
Recent advances in photoelectrochemical cells (PECs) for organic synthesis
Yan-Chen
Wu
a,
Ren-Jie
Song
 *a and
Jin-Heng
Li
*ab
*a and
Jin-Heng
Li
*ab
aKey Laboratory of Jiangxi Province for Persistent Pollutants Control and Resources Recycle, Nanchang Hangkong University, Nanchang 330063, China
bState Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha 410082, China. E-mail: srj0731@hnu.edu.cn; jhli@hnu.edu.cn
First published on 23rd June 2020
Abstract
Photoelectrochemical cells (PECs) have emerged as an environmentally friendly tool for fuel production, conversion of carbon dioxide, water splitting, and pollutant degradation in the past few years. Although PECs show advantages in these areas, their application in organic synthesis has just begun. Recently, oxidation, C–H functionalization and cross-coupling have been successfully achieved in PECs by the use of an oxidizing photoanode, such as BiVO4 or WO3, for the construction of C–N, C–O and C–P bonds. This highlight article focuses on the application of PECs in organic synthesis reactions and their reaction mechanisms, with a special emphasis on enantioselective conversions.
Introduction
The effective transformation of sunlight into chemical fuel and energy has continuously attracted the attention of chemists because sunlight is an inexpensive, abundant, non-polluting, and endlessly renewable source of clean energy.1 As a consequence, the development of green and effective methods for this conversion is an attractive research area in energy chemistry, synthetic chemistry and materials chemistry.Since the first photoelectrochemical cells (PECs) were reported in 1972, PEC-based solar water splitting was the most studied path in this promising area.2 As research progressed, scientists found that the potential of PECs was not limited to this simple field, and other applications of PECs were reported. Their applications were expanded to the fields of fuel production, conversion of carbon dioxide, and pollutant degradation.3,4 However, their application in organic synthesis was limited. Recently, much effort has been made to develop systems for this transformation through the use of numerous photoanode materials. Various oxidizing photoanodes, such as BiVO4 and WO3, have been used to realize organic redox reactions.5 Different reaction types, such as the oxidation of simple substrates, C–H functionalization and R1-H/R2-H cross coupling reactions, have been successfully achieved in PEC cell systems (Scheme 1). This organic transfer system in PEC cells has become a powerful strategy for the construction of high value-added chemicals, which has emerged as an atom-economical, environmentally-friendly, and energy saving method in organic chemistry.
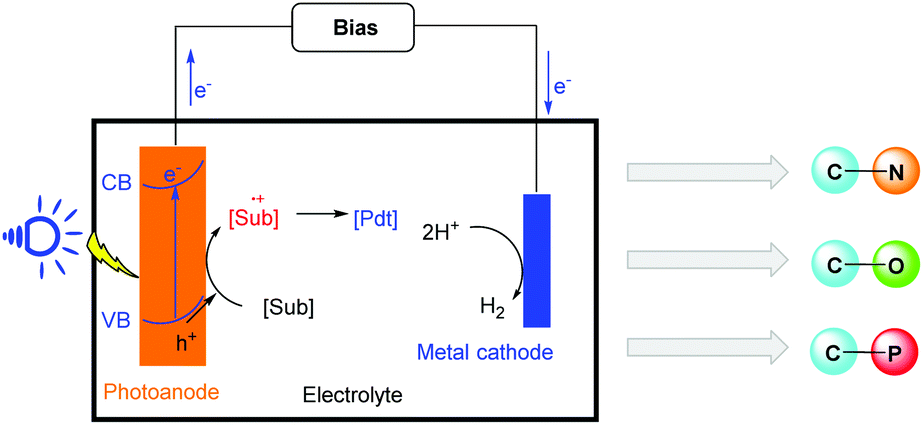 | ||
| Scheme 1 Photoelectrochemical cells (PECs) for oxidative transformations of organic substrates. | ||
Similarly to traditional photochemistry and electrochemistry,6,7 photoelectrochemistry is an environmentally friendly synthetic tool. Compared with these methods, the most obvious advantage of chemical reactions in PECs is the reduction of external bias, thereby saving electricity. Notably, organic system reactions in PECs mostly work under metal-free and chemical oxidant-free conditions, which will become an ideal research direction. With the use of a photoanode as the electrode material, the use of external power can be avoided and the applied voltage can be decreased. At present, PEC cells have no obvious advantage regarding the control of reactivity and selectivity. Organic transformation in PECs may avoid excessive oxidation and reduce side reactions to obtain better chemical selectivity due to lower external bias. We hope that these problems can be solved by designing different photoanode materials (doped with transition metal catalysts) in the future.
This highlight article aims to provide a concise overview of organic synthesis in photoelectrochemical cells for the formation of different chemical bonds and also emphasizes transformation pathways. The advantages and shortcomings of photoelectrochemical organic reactions using PEC cells are also discussed.
Photoelectrochemical simple oxidation in PEC cells
5-Hydroxymethylfurfural (HMF) is a key intermediate in biomass conversion, and 2,5-furandicarboxylic acid (FDCA) is an important monomer for the production of numerous polymer materials. Much effort has been made to achieve the conversion of HMF into FDCA. Although the methods are efficient, the requirements for transition-metal catalysts, high-pressure O2 or air (3–20 bar), an alkaline aqueous solution (pH ≥ 13), and elevated temperatures (30–130 °C) are drawbacks, which limit the applications of this transformation.8 Thus, it is attractive to find a mild, efficient synthetic strategy to complete this valuable transformation.In 2015, Choi’s group reported a TEMPO-mediated electrooxidative reaction of HMF for the synthesis of FDCA in a photoelectrochemical cell (Scheme 2a).9 Utilizing an n-type BiVO4 material as the oxidation photoanode and a platinum plate as the reduction cathode in the presence of TEMPO as a mediator, HMF can convert into FDCA with a near-quantitative yield and 100% faradaic efficiency at ambient conditions without the use of precious-metal catalysts. Meanwhile, the authors also demonstrated a novel strategy using PECs which utilizes solar energy for HMF oxidation as the anode reaction (Scheme 2b). The results suggest that PECs can not only be used for fuel production, but can also be used in producing high value-added chemicals from simple organic substrates by photoanode oxidation processes. More importantly, by using photogenerated holes in the VB of BiVO4 for oxidation, a lower applied potential could be used for TEMPO oxidation, which is about 0.1 V versus RHE.
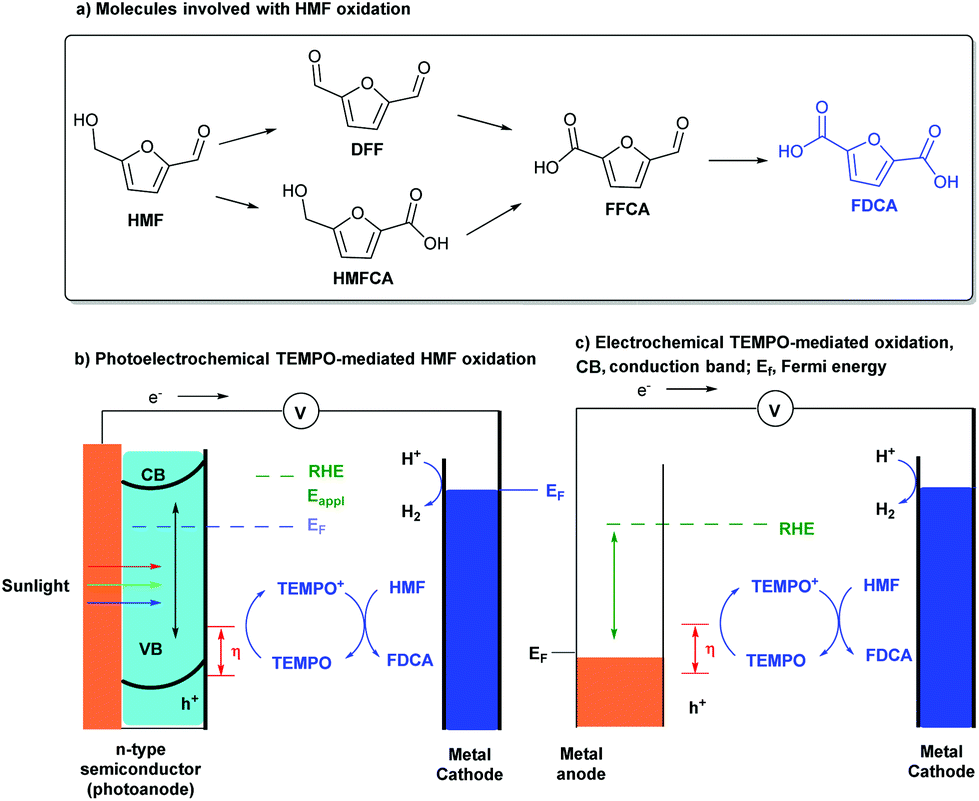 | ||
| Scheme 2 Schematic comparison of photoelectrochemical and electrochemical cells. | ||
Subsequently, organic transformation in photoelectrochemical cells has attracted a great deal of interest in organic synthetic chemistry because it avoids using expensive and toxic reagents or catalysts. In 2017, Sammis, Berlinguette and co-workers realized a photoelectrochemical alcohol oxidation and C–H functionalization in PEC cells. Employing a BiVO4 electrode as the photoanode, MeCN as the solvent, and NHPI as the electron transfer reagent, benzyl alcohol, cyclohexene and 1,2,3,4-tetrahydronaphthalene could furnish the corresponding ketones in moderate to good yields (Scheme 3).10 Compared with traditional chemical oxidation conditions, this method avoids the use of other additives, such as metal catalysts and strong oxidants, which may cause pollution problems. Moreover, this photoelectrochemical oxidative reaction was accomplished by using sunlight as the single energy source, which can reduce the applied voltage of an EC oxidation process by 1 V. Importantly, the results of the experiments showed that NHPI was necessary for this photoelectrochemical system.
 | ||
| Scheme 3 PEC cells for oxidation of organic substrates in organic media. | ||
Sayama and co-workers also successfully achieved a similar photoelectrochemical oxidation of benzylic alcohol derivatives on BiVO4/WO3 under visible light irradiation (Scheme 4).11a Employing a BiVO4/WO3 composite photoelectrode in aprotic organic media, the oxidation reaction proceeded smoothly and generated the corresponding ketones in up to 97% yield. The recyclability of this electrode material was also examined and the composite BiVO4/WO3 photoelectrode could be used at least three times for the photoelectrochemical oxidation of alcohols. It was noted that the TON of products to BiVO4 was approximately 1200.
 | ||
| Scheme 4 The photo-assisted electrochemical oxidation of benzylic alcohol derivatives and the proposed reaction mechanism. | ||
A similar simple oxidant reaction was reported by Yehezkeli, Goodwin, Cha and co-workers in the same year.11b They achieved the transformation from n-butanol to 2-ethylhexenal by tandem enzymatic oxidation and aldol condensation in a PEC system by employing BiVO4 as the anode material.
Direct C–H activation reactions have attracted widespread attention in organic synthesis because this reaction can often improve the atom- and step economies. Over the past decades, many fascinating methods have been developed, including transition-metal-catalyzed C–H activation and electrochemical C–H activation.12 In order to achieve specific reactivity for C–H activation under mild conditions, Sayama’s group also developed a novel photo-electrochemical C–H bond activation reaction of cyclohexane using a WO3 photoanode and visible light in air at room temperature and atmospheric pressure (Scheme 5).13 The oxidation ability of h+ at the valence band potential of WO3 is almost identical to that of TiO2 (+3.1 V vs. RHE), and the conduction band potential of WO3 is +0.5 V vs. RHE. In this strategy, excellent partial oxidation selectivity up to 99% and apparent faradaic efficiency (76%) were obtained and the IPCE at 365 and 420 nm were 57% and 24%, respectively.
 | ||
| Scheme 5 PEC cells for C–H bond activation of cyclohexane. | ||
A possible mechanism was also proposed in Scheme 5. Firstly, the single-electron transfer process of cyclohexane by the photoanode affords the cyclohexyl radical through C–H bond cleavage. Then the cyclohexyl radical is activated by O2 to give a cyclohexylperoxyl radical. Finally, the cyclohexylperoxyl radical is disproportionately transformed to the corresponding products cyclohexanol and cyclohexanone. Cyclohexanol can further convert to cyclohexanone via an additional two-electron oxidation process. However, another byproduct, namely dicyclohexyl, was often detected by coupling of a cyclohexyl radical in the absence of O2.
A peroxygenase catalyzed oxyfunctionalization of C–H bonds through photoelectrochemical H2O2 generation in PEC cells was successfully demonstrated by the Park group in 2019, in which FeOOH/BiVO4 was used as the photoanode, CN/rGO as the cathode and CIGS as the solar cell.14 It was noted that they also successfully achieved photoelectroenzymatic hydroxylation of ethylbenzene to give (R)-1-phenylethanol with a TTN of 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 and up to 99% ee via this system.
300 and up to 99% ee via this system.
Substituted furans are found as structural units in many natural products and bioactive molecules, while they are also important synthetic intermediates in organic synthesis.15 New and green synthetic investigations of furan derivatives have continuously attracted the attention of many organic chemists. In 2017, Sayama’s group reported a selective photoelectrochemical dimethoxylation reaction between furan and MeOH by using a composite n-type semiconductor BiVO4/WO3 photoanode and Br+/Br− as a mediator, with excellent faradaic efficiency up to 99% (Scheme 6).16 The utilization of photoelectrochemistry instead of an electrochemical method under dark conditions tactfully addressed the challenge of avoiding side reactions, such as the oxidation of MeOH and the methoxylated product. In addition, the applied potential in this system could be decreased by using solar energy. In order to prove that photoelectrolysis is a useful method for decreasing the applied potential, they tested the oxidation potential under dark and photoirradiation conditions, respectively. In the case of the BiVO4/WO3 photoelectrode, the oxidation potential shifted from 1.2 V to 0 V vs. SHE under photoirradiation. The bromide ion plays an important role in the transformation process; only a trace amount of product was detected in the absence of Et4NBr as a Br− source. The addition of Et4NBF4 as a co-supporting electrolyte also improved the efficiency of the dimethoxylation.
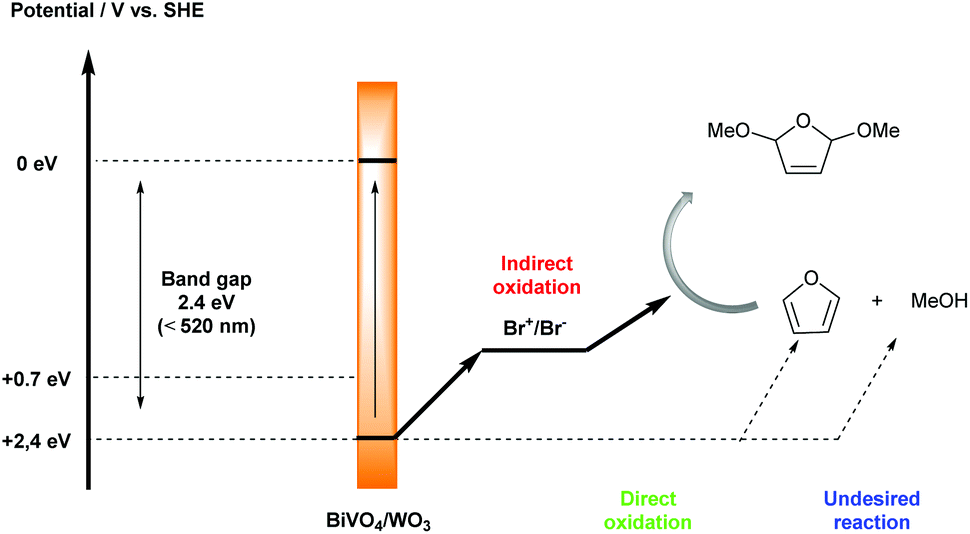 | ||
| Scheme 6 Plausible mechanism for photoelectrochemical dimethoxylation of furan with Br+/Br− as a mediator. | ||
Photoelectrochemical C–H functionalization and R1-H/R2-H cross coupling in PEC cells
Electrochemical organic synthesis methods have been recognized as some of the most environmentally friendly pathways for producing high value-added organic compounds.17 Recently, photoelectrochemical cells, an environmentally friendly synthetic tool, have been used to achieve the oxidation of simple substrates. Despite the conceptual advance, only simple organic substrates have been tested in PEC cell systems. The conversion of broad-scope organic molecules by the utilization of PEC cells is still a challenge.Recently, Hu and co-workers successfully realized a novel photoelectrocatalytic C–H amination of arenes and pyrazoles (Scheme 7).18 In this reaction, a haematite photoanode was used as the electrode due to its low cost, high stability and suitable bandgap of 2.1 eV for strong visible light absorption; compared to organic photoredox catalysts, such as acridinium, haematite (2.3 V versus the standard hydrogen electrode) has a similar oxidation potential, which suggests similar reactivity. Using LiClO4 as the supporting electrolyte, HFPI/MeOH (4:1) as the co-solvent, and blue LED as the light source, various arenes could be converted to the corresponding substituted N-heterocycle products in up to 89% yield.
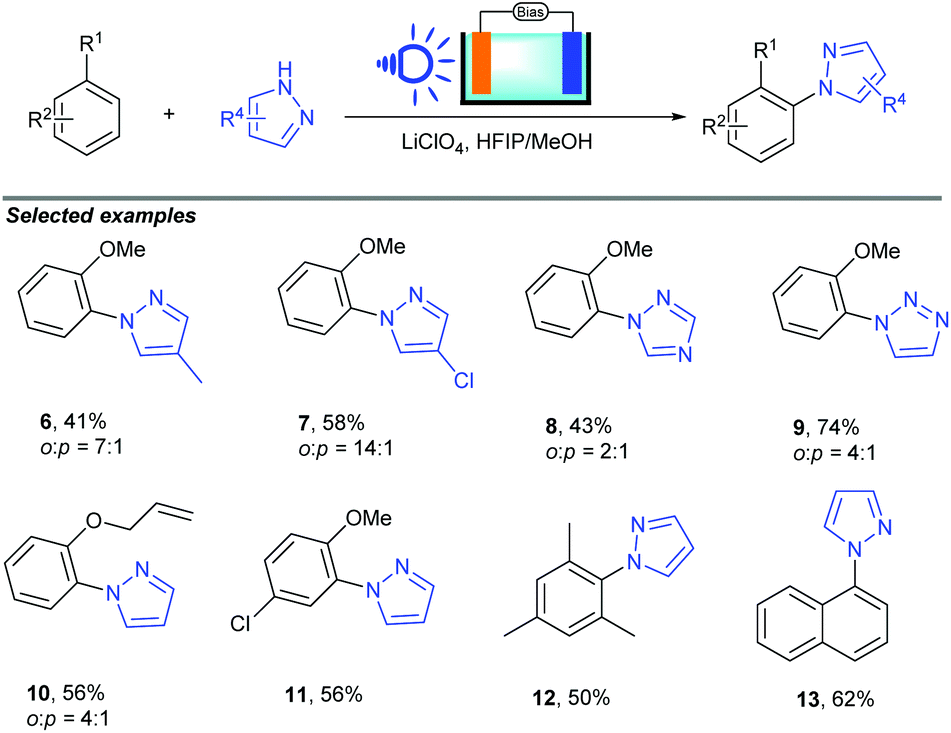 | ||
| Scheme 7 PEC cells for C–H amination of arenes. | ||
A plausible mechanism was also proposed by the authors; see Scheme 8. Unusual ortho selectivity was observed, possibly due to the formation of intermediate A, which possessed a hydrogen bond between hexafluoroisopropanol and pyrazole. Compared with the photoredox catalysed C–H/N–H cross coupling reaction, this method showed a different mechanism. The photogenerated redox process was localized in the valence band (VB) of the haematite photoanode and the PECs absorbed the light using a heterogeneous haematite semiconductor. It is worth noting that the charge in this photoelectrochemical reaction was transferred from the edge of the VB to the substrate for oxidation, which was different from the electrochemical reaction.
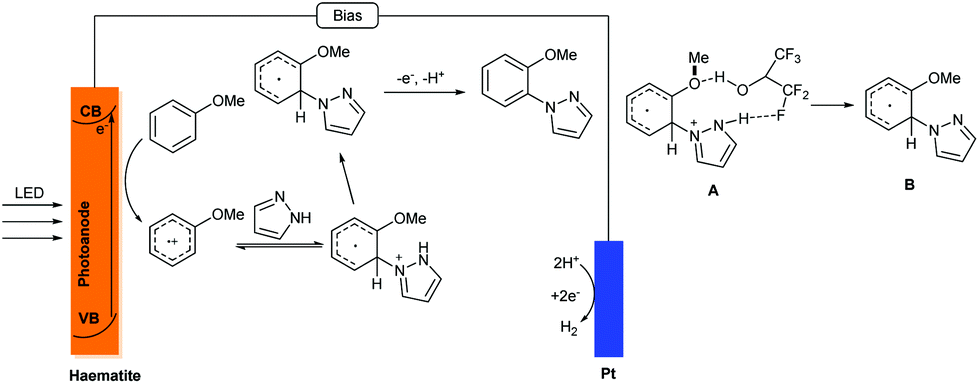 | ||
| Scheme 8 Possible mechanism of this photo-electrochemical C–H amination. | ||
Organophosphorus compounds, especially α-amino phosphonates, are a valuable skeleton widely present in materials chemistry, agrochemicals, and biochemistry.19 Most of the reported methods for their synthesis suffer from various disadvantages such as the need for metal catalysts, stoichiometric oxidants or directing groups. PEC cells, as a valuable electrochemical synthetic tool to save energy in organic synthesis, can solve these problems. In 2019, the Wu group also described an energy saving electrochemical synthesis method in PEC cells to achieve P–H/C–H cross coupling with a hydrogen evolution strategy between amines and P(O)H compounds (Scheme 9).20 By using BiVO4 as a photoanode and NHPI as a mediator, a series of organophosphorus compounds were produced in good to excellent yields with hydrogen evolution. In addition, this approach showed good functional-group tolerance and broad substrate scope. Importantly, nearly 90% external bias input was saved to achieve this conversion by the use of PEC cells as the reactor (0.1 V vs. 0.5 V for the BiVO4 PEC system and glassy carbon EC system). Generally, chemists are considering using PEC cells to achieve organic reactions because this method can avoid energy waste. In this transformation, they successfully proved that PEC systems can considerably decrease the applied voltage for C–P bond construction, and serve as viable methods to realize selective redox transformations under efficient and energy saving conditions through electron transfer on the electrode surface.
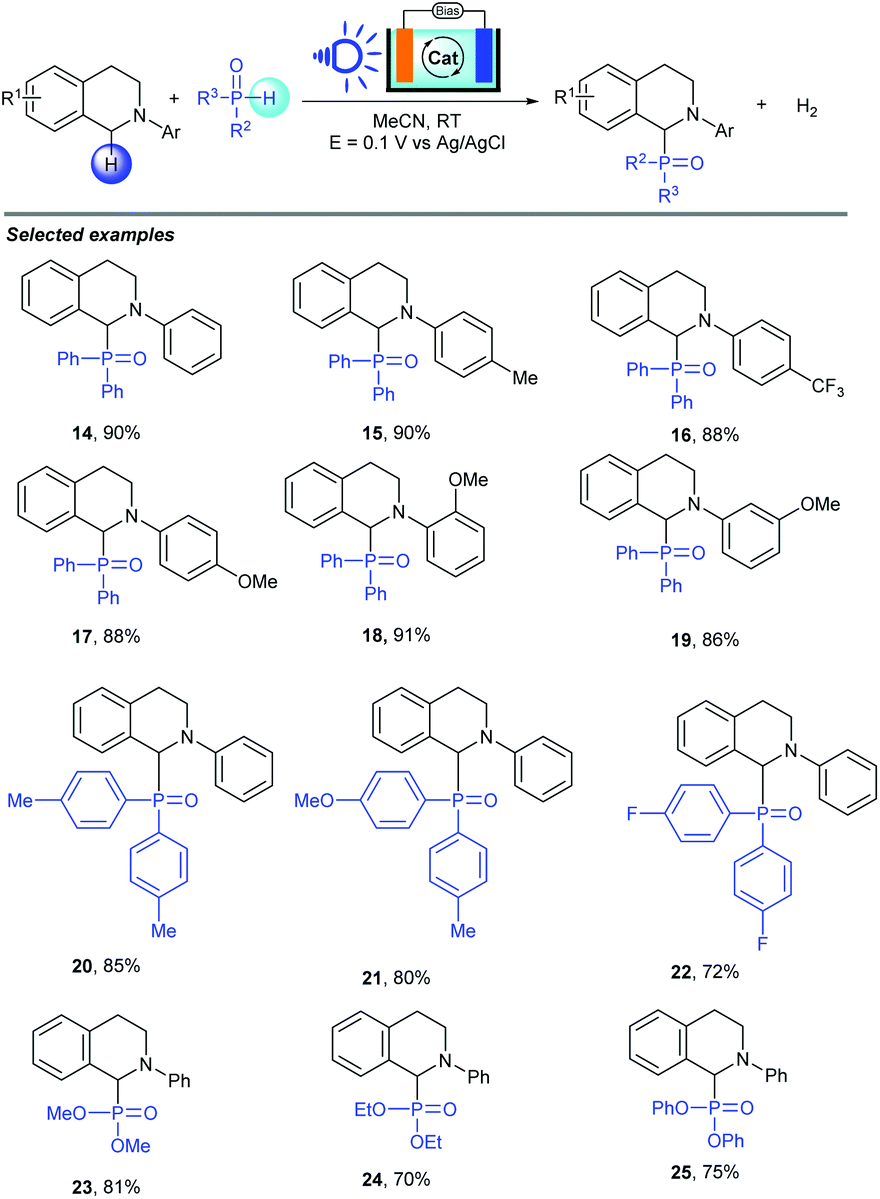 | ||
| Scheme 9 PEC cells for phosphorylation of C–H bond. | ||
A plausible mechanism for the P–H/C–H cross coupling reaction in PEC cells is outlined in Scheme 10. Firstly, high energy electron–hole pairs were generated from the BiVO4 photoelectrode once it was excited by the incident photons. Then, on the electrode surface, the radical cation intermediate D was formed from C when the holes reached the surface of BiVO4. At the same time, NHPI and 2,6-luttidine deprotonation obtained the corresponding PINO radicals by anodic oxidation, and the subsequent abstraction of a hydrogen atom (the PINO radical extracts a hydrogen atom from D to regenerate NHPI) proceeded to give the iminium ion intermediate C+, which reacted with diphenyl-phosphine oxide via nucleophilic attack to enable the formation of the final product with a C–P bond. On the cathode surface, the released protons could be reduced to release hydrogen gas during the reaction.
 | ||
| Scheme 10 The plausible mechanism for PEC cells for phosphorylation of C–H bond. | ||
As we know, photoelectrocatalysis occurring in dye-sensitized solar cell (DSSC) devices serves as a novel synthetic method which has successfully been used to achieve the conversion of light energy into electric and chemical energy. Recently, Huang, Wu and co-workers reported a visible-light-induced α-oxyamination between 1,3-dicarbonyls and TEMPO via a photo(electro)catalytic process using visible light as the energy source (Scheme 11).21 By using a DSSC anode or a DSSC system, the cross-coupling of 1,3-dicarbonyls and TEMPO occurred smoothly, leading to a series of α-oxyaminated carbonyl compounds in good to excellent yields (32–97%), featuring advantages such as easy separation, environmental protection, low energy consumption, and sustainability. Notably, the DSSC anode could be reused more than 8 times.
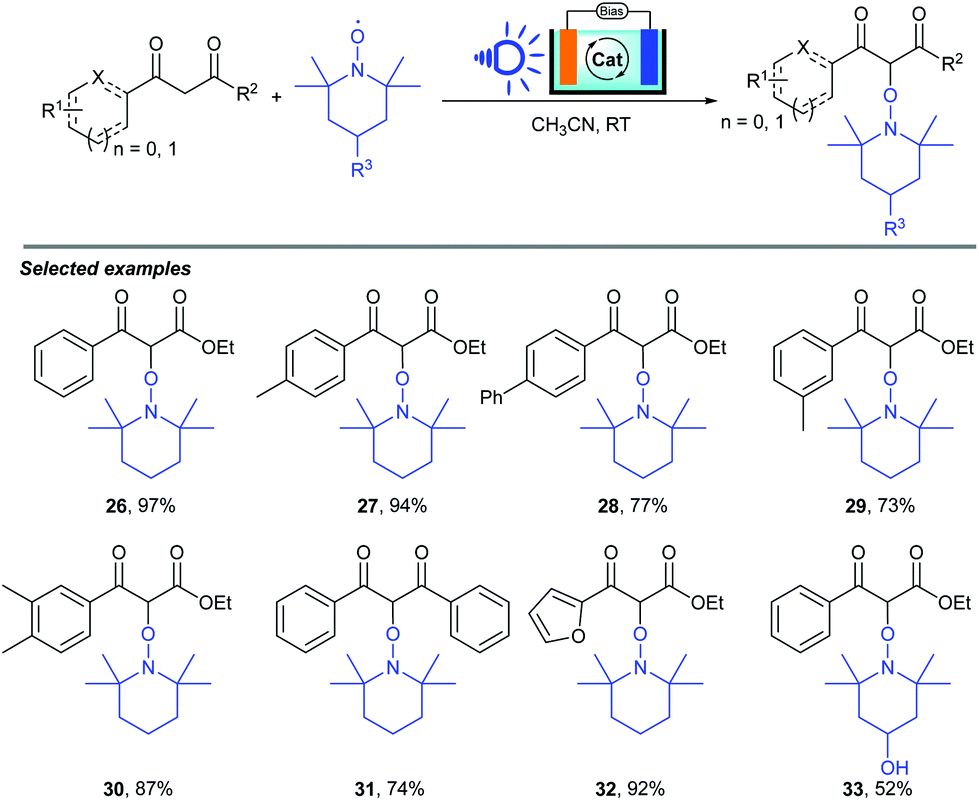 | ||
| Scheme 11 PEC cells for α-oxyamination of 1,3-dicarbonyls. | ||
The reaction mechanism for this visible-light-induced α-oxyamination of 1,3-dicarbonyls and TEMPO in PEC cells is shown in Scheme 12. Three different catalytic cycles with three types of catalysts (a trace amount of dissociated N719 in solution, unsensitized TiO2 film and N719-sensitized DSSC anode) were involved in the α-oxyamination reaction between 1,3-dicarbonyl and TEMPO. An excited state N719* [Ru(II)*] is formed from N719 under irradiation by blue light. Then, an electron is transferred from N719* to fill the conduction band (CB) of TiO2, which gives an oxidized Ru(III) complex. Subsequently, TEMPO is oxidized by Ru(III) to generate TEMPO+ (E) and the Ru(III) complex regains its ground state (N719), which undergoes single-electron transfer (SET) affording to the intermediate E. At the same time, a TEMPO anion (F) was formed with the aid of TEMPO oxidation. Moreover, the adsorption of TEMPO on the TiO2 film promotes the absorption of TiO2 under blue light. On the other hand, similarly to indirect electrolysis, TEMPO acted not only as coupling reagent but also as a catalyst in this transformation. Finally, the SET process and radical cross coupling happened to give the desired product L. Alternatively, the reaction between the dicarbonyl anion (G) and the TEMPO cation (E) could also afford the corresponding product L.
 | ||
| Scheme 12 The plausible mechanism for visible-light-induced α-oxyamination of 1,3-dicarbonyls and TEMPO in PEC cells. | ||
Conclusion and outlook
In view of the recent successful development of photoanode oxidation as a powerful tool in organic synthesis, a wide range of oxidative C–H/X–H (X = N, O, P, etc.) cross-coupling reactions have been reported. In this highlight, we summarized recent progress in photoelectrochemical oxidative conditions in PEC cells. A wide range of efficient and green methods for directly forming C–N, C–O, and C–P bonds have been developed. Generally, PEC cell photoelectrochemical systems use the oxidizing power of photoanodes, such as BiVO4 and WO3, for this transformation under metal- and chemical oxidant-free conditions. Compared with electrochemical systems, PEC systems save more energy. However, there are some limitations of the utilization of photoelectrochemical cells for organic synthesis, such as a more complex set-up, which is a major hurdle for reaction scale-up, and there is no uniform standard for the equipment. Although various photoelectrochemical organic reactions have been studied in recent years for the construction of carbon–heteroatom bonds, there are still some challenges: (i) methods for constructing carbon–carbon, other carbon–heteroatom and heteroatom–heteroatom bonds in PEC systems are relatively scarce; (ii) single electrode type and narrow substrate scope; (iii) low chemical- and region-selectivity in this transformation. Hopefully, future extensive studies of photoelectrochemical methods in PEC cells can enrich complicated reaction types by utilizing new electrode materials and/or the addition of transition metal catalysts or organic small molecule catalysts. We hope that this highlight will inspire chemists to take continued interest in the field of photoelectrochemical oxidative reactions with hydrogen evolution.Conflicts of interest
There are no conflicts of interest.Acknowledgements
We thank the National Natural Science Foundation of China (no. 21762030, 51878326, 21625203 and 21871126) for financial support. Ren-Jie Song was also supported by the China Scholarship Council (contract 201908360088).Notes and references
- (a) C. A. Grimes, O. K. Varghese and S. Ranjan, Light, Water, Hydrogen: The Solar Generation of Hydrogen by Water Photolysis, Springer Science, New York, 1st edn, 2008, pp. 1–33 CrossRef; (b) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Solar Water Splitting Cells, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed; (c) L. Vernon, Free radical chemistry of coal liquefaction: role of molecular hydrogen, Fuel, 1980, 59, 102 CrossRef CAS.
- (a) G. Ramila and P. Erb, Accessory cell-dependent selection of specific T-cell functions, Nature, 1983, 304, 442 CrossRef CAS PubMed; (b) H. M. Chen, C. K. Chen, R.-S. Liu, L. Zhang, J. Zhang and D. P. Wilkinson, Nano-architecture and material designs for water splitting photoelectrodes, Chem. Soc. Rev., 2012, 41, 5654 RSC; (c) J. R. Swierk and T. E. Mallouk, Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells, Chem. Soc. Rev., 2013, 42, 2357 RSC; (d) F. E. Osterloh, Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting, Chem. Soc. Rev., 2013, 42, 2294 RSC; (e) M. Liras, M. Barawi and V. A. de la Peña O'Shea, Hybrid materials based on conjugated polymers and inorganic semiconductors as photocatalysts: from environmental to energy applications, Chem. Soc. Rev., 2019, 48, 5454 RSC.
- (a) M. K. Debe, Electrocatalyst approaches and challenges for automotive fuel cells, Nature, 2012, 486, 43 CrossRef CAS PubMed; (b) S. Xu and E. A. Carter, Theoretical Insights into Heterogeneous (Photo)electrochemical CO2 Reduction, Chem. Rev., 2019, 119, 6631 CrossRef CAS PubMed; (c) S. Kment, F. Riboni, S. Pausova, L. Wang, L. Wang, H. Han, Z. Hubicka, J. Krysa, P. Schmuki and R. Zboril, Photoanodes based on TiO2 and α-Fe2O3 for solar water splitting – superior role of 1D nanoarchitectures and of combined heterostructures, Chem. Soc. Rev., 2017, 46, 3716 RSC; (d) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction, Chem. Rev., 2015, 115, 12936 CrossRef CAS PubMed; (e) M. A. Lumley, A. Radmilovic, Y. J. Jang, A. E. Lindberg and K.-S. Choi, Perspectives on the Development of Oxide-Based Photocathodes for Solar Fuel Production, J. Am. Chem. Soc., 2019, 141, 18358 CrossRef CAS PubMed.
- (a) Z. Wang, X. Mao, P. Chen, M. Xiao, S. A. Monny, S. Wang, M. Konarova, A. Du and L. Wang, Understanding the Roles of Oxygen Vacancies in Hematite-Based Photoelectrochemical Processes, Angew. Chem., Int. Ed., 2019, 58, 1030 CrossRef CAS PubMed; (b) Y. Xu, J. Jia, D. Zhong and Y. Wang, Degradation of dye wastewater in a thin-film photoelectrocatalytic (PEC) reactor with slant-placed TiO2/Ti anode, Chem. Eng. J., 2009, 150, 302 CrossRef CAS; (c) W.-W. Zhao, J.-J. Xu and H.-Y. Chen, Photoelectrochemical bioanalysis: the state of the art, Chem. Soc. Rev., 2015, 44, 729 RSC; (d) Y. Xu, Y. He, X. Cao, D. Zhong and J. Jia, TiO2/Ti Rotating Disk Photoelectrocatalytic (PEC) Reactor: A Combination of Highly Effective Thin-Film PEC and Conventional PEC Processes on a Single Electrode, Environ. Sci. Technol., 2008, 42, 2612 CrossRef CAS PubMed.
- (a) D. K. Lee, D. Lee, M. A. Lumley and K.-S. Choi, Progress on ternary oxide-based photoanodes for use in photoelectrochemical cells for solar water splitting, Chem. Soc. Rev., 2019, 48, 2126 RSC; (b) A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Electrochemistry of Graphene and RelatedMaterials, Chem. Rev., 2014, 114, 7150 CrossRef CAS PubMed; (c) Y.-C. Wu, S.-S. Jiang, R.-J. Song and J.-H. Li, A metal- and oxidizing-reagent-free anodic para-selective amination of anilines with phenothiazines, Chem. Commun., 2019, 55, 4371 RSC; (d) J. H. Kim and J. S. Lee, Elaborately Modified BiVO4 Photoanodes for Solar Water Splitting, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed; (e) X. Zhang, X. Wang, X. Yi, J. Ye and D. Wang, Alkali Treatment for Enhanced Photoelectrochemical Water Oxidation on Hematite Photoanode, ACS Sustainable Chem. Eng., 2019, 7, 5420 CrossRef CAS.
- (a) T. Bach and J. P. Hehn, Photochemical Reactions as Key Steps in Natural Product Synthesis, Angew. Chem., Int. Ed., 2011, 50, 1000 CrossRef CAS; (b) Y.-Q. Zou, F. M. Hörmann and T. Bach, Iminium and enamine catalysis in enantioselective photochemical reactions, Chem. Soc. Rev., 2018, 47, 278 RSC; (c) D. Ravelli, S. Protti and M. Fagnoni, Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates, Chem. Rev., 2016, 116, 9850 CrossRef CAS; (d) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Visible light-driven organic photochemical synthesis in China, Sci. China: Chem., 2019, 62, 24 CrossRef CAS.
- (a) P. Xiong and H.-C. Xu, Chemistry with Electrochemically Generated N-Centered Radicals, Acc. Chem. Res., 2019, 52, 3339 CrossRef CAS PubMed; (b) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Recent Advances in Oxidative R1-H/R2-H Cross-Coupling with Hydrogen Evolution via Photo-/Electrochemistry, Chem. Rev., 2019, 119, 6769 CrossRef CAS PubMed; (c) Y. Jiang, K. Xu and C. Zeng, Use of Electrochemistry in the Synthesis of Heterocyclic Structures, Chem. Rev., 2018, 118, 4485 CrossRef CAS PubMed; (d) K. D. Moeller, Using Physical Organic Chemistry To Shape the Course of Electrochemical Reactions, Chem. Rev., 2018, 118, 4817 CrossRef CAS PubMed; (e) C. Ma, P. Fang and T.-S. Mei, Recent Advances in C–H Functionalization Using Electrochemical Transition Metal Catalysis, ACS Catal., 2018, 8, 7179 CrossRef CAS; (f) Y.-C. Wu, S.-S. Jiang, S.-Z. Luo, R.-J. Song and J.-H. Li, Transition-metal- and oxidant-free directed anodic C–H sulfonylation of N,N-disubstituted anilines with sulfinates, Chem. Commun., 2019, 55, 8995 RSC; (g) Y. Yu, P. Guo, J.-S. Zhong, Y. Yuan and K.-Y. Ye, Merging photochemistry with electrochemistry in organic synthesis, Org. Chem. Front., 2020, 7, 131 RSC.
- (a) S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, How Catalysts and Experimental Conditions Determine the Selective Hydroconversion of Furfural and 5-Hydroxymethylfurfural, Chem. Rev., 2018, 118, 11023 CrossRef CAS PubMed; (b) X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Catalytic conversion of 5-hydroxymethylfurfural to some value-added derivatives, Green Chem., 2018, 20, 3657 RSC.
- H. G. Cha and K.-S. Choi, Combined biomass valorization and hydrogen production in a photoelectrochemical cell, Nat. Chem., 2015, 7, 328 CrossRef CAS PubMed.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Photoelectrochemical oxidation of organic substrates in organic media, Nat. Commun., 2017, 8, 390 CrossRef PubMed.
- (a) H. Tateno, Y. Miseki and K. Sayama, Photoelectrochemical Oxidation of Benzylic Alcohol Derivatives on BiVO4/WO3 under Visible Light Irradiation, ChemElectroChem, 2017, 4, 3283 CrossRef CAS; (b) A. W. Harris, O. Yehezkeli, G. R. Hafenstine, A. P. Goodwin and J. N. Cha, Light-Driven Catalytic Upgrading of Butanol in a Biohybrid Photoelectrochemical System, ACS Sustainable Chem. Eng., 2017, 5, 8199 CrossRef CAS.
- (a) S. Rej, Y. Ano and N. Chatani, Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed; (b) X.-H. Ouyang, R.-J. Song and J.-H. Li, Developments in the Chemistry of α-Carbonyl Alkyl Bromides, Chem. – Asian J., 2018, 13, 2316 CrossRef CAS PubMed.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Photo-Electrochemical C–H Bond Activation of Cyclohexane Using a WO3 Photoanode and Visible Light, Angew. Chem., Int. Ed., 2018, 57, 11238 CrossRef CAS PubMed.
- D. S. Choi, H. Lee, F. Tieves, Y. W. Lee, E. J. Son, W. Zhang, B. Shin, F. Hollmann and C. B. Park, Bias-Free In Situ H2O2 Generation in a Photovoltaic-Photoelectrochemical Tandem Cell for Biocatalytic Oxyfunctionalization, ACS Catal., 2019, 9, 10562 CrossRef CAS.
- P. Lenden, D. A. Entwistle and M. C. Willis, An Alkyne Hydroacylation Route to Highly Substituted Furans, Angew. Chem., Int. Ed., 2011, 50, 10657 CrossRef CAS PubMed.
- H. Tateno, Y. Miseki and K. Sayama, Photoelectrochemical dimethoxylation of furan via a bromide redox mediator using a BiVO4/WO3 photoanode, Chem. Commun., 2017, 53, 4378 RSC.
- (a) G. S. Sauer and S. Lin, An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes, ACS Catal., 2018, 8, 5175 CrossRef CAS; (b) C. Wan, R.-J. Song and J.-H. Li, Electrooxidative 1,2-Bromoesterification of Alkenes with Acids and N-Bromosuccinimide, Org. Lett., 2019, 21, 2800 CrossRef CAS PubMed; (c) M. D. Kärkäs, Electrochemical strategies for C–H functionalization and C–N bond formation, Chem. Soc. Rev., 2018, 47, 5786 RSC.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Photoelectrocatalytic arene C–H amination, Nat. Catal., 2019, 2, 366 CrossRef CAS PubMed.
- (a) M. A. Shameem and A. Orthaber, Organophosphorus Compounds in Organic Electronics, Chem. – Eur. J., 2016, 22, 10718 CrossRef CAS PubMed; (b) L. Chen, Y.-X. Zou, X.-Y. Liu and X.-J. Gou, Dehydrative Cross-Coupling and Related Reactions between Alcohols (C−OH) and P(O)−H Compounds for C–P Bond Formation, Adv. Synth. Catal., 2019, 361, 3490 CrossRef CAS.
- J.-H. Wang, X.-B. Li, J. Li, T. Lei, H.-L. Wu, X.-L. Nan, C.-H. Tung and L.-Z. Wu, Photoelectrochemical cell for P–H/C–H cross-coupling with hydrogen evolution, Chem. Commun., 2019, 55, 10376 RSC.
- M. Gong, J. K. Kim, X. Zhao, Y. Li, J. Zhang, M. Huang and Y. Wu, Visible-light-induced α-oxyamination of 1,3-dicarbonyls with TEMPO via a photo(electro)catalytic process applying a DSSC anode or in a DSSC system, Green Chem., 2019, 21, 3615 RSC.
| This journal is © the Partner Organisations 2020 |