
Anthranils: versatile building blocks in the construction of C–N bonds and N-heterocycles
Yang
Gao
 *a,
Jianhong
Nie
a,
Yanping
Huo
a and
Xiao-Qiang
Hu
*b
*a,
Jianhong
Nie
a,
Yanping
Huo
a and
Xiao-Qiang
Hu
*b
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, 510006, China. E-mail: gaoyang@gdut.edu.cn
bKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education & Hubei Key Laboratory of Catalysis and Materials Science, School of Chemistry and Materials Science, South-Central University for Nationalities, Wuhan 430074, China. E-mail: huxiaoqiang@mail.scuec.edu.cn
First published on 9th April 2020
Abstract
Anthranils are readily available and have recently emerged as versatile building blocks in the assembly of various C–N bonds and medicinally active heterocyclic systems. A great variety of amination reactions, including cross-coupling reactions, C–H aminations and annulation reactions, have been intensively developed with the use of anthranils as aminating synthons. The success of these impressive reactions demonstrates the great synthetic potential of anthranils, which provides a powerful platform for C–N bond formation and heterocycle synthesis. In this review, we mainly summarize the recent progress in the representative transformations of anthranils with special emphasis on mechanistic aspects. We hope that a deep understanding of the unique properties of anthranils and the underlying working mechanism can provide a guideline for researchers who are interested in anthranil chemistry, leading to further exploration of novel and efficient catalytic systems for C–N bond construction.
 Yang Gao | Yang Gao received his PhD from South China University of Technology (SCUT) under the supervision of Prof. Huanfeng Jiang (2011–2017). He also studied as a graduate trainee at McGill University under the supervision of prof. Chao-jun Li (2016–2017). Then, Dr Gao did his postdoctoral research with Prof. Lukas J Goossen at Ruhr-Universität Bochum (2018–2019). Since 2017, Dr Gao has been working in Guangdong University of Technology (GDUT) as an associate professor, and his research interest focuses on transition metal-catalyzed C–N coupling and metallaphotocatalysis. |
 Jianhong Nie | Jianhong Nie received his B.E. degree in architecture from Tian College of Guangdong Polytechnic Normal University in 2014. He then started his graduate research in Guangdong University of Technology as a M.E. candidate under the supervision of Prof. Yang Gao and Prof. Xianwei Li. His research topic focused on catalytic C–H functionalization. |
 Yanping Huo | Yanping Huo is a Professor of the School of Chemical Engineering and Light Industry, Guangdong University of Technology (GDUT), China. He received his Ph.D. from Guangzhou Institute of Chemistry, Chinese Academy of Science, China (2006). He had been a visiting scholar in Shanghai Institute of Organic Chemistry, Chinese Academy of Science (SIOC) (2009) and a research associate in the Department of Chemistry, The University of Hong Kong (2010–2011). His research interests include the design, synthesis, and characterization of organic light-emitting materials. |
 Xiao-Qiang Hu | Xiao-Qiang Hu was born in 1988. He received his B.S. from Wuhan Polytechnic University in 2011. In 2017, he received his Ph.D. under the direction of Prof. Wen-Jing Xiao and Jia-Rong Chen at Central China Normal University (CCNU). After his postdoctoral research (Ruhr Explores Solvation) with Prof. Lukas J. Goossen at Ruhr-Universität Bochum (2017–2019), Dr Hu became a full professor at the School of Chemistry and Materials Science, South-Central University for Nationalities (SCUEC), China. His research interests include inert C–H bond activation, radical cascade reaction and the synthesis of biologically active heterocycles. |
1. Introduction
The C–N bond constitutes one of the most abundant and important chemical bonds in myriads of natural products, and pharmaceutical and materials molecules.1 The development of novel catalytic systems and new aminating reagents for efficient C–N bond formation has attracted considerable attention of synthetic chemists. Anthranils, also known as 2,1-benzisoxazoles, are an important class of easily accessible and highly versatile synthons, which exhibit rich and tunable reactivities especially in transition metal-catalysed processes. They are generally bench stable because they satisfy the Hückel rule. However, at a relatively high temperature, anthranils could be transferred to aryl nitrene or ketene species through a thermolytic N–O bond cleavage (Scheme 1A).2 The thermolysis of anthranils was generally controlled by the substituents at the 3-position of anthranils. Anthranils without any substituents at the 3-position give ketene species and 3-aryl substituted anthranils afford aryl nitrene intermediates. In recent years, the development of transition metal catalysis (Au, Rh, Co, Cu, Ni etc.) has been providing a powerful tool for the activation of anthranils and enabling the conversion of these useful synthons into highly reactive metal nitrene species for the rapid synthesis of various promising high-value N-containing molecules (Scheme 1B). In this context, anthranils could serve as efficient electrophilic aminating reagents in transition metal-catalysed cross-coupling reactions with organometallic reagents or C–H amination reactions. Additionally, in the presence of a gold catalyst, anthranils could act as N-nucleophiles to react with alkynes to in situ generate α-imino gold carbene complexes for further amination transformations (Scheme 1C). Moreover, anthranils are also good 1,3-dipolar synthons because of the incorporation of a nucleophilic amino group and an electrophilic formyl group in one molecule. Accordingly, various important [4 + 3] and [4 + 2] annulations of anthranils have been developed towards the synthesis of diversely functionalized heterocycles (Scheme 1D).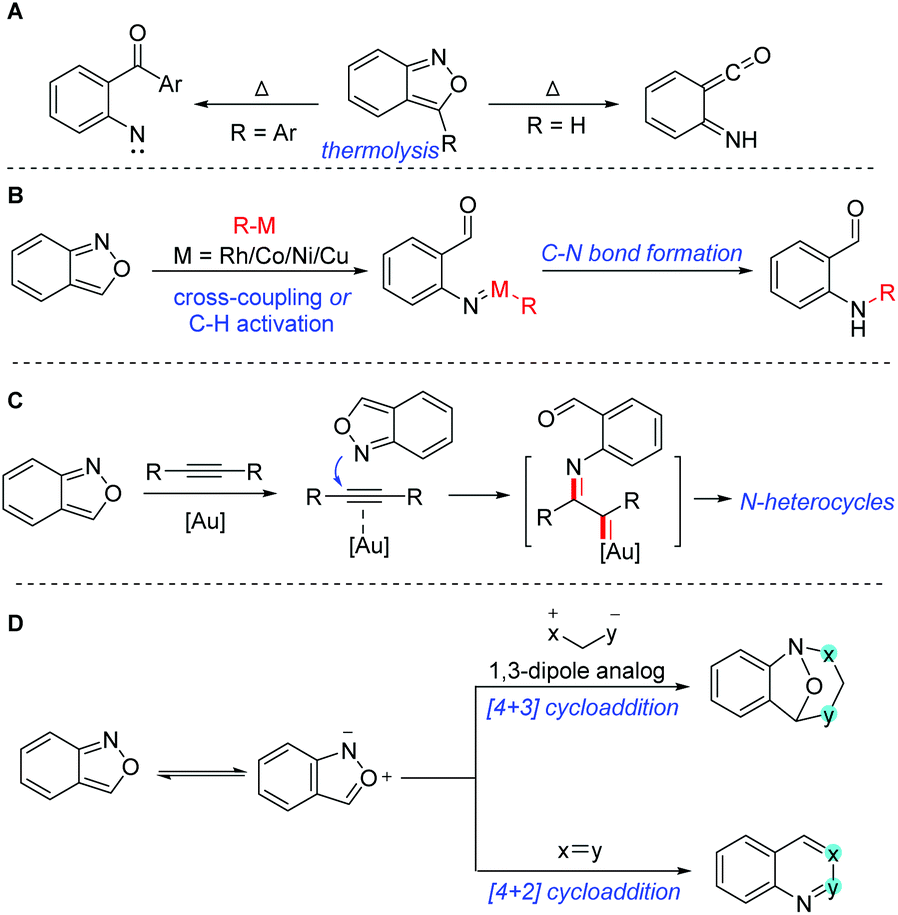 | ||
| Scheme 1 General reactivity of anthranils. | ||
To the best of our knowledge, there is still no review devoted to this fantastic area so far. Herein we highlight the recent advances in the representative transformations of anthranils with particular emphases on mechanistic aspects. To better calibrate the scope of this topic, this review is mainly organized according to the different reaction modes of anthranils.
2. Preparation of anthranils
Anthranils 1 are easily available in great structural diversity using some well-established synthetic routes (Scheme 2), which make them extremely promising raw materials in organic synthesis. Traditionally, anthranils were generated from the semi-reduction of O-nitrobenzaldehyde 2 by SnCl2.3 However, this method is not sustainable due to the use of toxic SnCl2 as a stoichiometric reductant. Hence, H2 as a green reductant has been used for the semi-reduction of O-nitrobenzaldehydes. In 2019, Climent and co-workers reported a Pt-supported nanoparticle catalysed hydrogen reduction of O-nitrobenzaldehyde for the selective formation of 2,1-benzisoxazoles in generally good yields.4 Besides, the selective oxidation of 2-aminobenzophenone 3 provides an alternative approach towards anthranils.5 The thermolysis or metal-catalysed decomposition of 2-azidobenzaldehyde 4 could also afford anthranils.6 Moreover, palladium-catalysed direct C–H arylation could be applied for the further functionalization of anthranils to synthesize 3-aryl substituted anthranils 5.7 These practical and efficient methods make anthranils cheap and readily available feedstocks.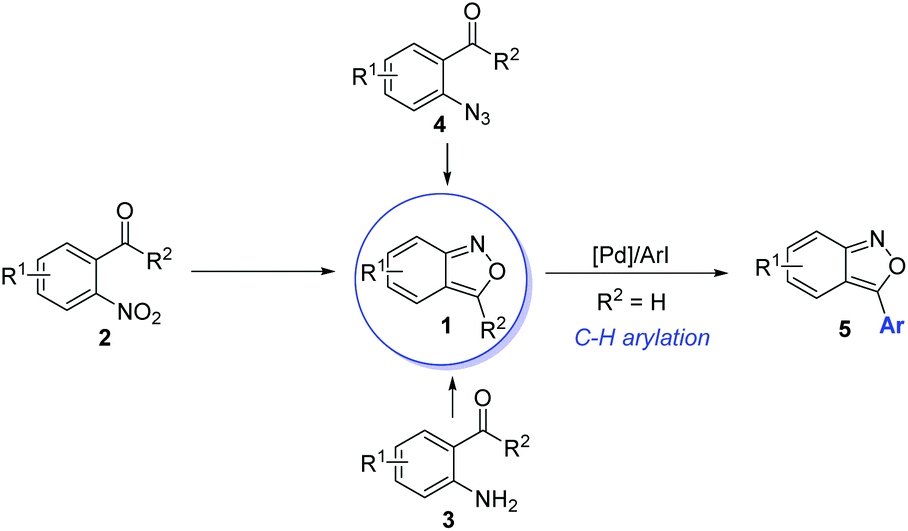 | ||
| Scheme 2 Methods for the preparation of anthranils. | ||
3. Transition metal-catalysed C–N bond formation through electrophilic aminations with anthranils
Transition metal-catalysed cross-coupling reactions of anthranils with organometallic reagents have been developed as an efficient route to construct C–N bonds. Anthranils served as electrophilic aminating reagents in these reactions through a selective N–O bond cleavage. These reactions usually feature mild reaction conditions and high efficiency. The first example could be tracked back to 1987.8 Baum and co-workers disclosed that anthranils could react with RZnCl 6 for the construction of aniline derivatives 7 in the presence of a nickel catalyst (Scheme 3).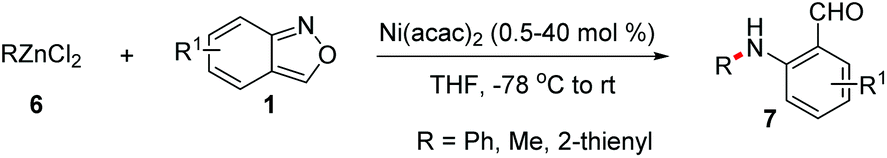 | ||
| Scheme 3 Ni-Catalysed amination of RZnCl with anthranils. | ||
In 2019, this reaction was further improved by Knochel and co-workers by using more stable organozinc pivalates instead of RZnCl and CoCl2 as the catalyst under mild reaction conditions (Scheme 4).9 A series of organozinc pivalates 8 and anthranils 1 bearing different functional groups could take part in the reaction and various aniline derivatives 10 were obtained in good yields. Significantly, a cascade intramolecular condensation can be achieved for the formation of various useful quinoline derivatives 11 and 12, when using alkenylzinc pivalates, electron-rich arylzinc pivalates or heterocyclic zinc pivalates as substrates. In addition, the photophysical properties of the products were tested. It was found that these N-heterocycles are of particular interest for organic light emitting diodes, because of their high photoluminescence quantum yields and long exciton lifetimes.
 | ||
| Scheme 4 Co-Catalysed amination of organozinc pivalates with anthranils. | ||
In 2018, the group of Li developed an elegant asymmetric hydroamination of olefins with the use of anthranils as electrophilic aminating reagents (Scheme 5).10 In this transformation, CuOAc and PhSiH3 were used to generate the highly reactive CuH species and (S,S)-Ph-BPE proved to be the best ligand. This protocol provided an efficient and highly enantioselective method to construct chiral secondary arylamines tethered to a benzylic alcohol 14. A plausible mechanism is outlined in Scheme 5. Initially, the active L*CuH species 13a was generated from CuOAc bearing a chiral phosphine ligand and PhSiH3via σ-bond metathesis. Styrene then readily inserted into the Cu–H bond of 13a in a highly enantioselective and regioselective fashion, providing the alkylcopper intermediate 13b. An oxidative insertion of the N–O bond of anthranil by intermediate 13b afforded 13c, which was essentially a copper nitrene species. Intermediate 13c underwent a sequential migratory insertion and protonation process to generate the aldehyde intermediate 13e and the L*CuOtBu species. The further reduction of aldehyde 13e by L*CuH gave the final products.
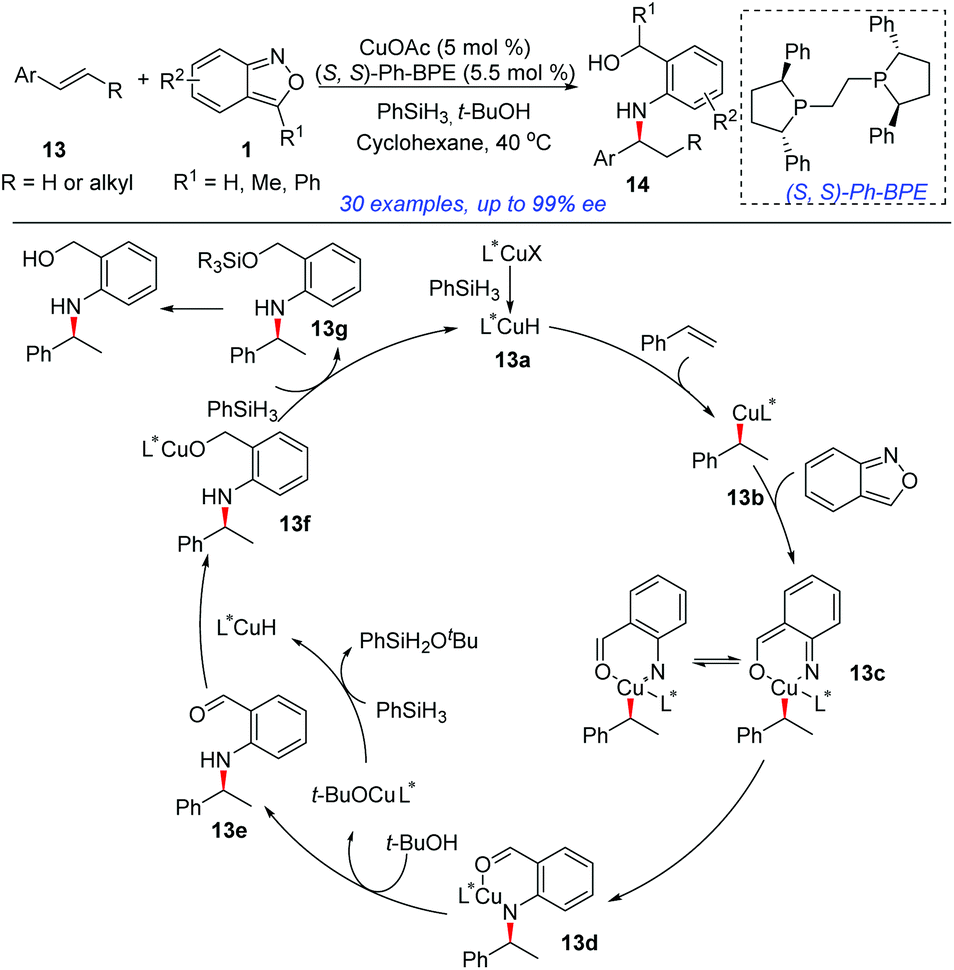 | ||
| Scheme 5 Cu-Catalysed asymmetric hydroamination of olefins with anthranils. | ||
4. Transition metal-catalysed C–H aminations with anthranils
Transition metal-catalysed C–H bond aminations have been established as a straightforward approach to construct C–N bonds.11 With the assistance of directing groups, the direct amination of both C(sp2)–H and C(sp3)–H bonds has been achieved with anthranils. Notably, the C–H aminations involving anthranils generally result in arylamine products bearing an electrophilic formyl group, which may undergo a further intramolecular annulation to construct highly functionalized N-heterocycles. These C–H amination reactions are usually achieved by using Rh and Co catalysts. However, other common transition metal catalysts such as [Cp*lrCl2]2, [{Ru(p-cymene)Cl2}]2, and Pd(OAc)2 have been proved to be unsuitable for this type of reaction at the current stage.4.1 Rh-Catalysed C–H amination with anthranils
In 2016, Li and Jiao's group independently disclosed a Rh-catalysed benzylic C(sp3)–H amination of 8-methylquinoline 15 with anthranils 1 for the first time (Scheme 6).12 This transformation is compatible with a variety of functional groups and various aminated products tethered with a formyl group were obtained in good yields. The large KIE value (parallel 7.3 and competitive 8.1) indicated that the C–H/C–D dissociation was involved in the rate-determining step. Based on the results of mechanistic studies, a plausible mechanism was proposed. Firstly, the rhodacycle intermediate 15a was formed through the C(sp3)–H activation of 8-methylquinoline with the reactive Rh(III) catalyst, and then the coordination of anthranil took place to afford 15b. The N–O bond cleavage of anthranil occurred to generate Rh(V)-nitrenoid species 15c. Subsequent nitrenoid insertion in the Rh–C bond led to amido rhodium complex 15d. Finally, the expected amination product was released via a protodemetalation process of intermediate 15d with the regeneration of the reactive Rh(III) species.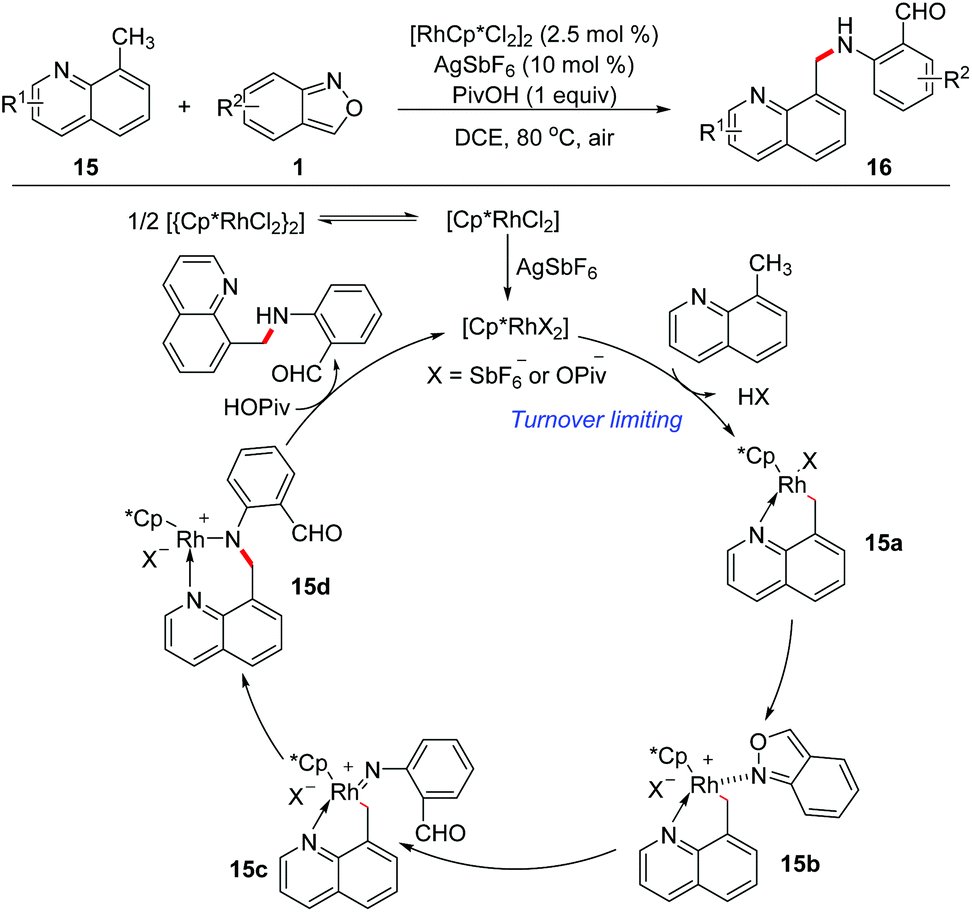 | ||
| Scheme 6 Rh-Catalysed C(sp3)–H amination of 8-methylquinoline with anthranils. | ||
The C(sp2)–H amination of arenes has also been achieved with anthranils as aminating reagents. Oxime ether and other N-containing heterocycles such as pyridine, quinoline, purine, and pyrazole derivatives could act as directing groups in this Rh-catalysed C(sp2)–H amination reaction (Scheme 7).13 A series of amination products, which are useful precursors for various important N-heterocycles, were obtained in moderate to good yields.
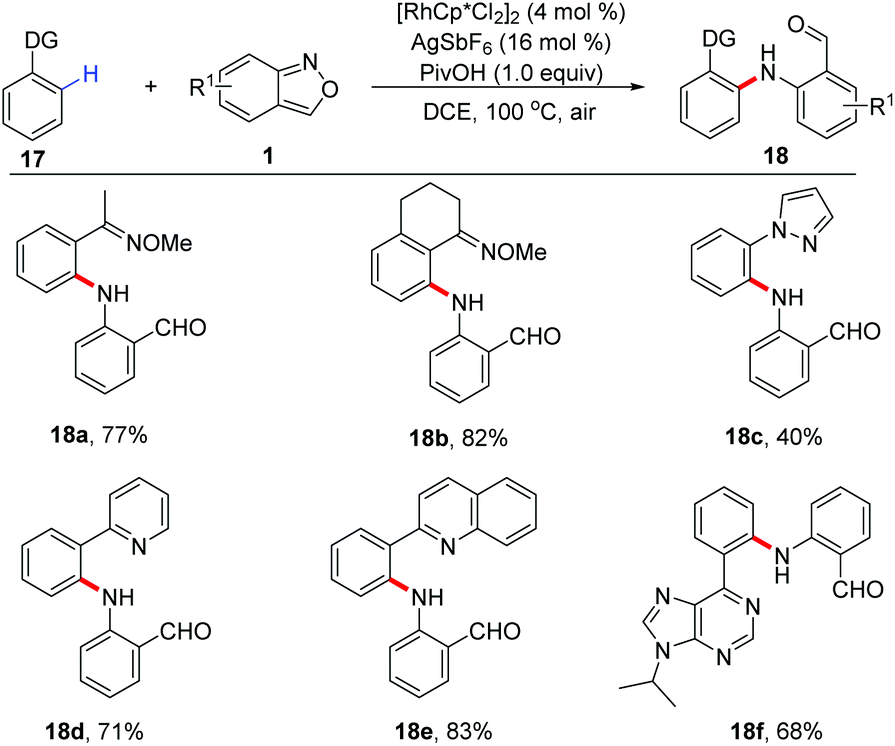 | ||
| Scheme 7 Rh-Catalysed sp2 C–H amination of arenes with anthranils. | ||
In 2017, a Rh(III)-catalysed C–H amination of benzamides and isoquinolones 19 with anthranils has been realized by Li and co-workers.14 The weakly coordinated tertiary amide served as a directing group in this transformation. This C–H amination protocol provided an efficient method to synthesize various bifunctionalized amination products 20, which can further cyclize to give acridines by treating with TFA. Moreover, when 3-unsubstituted anthranils were used in this reaction, the corresponding acridine derivatives 21 were obtained in one pot through a subsequent C–H amination/condensation process (Scheme 8).
 | ||
| Scheme 8 Rh-Catalysed C–H amination of arenes with anthranils using amides as directing groups. | ||
The site-selective C–H amination of 7-azaindoles 22 with anthranils 1 was reported by Kim and co-workers (Scheme 9).15 In the presence of a Rh(III) catalyst, ortho-amination of 7-azaindoles can be successfully achieved. This transformation efficiently provided a range of ortho-aminated N-aryl-7-azaindoles 23 with excellent site-selectivity and functional group compatibility. The ortho-aminated 7-azaindoles formed were readily transformed into biologically relevant heterocycles such as azaindoloacridine, azaindoloacridone and bis-indole compounds. More importantly, the synthetic potential of this methodology was further demonstrated by its good performance in in vitro anticancer activity against human breast adenocarcinoma cells (MCF-7), human renal carcinoma cells (786-O), and human prostate adenocarcinoma cells (DU145).
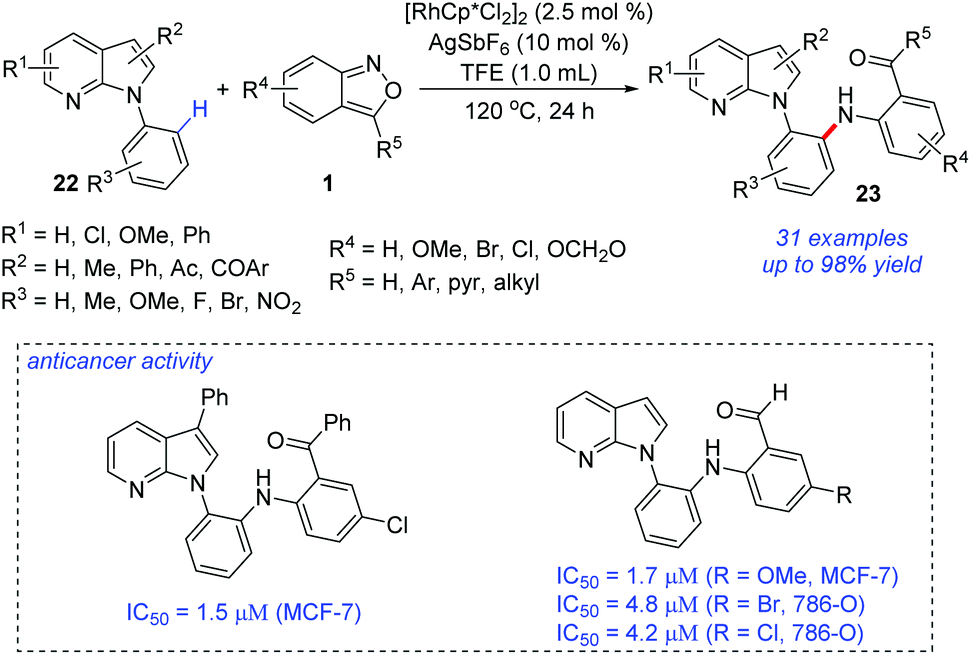 | ||
| Scheme 9 Rh-Catalysed site-selective C–H amination of 7-azaindoles with anthranils. | ||
Likewise, Yang and Kim independently reported a Rh(III)-catalysed C-7 selective amination of indolines (Scheme 10).16 Pyrimidinyl served as a directing group in this reaction and various C-7 aminated indolines 25 bearing different functional groups were obtained in generally good yields with excellent site-selectivity. The incorporation of amino and carbonyl groups into the resulting framework provided opportunities for further derivatization.
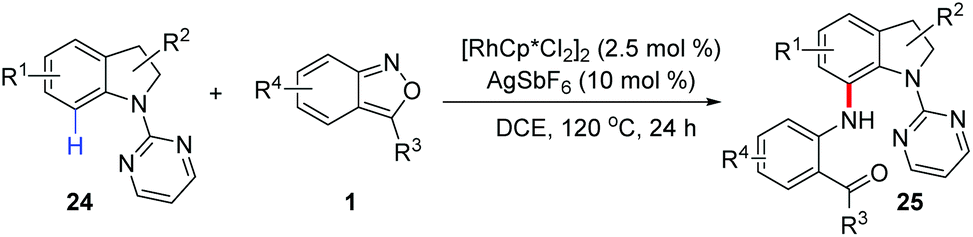 | ||
| Scheme 10 Rh-Catalysed C-7 selective amination of indolines with anthranils. | ||
In 2018, a rhodium-catalysed regioselective C–H amination of azobenzenes 26 with anthranils 1 was developed by Zhou and co-workers (Scheme 11).17 This reaction showed excellent functional tolerance and a range of aminated products were obtained in good yields (up to 92%). Moreover, this transformation can be further applied to the synthesis of various N-heterocycles such as 5H-dibenzo[b,e][1,4]diazepine, acridine and indole derivatives, which are biologically interesting compounds.
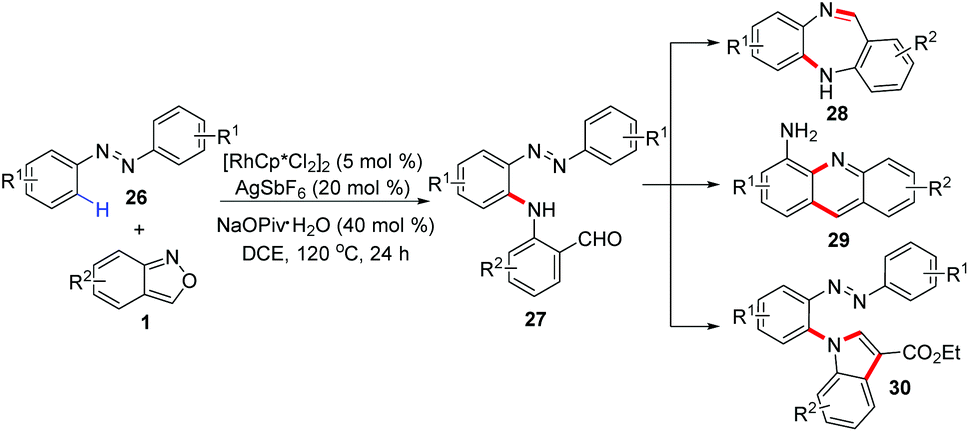 | ||
| Scheme 11 Rh-Catalysed regioselective C–H amination of azobenzenes with anthranils. | ||
Amide bonds are ubiquitous in a wide range of pharmaceuticals, agrochemicals, and natural products. In 2017, Maji and co-workers explored a Cp*RhIII-catalysed directed amidation of N-sulfonyl-2-aminobenzaldehyde and salicylaldehyde with anthranils for the synthesis of amide derivatives 32 (Scheme 12).18 This transformation went through a C–H activation of the aldehyde by using –NHTs or –OH as the directing group. A wide range of substrates bearing various functional groups were well-tolerated under the mild reaction conditions. Significantly, the synthesized amides can be further used as important precursors for the preparation of benzoxazinone derivatives, which are bioactive natural products.
 | ||
| Scheme 12 RhIII-Catalysed directed amidation of N-sulfonyl-2-aminobenzaldehyde and salicylaldehyde with anthranils. | ||
Anthranils could serve as bifunctional reagents due to their ability to deliver a nucleophilic amino group and an electrophilic formyl group. Accordingly, the C–H amination/annulation cascade reactions involving anthranils have been developed for the efficient construction of various useful N-heterocycles. In 2016, Li and Wang et al. independently reported a Rh(III)-catalysed C–H amination/annulation of indoles 33 with anthranils (Scheme 13).19 This reaction provided a facile access to quinoline-fused heterocycles 35 and 36 under redox-neutral conditions. The direct C–H amination was a key step for this transformation to generate diarylamines bearing a proximal carbonyl group, which was followed by a Friedel–Crafts-type addition/dehydration cascade to give the corresponding bicyclic heterocycles.
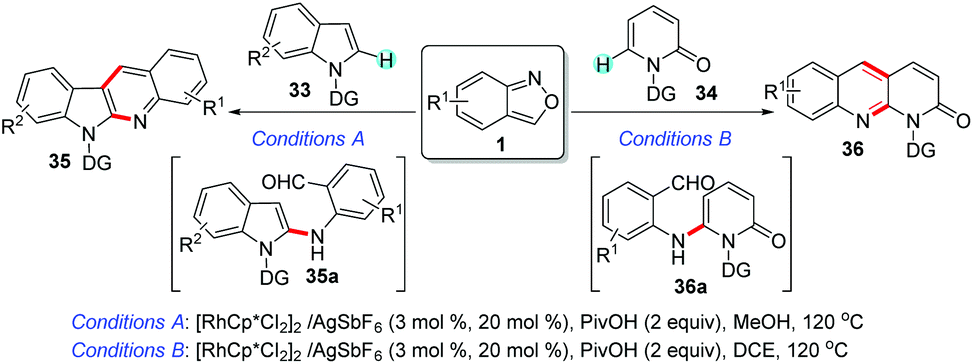 | ||
| Scheme 13 Rh(III)-Catalysed C–H activation/annulation of indoles with anthranils. | ||
Recently, Samanta and co-workers developed a RhIII-catalysed C4-selective amination/annulation of indole derivatives 37 with anthranils for the rapid synthesis of indoloquinoline derivatives 38 (Scheme 14).20 In this reaction, aldoximes were used as the directing group, which could be converted to cyano groups at the end of the reaction. Mechanistic studies revealed the important role of the newly generated azacycle in the transformation of O-protected aldoximes into cyano derivatives. Additionally, the photophysical properties of the indoloquinoline derivatives obtained were also studied.
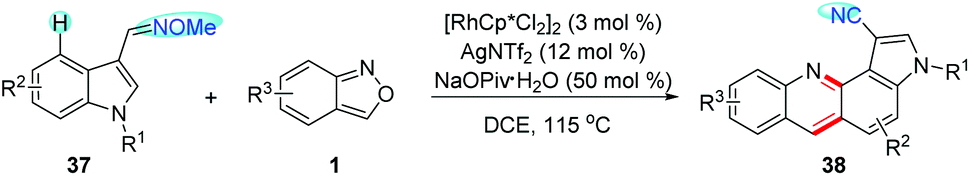 | ||
| Scheme 14 Rh(III)-Catalysed C4-selective amination/annulation of indole derivatives with anthranils. | ||
Using a similar C–H amination/annulation strategy, an efficient rhodium-catalysed method for the synthesis of benzophenanthroline 40 and benzophenanthrolinone 41 derivatives from quinoline N-oxides 39 and anthranils have been developed by the same group (Scheme 15a and b).21 This approach was achieved in a two-step one-pot manner and a broad substrate scope was obtained. For the synthesis of benzophenanthrolines 40, TFA or PCl3 was added to induce the intramolecular condensation of firstly generated C–H aminated products. In addition, benzophenanthrolinones 41 were synthesized through a TBHP-promoted intramolecular cross dehydrogenation coupling (CDC) reaction. Importantly, this method could be used in late-stage modifications of complex bioactive molecules such as cinchonidine 39a and cinchophen N-oxide 39b (Scheme 15c and d). Notably, a drug candidate for NHE related disease was obtained based on this method (Scheme 15e). Later, they achieved a similar C–H amination of quinoline N-oxides by using a cheap copper catalyst.22
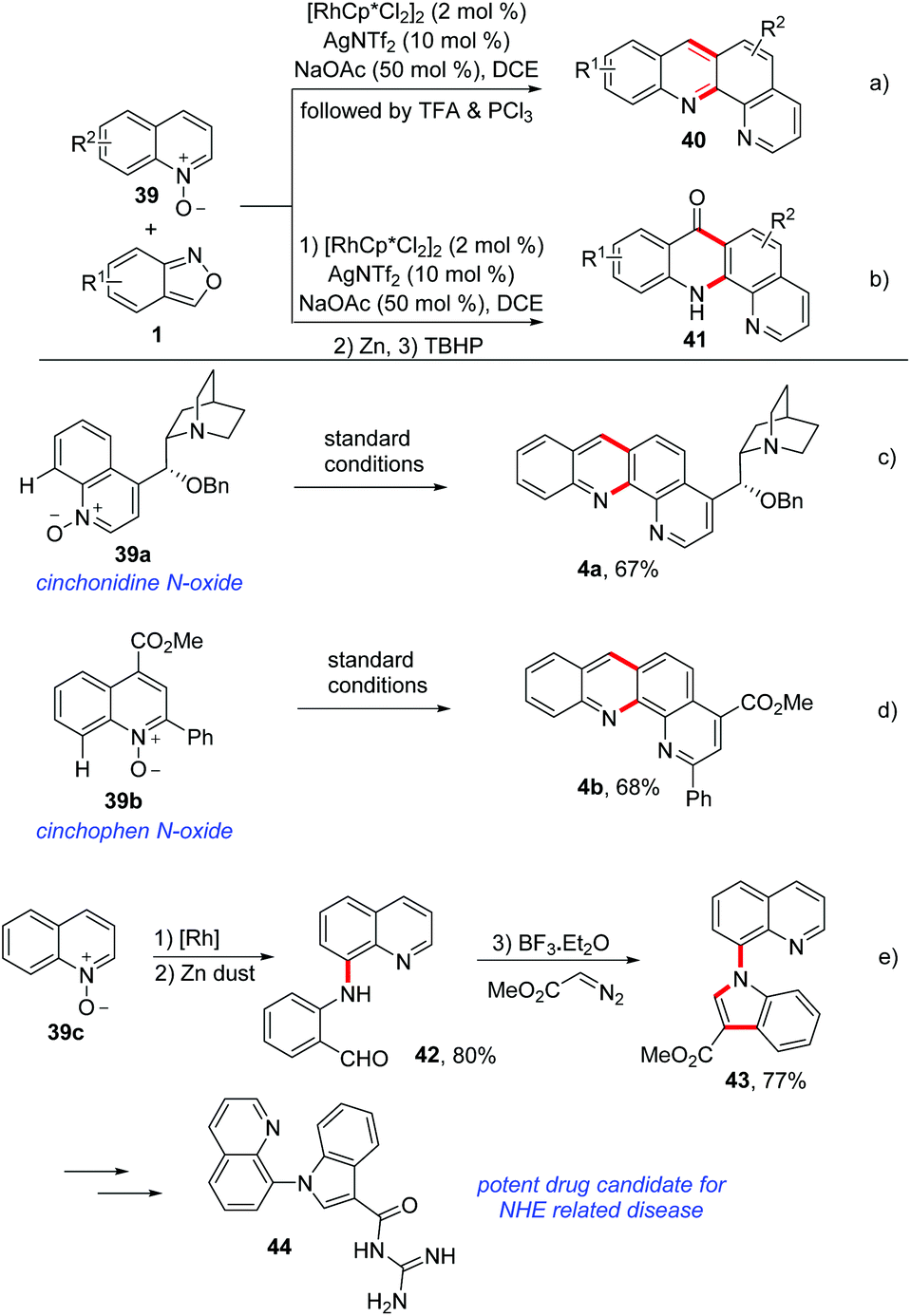 | ||
| Scheme 15 Rh(III)-Catalysed C–H activation/annulation of quinoline N-oxides with anthranils. | ||
The application of a transient directing group is an attractive strategy in metal-catalysed C–H activation, since it avoids the additional steps to remove the directing group and increases the synthetic practicality. In 2017, the group of Kim reported a Rh-catalysed C(sp2)–H amination of benzaldehydes 45 with anthranils (Scheme 16).23 Notably, the in situ-generated O-aminophenyl ketones from anthranils served as transient directing groups in this reaction. As a result, a series of 2-acyl acridines 46 were obtained through a cascade C–H amination and acid-mediated intramolecular cyclization. Later, by using sulfonimide as the directing group, they also completed the synthesis of 2-acyl acridines from aldimines and anthranils.24
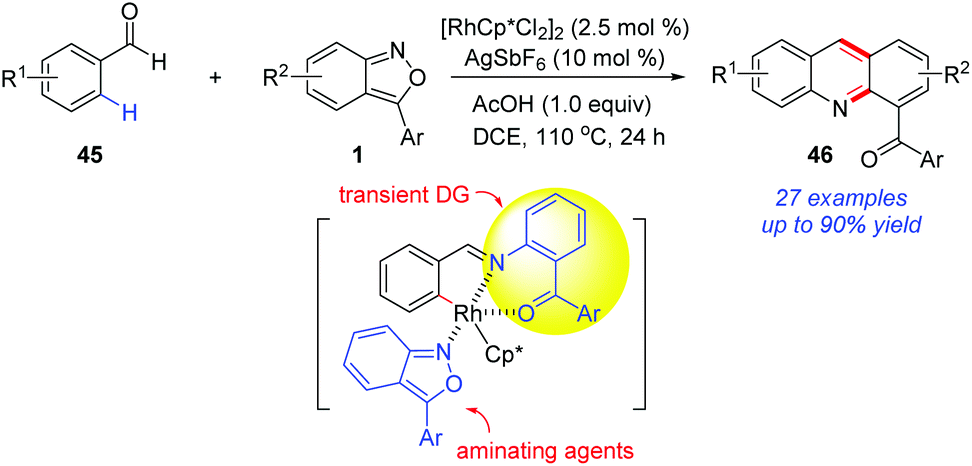 | ||
| Scheme 16 Rh-Catalysed C(sp2)–H amination of benzaldehydes with anthranils using a transient directing group strategy. | ||
Very recently, Cui et al. disclosed a Rh(III)-catalysed C–H amination and cascade annulation reaction for the synthesis of benzimidazole derivatives (Scheme 17).25 In this protocol, imidamide acted as the directing group and enabled further intramolecular annulation to construct the benzimidazoles. When para-substituted benzimidazoles were used, the diamination products 48 were obtained, which could be further converted into imidazo[4,5-c]acridines 49 with unique fluorescence properties (Scheme 17a). When ortho- or meta-substituted benzimidazoles were used in this catalytic system, the second amination process was totally blocked and the corresponding 1,2-disubstituted benzimidazoles 50 were obtained in good yields (Scheme 17b).
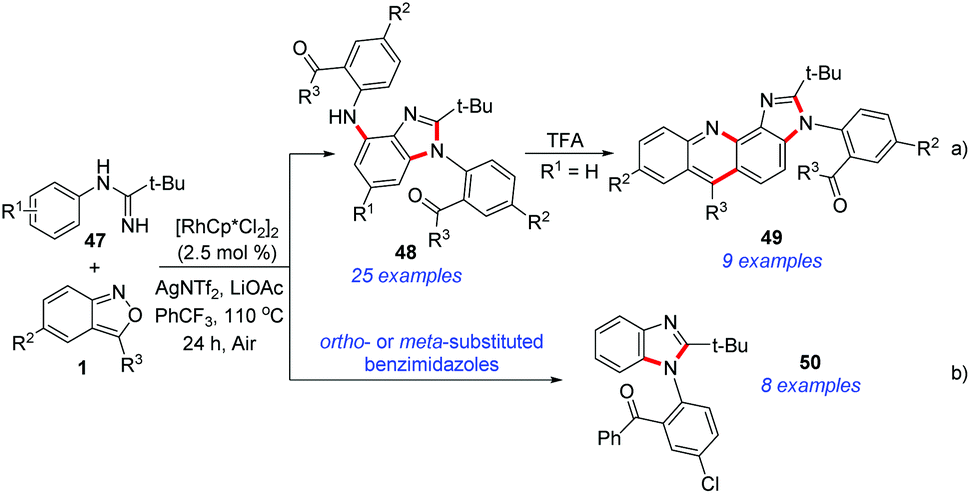 | ||
| Scheme 17 Rh-Catalysed C–H amination/annulation of N-phenylpivalimidamide and anthranils. | ||
Similarly, Wu and co-workers found that when the imidamide directing group bearing a methylene moiety was used, the 1,2-disubstituted benzimidazoles 53 formed could further undergo an intramolecular cyclization to construct benzimidazo[1,2-a]quinolines 52 (Scheme 18).26 This cascade reaction proceeded via a C–H amination/cyclization/cyclization process in an ionic liquid without any additives. A wide range of benzimidazo[1,2-a]quinolines bearing different functional groups were synthesized from readily available imidamides 51 and anthranils. This reaction features simple operation, moderate-to-high yield, and broad substrate scope.
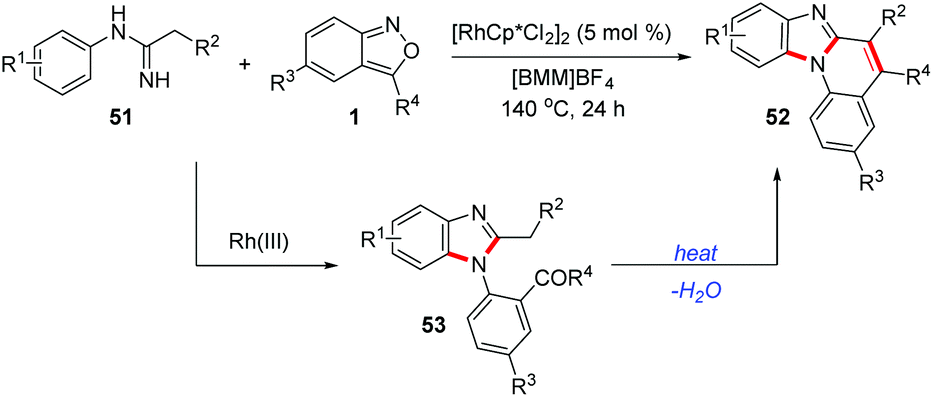 | ||
| Scheme 18 Rh-Catalysed synthesis of benzimidazo[1,2-a]quinolones from imidamides and anthranils. | ||
In 2019, a Rh(III)-catalysed C–H amination/annulation protocol of aroyl sulfoxonium ylides 54 and anthranils has been developed for the synthesis of 10H-indolo[1,2-a]indol-10-one derivatives 55 (Scheme 19).27 This transformation was initiated by an ortho-C–H amination of α-aroyl sulfoxonium ylides with anthranils to generate 54d. Subsequently, the insertion of the N–H bond to the in situ-formed Rh carbene species 54f led to an unprecedented [4 + 1] annulation toward N-(2-formylphenyl) indolones 54h. Finally, the aldol condensation of 54h constructed the second indole ring. A series of 10H-indolo[1,2-a]indol-10-one derivatives, which are difficult to be obtained by other means, were synthesized using this novel method.
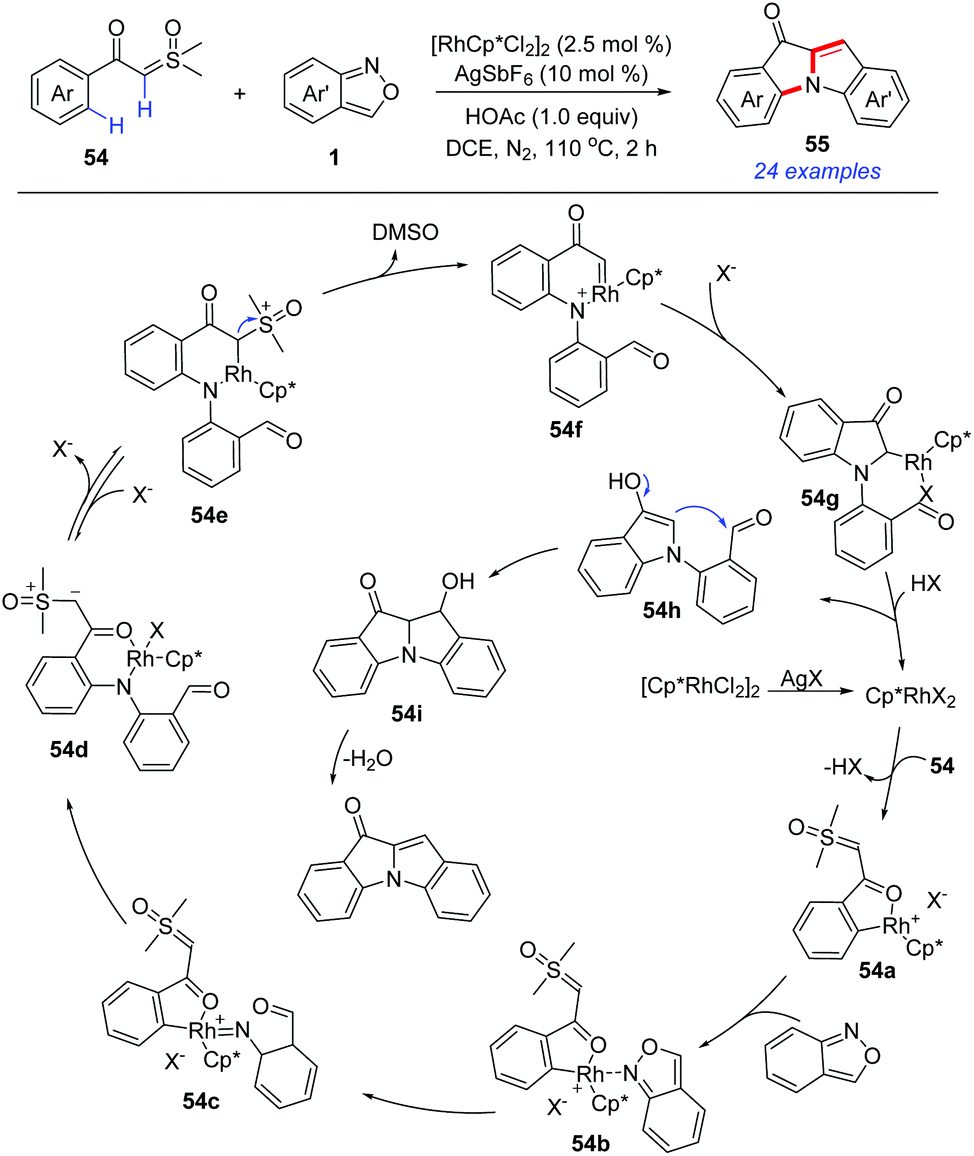 | ||
| Scheme 19 Rh-Catalysed C–H annulation of α-aroyl sulfoxonium ylides with anthranils. | ||
4.2 Co-Catalysed C–H amination with anthranils
The applications of cost-effective, Earth-abundant, and functionally unique first-row metal catalysts in C–H activation have attracted much attention. Pioneered by Kanai and Matsunaga, Cp*Co(III) complexes have allowed the development of new catalytic systems of C–H activation.28 In 2016, a cooperative cobalt- and copper-catalysed oxidative coupling of imidate esters 56 with anthranils was reported by Li and co-workers (Scheme 20).29 In the presence of [Cp*Co(MeCN)3](SbF6)2, the selective ortho-C–H amination of imidate esters could be achieved smoothly with the use of anthranils as a convenient aminating reagent. Subsequently, a copper catalysed intramolecular N–N bond formation occurred rapidly to deliver the final 1H-indazoles products 57. The current reaction provides an efficient method for the synthesis of various 1H-indazoles in moderate to good yields. Anthranils proved to be the terminal organic oxidant in this transformation.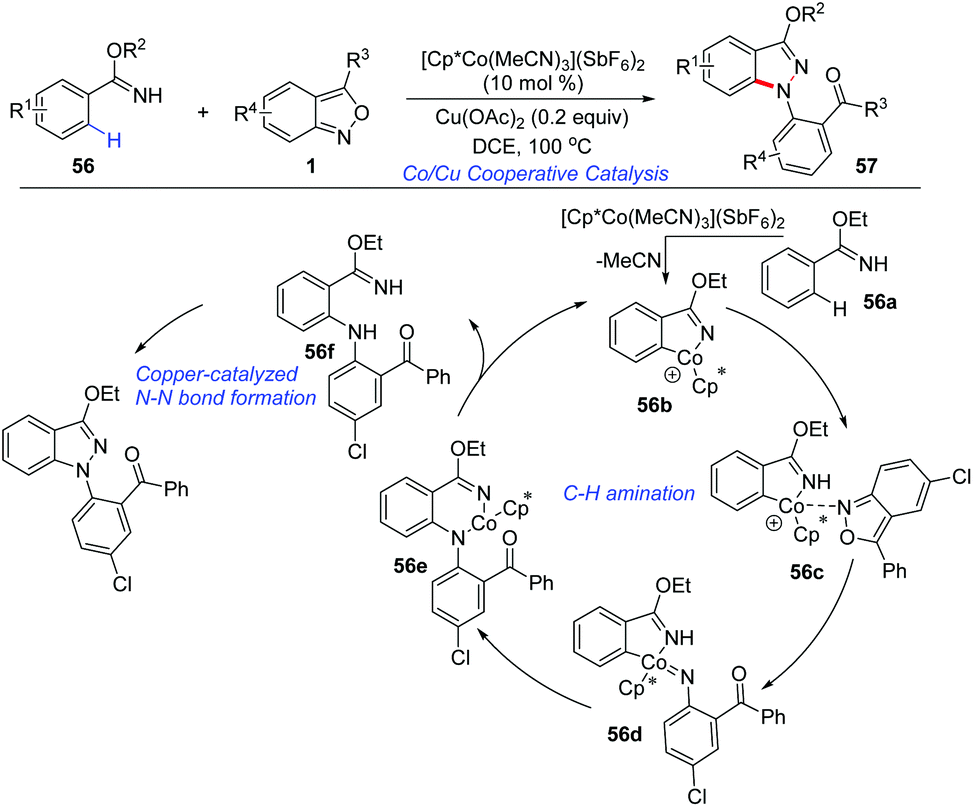 | ||
| Scheme 20 Co-Catalysed C–H amination of imidate esters with anthranils. | ||
In addition, Co-catalysed C(sp3)–H bond amination has also been achieved (Scheme 21). In 2019, Loh and co-workers reported an interesting [Cp*Co(MeCN)3](SbF6)2 catalysed C(sp3)–H amination of thioamides 58 with anthranils.30 The excellent site-selectivity on primary β C(sp3)–H bonds was observed for a diverse array of thioamides with high functional group tolerance.
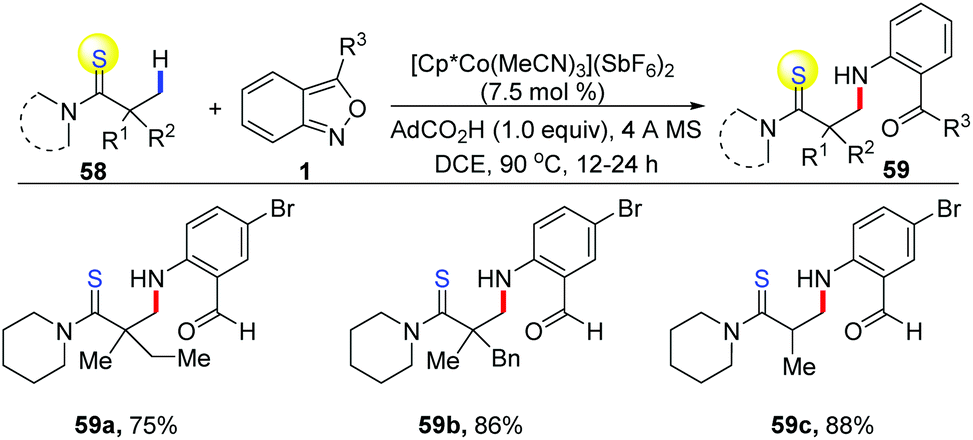 | ||
| Scheme 21 Co-Catalysed C(sp3)–H bond amination with anthranils. | ||
5. Annulation reaction of anthranils
Anthranils are also good 1,3-dipolar reagents for various annulation reactions because of the incorporation of a nucleophilic amino group and an electrophilic formyl group in one molecule (Scheme 22). The efficient [4 + 3] cycloaddition of anthranils with 1,3-dipole substrates has been developed for the synthesis of biologically active benzazepine and benzodiazepine derivatives. Moreover, anthranils could participate in the cascade [4 + 2] annulation for the rapid synthesis of quinoline derivatives and various benzoheterocycles.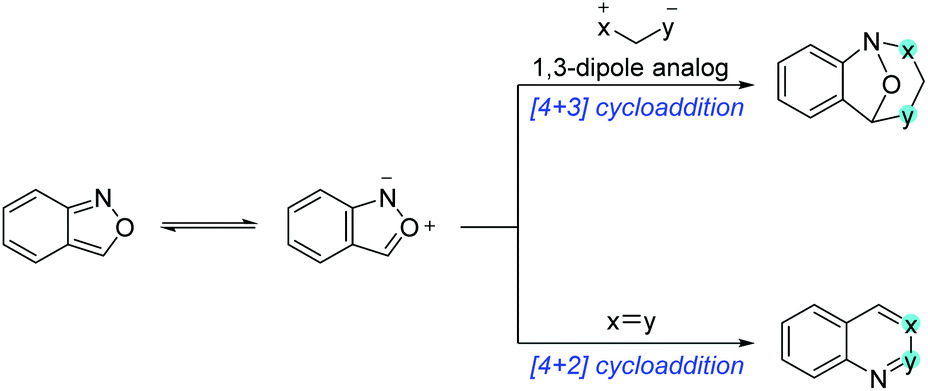 | ||
| Scheme 22 Annulation reaction of anthranils. | ||
5.1 [4 + 3] cycloaddition of anthranils with 1,3-dipole analogs
Seven-membered N-heterocyclic compounds, such as benzodiazepine and tetrahydro-1-benzazepine, are very important in medical chemistry and widely present in a variety of drugs and other bioactive molecules (Fig. 1).31 The development of a novel and efficient method for the facile synthesis of benzodiazepine and related scaffolds is highly desirable.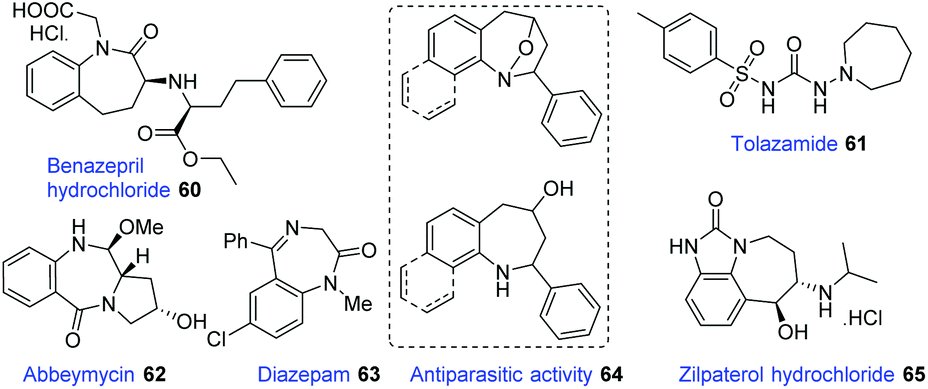 | ||
| Fig. 1 Selected biologically active molecules. | ||
In 2017, Luo and co-workers reported a Sc(OTf)3-catalysed [4 + 3]-annulation reaction of cyclopropane 1,1-diesters 66 and anthranils under mild reaction conditions (Scheme 23).32 This protocol provides a practical and straightforward access to biologically important tetrahydro-1-benzazepine scaffolds and a wide range of tetrahydro-1-benzazepine derivatives 67 can be obtained in good yields under the standard conditions.
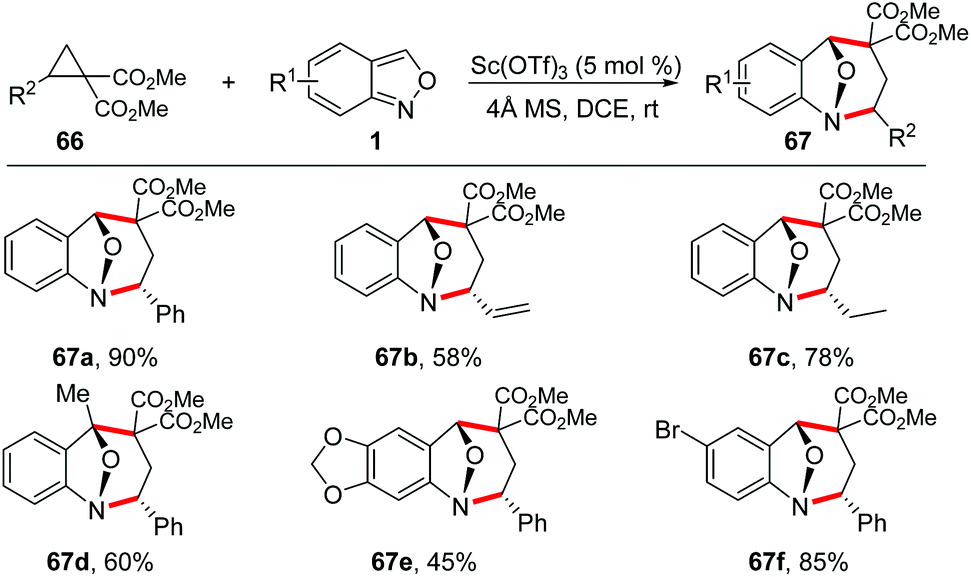 | ||
| Scheme 23 [4 + 3]-annulation reaction between cyclopropane 1,1-diesters and anthranils. | ||
The stereospecificity of this [4 + 3]-annulation reaction was explored by employing enantioenriched phenyl-substituted cyclopropane 66a (99% ee) as the substrate. As a result, anthranil reacted with 66a smoothly to give (1R,2R,5R)-67a (94% ee) with an almost complete stereospecificity. Notably, the configuration inversion of the stereocenter in 66a was observed in this transformation. Therefore, a plausible step-wise mechanism was proposed to explain this observation. As shown in Scheme 24, firstly, an SN2-like nucleophilic attack of anthranil to the Lewis acid activated D–A cyclopropane gave a zwitterion intermediate 68, which was followed by an immediate aromatization process to give oxonium ion species. The annulation was accomplished by an intramolecular nucleophilic addition of a carbanion to the oxonium ion intermediate via transition state TS-1 or TS-2. It is likely that the π–π interaction between the two aromatic rings in TS-1 plays a critical role for this step, leading to the endo-product with excellent diastereoselectivity.
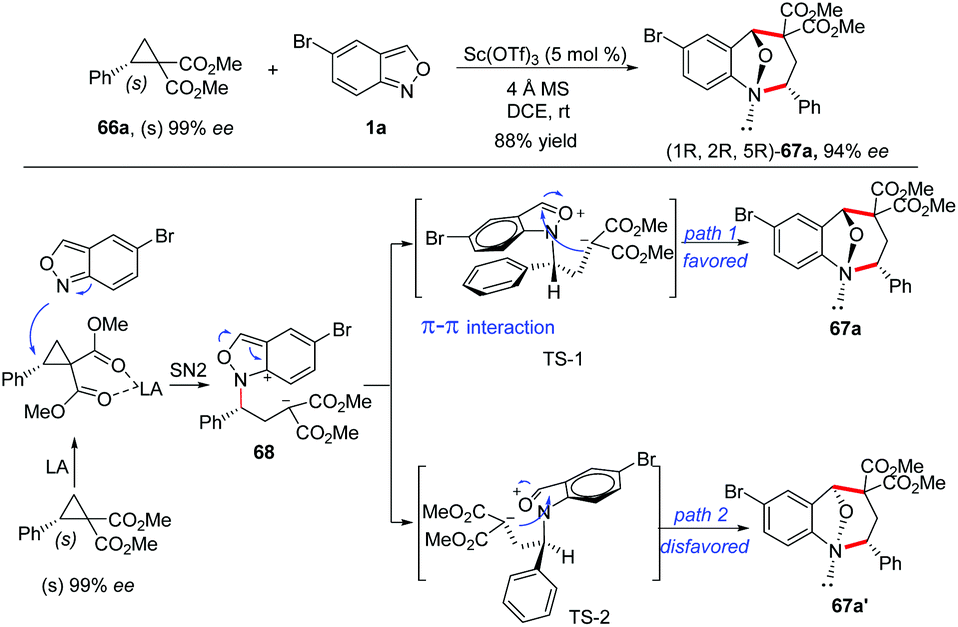 | ||
| Scheme 24 [4 + 3]-annulation reaction between cyclopropane 1,1-diesters and anthranils. | ||
Similarly, the Sc(OTf)3-catalysed annulations between anthranils and γ-butyrolactone-fused donor–acceptor cyclopropanes 69 have been developed by the group of Yang (Scheme 25).33a Depending on the structure of anthranils, this reaction could undergo a chemodivergent [4 + 3] annulation or an efficient cascade process, producing the corresponding bridged cyclic products 70 and γ-butyrolactone-fused tetrahydroquinolines 71 in moderate to high yields, respectively. A plausible mechanism for the formation of γ-butyrolactone-fused tetrahydroquinoline is proposed in Scheme 25. The cyclopropanes were firstly activated by Sc(OTf)3, which was then attacked by anthranils to produce intermediate 69b. When R1 is a phenyl group, the intermediate 69b might undergo a fragmentation pathway to provide intermediates 69c and 69d, which is followed by a Sc(OTf)3-catalysed Povarov-type [4 + 2] annulation to give the final tetrahydroquinoline products. Very recently, a PTSA-catalysed annulation of N′-aryl anthranil hydrazides with cyclopropane aldehydes has been demonstrated for the synthesis of various pyrroloquinazolinones.33b
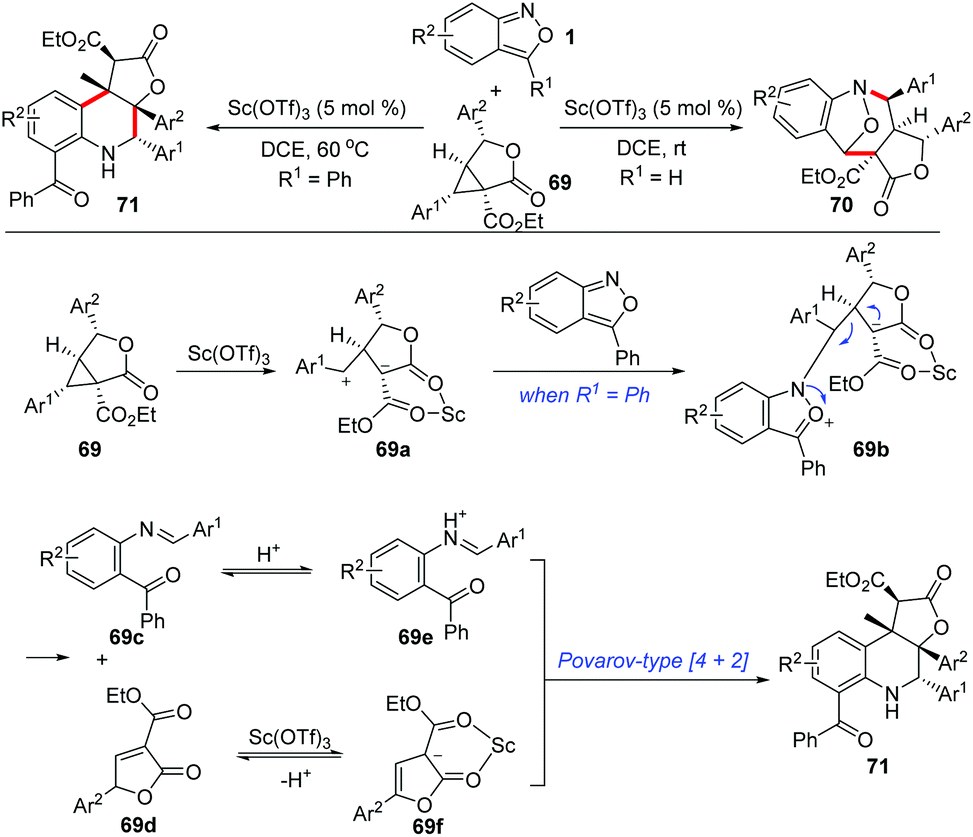 | ||
| Scheme 25 Sc(OTf)3-Catalysed chemodivergent annulations of γ-butyrolactone-fused cyclopropanes with anthranils. | ||
In 2019, a Pd-catalysed [4 + 3] cyclization reaction between anthranils and vinylcyclopropanes (VCPs) 72 was realized by You and co-workers (Scheme 26).34 In the presence of Pd2dba3 as the catalyst and a catalytic amount of borane as an activator, various bridged cyclic products 73 were obtained in good to excellent yields with excellent stereoselectivities. By introducing a chiral PHOX ligand (L), asymmetric dearomatization of anthranils with vinylcyclopropanes can be successfully achieved in a highly enantioselective fashion. The authors found that borane played a key role in the reaction efficiency. The results of NMR experiments indicated the intermediacy of a borane–anthranil complex in this transformation.
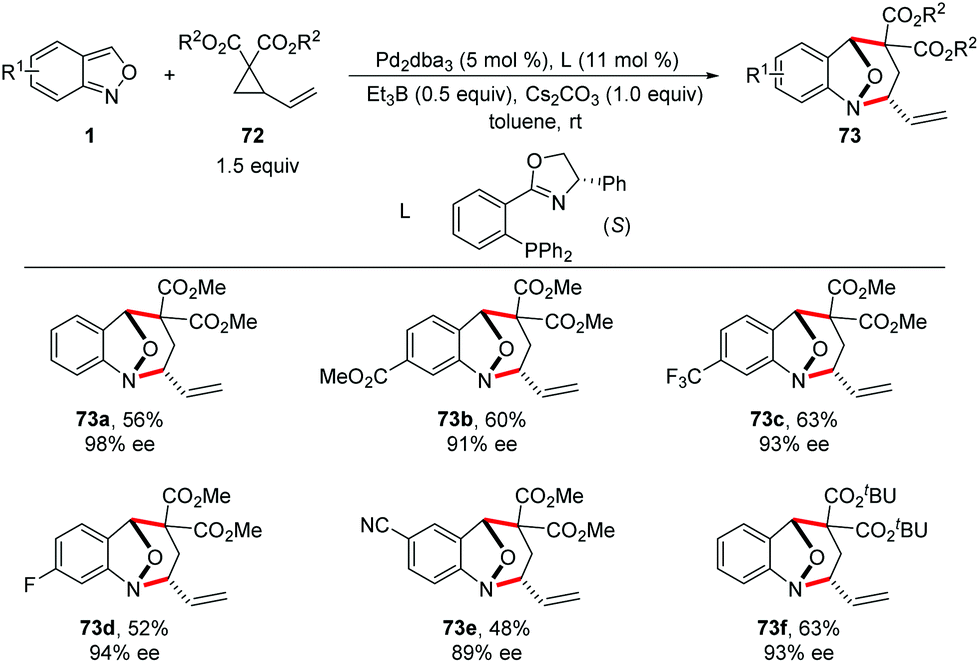 | ||
| Scheme 26 Pd-Catalysed [4 + 3] cyclization reaction between anthranils and vinylcyclopropanes. | ||
A plausible catalytic cycle is proposed in Scheme 27. Firstly, the coordination and oxidative addition of a palladium catalyst to VCP (72a) led to the ring-opening intermediate 72b. At the same time, the anthranil substrate was activated by Et3B to form borane–anthranil complex 74. Next, a catalyst-controlled dearomative addition of 72b to 74 delivered intermediate 72c. The subsequent intramolecular allylic cyclization afforded the desired annulation products. Then, both the borane and palladium catalysts were released to complete the catalytic cycle.
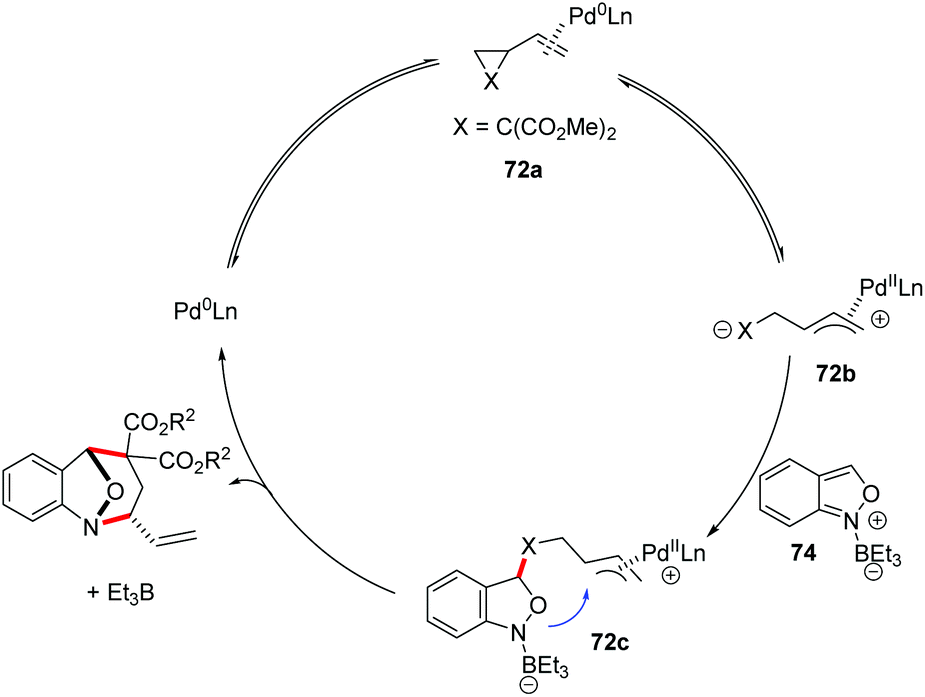 | ||
| Scheme 27 Plausible mechanism of Pd-catalysed [4 + 3] annulation between anthranils and vinylcyclopropanes. | ||
Recently, a base-promoted [4 + 3] cycloaddition of azaoxyallyl cations and anthranils has been developed for rapid access to multisubstituted benzodiazepines (Scheme 28).35 A variety of anthranils and α-halo hydroxamates 75 participated in this reaction very well under transition metal-free conditions. The in situ-generated azaoxyallyl cation was considered to be a key intermediate for this transformation. A plausible mechanism of this transformation is proposed in Scheme 28. The azaoxyallyl cation intermediate 75b was initially formed from α-halohydroxamate in the presence of K2CO3. Next, anthranil reacted with intermediate 75b to generate a zwitterionic intermediate 75c. Finally, a fast intramolecular nucleophilic addition of intermediate 75c produces the corresponding [4 + 3] cycloadducts 76.
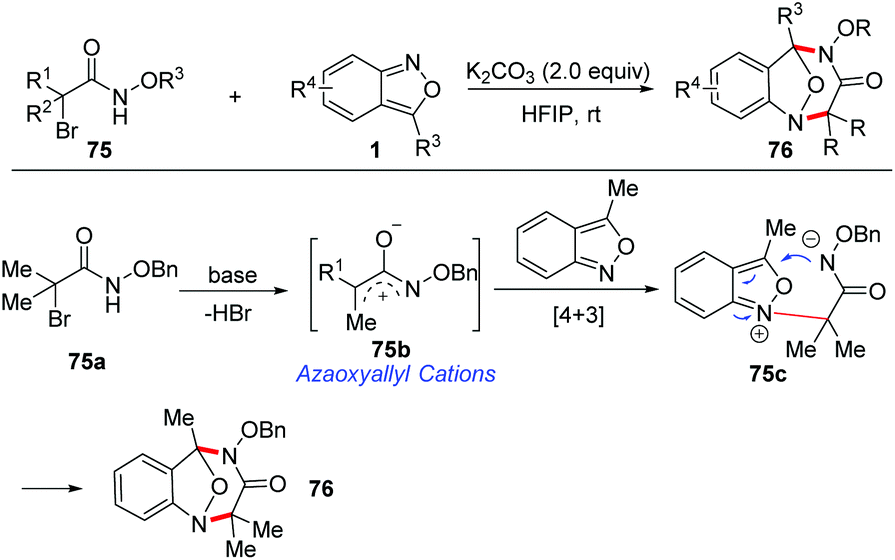 | ||
| Scheme 28 [4 + 3]-cycloaddition of azaoxyallyl cations and anthranils. | ||
5.2 Formal [4 + 2] cycloaddition of anthranils
In 1989, Yujiro and co-workers disclosed a titanium salt-promoted [4 + 2] cycloaddition reaction of anthranils with enamines 77 for the formation of quinoline derivatives 78 (Scheme 29).36 In the presence of TiCl4, the [4 + 2] cycloaddition reaction of anthranils and enamines firstly occurred to give the cycloadduct intermediate 77a, followed by a cascade deamination and N–O bond cleavage process to give quinine N-oxides 77b. Then, the reduction of quinine N-oxides by zinc dust generated the final quinoline products 78.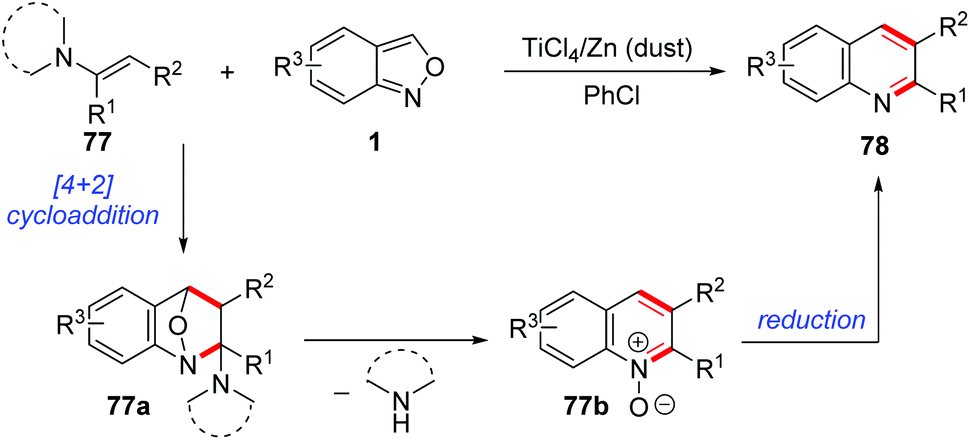 | ||
| Scheme 29 [4 + 2] cycloaddition reaction of anthranils and enamine. | ||
In 2017, an interesting copper-catalysed α,β-functionalisation of saturated ketones with anthranils was reported by Tiwari and co-workers (Scheme 30).37 This reaction provides an efficient approach to synthesize quinoline derivatives from the readily available saturated ketones 79 and anthranils. The in situ-generated α,β-unsaturated ketone 79a was a key intermediate for this reaction, which was demonstrated by control experiments. When the α,β-unsaturated ketone was subjected to the standard conditions, the desired quinoline product was obtained as expected. Accordingly, a plausible mechanism is proposed in Scheme 30. In this reaction, anthranils could undergo an aza-Michael addition to the in situ-generated α,β-unsaturated ketone 79a to give an aldehyde intermediate 79b, which underwent a rapid intramolecular annulation process to afford the final products 80. TEMPO is a crucial additive for Cu-catalysed oxidative dehydrogenation of saturated ketones.
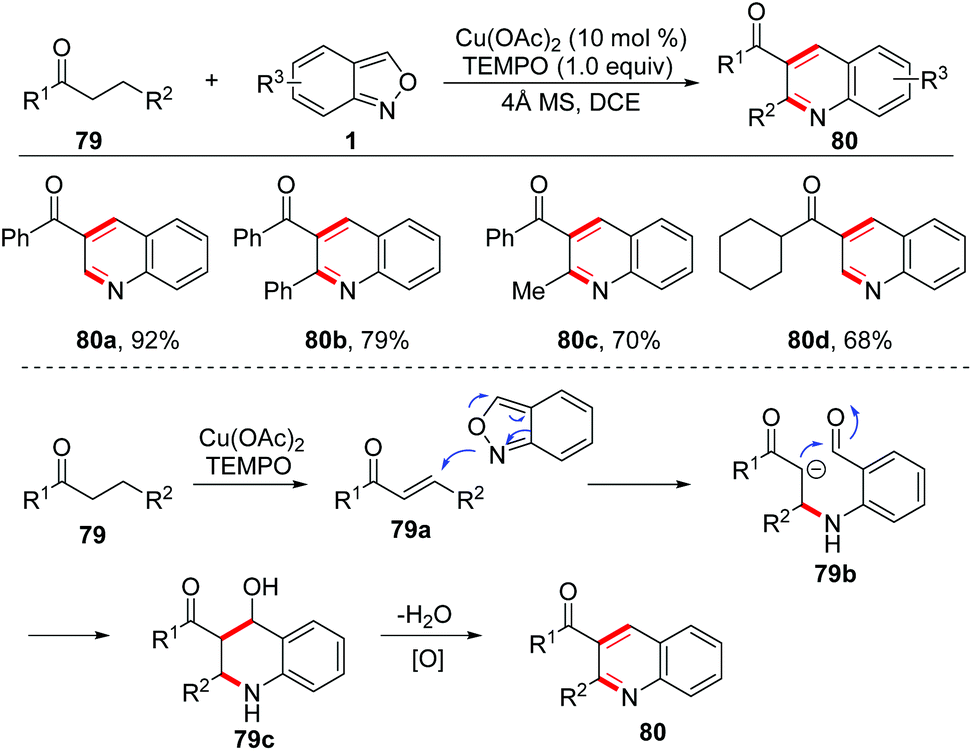 | ||
| Scheme 30 Copper-catalysed α,β-functionalization of saturated ketones with anthranils. | ||
A similar strategy has been further applied in a three-component annulation of acetophenone 81, anthranils and DMSO (Scheme 31).38 In this reaction, the α,β-unsaturated ketones were generated from acetophenone and DMSO through a one-carbon homologation of DMSO with the use of K2S2O8 as the oxidant.
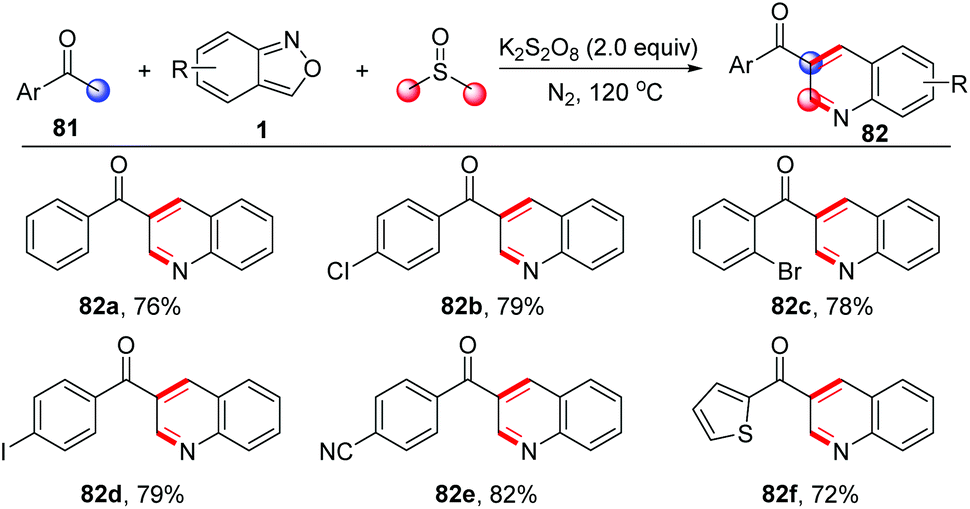 | ||
| Scheme 31 Metal-free annulation of acetophenones and anthranils for the synthesis of functionalized quinolines. | ||
In 2019, Cui and co-workers reported an efficient and atom-economical [4 + 2] cyclisation of glycine derivatives 83 and anthranils for the synthesis of 3,4-dihydroquinazolines 84 under CuCl2/BPO oxidative conditions (Scheme 32).39 A plausible mechanism is proposed in Scheme 32. The glycine ester was firstly oxidized to imine intermediate 83a in the presence of a copper catalyst and BPO. Subsequently, anthranil attacked the reactive imine species to afford the intermediate 83c. Then, a selective N–O bond cleavage of 83c occurred to generate a nitrene intermediate 83d. The final product 84 could be obtained through an intramolecular nucleophilic addition of the amine to aldehyde. In addition, a control experiment indicated that the in situ-generated PhCOOH from BPO might serve as a proton donor to improve the electrophilicity of the imine intermediate and facilitate the following nucleophilic addition of anthranil to the imine intermediates.
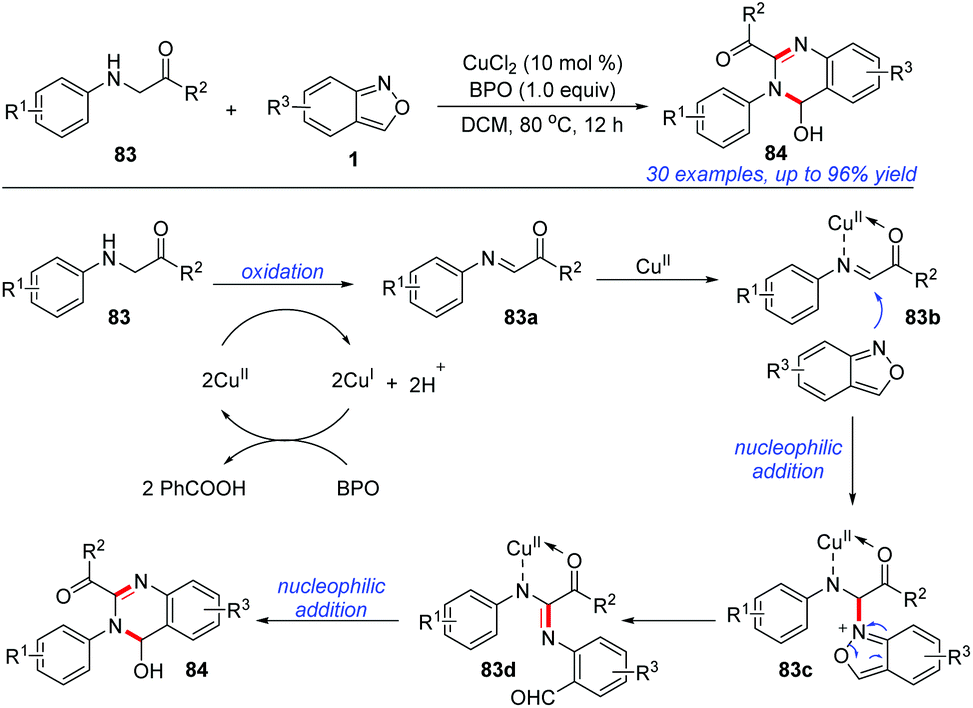 | ||
| Scheme 32 Synthesis of 3,4-dihydroquinazolines from glycine derivatives and anthranils. | ||
In 2018, a novel Cu(II)/Ag(I)-catalysed cascade reaction of anthranils with sulfonylhydrazones 85 to construct 2-aryl-3-sulfonyl substituted quinolines 86 was reported by Ji's group (Scheme 33).40 Various poly-substituted quinolines were obtained in moderate to good yields under the cooperative catalytic system. Preliminary mechanistic exploration indicated that a sulfonyl radical might be involved in this transformation.
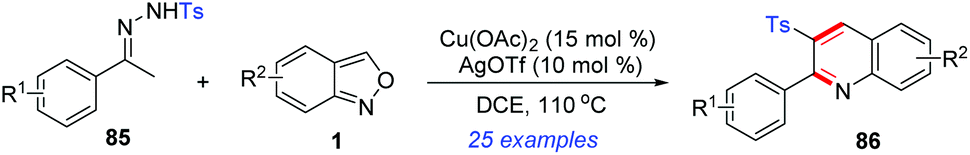 | ||
| Scheme 33 Synthesis of 2,3-disubstituted quinoline derivatives from p-toluenesulfonylhydrazones and anthranils. | ||
Recently, Zou and co-workers reported a copper-catalysed [4 + 1 + 1] annulation reaction of sulfoxonium ylides 87 and anthranils for the rapid synthesis of 2,3-diaroylquinolines 88 (Scheme 34).41 This transformation was initiated by a copper-catalysed homocoupling of sulfoxonium ylides to provide α,α,β-tricarbonyl sulfoxonium ylides 87b, which then converted to copper carbene species 87c with the release of DMSO. The reaction of 87c with anthranils afforded imine intermediates 87e. Finally, 2,3-diaroylquinoline products 88 were formed through an intramolecular dehydration process. It was found that AgOTf and dioxygen were also critical to this reaction.
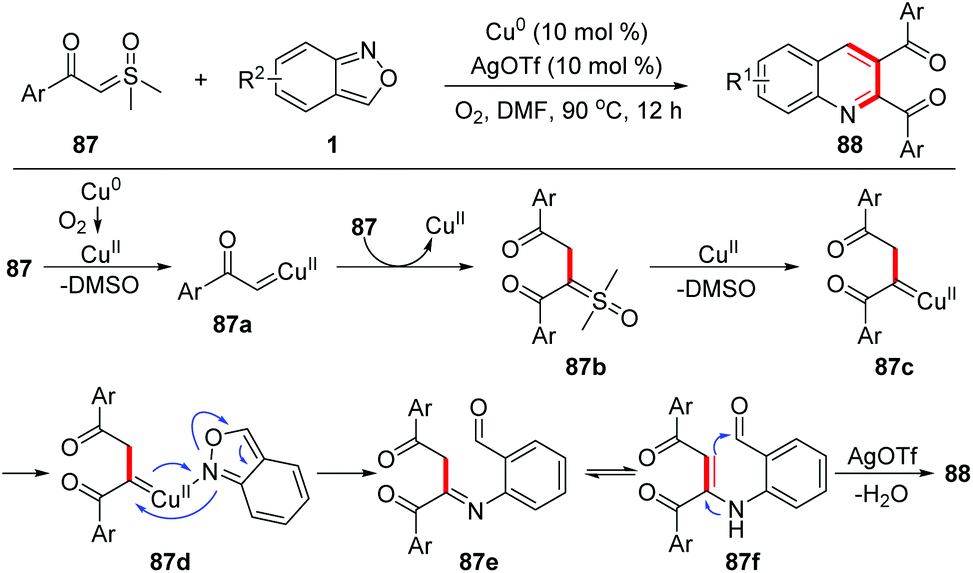 | ||
| Scheme 34 Copper-catalyzed [4 + 1 + 1] annulation reaction of sulfoxonium ylides and anthranils. | ||
The thermolytic N–O bond cleavage of anthranils can generate highly reactive ketene species 88. In 2019, Zhang and co-worker disclosed a series of nucleophilic additions of the in situ-generated ketene intermediates from anthranils (Scheme 35).42 In this reaction, various commonly used nucleophiles proved to be suitable substrates. For example, anilines and phenols were verified as good reaction partners with anthranils, affording the corresponding anthranilamide 89 and anthranilate 90 products in moderate to good yields, respectively. Notably, the high-value benzoxazinone heterocycle 91 was obtained by using benzoic acids as the nucleophiles. Moreover, malononitrile could react smoothly with anthranils to efficiently produce quinoline derivatives 92 under catalyst-free conditions.
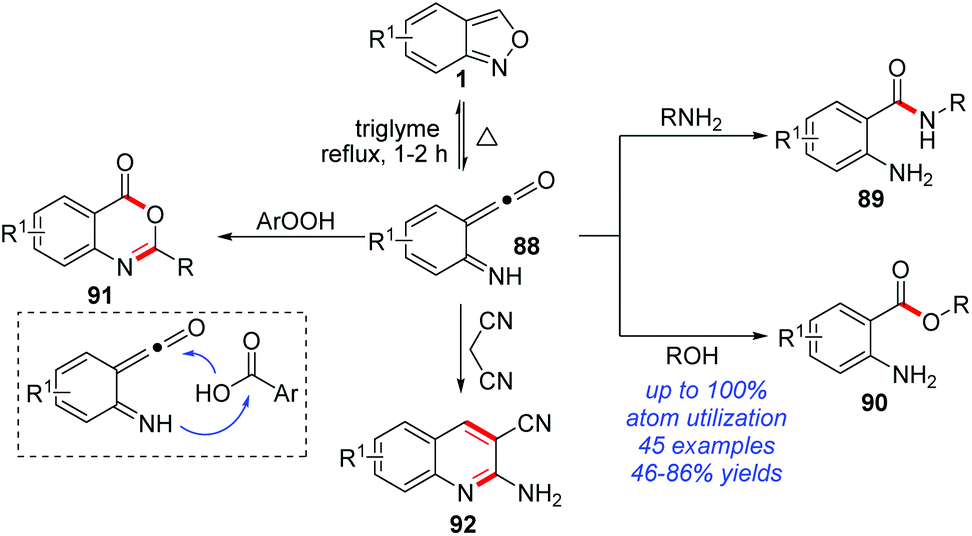 | ||
| Scheme 35 Thermolytic reaction of anthranils with nucleophiles. | ||
Early in 1967, a piperidine catalysed cyclization of anthranils with some active methylene reagents, such as malononitrile, dimethyl malonate and ethyl cyanoacetate, has been developed for the rapid synthesis of quinoline oxide 93 (Scheme 36).43 This annulation went through a different pathway from the one mentioned above. With piperidine as a proton shuttle, the active methylene attacked at the 3-position of anthranils to give the adduct 93a. Then, a proton transfer from the acidic methylene to the O-atom generated O-hydroxylaminobenzylidene intermediate 93b through a C–O bond cleavage process. Subsequently, the ring closure of 93b by a rapid intramolecular addition of the –NHOH group to a CN motif gave the final quinoline 1-oxide 93.
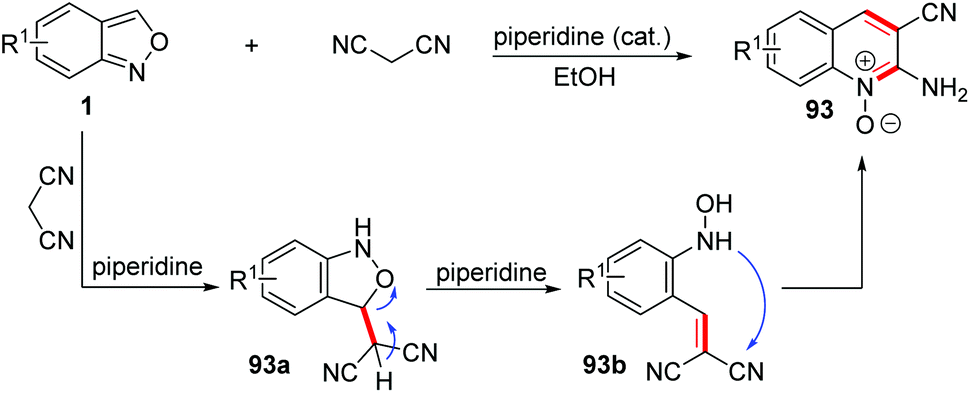 | ||
| Scheme 36 Piperidine catalysed annulation of anthranils and malononitrile. | ||
A catalyst-free cyclisation of anthranils and 1,2,3,4-tetrahydroisoquinoline (THIQ) has been described by Li and co-workers (Scheme 37).44 Various quinazolinone 95 and 3,4-dihydroquinazoline 96 derivatives bearing different functional groups were obtained under catalyst-free conditions. Significantly, by using this method, the important rutaecarpine 95d could be obtained in good yield and it was further applied to the synthesis of valeric acid-rutaecarpine, which was investigated as an antiplatelet inhibitor. The author believed that an active aryl nitrene species might be an intermediate for this reaction, which was generated through the N–O bond cleavage of anthranils at relatively high temperature (130 °C).
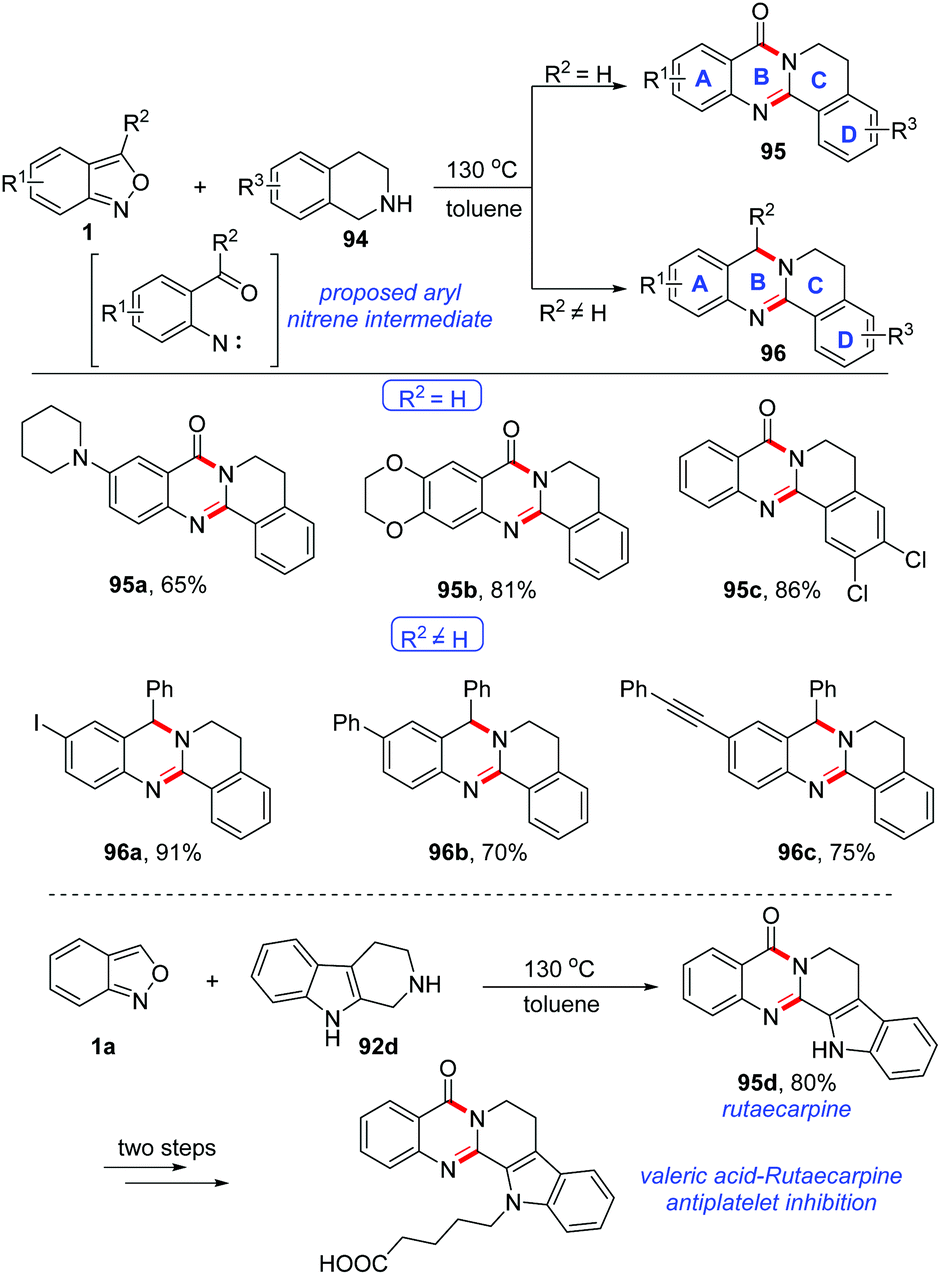 | ||
| Scheme 37 A catalyst-free reaction of anthranils and 1,2,3,4-tetrahydroisoquinoline (THIQ). | ||
Recently, a copper-catalysed protocol for the synthesis of quinoline derivatives from the readily available anthranils and 1,3-diketones/aldehydes 97 was disclosed by the group of Li (Scheme 38).45 In this reaction, 2-aminobenzaldehyde proved to be a key intermediate on the basis of control experiments, which was generated from a copper-catalysed N–O bond cleavage procedure. The final quinoline products were obtained through a subsequent Friedlander reaction of 2-aminobenzaldehyde and 1,3-diketones.46
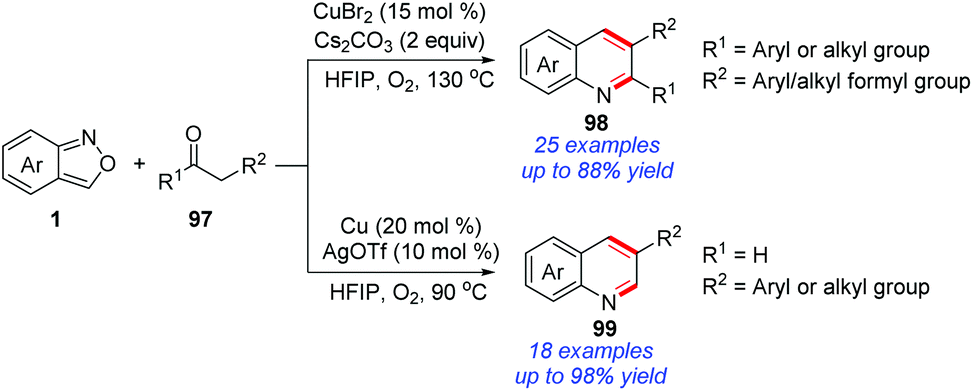 | ||
| Scheme 38 Cascade annulation reaction of anthranils with 1,3-diketones/aldehydes. | ||
6. Gold-catalysed annulation of anthranils with alkynes
Gold carbene complexes47 have been recognized as versatile and powerful intermediates in gold catalysed organic synthesis. In this field, α-imino gold carbene complexes48 have emerged as useful intermediates towards the synthesis of N-heterocycles. In the presence of gold catalysts, anthranils could serve as nucleophiles to attack the alkynes to form active α-imino gold carbene species and various novel annulation reactions have been developed. A recent review on the gold-catalysed heterocyclic synthesis partially summarized the progress of the gold-catalysed annulations of anthranils and alkynes.49 Therefore, in this part, we mainly focus on updating contributions in this field.In 2016, the Hashmi group reported a pioneering gold-catalysed C–H annulation of anthranils and alkynes 99 for the construction of unprotected 7-acylindoles 100. Various alkynes including ynamides, non-polarized alkynes and non-terminal alkynes could participate in this reaction very well and a wide range of 7-acylindoles were obtained in good yields (Scheme 39).50 A tentative mechanism for this transformation is proposed in Scheme 39. Initially, the intermolecular addition of anthranils to the gold activated alkyne species produced the intermediate 100b. Subsequently, the α-imino gold carbene complex 100c was formed through the breakage of the labile N–O bond. Finally, intermediate 100c underwent a fast intramolecular ortho-aryl C–H insertion to give the desired 7-formyl indoles by taking advantage of the high electrophilicity of the gold carbenoid.
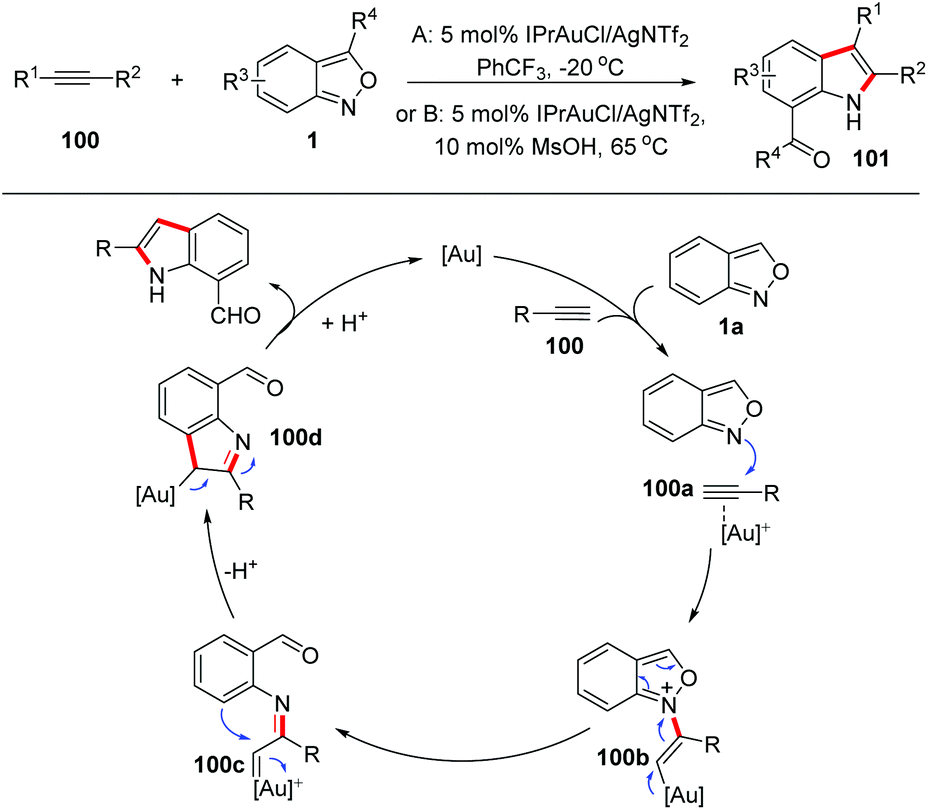 | ||
| Scheme 39 Gold-catalysed C–H annulation of anthranils with alkynes for the synthesis of indole derivatives. | ||
Later, a relevant transformation was reported by the same group with propargylic silyl ethers 102 as the starting materials. Under similar gold-catalysed conditions, propargylic silyl ethers reacted smoothly with anthranils to produce quinoline derivatives 103 as the only products instead of the above-mentioned 7-acylindoles (Scheme 40).51 The difference was caused by the adjacent methylene moiety of the α-imino gold carbene complex 103a. In this regard, a 1,2-hydrogen shift took place to form an N-aryl-α,β-unsaturated imine 103b. Then, an intramolecular Mukaiyama-type nucleophilic addition of silyl enol ether to the carbonyl functional group led to the expected 2-aminoquinoline products 103. The proposed mechanism was further verified by an 18O-labelling experiment. The presence of the ynamide moiety is not a requisite for the reaction and less polarized propargyl silyl ethers can also react with anthranils under modified reaction conditions, furnishing various quinoline derivatives in good yields.
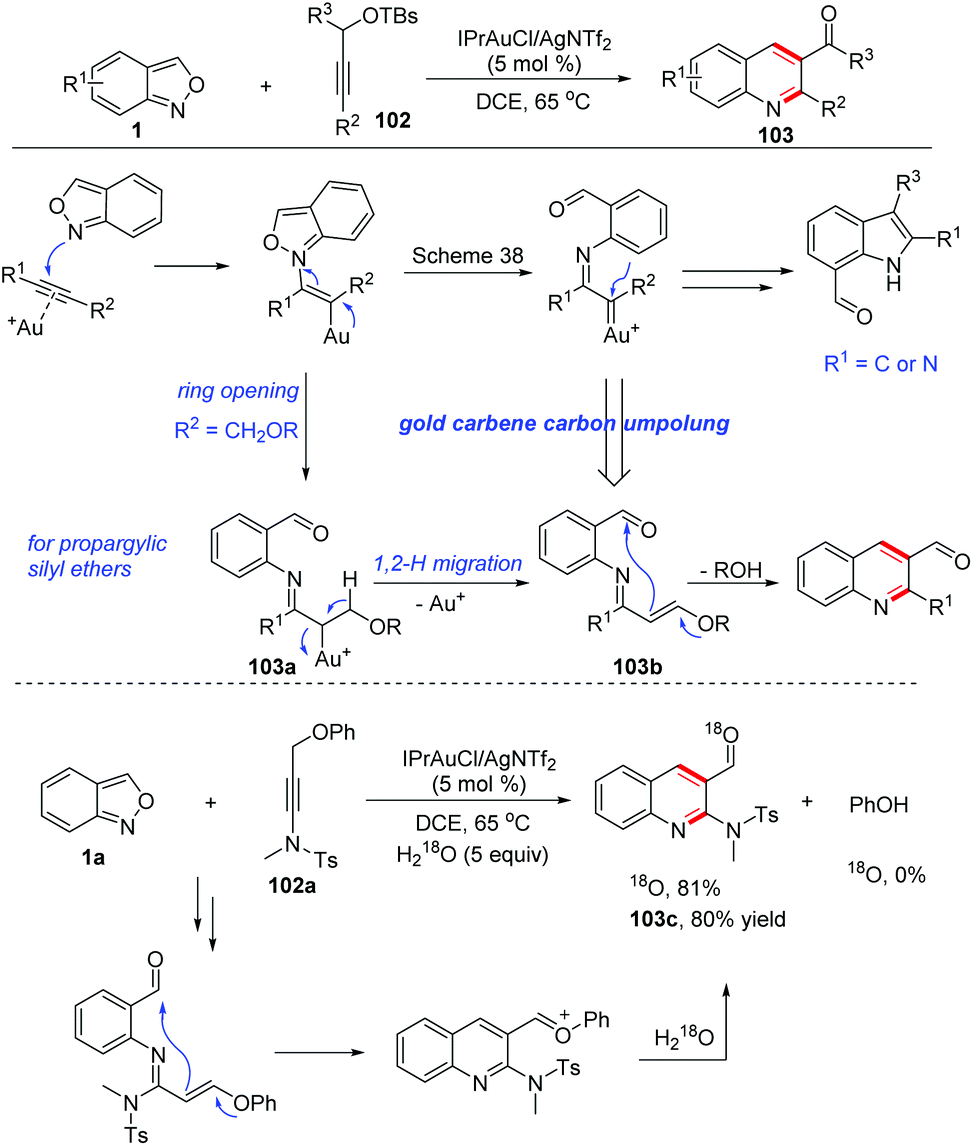 | ||
| Scheme 40 Gold-catalysed C–H annulation of anthranils for the synthesis of quinoline derivatives. | ||
Likewise, Liu and co-workers demonstrated that the 1,2-alkyne migration could occur in the related gold-catalysed [4 + 1]-annulation reactions between anthranils and 1,4-diyn-3-ols 104 for the synthesis of tetrasubstituted pyrroles 105 (Scheme 41a).52 Additionally, they also found that an α-imino gold carbene species 106a could undergo a 1,2-allene shift to form (pyrrol-2-yl) methylgold intermediates 106b in gold-catalysed [4 + 1]-annulations of 4-methoxy-1,2-dienyl-5-ynes 106 with anthranils (Scheme 41b).53
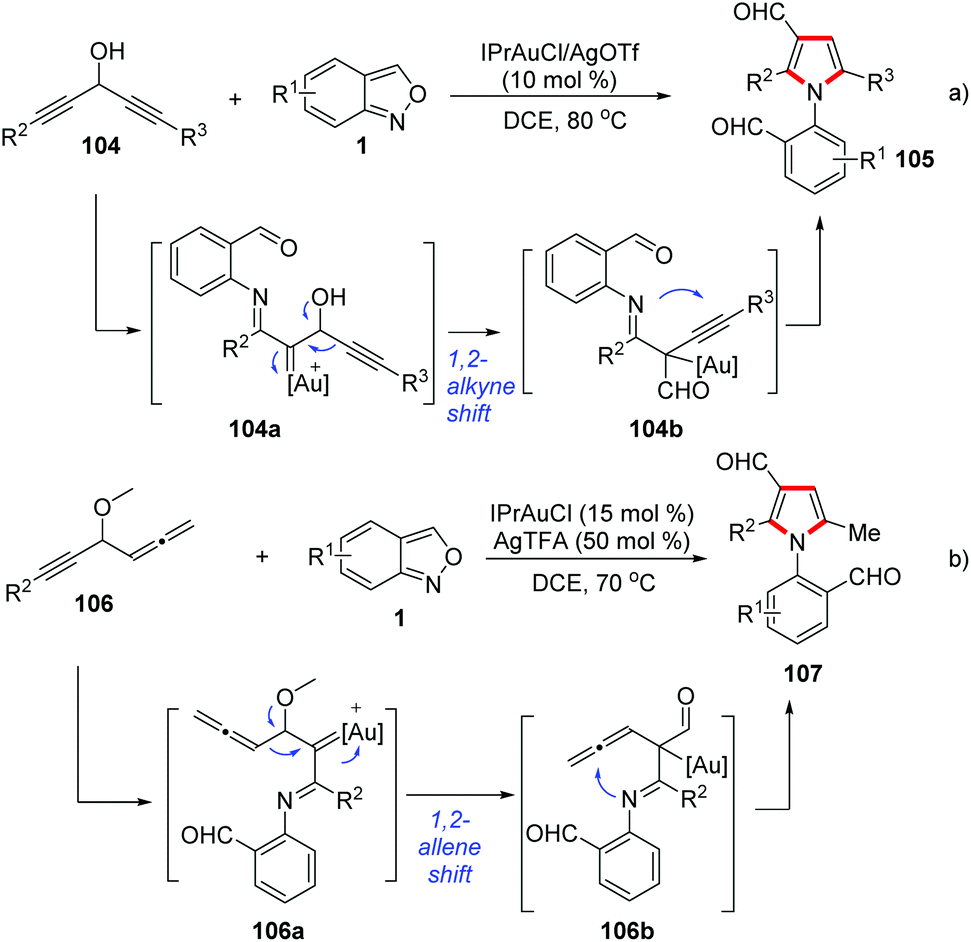 | ||
| Scheme 41 Functional group migration in α-imino gold carbene species. | ||
In 2018, Liu and co-workers discovered a 1,2-carbon migration of gold carbene species in gold-catalysed iminations of terminal propargyl alcohols 108 with anthranils.54 This transformation provided a facile access to E-configurated α-amino-2-en-1-ones and α-amino-2-en-1-als 109 with complete chemoselectivity (Scheme 42). In contrast to the typical C(2)-addition pathway, an exclusive C(1)-nucleophilic addition of anthranils to terminal alkynes was observed. For 3,3-dialkylprop-1-yn-3-ols, the methyl substituent as a 1,2-migration group is superior to other long alkyl chains towards α-imino gold carbenes. For secondary prop-1-yn-3-ols, phenyl, vinyl and cyclopropyl substituents are faster migrating groups than hydrogen (Scheme 42b). It should be noted that the steric hindrance of propargyl alcohols played an important role in the selective C(1)-nucleophilic additions, since the simple unsubstituted propargyl alcohol 108a mainly underwent the C(2)-imination route to give the corresponding indole product 110 (Scheme 42c).
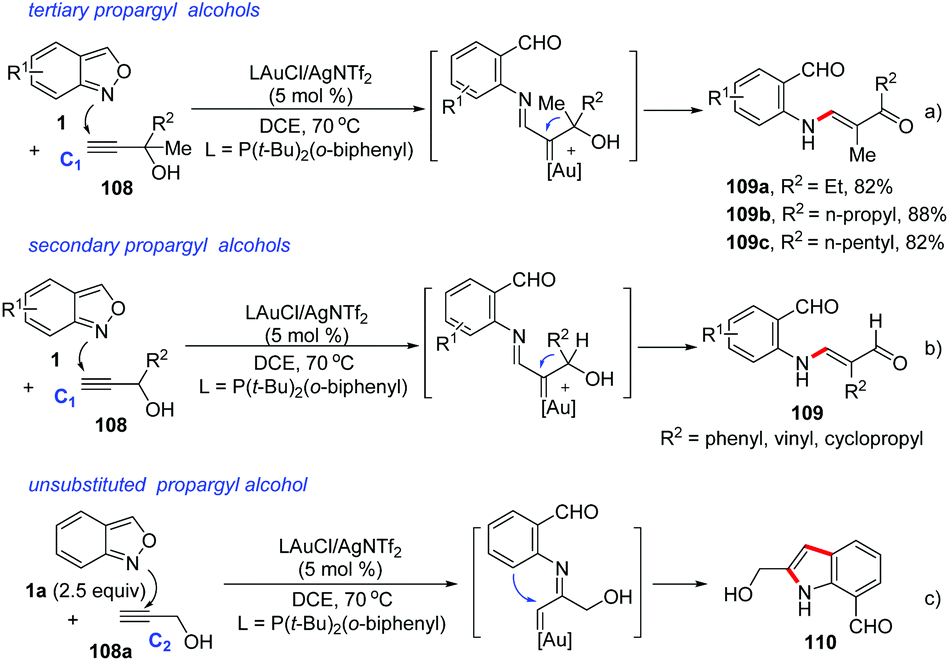 | ||
| Scheme 42 1,2-Carbon migration in gold-catalysed iminations of terminal propargyl alcohols with anthranils. | ||
Depending on the surrounding functional groups, the α-imino gold carbene intermediate may evolve to different complex structures. For example, the Hashmi group disclosed that anthranils could react with o-ethynylbiaryls 111 in the presence of a phosphite gold catalyst (ArO)3PAuCl for the synthesis of N-doped polycyclic aromatic hydrocarbons (PAH) 112 in moderate to good yields (Scheme 43).55 In this transformation, the carbene complex 111a underwent a regioselective insertion into the C–H bond of the o-aryl group to generate the iminophenanthrene intermediate 111b, which evolved towards the final products via a Friedel–Crafts-type cyclization. In addition, the annulation reactions of anthranils with N-benzyl ynamides,56N-aryl ynamides,57 aryloxyethynes and aryl propargyl ethers58 have been explored for the construction of various useful condensed N-heterocycles.
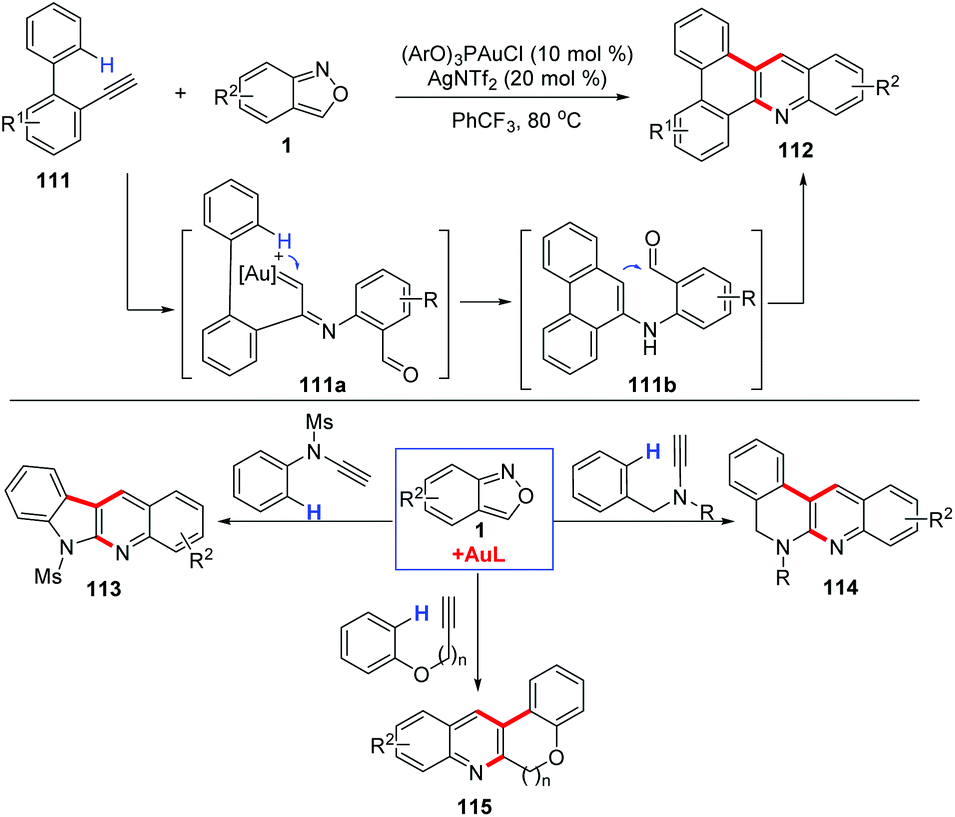 | ||
| Scheme 43 Gold-catalysed C–H annulation of anthranils with alkynes for the synthesis of N-doped polycyclic aromatic hydrocarbons (PAH). | ||
Pyrrolo[2,3-b]quinoline cores are found in several bioactive and naturally occurring molecules,59 such as PGP-4008 (116), neocryptofelepine (117), and perophoramidine (118). Compound 116 is effective at inhibiting Pgp (P-glycoprotein) in vitro; the other two species (117 and 118) are natural alkaloids (Fig. 2).
 | ||
| Fig. 2 Representative bioactive and natural products. | ||
In 2019, Liu and co-workers reported a ready access to pyrrolo[2,3-b]quinolones 120 by the reaction of terminal N-propargyl ynamides 119 and anthranils with a dual gold(III) and Brønsted acid catalysis. Various bioactive pyrrolo[2,3-b]quinolone derivatives were obtained in one-pot under mild reaction conditions (Scheme 44).60 A tentative mechanism for this transformation was proposed. Initially, the α-imino gold carbene complex 119a was generated through the abovementioned process, and was attacked by a tethered internal alkyne to generate the alkenyl cation 119b. Subsequently, a hydration of 119b resulted in dihydropyrrole intermediate 119c, which was further oxidized to 4-aminopyrroles 119d. Finally, under acidic conditions, 4-aminopyrroles underwent an intramolecular condensation to give the desired pyrrolo[2,3-b]quinolone products 120 catalysed by TsOH (20 mol%).
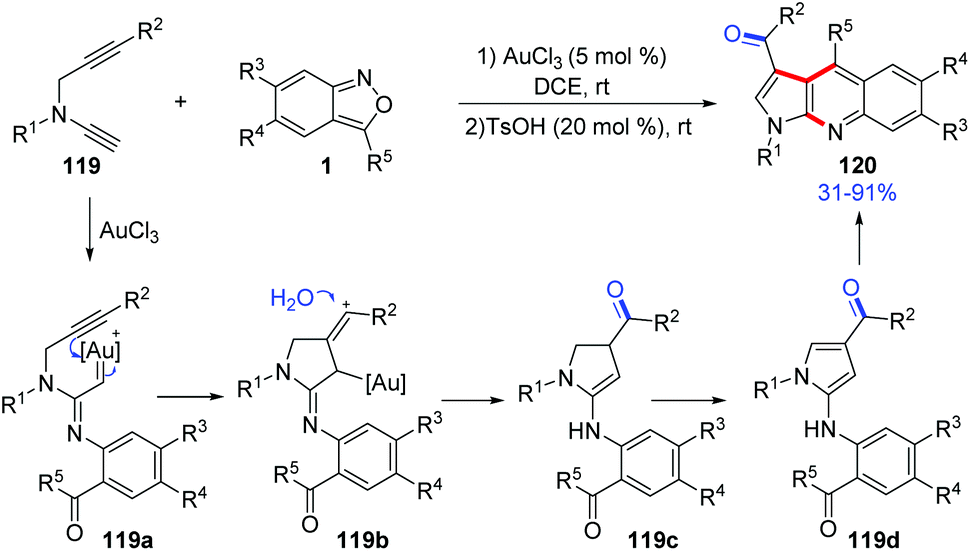 | ||
| Scheme 44 Gold-catalysed annulation of terminal N-propargyl ynamides and anthranils. | ||
A relevant annulation reaction of anthranils with N-allyl ynamides for the synthesis of azabicyclo[3.1.0]hexane-2-imines 122 was reported by the group of Hashmi (Scheme 45).61 With NaAuCl4·2H2O as the catalyst, various azabicyclo[3.1.0]hexane-2-imines bearing different functional groups were obtained with high synthetic efficiency. The in situ-generated α-imino gold carbine 121b was a key intermediate for this reaction, which could be converted to the final product through an intramolecular trapping of gold carbene by olefins.
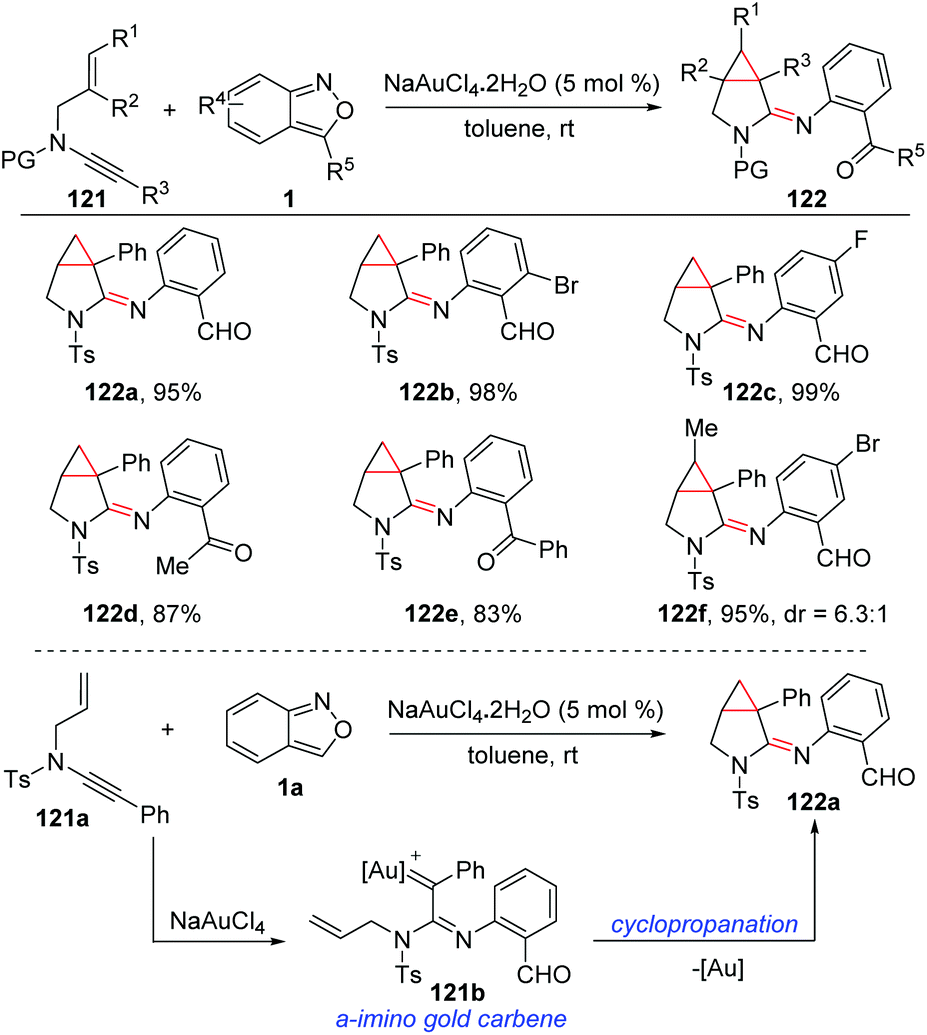 | ||
| Scheme 45 Gold-catalysed annulation of anthranils with N-allyl ynamides. | ||
The group of Liu reported a gold-catalysed 1,3-carbofunctionalisation of anthranils with vinyl propargyl esters 123 for the formation of 1,3-dihydrobenzo[c]-isoxazoles 124 (Scheme 46).62 As proposed in the mechanism, a 1,2-acyloxy shift was expected to form the 4-vinyl alkenylgold intermediate 123b, which was attacked by anthranil at the oxonium center to produce intermediate 123c. The intermediate 123c bearing an acid/base pair underwent a subsequent cyclization to give complex 123d. The phenyl group of 123d enhanced self-ionization to form a benzylic cation 123e, which was followed by an intramolecular cyclization to afford the final product 124.
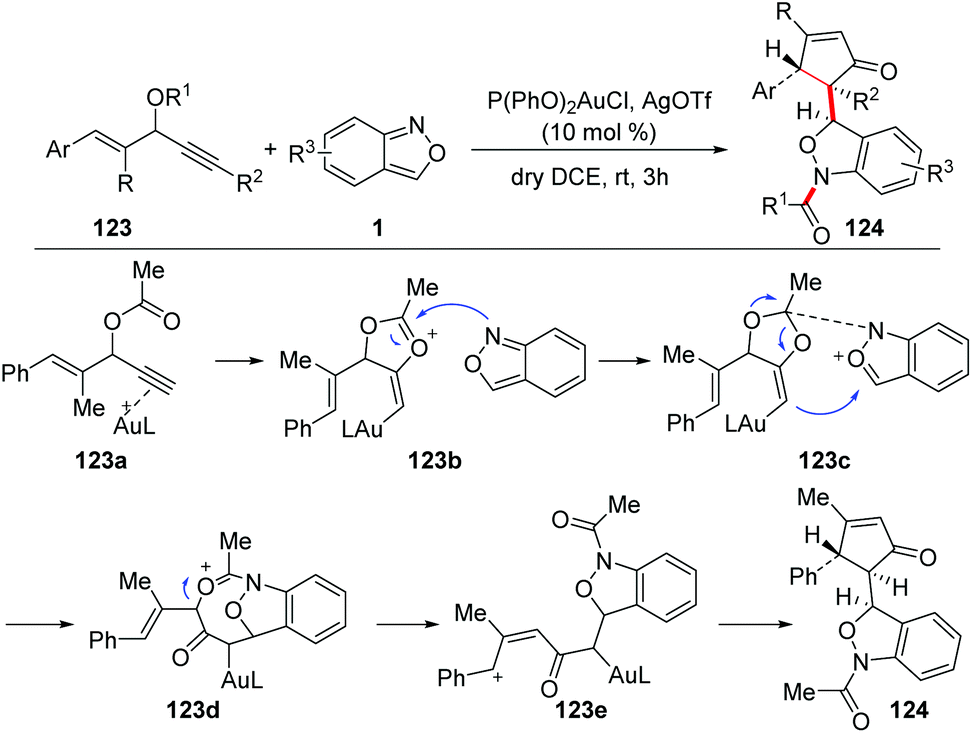 | ||
| Scheme 46 Gold-catalysed 1,3-carbofunctionalisations of anthranils with vinyl propargyl esters. | ||
As described above, anthranils could react with 1,3-dipole analogs such as cyclopropane 1,1-diesters to construct the tetrahydro-1-benzazepine derivatives through Lewis acid-catalysed [4 + 3] annulations. In 2019, Liu and co-workers developed a novel annulation of anthranils with alkyne-based 1,3-dipoles for the first time.63 Two distinct [4 + 3]-nitroxy annulations between 1,5-enynes 125 and anthranils have been developed to access different tetrahydro-1H-benzo[b]azepine derivatives. The chemoselectivity was varied with the types of alkynes. Terminal alkyne substrates delivered benzo[b]azepines 126via a novel skeletal rearrangement (Scheme 47a), while internal 1,5-enynes directly afforded products 127 (Scheme 47b).
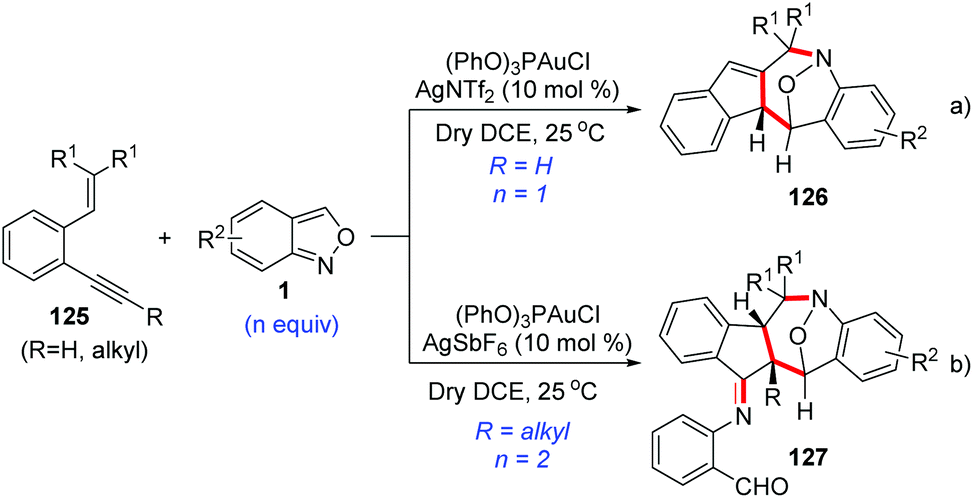 | ||
| Scheme 47 Gold-catalysed [4 + 3]-nitroxy annulations between 1,5-enynes and anthranils. | ||
A plausible mechanism is proposed in Scheme 48. Initially, internal 1,5-enyne reacted with LAu+ to form a cyclopropyl gold carbene 125b, which was a 1,3-dipole analog. The [4 + 3] annulation of the 1,3-dipole species 125c with anthranil yielded a new gold carbene species 125d, which was subsequently captured by a second anthranil to give product 127. For terminal 1,5-enynes, the cyclopropylgold carbene 125f was also initially generated and then underwent a deprotonation procedure to deliver cyclopropylidenylgold species 125g, which underwent a “methylenecyclopropane-trimethylenemethane” rearrangement to give a gold-containing isobenzofulvene species 125h. An exo-[4 + 3]-annulation between fulvene intermediate 125h and anthranil afforded the observed product 126.
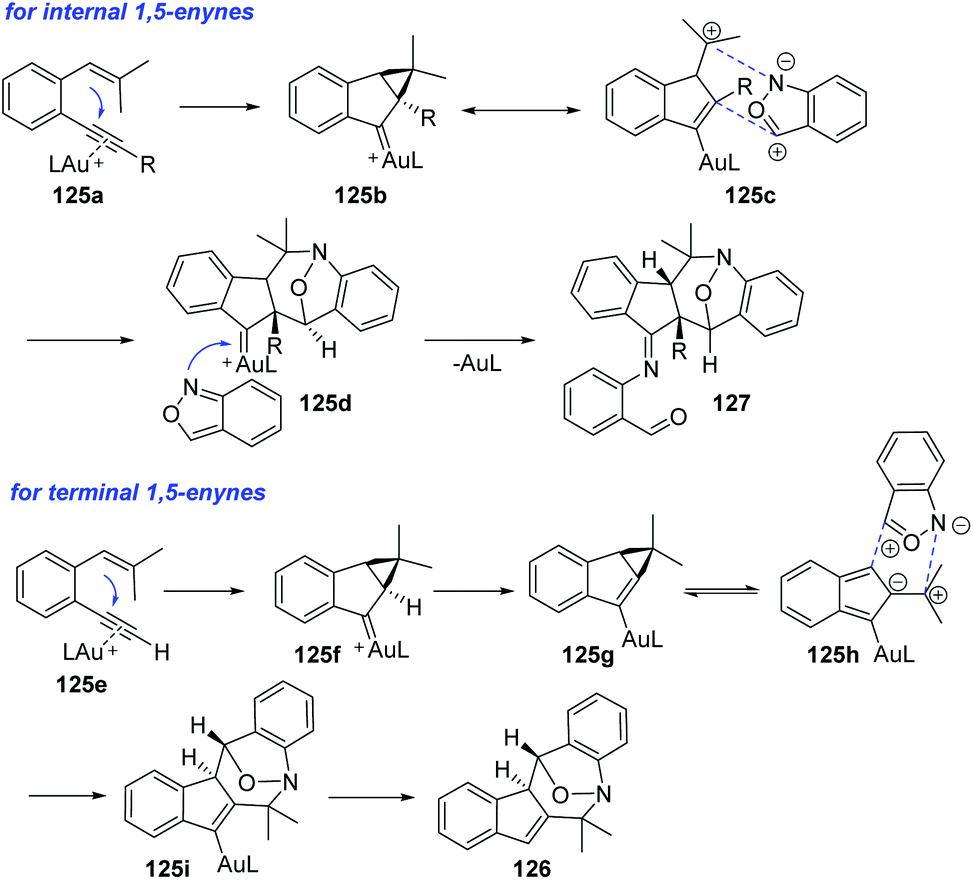 | ||
| Scheme 48 Proposed mechanism for gold-catalysed [4 + 3]-nitroxy annulations between 1,5-enynes and anthranils. | ||
Liu et al. described a gold-catalysed [4 + 2]-annulation of α-alkyl alkenylgold carbenes and anthranils. This [4 + 2]-annulation provides an efficient method to synthesize 3,4-dihydroquinoline derivatives with high anti-stereoselectivity (Scheme 49).64 In the presence of gold catalysts, vinylallene 128, enynyl acetates 130 and cyclopropane 132 derivatives could be employed as precursors for the in situ generation of alkylgold carbenes. Subsequently, the anthranil attacked the alkylgold carbene species 128a to form the imine intermediate 128c. Finally, an intramolecular carbonyl-enamine reaction occurred and the desired 3,4-dihydroquinolines 129 could be obtained.
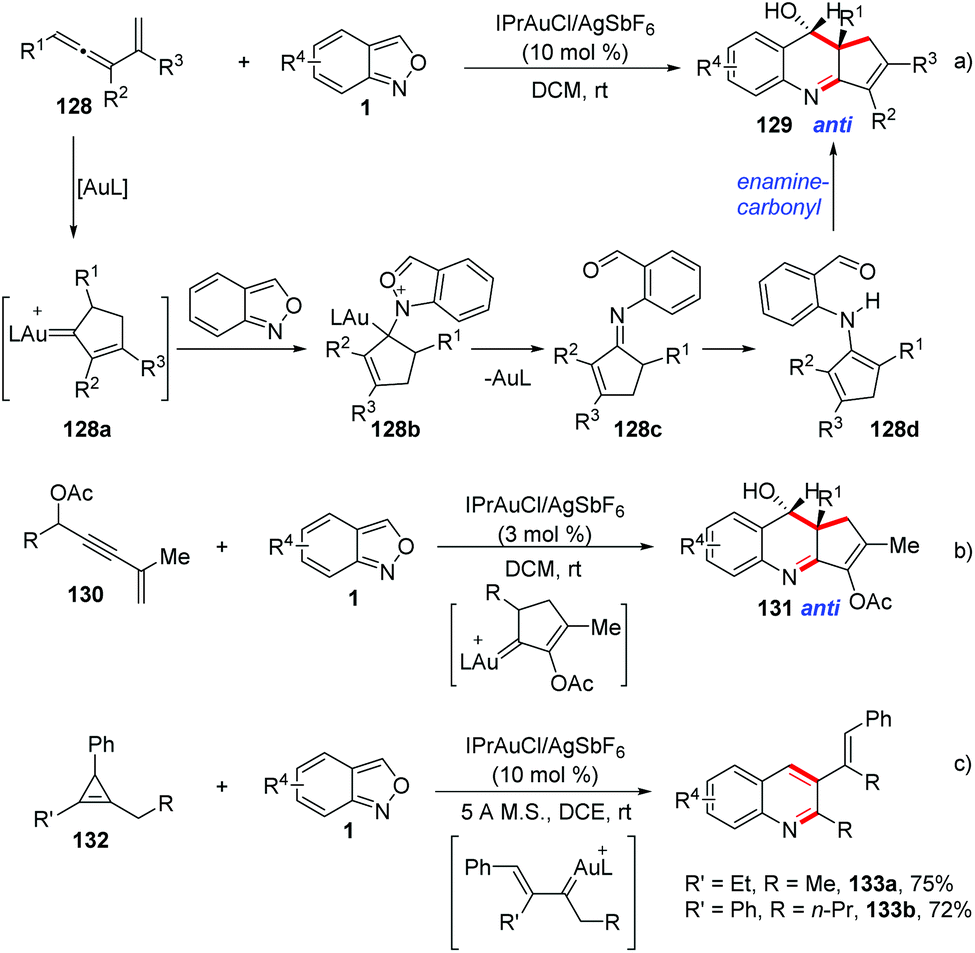 | ||
| Scheme 49 Gold-catalysed [4 + 2]-annulations between α-alkyl alkenylgold carbenes and anthranils. | ||
In addition, Liu and co-workers described an elegant gold-catalysed chemoselective annulation of electron-deficient alkynes 134 with anthranils to yield quinolone oxides (Scheme 50).65 This annulation was compatible with substrates over a wide scope. It was found that the electron density of ligands has a significant impact on the chemoselectivity of this reaction. When an electron-deficient phosphite ligand was used, the O-atom of anthranils could selectively attack the gold activated alkyne to give the final quinolone oxides. In contrast, an electron-rich ligand was beneficial for the N-attack pathway, affording the corresponding indole products as described in Hashmi's work (Scheme 39). An unusual O-attack mechanism was proposed for this annulation. Initially, the O-atom of anthranils attacked the gold activated alkyne to form the species 134b, which evolved to the gold carbene 134c through N–O bond cleavage. Then, a rapid cyclization of 134c formed the seven-membered heterocycle 134d, which underwent an oxoninum-induced dissociation of LAu+ to generate the species 134e. The final product 135 was obtained through a subsequent 6π electrocyclization. When propiolates bearing R = 4-MeOC6H4 and styryl groups were subjected in this reaction, the corresponding quinolin-4(1H)-one derivatives 136 were obtained through a facile epoxide rearrangement. Additionally, zinc(II)-catalysed hydrative lactonizations of quinolone oxides provided a facile access to the highly oxygenated tetrahydroquinoline derivatives 137, and these processes are unprecedented in the literature.
 | ||
| Scheme 50 Gold-catalysed chemoselective annulation of electron-deficient alkynes with anthranils. | ||
7. Summary
The reaction of readily available anthranils represents a powerful strategy to construct various C–N bonds, which has wide applications in the synthesis of high-value N-containing heterocycles. By taking advantage of the rich and tunable reactivities of anthranils, a series of reaction modes and unprecedented transformations have been developed over the past decades. As a result, diverse C–N bonds and bioactive scaffolds, which are difficult to be obtained through other means, have been successfully synthesized in a catalytic and selective manner with the use of anthranils as building blocks. Moreover, additional efforts have been made in elucidating the mechanistic pathways, which provide us valuable insights into the reaction mechanism and further promote chemists to develop more efficient and atom-economical methodologies.Despite notable recent advances, there are still some challenging problems in this field. The catalytic asymmetric conversion of anthranils remains elusive and the application of these methods into practical synthesis is still highly desirable. The further design of new and selective dual catalytic systems will provide a general solution to these challenges. This review is meant to stimulate continued interest of the synthetic community in anthranil chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (21901045 and 21901258), the Guangdong University of Technology (220413199 and xj201811845075) and the Guangdong Natural Science Foundation (2018A030310570) for financial support.Notes and references
-
(a) U. H. F. Bunz, The Larger Linear N-Heteroacenes, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed
; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed
-
(a) R. Y. Ning, J. F. Blount, P. B. Madan and R. I. Fryer, Intramolecular nitrene insertion into nitrogen containing rings. Pyrolyses of 3-(1-methyl-2-imidazolyl)- and 3-(1-methyl-5-pyrazolyl)-2,1-benzisoxazole (anthranils), J. Org. Chem., 1977, 42, 1791–1794 CrossRef CAS
- J. Chauhan and S. Fletcher, One-pot synthesis of 2,1-benzisoxazoles (anthranils) by a stannous chloride-mediated tandem reduction–heterocyclization of 2-nitroacylbenzenes under neutral conditions, Tetrahedron Lett., 2012, 53, 4951–4954 CrossRef CAS
- L. Marti, L. M. Sanchez, M. J. Climent, A. Corma, S. Iborra, G. P. Romanelli and P. Concepcion, Chemoselective Reductive Heterocyclization by Controlling the Binomial Architecture of Metal Particles and Acid–Base Properties of the Support, ACS Catal., 2017, 7, 8255–8262 CrossRef CAS
- D. Zhao, Q. Shen and J.-X. Li, Potassium Iodide-Catalyzed Three-Component Synthesis of 2-Arylquinazolines via Amination of Benzylic C–H Bonds of Methylarenes, Adv. Synth. Catal., 2015, 357, 339–344 CrossRef CAS
-
(a) B. J. Stokes, C. V. Vogel, L. K. Urnezis, M. Pan and T. G. Driver, Intramolecular Fe(II)-Catalyzed N–O or N–N Bond Formation from Aryl Azides, Org. Lett., 2010, 12, 2884–2887 CrossRef CAS PubMed
- M. Aidene, F. Belkessam, J.-F. Soulé and H. Doucet, Reactivity of 2,1-Benzisoxazole in Palladium-Catalyzed Direct Arylation with Aryl Bromides, ChemCatChem, 2016, 8, 1583–1590 CrossRef CAS
- J. S. Baum and M. E. Condon, Nickel-Catalyzed Transformations of 2,1-Benzisoxazoles with Organozinc Reagents, J. Org. Chem., 1987, 52, 2983–2988 CrossRef CAS
- J. Li, E. Tan, N. Keller, Y.-H. Chen, P. M. Zehetmaier, A. C. Jakowetz, T. Bein and P. Knochel, Cobalt-Catalyzed Electrophilic Aminations with Anthranils: An Expedient Route to Condensed Quinolines, J. Am. Chem. Soc., 2019, 141, 98–103 CrossRef CAS PubMed
- F. Xie, B. Shen and X. Li, Enantioselective Copper-Catalyzed Hydroamination of Vinylarenes with Anthranils, Org. Lett., 2018, 20, 7154–7157 CrossRef CAS PubMed
-
(a)
F. Xie and X. Li, in Rhodium Catalysis in Organic Synthesis, ed. K. Tanaka, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019, pp. 521–592 Search PubMed
-
(a) S. Yu, G. Tang, Y. Li, X. Zhou, Y. Lan and X. Li, Anthranil: An Aminating Reagent Leading to Bifunctionality for Both C(sp3)-H and C(sp2)-H under Rhodium(III) Catalysis, Angew. Chem., Int. Ed., 2016, 55, 8696–8700 CrossRef CAS PubMed
- M. Zou, J. Liu, C. Tang and N. Jiao, Rh-Catalyzed N–O Bond Cleavage of Anthranil: A C–H Amination Reagent for Simultaneous Incorporation of Amine and a Functional Group, Org. Lett., 2016, 18, 3030–3033 CrossRef CAS PubMed
- M. Wang, L. Kong, F. Wang and X. Li, Rhodium-Catalyzed Amination and Annulation of Arenes with Anthranils: C-H Activation Assisted by Weakly Coordinating Amides, Adv. Synth. Catal., 2017, 359, 4411–4416 CrossRef CAS
- M. Jeon, J. Park, P. Dey, Y. Oh, H. Oh, S. Han, S. H. Um, H. S. Kim, N. K. Mishra and I. S. Kim, Site-Selective Rhodium(III)-Catalyzed C–H Amination of 7-Azaindoles with Anthranils: Synthesis and Anticancer Evaluation, Adv. Synth. Catal., 2017, 359, 3471–3478 CrossRef CAS
-
(a) H. Li, J. Jie, S. Wu, X. Yang and H. Xu, Rh(III)-Catalyzed direct C-7 amination of indolines with anthranils, Org. Chem. Front., 2017, 4, 250–254 RSC
- T. Fu, J. Yang, H. Sun, C. Zhang, H. Xiang and X. Zhou, Rh(III)-Catalyzed C-H Amination of Azobenzenes with Anthranils, Asian J. Org. Chem., 2018, 7, 1844–1848 CrossRef CAS
- S. Debbarma and M. Sudan Maji, Cp*RhIII-Catalyzed Directed Amidation of Aldehydes with Anthranils, Eur. J. Org. Chem., 2017, 3699–3706 CrossRef CAS
-
(a) S. Yu, Y. Li, X. Zhou, H. Wang, L. Kong and X. Li, Access to Structurally Diverse Quinoline-Fused Heterocycles via Rhodium(III)-Catalyzed C–C/C–N Coupling of Bifunctional Substrates, Org. Lett., 2016, 18, 2812–2815 CrossRef CAS PubMed
- A. Biswas, S. Bera, P. Poddar, D. Dhara and R. Samanta, Rh(III)-Catalyzed Tandem Indole C4-Arylamination/Annulation with Anthranils: Access to Indoloquinolines and Their Application in Photophysical Studies, . Chem. Commun., 2020, 56, 1440–1443 RSC
- A. Biswas, S. Sarkar and R. Samanta, Rh(III)-Catalyzed Straightforward Synthesis of Benzophenanthroline and Benzophenanthrolinone Derivatives using Anthranils, Chem. – Eur. J., 2019, 25, 3000–3004 CAS
- A. Biswas, U. Karmakar, S. Nandi and R. Samanta, Copper Catalyzed Direct, Regioselective Arylamination of N-Oxides: Studies to Access Conjugated π-Systems, J. Org. Chem., 2017, 82, 8933–8942 CrossRef CAS PubMed
- S. Kim, S. H. Han, N. K. Mishra, R. Chun, Y. H. Jung, H. S. Kim, J. S. Park and I. S. Kim, Dual Role of Anthranils as Amination and Transient Directing Group Sources: Synthesis of 2-Acyl Acridines, Org. Lett., 2018, 20, 4010–4014 CrossRef CAS PubMed
- S. Kim, A. Kundu, R. Chun, S. H. Han, A. K. Pandey, S. Yoo, J. Park, H. S. Kim, J.-M. Ku and I. S. Kim, Direct Synthesis of 2-Acyl Acridines Using Aldimines and Anthranils: Evaluation of Cytotoxicity and Anti-Inflammatory Activity, Asian J. Org. Chem., 2018, 7, 2069–2075 CrossRef CAS
- S. Huang, H. Li, X. Sun, L. Xu, L. Wang and X. Cui, Rh(III)-Catalyzed Sequential C–H Amination/Annulation Cascade Reactions: Synthesis of Multisubstituted Benzimidazoles, Org. Lett., 2019, 21, 5570–5574 CrossRef CAS PubMed
- Y. Hu, T. Wang, Y. Liu, R. Nie, N. Yang, Q. Wang, G.-B. Li and Y. Wu, Practical Synthesis of Benzimidazo[1,2-a]quinolines via Rh(III)-Catalyzed C–H Activation Cascade Reaction from Imidamides and Anthranils, Org. Lett., 2019, 22, 501–504 CrossRef PubMed
- X. Wu, Y. Xiao, S. Sun, J.-T. Yu and J. Cheng, Rhodium-Catalyzed Reaction of Sulfoxonium Ylides and Anthranils toward Indoloindolones via a (4+1) Annulation, Org. Lett., 2019, 21, 6653–6657 CrossRef CAS PubMed
-
(a) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Dehydrative Direct C–H Allylation with Allylic Alcohols under [Cp*CoIII] Catalysis, Angew. Chem., Int. Ed., 2015, 54, 9944–9947 CrossRef CAS PubMed
- L. Li, H. Wang, S. Yu, X. Yang and X. Li, Cooperative Co(III)/Cu(II)-Catalyzed C–N/N–N Coupling of Imidates with Anthranils: Access to 1H-Indazoles via C–H Activation, Org. Lett., 2016, 18, 3662–3665 CrossRef CAS PubMed
- R.-H. Liu, Q.-C. Shan, X.-H. Hu and T.-P. Loh, Site-Selective C(sp3)–H Amination of Thioamide with Anthranils under Cp*CoIII Catalysis, Chem. Commun., 2019, 55, 5519–5522 RSC
-
(a) E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed
- Z.-H. Wang, H.-H. Zhang, D.-M. Wang, P.-F. Xu and Y.-C. Luo, Lewis acid catalyzed diastereoselective [3+4]-annulation of donor–acceptor cyclopropanes with anthranils: synthesis of tetrahydro-1-benzazepine derivatives, Chem. Commun., 2017, 53, 8521–8524 RSC
-
(a) X. Zhang, M. Feng, G. Yang and Z. Chai, Sc(OTf)3-Catalyzed Chemodivergent Annulations of γ-Butyrolactone-Fused Cyclopropanes with Anthranils, J. Org. Chem., 2019, 85, 430–440 CrossRef PubMed
- Q. Cheng, J.-H. Xie, Y.-C. Weng and S.-L. You, Pd-Catalyzed Dearomatization of Anthranils with Vinylcyclopropanes via [4+3] Cyclization Reaction, Angew. Chem., Int. Ed., 2019, 58, 5739–5743 CrossRef CAS PubMed
- J. Feng, M. Zhou, X. Lin, A. Lu, X. Zhang and M. Zhao, Base-Mediated [3+4]-Cycloaddition of Anthranils with Azaoxyallyl Cations: A New Approach to Multisubstituted Benzodiazepines, Org. Lett., 2019, 21, 6245–6248 CrossRef CAS PubMed
- O. Kazuko, S. Hiroko and N. Yujiro, Reaction of Anthranils with Enamines. Novel Synthesis of Quinolines, Nippon Kagaku Kaishi, 1989, 5, 846–854 Search PubMed
- D. K. Tiwari, M. Phanindrudu, S. B. Wakade, J. B. Nanubolu and D. K. Tiwari, α,β-Functionalization of saturated ketones with anthranils via Cu-catalyzed sequential dehydrogenation/aza-Michael addition/annulation cascade reactions in one-pot, Chem. Commun., 2017, 53, 5302–5305 RSC
- S. B. Wakade, D. K. Tiwari, P. S. K. P. Ganesh, M. Phanindrudu, P. R. Likhar and D. K. Tiwari, Transition Metal-Free Quinoline Synthesis from Acetophenones and Anthranils via Sequential One-Carbon Homologation/Conjugate Addition/Annulation Cascade, Org. Lett., 2017, 19, 4948–4951 CrossRef CAS PubMed
- J. Ren, C. Pi, Y. Wu and X. Cui, Copper-Catalyzed Oxidative [4+2]-Cyclization Reaction of Glycine Esters with Anthranils: Access to 3,4-Dihydroquinazolines, Org. Lett., 2019, 21, 4067–4071 CrossRef CAS PubMed
- F. Wang, P. Xu, S.-Y. Wang and S.-J. Ji, Cu(II)/Ag(I)-Catalyzed Cascade Reaction of Sulfonylhydrazone with Anthranils: Synthesis of 2-Aryl-3-sulfonyl Substituted Quinoline Derivatives, Org. Lett., 2018, 20, 2204–2207 CrossRef CAS PubMed
- S. Zhu, K. Shi, H. Zhu, Z.-K. Jia, X.-F. Xia, D. Wang and L.-H. Zou, Copper-Catalyzed Annulation or Homocoupling of Sulfoxonium Ylides: Synthesis of 2,3-Diaroylquinolines or α,α,β-Tricarbonyl Sulfoxonium Ylides, Org. Lett., 2020, 22, 1504–1509 CrossRef CAS PubMed
- J. Jiang, X. Cai, Y. Hu, X. Liu, X. Chen, S.-Y. Wang, Y. Zhang and S. Zhang, Thermo-promoted Reactions of Anthranils with Carboxylic Acids, Amines, Phenols and Malononitrile under Catalyst-free Conditions, J. Org. Chem., 2019, 84, 2022–2031 CrossRef CAS PubMed
- E. C. Taylor and J. Sartulin, Ring Expansion of Anthranils to Quinoline-1-oxides, Tetrahedron Lett., 1967, 25, 2337–2339 CrossRef
- J. Li, Z.-B. Wang, Y. Xu, X.-C. Lu, S.-R. Zhu and L. Liu, Catalyst-free cyclization of anthranils and cyclic amines: one-step synthesis of rutaecarpine, Chem. Commun., 2019, 55, 12072–12075 RSC
- L.-H. Zou, H. Zhu, S. Zhu, K. Shi, C. Yan and P.-G. Li, Copper-Catalyzed Ring-Opening/Reconstruction of Anthranils with Oxo-Compounds: Synthesis of Quinoline Derivatives, J. Org. Chem., 2019, 84, 12301–12313 CrossRef CAS PubMed
-
(a) L. Nunes dos Santos Comprido, J. E. M. N. Klein, G. Knizia, J. Kästner and A. S. K. Hashmi, Gold(I) Vinylidene Complexes as Reactive Intermediates and Their Tendency to π-Backbond, Chem. – Eur. J., 2016, 22, 2892–2895 CrossRef CAS PubMed
- For alternative entries to α-imino gold carbene complexes, see: X. Tian, L. Song and A. S. K. Hashmi, α-Imino Gold Carbene Intermediates from Readily Accessible Sulfilimines: Intermolecular Access to Structural Diversity, Chem. – Eur. J., 2020, 26, 3197–3204 CrossRef CAS PubMed
- J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. do C. Carreiras and E. Soriano, Recent Advances in the Friedländer Reaction, Chem. Rev., 2009, 109, 2652–2671 CrossRef CAS PubMed
-
(a) E. Aguilar and J. Santamaría, Gold-catalyzed heterocyclic syntheses through α-imino gold carbene complexes as intermediates, Org. Chem. Front., 2019, 6, 1513–1540 RSC
- H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold-Catalyzed C-H Annulation of Anthranils with Alkynes: A Facile, Flexible, and Atom-Economical Synthesis of Unprotected 7-Acylindoles, Angew. Chem., Int. Ed., 2016, 55, 794–797 CrossRef CAS PubMed
- H. Jin, B. Tian, X. Song, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold-Catalyzed Synthesis of Quinolines from Propargyl Silyl Ethers and Anthranils through the Umpolung of a Gold Carbene Carbon, Angew. Chem., Int. Ed., 2016, 55, 12688–12692 CrossRef CAS PubMed
- R. D. Kardile, B. S. Kale, P. Sharma and R.-S. Liu, Gold-Catalyzed [4+1]-Annulation Reactions between 1,4-Diyn-3-ols and Isoxazoles To Construct a Pyrrole Core, Org. Lett., 2018, 20, 3806–3809 CrossRef CAS PubMed
- H.-C. Hsieh, K.-C. Tan, A. S. Kulandai Raj and R.-S. Liu, Gold-catalyzed [4+1]-Annulation Reactions between Anthranils and 4-Methoxy-1,2-dienyl-5-ynes Involving a 1,2-Allene Shift, Chem. Commun., 2019, 55, 1979–1982 RSC
- R.-S. Liu, M. Skaria, S. A. More, M.-J. Cheng and T.-C. Kuo, Gold-catalyzed Iminations of Terminal Propargyl Alcohols with Anthranils with Atypical Chemoselectivity for C(1)-Additions and 1,2-Carbon Migration, Chem. – Eur. J., 2019, 26, 3600–3608 Search PubMed
- Z. Zeng, H. Jin, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold-Catalyzed Regiospecific C–H Annulation of o-Ethynylbiaryls with Anthranils: π-Extension by Ring-Expansion En Route to N-Doped PAHs, Angew. Chem., Int. Ed., 2018, 57, 6935–6939 CrossRef CAS PubMed
- Z. Zeng, H. Jin, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold(III)-Catalyzed Site-Selective and Divergent Synthesis of 2-Aminopyrroles and Quinoline-Based Polyazaheterocycles, Angew. Chem., Int. Ed., 2018, 57, 16549–16553 CrossRef CAS PubMed
- M.-H. Tsai, C.-Y. Wang, A. S. Kulandai Raj and R.-S. Liu, Gold(III)-Catalyzed Site-Selective and Divergent Synthesis of 2-Aminopyrroles and Quinoline-Based Polyazaheterocycles, Chem. Commun., 2018, 54, 10866–10869 RSC
- M. D. Patil and R.-S. Liu, Direct access to benzofuro[2,3-b]quinoline and 6H-chromeno[3,4-b]quinoline cores through gold-catalyzed annulation of anthranils with arenoxyethynes and aryl propargyl ethers, Org. Biomol. Chem., 2019, 17, 4452–4455 RSC
-
(a) H. Zhao, Y. Xing, P. Lu and Y. Wang, Synthesis of 2,3-Disubstituted Quinolines via Ketenimine or Carbodiimide Intermediates, Chem. – Eur. J., 2016, 22, 15144–15150 CrossRef CAS PubMed
- Y. Hsu, S. Hsieh and R. Liu, Gold-catalyzed Annulations of N-Propargyl Ynamides with Anthranils With Two Distinct Chemoselectivities, Chem. – Eur. J., 2019, 25, 5288–5297 CrossRef CAS PubMed
- L. Song, X. Tian, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold(III)-catalyzed chemoselective annulations of anthranils with N-allylynamides for the synthesis of 3-azabicyclo[3.1.0]hexan-2-imines, Chem. Commun., 2019, 55, 9007–9010 RSC
- M. Skaria, P. Sharma and R.-S. Liu, Gold(I)-Catalyzed 1,3-Carbofunctionalizations of Anthranils with Vinyl Propargyl Esters To Yield 1,3-Dihydrobenzo[c]-isoxazoles, Org. Lett., 2019, 21, 2876–2879 CrossRef CAS PubMed
- R. R. Singh, M. Skaria, L.-Y. Chen, M.-J. Cheng and R.-S. Liu, Gold-catalyzed (4+3)-annulations of 2-alkenyl-1-alkynylbenzenes with anthranils with alkyne-dependent chemoselectivity: skeletal rearrangement versus non-rearrangement, Chem. Sci., 2019, 10, 1201–1206 RSC
- B. D. Mokar, P. D. Jadhav, Y. B. Pandit and R.-S. Liu, Gold-catalyzed (4+3)-annulations of 2-alkenyl-1-alkynylbenzenes with anthranils with alkyne-dependent chemoselectivity: skeletal rearrangement versus non-rearrangement, Chem. Sci., 2018, 9, 4488–4492 RSC
- R. L. Sahani and R.-S. Liu, Gold-Catalyzed [4+2] Annulation/Cyclization Cascades of Benzisoxazoles with Propiolate Derivatives to Access Highly Oxygenated Tetrahydroquinolines, Angew. Chem., Int. Ed., 2017, 56, 12736–12740 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2020 |