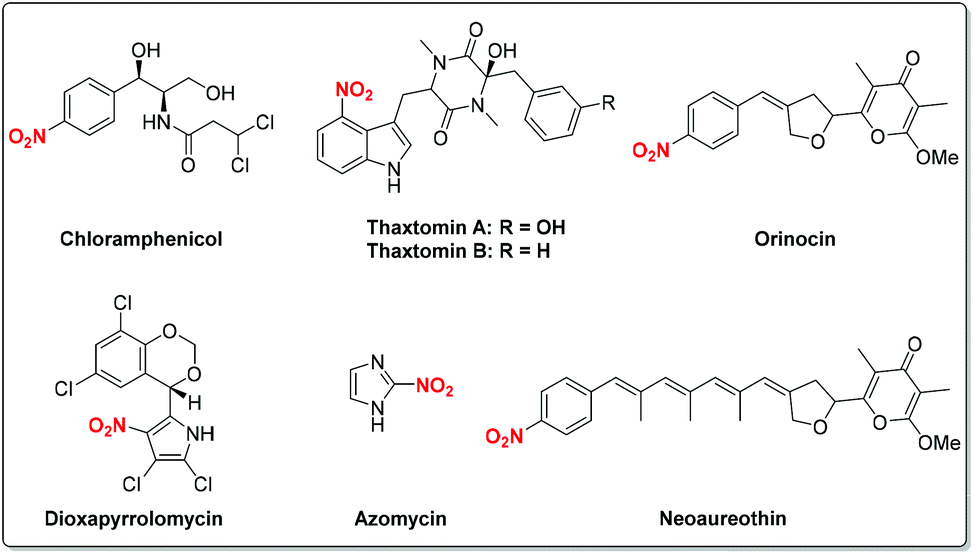
Nitro-containing compounds are important constituents of plastics, explosives, dyes, perfumes, pharmaceuticals and industrial chemicals (Fig. 1).1 Moreover, nitro compounds have been extensively explored as valuable synthetic intermediates in various organic transformations.2 For instance, the radical-initiated reductive coupling of nitro compounds with alkenes provides a concise way to produce amines under transition metal catalysis. On the other hand, for the traditional nitration process, a nitronium ion (NO2+) is generated from mixed strong acidic systems (H2SO4/HNO3) which then undergoes an electrophilic substitution to deliver the nitrated products. This process often suffers from isomeric mixtures and poor functional group tolerance.3 Therefore, the development of efficient and novel methods for the synthesis of nitro compounds has attracted great attention. To this end, a variety of nitrating reagents including AgNO2, NaNO2, Fe(NO3)3·9H2O, tBuONO, etc. have been well explored as sources of nitro radicals for C–N bond-forming reactions. As a result, with the rapid development of free radical chemistry, continuous efforts have been devoted to the radical reactions involving the nitro group. Despite tremendous achievements in this area, to the best of our knowledge, there is still no comprehensive review devoted to this impressive topic.
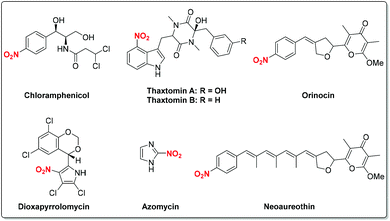 |
| | Fig. 1 Selected examples of nitro-containing compounds in natural products and bioactive molecules. | |
The main focus of this review is on radical reactions involving the nitro group for various transformations. Accordingly, the chemistry of the nitro radical generated from different nitrating reagents is discussed in detail, thus giving rise to nitro-containing compounds as well as isoxazoline derivatives. Additionally, we will summarize recent progress on the radical-initiated conversion of nitro compounds into amines, oximes, alkenes, nitrones and alcohols under appropriate conditions.
2. Transformations involving nitro radicals
The nitro group is a valuable building block in the preparation of pharmaceuticals, materials and dyes. Traditional methods for the synthesis of nitro compounds involving nitronium cation (NO2+) or nitrite anion (NO2−) intermediates often suffer from isomeric mixtures and poor functional group tolerance. Over the past few years, considerable efforts have been devoted to the chemistry of the nitrogen dioxide radical (˙NO2), providing an efficient and powerful strategy to access nitro compounds.
2.1. Fe-Catalyzed or mediated transformations
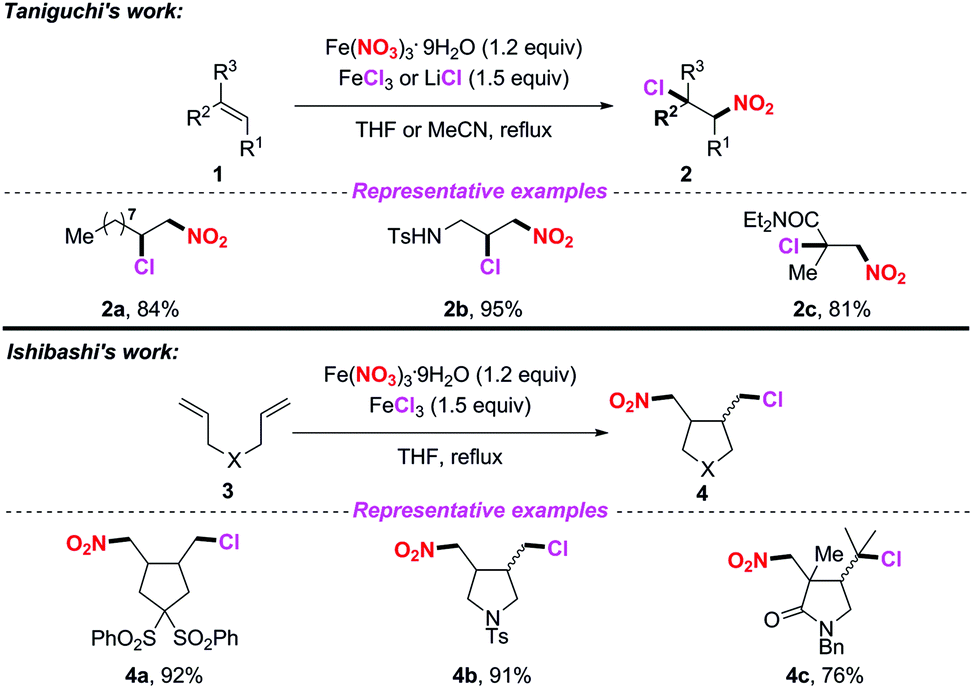
Iron(III) nitrate nonahydrate has been frequently used as a NO2 radical source in radical-mediated nitration reactions. In 2010, the groups of Taniguchi4 and Ishibashi5 independently developed the iron-mediated halo-nitration reaction of alkenes to obtain nitro compounds (Scheme 1). Accordingly, both the reactions, involving the addition of the NO2 radical to alkenes and the resulting terminal radicals, were trapped by a chlorine atom in the presence of a chloride salt. Both methods featured a broad substrate scope, providing a practical way to access α-chloronitroalkanes or chloro-cyclonitroalkanes in good yields. The use of iron nitrate as the nitro source in these processes further highlights its applicability, which avoids the inconvenience of using nitrogen dioxide.
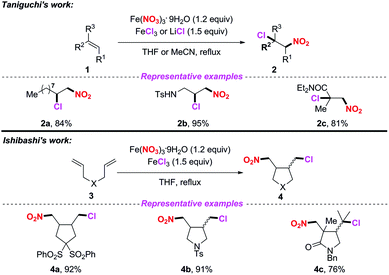 |
| | Scheme 1 Iron-mediated radical halo-nitration of alkenes and 1,6-dienes. | |
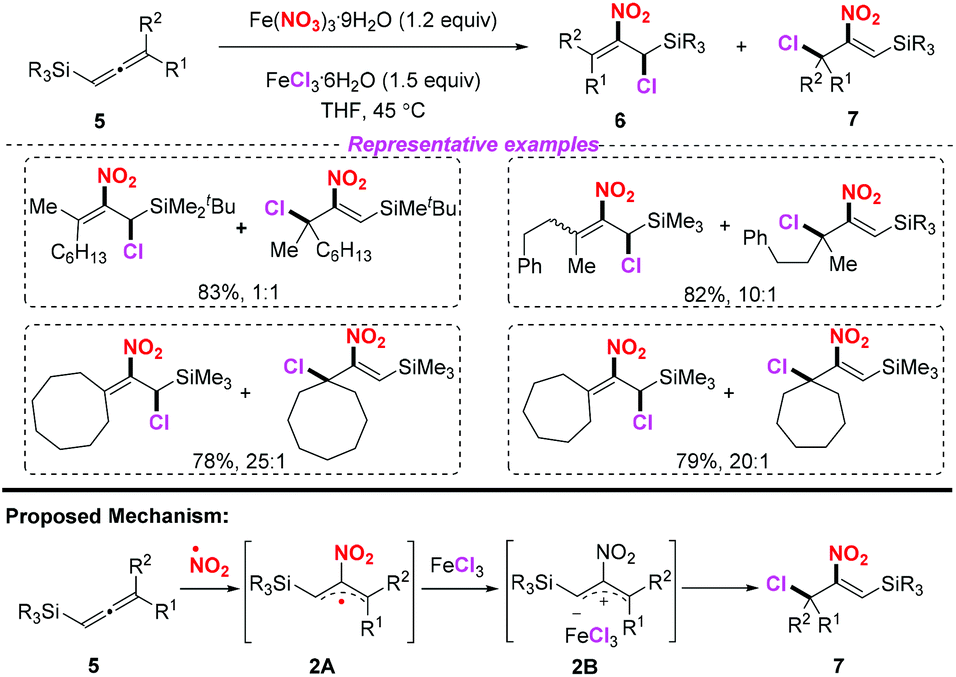
In 2013, Lee and co-workers realized an efficient nitration of silyl allenes with nitrogen dioxide radical (Scheme 2).6 Accordingly, Fe(NO3)3·9H2O was employed as the nitro radical source, providing rapid access to the formation of halo-nitroalkenes. This method exhibited a broad substrate scope, giving rise to various halo-nitroalkenes in good yields. However, in some cases, this method suffered from poor regioselectivity, which limited its further application.
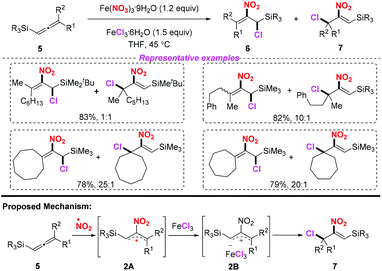 |
| | Scheme 2 Iron-mediated radical halo-nitration of silyl allenes to form halo-nitroalkenes. | |
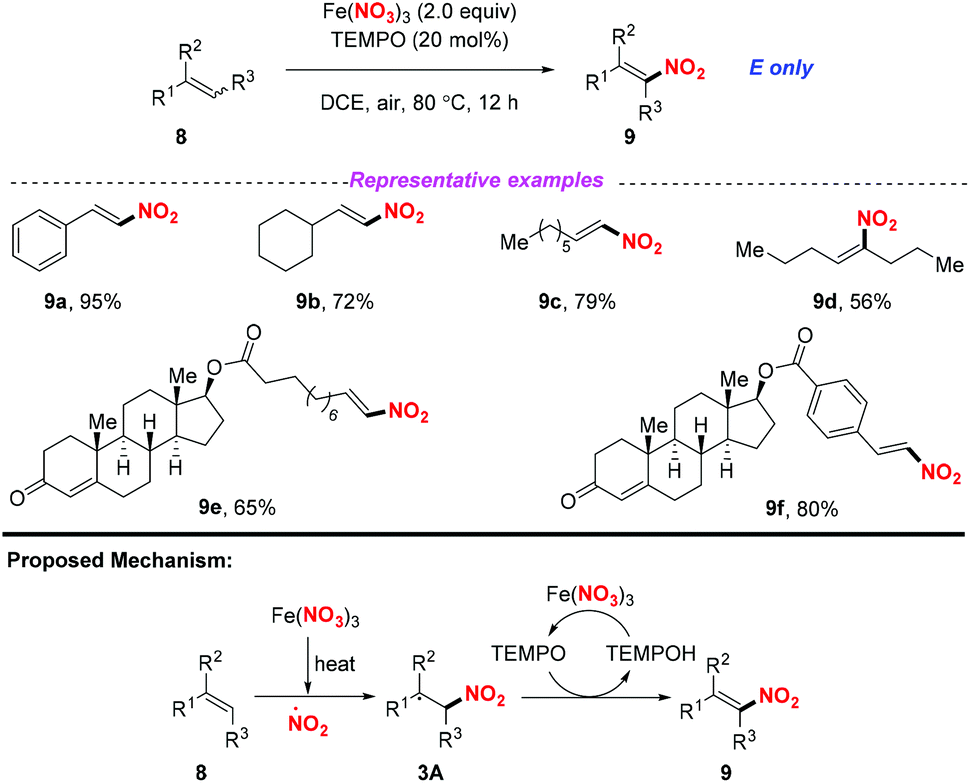
At the same time, Maiti and co-workers reported a selective nitration of olefin with Fe(NO3)3 and TEMPO (Scheme 3).7 As for the possible mechanism, the generation of the nitro radical from Fe(NO3)3 would occur under thermal conditions. Then, the addition of the nitro radical to olefin would result in a secondary or benzylic radical, followed by the reception of a hydrogen radical with TEMPO to obtain nitroalkene compounds 9. By employing this method, a variety of aromatic, aliphatic, and heteroaromatic olefins were applicable for this transformation, providing the corresponding products in good yields with excellent E-selectivity. This method exhibited a broad substrate scope, which could be further applied to the late-stage functionalization of drug-like molecules.
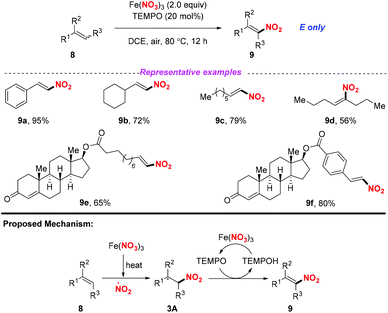 |
| | Scheme 3 Iron-mediated selective nitration of olefins. | |
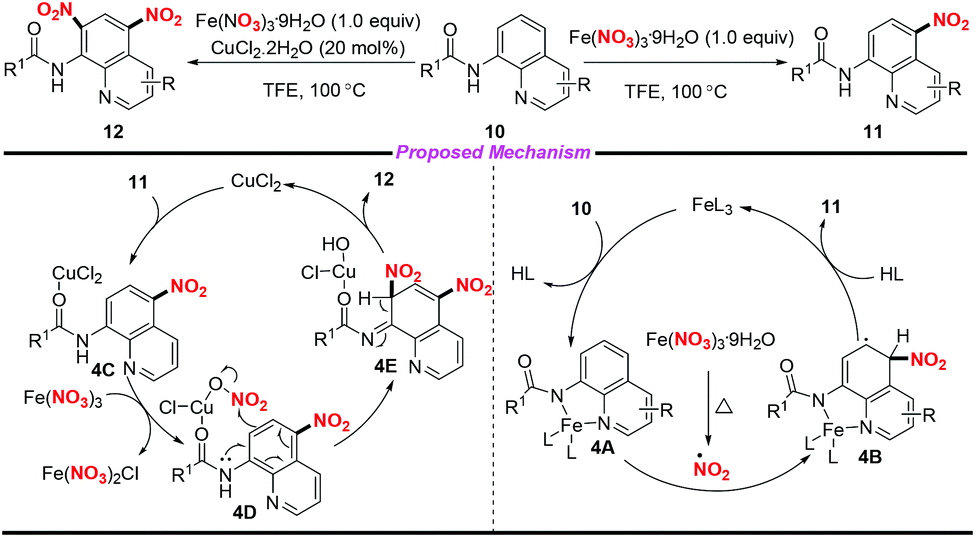
In 2016, Fan and co-workers demonstrated an efficient and regioselective remote C(5)–H nitration of 8-aminoquinoline amides by using Fe(NO3)3·9H2O as a promoter and nitro source (Scheme 4).8 Interestingly, the reaction could undergo bisnitration to afford 5,7-dinitro-8-aminoquinoline amides when CuCl2·2H2O was used as a catalyst. On the basis of several controlled experiments, two possible mechanisms were presented by the authors to clarify the mono- and bisnitration pathways. In the mononitration process, the coordination of 10 with Fe(III) led to a chelated intermediate 4A, followed by the addition of a NO2 radical at the C-5 position of the quinoline unit to form intermediate 4B. The mononitrated product 11 was obtained via aromatization and decomposition of 4B. Moreover, intermediate 4D could be generated from 11 in the presence of CuCl2·2H2O and Fe(NO3)3·9H2O, which would undergo a possible electrophilic substitution and aromatization sequence to afford the bisnitrated product 12.
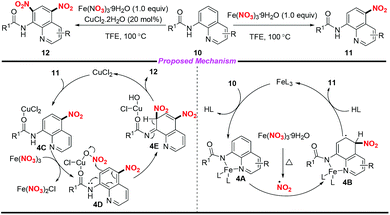 |
| | Scheme 4 Iron-promoted mono- and bisnitration of 8-aminoquinolinamides. | |
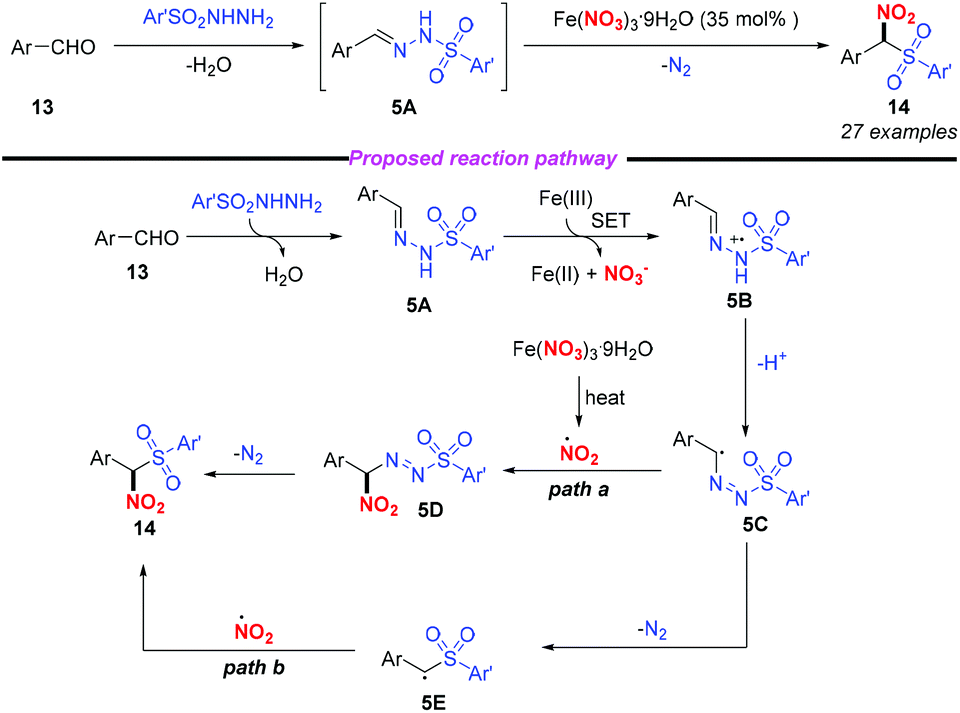
In 2015, Punniyamurthy and co-workers developed an iron(III)-mediated radical–radical coupling strategy for the generation of biarylsulfonylnitromethane to further transformations (Scheme 5).9 This protocol featured mild reaction conditions and a broad substrate scope. Based on the mechanistic studies, the authors proposed that the condensation of aldehyde 13 combined with arylsulfonyl hydrazide could give sulfonyl hydrazine 5A, which would undergo a single electron transfer by Fe(NO3)3 to form intermediate 5B. The subsequent isomerization of 5B gave the radical intermediate 5C that could couple with the nitro radical to give 5D and the target product 14 (path a). Alternatively, the formation of 14 could occur through the radical coupling of the nitro radical and the radical intermediate 5E (path b). In this reaction, the simple and commercially available aldehyde and arylsulfonyl hydrazide were used as the starting materials, and water and nitrogen were produced as the only by-products, showing promising potential for further applications.
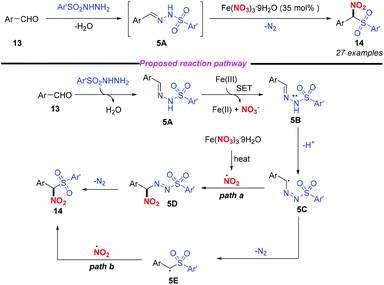 |
| | Scheme 5 Fe(III)-Mediated radical nitration of bisarylsulfonyl hydrazones for the synthesis of biarylsulfonylnitromethane. | |
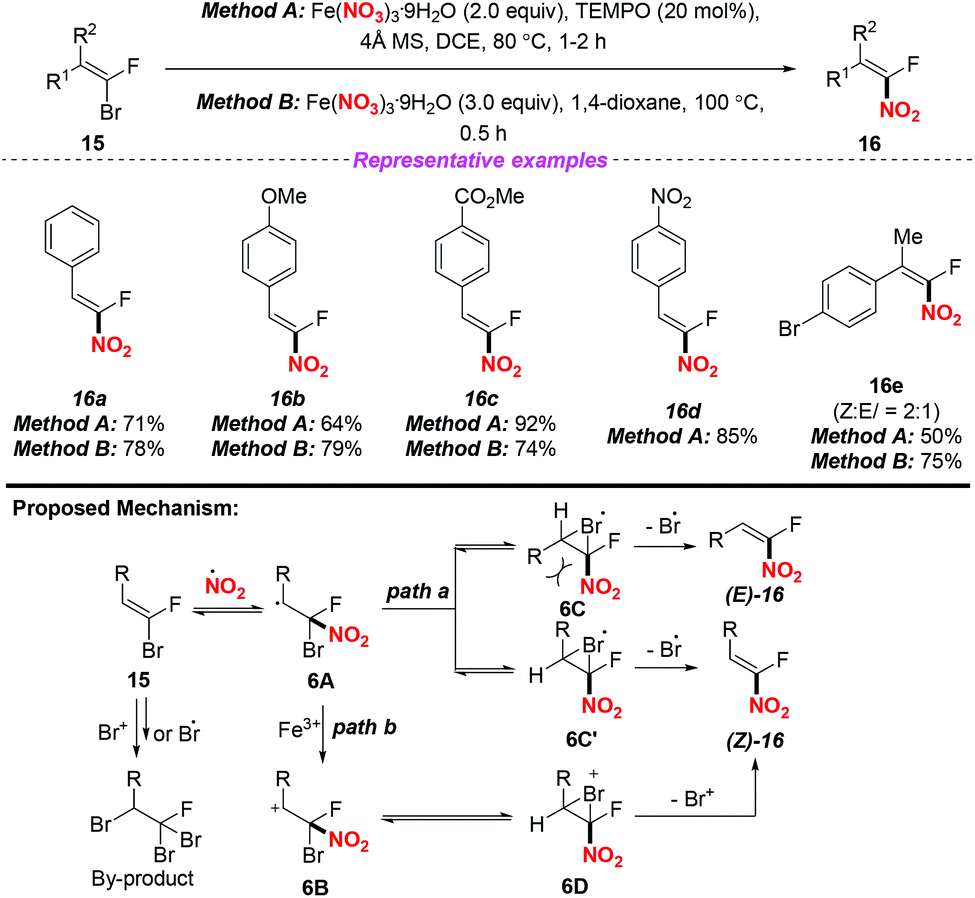
In 2017, Nenajdenko and co-workers described an efficient method for the stereoselective synthesis of β-fluoro-β-nitrostyrenes via a radical nitration–debromination sequence (Scheme 6).10 The reaction proceeded readily either by employing method A with the use of the Fe(NO3)3/TEMPO system at 80 °C or by applying method B with Fe(NO3)3 in 1,4-dioxane at 100 °C, giving the corresponding α-fluoro-nitroalkenes in good yields. A possible mechanism for clarifying the stereoselectivity of the product was proposed, as shown in Scheme 6. The stereoselective formation of 1-fluoro-1-nitroolefin 15 could be explained by the formation of intermediate 6A, which would exist preferentially in the form of closed 3-membered bromo radicals 6C or 6C′. Owing to the steric repulsion between its large R-group and NO2 moiety, 6C should be less favoured than 6C′. Accordingly, the final products (Z)-16 could be generated by the elimination of bromine from the conversion of the more stable cyclic intermediate 6C′ (path a). Besides, the elimination of Br+ from cation 6B and its cyclic form 6D for accessing (Z)-16 was also considered (path b).
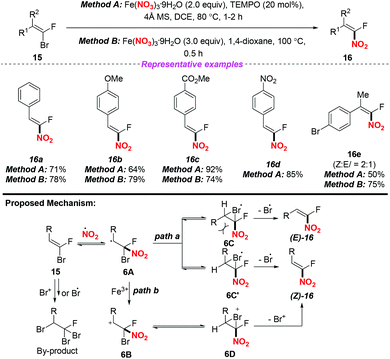 |
| | Scheme 6 Fe(III)-Mediated stereoselective synthesis of β-fluoro-β-nitrostyrenes via radical nitration debromination. | |
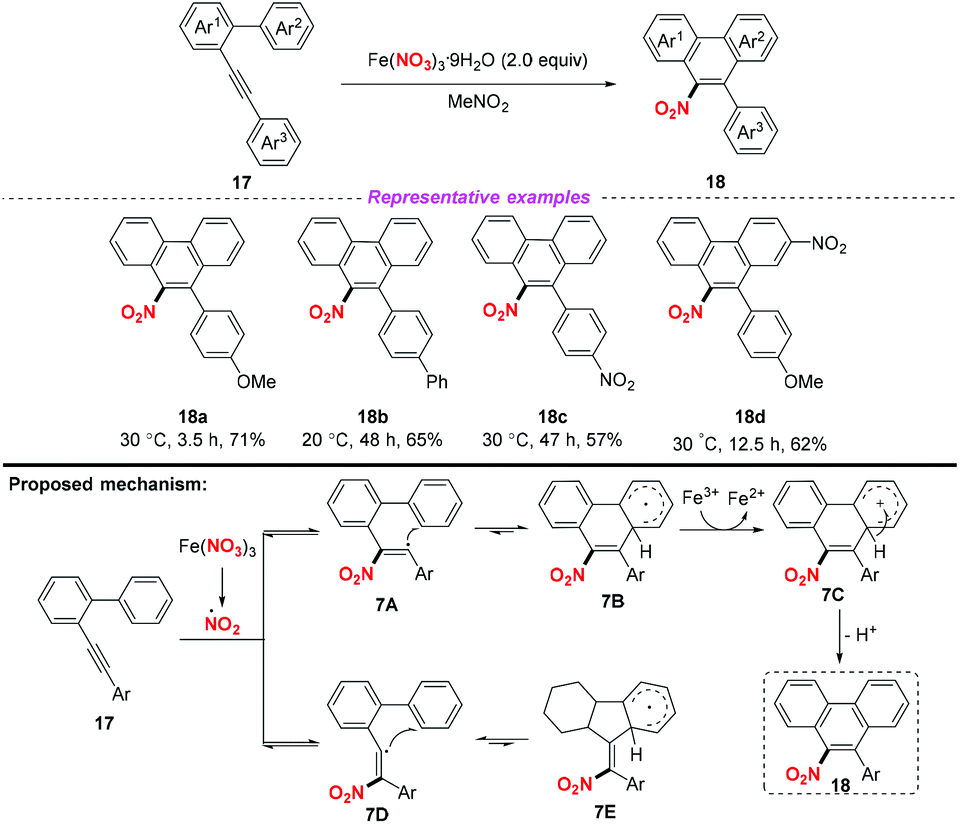
Subsequently, Guo and co-workers demonstrated an iron-mediated nitration and cyclization of arene–alkynes, providing a practical protocol for the synthesis of 9-nitrophenathrenes (Scheme 7).11 This methodology showed a broad substrate scope and good functional group tolerance, affording the corresponding products in good yields. According to the proposed mechanism, the reaction was initiated by the addition of the nitro radical to the alkyne moiety. Notably, Fe(NO3)3·9H2O was used as both a nitro source and an oxidant in this transformation.
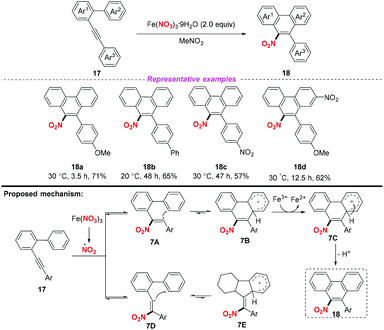 |
| | Scheme 7 Iron-mediated nitration and cyclization of arene–alkynes. | |
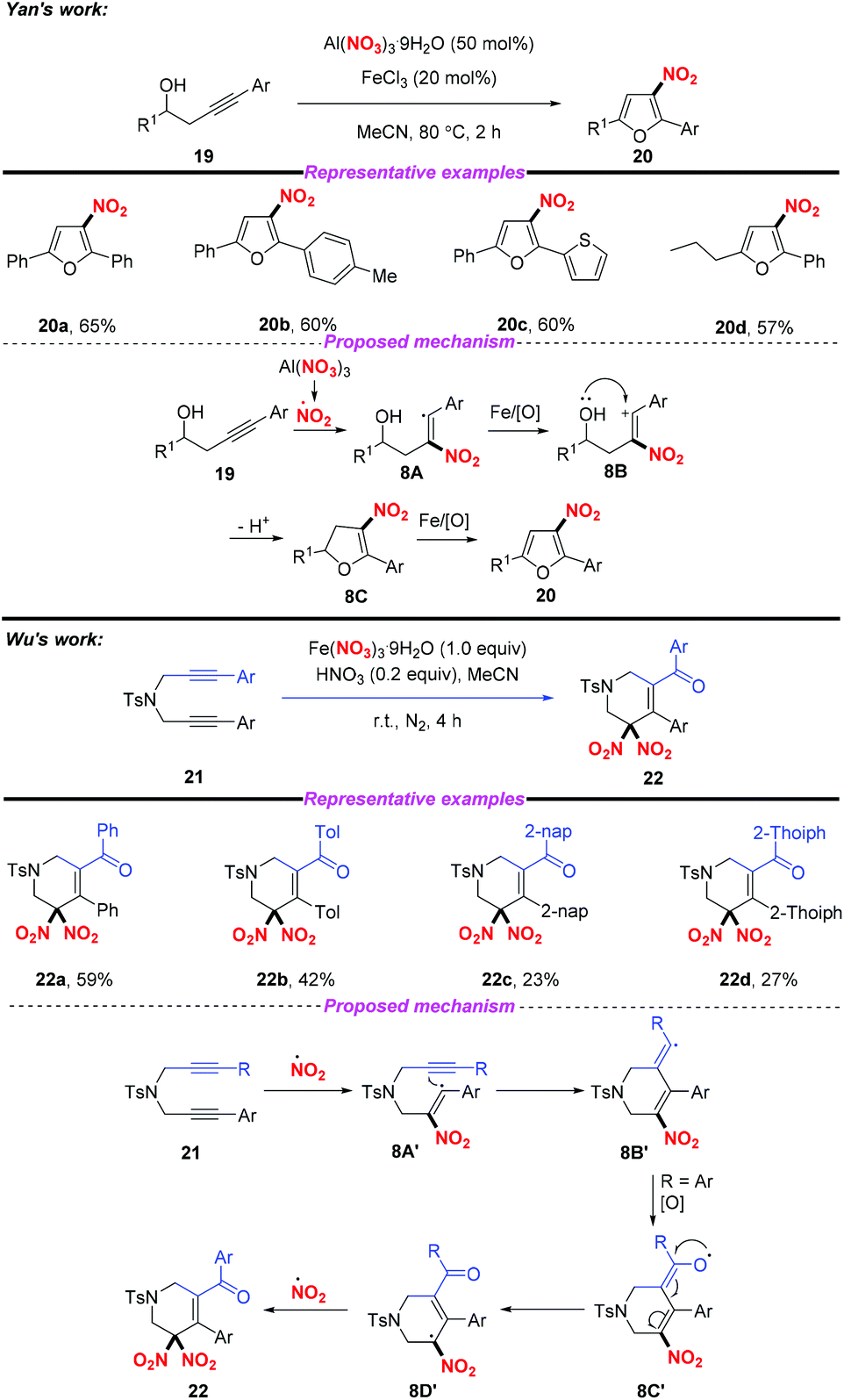
Similarly, the groups of Yan12 and Wu13 independently described the Fe-catalyzed or Fe-mediated tandem radical nitration/cyclization of alkynes to afford heterocyclic compounds containing a nitro group (Scheme 8). In Yan's work, they employed Al(NO3)3·9H2O as a nitro radical source and the homopropargylic alcohols as receptors for the synthesis of 3-nitrofurans. Moreover, Wu and co-workers found that cyclic gem-dinitro compounds could be obtained through the nitration of 1,6-diynes using Fe(NO3)3·9H2O as the nitro radical source. Notably, the electrophilic cyclization of alkynes could proceed in both reactions without the use of transition metals, leading to the corresponding nitro-containing heterocyclic compounds albeit with low yields.
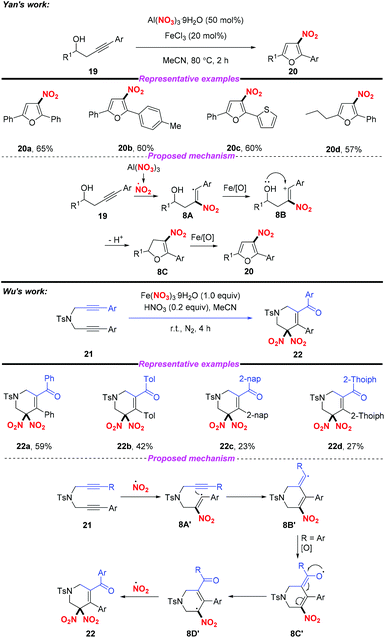 |
| | Scheme 8 Iron-catalyzed/iron-mediated tandem cyclization via radical nitration. | |
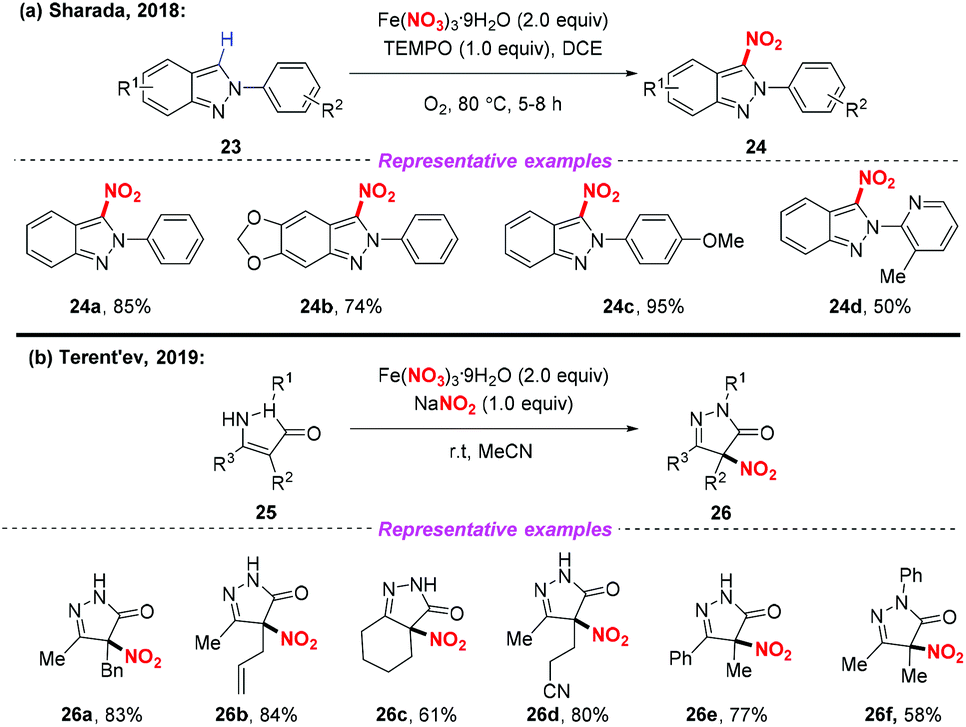
Recently, the radical nitration of heteroarenes was achieved by Sharada and co-workers, using Fe(NO3)3·9H2O as the nitro radical source (Scheme 9a).14 This strategy opened the door to the introduction of the nitro group into heteroarenes, providing the corresponding 3-nitro-2H-indazoles in good yields. Later, Terent'ev and co-workers also disclosed a mild nitration of pyrazolin-5-ones for the synthesis of 4-nitropyrazolin-5-ones by employing the Fe(NO3)3/NaNO2 system. It was demonstrated that the reaction was compatible with functional groups such as N-phenyl and allyl groups, which are usually sensitive under nitration conditions (Scheme 9b).15
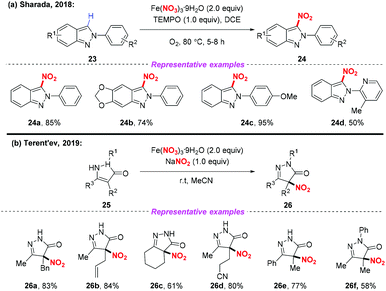 |
| | Scheme 9 Iron(III)-mediated radical nitration of heterocyclic compounds. | |
2.2. Cu-Catalyzed transformations
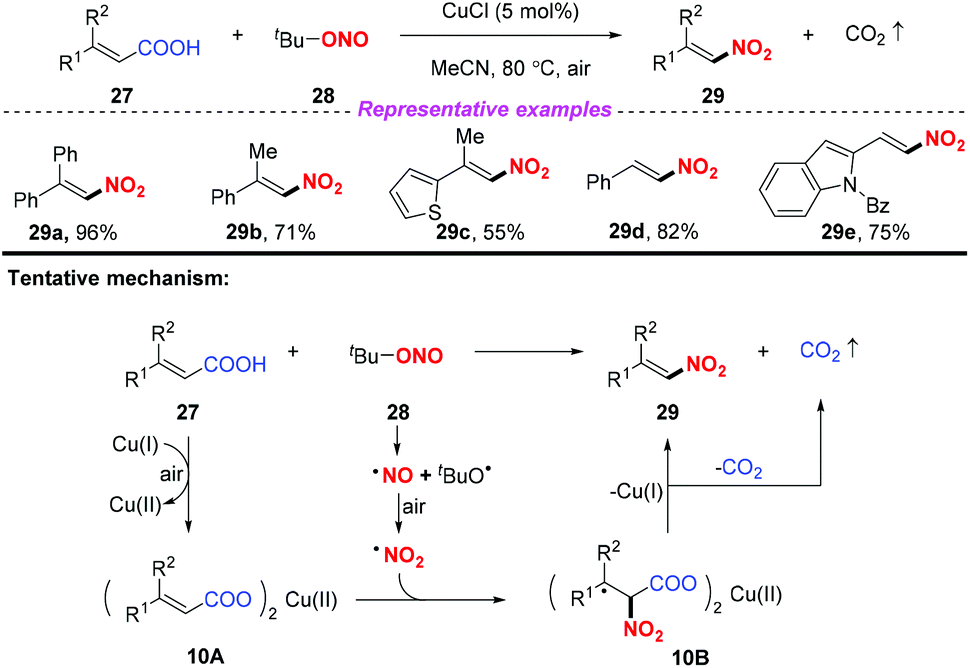
Due to its advantages of low cost, rich abundance in nature, and low toxicity, copper has received considerable attention in the development of various transformations involving nitro radicals. In 2013, Prabhu and co-workers developed a copper-catalyzed nitrodecarboxylation of unsaturated carboxylic acids for the synthesis of substituted nitroolefins (Scheme 10).16 The protocol exhibited a broad substrate scope and good functional group tolerance and the corresponding products were obtained in moderate to good yields. Accordingly, the combination of α,β-unsaturated acid 27 with CuCl would provide the corresponding Cu(II) salt 10A, which on further reaction with the nitro radical formed intermediate 10B. This intermediate 10B would undergo decarboxylation to deliver the corresponding nitroolefin 29.
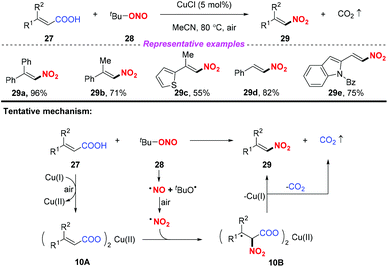 |
| | Scheme 10 Copper-catalyzed nitrodecarboxylation of unsaturated carboxylic acids for the synthesis nitroolefins. | |
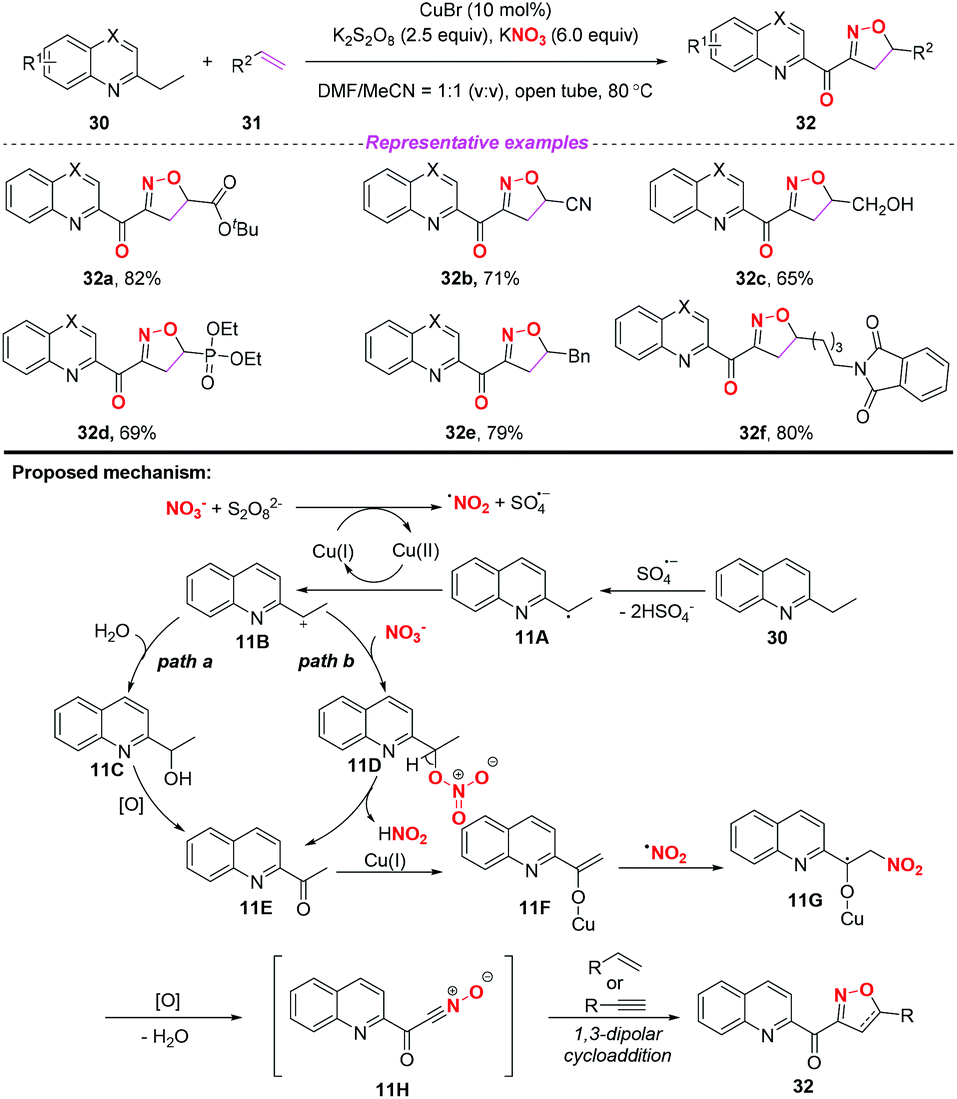
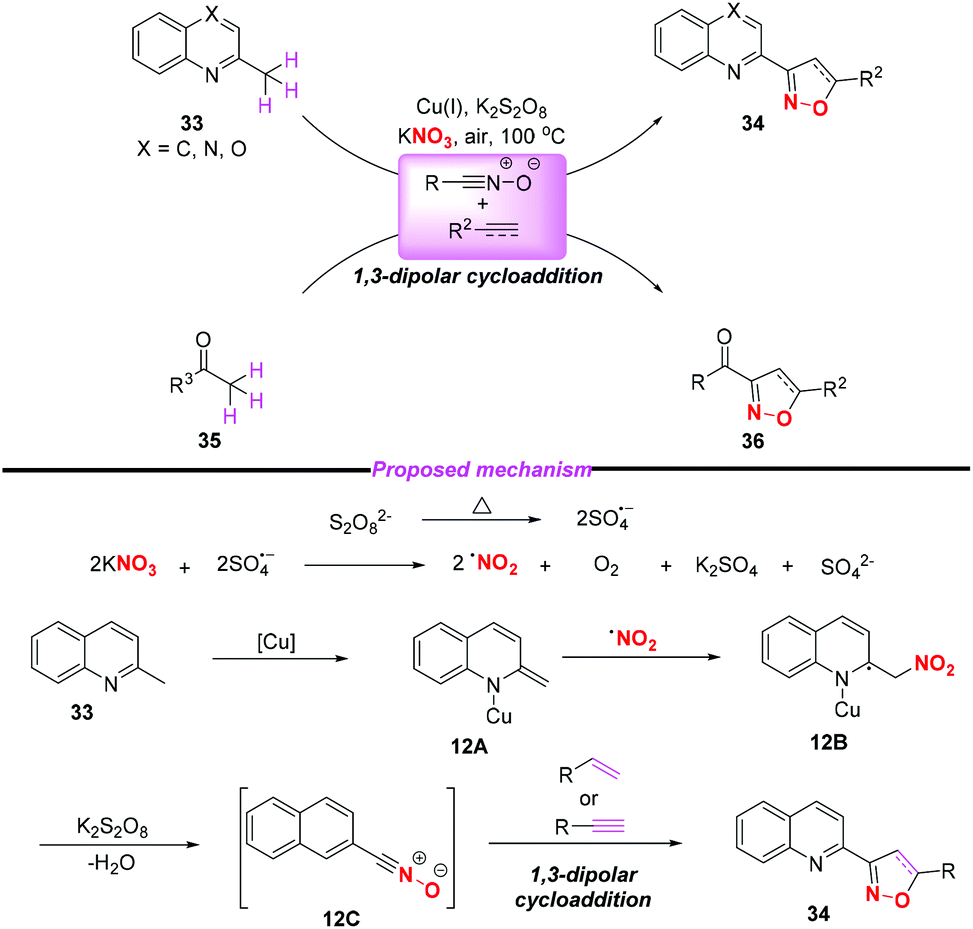
In 2015, the group of Yang developed a copper-catalyzed selective cascade sp3 C–H bond oxidative functionalization of 2-ethylazaarenes toward the synthesis of isoxazoline derivatives (Scheme 11).17 The scope of the copper catalyzed sp3 C–H bond oxidative functionalization was investigated and the corresponding isoxazolines were obtained in moderate to good yields. According to the proposed mechanism, the nitro radical, sulfate anion radical and Cu(II) were initially generated through the reaction of KNO3 with K2S2O8 in the presence of the Cu(I) catalyst. Then, 2-ethylquinoline 30 would undergo a single electron transfer process by a sulfate anion radical to give intermediate 11A,18 which was oxidized by Cu(II) to produce the benzylic cation 11B. Subsequently, the key intermediate 11E might be generated from 11Bvia path a or path b, followed by the conversion to active metal enol species 11F. After the addition of the nitro radical to the reactive species 11F, the oxidation and dehydration sequence occurred to give nitrile oxides 11H. The formation of isoxazolines 32 would occur through intramolecular 1,3-dipolar cycloaddition of nitrile oxides 11H with alkenes or alkynes. The potential application of isoxazolines further increased the practicality of the present method, with the combination of simple substrates and cheap catalytic systems.
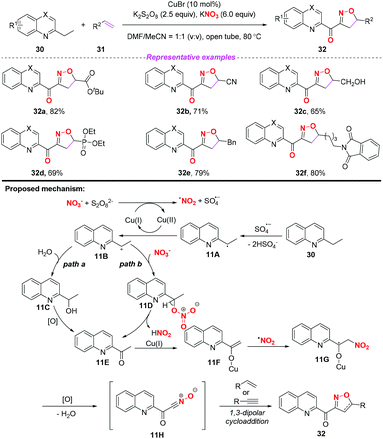 |
| | Scheme 11 Copper-catalyzed selective cascade sp3 C–H bond oxidative functionalization of 2-ethylazaarenes. | |
Inspired by the above achievement, the same group expanded this strategy to the construction of isoxazoline derivatives using alkylazaarenes and substituted ethanones as the starting materials (Scheme 12).19 Similarly, the mechanistic studies revealed that a nitro radical single electron transfer process might be involved as well. As important and versatile synthons, alkynes or alkenes were commercially available in large quantities, greatly increasing the scope and application of the current method.
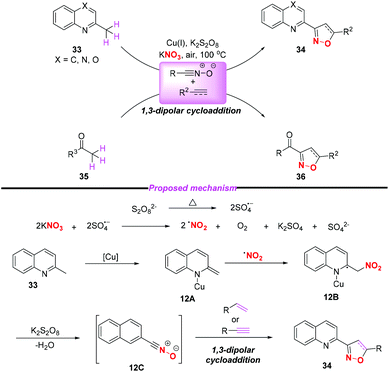 |
| | Scheme 12 Copper-catalyzed sp3 C–H bond oxidative functionalization of alkylazaarenes. | |
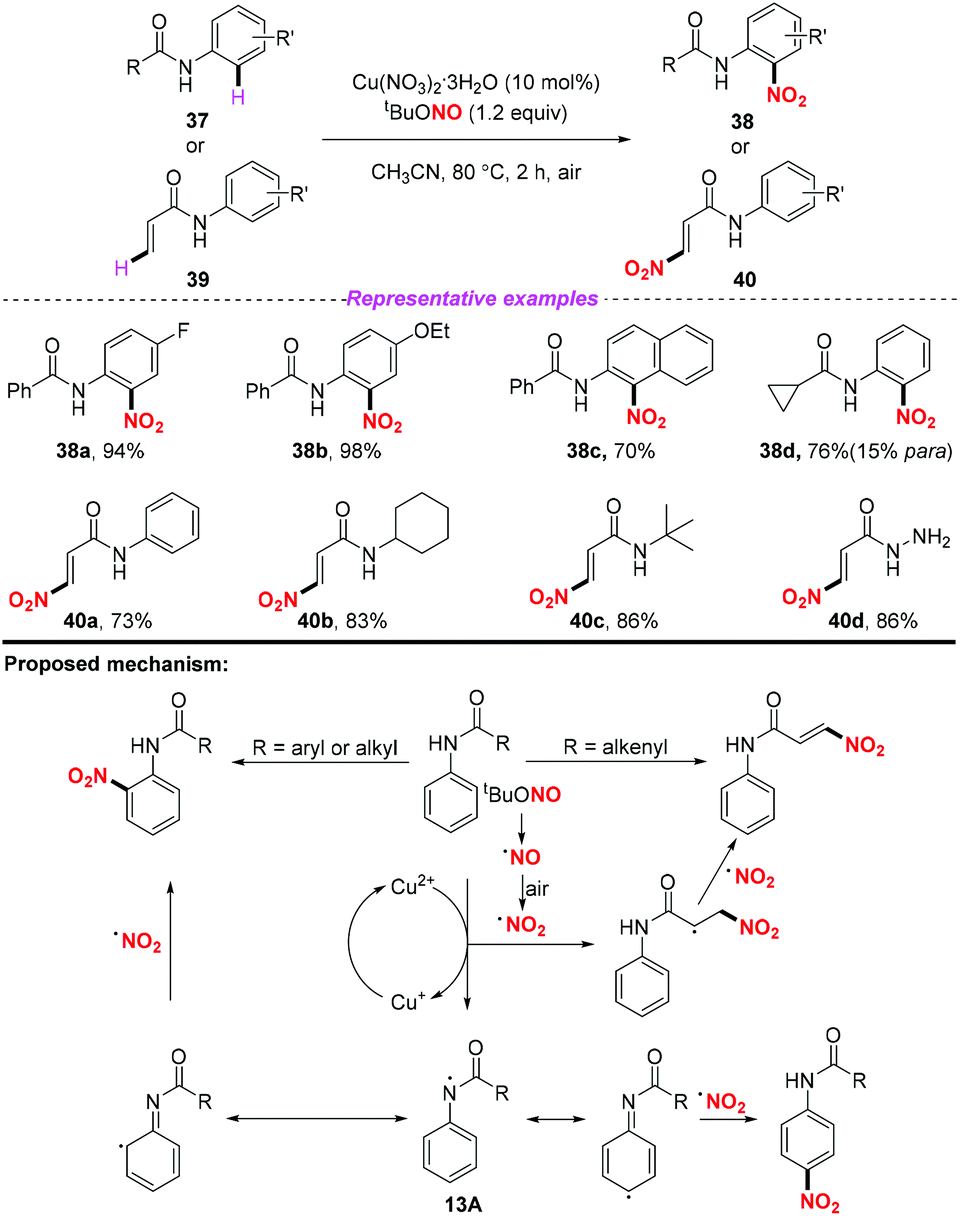
At the same time, Jiang and co-workers described a copper promoted nitration of anilides and acrylamides by using tert-butyl nitrite as a metal-free nitrating reagent (Scheme 13).20 According to the putative mechanism, firstly, tBuONO underwent a thermal homolysis and oxidation sequence to form NO2˙. In the arene nitration process, a single electron oxidation of the anilide by the copper(II) salt resulted in the formation of intermediate 13A. Then, 13A underwent a reversible conversion followed by the coupling of the nitro radical at the ortho- or para-positions of the aromatic ring to afford the corresponding nitration products 38. As for the olefin nitration process, the addition of the nitro radical to the alkenyl moieties followed by an anti-elimination in the presence of another nitro radical would give rise to the nitro substituted olefin stereoselectively. This reaction could not only realize the activation of the aryl C–H bond, but also demonstrated that the alkenyl C–H bond could be subjected to the nitration process. However, when the anilides had no substituent at the para position, the nitro-containing product was obtained with poor regioselectivity.
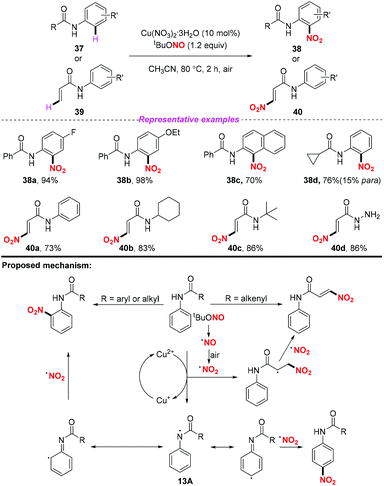 |
| | Scheme 13 Copper-promoted nitration of anilides and acrylamides. | |
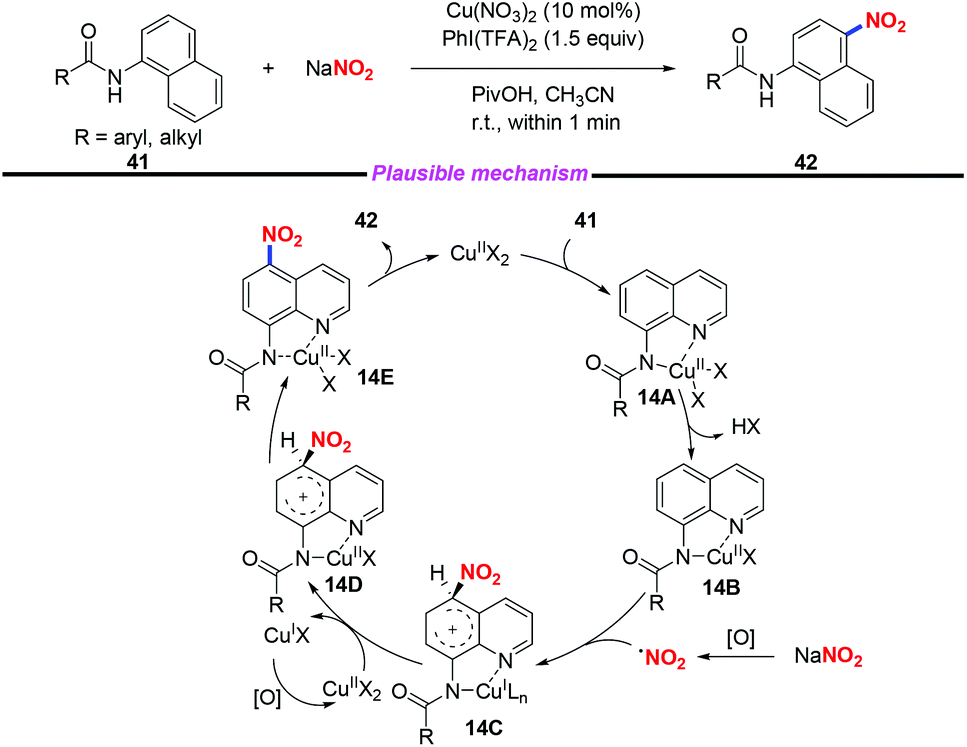
Additionally, a rapid and efficient method for copper-catalyzed highly regioselective C–H nitration of quinolines was achieved by Zhang and co-workers (Scheme 14).21 In this transformation, by employing sodium nitrite as the nitro source, a variety of nitrated quinoline derivatives were obtained in moderate to good yields. Based on the experimental results and their previous reports, a plausible mechanism was proposed for this transformation. Initially, the chelated complex 14A was formed through the coordination of substrate 41 with Cu(II), followed by the deprotonation of the amide group to give complex 14B. The nitro radical was generated in the presence of PhI(TFA)2 and NaNO2. Then, a single electron transfer would occur between the nitro radical and complex 14B to produce the Cu(I) intermediate 14C, followed by oxidation, proton transfer and ligand dissociation sequence to release the desired product and regenerate the Cu(II) species.
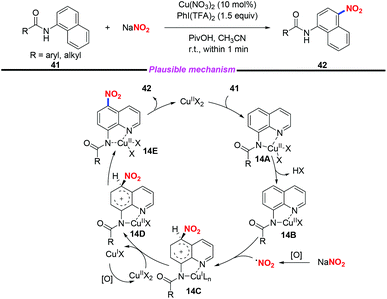 |
| | Scheme 14 Copper-catalyzed direct C–H nitration of quinolines. | |
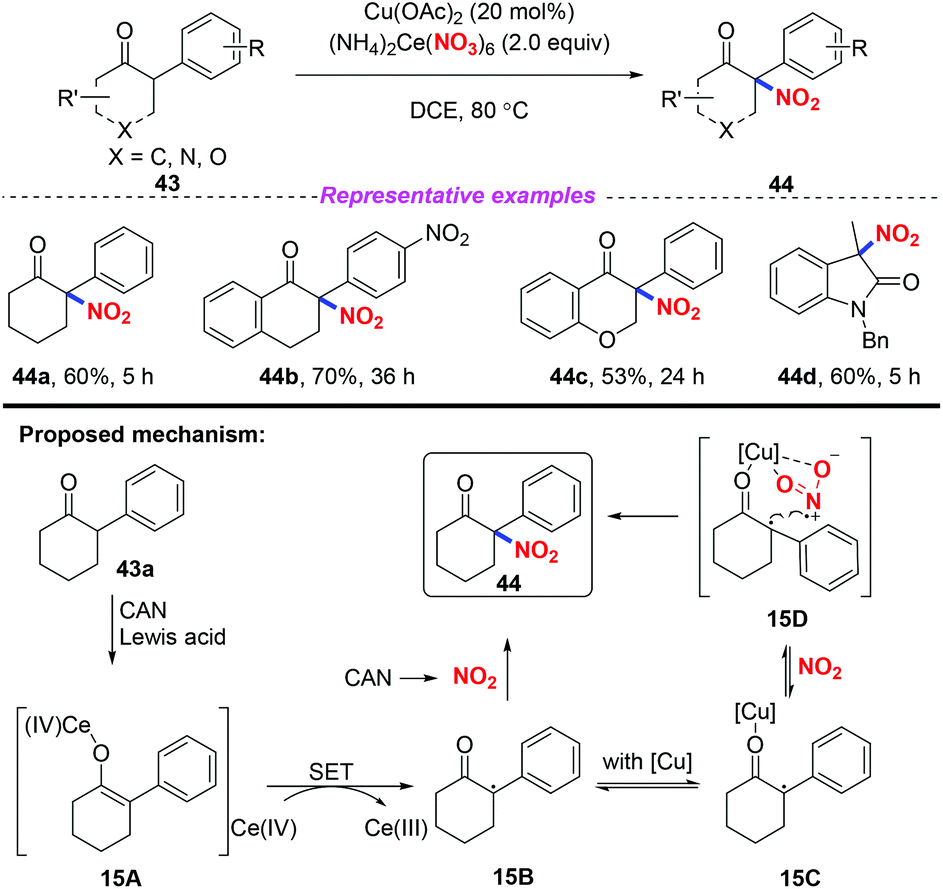
In 2017, Zhang and co-workers developed an efficient and direct copper-assisted nitrating approach for the synthesis of tertiary α-nitro-α-substituted scaffolds by using ceric ammonium nitrate as a nitrating reagent, oxidant, and Lewis acid, using copper salt as a catalyst (Scheme 15).22 Additionally, this method was successfully applied to the efficient synthesis of commonly used clinical drug ketamine in four steps. Based on the experimental results, a possible mechanism was proposed, as shown in Scheme 15. Initially, CAN served as a Lewis acid to promote the formation of the enolized intermediate 15A, which was oxidized by CAN to produce radical species 15B. Then, radical 15B was either trapped by nitrogen dioxide derived from CAN without Cu species or reacted through intermediate 15D stabilized by Cu species to afford product 44.
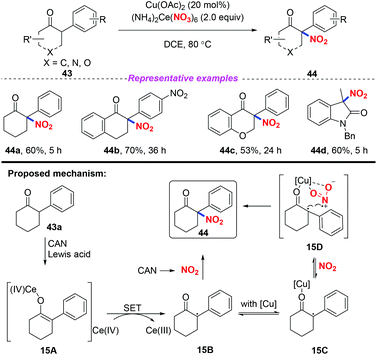 |
| | Scheme 15 Copper-assisted nitration of cyclic ketones with ceric ammonium nitrate. | |
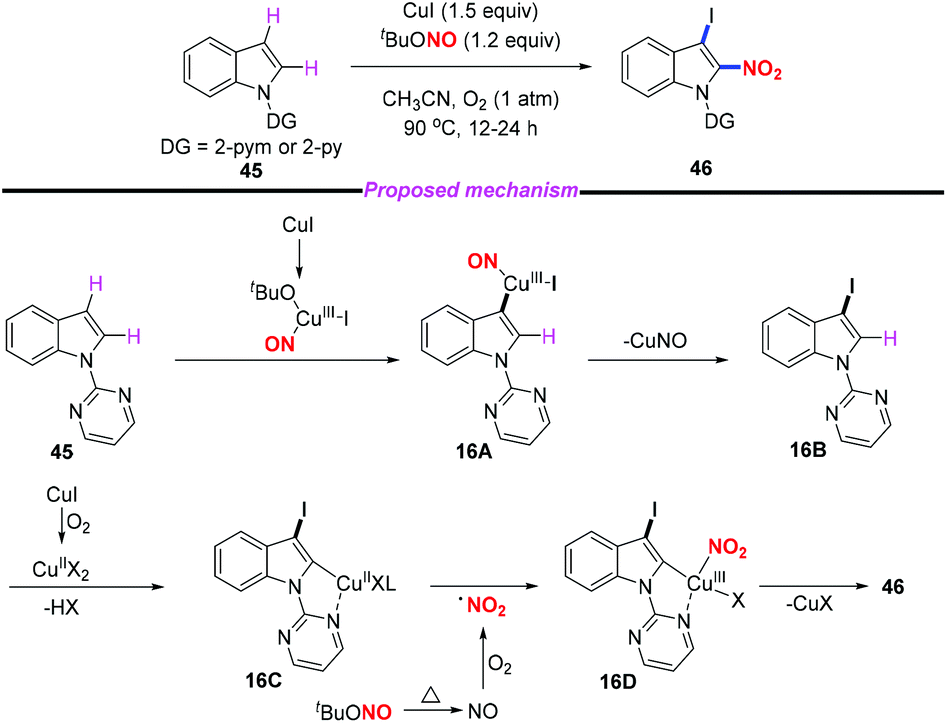
Regioselective nitration in the indole skeleton molecule remains a challenge and the development of efficient and green nitration methods under mild reaction conditions is highly desirable. In 2018, Jiang and co-workers reported an efficient copper-mediated aerobic oxidative C–H iodination and nitration of indoles through double C–H functionalization (Scheme 16).23 This method provided rapid access to 3-iodo-2-nitroindoles in good yields with high regioselectivity under mild conditions. Moreover, the utility of this method was further demonstrated through the derivatization of the iodination and nitration products. According to the proposed mechanism, the iodination step was suggested to proceed through the electrophilic addition of a Cu(III)-iodide species to the C3 position of the indole motif. The subsequent nitration step involved C–H activation, nitro radical oxidative addition, and reductive elimination sequence. In this reaction, cuprous iodide acted both as a catalyst and an iodine source. The resulting 2-nitro-3-iodoindoles could undergo Heck, Suzuki, Negishi, Sonogashira and other coupling reactions to obtain 2-nitro-3-substituted indole compounds, which have potential biological activity.
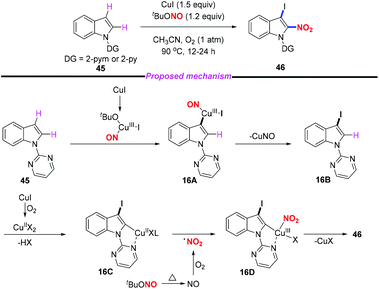 |
| | Scheme 16 Copper-mediated C–H iodination and nitration of indoles. | |
2.3. Ag-Catalyzed or mediated transformations
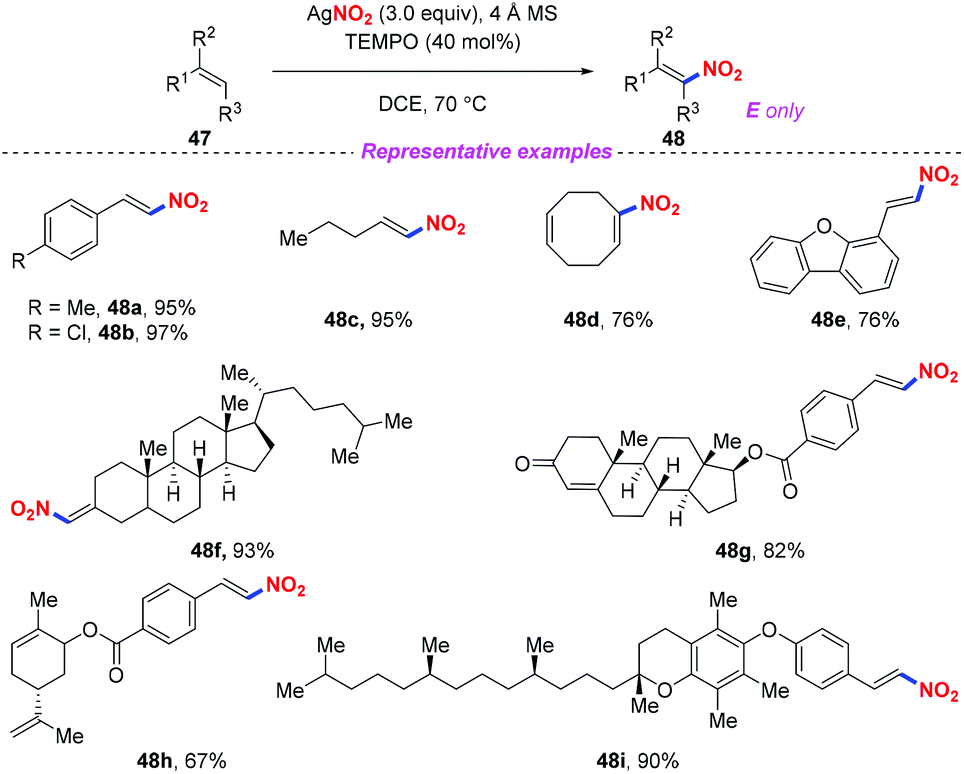
Nitroolefins are valuable building blocks, and have been widely applied in various carbon–carbon bond-forming transformations. Thus, the incorporation of a nitro group into the olefins has become an efficient and straightforward method to furnish nitroolefins. In 2013, Maiti and co-workers reported an efficient and stereoselective nitration of mono- and disubstituted olefins with silver nitrite and TEMPO under ambient conditions (Scheme 17).24 This protocol featured a broad substrate scope and high stereoselectively, which could be further applied to the late-stage site-selective functionalization of complex molecule synthesis.
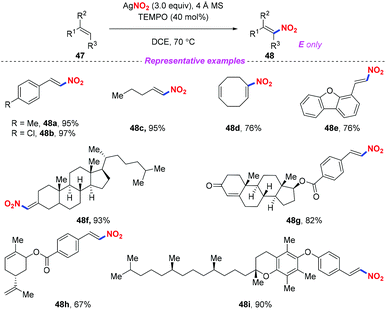 |
| | Scheme 17 Silver-mediated efficient and stereoselective nitration of mono- and disubstituted olefins. | |
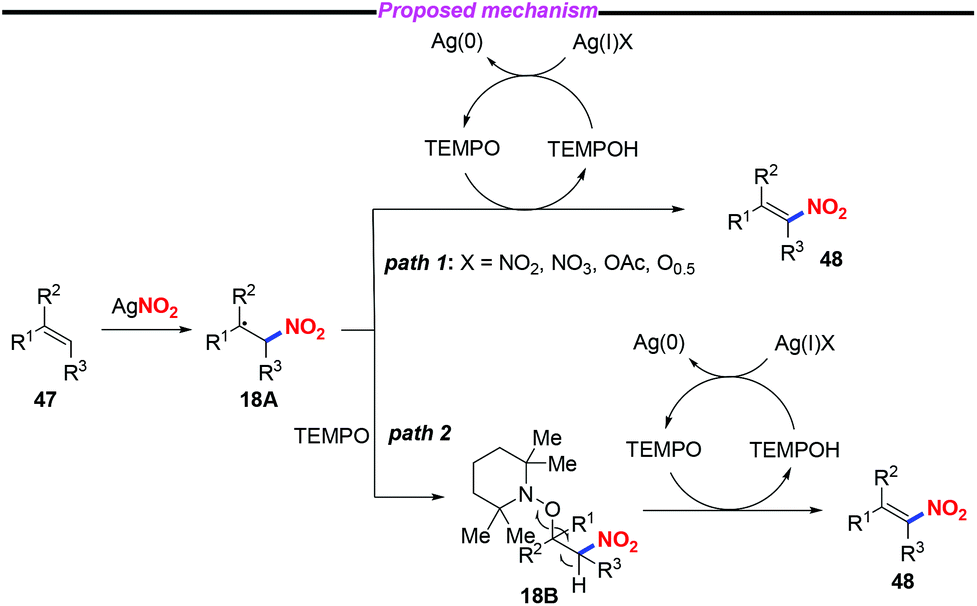
A plausible mechanism for the nitration of olefin has been outlined in Scheme 18. Initially, a nitro radical might be generated from AgNO2 under thermal conditions. The addition of the nitro radical to olefin 47 would give rise to the carbon-centered radical 18A. Subsequently, the nitroolefin product 48 could be formed via H-atom abstraction either from intermediate 18A (path 1) or from intermediate 18B (path 2).
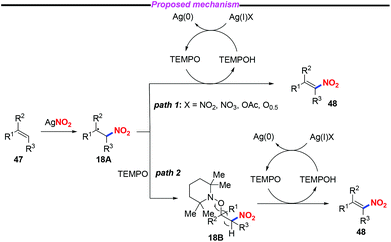 |
| | Scheme 18 Proposed mechanism for the nitration of olefins. | |
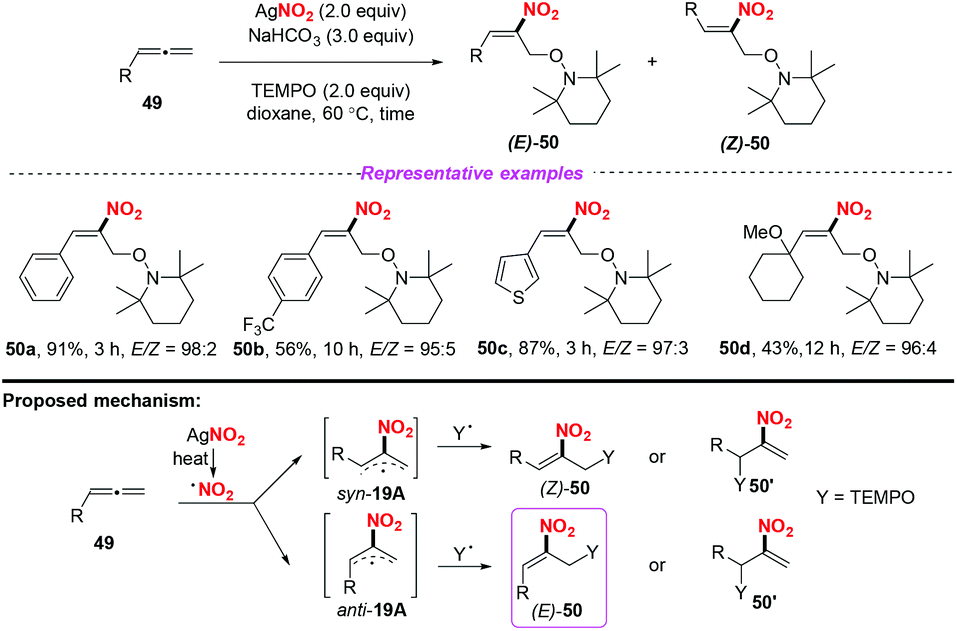
Later, Ma and co-workers reported a highly regio- and stereoselective nitro-oxoamination reaction of mono-substituted allenes, using AgNO2 as the nitro radical source (Scheme 19).25 The reaction proceeded smoothly to access various nitroolefins 50 with good functional group compatibility. The reaction was proposed to occur through the addition of nitro radical species to allenes giving rise to the stable allylic radical intermediate anti-19A or syn-19B, which could be directly captured by a TEMPO to form C–N and C–O bonds in one step.
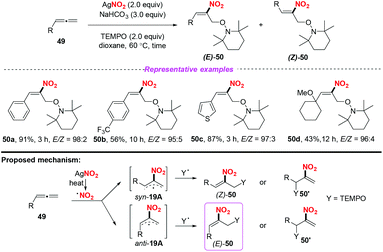 |
| | Scheme 19 Silver-mediated regio- and stereoselective nitro-oxoamination reaction of mono-substituted allenes. | |
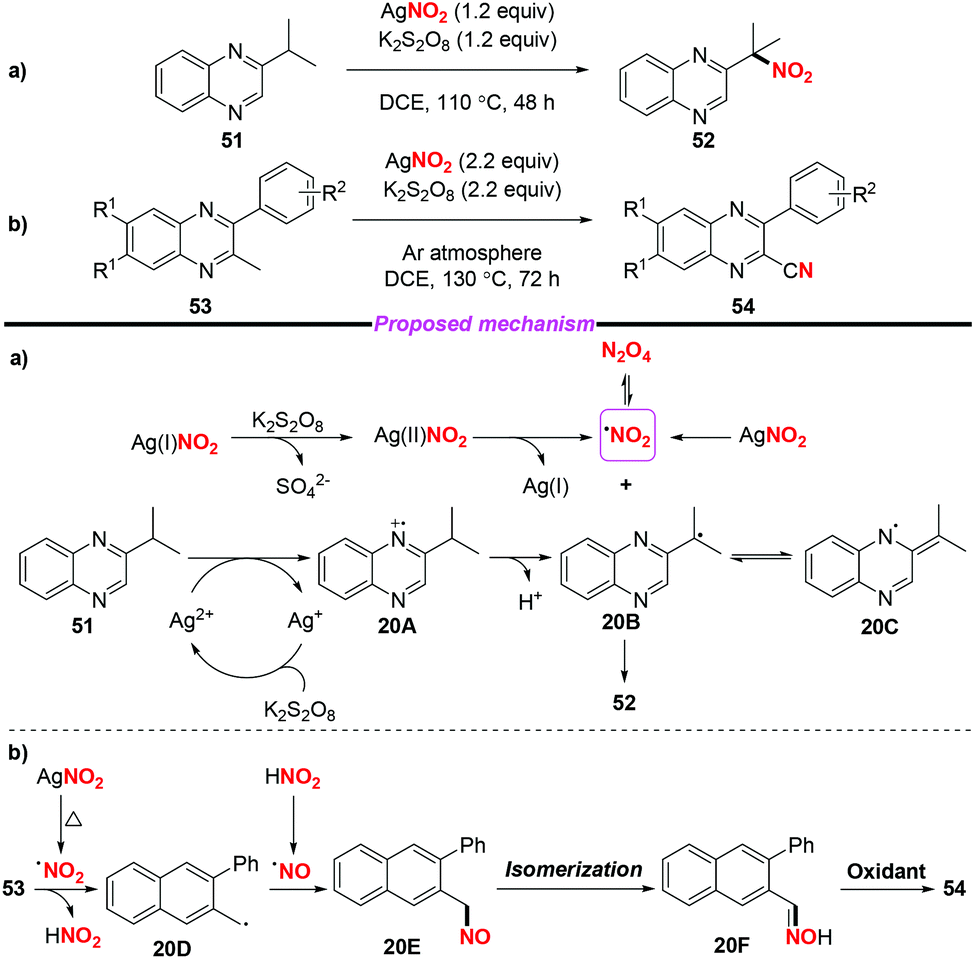
At the same time, Liu and co-workers described the AgNO2-mediated nitration of the quinoxaline tertiary C–H bond and conversion of 2-methyl quinoxalines into 2-quinoxaline nitriles (Scheme 20).26 To gain insight into the mechanism of the nitration process, the authors conducted some control experiments to confirm that the reaction might involve a free radical process. According to the proposed mechanism (shown in Scheme 20a) the nitro radical might be generated from AgNO2 upon treatment with or without K2S2O8. A single-electron transfer between the Ag(II) species and 51 would deliver radical intermediate 20B (or 20C), followed by the combination with the nitro radical to afford nitroalkane 52. On the other hand, the direct conversion of 53 to 54 might involve the generation of nitroso intermediate 20E and oxime 20F, which could be further oxidized into nitrile 54.
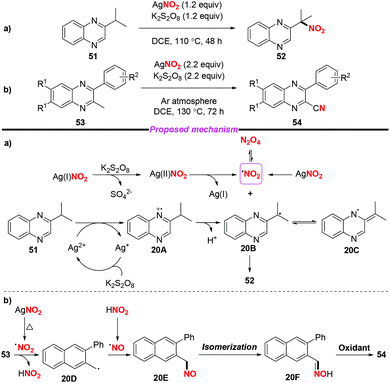 |
| | Scheme 20 Silver-mediated nitration of the quinoxaline benzylic C–H bond. | |
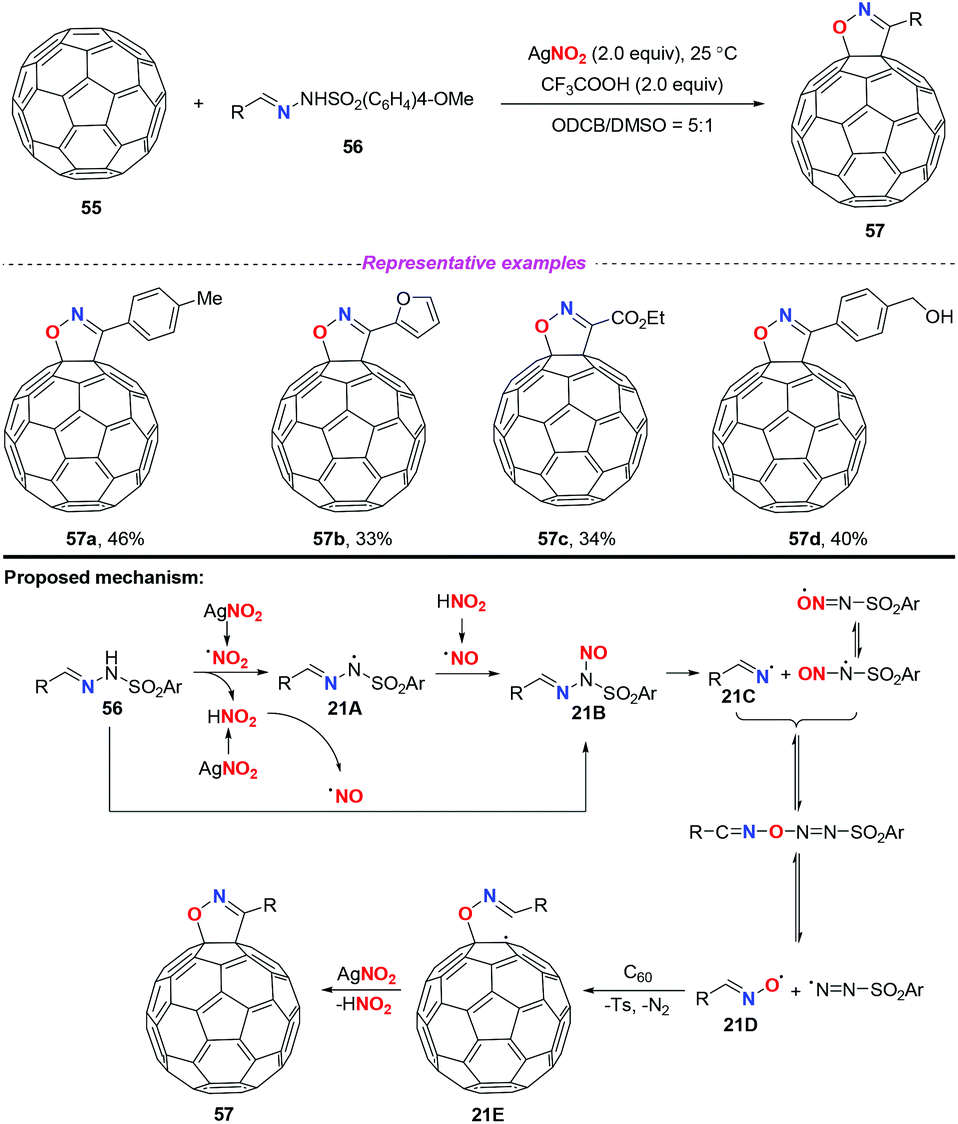
In 2015, Zhang and co-workers developed a novel AgNO2-mediated transformation of sulfonylhydrazones with [60]fullerene to afford fulleroisoxazolines (Scheme 21).27 It should be noted that the cleavage of the N–N bond of sulfonylhydrazones in synthetic chemistry was first disclosed. In this transformation, silver nitrite played dual roles, as a reaction initiator and as an oxygen source. Similar to previous reports, a nitro radical or a nitrosonium ion might be generated under the applied reaction conditions. Subsequently, the active species 21B could be produced either through the coupling of the nitro radical with the nitrogen-centered radical 21A or through the nucleophilic attack of the nitrosonium ion to sulfonylhydrazones 56. The decomposition of the unstable species 21B would generate the radical intermediate 21Dvia fast equilibrium conversions. As a result, the addition of radical 21D to [60]fullerene led to fullerenyl radical 21E, followed by an intramolecular radical cyclization to give the final product 57. Since the functionalization of fullerene is challenging, this work represented a rare example of free radical initiated transformation of fullerene, although the results were not satisfactory.
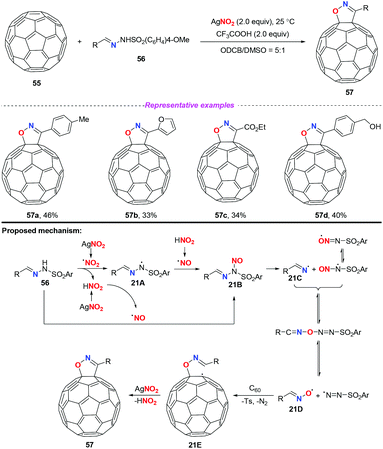 |
| | Scheme 21 Silver-mediated transformation of sulfonylhydrazones with [60]fullerene. | |
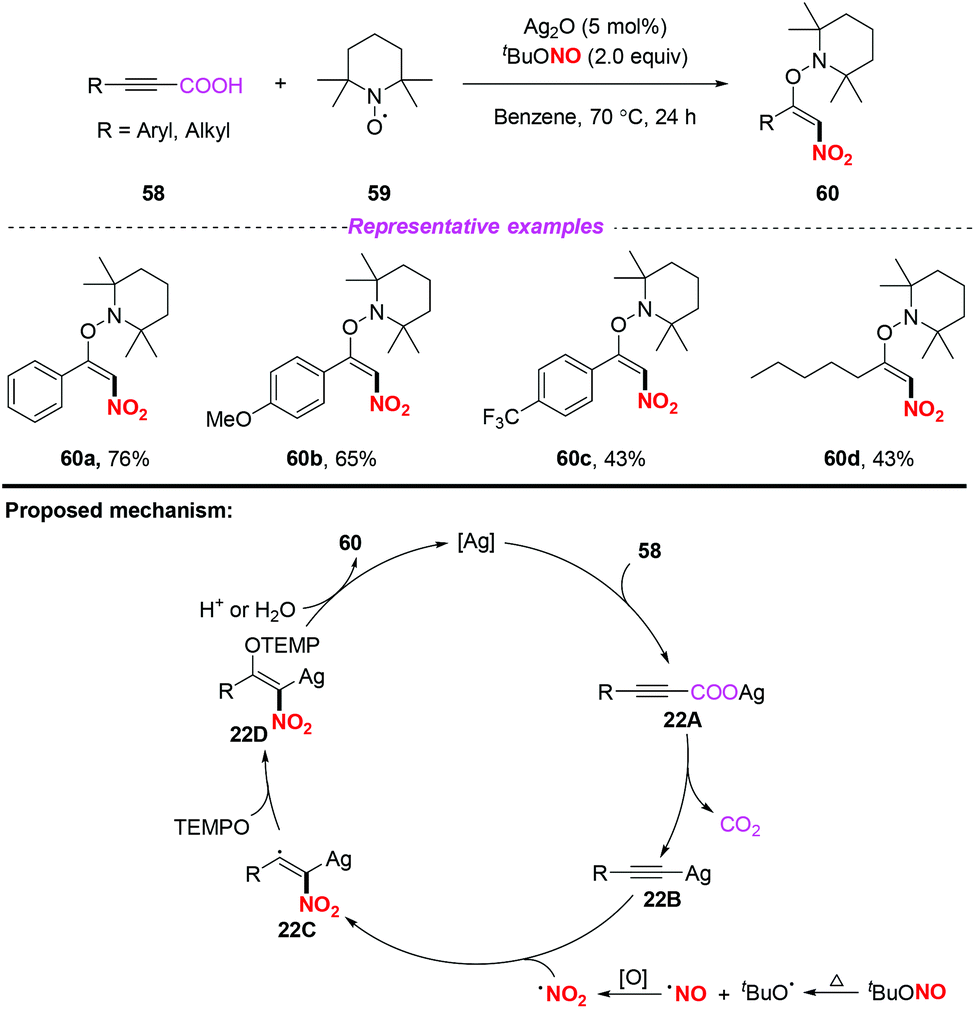
In the same year, Mao and co-workers developed a simple and efficient silver-catalyzed decarboxylative nitroaminoxylation of phenylpropiolic acids, resulting in the synthesis of (E)-β-nitroolefinic alkoxyamines with satisfactory yields (Scheme 22).28 The mechanism for the formation of product 60 was proposed as follows: initially, the reaction of phenylpropiolic acid 58 with silver oxide would provide silver carboxylate 22A, which could be converted into the Ag-acetylide intermediate 22Bvia decarboxylation. Additionally, the nitro radical was generated from tBuONO through the oxidation of a NO radical. Subsequently, the addition of the nitro radical to the Ag-acetylide intermediate 22B resulted in the radical intermediate 22C, which was trapped by TEMPO to give 22D. Finally, the protonation of 22D would release the desired product 60 and regenerate the silver catalyst. Similar to Ma's work,25 this reaction also illustrated the process of nitro radical addition to unsaturated bonds.
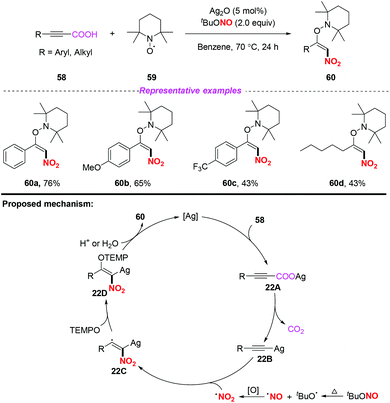 |
| | Scheme 22 Silver-catalyzed decarboxylative nitroaminoxylation reaction of phenylpropiolic acids. | |
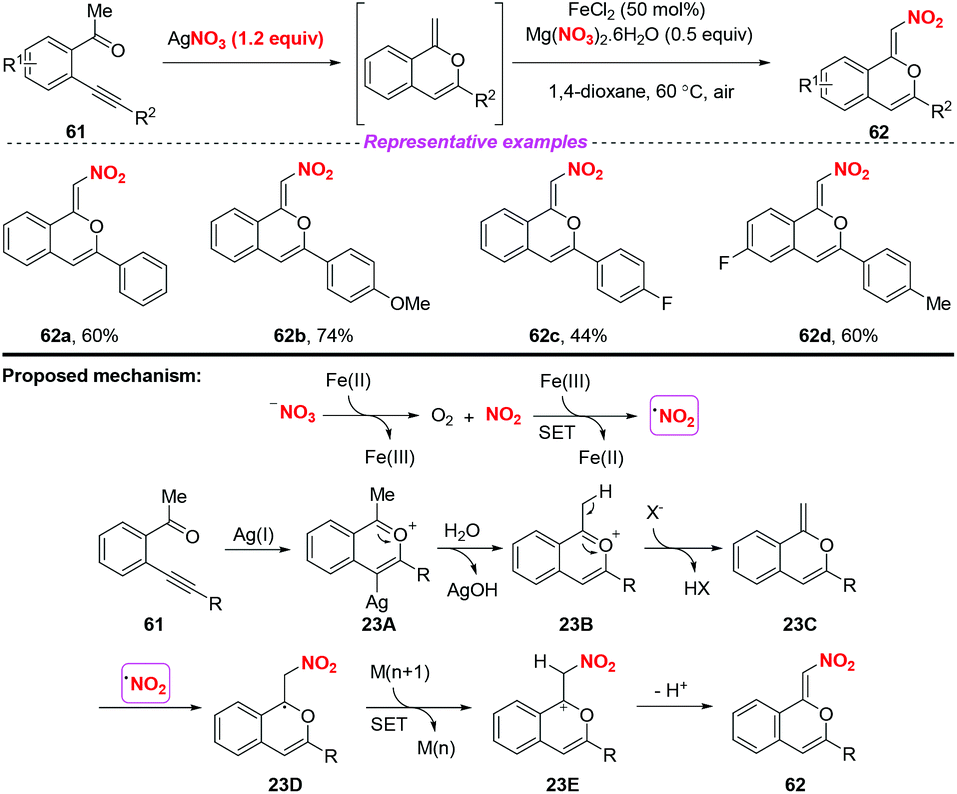
In 2017, Jiang and co-workers established a silver-mediated C(sp3)–H functionalization and 6-endo-dig oxo-cyclization of conjugated β-alkynyl ketones under oxidative conditions (Scheme 23).29 By employing AgNO3 as a nitrating reagent, this method enabled the synthesis of nitro-branched (Z)-isochromenes in moderate to good yields. On the basis of radical trapping studies and previous reports, a possible mechanism involving the formation of the nitro radical by the SET process between Fe(II) salts and activated NO3− was proposed. The subsequent transformation steps, Ag-catalyzed 6-endo-dig oxo-cyclization/H-transfer/SET/radical addition afforded isochromenes 62. This reaction showed that not only the nitrite group but also the nitrate group could be used as the source of nitro radicals for accessing nitro-containing products.
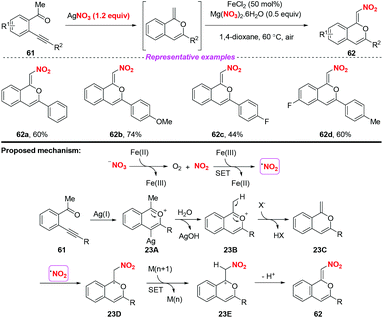 |
| | Scheme 23 Silver-catalyzed C(sp3)–H nitration of β-alkynyl ketones for accessing functionalized isochromenes. | |
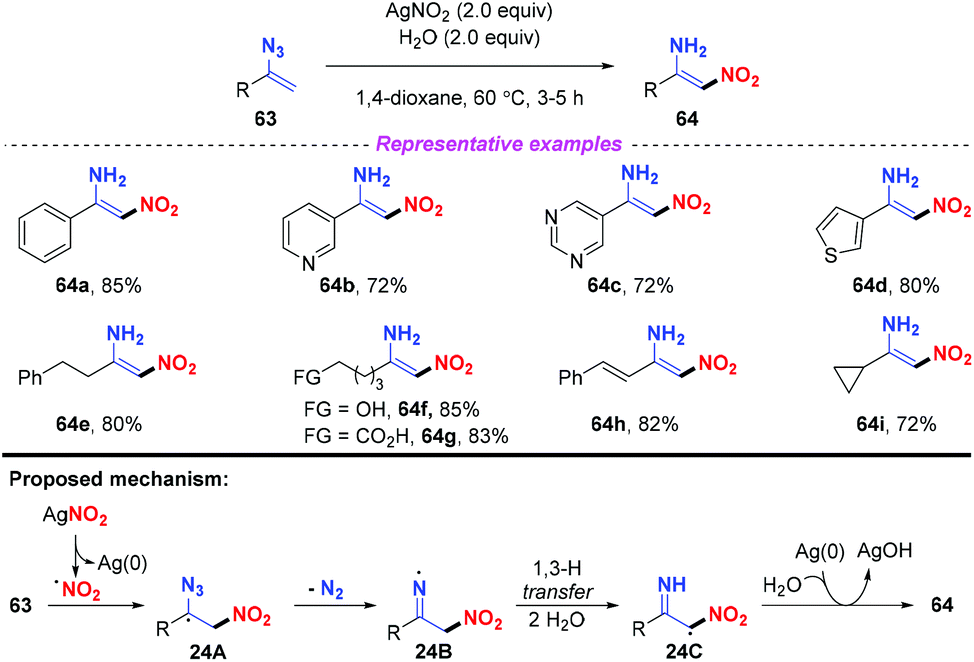
The nitrification of vinyl azides into N-unprotected enamines was achieved by Bi and co-workers (Scheme 24).30 In general, various (hetero)arenes or alkanes were compatible in this reaction, furnishing the corresponding products 64 in good yields. Notably, by employing this strategy, alkyl vinyl azides bearing OH and COOH groups were well-tolerated to furnish the functionalized nitroenamine products. On the basis of experimental results and DFT calculations, a plausible mechanism was proposed, as depicted in Scheme 24. The nitro radical, which was firstly generated from AgNO2 with the release of Ag(0), would attack the terminal carbon of vinyl azide 63, leading to intermediate 24A. The iminyl radical 24B was subsequently formed by releasing N2 from 24A, which would undergo a 1,3-H transfer with the assistance of water to give intermediate 24C. Finally, the resulting radical 24C could be converted to 64 by the transfer of an H atom from water to the iminyl N-site.
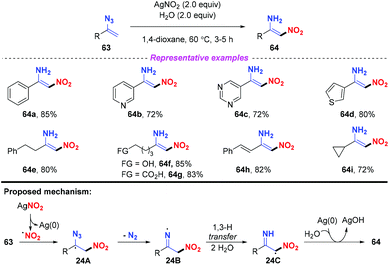 |
| | Scheme 24 Silver-mediated nitrification of vinyl azides into nitroenamines. | |
2.4. Pd-Catalyzed transformations
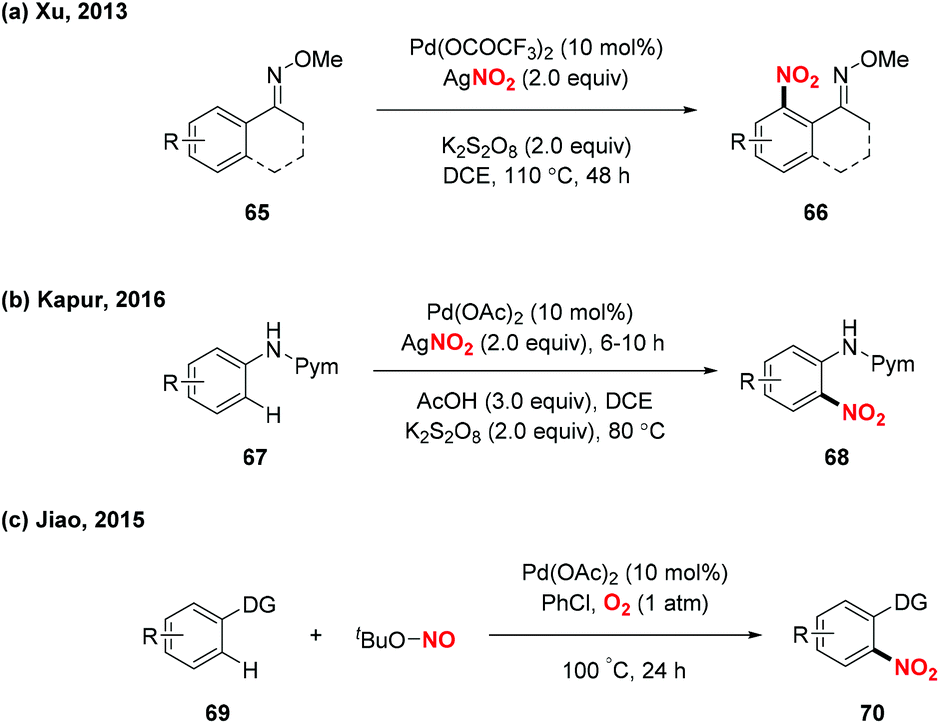
Over the past decades, palladium-catalyzed C–H functionalizations of organic compounds have been widely applied in synthetic organic chemistry. In 2013, Liu and co-workers developed a novel palladium-catalyzed chelation-assisted site-regiospecific nitration of aromatic C–H bonds, using AgNO2 as a nitro radical source (Scheme 25a).31 This protocol exhibited excellent mononitration selectivity, broad functional group and substrate tolerance, providing the corresponding nitroarenes in good yields. Later, Kapur and co-workers described a palladium-catalyzed regioselective C–H nitration of anilines using pyrimidine as a removable directing group (Scheme 25b).32 Additionally, Jiao and co-workers demonstrated Pd-catalyzed aerobic oxidative C–H nitration of arenes with tert-butyl nitrite (TBN) as the radical precursor. In this reaction, molecular oxygen was employed as the terminal oxidant and oxygen source to initiate the active radical reactants (Scheme 25c).33
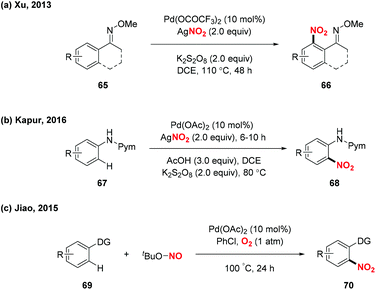 |
| | Scheme 25 Palladium-catalyzed nitration of aromatic C–H bonds (Xu, Kapur and Jiao). | |
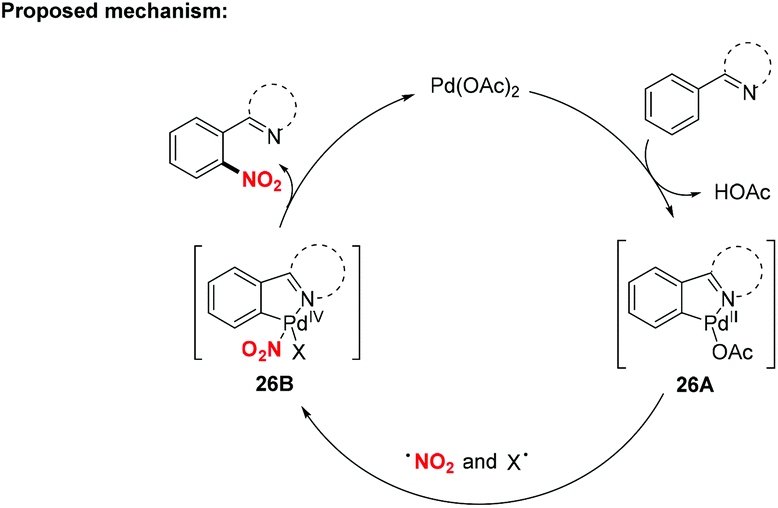
According to the mechanism proposed by Jiao and co-workers, the palladacycle intermediate 26A was firstly formed by directing group assisted ortho-selective cyclometallation on the benzene. Then, oxidative addition of active radicals (such as NO2, tBuO) to 26A provided Pd(IV) intermediate 26B, which would undergo reductive elimination to afford the corresponding products (Scheme 26).
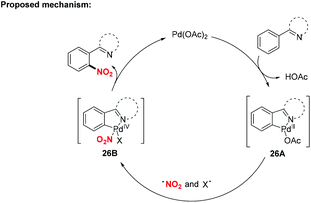 |
| | Scheme 26 Proposed mechanism for the palladium-catalyzed nitration of aromatic C–H bonds. | |
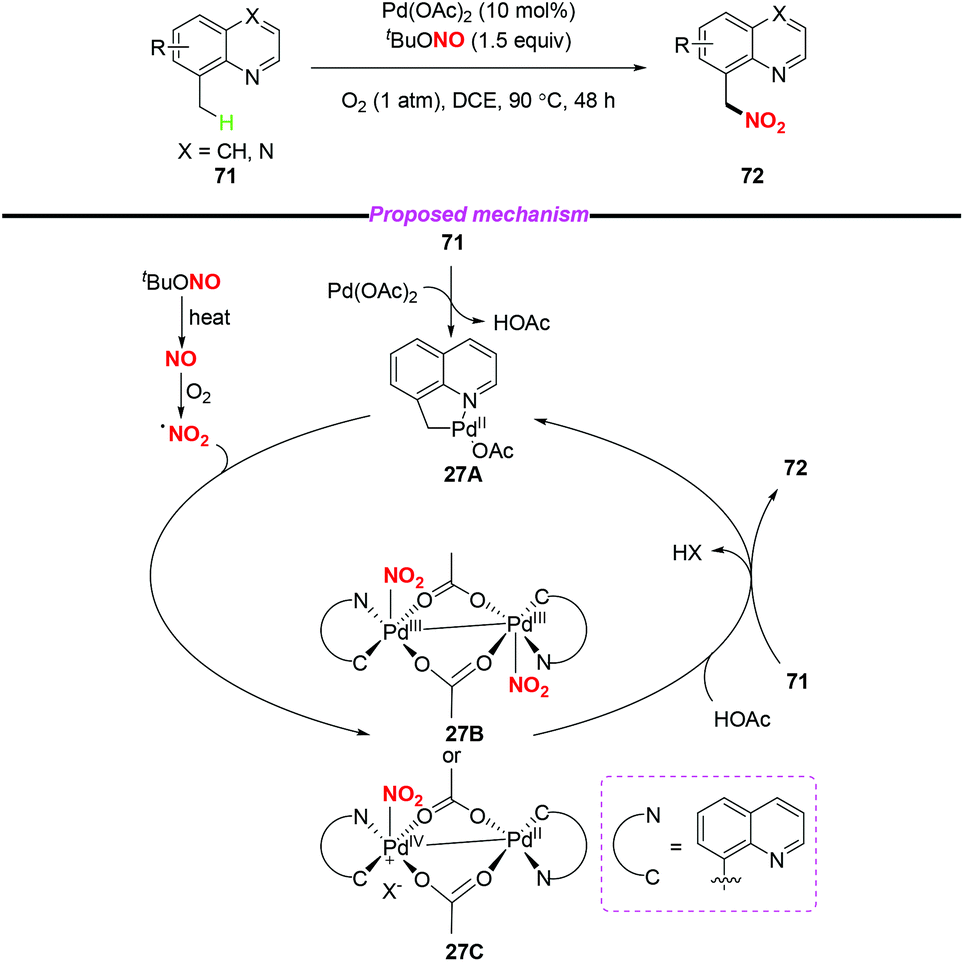
In 2015, Liu and co-workers described a novel palladium-catalyzed sp3 C–H nitration of 8-methylquinolines, which produces 8-(nitromethyl)quinolines in moderate to excellent yields (Scheme 27).34 During the reaction process, tBuONO was used as a NO2 radical source under mild conditions. To gain insight into the mechanism of the sp3 C–H nitration, the authors carried out several mechanistic experiments, and proposed a possible mechanism to clarify the reaction pathway. Firstly, palladacycle intermediate 27A was formed through a reaction of Pd(OAc)2 with 8-methylquinoline, and the NO2 radical was generated from tert-butyl nitrite via a NO radical intermediate. Then, the addition of the NO2 radical to the palladium center of 27A delivered Pd(III)–Pd(III) species 27B or Pd(IV)–Pd(II) species 27C. Finally, the reductive elimination of 27B or 27C in the presence of another molecule of 71 would release the desired product 72 and regenerate 27A.
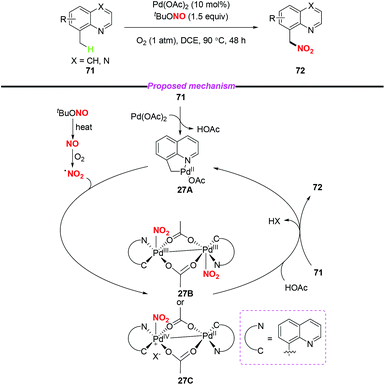 |
| | Scheme 27 Palladium-catalyzed sp3 C–H nitration of 8-methylquinolines. | |
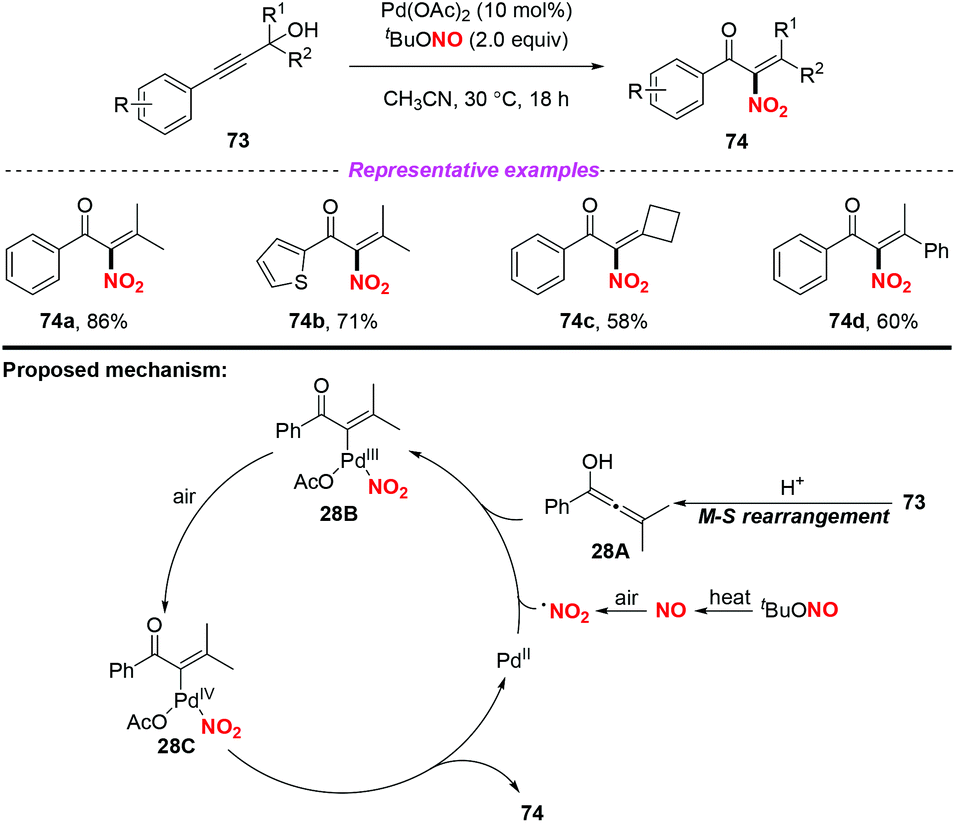
A palladium-catalyzed Meyer–Schuster/nitration of propargylic alcohols to access α-nitroenones under aerobic conditions was developed by Song and co-workers in 2016 (Scheme 28).35 Similarly, the NO2 radical was generated from tBuONO (TBN) via the NO radical intermediate. On the basis of several control experiments, a tentative reaction mechanism is depicted in Scheme 28. Firstly, propargylic alcohol 73 was converted into the allenol intermediate 28Avia the Meyer–Schuster rearrangement. The addition of the NO2 radical to intermediate 28A led to the formation of intermediate 28B in the presence of Pd(OAc)2, which was further oxidized into intermediate 28C. Finally, the desired product 74 was formed via reductive elimination along with the regeneration of the palladium catalyst. When α-nitroenone was subjected to the subsequent hydrogenation reduction, α-amino alcohol was obtained, which could be used as a raw material to synthesize various oxazoline ligands.
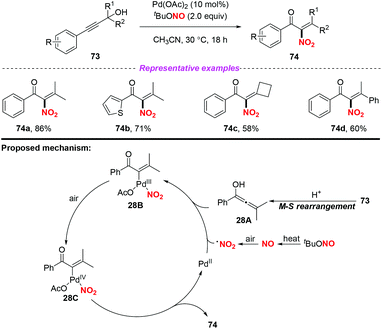 |
| | Scheme 28 Palladium-catalyzed nitration of Meyer–Schuster intermediates with TBN as a nitrogen source at ambient temperature. | |
2.5. Mn-Catalyzed transformations
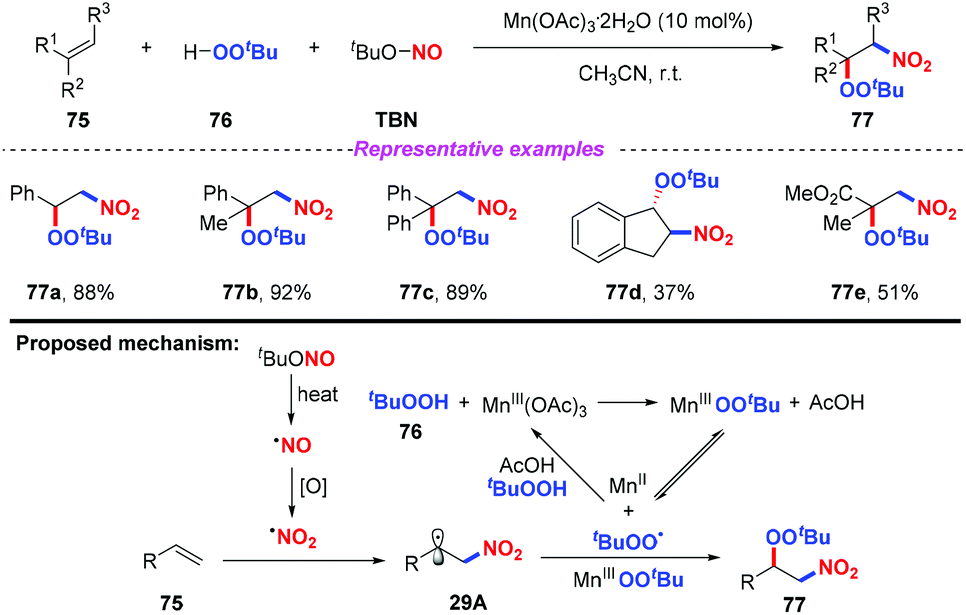
Recently, Li and co-workers reported a novel and selective three-component nitration–peroxidation of alkenes with tBuONO (TBN) and tBuOOH (TBHP) to afford β-peroxy nitro compounds in moderate to good yields (Scheme 29).36 The reaction proceeded readily at room temperature by employing Mn(OAc)3·2H2O as the catalyst. Based on the experimental results, a possible mechanism for the nitration–peroxidation of alkenes was proposed, as shown in Scheme 29. Initially, the nitro radical and peroxyl radical (˙OOtBu or Mn-OOtBu) were generated from TBN and tBuOOH, respectively. Then, the addition of the nitro radical to alkene 75 would result in the formation of key intermediate 29A, followed by the radical cross-coupling with a peroxyl radical (˙OOtBu or Mn-OOtBu) to give the final product 77.
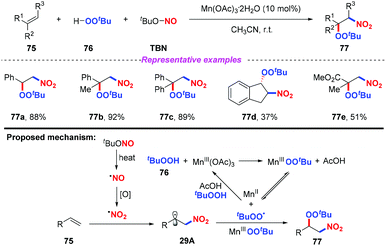 |
| | Scheme 29 Manganese-catalyzed nitration peroxidation of alkenes. | |
2.6. Ru-Catalyzed transformations
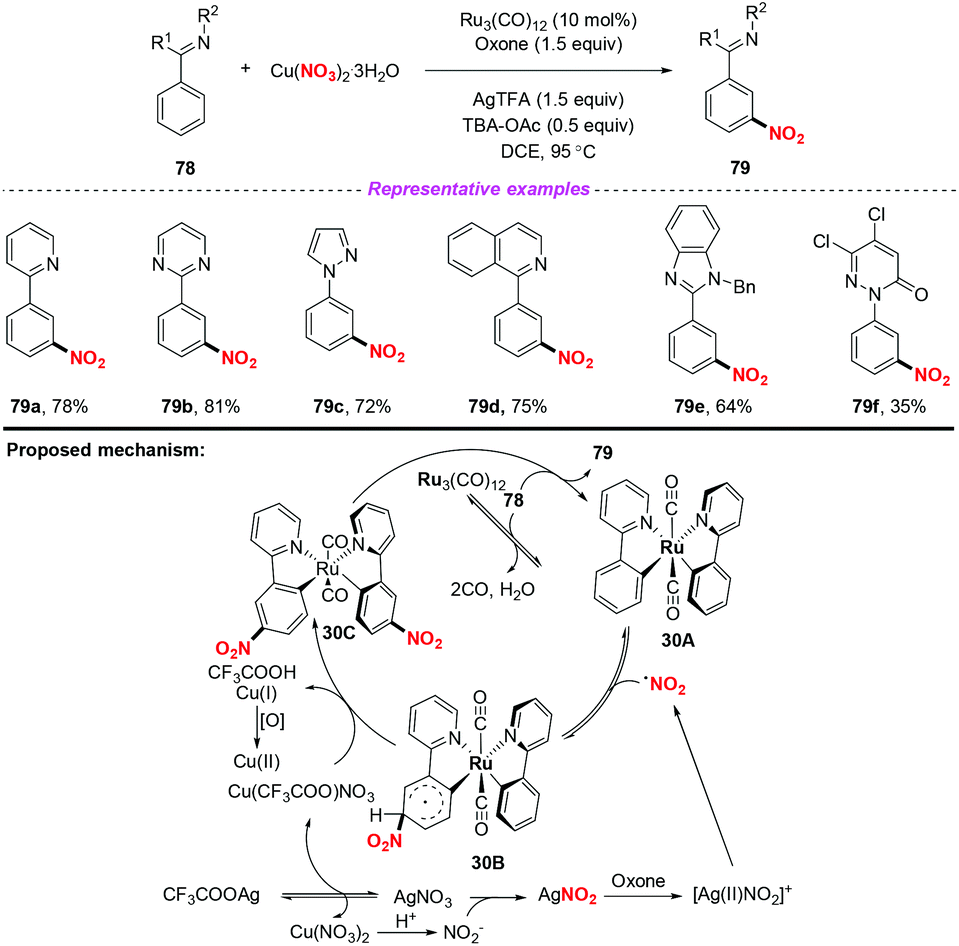
Over the past few years, significant progress in Ru-catalyzed meta-CAr–H functionalization has been achieved by using the ortho-metalation strategy. In 2016, Zhang and co-workers reported the first example of Ru-catalyzed meta-selective CAr–H nitration of 2-aryl N-aromatics using Cu(NO3)2·3H2O as the nitro radical source (Scheme 30).37 Moreover, by using the nitration products as the starting materials, this methodology was further applied to the synthesis of pharmaceutical intermediates, clinical candidates and marketed drugs. On the basis of the detailed mechanistic investigation, a plausible catalytic cycle is depicted in Scheme 30. Firstly, the active Ru complex 30A was formed through the CAr–H activation of substrate 78 with Ru3(CO)12, which was confirmed by X-ray diffraction analysis. Subsequently, electrophilic addition of the nitrogen dioxide radical to the para-carbon of the Ru–CAr σ-bond would deliver species 30B, followed by deprotonation to give a more stable intermediate 30C with the assistance of a new copper(II) salt, Cu(CF3COO)NO3. In this process, the new copper(II) salt was derived from an anion exchange between Cu(NO3)2 and AgTFA. Finally, ligand exchange of complex 30C with 78 would provide the nitration product 79, and regenerate complex 30A.
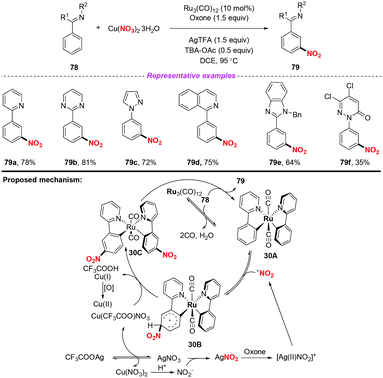 |
| | Scheme 30 Ruthenium-catalyzed meta-selective CAr–H nitration of 2-aryl N-aromatics. | |
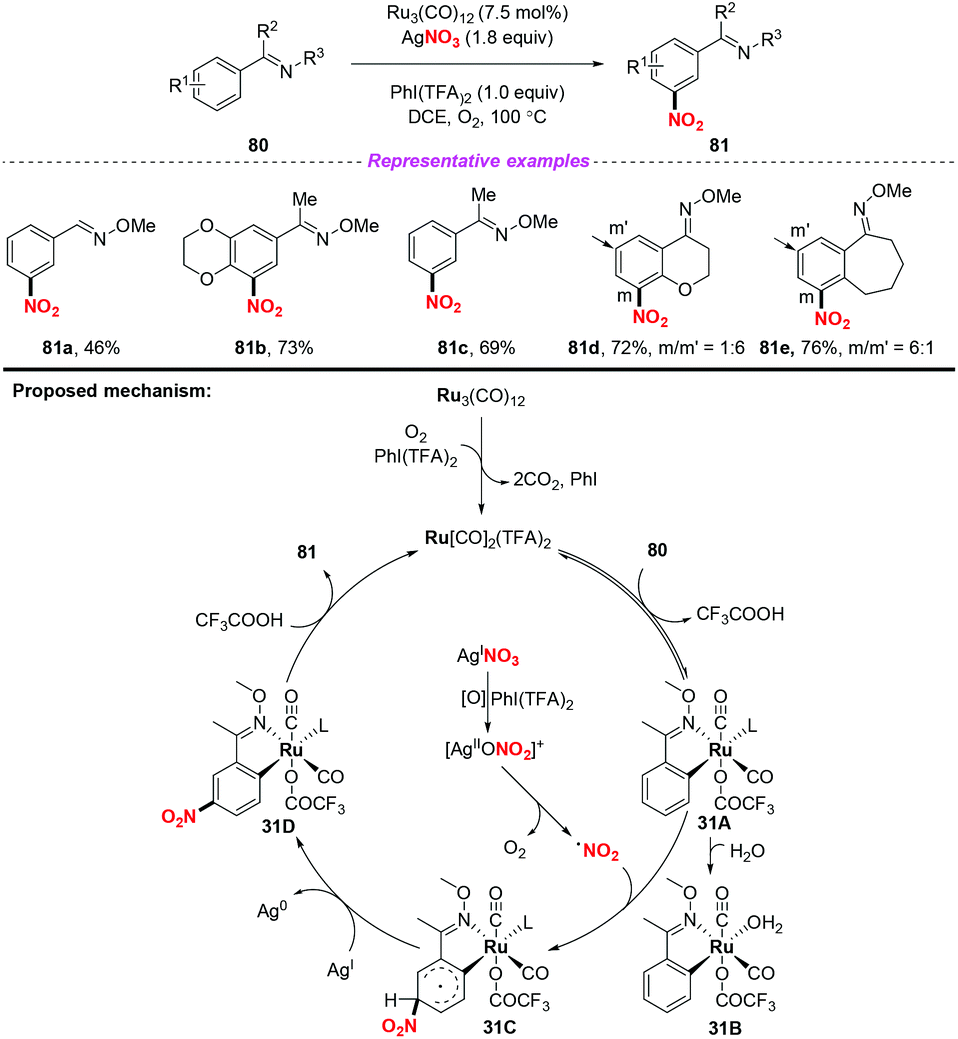
The same group later developed an oxime-directed meta-CAr–H nitration of arenes by using AgNO3 as the nitro radical source (Scheme 31).38 Mechanistic studies revealed that a new ortho-ruthenated monomeric octahedral complex was involved. On the basis of the experimental results, a possible mechanism for the meta-CAr–H nitration process was proposed, as shown in Scheme 46. Initially, the active Ru(II) catalyst was formed through the oxidation of the Ru(0) precatalyst using PhI(TFA)2 and O2 as the oxidant. Then, the ortho-C–H bond cleavage of 80 led to Ru(II) complex 31A, which would further undergo a water-ligand exchange to give complex 31B. On the other hand, AgNO3 was oxidized by PhI(TFA)2 to yield a Ag(II) salt, followed by the release of the nitrogen dioxide radical. The resulting nitrogen dioxide radical would attack the para position of the C–Ru bond to afford RuII species 31C. The reductive deprotonation of 31C would result in the formation of intermediate 31D. Finally, ligand exchange of 31D with CF3COOH would release the meta-nitrated product 81 and regenerate the active Ru(II) catalyst.
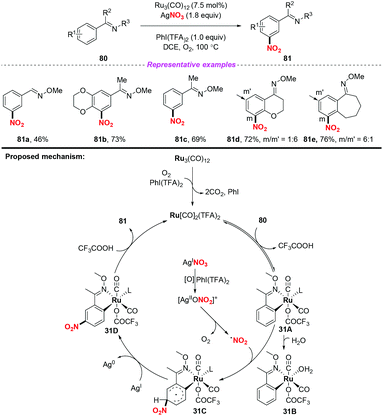 |
| | Scheme 31 Ruthenium-catalyzed oxime-directed meta-CAr–H nitration of arenes. | |
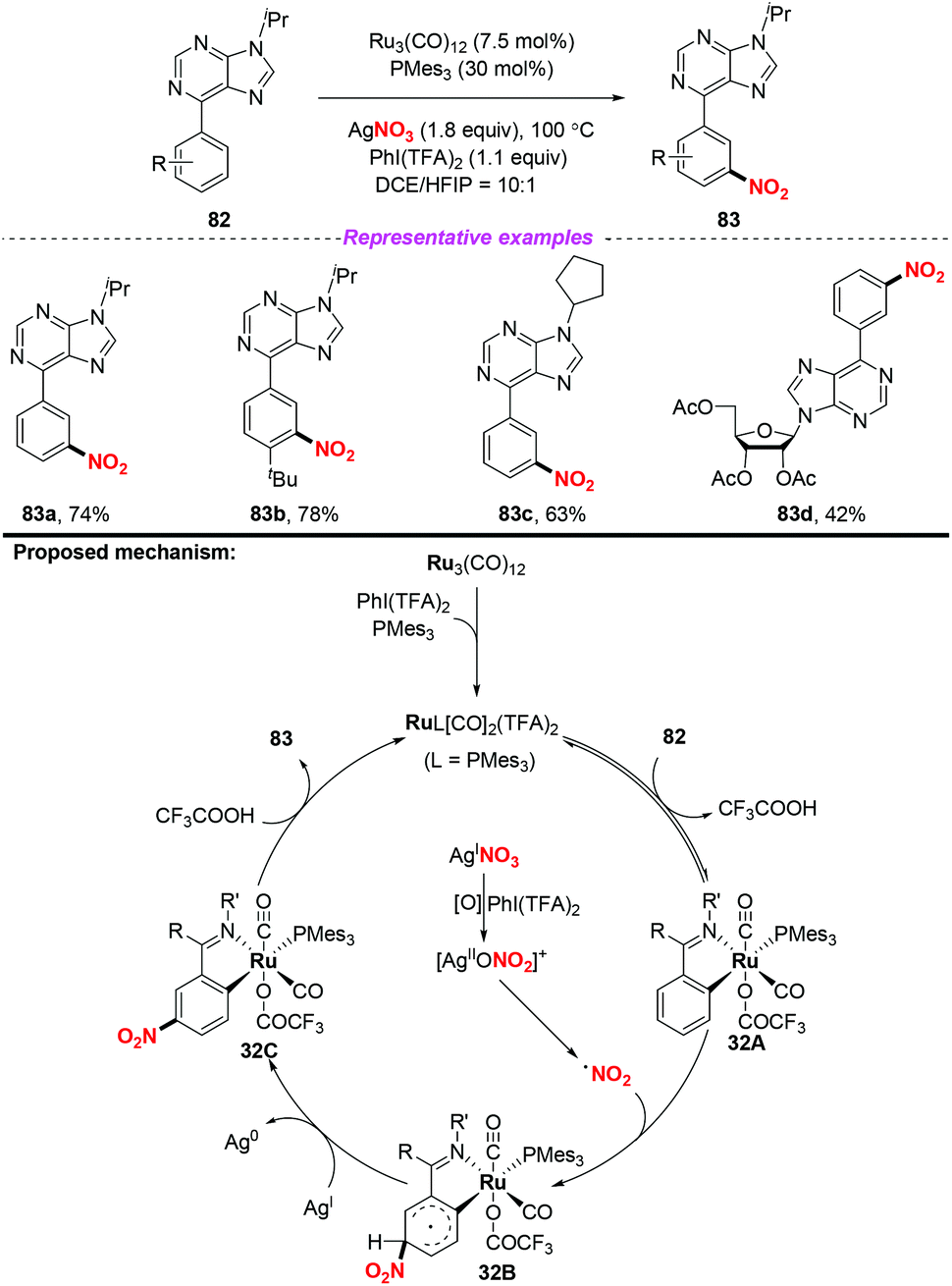
In 2018, the same group extended the above strategy to the ruthenium-catalyzed tertiary phosphine ligand promoted meta-C–H nitration of 6-arylpurines and nucleosides (Scheme 32).39 The authors found that the sterically hindered phosphine ligand played a crucial role in catalytic efficiency. By employing this method, a series of meta nitrated products were obtained in good yields, showing a broad substrate scope and high functional group tolerance. The possible reaction pathway was similar to the previous reports, and is shown in Scheme 32.
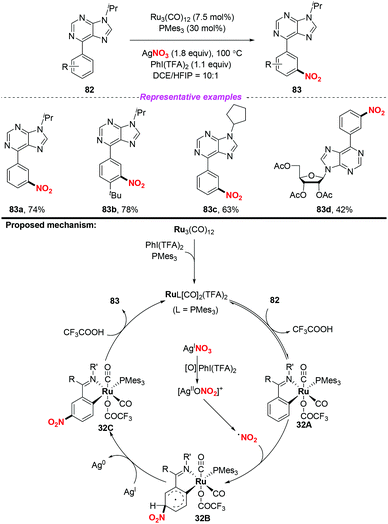 |
| | Scheme 32 Ruthenium-catalyzed PMes3-promoted meta-C–H nitration of arenes. | |
Recently, the group of Ding developed an efficient method for the synthesis of meta-nitrated 2-arylbenzothiazole 85 through the ruthenium-catalyzed meta-selective C–H nitration pathway (Scheme 33a). Similarly, Maiti and co-workers also developed a directing group assisted ruthenium catalyzed approach to produce meta-nitrated phenols 86 (Scheme 33b).40b Both the reactions featured a broad substrate scope and gave rise to meta-nitrated benzothiazole and phenol derivatives with structural complexity that could not be readily prepared by other methods.
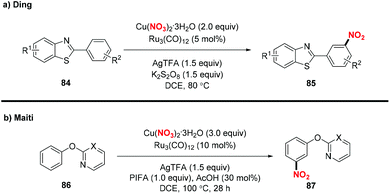 |
| | Scheme 33 Ruthenium-catalyzed meta-C–H nitration of arylbenzothiazole. | |
2.7. Under transition metal free conditions
2.7.1 TBN as a nitro radical source.
Except for the abovementioned approaches employing nitro radicals under transition metal catalysis, the development of a metal-free nitration process is promising for the formation of nitro compounds through a radical process. For instance, in 2013, Maiti and co-workers developed a mild, metal-free decarboxylative nitration protocol for the synthesis of nitroolefins from α,β-unsaturated carboxylic acids using tBuONO and TEMPO under aerobic conditions (Scheme 34).41 The substrate scope of this reaction was quite broad, and α,β-unsaturated carboxylic acids bearing aromatic and heterocyclic groups were well-tolerated. According to the proposed mechanism, the nitro radical was generated from TBN via the homolytic cleavage of O–NO bonds and subsequent oxidation, which would attack the double bond of olefins to form the benzylic radical 34A. Then, the radical coupling of intermediate 34A with TEMPO would lead to the preferential formation of energetically more favorable intermediate 34B over 34C, followed by anti-elimination to afford E-nitroolefins.
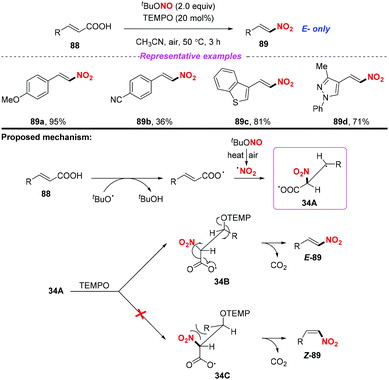 |
| | Scheme 34 Synthesis of (E)-nitroolefins via decarboxylative nitration using TBN and TEMPO. | |
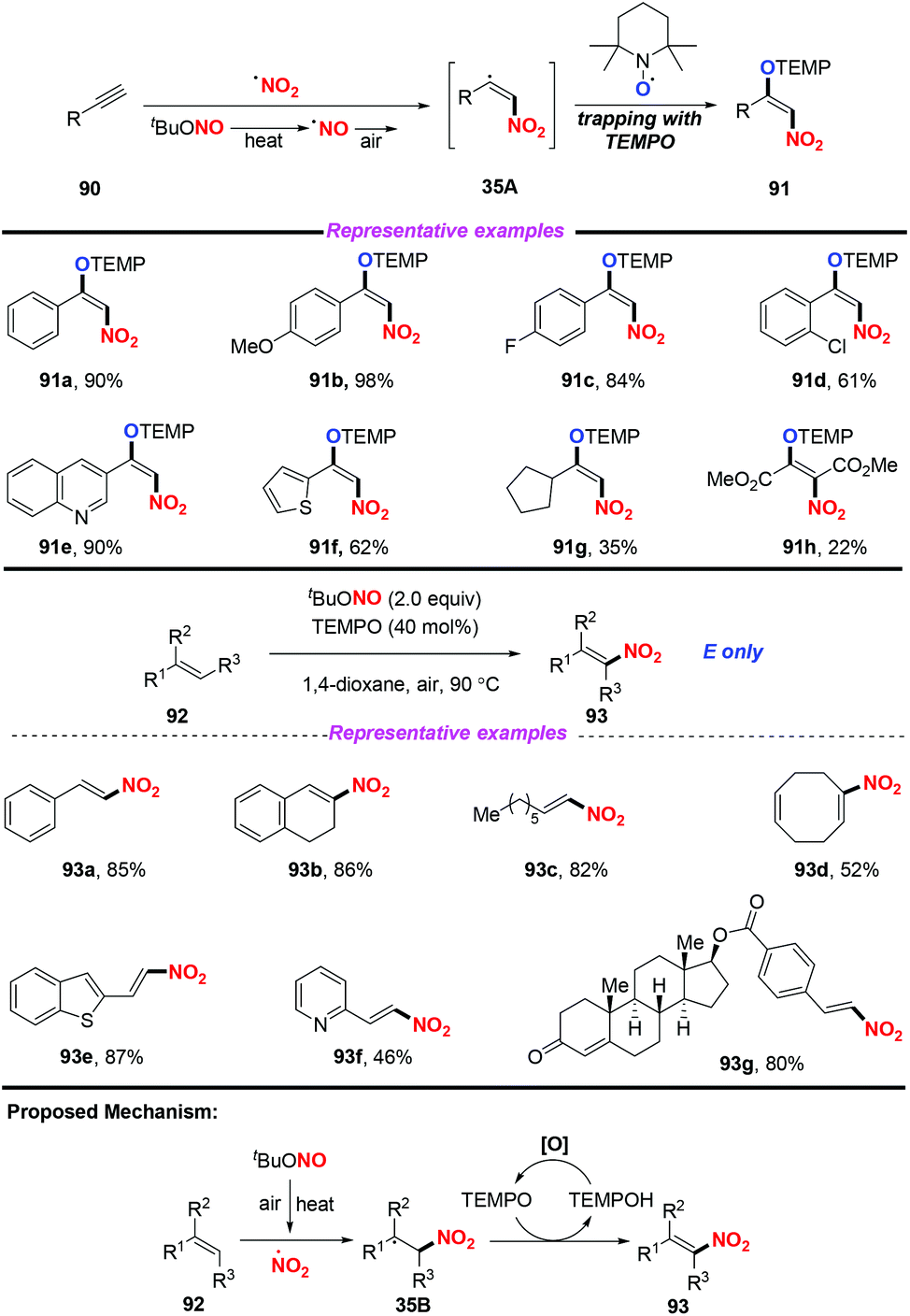
Additionally, the same group also extended this strategy for the stereoselective nitro-aminoxylation of alkynes with TBN and TEMPO (Scheme 35a).42a The reaction featured a wide substrate scope and good functional group tolerance, leading to (E)-β-nitroolefinic alkoxyamines in high yields. Meanwhile, by employing the TBN and TEMPO system, the authors found that alkenes were suitable for the stereoselective nitration process, providing direct access to nitroolefins with excellent E-selectivity (Scheme 35b).42b
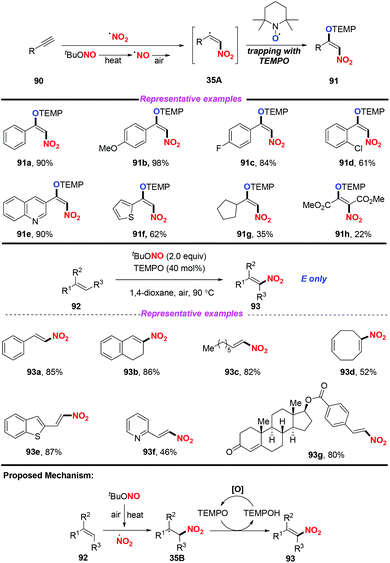 |
| | Scheme 35 Aerobic oxynitration of alkynes and alkenes with TBN and TEMPO. | |
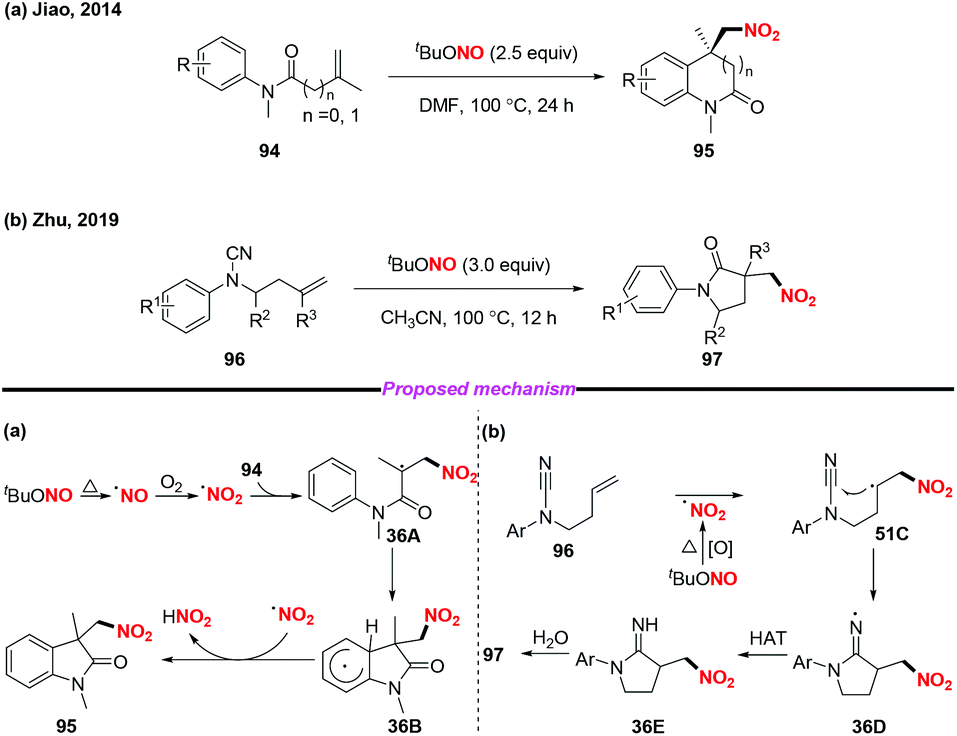
In 2014, the group of Jiao reported a highly efficient metal-free nitro-carbocyclization of activated alkenes, giving rise to various nitro-containing oxindoles via tandem C–N and C–C bond formation (Scheme 36a).43 Additionally, Zhu and co-workers recently developed a novel method for the preparation of γ-lactams through radical-mediated nitration–aminocarbonylation of unactivated olefins under metal-free conditions (Scheme 36b).44 Notably, the nitro radical was generated from the homolysis of tert-butyl nitrite in both reactions for further transformations.
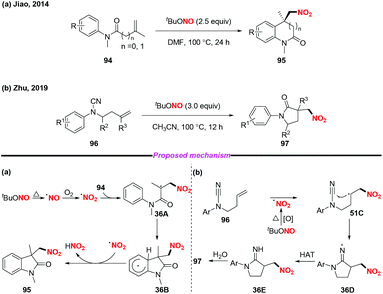 |
| | Scheme 36 Synthesis of nitro-containing oxindoles and γ-lactams under metal-free conditions. | |
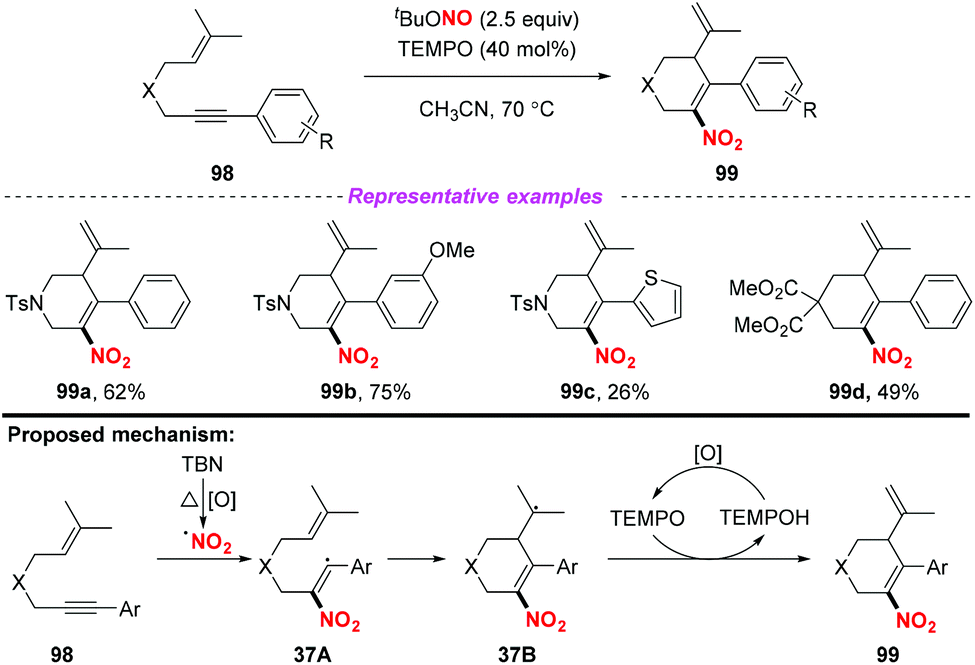
In 2015, Liang and co-workers demonstrated a novel and convenient metal-free nitration and cyclization of 1,6-enynes (Scheme 37).45 This transformation exhibited good functional-group tolerance and was amenable to scale up. Mechanistic investigations revealed that the reaction was initiated via the addition of the nitro radical to the C–C triple bond of the 1,6-enynes 98 to generate intermediate 37A, which subsequently underwent intramolecular carbocyclization to give radical intermediate 37B. Finally, the desired product 99 was formed through the oxidative dehydrogenation of intermediate 37B with TEMPO. In contrast to the traditional enyne ring-closing reaction using precious transition metal catalysts such as rhodium, gold and platinum, this reaction proceeded smoothly under metal-free conditions, affording six-membered (hetero)cyclic compounds containing nitro groups in moderate to good yields.
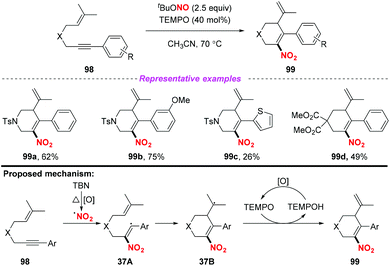 |
| | Scheme 37 Metal-free nitro-carbocyclization of 1,6-enynes with TBN and TEMPO. | |
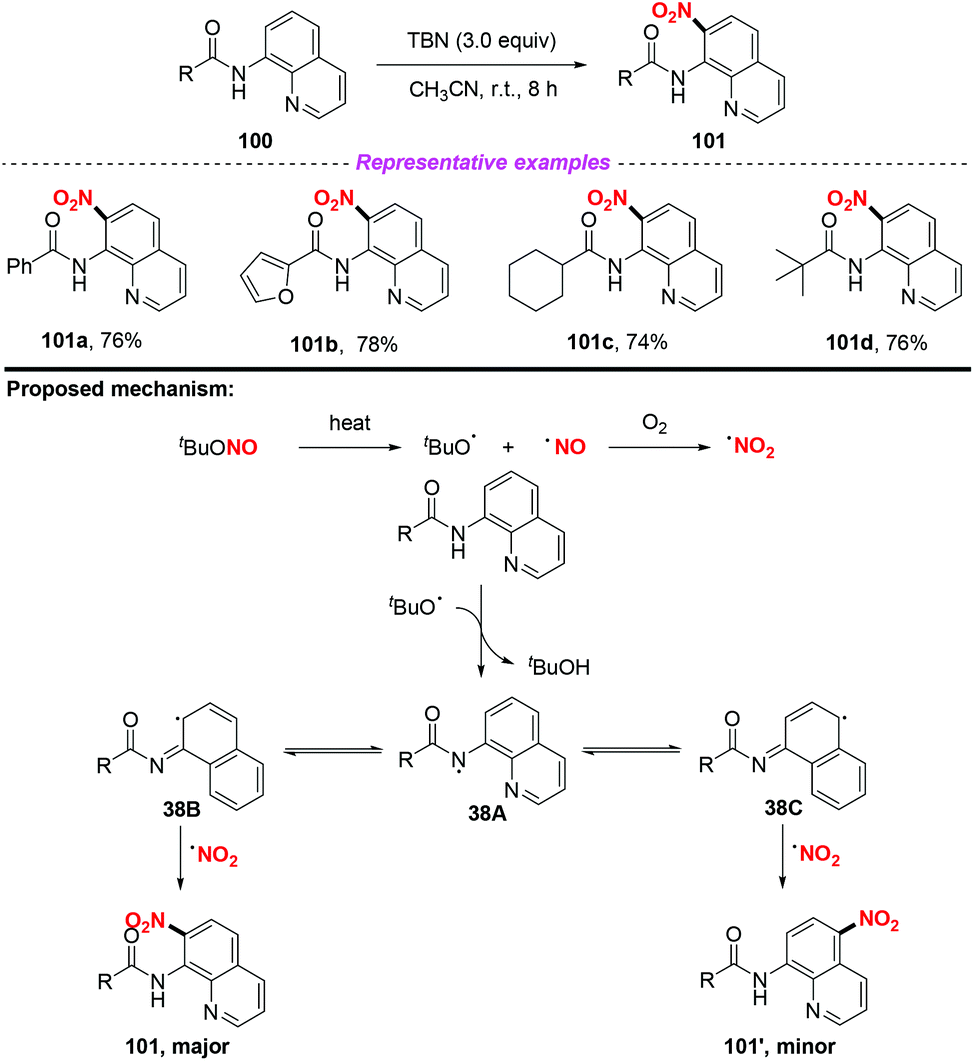
Furthermore, the group of Hajra developed a regioselective nitration at the C-7 position of quinoline compounds using TBN under metal-free conditions in 2018 (Scheme 38).46 Preliminary mechanistic studies involved the radical trapping experiments, according to which a radical pathway was involved in this transformation. Firstly, TBN would undergo thermal homolysis to generate an alkoxy radical and nitric oxide, followed by oxidation to form a nitro radical in the presence of molecular oxygen. Then, carboxamide would undergo an alkoxy radical mediated intermolecular hydrogen atom transfer process to give nitrogen-centered radical 38A, which would isomerise to quinoline radical 38B or 38C. Finally, the major product 101 was delivered via the coupling of the nitro radical with quinoline radical 38B, accompanied by the formation of a minor amount of the C-5 nitrated product 101′.
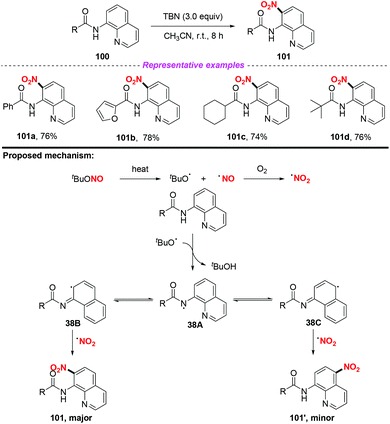 |
| | Scheme 38 Metal-free selective nitration at the C-7 position of quinoline compounds using TBN. | |
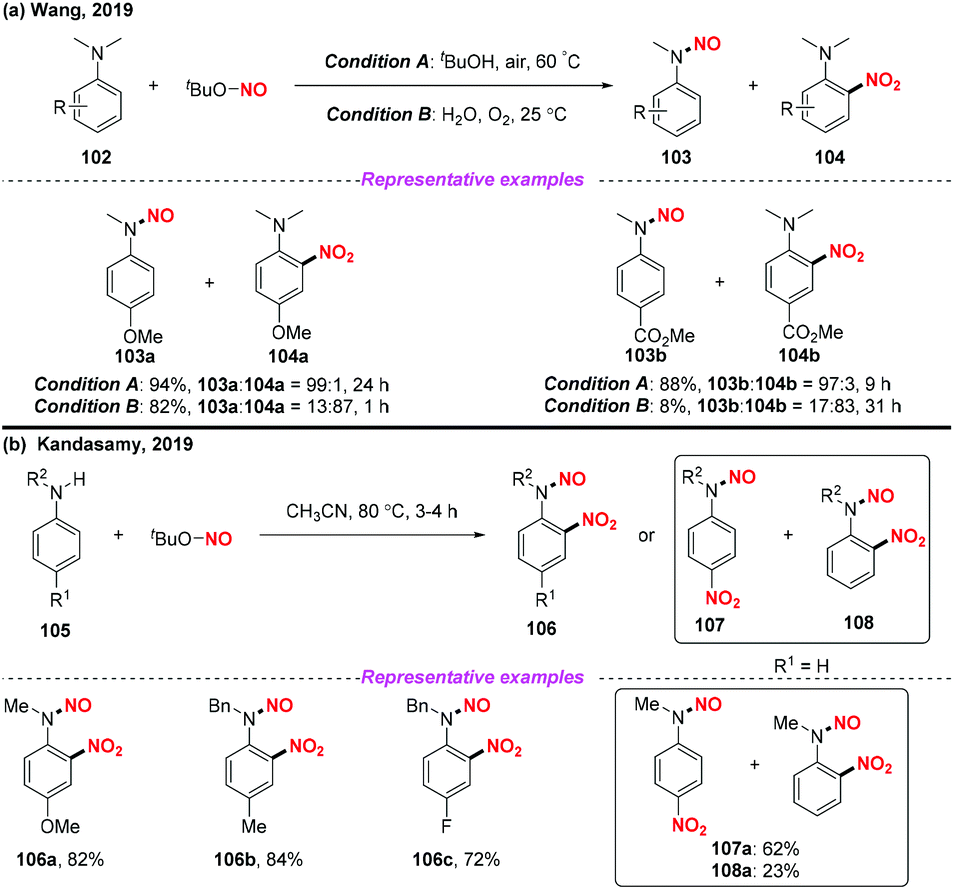
Recently, Wang and co-workers presented a solvent-controlled chemoselective N-dealkylation-N-nitrosation or nitration of N-alkyl anilines utilizing TBN as a nitro radical source (Scheme 39a).47 Similarly, the Kandasamy group developed a practical protocol for the regioselective nitration of N-alkyl anilines using TBN under metal free conditions (Scheme 39b).48 In both reactions, the nitro radical was generated from the homolysis of tert-butyl nitrite for the synthesis of N-nitrosoanilines, nitroanilines and N-nitroso-nitroanilines. It was interesting that three different products could be generated from N-alkyl anilines under different reaction conditions.
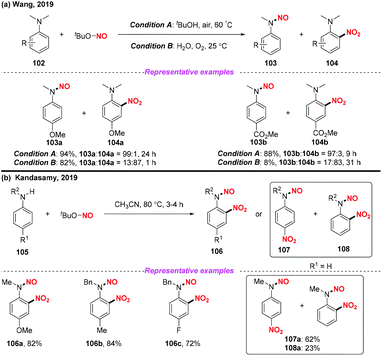 |
| | Scheme 39 Metal-free nitration of N-alkyl anilines. | |
2.7.2. Other nitro radical sources.
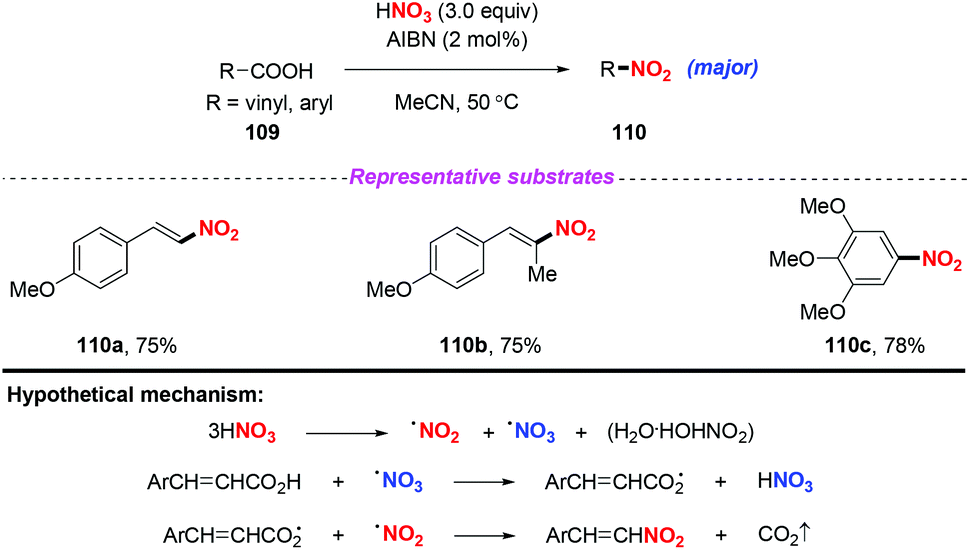
In 2002, Roy and co-workers found that nitric acid could be employed as the nitro radical source for the nitrodecarboxylation of alkenyl and aryl carboxylic acids, giving rise to nitrostyrenes and nitroarenes (Scheme 40).49 In this transformation, AIBN played a dual role as both a free radical initiator and a catalyst. A proposed mechanism for this transformation is depicted in Scheme 40. Initially, HNO3 could undergo thermal homolysis to generate NO2˙, NO3˙, and H2O·HONO2. Then, the combination of the nitro radical with the acyloxy radical followed by subsequent nitrodecarboxylation would furnish the desired product. Interestingly, in this reaction, the cheap and commercially available nitric acid was employed as the source of the nitro radical. It should be noted that the reaction could only tolerate electron-rich alkenyl and aryl carboxylic acids, resulting in limited substrate scopes.
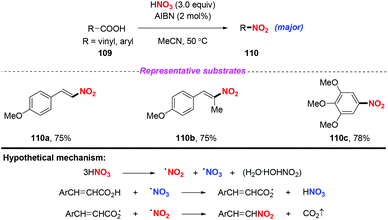 |
| | Scheme 40 AIBN-catalyzed nitrodecarboxylation of aromatic α,β-unsaturated carboxylic acids and ring-activated benzoic acids. | |
In the same year, the Koomen group developed an efficient and selective nitration of purine at the C-2 position using a mixture of tetrabutylammonium nitrate (TBAN) and trifluoroacetic anhydride (TFAA).50 In 2005, the same group presented an extensive NMR study to demonstrate that the reaction occurred in a three-step process (Scheme 41).51 Initially, the homolytic cleavage of CF3CO2NO2 generated the trifluoroacetoxyl radical and nitro radical. The subsequent addition of the reactive CF3CO2˙ to purine 111 at the C-8 position would form a highly delocalized radical 41A. Then the combination of 41A with the nitro radical would give rise to intermediate 41B followed by the elimination of CF3CO2H, leading to the nitrated product 112.
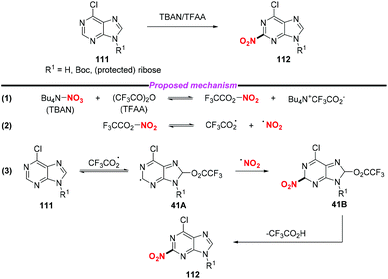 |
| | Scheme 41 Metal-free selective purine C2 nitration reaction using a mixture of TBAN and TFAA. | |
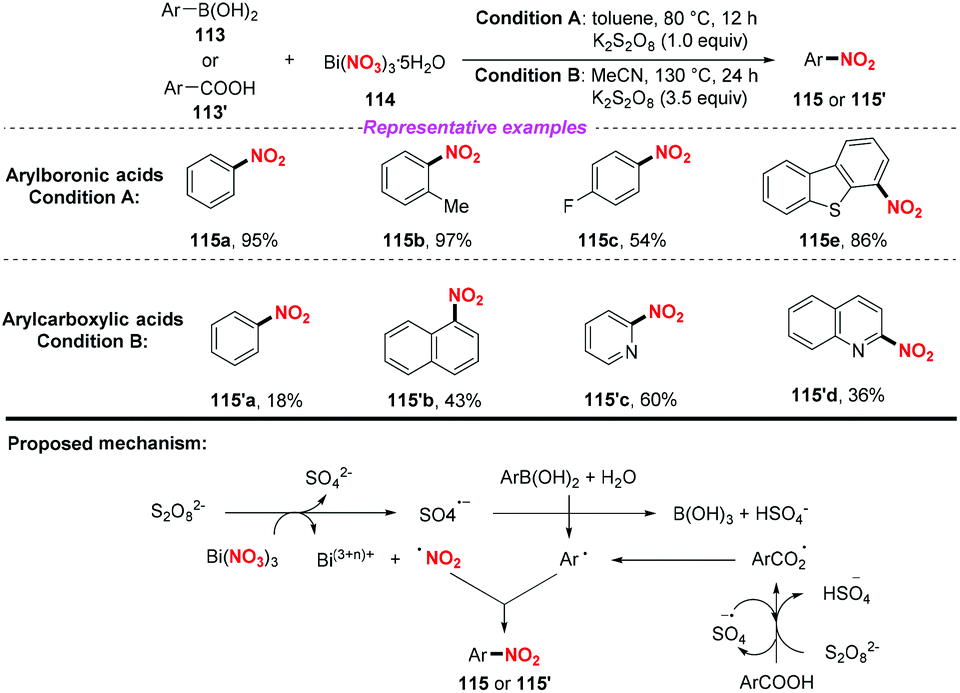
In 2015, the Maiti group disclosed a novel and one pot synthetic method of ipso-nitration of arylboronic acids by using bismuth nitrate as a nitro radical source.52a In the conversion of arylboronic acid, this protocol exhibited a broad substrate scope and good functional group tolerance, affording various nitroarenes in good to excellent yields. In 2019, the same group demonstrated that aromatic carboxylic acids could be converted into nitroarenes by using Bi(NO3)3/K2S2O8 under acid-free conditions.52b However, compared to their previous work, this method suffered from high temperature, narrow substrate scope and poor yield. On the basis of the control experiments, a radical–radical coupling pathway was proposed to be involved in both reactions (Scheme 42).
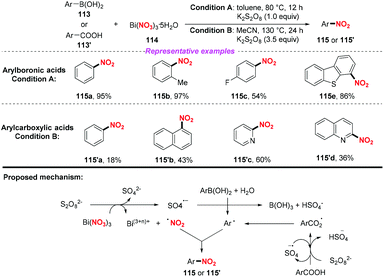 |
| | Scheme 42 Metal-free ipso-nitration of arylboronic acids and aromatic carboxylic acids. | |
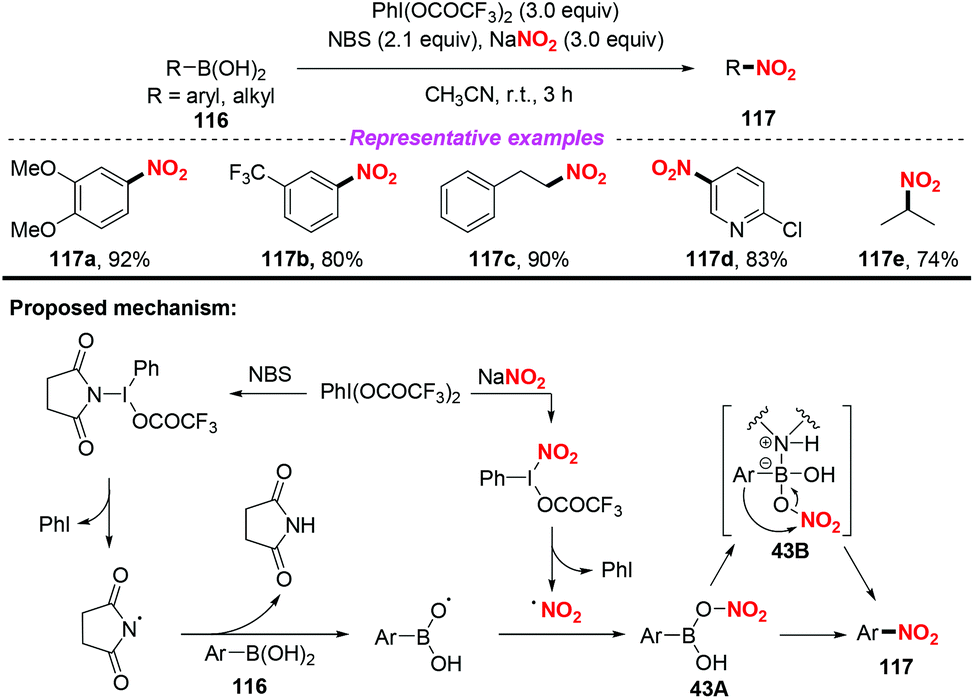
In 2015, Goswami and co-workers developed a novel transition metal free method for the oxidative ipso-nitration of organoboronic acids, which was the complement of Maiti's work (Scheme 43).53 In this transformation, a combination of PIFA-NBS and sodium nitrite was used as the nitro source. Based on the control experimental results, oxygen-centred radicals could be generated from organoboronic acids in the presence of NBS and PIFA. On the other hand, the nitro radical was released through one electron oxidation of NaNO2 by PIFA, which reacted with the oxygen-centred radical to form the stable species 43A. Finally, nitro transfer to the aryl group via 1,3-aryl migration would enable the coordinated species 43B to afford nitroarenes 117. This reaction featured good to excellent yields and broad substrate scope, which could realize the nitration of arylboronic acids, alkenylboronic acids and alkylboronic acids.
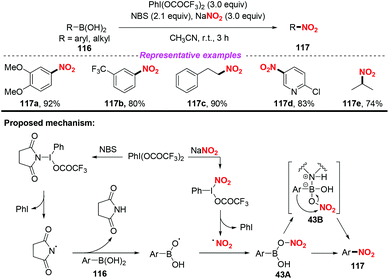 |
| | Scheme 43 Metal-free oxidative ipso-nitration of diversely functionalized organoboronic acids. | |
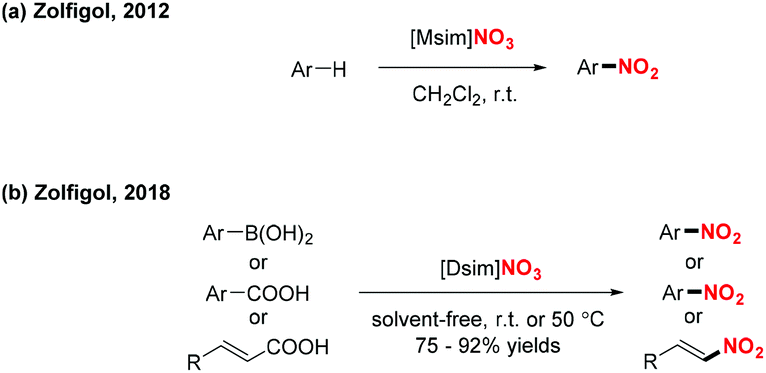
In 2012 and 2018, the group of Zolfigol designed two novel Brønsted imidazolium nitrates ([Msim]-NO3 and [Dsim]-NO3), which served as an ionic liquid and a nitrating agent for the efficient nitration of aromatic compounds, arylboronic acids, α,β-unsaturated acids and benzoic acid derivatives, respectively (Scheme 44).54 These transformations exhibited a broad substrate scope and good functional group tolerance, affording various nitro-containing products in good to excellent yields.
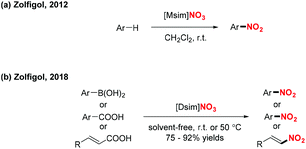 |
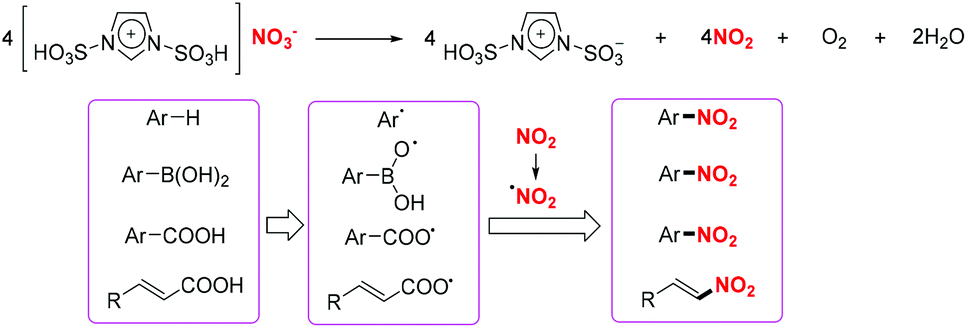
| | Scheme 44 Ionic liquid as nitrating reagent for the nitration of arylboronic acids, α,β-unsaturated acids and benzoic acid derivatives. | |
A plausible mechanism for these transformations involving the release of nitrogen dioxide from [Msim]NO3 was proposed. According to this, the combination of aryl radicals or oxygen-centred radicals with nitro radicals leads to the synthesis of nitro-containing compounds (Scheme 45).
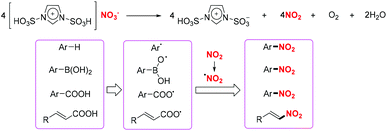 |
| | Scheme 45 Proposed mechanism for the synthesis of nitroolefins and nitroarenes. | |
In 2019, Zou and co-workers developed a mild and selective radical nitration/nitrosation of indoles with NaNO2 using K2S2O8 as an oxidant (Scheme 46).55 The reaction worked well under thermal conditions to provide 3-nitro- or 3-nitrosoindoles, showing broad substrate scope compatibility and operational simplicity. A possible mechanism was depicted in Scheme 46. Under thermal conditions, sulphate radical anions were generated from K2S2O8via homolytic decomposition, which subsequently reacted with NaNO2 to produce a nitro radical. The nitro radical generated might exist in the dimeric forms 46A, 46B and 46C. The nitrosyl N-atom of 46B or 46C would be attacked by the nucleophilic indole to give the nitroso compound 46D. The 3-nitrosoindole 46D was remarkably stable when the 2-phenyl substituted indole substrate was protected by a methyl group. Nevertheless, if the indole was not protected, 46D would be further oxidized to the 3-nitroindoles 119via oxime 119′.
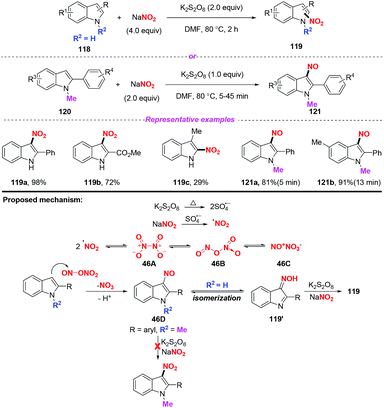 |
| | Scheme 46 Radical nitration or nitrosation of indoles by the NaNO2/K2S2O8 system. | |
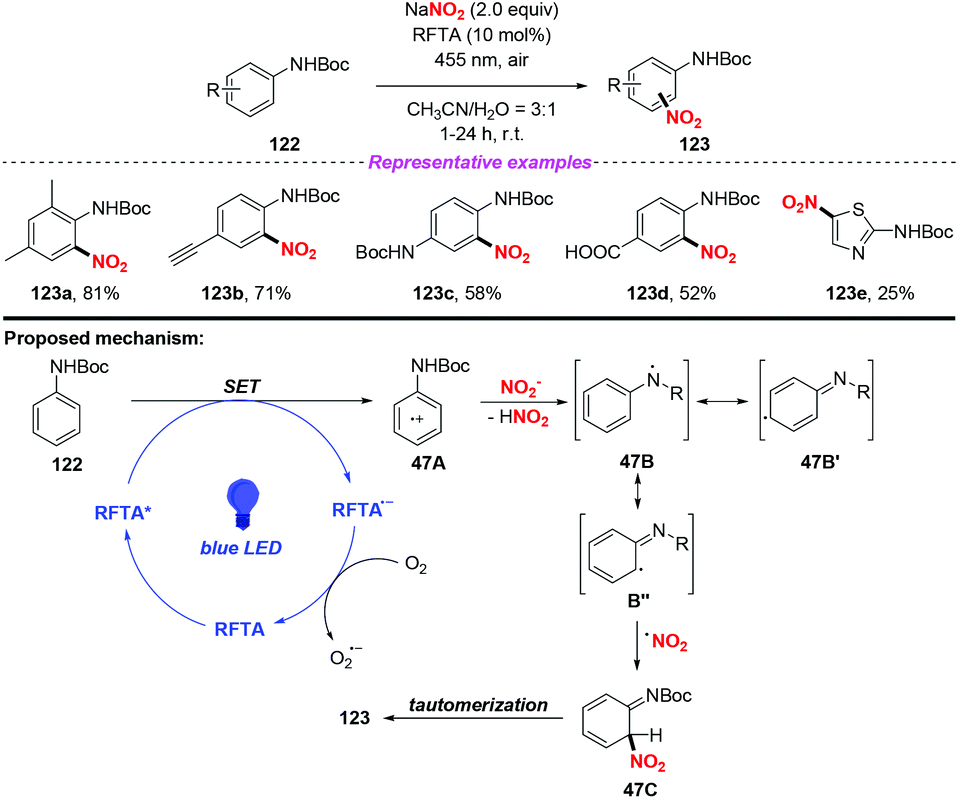
The photocatalytic nitration of protected anilines by using riboflavin tetraacetate as an organic photoredox catalyst was achieved by König and co-workers (Scheme 47).56 In this reaction, NaNO2 was utilized as the nitro radical source under ambient conditions, providing various nitroanilines in moderate to high yields without the need for acid or stoichiometric oxidation agents. A possible mechanism of the photonitration was proposed as follows: initially, the radical cation 47A was generated from aniline 122via a single-electron transfer of the photoexcited catalyst, followed by the loss of a proton to form stabilized radical 47B′′. Then, the nitro radical could react with 47B′′ to give 47C, which underwent rearomatization to afford the para- and ortho-regioisomeric substitution product 123. This protocol exhibited mild reaction conditions, broad substrate scope and good functional group compatibility, affording various nitroaromatic in good yields. However, when the aromatic ring was replaced by the heterocyclic ring, such as thiazole, the reaction efficiency was remarkably decreased.
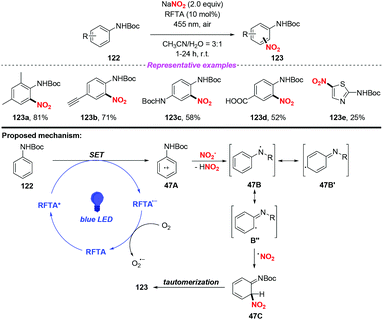 |
| | Scheme 47 Visible-light-mediated nitration of protected anilines. | |
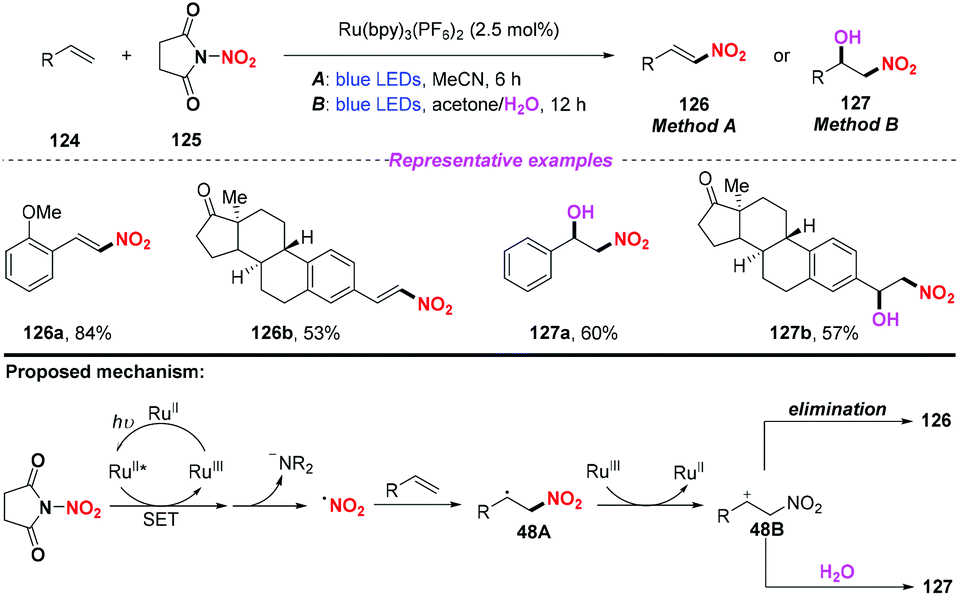
Recently, the group of Katayev designed and developed a practical and bench-stable succinimide-derived nitrating reagent for C–NO2 bond formation in the presence of a photocatalyst under visible light (Scheme 48).57 This protocol not only enabled the direct nitration of the sp2 C–H bond of alkenes, but also provided rapid access to valuable nitrohydrin building blocks and functionalized isoxazole-motifs in one-step. According to the proposed mechanism, the generation of the nitro radical through photoinduced single-electron transfer and subsequent N–N bond fragmentation of the reagent were the key to promote the reaction.
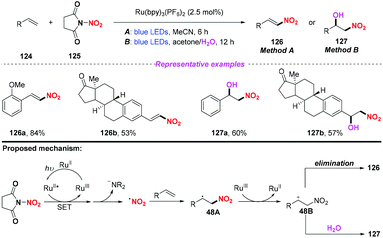 |
| | Scheme 48 Visible-light-mediated nitration of olefins. | |
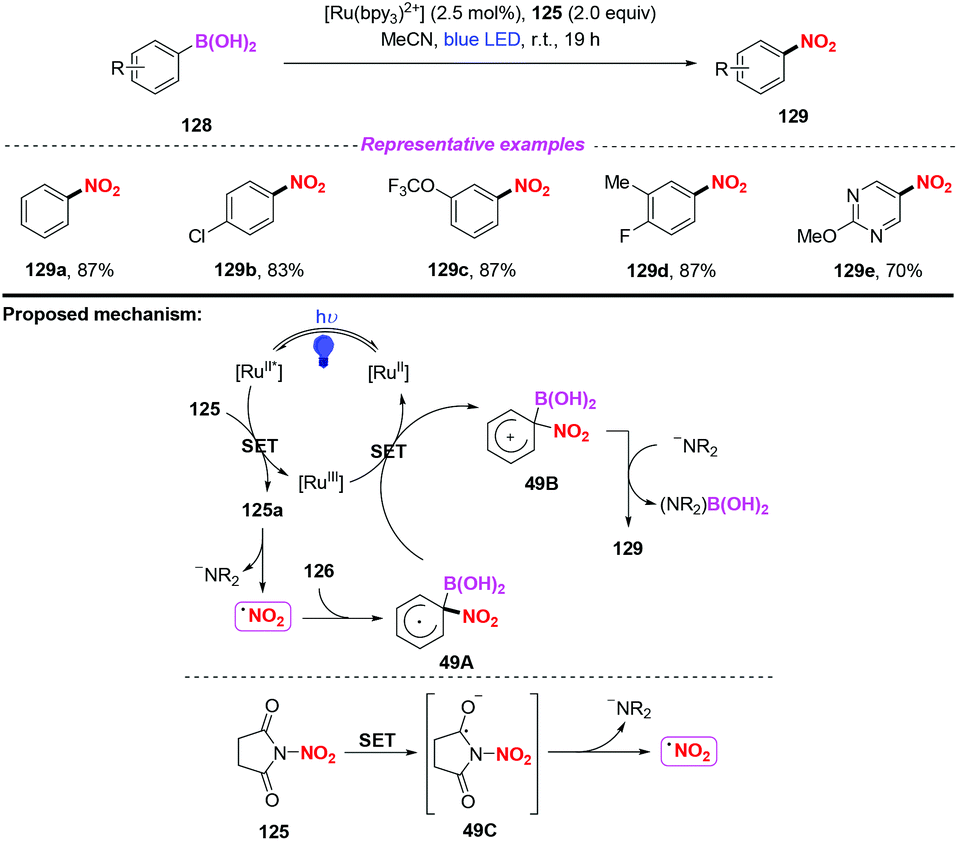
After this, the same group further developed a photocatalytic and metal-free ipso-nitration of aryl- and heteroarylboronic acids 128 by using this newly nitrating reagent (Scheme 49).58 The reaction featured operational simplicity, mild conditions, and excellent functional group compatibility, delivering nitro(hetero)aromatic compounds 129 in good yields. Similar to their previous report, the ipso-nitration process included the formation of the NO2 radical, radical addition, single-electron transfer oxidation and deboronylation.
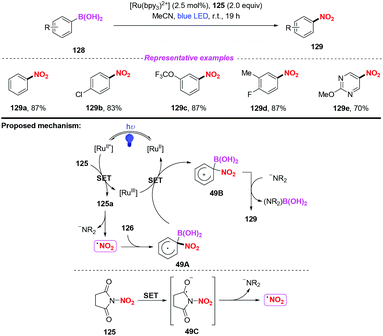 |
| | Scheme 49 Visible-light-mediated nitration of heteroarylboronic acids. | |
3. Nitro group participated in radical reactions
3.1. Fe-Catalyzed or mediated transformations
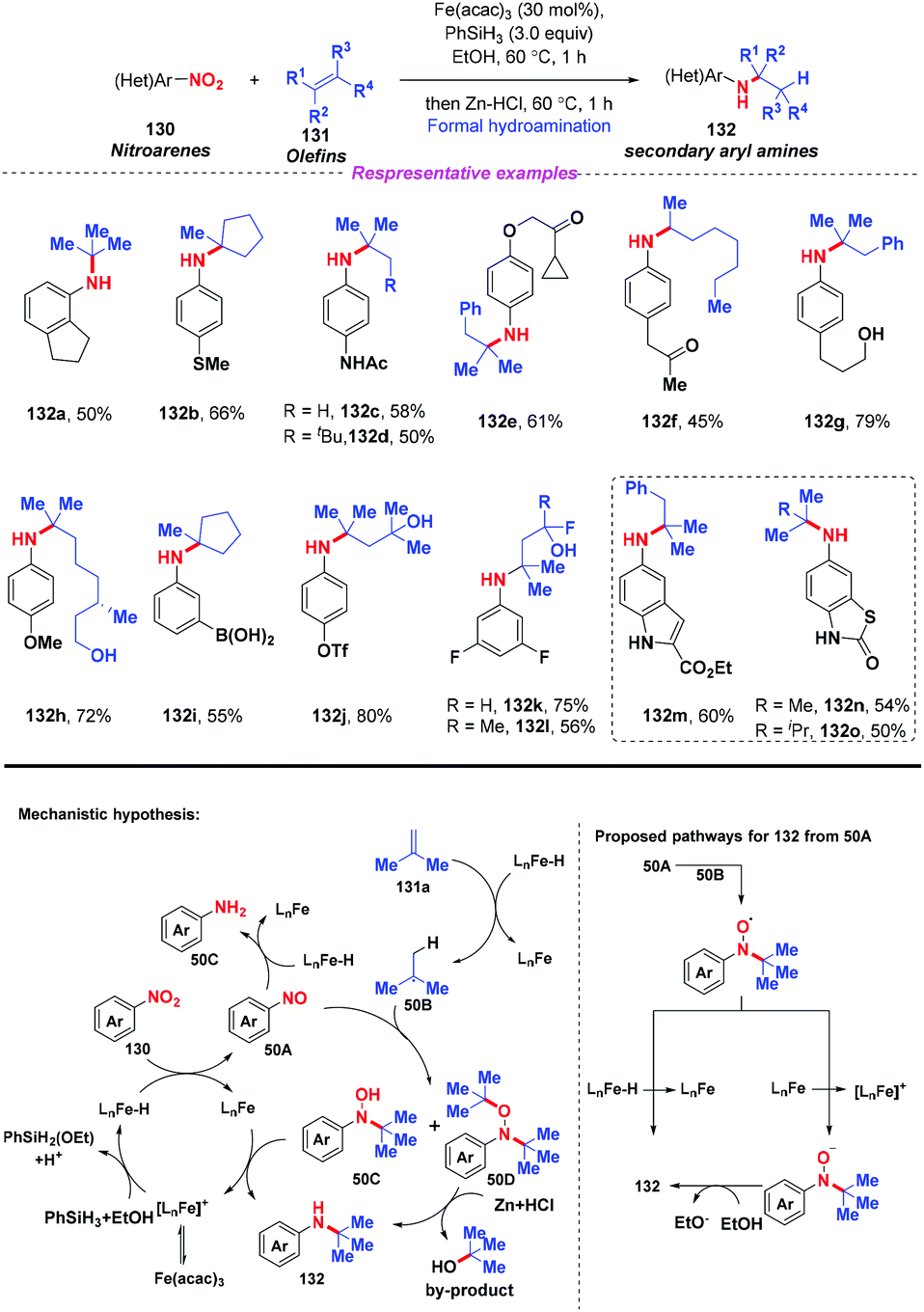
Given the prevalence of amines in pharmaceutical chemistry and some limitations of the current method for amine synthesis, Baran and co-workers59 developed a practical method for direct C–N bond formation between nitro(hetero)arenes and simple olefins in 2015 (Scheme 50). In this pioneering report, the authors employed aliphatic olefins as coupling-partners, nitroarenes as radical pro-receptors, cheap silane and zinc metals as reducing agents and abundant iron salts as catalysts, furnishing various secondary amines in moderate to good yields. A possible mechanism for the formation of hydroamine products was described as follows: in accordance with previous studies,60 iron hydride species was initially formed in situ and it reacted with olefin 131 to give the alkyl radical 50B. Additionally, the iron hydride species would reduce the nitroarenes to the corresponding nitrosoarenes, which could either undergo further reduction to the aniline side product 50C, or coupling with either one or two equivalents of alkyl radical 50B to afford intermediate 50C or 50D, respectively. Finally, the reduction of hydroxylamine 50C by the LnFe species would release the desired secondary amine 132 and regenerate the iron catalyst. The desired product 132 might also be generated by the reduction of intermediate 50D in the presence of Zn and HCl.
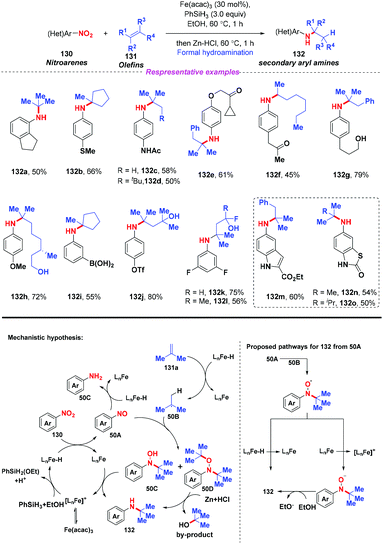 |
| | Scheme 50 Iron-mediated olefin hydroamination of nitroarenes. | |
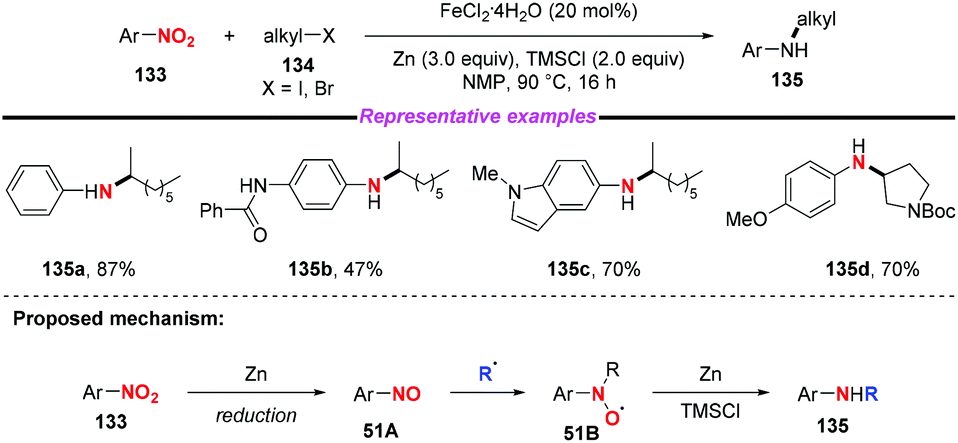
Subsequently, Hu and co-workers developed an iron-catalyzed reductive coupling of nitro(hetero)arenes with alkyl halides to synthesize (hetero)aryl amines using a FeCl2·4H2O/TMSCl system (Scheme 51).61 This approach tolerated various functional groups. Remarkably, primary, secondary and tertiary alkyl halides could be coupled, showing broad substrate scope compatibility. On the basis of mechanistic studies, it was suggested that the addition of alkyl radicals to nitrosoarene was the major reaction pathway.
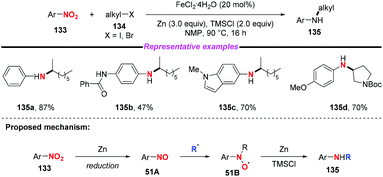 |
| | Scheme 51 Iron-catalyzed reductive coupling of nitro(hetero)arenes with alkyl halides. | |
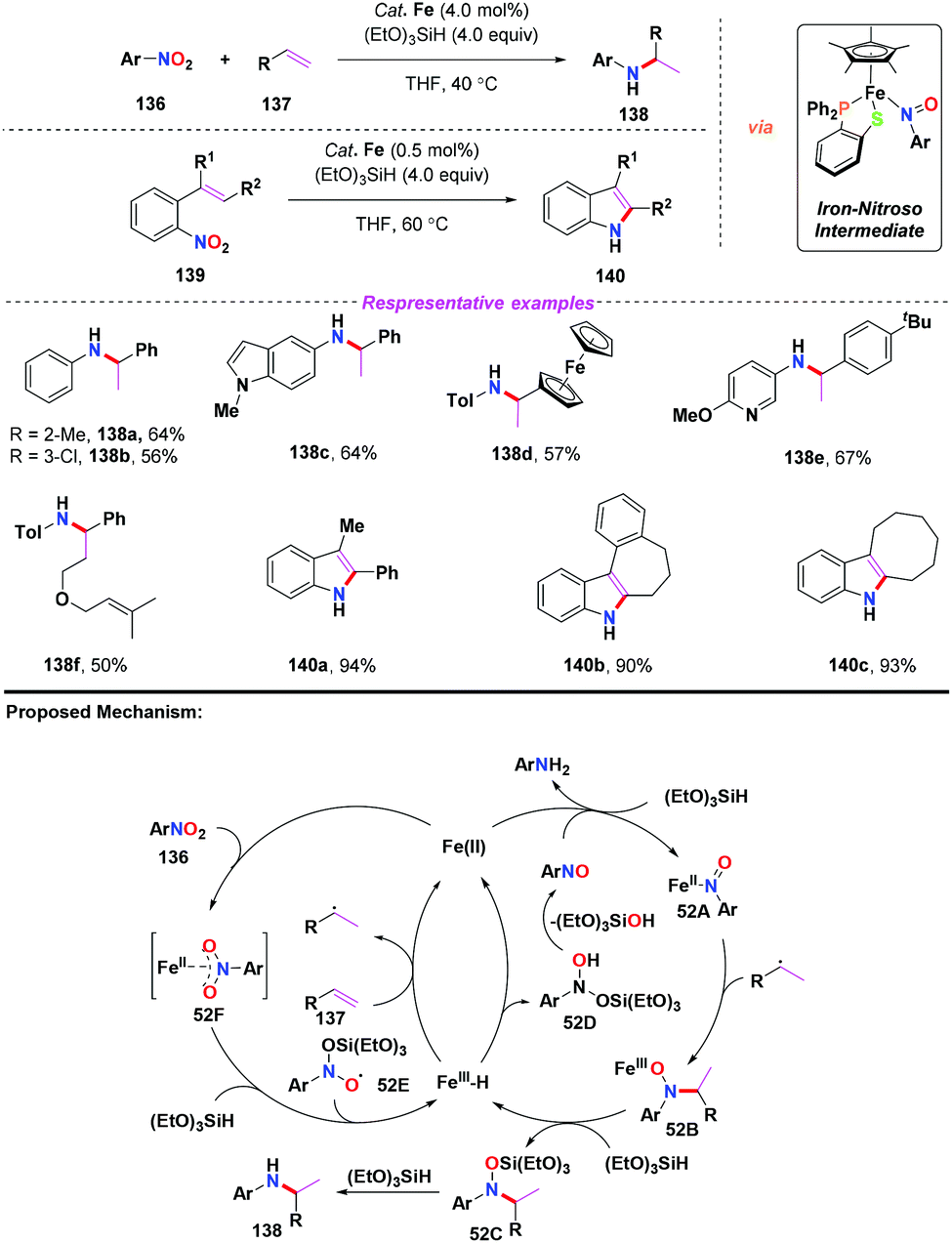
Recently, Wang and co-workers further applied the iron(II)/(EtO)3SiH system for the reductive coupling of nitroarenes with olefins via an iron-nitroso intermediate (Scheme 52).62 By employing a single half-sandwich iron(II) compound as the catalyst, a variety of branched amines and indole derivatives were obtained in moderate to good yields under mild conditions. To gain more insights into the mechanism, several control experiments were carried out to confirm the formation of radical intermediates. Accordingly, similar to Baran's work, a radical mechanism was proposed for the C–N bond coupling (Scheme 52). Compared with Baran's work, this reaction worked well via the intramolecular reductive coupling process to enable the synthesis of indoles in excellent yields.
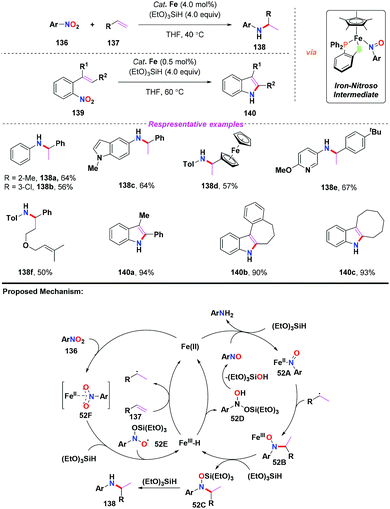 |
| | Scheme 52 Iron-catalyzed olefin hydroamination of nitroarenes. | |
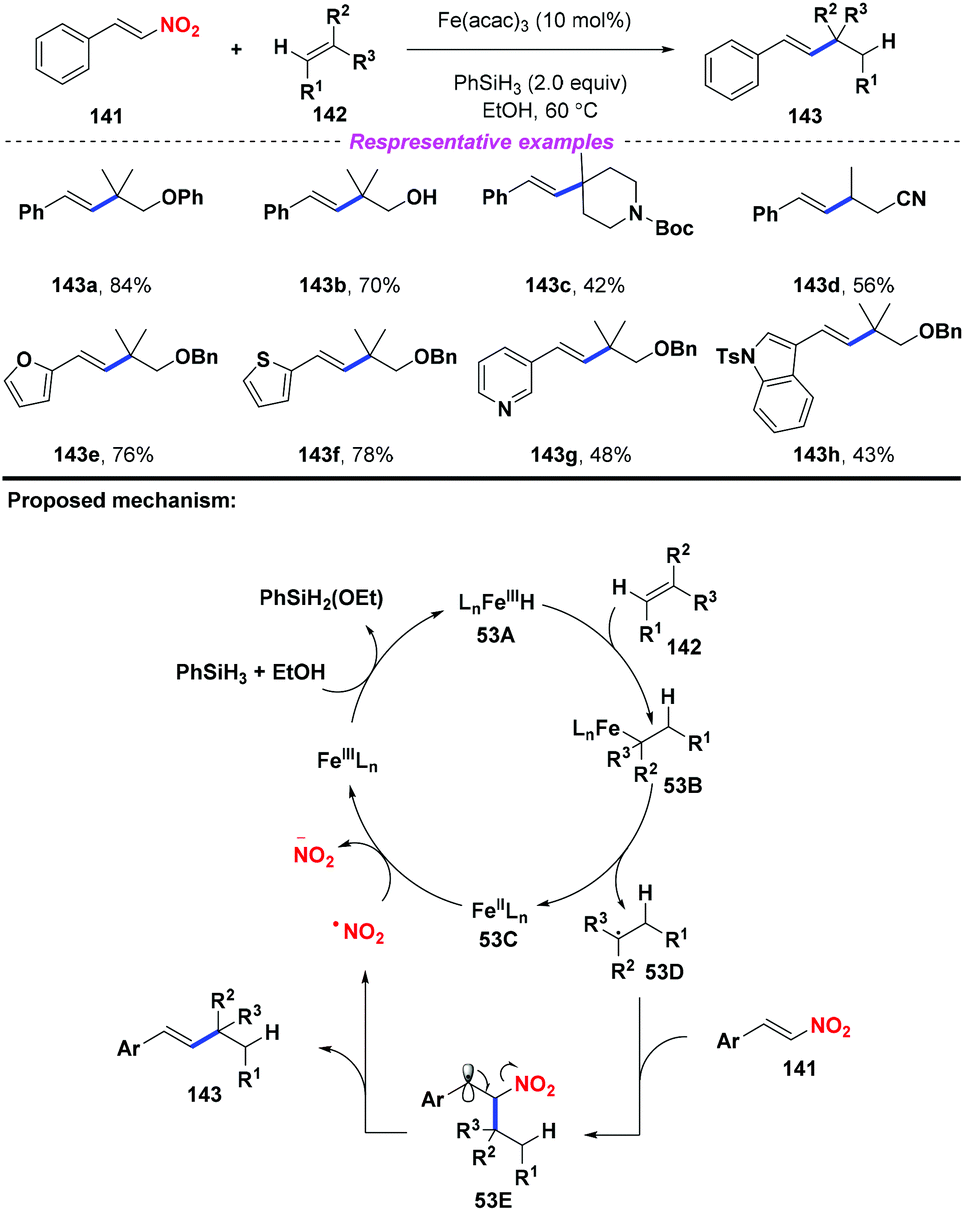
In 2015, Cui and co-workers developed an iron-catalyzed reductive coupling of unactivated alkenes with nitroalkenes (Scheme 53).63 According to the proposed mechanism, initially, the Fe(III)-catalyst was converted into Fe hydride species 53A in the presence of silane and alcohol. Then, the addition of 53A to alkene 142 would produce intermediate 53B, followed by disintegration to give Fe(II) species 53C and alkyl radical 53D. The resulting alkyl radical 53D was captured by nitroalkene 141 to generate the β-nitro radical 53E. Finally, the continuous elimination of 53E would furnish the alkylated styrene product 143 with the release of a nitro radical, which would undergo reduction by Fe(II) species 53C to complete the catalytic cycle. This reaction exhibited good functional group compatibility, and many sensitive groups such as hydroxyl, cyano, ether, and heterocycle groups were compatible in this transformation.
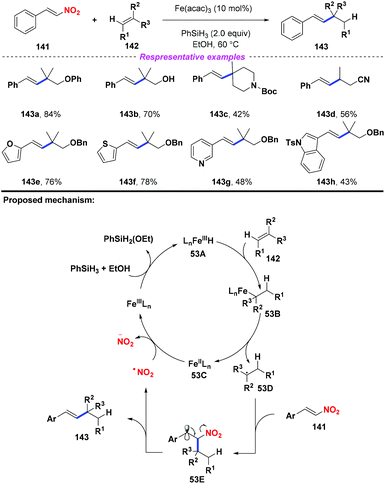 |
| | Scheme 53 Fe-Catalyzed reductive coupling of unactivated alkenes with nitroalkenes. | |
3.2. Ni-Catalyzed transformations
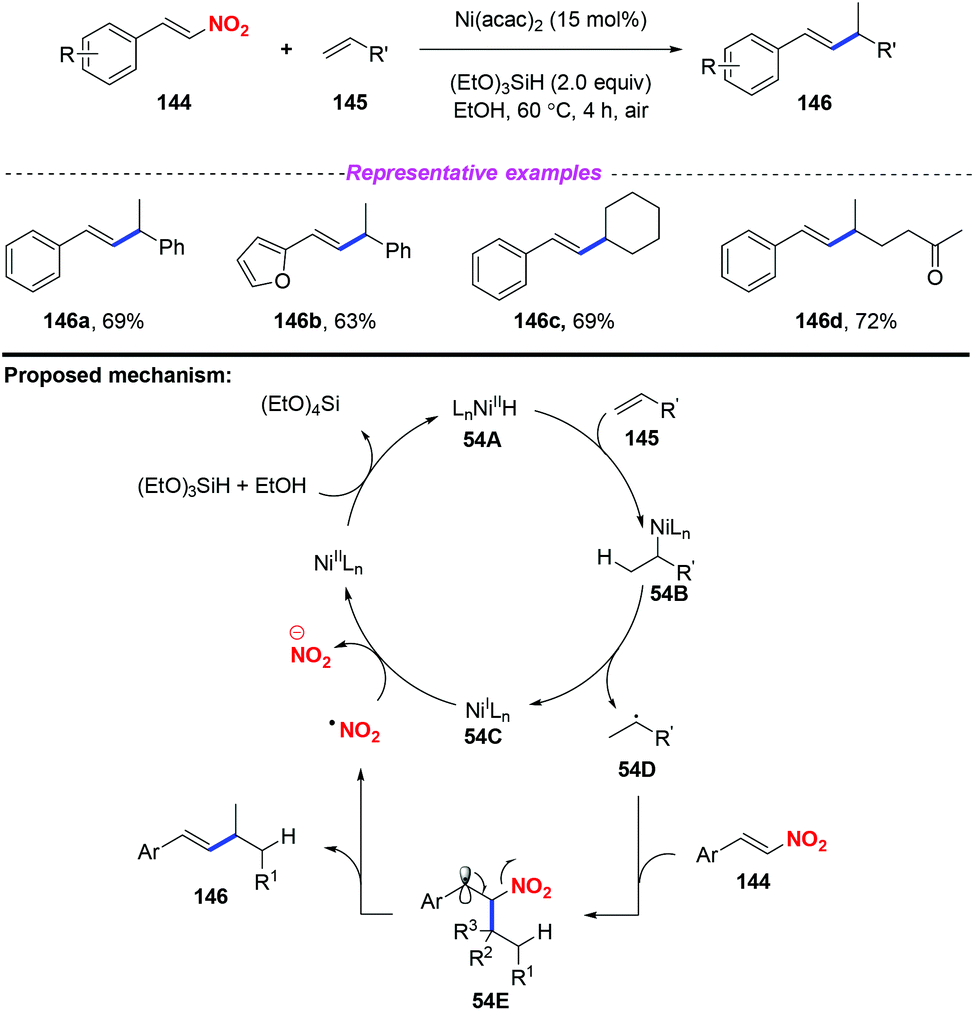
Given the importance of alkylated styrenes in natural products and pharmaceuticals, the group of Wang developed a nickel-catalyzed denitrated coupling reaction of nitroalkenes with aliphatic and aromatic alkenes in 2016 (Scheme 54).64 On the basis of mechanistic investigations and previous reports, a plausible mechanism for this reaction was proposed, as depicted in Scheme 54. Initially, Ni hydride 54A was formed from Ni(II) and (EtO)3SiH, which was regioselectively added to alkene 145 and resulted in intermediate 54B. The dissociation of 54B delivered Ni(I) species 54C and alkyl radical 54D, which were trapped by nitroalkene 144 to form the radical 54E. Subsequently, the elimination of 54E would give the alkylated styrene product 146 along with the release of a nitro radical.
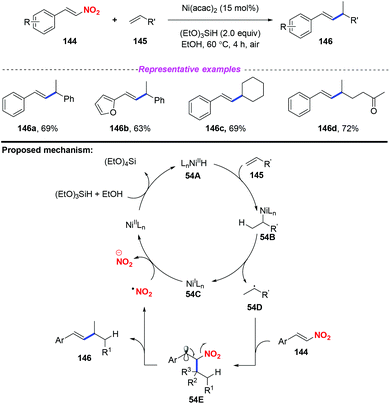 |
| | Scheme 54 Nickel-catalyzed denitrated coupling reaction of nitroalkenes with aliphatic and aromatic alkenes. | |
3.3. Ag- or Mn-Catalyzed transformations
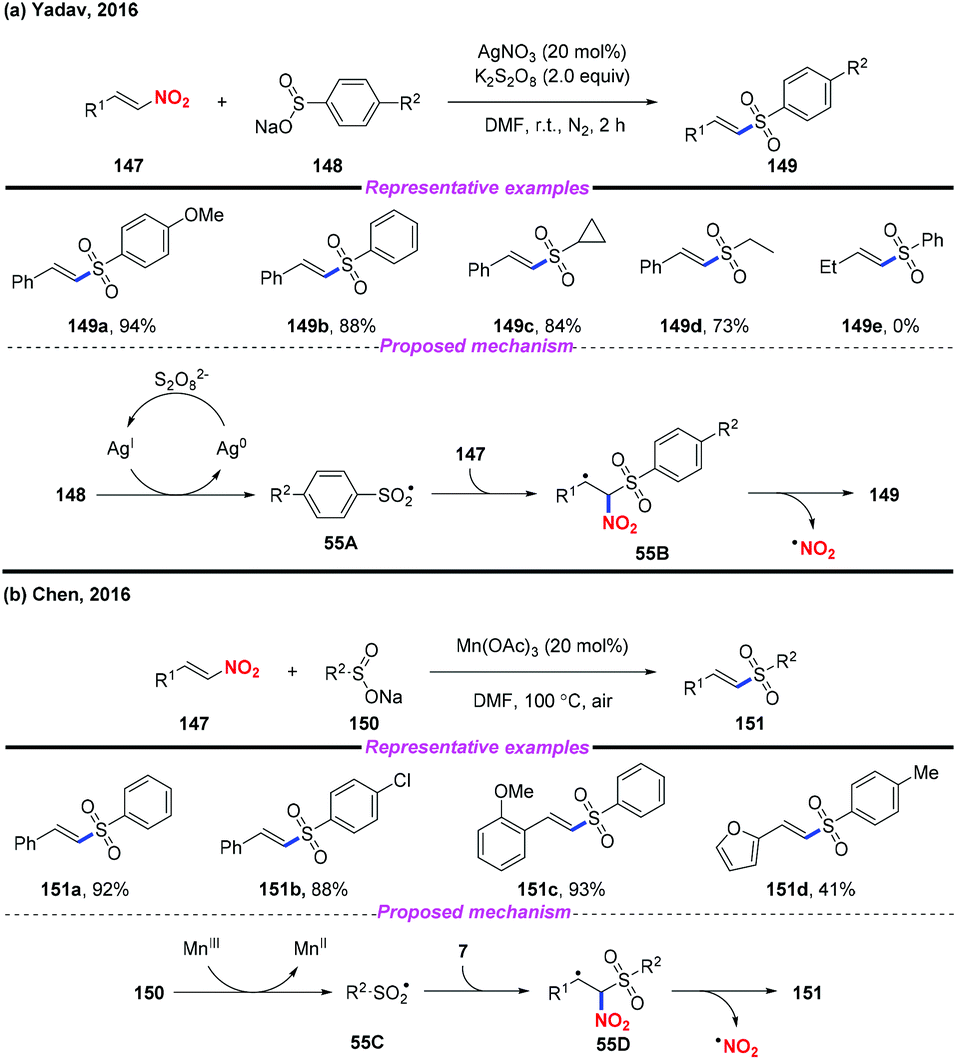
Vinyl sulfones are valuable building blocks in synthetic organic chemistry, and have been widely used in Michael addition and cycloaddition reactions. In 2016, Yadav and co-workers reported a silver-catalyzed approach for the highly stereoselective synthesis of (E)-vinyl sulfones by denitrative coupling of nitroolefins and sodium sulfinates in the presence of K2S2O8 (Scheme 55a).65 Additionally, Chen and co-workers described a Mn(III)-mediated regioselective synthesis of (E)-vinyl sulfones from nitroolefins and sodium sulfinates at the same time (Scheme 55b).66 According to the mechanism proposed by both groups, a sulfonyl radical was initially generated from sodium sulfonate, followed by radical addition and elimination sequence to give the desired product.
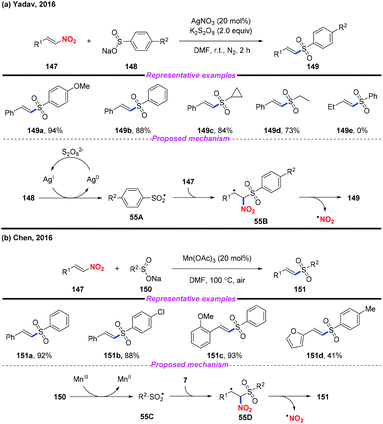 |
| | Scheme 55 Silver-catalyzed coupling reaction of nitroolefins with sodium sulifinates. | |
3.4. Under transition metal free conditions
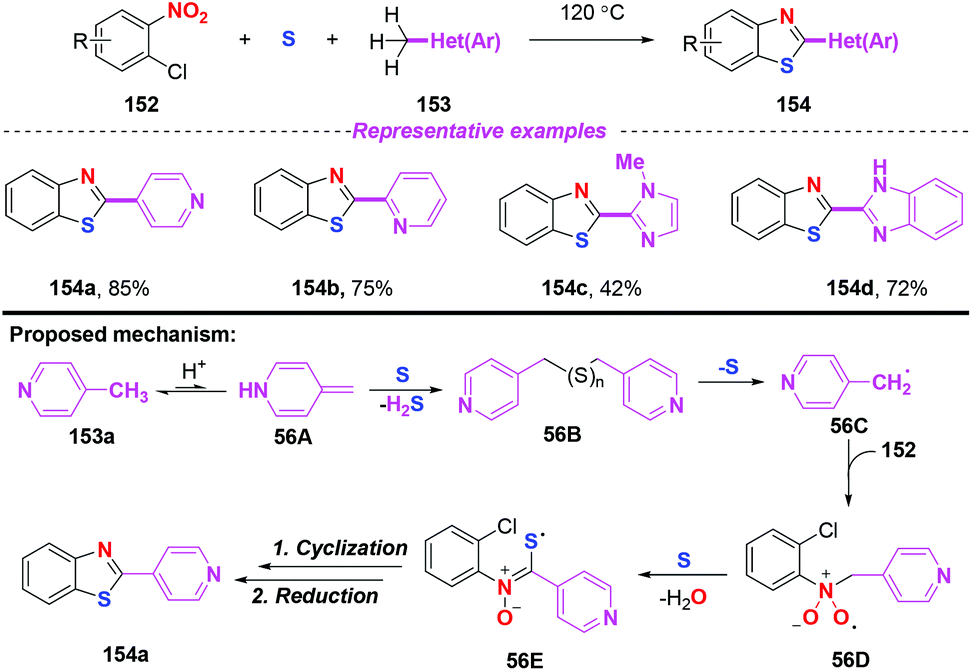
In 2013, a straightforward and atom economical approach to 2-hetarylbenzothiazoles starting from 2-halonitroarenes, methylhetarenes, and elemental sulfur was achieved by Nguyen and co-workers (Scheme 56).67 By the combination of 4-picoline and sulfur, benzyl radicals were generated under mild conditions without the need for additional radical initiators. As disclosed by their previous work, the mechanism for the outcome involved single electron transfer, radical addition and cyclization. In particular, sulfur-centered radical 56E was generated from nitro radical species 56D and elemental sulfur via a single electron transfer process.
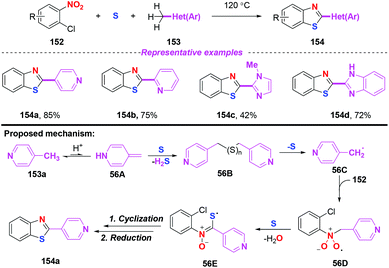 |
| | Scheme 56 Synthesis of 2-hetarylbenzothiazoles from 2-halonitroarenes, methylhetarenes, and elemental sulfur. | |
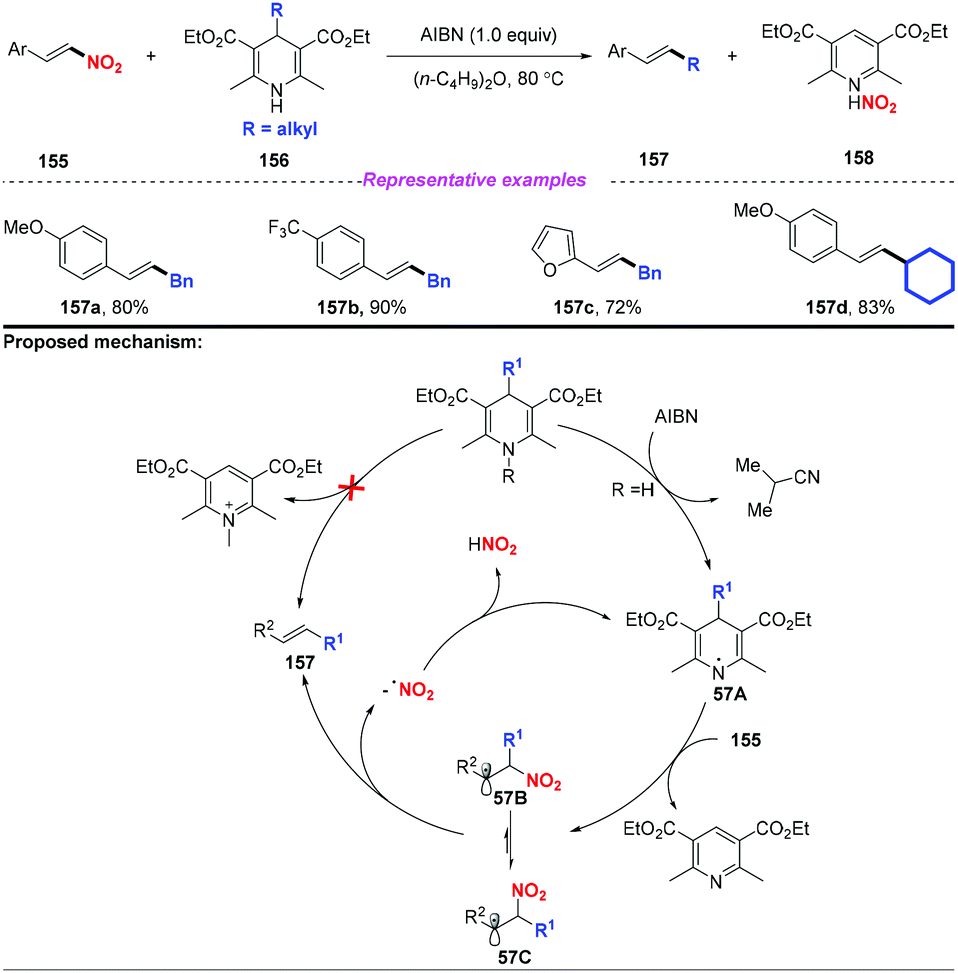
Subsequently, Tang and co-workers disclosed an efficient alkyl transfer reaction that enabled the formation of C–C bonds through C–C bond cleavage (Scheme 57).68 The scope of this reaction was quite broad, and a range of alkyl substituted Hantzsch esters were applied as alkylation reagents to afford nitroolefins in good yields. The proposed mechanism for this transformation was considered to proceed through a free radical-chain pathway. Initially, the H radical is extracted from dihydropyridine 156 to generate the DHP radical 57A, followed by fragmentation to provide the alkyl radical. Subsequently, the alkyl radical was added to the nitroolefin, resulting in the formation of radical 57B or 57C interconverted by internal rotation. Finally, the resulting radical would undergo β-elimination to give the thermodynamically controlled product with the release of a nitro radical.
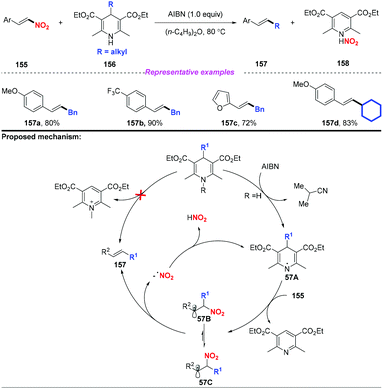 |
| | Scheme 57 AIBN-mediated coupling of nitroolefins with Hantzsch esters. | |
In 2018, Chen and co-workers reported a metal-free sulfonylation of nitroalkenes to furnish β,β-disubstituted nitroalkenes (Scheme 58).69 Accordingly, the putative mechanism revealed that the Lewis base-promoted equilibrium between nitroalkenes and allylic nitro compounds was the key to complete the process.
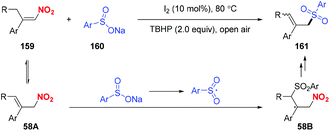 |
| | Scheme 58 I2-Catalyzed sulfonylation of nitroalkenes for the synthesis of allyl sulfones. | |
3.5. Under photocatalysis
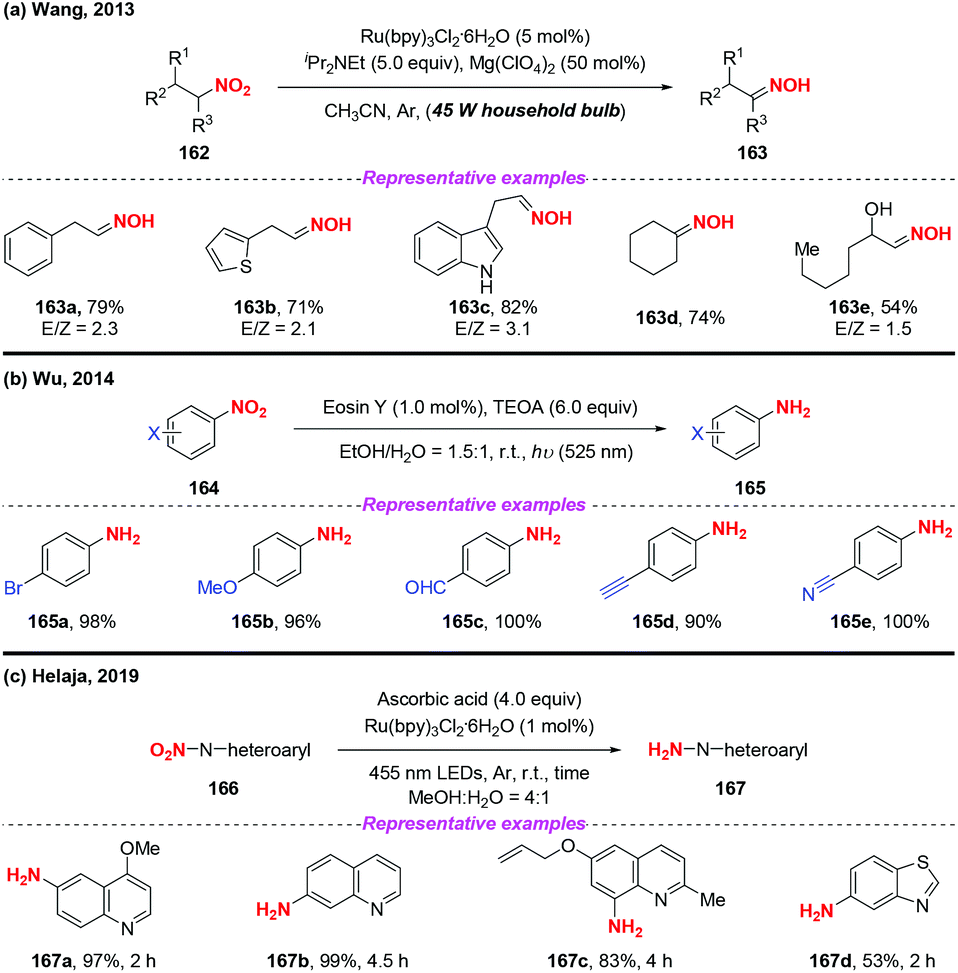
Over the past few years, visible-light-promoted organic transformations have made impressive progress in synthetic chemistry with the advantages of operational simplicity and mild conditions. In this case, the visible-light photocatalyzed reduction of nitro compounds has been independently developed by the groups of Wang70 (Scheme 59a), Wu71 (Scheme 59b) and Helaja72 (Scheme 59c). For instance, Wang and co-workers found that nitroalkanes could be smoothly reduced to the corresponding oximes under the synergistic effects of visible light irradiation. Moreover, Wu and co-workers employed visible light in nitroarene photoreduction using eosin Y as a photosensitizer and triethanolamine (TEOA) as a reductant. To address the issue that the eosin Y/TEOA photoreduction protocol was incompatible with nitroquinolines, Helaja and co-workers recently utilized a chemoselective strategy for reduction of nitro N-heteroaryls by employing ascorbic acid (AscH2) as hydrogen source.
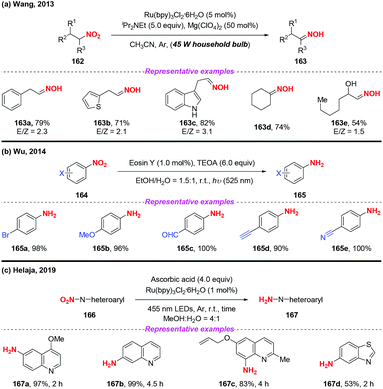 |
| | Scheme 59 Photoinduced reduction of nitro-containing compounds. | |
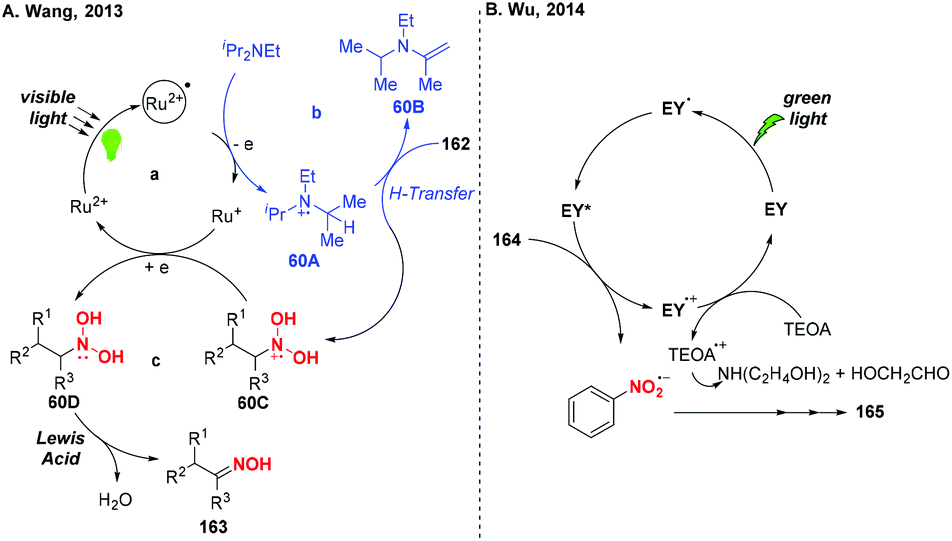
According to the mechanism proposed by Wang and co-workers, the amine radical cation 60A was initially formed via a single-electron transfer oxidation by the excited Ru(II)* and then underwent a hydrogen atom transfer process with substrate 162 to give a reduced cationic intermediate 60C (Scheme 60). Subsequently, 60C would proceed through a single-electron reduction by Ru(I) to produce intermediate 60D, which would undergo Lewis acid promoted dehydration to give oxime 163. In Wu's protocol, they suggested that the nitro group has much greater ability of accepting electrons than the other reducible groups, resulting in the selective reduction of nitrobenzenes under visible light.
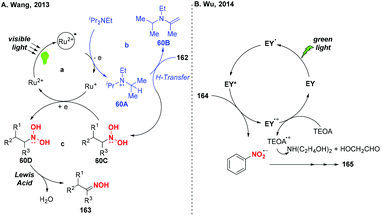 |
| | Scheme 60 Possible mechanism for the photoinduced reduction of nitroalkanes and nitroarenes. | |
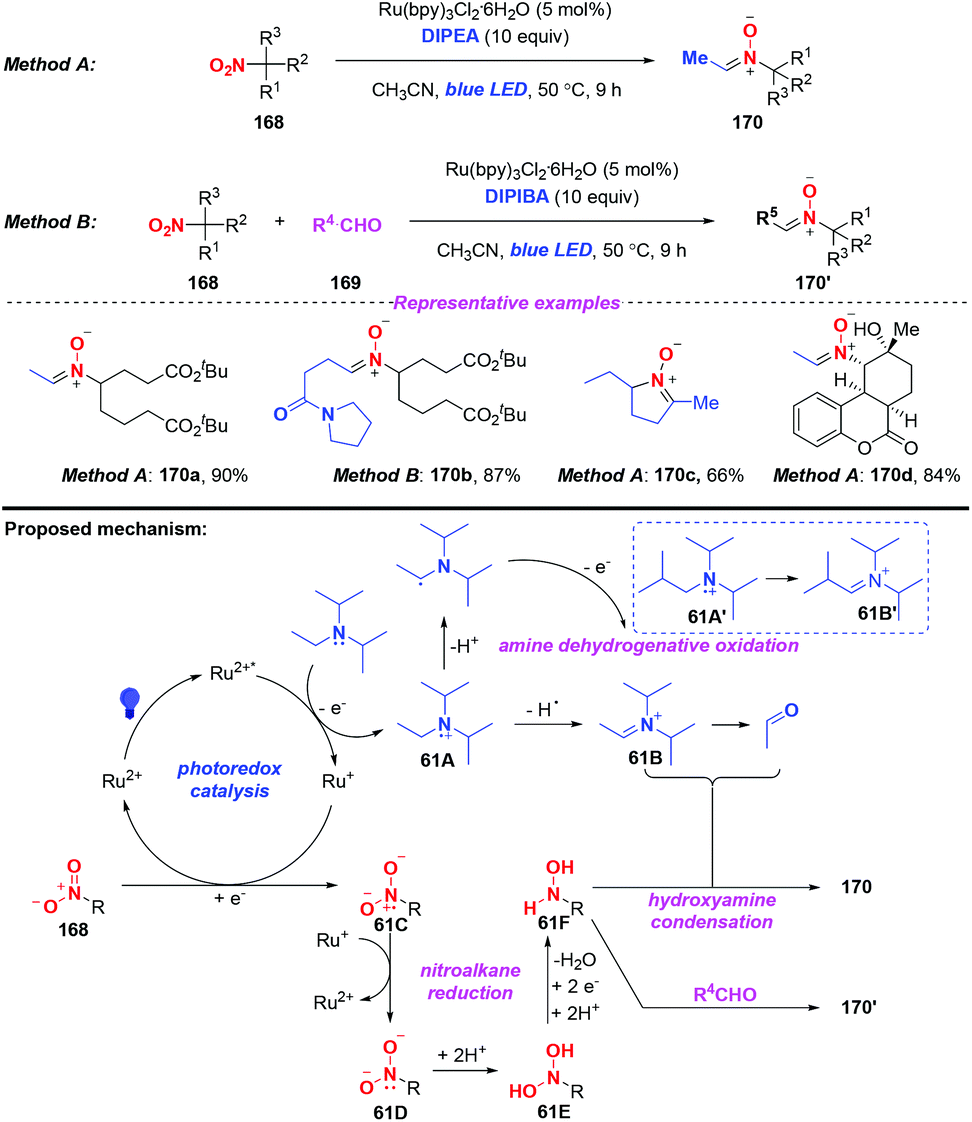
Nitrones are considered as versatile intermediates for the synthesis of naturally occurring compounds and bioactive molecules. In 2015, the group of Hong presented a concise visible-light-induced photocatalytic transformation of nitroalkanes to nitrones in good yields (Scheme 61).73 On the basis of the experimental results, a possible mechanism was proposed for this transformation. Initially, the excited Ru(bpy)32+* was quenched by DIPEA via a single-electron transfer oxidation to generate the radical cation of DIPEA. The radical cation 61A would be subsequently converted into iminium species 61B or acetaldehyde either through the release of a hydrogen radical or a proton. Alternatively, nitroalkane 168 would undergo a single-electron transfer reduction by Ru+ to afford 61C and 61D, respectively. The subsequent protonation of 11D would afford the dihydroxyamine 61E and the hydroxylamine species 61F. Finally, the alkylnitrone product 170 was produced through the condensation of the hydroxylamine species 61F with iminium species 61B or acetaldehyde. On the other hand, the hydroxylamine 61F would react with the external alkylaldehyde when DIPEA was replaced by DIPIBA, giving rise to the cross alkylated nitrone 170′ in good yield.
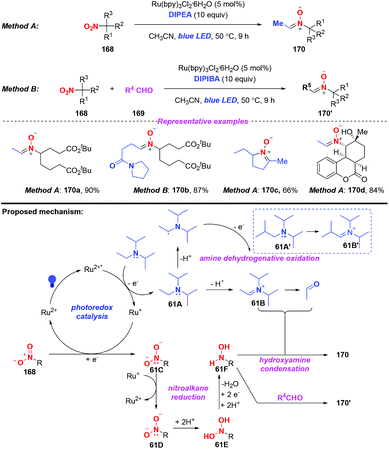 |
| | Scheme 61 Visible-light-promoted reduction of nitroalkanes to access nitrones. | |
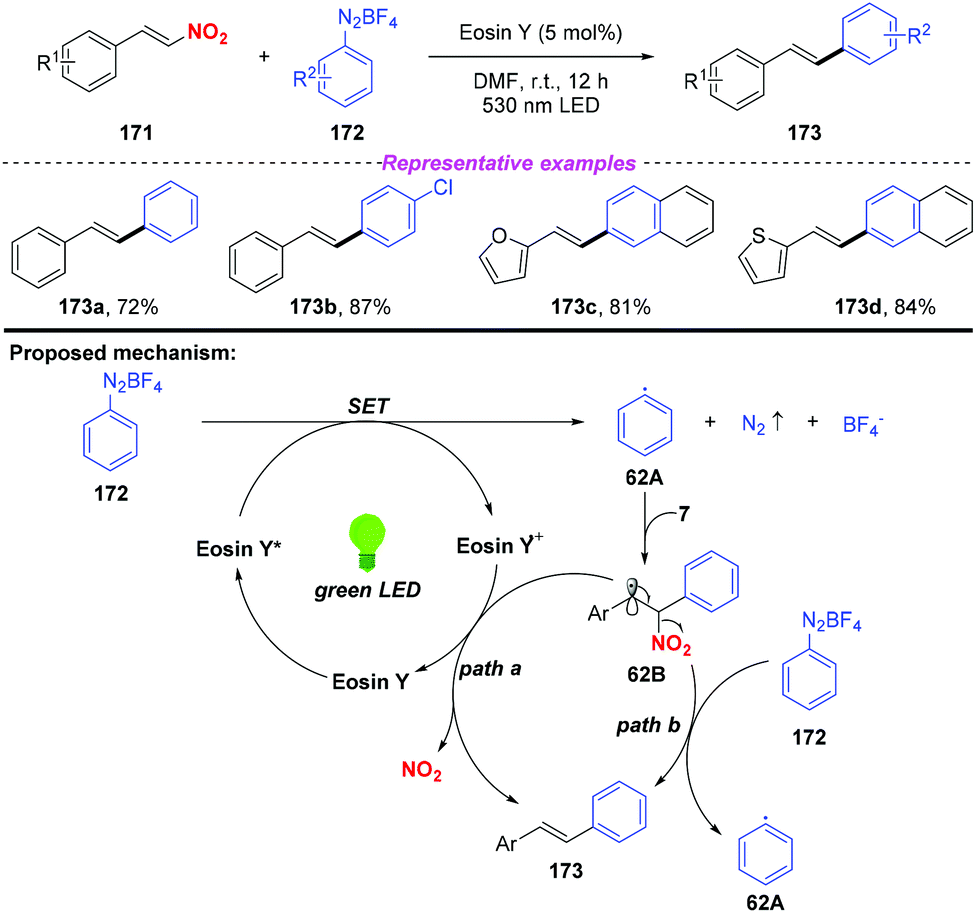
In 2016, Wang and co-workers reported a visible-light-induced cross-coupling of aryl diazonium salts with nitroalkenes for the synthesis of stilbene derivatives (Scheme 62).74 This reaction features high stereoselectivity, providing various (E)-stilbene compounds in good yields. To gain insight into the reaction mechanism, several mechanistic investigations have been conducted. According to the proposed mechanism, the aryl radical 62A was produced by a single electron transfer between the excited state of Eosin Y and aryl diazonium salt 172, which was trapped by nitroalkene 171 to give the β-nitro radical 62B. Subsequently, the radical intermediate 62B could be converted into coupling product 173 either through a single-electron oxidation by the Eosin Y radical cation or a radical chain-transfer process by the aryl diazonium salt 172 along with the elimination of the nitro group.
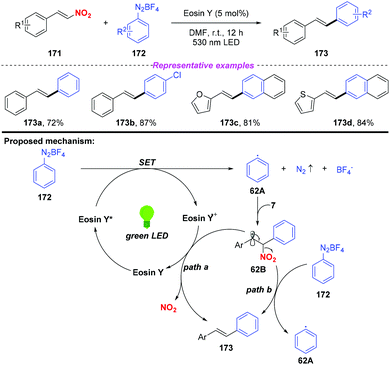 |
| | Scheme 62 Photoinduced synthesis of stilbene derivatives via cross-coupling of aryl diazonium salts with nitroalkenes. | |
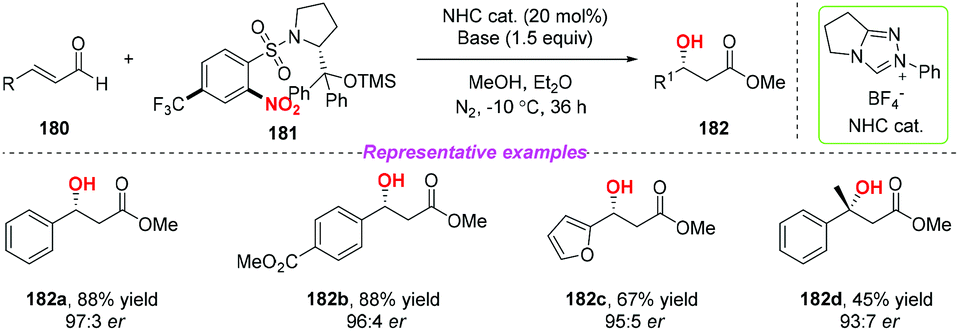
3.6. NHC-catalyzed transformations
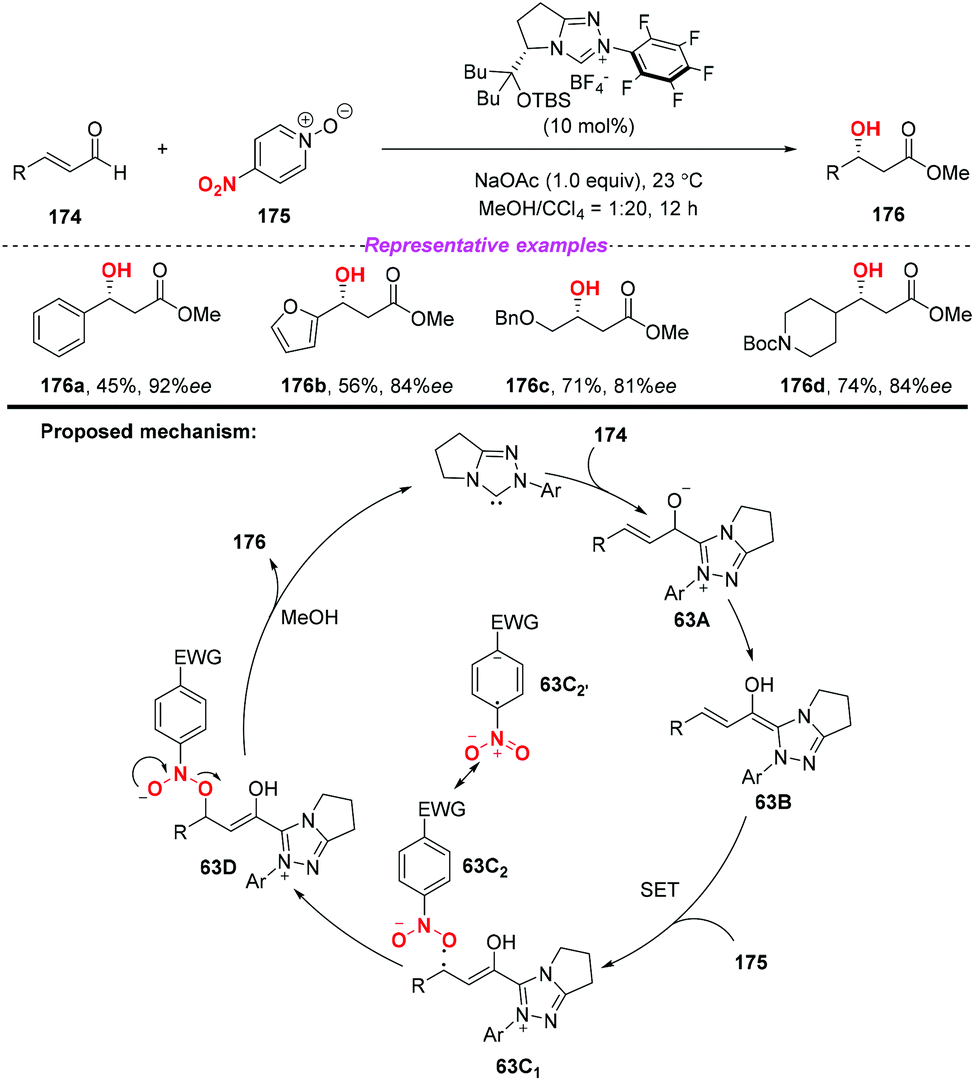
During the past two decades, N-heterocyclic carbene (NHC)-catalyzed reactions have achieved considerable progress in organic synthesis. In particular, it has been proved that NHCs could be applied to the development of single electron transfer (SET) reactions. In 2014, Rovis and co-workers developed the first example of the NHC-catalyzed oxidation reaction using electron-deficient nitrobenzenes as an oxidant (Scheme 63).75 A proposed mechanism for the reaction has been shown in Scheme 63. Initially, the combination of substrate 174 with a catalyst would give intermediate 63A, which subsequently underwent a single-electron oxidation by the nitrobenzene to afford Breslow-centered radical cation 63C1 and nitrobenzene-derived radical anion 63C2. After this, the cross-coupling of a homoenolate-centered radical and an O-centered radical led to species 63D. Finally, NHC-bound alkoxide 63D would react with MeOH to deliver the product 176 and release the catalyst.
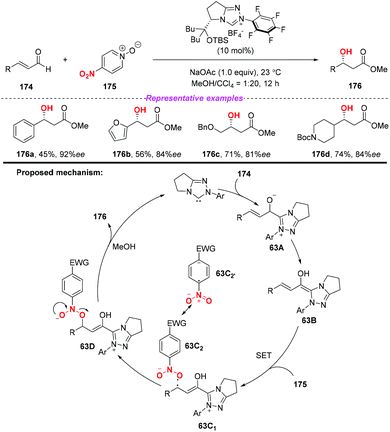 |
| | Scheme 63 NHC-catalyzed β-hydroxylation of enals using nitroarenes as an oxidant. | |
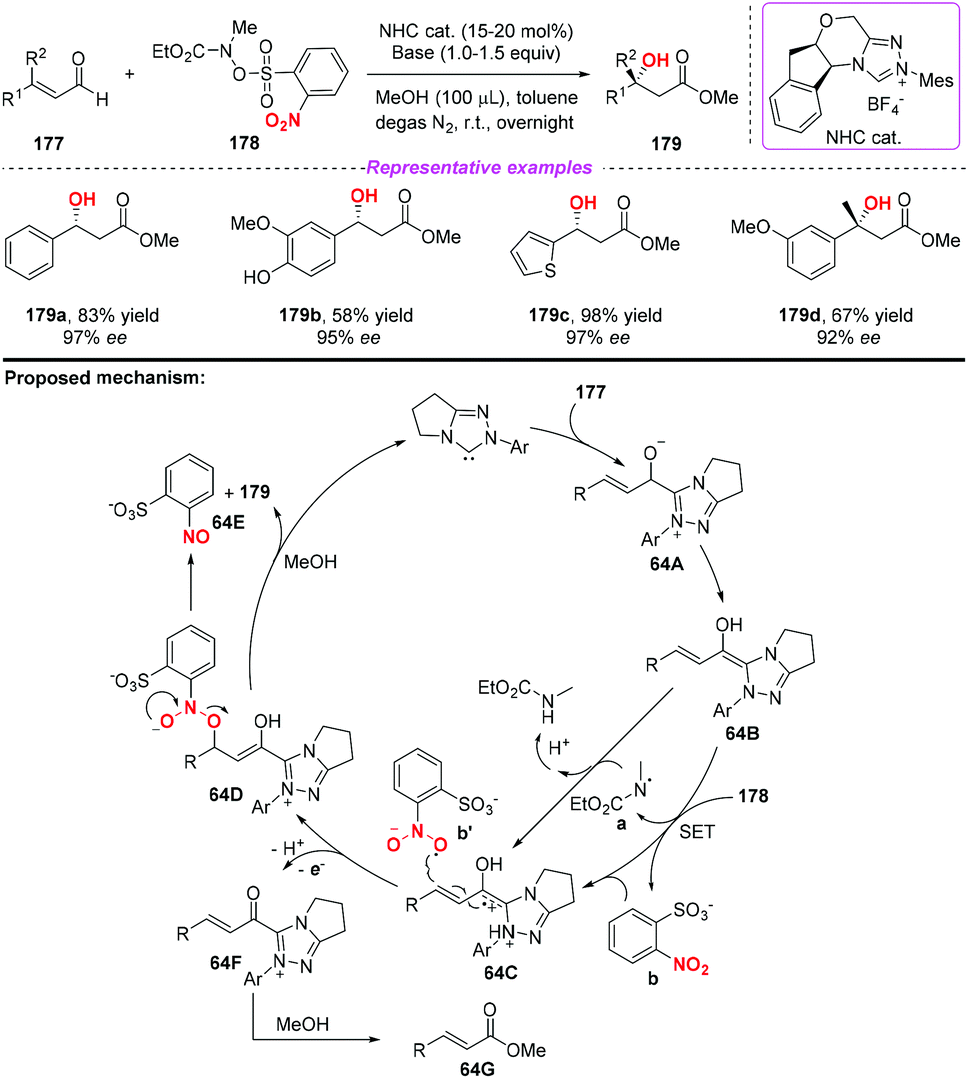
At almost the same time, the group of Chi also reported a similar N-heterocyclic carbene-catalyzed β-hydroxylation of enals by using nitrobenzenesulfonic carbamate 178 as the oxidant (Scheme 64).76 This oxidative single-electron-transfer reaction provided highly enantioselective access to β-hydroxyl esters in good to excellent yields. Similar to Rovis's work, the authors presented a plausible mechanism including the formation of Breslow intermediate 64A, radical coupling, single electron oxidation and deprotonation processes.
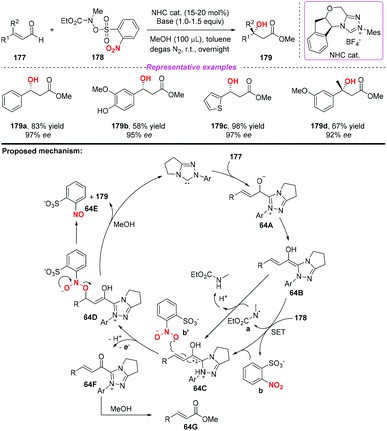 |
| | Scheme 64 Chiral NHC-catalyzed β-hydroxylation of enals using nitroarenes as an oxidant. | |
In addition, the same group developed a new class of chiral nitroarenes as enantioselective single-electron-transfer oxidants for carbene-catalyzed radical reactions in 2019 (Scheme 65).77 Notably, the high enantioselectivity control in carbene-catalyzed β-hydroxylation of enals was realized by chiral transfer from the oxidants to the substrates. By using this method, a variety of chiral β-hydroxyl esters were obtained in moderate to good yields with high enantioselectivities.
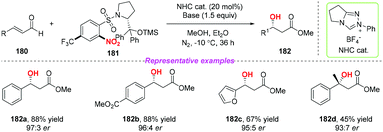 |
| | Scheme 65 NHC-catalyzed β-hydroxylation of enals using chiral nitroarenes as an oxidant. | |
4. Conclusion and outlook
In conclusion, this review summarizes the recent advances in radical reactions involving the nitro group. Overall, approaches for the synthesis of amines, oximes, alkenes, nitrones and alcohols through the radical-initiated pathway have been developed. Besides, a variety of readily available nitrating reagents including AgNO2, NaNO2, Fe(NO3)3·9H2O, tBuONO, etc. have been well explored as the source of the nitro radical. Usually, the nitro radical can be readily generated in the presence of a transition metal catalyst or under photocatalysis for further transformations. The transformations usually feature a broad substrate scope and good functional group tolerance under mild conditions.
Despite the remarkable achievements in the radical chemistry of the nitro group, there are still many issues that need to be addressed. It is anticipated that the asymmetric conversion of functionalized nitro compounds into optically pure molecules will be continually developed, since only a few examples have been reported so far. Furthermore, the discovery of new reactivities with the nitro radical through sustainable processes based on electrochemistry or flow chemistry can be expected in the near future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21871053 and 21532001) and the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005) is gratefully acknowledged. We thank Dr Sarita Yadav for reviewing the English.
Notes and references
-
(a) A. G. M. Barrett and G. G. Graboski, Conjugated Nitroalkenes: Versatile Intermediates in Organic Synthesis, Chem. Rev., 1986, 86, 751 CrossRef CAS;
(b) K.-S. Ju and R. E. Parales, Nitroaromatic Compounds, from Synthesis to Biodegradation, Microbiol. Mol. Biol. Rev., 2010, 74, 250 CrossRef CAS PubMed;
(c) M. A. Reddy, N. Jain, D. Yada, C. Kishore, V. J. Reddy, P. S. Reddy, A. Addlagatta, S. V. Kalivendi and B. Sreedhar, Design and Synthesis of Resveratrol-Based Nitrovinylstilbenes as Antimitotic Agents, J. Med. Chem., 2011, 54, 6751 CrossRef CAS PubMed.
-
(a)
N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 CrossRef;
(b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Recent Contributions from the Baylis−Hillman Reaction to Organic Chemistry, Chem. Rev., 2010, 110, 5447 CrossRef CAS PubMed;
(c) L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Development of Cascade Reactions for the Concise Construction of Diverse Heterocyclic Architectures, Acc. Chem. Res., 2012, 45, 1278 CrossRef CAS PubMed.
-
(a)
K. Schofield, Aromatic Nitrations, Cambridge University Press, Cambridge, 1980 Search PubMed;
(b)
G. A. Olah, R. Malhorta and S. C. Narang, Nitration: Methods and Mechanisms, VCH, New York, 1989 Search PubMed.
- T. Taniguchi, T. Fujii and H. Ishibashi, Iron-Mediated Radical Halo-Nitration of Alkenes, J. Org. Chem., 2010, 75, 8126 CrossRef CAS PubMed.
- T. Taniguchi and H. Ishibashi, Iron-Mediated Radical Nitro-Cyclization Reaction of 1, 6-Dienes, Org. Lett., 2010, 12, 124 CrossRef CAS PubMed.
- V. R. Sabbasani and D. Lee, Nitration of Silyl Allenes to Form Functionalized Nitroalkenes, Org. Lett., 2013, 15, 3954 CrossRef CAS PubMed.
- T. Naveen, S. Maity, U. Sharma and D. Maiti, A Predictably Selective Nitration of Olefin with Fe(NO3)3 and TEMPO, J. Org. Chem., 2013, 78, 5949 CrossRef CAS PubMed.
- Y. He, N. Zhao, L. Qiu, X. Zhang and X. Fan, Regio- and Chemoselective Mono- and Bisnitration of 8-Aminoquinoline Amides with Fe(NO3)3 ·9H2O as Promoter and Nitro Source, Org. Lett., 2016, 18, 6054 CrossRef CAS PubMed.
- D. Sar, R. Bag, D. Bhattacharjee, R. C. Deka and T. Punniyamurthy, Iron(III)-Mediated Radical Nitration of Bisarylsulfonyl Hydrazones: Synthesis of Bisarylnitromethyl Sulfones, J. Org. Chem., 2015, 80, 6776 CrossRef CAS PubMed.
- V. A. Motornov, V. M. Muzalevskiy, A. A. Tabolin, R. A. Novikov, Y. V. Nelyubina, V. G. Nenajdenko and S. L. Ioffe, Radical Nitration-Debromination of α-Bromo-α-fluoroalkenes as a Stereoselective Route to Aromatic α-Fluoronitroalkenes-Functionalized Fluorinated Building Blocks for Organic Synthesis, J. Org. Chem., 2017, 82, 5274 CrossRef CAS PubMed.
- W. Liu, Y. Zhang and H. Guo, Nitration and Cyclization of Arene-Alkynes: An Access to 9-Nitrophenathrenes, J. Org. Chem., 2018, 83, 10518 CrossRef CAS PubMed.
- T. Wang, Y. Jiang, Y. Wang and R. Yan, Fe-Catalyzed Tandem Cyclization for the Synthesis of 3-Nitrofurans from Homopropargylic Alcohols and Al(NO3)3·9H2O, Org. Biomol. Chem., 2018, 16, 5232 RSC.
- X. Yi, K. Chen, W. Chen, W. Chen, M. Liu and H. Wu, Synthesis of Cyclic gem-Dinitro Compounds via Radical Nitration of 1,6-Diynes with Fe(NO3)3·9H2O, Org. Biomol. Chem., 2019, 17, 4725 RSC.
- A. Murugan, K. G. Gorantla, B. S. Mallik and D. S. Sharada, Iron Promoted C3−H Nitration of 2H-Indazole: Direct Access to 3-Nitro-2H-Indazoles, Org. Biomol. Chem., 2018, 16, 5113 RSC.
- I. B. Krylov, A. S. Budnikov, E. S. Lopat'eva, G. I. Nikishin and A. O. Terent'ev, Mild Nitration of Pyrazolin-5-ones by a Combination of Fe(NO3)3 and NaNO2: Discovery of a New Readily Available Class of
Fungicides, 4-Nitropyrazolin-5-ones, Chem. – Eur. J., 2019, 25, 5922 CrossRef CAS PubMed.
- B. V. Rokade and K. R. Prabhu, Synthesis of Substituted Nitroolefins: A Copper Catalyzed Nitrodecarboxylation of Unsaturated Carboxylic acids, Org. Biomol. Chem., 2013, 11, 6713 RSC.
- G.-W. Wang, M.-X. Cheng, R.-S. Ma and S.-D. Yang, Cu-Catalyzed Selective Cascade sp3 C–H Bond Oxidative Functionalization towards Isoxazoline Derivatives, Chem. Commun., 2015, 51, 6308 RSC.
-
(a) S. Manna, S. Maity, S. Rana, S. Agasti and D. Maiti,
ipso-Nitration of Arylboronic Acids with Bismuth Nitrate and Perdisulfate, Org. Lett., 2012, 14, 1736 CrossRef CAS PubMed;
(b) Y.-M. Li, X.-H. Wei, X.-A. Li and S.-D. Yang, Metal-Free Carbonitration of Alkenes Using K2S2O8, Chem. Commun., 2013, 49, 11701 RSC;
(c) D.-G. Wu, J. Zhang, J. H. Cui, W. Zhang and Y.-K. Liu, AgNO2-Mediated Direct Nitration of the Quinoxaline Tertiary Benzylic C–H Bond and Direct Conversion of 2-Methyl Quinoxalines into Related Nitriles, Chem. Commun., 2014, 50, 10857 RSC.
- G.-W. Wang, S.-X. Li, Q.-X. Wu and S.-D. Yang, Cu-catalyzed sp3 C–H Bond Oxidative Functionalization of Alkylazaarenes and Substituted Ethanones: An Efficient Approach to Isoxazoline Derivatives, Org. Chem. Front., 2015, 2, 569 RSC.
- Y.-F. Ji, H. Yan and Q.-B. Jiang, Effective Nitration of Anilides and Acrylamides by tert-Butyl Nitrite, Eur. J. Org. Chem., 2015, 2051 CrossRef CAS.
- X. Zhu, L. Qiao, P. Ye, B. Ying, J. Xu, C. Shen and P. Zhang, Copper-Catalyzed Rapid C–H Nitration of 8-Aminoquinolines by Using Sodium Nitrite as the Nitro Source Under Mild Conditions, RSC Adv., 2016, 6, 89979 RSC.
- Z.-Q. Zhang, T. Chen and F.-M. Zhang, Copper-Assisted Direct Nitration of Cyclic Ketones with Ceric Ammonium Nitrate for the Synthesis of Tertiary α-Nitro-α-substituted Scaffolds, Org. Lett., 2017, 19, 1124 CrossRef CAS PubMed.
- D. Tu, J. Luo and C. Jiang, Copper-Mediated Domino C–H Iodination and Nitration of Indoles, Chem. Commun., 2018, 54, 2514 RSC.
- S. Maity, S. Manna, S. Rana, T. Naveen, A. Mallick and D. Maiti, Efficient and Stereoselective Nitration of Mono- and Disubstituted Olefins with AgNO2 and TEMPO, J. Am. Chem. Soc., 2013, 135, 3355 CrossRef CAS PubMed.
- C. Xue, C. Fu and S. Ma, Highly Regio- and Stereoselective Nitro-Oxoamination of Mono-Substituted Allenes, Chem. Commun., 2014, 50, 15333 RSC.
- D. Wu, J. Zhang, J. Cui, W. Zhang and Y. Liu, AgNO2-Mediated Direct Nitration of the Quinoxaline Tertiary Benzylic C–H Bond and Direct Conversion of 2-Methyl Quinoxalines into Related Nitriles, Chem. Commun., 2014, 50, 10857 RSC.
- T.-X. Liu, J. Ma, D. Chao, P. Zhang, Q. Liu, L. Shi, Z. Zhang and G. Zhang, AgNO2-Mediated Cleavage of the N–N Bond of Sulfonylhydrazones and Oxygen Transfer: Access to Fulleroisox zolines via Radical Cyclization with [60]Fullerene, Chem. Commun., 2015, 51, 12775 RSC.
- H. Yan, J. Mao, G. Rong, D. Liu, Y. Zheng and Y. He, Facile Synthesis of (E)-β-Nitroolefinic Alkoxyamines via Silver-Catalyzed Decarboxylative Nitroaminoxylation of Phenylpropiolic Acids, Green Chem., 2015, 17, 2723 RSC.
- J. Sun, J.-K. Qiu, Y.-N. Wu, W.-J. Hao, C. Guo, G. Li, S.-J. Tu and B. Jiang, Silver-Mediated Radical C(sp3)−H Biphosphinylation and Nitration of β-Alkynyl Ketones for Accessing Functional Isochromenes, Org. Lett., 2017, 19, 754 CrossRef CAS PubMed.
- Y. Ning, X.-F. Zhao, Y.-B. Wu and X. Bi, Radical Enamination of Vinyl Azides: Direct Synthesis of N-Unprotected Enamines, Org. Lett., 2017, 19, 6240 CrossRef CAS PubMed.
- W. Zhang, S. Lou, Y. Liu and Z. Xu, Palladium-Catalyzed Chelation-Assisted Aromatic C−H Nitration: Regiospecific Synthesis of Nitroarenes Free from the Effect of the Orientation Rules, J. Org. Chem., 2013, 78, 5932 CrossRef CAS PubMed.
- G. G. Pawar, A. Brahmanandan and M. Kapur, Palladium(II)-Catalyzed, Heteroatom-Directed, Regioselective C−H Nitration of Anilines Using Pyrimidine as a Removable Directing Group, Org. Lett., 2016, 18, 448 CrossRef CAS PubMed.
- Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, Aerobic Oxidation of PdII to PdIV by Active Radical Reactants: Direct C−H Nitration and Acylation of Arenes via Oxygenation Process with Molecular Oxygen, ACS Catal., 2015, 5, 1956 CrossRef CAS.
- W. Zhang, S. Ren, J. Zhang and Y. Liu, Palladium-Catalyzed sp3 C−H Nitration of 8-Methylquinolines, J. Org. Chem., 2015, 80, 5973 CrossRef CAS PubMed.
- Y. Lin, W. Kong and Q. Song, Palladium-Catalyzed Nitration of Meyer−Schuster Intermediates with tBuONO as Nitrogen Source at Ambient Temperature, Org. Lett., 2016, 18, 3702 CrossRef CAS PubMed.
- Y. Chen, Y. Ma, L. Li, H. Jiang and Z. Li, Nitration-Peroxidation of Alkenes: A Selective Approach to β-Peroxyl Nitroalkanes, Org. Lett., 2019, 21, 1480 CrossRef CAS PubMed.
- Z. Fan, J. Ni and A. Zhang, Meta-Selective CAr−H Nitration of Arenes through a Ru3(CO)12-Catalyzed Ortho-Metalation Strategy, J. Am. Chem. Soc., 2016, 138, 8470 CrossRef CAS PubMed.
- Z. Fan, J. Li, H. Lu, D.-Y. Wang, C. Wang, M. Uchiyama and A. Zhang, Monomeric Octahedral Ruthenium(II) Complex Enabled meta-C−H Nitration of Arenes with Removable Auxiliaries, Org. Lett., 2017, 19, 3199 CrossRef CAS PubMed.
- Z. Fan, H. Lu and A. Zhang, PMes3-Promoted Ruthenium-Catalyzed Meta C-H Nitration of 6-Arylpurines, J. Org. Chem., 2018, 83, 3245 CrossRef CAS PubMed.
-
(a) D. Liu, P. Luo, J. Ge, Z. Jiang, Y. Peng and Q. Ding, Synthesis of 2-Arylbenzothiazole and 2-Arylthiazole Derivatives via a Ru-Catalyzed meta-Selective C−H Nitration Reaction, J. Org. Chem., 2019, 84, 12784 CrossRef CAS PubMed;
(b) S. Sasmal, S. K. Sinha, G. K. Lahiri and D. Maiti, A Directing Group-Assisted Ruthenium-Catalyzed Approach to Access meta-Nitrated Phenols, Chem. Commun., 2020, 56, 7100 RSC.
- S. Manna, S. Jana, T. Saboo, A. Maji and D. Maiti, Synthesis of (E)-Nitroolefins via Decarboxylative Nitration Using t-Butylnitrite (t-BuONO) and TEMPO, Chem. Commun., 2013, 49, 5286 RSC.
-
(a) U. Dutta, S. Maity, R. Kancherla and D. Maiti, Aerobic Oxynitration of Alkynes with tBuONO and TEMPO, Org. Lett., 2014, 16, 6302 CrossRef CAS PubMed;
(b) S. Maity, T. Naveen, U. Sharma and D. Maiti, Stereoselective Nitration of Olefins with tBuONO and TEMPO: Direct Access to Nitroolefins under Metal-free Conditions, Org. Lett., 2013, 15, 3384 CrossRef CAS PubMed.
- T. Shen, Y. Yuan and N. Jiao, Metal-Free Nitro-Carbocyclization of Activated Alkenes: A Direct Approach to Synthesize Oxindoles by Cascade C–N and C–C Bond Formation, Chem. Commun., 2014, 50, 554 RSC.
- D. Chen, M. Ji and C. Zhu, Intramolecular Nitration–Aminocarbonylation of Unactivated Olefins: Metal-Free Synthesis of γ-Lactams, Chem. Commun., 2019, 55, 7796 RSC.
- X.-H. Hao, P. Gao, X.-R. Song, Y.-F. Qiu, D.-P. Jin, X.-Y. Liu and Y.-M. Liang, Metal-Free Nitro-Carbocyclization of 1, 6-Enynes with tBuONO and TEMPO, Chem. Commun., 2015, 51, 6839 RSC.
- S. Mondal, S. Samanta and A. Hajra, Regioselective C-7 Nitration of 8-Aminoquinoline Amides Using tert-Butyl Nitrite, Adv. Synth. Catal., 2018, 360, 1026 CrossRef CAS.
- X. Guo, C. Lv, Q. Mahmood, L. Zhou, G. Xu and Q. Wang, Solvent-Controlled Chemoselective N-Dealkylation-N-Nitrosation or C-Nitration of N-Alkyl Anilines with tert-butyl Nitrite, Org. Chem. Front., 2019, 6, 3401 RSC.
- P. Chaudhary, S. Gupta, N. Muniyappan, S. Sabiah and J. Kandasamy, Regioselective Nitration of N-Alkyl Anilines using tert-Butyl Nitrite under Mild Condition, J. Org. Chem., 2019, 84, 104 CrossRef CAS PubMed.
- J. P. Das, P. Sinha and S. Roy, A Nitro-Hunsdiecker Reaction: From Unsaturated Carboxylic Acids to Nitrostyrenes and Nitroarenes, Org. Lett., 2002, 4, 3055 CrossRef CAS PubMed.
-
(a) P. Y. F. Deghati, M. J. Wanner and G.-J. Koomen, Regioselective Nitration of Purine Nucleosides: Synthesis of 2-Nitroadenosine and 2-Nitroinosine, Tetrahedron Lett., 2000, 41, 1291 CrossRef CAS;
(b) P. Y. F. Deghati, H. Bieravgel, M. J. Wanner and G.-J. Koomen, Mild and Regioselective Nitration of 1-Deazapurine Nucleosides Using TBAN/TFAA, Tetrahedron Lett., 2000, 41, 569 CrossRef CAS.
- B. Rodenko, M. Koch, A. M. Burg, M. J. Wanner and G.-J. Koomen, The Mechanism of Selective Purine C-Nitration Revealed: NMR Studies Demonstrate Formation and Radical Rearrangement of an N7-Nitramine Intermediate, J. Am. Chem. Soc., 2005, 127, 5957 CrossRef CAS PubMed.
-
(a) S. Manna, S. Maity, S. Rana, S. Agasti and D. Maiti,
ipso-Nitration of Arylboronic Acids with Bismuth Nitrate and Perdisulfate, Org. Lett., 2012, 14, 1736 CrossRef CAS PubMed;
(b) S. Agasti, S. Maiti, S. Maity, M. Anniyappan, M. B. Talawar and D. Maiti, Bismuth nitrate as a source of nitro radical in ipso-nitration of carboxylic acids, Polyhedron, 2019, 172, 120 CrossRef CAS.
- N. Chatterjee, D. Bhatt and A. Goswami, A Novel Transition Metal Free [bis-(Trifluoroacetoxy)-Iodo]Benzene (PIFA) Mediated Oxidative ipso Nitration of Organoboronic Acids, Org. Biomol. Chem., 2015, 13, 4828 RSC.
-
(a) M. A. Zolfigol, A. Khazaei, R. M.-Z. Ahmad, A. Zare, H. G. Kruger, Z. Asgari, V. Khakyzadeh and K.-R. Masoud, Design of Ionic Liquid 3-Methyl-1-sulfonic Acid Imidazolium Nitrate as Reagent for the Nitration of Aromatic Compounds by in Situ Generation of NO2 in Acidic Media, J. Org. Chem., 2012, 77, 3640 CrossRef CAS PubMed;
(b) M. Zarei, E. Noroozizadeh, R. M.-Z. Ahmad and M. A. Zolfigol, Synthesis of Nitroolefins and Nitroarenes under Mild Conditions, J. Org. Chem., 2018, 83, 3645 CrossRef CAS PubMed.
- A. Shoberu, C.-K. Li, Z.-K. Tao, G.-Y. Zhang and J.-P. Zou, NaNO2/K2S2O8-mediated Selective Radical Nitration/Nitrosation of Indoles: Efficient Approach to 3-Nitro- and 3-Nitrosoindoles, Adv. Synth. Catal., 2019, 361, 2255 CrossRef CAS.
- S. J. S. Düsel and B. König, Visible-Light-Mediated Nitration of Protected Anilines, J. Org. Chem., 2018, 83, 2802 CrossRef PubMed.
- K. Zhang, B. Jelier, A. Passera, G. Jeschke and D. Katayev, Synthetic Diversity from a Versatile and Radical Nitrating Reagent, Chem. – Eur. J., 2019, 25, 12929 CrossRef CAS PubMed.
- K. Zhang, A. Budinská, A. Passera and D. Katayev, N-Nitroheterocycles: Bench-Stable Organic Reagents for Catalytic Ipso-Nitration of Aryl- and Heteroarylboronic Acids, Org. Lett., 2020, 22, 2714 CrossRef CAS PubMed.
- J.-H. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. L. Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Practical Olefin Hydroamination with Nitroarenes, Science, 2015, 348, 886 CrossRef CAS PubMed.
-
(a) H. Gao, D. H. Ess, M. Yousufuddin and L. Kürti, Transition-Metal-Free Direct Arylation: Synthesis of Halogenated 2-Amino-2′-Hydroxy-1, 1′-Biaryls and Mechanism by DFT Calculations, J. Am. Chem. Soc., 2013, 135, 7086 CrossRef CAS PubMed;
(b) J. C. Lo, Y. Yabe and P. S. Baran, A Practical and Catalytic Reductive Olefin Coupling, J. Am. Chem. Soc., 2014, 136, 1304 CrossRef CAS PubMed.
- C. W. Cheung and X. Hu, Amine Synthesis via Iron-Catalysed Reductive Coupling of Nitroarenes with Alkyl Halides, Nat. Commun., 2016, 7, 12494 CrossRef PubMed.
- H. Song, Z. Yang, C.-H. Tung and W. Wang, Iron-Catalyzed Reductive Coupling of Nitroarenes with Olefins: Intermediate of Iron−Nitroso Complex, ACS Catal., 2020, 10, 276 CrossRef CAS.
- J. Zheng, D. Wang and S. Cui, Fe-Catalyzed Reductive Coupling of Unactivated Alkenes with β-Nitroalkenes, Org. Lett., 2015, 17, 4572 CrossRef CAS PubMed.
- N. Zhang, Z.-J. Quan and X.-C. Wang, Nickel-Catalyzed Denitrated Coupling Reaction of Nitroalkenes with Aliphatic and Aromatic Alkenes, Adv. Synth. Catal., 2016, 358, 3179 CrossRef CAS.
- T. Keshari, R. Kapoorr and S. L. D. Yadav, Silver-Catalyzed Denitrative Sulfonylation of Nitrostyrenes: A Convenient Approach to (E)-Vinyl Sulfones, Eur. J. Org. Chem., 2016, 2695 CrossRef CAS.
- G. Nie, X. Deng, X. Lei, Q. Hu and Y. Chen, Mn(III)-Mediated Regioselective Synthesis of (E)-Vinyl Sulfones from Sodium Sulfinates and Nitroolefins, RSC Adv., 2016, 6, 75277 RSC.
- T. B. Nguyen, L. Ermolenko and A.-M. Ali, Nitro-Methyl Redox Coupling: Efficient Approachto2-Hetarylbenzothiazolesfrom 2-Halonitroarene, Methylhetarene, and Elemental Sulfur, Org. Lett., 2013, 15, 4218 CrossRef CAS PubMed.
- G. Li, L. Wu, G. Lv, H. Liu, Q. Fu, X. Zhang and Z. Tang, Alkyl Transfer from C–C Cleavage: Replacing the Nitro Group of Nitro-Olefins, Chem. Commun., 2014, 50, 6246 RSC.
- X. Lei, L. Zheng, C. Zhang, X. Shi and Y. Chen, Allylic C−S Bond Construction through Metal-Free Direct Nitroalkene Sulfonation, J. Org. Chem., 2018, 83, 1772 CrossRef CAS PubMed.
- S. Cai, S. Zhang, Y. Zhao and D. Z. Wang, New Approach to Oximes through Reduction of Nitro Compounds Enabled by Visible Light Photoredox Catalysis, Org. Lett., 2013, 15, 2660 CrossRef CAS PubMed.
- X.-J. Yang, B. Chen, L.-Q. Zheng, L.-Z. Wu and C.-H. Tung, Highly Efficient and Selective Photocatalytic Hydrogenation of Functionalized Nitrobenzenes, Green Chem., 2014, 16, 1082 RSC.
- A. R. Todorov, S. Aikonen, M. Muuronen and J. Helaja, Visible-Light-Photocatalyzed Reductions of N-Heterocyclic Nitroaryls to Anilines Utilizing Ascorbic Acid Reductant, Org. Lett., 2019, 21, 3764 CrossRef CAS PubMed.
- C.-W. Lin, B.-C. Hong, W.-C. Chang and G.-H. Lee, A New Approach to Nitrones through Cascade Reaction of Nitro Compounds Enabled by Visible Light Photoredox Catalysis, Org. Lett., 2015, 17, 2314 CrossRef CAS PubMed.
- N. Zhang, Z.-J. Quan, Z. Zhang, Y.-X. Da and X.-C. Wang, Synthesis of Stilbene Derivatives via Visible-Light-Induced Cross-Coupling of Aryl Diazonium Salts with Nitroalkenes Using –NO2 as a Leaving Group, Chem. Commun., 2016, 52, 14234 RSC.
- N. A. White and T. Rovis, Enantioselective N-Heterocyclic Carbene-Catalyzed β-Hydroxylation of Enals Using Nitroarenes: An Atom Transfer Reaction That Proceeds via Single Electron Transfer, J. Am. Chem. Soc., 2014, 136, 14674 CrossRef CAS PubMed.
- Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, N-Heterocyclic Carbene-Catalyzed Radical Reactions for Highly Enantioselective β-Hydroxylation of Enals, J. Am. Chem. Soc., 2015, 137, 2416 CrossRef CAS PubMed.
- H. Wang, Y. Wang, X. Chen, C. Mou, S. Yu, H. Chai, Z. Jin and Y. R. Chi, Chiral Nitroarenes as Enantioselective Single-Electron-Transfer Oxidants for Carbene-Catalyzed Radical Reactions, Org. Lett., 2019, 21, 7440 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Fu-Sheng
He
*a and
Jie
Wu
b,
Fu-Sheng
He
*a and
Jie
Wu