
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in carbon-based supercapacitors
Ling
Miao
a,
Ziyang
Song
a,
Dazhang
Zhu
a,
Liangchun
Li
 *a,
Lihua
Gan
*a and
Mingxian
Liu
*ab
*a,
Lihua
Gan
*a and
Mingxian
Liu
*ab
aShanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai 200092, P. R. China. E-mail: lilc@tongji.edu.cn; ganlh@tongji.edu.cn; liumx@tongji.edu.cn
bCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China
First published on 7th July 2020
Abstract
Rapid advancements in modern electronics have been starved of further breakthroughs to achieve high-energy, large-power, and long-running energy storage devices. Carbon-based supercapacitors (CSs) are promising large-power systems that can store electrical energy at the interface between the carbonaceous electrode surface and adsorbed electrolyte layer. However, commercial CSs using activated carbons suffer from limited energy densities in the organic electrolytes owing to sluggish mass diffusion and restricted charge accumulation. To overcome these issues, significant efforts have been devoted toward increasing the energy storage of CSs by the exploration of both large-capacitance electrodes and high-potential electrolytes. This paper reviews the recent advances made in the two core components (i.e., electrodes and electrolytes) of CSs. Firstly, we describe the involved energy storage mechanisms of CSs, followed by a brief overview of the key factors affecting the electrochemical performances. Then, novel design concepts have been summarized that can be used to fabricate carbon-based electrodes, such as microporous carbons, mesoporous carbons, hierarchical porous carbons, functionalized carbons, and carbon composites. Further, tailoring of the geometrical morphologies, pore structures, and surface functionalities can be discussed in pursuit of large-capacitance electrodes. Furthermore, various types of aqueous and nonaqueous electrolytes, such as water-in-salt, organic media, ionic liquids, and (quasi-)solid-state electrolytes, have been systematically investigated to settle the water hydrolysis concern of traditional electrolytes. Finally, the challenges in maximizing the synergistic effect between the electrodes and electrolytes are summarized, and we have proposed the future outlook for the development of advanced CSs.
 Ling Miao | Ling Miao is a Postdoctoral Fellow in Prof. Mingxian Liu's group at Tongji University. She received her PhD from the School of Chemical Science and Engineering, Tongji University (2018). Her research interests focus on the design and tailored synthesis of ionic-liquid-derived porous carbons for energy-related applications. |
 Ziyang Song | Ziyang Song received his BS in 2016 from Jinggangshan University, China. He currently continues to pursue his PhD under the supervision of Prof. Mingxian Liu at Tongji University. His research currently focuses on the design and synthesis of advanced carbon materials, as well as exploring new solid gel electrolytes for energy conversion and storage. |
 Dazhang Zhu | Dazhang Zhu received his PhD in Environmental Science from Tongji University (2007) and became a Professor in the School of Chemical Science and Engineering, Tongji University (2017). His research interests include the controllable preparation of nanocrystals/nanostructures and their applications in supercapacitors and environmental protection. |
 Liangchun Li | Liangchun Li received his PhD in 2009 at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), China. In 2009–2012, he was a Special Postdoctoral Researcher at RIKEN, Japan. In 2012, he joined the group of Prof. S. Kitagawa in Kyoto University. In 2013, he obtained his JSPS Fellowship. After his post doc research, he became a Professor in the School of Chemical Science and Engineering (2015), Tongji University. His current research efforts are focused on the development of porous coordination polymers for catalysts and derived porous carbons for energy storage. |
 Lihua Gan | Lihua Gan received his PhD in Materials Physics and Chemistry from Tongji University and became a Professor at the School of Chemical Science and Engineering in Tongji University (2005). His research interests include the design and application of nanomaterials, which cover porous metal oxides, various carbonaceous materials, and carbon-based materials. These nanomaterials are used in supercapacitors and lithium-ion batteries, and they are used as adsorbents for the treatment of pollutants. |
 Mingxian Liu | Mingxian Liu received his PhD in Physical Chemistry from Tongji University (2009). He started his postdoctoral research in East China University of Science and Technology (2009–2011). After that, he joined the School of Chemical Science and Engineering at Tongji University, and he became a Professor in 2016. His current research interests focus on the structural design and controllable synthesis of carbon-based materials for energy conversion and storage. |
1. Introduction
Energy conversion and storage is the fundamental pillar of modern society driven by the mutual promotion between environmental consciousness and economic development.1 The diversification of portable electronic systems and the ongoing spread of electronics have been starved of further breakthroughs to achieve high-energy, large-power, and long-running energy storage devices.1–3 Carbon-based supercapacitors (CSs) are promising large-power devices that keep on dominating the commercial market, affording fascinating features such as fast pulses of energy, millions of charge/discharge cycles, wide operating temperatures, and high Coulombic efficiencies.4,5 CSs consist of two carbonaceous electrodes soaked in an aqueous/nonaqueous electrolyte, and a porous membrane separator allowing electrolyte permeation (Fig. 1). Their energy storage mechanism is fundamentally derived from reversible charge separation at each interface of the electrolyte with the cathode/anode surface to form two closely spaced charge layers.5 The corresponding “double-layer capacitance” (CD = εA/d, where ε is the dielectric constant) arises from the minimal separation distance (d, down to a few angstroms) and large interfacial area (A) between the intrinsic double-layer configuration. Such physical interfacial contact makes CSs fully charged/discharged within seconds with large power delivery/uptake. However, commercial CSs using activated carbons suffer from limited energy densities (<10 W h kg−1) in organic electrolytes owing to the sluggish mass diffusion and restricted charge accumulation. | ||
| Fig. 1 Schematic representation of CSs. | ||
To overcome this issue, significant efforts have been devoted toward increasing the energy storage (E = 0.5CV2) of CSs by the exploration of two core components, i.e., large-capacitance (C) electrodes and high-potential (V) electrolytes.5,6 Regarding the role of carbon-based electrodes, the design of large-surface-area carbon materials with engineered surface topography/pore feature or doping defects/functionalities to optimize the electrochemical activity, surface polarization, and electrical conductivity has become intensive research realms. On the other hand, with regard to exploiting electrolytes, particularly aqueous electrolytes with wider potential windows and nonaqueous electrolytes with higher ionic conductivities, electrochemical inertness and operation safety have become highly desirable for advanced CSs. Besides, pseudocapacitors or asymmetric/hybrid capacitors are also involved in the pursuit of high-performance CSs: the modification of pseudo-active substances endows bare carbons with supplementary faradaic pseudocapacitance based on the electron transfer mechanism, while the hybrid of capacitive carbon and pseudocapacitive/battery-type electrode can bridge the voltage gap between the two electrodes to increase the device potential.6,7 However, these upgraded device configurations generally come with undesired decreases in the power/cycling capabilities caused by the limited kinetics of redox reactions or phase transformation of active materials.8 Therefore, the fundamental challenge for advanced CSs is how to maximize the role of the carbon/electrolyte in an ideal configuration to achieve high-energy, large-power, and long-running devices.
Porous carbons have been sustainably popularized as electrode materials in CSs, with the help of a representative report on anomalously increased capacitance in the subnanopores and other overwhelming theoretical/experimental studies.9–12 Besides the common physicochemical stability and electronic properties, porous carbons stand out because of their versatile syntheses. Alternative carbon precursors encompass conventional biomasses and phenolic resins as well as novel organic salts/polymers such as metal salts, metal–organic frameworks (MOFs), covalent organic frameworks, conjugated microporous polymers, hypercrosslinked polymers, ionic liquids and poly(ionic liquid)s, etc.13–16 High flexibility in the precursor choice and condition regulation can customize ideal porous carbon electrodes with the following structural superiorities: (a) large surface area to guarantee a vast accumulation platform for high energy storage, coupled with abundant adsorbing sites exposed on the surface topography to improve the accessible surface area;17,18 (b) tunable pore structures to ensure the rapid transmission of electrolyte ions and continuous infiltration into the electrode surface, thereby enabling higher power delivery;11,19,20 (c) surface wettability/activity to boost the electrolyte/electrode interfacial contact and afford more active sites for improved charge accumulation and ion kinetics;21–23 and (d) stable conductive frameworks for long-term operation.7,24,25 In the following sections, we focus on the recent advances in the two core components (i.e., electrodes and electrolytes) of CSs. Novel fabrication strategies of carbon-based materials are highlighted with tailoring the geometrical morphologies, pore structures, and surface functionalities in pursuit of large-capacitance electrodes. Furthermore, a brief overview of emerging high-potential electrolytes is presented, such as water-in-salt (WIS), organic media, ionic liquids, and (quasi-)solid-state electrolytes. Finally, the challenges in realizing the electrode/electrolyte synergy are summarized, with future outlooks for the advancement of next-generation CSs.
2. Carbon-based electrodes
2.1. Large surface area
The International Union of Pure and Applied Chemistry (IUPAC) categorized pores according to their diameters into three types: micropore (<2 nm), mesopore (2–50 nm), and macropore (>50 nm).26 The above formula of CD states that the large surface area is a priority in achieving the large capacitance of carbon-based electrodes (large amounts of charged ions on the pore surface). Micropores perform a vital function in supplying a vast accumulation platform for high energy storage via molecular sieving and controlled diffusion effects.9,17,27,28 As such, activated carbons with predominant microporosity have long been the most popular as supercapacitor electrodes. These materials are usually produced by the pre-carbonization of carbon-rich precursors (particularly biomass sources) and subsequent physical/chemical activation to afford microporous structures.13 Physical activation is conducted in an oxidative atmosphere (e.g., CO2, O2, or steam) at higher temperatures of 700–1200 °C, whereas chemical activation refers to heat treatment with chemicals (KOH, NaOH, H3PO4, ZnCl2, K2CO3, etc.) at relatively lower temperatures. For instance, Wang's group prepared wrinkled porous carbon nanosheets through the carbonization and alkali activation of biomass sugarcane bagasse pith/chitosan, and the resulting typical carbon with a high surface area of 1786.1 m2 g−1 exhibited electrode capacitances of 339/280 F g−1 at current densities of 0.25/100 A g−1 in 6 M KOH electrolyte.29 In another example, Gao's group reported a dual-activator approach with egg yolk as the precursor and KOH/C3N3Na3S3 as the activators (Fig. 2a).30 By regulating the mass ratio of egg yolk/C3N3Na3S3, as-obtained carbon materials afforded a tunable microporous area (26.26–74.69%) with the largest surface area (3519.50 m2 g−1) and pore volume (2.68 cm3 g−1), contributing to the maximum energy density at 12.7 W h kg−1 in H2SO4 (Fig. 2b and e). | ||
| Fig. 2 Schematic of the preparation of egg yolk-derived activated carbons (a). Influence among the structural parameters, i.e., SBET (b); Smicro/SBET (c); Vtot (d); and electrochemical performances such as specific capacitance, power density, energy density, and rate density. Ragone plots (e). (Reproduced with permission.30 Copyright 2018, The Royal Society of Chemistry.) | ||
2.2. Morphology control
Large surface area is always desirable for CSs, but the electrode capacitance does not necessarily increase along with the surface area because the entire area is not electrochemically accessible to the electrolyte ions, particularly in excess of 1500 m2 g−1.31 Creating abundant adsorbing sites exposed on the surface topography helps to relieve the barrier toward surface accessibility for improving the storage capacity.18 Therefore, considerable research efforts have focused on fabricating diverse carbon morphologies with refined control over the surface topography and interior texture.Spherical carbon nanoarchitectures featuring the smallest surface-to-volume ratio and close-packing nature can be constructed by various synthesis methods such as emulsion polymerization, modified Stöber synthesis, self-assembly, and hydrothermal carbonization.18,32 Recently, uniform mesoporous carbon nanospheres with large accessible pores (5–37 nm) were prepared through a versatile nanoemulsion polymerization process with dopamine as the precursor and PEO106–PPO70–PEO106 (F127) as the template in a water/ethanol system (Fig. 3a).33 By adjusting the mediator (1,3,5-trimethylbenzene, TMB) content, the resulting modification of the interfacial interactions between dopamine and F127 afforded the evolution of tunable pore diameters and various morphologies. Yu's group obtained hollow carbon spheres with controllable pore sizes (from micropores to 13.9 nm) relying on the steric effect of the propoxy of tetrapropyl orthosilicate (TPOS) using modified Stöber synthesis (Fig. 3b).34 In contrast to traditional tetraethyl orthosilicate (TEOS), the longer hydrolysis and condensation of TPOS offered subtler monitoring ability over the SiO2 core and oligomer formation (Fig. 3c and d), and the superior capacitive performances of 310 and 157 F g−1 could be realized at a current density from 1 to 50 A g−1 in a basic electrolyte. Further, Wei's group monitored the interfacial capillary force and polymer nanoshell thickness in the TPOS-involved Stöber process to achieve the transformation from nanoball-like architecture to carbon nanobowls with enhanced mass loading.35 Furthermore, recently, Qiao's group designed a surfactant-assisted assembly approach to fabricate a series of multi-chamber carbon microspheres derived from 2,6-diaminopyridine.36 A dual-surfactant system (F127 and sodium dodecylbenzenesulfonate) could induce the establishment of refined hierarchical architectures as well as tune the microsphere size in the scale of 250–3091 nm, and therefore, the as-synthesized electrode with a multi-chamber core and microporous shell functioned as a competitive supercapacitor material.
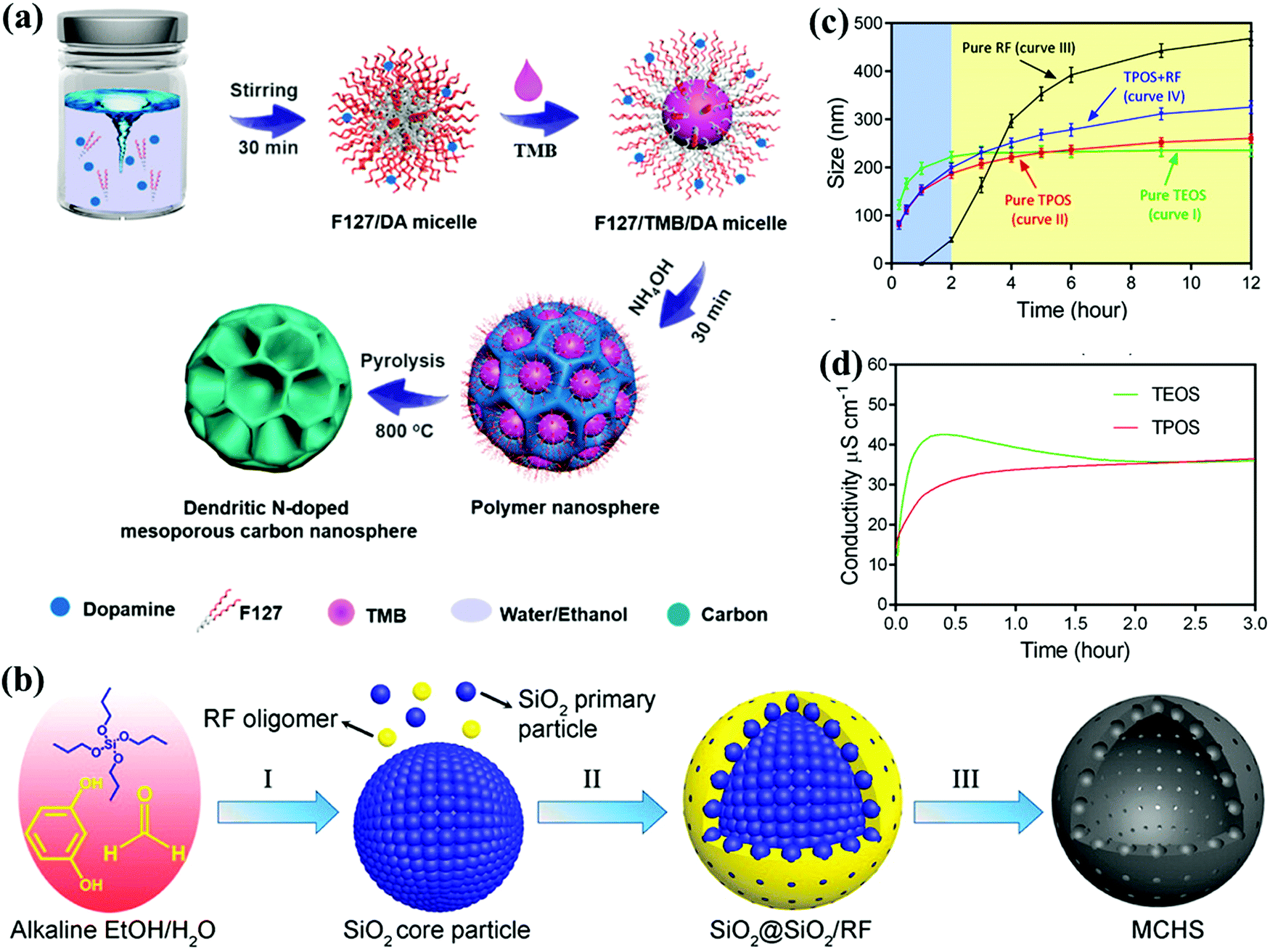 | ||
| Fig. 3 Synthesis of the procedure for fabricating uniform mesoporous carbon nanospheres (a). (Reproduced with permission.33 Copyright 2019, American Chemical Society.) Schematic of the synthesis of hollow carbon spheres (b). Particle sizes (c) and conductivity evolution (d) in TPOS/TEOS systems with reaction time. (Reproduced with permission.34 Copyright 2016, American Chemical Society.) | ||
As promising supercapacitor electrodes, porous carbon nanosheets possess high aspect ratios of 2D sheet-like morphologies for short ion-transfer channels; meanwhile, their porous architectures effectively prevent the layer overlapping/restacking of the rich interfacial accessible sites.37 Recently, the fabrication of carbon nanosheets has been performed by means of exfoliating carbon nanorods/nanotubes, lithographic patterning, chemical vapor deposition, thermal conversion, etc.37–42 For example, Fischer's group developed 2D carbon nanosheets via the controlled pyrolysis of rod-shaped MOFs for fabricating a high-potential (1.6 V) supercapacitor (Fig. 4a).43 Zhang's group proposed a top-down exfoliation–chlorination approach to synthesize ultrathin carbon nanosheets derived from the multilayered carbide precursor, Ti3AlC2.44 The inherent 2D intersheet space in exfoliated metal carbides/nitrides (MXenes) affords graphitized carbon sheets (thickness: 3–4 nm), large accessible interface area (1766 m2 g−1), and reduced diffusion distances in the thin dimension. Besides, natural layered shrimp shells were employed as a 2D hard template to assemble graphene-like carbon nanostructures exhibiting high conductivity (7.8 S cm−1) and outstanding capacitive performance of 322 F g−1 at 0.5 A g−1 in an aqueous electrolyte.45 Recently, Qiu's group established the correlation between sheet thickness and ion transport behavior (ion diffusion coefficient and Warburg coefficient) inside the 2D carbon electrodes (Fig. 4b).46 By using dopamine and MgAl-layered double hydroxides as the carbon suppliers and 2D structure-directing templates, carbon sheets with controllable thickness could be obtained by simply varying the reaction time, and the fabricated electrode with an optimal thickness of ∼99.23 nm could be fabricated, delivering an outstanding retention rate of 81% at 100 A g−1 and high energy output of 94 W h kg−1 at 1.8 kW kg−1 in an ionic liquid device.
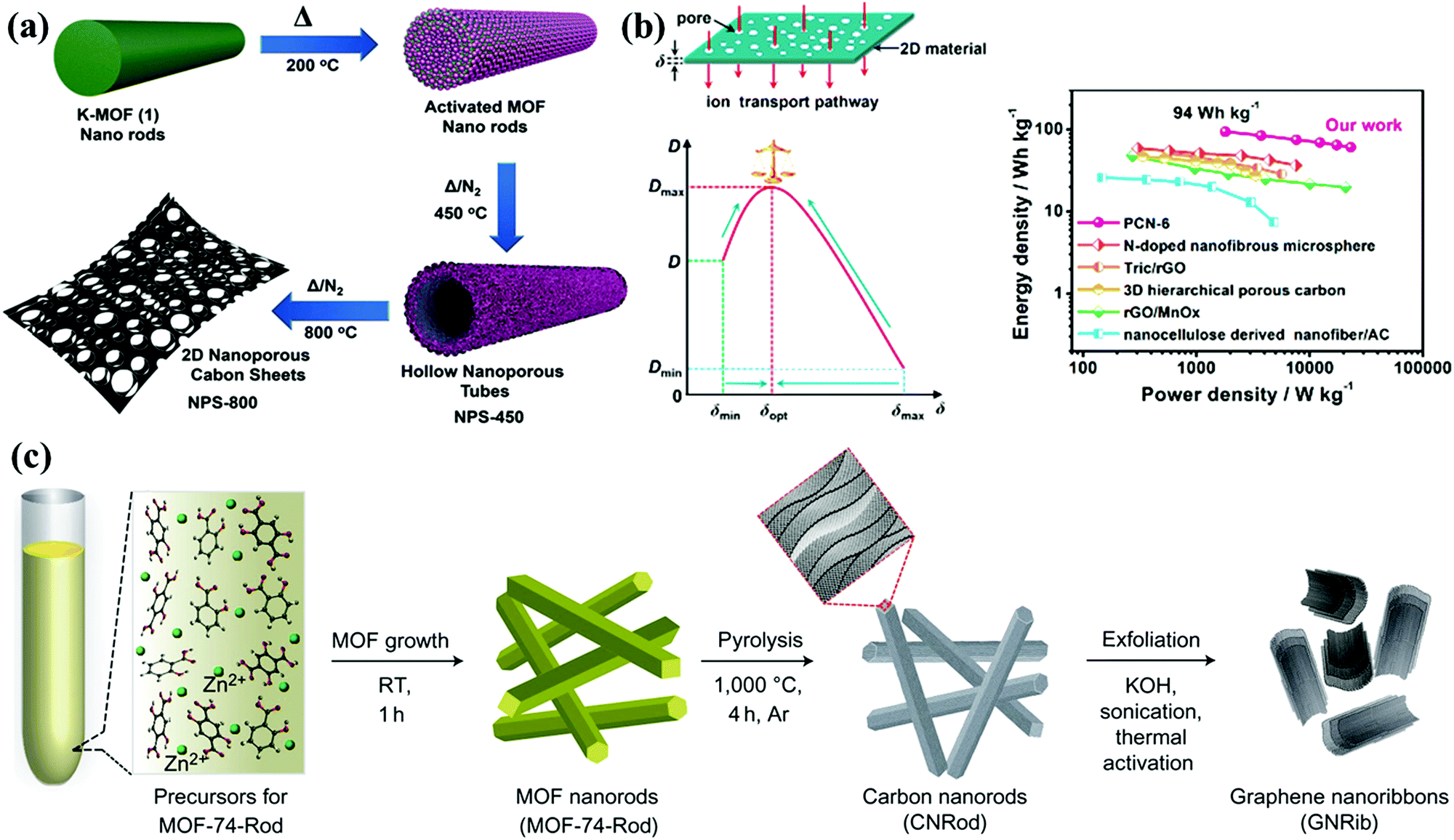 | ||
| Fig. 4 Synthesis of MOF-based 2D carbon nanosheets (a). (Reproduced with permission.43 Copyright 2018, Wiley-VCH.) Schematic illustration of ion transfer inside a 2D carbon electrode; correlation between the sheet thickness and ion transport behavior; and Ragone plots (b). (Reproduced with permission.46 Copyright 2019, Elsevier.) | ||
Further, 1D carbon nanofibers and nanorods could afford shortcut channels for electron transfer, and they can be readily shaped into relevant geometries or assembled with functionalized modules for preparing advanced CSs.47–52 For instance, Xu's group demonstrated the preparation of carbon nanorods via the self-sacrificial thermal conversion of rod-like MOF precursors that were prepared by the salicylic-acid-modulated hydrothermal reaction of 2,5-dihydroxyterephthalic acid and zinc nitrate, and the as-obtained carbon nanorods could be further transformed into graphene nanoribbons featuring comparable electronic properties via the sonication–activation treatment.53 Zhang's group utilized the self-assembly of chitin fibers in urea/NaOH solution to fabricate unique elastic microspheres with a carbon nanofiber framework and high surface area of over 1000 m2 g−1.54 When applied as a supercapacitor electrode, this carbon material exhibited high loading intensity, distinguishing elastic recovery property (over 5 cyclic compressions at strain of 75%), and high energy delivery of 58.7 W h kg−1 in organic media. Chen's group synthesized a novel self-standing composite electrode (hexagonal nanoprism carbon arrays supported on carbon fiber substrates) by chemical vapor deposition using ethylenediamine precursor and ZnO sacrifice template.55 The in situ growth on carbon fiber lowered the interfacial resistance for rapid electron transport, which effectively contributed toward the superior rate performance with sweep rate/current load increasing to 20 V s−1/300 mA cm−2 in a bind-free supercapacitor.
Generally, external-templating strategies offer precise control over diverse morphologies as mentioned above, and they can be further extended to build core–shell/hollow and other complex nanoarchitectures that synergize the accessible interior spaces/active interfaces into a single system.18,56,57 For example, Yu's group reported a new rigid-interface-induced outward contraction approach to fabricate hollow mesoporous carbon nanocubes with MOF as the precursor (Fig. 5a).58 A thick mesoporous SiO2 layer on MOF nanocubes could offset the inward contraction of the MOF@SiO2 core–shell architecture at higher temperatures, and the subsequent outward contraction of the surplus frameworks induced the formation of carbon materials in the mesoporous walls and hollow cavities after template removal. Yang's group constructed a spheres-in-tube hierarchical porous nanostructure by filling anodic aluminum oxide channels with SiO2 nanoparticles and subsequent polypyrrole coating, and the resultant carbon electrode exhibited superior capacitive performances than those of pure nanotubes/hollow spheres.59 Alternatively, solvent-induced/template-free approaches allow the straightforward and scalable assembly of advanced carbon nanoarchitectures.37,60–64 Feng's group reported the comprehensive construction of porous carbon superstructures via the hierarchical assembly of polyimide precursor sheets (Fig. 5b).65 By changing the monomer concentrations in an aprotic solvent, the assembly of linear polymer chains could be easily monitored, yielding a class of unprecedented hierarchical superstructures such as lantern-shaped/flower-like nanospheres. Kim's group developed a Lewis acid/base-interaction-mediated self-assembly strategy on the basis of classic Friedel–Crafts crosslinking reactions to prepare functionalized carbon materials with well-engineered morphologies.66 Layered, tubular, or hollow spherical assemblies were readily accessible via the rational combination between aromatic hydrocarbon monomers (base) and Lewis acid catalysts in common solvents. These inspiring findings have put forward new insights into the facile fabrication of alternative carbon nanoarchitectures, and in-depth efforts are still underway to realize fine control over the geometric parameters in solvent-induced/template-free processes.
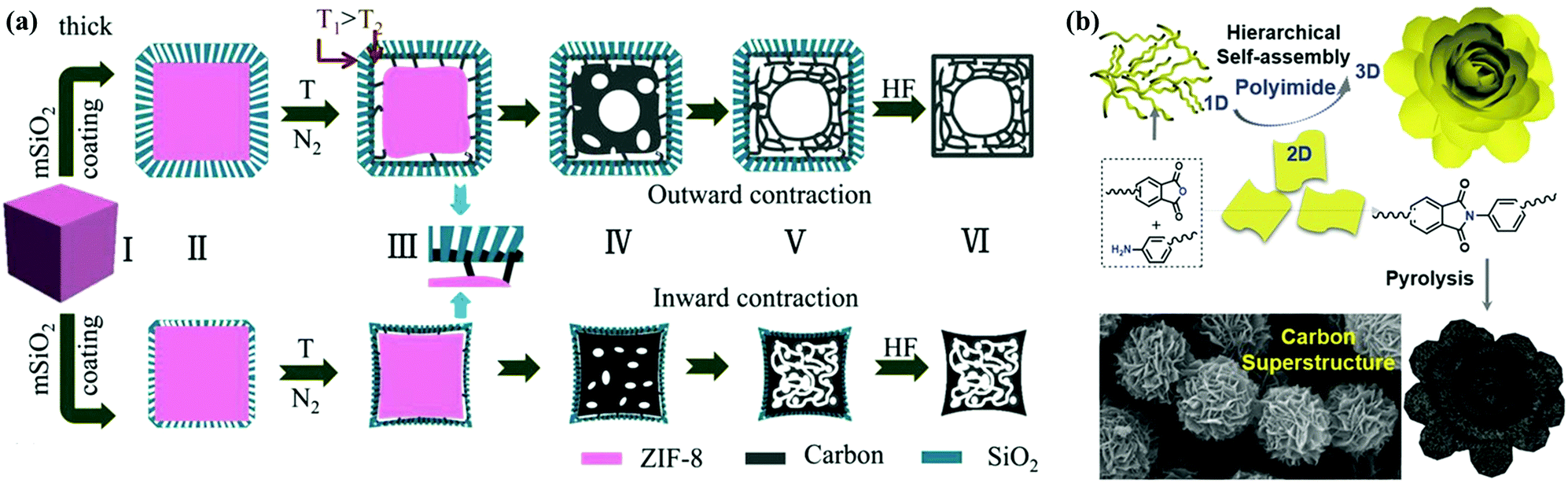 | ||
| Fig. 5 Schematic illustration of hollow mesoporous carbon nanocubes (a). (Reproduced with permission.58 Copyright 2017, Wiley-VCH.) Fabrication scheme of porous carbon superstructures (b). (Reproduced with permission.65 Copyright 2016, Wiley-VCH.) | ||
2.3. Pore structure regulation
Appropriate regulation of pore diameters in carbon materials can be used to transport sufficient electrolyte ions throughout the electrodes, consequently allowing carbon surfaces to be fully utilized. Micropores mainly contribute toward a vast accumulation platform through molecular sieving and controlled diffusion effects as mentioned above, while meso-/macropores can shorten the transportation route and reduce the interfacial resistance of the electrolytes as ion-diffusion shortcuts to the fine micropores.13,19 Moreover, hierarchical porous structures consisting of interconnected micro-, meso-, and macropores are particularly interesting for high-performance CSs in consideration of the synergistic effects among the abundant micropores as ion-adsorbing sites and interconnected meso-/macropores ensuring rapid ion transmission, substantially achieving mutual enhancements in energy/power densities.11,19,20 Furthermore, open and interconnected pore architectures accelerate continuous ion-diffusion kinetics within the pores, and they subsequently offer an easy approach to fabricate active electrode surfaces for compact devices particularly with large volumetric capacitances.11 Therefore, considerable research efforts have been focused on developing facile and efficient fabrication methods to realize the integral regulation of controlling the pore size distribution within the carbon materials.Activated carbons with a tiny micropore diameter (<0.5 nm) and tortuous pore alleys are not adequately accessible in commercial organic electrolytes owing to hampered or even blocked mass diffusion.5 Tailoring the micropore diameter and shape is essential for enhanced surface accessibility, particularly in ionic liquids/organic electrolytes, to maximize the double-layer capacitances.9,17,27,67–69 As two pioneering classes, micropore textures of zeolite-templated carbons could recently be customized according to different diameters/shapes of zeolite pores where the lanthanum-catalyzed carbonization of acetylene/ethylene occurred.70 Chlorine treatment and physical activation of crosslinked polycarbosilane aerogels endowed the carbide-derived carbon with enlarged surface area/micropore volume (2410 m2 g−1/0.63 cm3 g−1) and therefore high capacitance of 170 F g−1 at 0.1 A g−1 in EMIMBF4 and stable operation even at 60 A g−1 in TEABF4/ACN.71 Beyond these, elaborately designed microporous carbons are mainly fabricated based on the similar self-porogen/template role of novel metal complexes.72–75 Another highlighted fabrication strategy is the direct pyrolysis of emerging organic polymers (e.g., covalent organic frameworks, conjugated microporous polymers, and hypercrosslinked polymers) assembled by building blocks with intrinsic microporosity and periodic skeleton.76 Recently, Shim's group constructed a triazine-based covalent organic polymer with predominant micropores as the carbon precursor via the Friedel–Crafts reaction.77 Upon further physical activation, the surface area increased to 2003 m2 g−1 without sacrificing the narrow micropore diameter distribution (0.5–1.5 nm). Wang's group reported a novel hypercrosslinked polymerization route to obtain a stable microporous precursor frame as the growing polymer chains were crosslinked with adjacent rigid bridges to avoid frame mobilization/collapse (Fig. 6a).78 As expected, the carbon electrode with well-retained micropore textures delivered a high energy density of 16.2 W h kg−1 at a power density of 50 W kg−1 and capacitance retention of 92% over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (Fig. 6b and c).
000 cycles (Fig. 6b and c).
 | ||
| Fig. 6 Synthesis of hypercrosslinked polymer-derived carbons (a). Ragone plots (b). Long-term cycling performance (c). (Reproduced with permission.78 Copyright 2019, Elsevier.) | ||
Ordered mesopore paths in carbon materials can be introduced to improve the utilization of micropores since they can create express entries for electrolyte transmission into the inner electrode surface even at high currents, thereby enabling remarkable rate/power performances.19 During the past few decades, the hard-template nanocasting method has become mature and versatile for the assembly of the required mesoporous textures.19,79,80 The applied porous solid templates (SiO2, MgO, Fe3O4, etc.) inversely replicated the interior structures into ultimate carbons.81–84 For instance, Chen's group synthesized monodispersed carbon spheres with tunable mesopore size (3.1–10.0 nm) based on the self-activation calcination of phenolic resin encapsulated in the in situ generated silica nanoreactors from TEOS (Fig. 7a).85 Choy's group recently proposed a cost-effective geopolymer approach to obtain an appropriate mesopore ratio in carbons, and natural clay as the silica supplier for lab-made geopolymer templates was ∼400 times cheaper as compared to TEOS.86
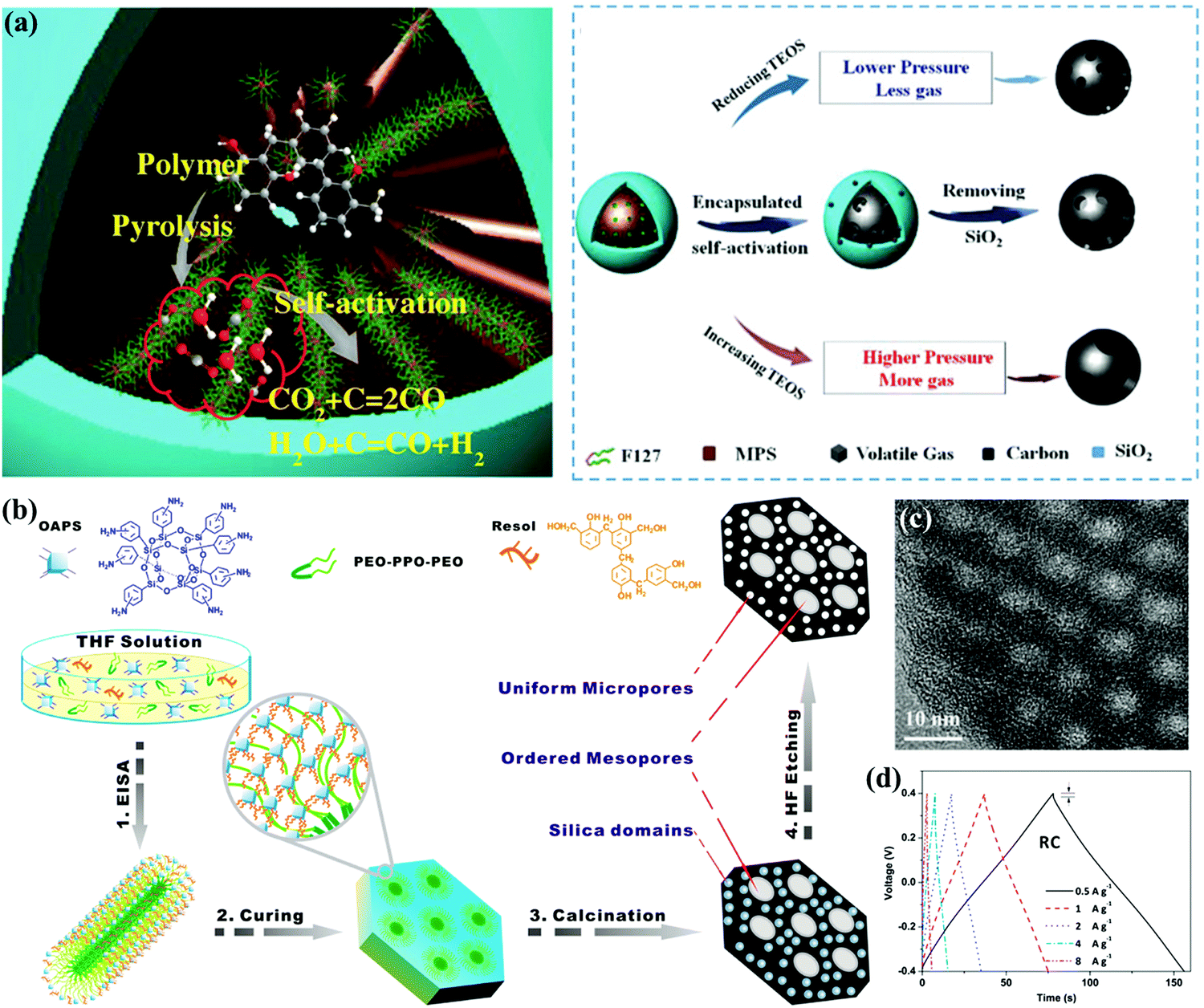 | ||
| Fig. 7 Schematic illustration of monodispersed mesoporous carbon spheres (a). (Reproduced with permission.85 Copyright 2018, Wiley-VCH.) Schematic illustration of ordered mesoporous carbons (b). TEM image (c). Galvanostatic charge/discharge curves (d). (Reproduced with permission.98 Copyright 2016, Elsevier.) | ||
With regard to direct soft-template strategies, tunable mesostructures can be generated based on the cooperative assembly between the precursor molecules and single-chain surfactants/block copolymers as sacrificial organic templates.11,87,88 Suitable pyrolysis/extraction processes have been developed for the effective elimination of surfactant templates; nowadays, a big family of block copolymer templates such as PS–P4VP/PEO, PEO–PPO–PEO, PPO–PEO–PPO, and so on (PS: polystyrene; P4VP: poly(4-vinyl pyridine); PEO: polypropylene oxide; and PPO: polyethylene oxide) have been successfully utilized in these strategies.89–93 For instance, Zhang's group developed a confined self-assembly process to synthesize MXene-derived mesoporous carbon layers with phenolic resol as an additional carbon precursor in the presence of the F127 template.94 F127 with hydrophilic PEO segments can easily interact with the precursor molecules, and similar mesopore size distributions centered at 3.1 and 7–12 nm can be obtained within the carbon materials after thermosetting at 100 °C for a rigid polymer structure and sequential two-step pyrolysis for simultaneous template removal. Gao's group prepared interconnected mesoporous carbon spheres with a large pore volume of 1.12–3.22 cm3 g−1 and tunable pore size of 5–20 nm via a sol–gel process by regulating the volume ratio of ethanol/water and the dosage of a cationic Gemini surfactant template, i.e., pentane-1,5-bis(dimethylcetyl ammonium bromide).95 Besides, combined hard–soft templating approaches have been adopted to generate multi-model pore sizes.96,97 As a representative example, Su's group described a block-copolymer-assisted approach based on aminophenyl-functionalized polyhedral oligosilsesquioxanes as the carbon source as well as soft template to prepare carbon electrodes with large pore volume of 1.19 cm3 g−1, highly ordered mesopores (∼4 nm), and uniform micropores (∼1 nm) (Fig. 7b and c).98 The unique nanostructure could be attributed to the molecule-level role of the template in the inherent siloxane cages, and the resultant electrode exhibited an outstanding rate ability with 97/94% capacitance preservation in a H2SO4/ionic liquid electrolyte (Fig. 7d).
Hierarchical porous carbons can be fabricated by modifying the hard-/soft-templating protocols to realize integral regulation over the pore size distribution.16,92,99 Salt-template techniques using soluble/decomposable/reactive salts have emerged as a simple and sustainable branch of hard-templating strategies for the generation of hierarchical porosity.16,100–102 Through this technique, certain disadvantages of other templating approaches such as the use of hazardous etchants or low-temperature restriction can be compensated, while diverse salts can interact differently with most precursor molecules during annealing when compared with SiO2/metal oxide templates.103 Recently, Chu's group proposed a facile and ecofriendly route from a gelatin–NaNO3 biopolymer precursor to design hierarchical porous carbons with a large adsorbing surface of 2872.2 m2 g−1 and mesopores/macropores centered at 2–4 and 50–150 nm, respectively (Fig. 8a).104 The decomposition of NaNO3 above 600 °C released activating gases (CO2/N2/O2/NO) to yield a micro-/mesoporous structure, and vacancies/macropores formed by the water removal of Na2O depended on the hard-templating effect of nanoconfined NaNO3. Gan's group fabricated unique hollow carbon nanorods via the direct carbonization of an all-in-one deep-eutectic-solvent source, wherein ZnCl2 was involved in the formation of the polymer shell as a raw reactant and also hydrolyzed for forming the final ZnCl2/Zn2OCl2·2H2O/ZnO core.105 The combination of micro-/mesoporous shells (thickness: 100 nm) and open hollow nanorods (width: 400 nm) within the carbons endowed the ionic-liquid-based device with long-term operation at 4 V and a three-fold energy density value (116.5 W h kg−1) when compared with commercial activated carbon tested under the same configuration.
 | ||
| Fig. 8 Schematic illustration of the synthesis of hierarchical porous carbons from a gelatin–NaNO3 biopolymer (a). (Reproduced with permission.104 Copyright 2018, Wiley-VCH.) Examples of porous carbon structures after the calcination of block copolymers (b). (Reproduced with permission.92 Copyright 2019, The Royal Society of Chemistry.) | ||
With regard to modified soft-template constituents, the direct fabrication of hierarchical porous carbons from block copolymers becomes an alternate template-free strategy if the precursors consist of one polymer block with high carbon residue and the other block as the self-porogen during high-temperature calcination (Fig. 8b).92,106–108 Chen's group recently reported a self-template approach to synthesize N-doped hierarchical porous carbons by calcining polyacrylonitrile-block-PS copolymers.109 PS, as the self-template block, promoted the generation of mesopores (6–11 nm), while micropores developed by the activation of the carbon-supply block (polyacrylonitrile) contributed to a large surface area of 2104.5 cm2 g−1. Inspired by such a design concept, multiple physically interlaced polymer networks with different thermostabilities (i.e., interpenetrating polymer networks) have been employed to obtain multiscale pore distributions.110–113 Instead of covalently bonded chains, molecule-level interlaced networks tend to generate highly interconnected hierarchical porous carbon foam as the macro-phase separation between the carbon sources/pore formers can be avoided during annealing. Besides, the simultaneous carbonization–activation process was considered to be another noteworthy template-free approach for the construction of 3D interconnected macrostructures because of the high-temperature decomposition of organics in alkaline media.114–118 As a typical example, Fan's group reported the one-step pyrolysis of KOH-assisted wheat flour to obtain interconnected honeycomb-like porous carbons.119 The alkali-treated flour gelatin as the self-porogen could undergo a series of stages such as framework decomposition, volume expansion, and intermediate foaming during annealing to form hierarchical porous structures. The 3D interconnected honeycomb-like architecture facilitated charge transfer and storage within the carbon, demonstrating the development of a simple and cost-effective strategy for fabricating high-performance carbon electrode materials.
2.4. Heteroatom functionalization
Doping carbon materials with heteroatom (N, O, S, B, P, etc.) functionalities has considerably improved the electrochemical performances of carbon-based electrodes.22,120 Firstly, binding heterogeneous atoms into the carbon backbone induces positive partial charges on the neighboring carbon atoms/heteroatoms with lower electronegativity to present a polarized electrode surface, and therefore, these polarized sites enhance the interactions with electrolyte ions to facilitate adsorption.20,23 Secondly, the substitutive incorporation of electron-donor heteroatoms supplements the lone pair of electrons to the delocalized carbon framework for improved intrinsic conductivity, and the decreased valence band contributes toward the long-term running and high electron density at the Fermi level.25 Thirdly, the embedment of pseudo-active heteroatoms can facilitate proton adsorption and endow bare carbons with remarkable faradaic pseudocapacitance based on electrochemical redox reactions.22,121 Additionally, the co-/multi-doping of diverse heteroatoms probably produces synergistic contributions induced by spin/charge redistribution, consequently conducive toward affording dense electroactive density and improved overall electrode performance.120 However, excessive/highly active heteroatoms doped by various fabrication methods and conditions could lead to undeveloped porosity or framework collapse in functionalized carbons and render CSs with decayed performances. Therefore, considerable research effort has been focused on achieving synergy between heteroatom doping and porous architecture by means of suitable fabrication methods and conditions (Table 1).| Material | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Heteroatom content | Measurement system | Capacitance (F g−1) | Energy density (W h kg−1) | Power density (W kg−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| N-doped porous carbon nanosheets | 1786.1 | 0.8157 | N: 2.10 at%, O: 7.11 at% | 6 M KOH, 0.25 A g−1 | 339 | 11.77 | 34.11 | 29 |
| N/O/S-doped hierarchical porous carbon materials | 3519.50 | 2.68 | — | 1 M H2SO4, 1 A g−1 | 549 | 12.7 | — | 30 |
| Mesoporous hollow carbon spheres | 1582 | 2.45 | — | 6 M KOH, 1 A g−1 | 310 | — | — | 34 |
| N/S-co-doped carbon nanobowls | 1567 | 2.25 | N: 3.3 wt%, S: 1.7 wt% | 6 M KOH, 0.1 A g−1 | 279 | 9.6 | 25 | 35 |
| N-doped multi-chamber carbon microspheres | 1797 | 0.96 | N: 4.58 wt%, O: 2.12 wt% | 6 M KOH, 0.2 A g−1 | 301 | — | — | 36 |
| N-rich porous carbon nanosheets | 2406 | — | N: 9.4 at%, O: 4.7 at% | EMIMBF4, 0.5 A g−1 | 250 | 139 | 500 | 40 |
| 2D porous carbon nanosheets | 1907 | 0.77 | N: 1.54 at%, O: 6.59 at% | EMIMBF4, 1 A g−1 | 221 | 94 | 1800 | 46 |
| Porous carbon nanorods | 1559 | — | N: 1.47 at%, O: 0.62 at% | 1 M H2SO4, 0.05 A g−1 | 187 | — | — | 53 |
| N-doped porous carbon nanofibrous microspheres | 1147 | 2.12 | N: 2.4 at%, O: 6.1 at% | EMIMTFSI, 5 mV s−1 | 113 | 58.7 | 300 | 54 |
| Spheres-in-tube hierarchical porous carbon | 318 | 0.78 | N: 8.74 at%, O: 3.39 at% | 1 M Na2SO4, 0.2 A g−1 | — | 29.5 | 401 | 59 |
| N-doped porous carbon superstructures | 1375 | 0.996 | N: 3.46 wt%, O: 7.99 wt% | 6 M KOH, 0.6 A g−1 | ∼364 | — | — | 65 |
| Porous interconnected carbon nanosheets | 2220 | 1.11 | — | 1 M TEABF4/ACN, 0.1 A g−1 | 150 | ∼30 | 13000 |
72 |
| Ultramicroporous carbon materials | 1312 | 0.67 | — | EMIMBF4, 0.2 A g−1 | 223 | 32.5 | — | 75 |
| N/S-doped porous carbon | 1339 | 0.96 | N: 4.5 at%, S: 5.8 at% | 6 M KOH, 0.2 A g−1 | 464 | 16.2 | 50 | 78 |
| Order mesoporous carbon spheres | 1186 | 0.27 | — | 6 M KOH, 1 A g−1 | 226.1 | 27 | 980 | 85 |
| N-rich mesoporous carbons | 458 | 0.42 | N: 19.10 wt% | 1 M H2SO4, 0.2 A g−1 | 252 | — | — | 91 |
| N/O-doped hierarchical porous carbon nanorods | 1882 | — | N: 8.1 wt%, O: 10.0 wt% | EMIMBF4, 0.2 A g−1 | 214 | 116.5 | 472 | 105 |
| Honeycomb-like porous carbon foam | 1313 | 0.716 | N: 1.1 at%, O: 11.2 at% | 1 M Na2SO4, 2 mV s−1 | 260 | 29.3 | — | 119 |
| B/N-codoped carbon nanosheets | 2362 | 1.448 | N: 3.1 at%, B: 0.5 at% | 1 M Na2SO4, 0.5 A g−1 | 235.6 | 30.1 | 225.1 | 124 |
| N-doped microporous carbon spheres | 1478 | 0.76 | N: 8.71 at%, O: 7.89 at% | 6 M KOH, 1 A g−1 | 292 | 8.75 | 500 | 130 |
Functionalized carbons are generally fabricated by posttreatment procedures of the as-prepared carbon materials at elevated temperatures or self-doping by pyrolyzing heteroatom-rich precursors (e.g., organic salts, synthetic polymers, and biomass sources).122 The highly praised self-doping strategy allows the homogenous distribution of heteroatoms and multispecies, but it is relatively confined to the chemical stability of reaction systems and the precursor thermostability when compared with the widely applicable posttreatment approach. With an increase in the number of comprehensive studies, the self-doping strategy has been recently subdivided into adding a supplementary dopant (e.g., ethanediamine or phosphoric/boric acid) to the carbon precursors before pyrolyzing or the direct pyrolysis of organic salt/polymer precursors assembled by heteroatom-containing building blocks.123 The supplementary dopant can not only provide plentiful heteroatoms, but also serve as a catalyst, stabilizer, activator, or structure-directing agent. For example, Jia's group synthesized chitosan-derived N/B-codoped porous carbon nanosheets with three amino acids and boric acid as the N/B source.124 Additionally, amino acids interacted with chitosan as the structure-directing agent to induce architecture evolution, and boric acid and KOH acted as the reactive templates for creating micropores and mesopores (2–4 nm) in the final carbon. Wang's group demonstrated that the presence of poly(ionic liquid)s was beneficial for optimizing the hydrothermal carbonization of sugar precursors for N-doped porous carbon nanoparticles, wherein the poly(ionic liquid) chains introduced electrostatic repulsion for minimized particle agglomeration and unexpectedly functioned as the pore former and N supply.125 As an extension of the post-doping strategy, the one-step doping activation is frequently applied via directly calcining the mixtures of carbon precursors and heteroatom-doped activators,123 and the resultant N-rich microporous carbons activated by NaNH2 afforded outstanding capacitance of 350.2 F g−1 at 1 A g−1 in 6 M KOH with excellent rate retention of 74.2% at 10 A g−1.126
Another desirable route toward fabricating functionalized carbons at a higher doping level is the direct pyrolysis of organic salt/polymer precursors (e.g., metal complexes, conjugated polymer networks, ionic liquids, and poly(ionic liquid)s).14–16 Through the judicious assembly of molecular building blocks, heterogeneous/crosslinkable/decomposable modules can be introduced into organic salt/polymer precursors to simultaneously optimize the desired functionality/porosity. Recently, 3D hierarchical porous carbon with large interlayer space and rich N content (5.74 at%) was fabricated by the direct calcination of tetrasodium ethylenediamine tetraacetic acid, wherein the decomposed C/N-containing skeleton of the salt precursor transformed into a N-doped carbon product coated on the self-generated Na2CO3 template (Fig. 9a).127 Similarly, MOFs assembled by versatile organic ligands and metal ions enriched the possibilities for designed compositions/structures in the final products, and metal species could be used to obtain carbon composites or as a hard template for additional porosities.14,128 Conjugated polymer networks assembled via the covalent bonding of multifunctional aromatic building blocks allow a wealth of choices of heteroatom incorporations with targeted locations, and the intensive inter-/intrachain π–π interactions of the robust conjugated skeleton guarantee smooth conversion into porous carbon nanoarchitectures by direct calcination.15,65,129–132 Wang's group fabricated a novel class of multi-heteroatom-doped porous carbons based on the in situ dehalogenation of heteroatom-containing nucleophiles with halogenated conjugated dienes (Fig. 9b).133 As-obtained carbons with various P/N/O and abundant multimodal pores afforded a high packing density (2.13 gcm−3) and remarkable gravimetric/volumetric energy densities of 17.3 W h kg−1/36.8 W h L−1 in an alkaline electrolyte, even approaching those of Ni-MH cells (Fig. 9c and d). Further, Liu's group developed a universal amine/benzoquinone-derived strategy to prepare high redox-active N/O-codoped porous carbons with various geometries and high surface areas to support the double-layer capacitance.134 Moreover, the pseudocapacitive contribution of the electroactive species was well illuminated due to the fast redox reactions of quinone/hydroquinone shifts and pyrrolic/pyridinic-N responses in acid electrolytes, and the assembled supercapacitor afforded exceptional energy delivery of 18.2 W h kg−1 at 300 W kg−1. Following this direction, poly(ionic liquid)s as an emerging carbon precursor class not only inherit the composition designability of the final carbons via matching the cations/anion pairs of monomeric ionic liquids, but also enable the flexible tuning of their structures/morphologies owing to versatile polymeric chains.16,135 Furthermore, the poly(ionic liquid) matrix can readily crosslink the templates for targeted porous nanoarchitectures74,136,137 or be shaped/processed via classical polymer operations (electrospinning, spin-coating, embossing, etc.) for solid-state/flexible CSs.16,138–140
 | ||
| Fig. 9 Schematic illustration of N-rich 3D hierarchical porous carbons (a). (Reproduced with permission.127 Copyright 2019, Elsevier.) Synthesis of multi-heteroatom-doped porous carbons (b). Packing density (c). Ragone plots (d). (Reproduced with permission.133 Copyright 2019, Wiley-VCH.) | ||
2.5. Carbon composites
The modification of pseudo-active substances (e.g., metal species and conducting polymers) endows bare carbons with supplementary pseudocapacitance based on the electron transfer mechanism.6,7 Meanwhile, fabricating composite materials by employing carbon as the support boosts the electrochemical utilization of pseudo-active sites and simultaneously serves as efficient conductive pathways for improved electron transfer and mechanical stability.8 Moreover, the hybrid of capacitive carbon and pseudocapacitive/battery-type electrode can bridge the voltage gap between the two electrodes to increase the device potential.7 Therefore, considerable research efforts have been devoted toward fabricating optimal architectures to completely utilize the structural superiorities and electroactivities of each substance toward realizing competitive volumetric performances within dense composite electrodes (Table 2).| Positive//negative material | Classification | Potential (V) | Electrolyte | Capacitance | Energy density | Power density | Ref. |
|---|---|---|---|---|---|---|---|
| Porous carbon fibers@MnO2 | Symmetric | 0.8 | 6 M KOH | 1148 F g−1 | 10.3 W h kg−1 | — | 141 |
| Hierarchical porous N-doped carbon@Fe/Fe3C | Symmetric | 1.0 | 6 M KOH | 246 F g−1 | — | — | 142 |
| Nanoporous duct-like graphene@MnO2//nanoporous duct-like graphene | Asymmetric | 2.0 | 2 M LiNO3 | — | 28.2 mW h cm−3 | 55.7 W cm−3 | 143 |
| N-doped carbon nanowires@MnO2//N-doped carbon nanowires@Fe2O3 | Asymmetric | 1.6 | 5 M LiCl | 72 mF cm−2 | — | — | 147 |
| N,S-doped mesoporous carbon@MnO2 | Symmetric | 3.5 | EMIMBF4 | 200 F g−1 | — | — | 148 |
| Fe2O3@P-doped mesoporous carbon | Symmetric | 3.5 | EMIMBF4 | 179 F g−1 | — | — | 149 |
| Na0.5MnO2//reduced graphene oxide@porous carbon/Fe3C | Hybrid | 2.4 | 1 M Na2SO4 | — | 46.2 W h kg−1 | 1.2 kW kg−1 | 151 |
| Mn3O4/porous carbon rods//porous carbon rods | Hybrid | 4.0 | 1 M LiPF6 | — | 174 W h kg−1 | 200 W kg−1 | 152 |
| NiSx@porous carbon matrix//AC | Hybrid | 2.8 | 1 M NaClO4 | — | 99.3 W h kg−1 | 140 W h kg−1 | 153 |
| SnS/porous carbon microtubes//porous carbon microtubes | Hybrid | 3.5 | 1 M NaClO4 | — | 115 W h kg−1 | — | 154 |
| Nb2O5@carbon core–shell nanoparticles/reduced graphene oxide//AC | Hybrid | 3.3 | 1 M NaPF6 | — | 76 W h kg−1 | 80 W kg−1 | 156 |
| FeSe2/N-doped carbon//AC | Hybrid | 2.5 | 3 M KFSI | — | 230 W h kg−1 | — | 157 |
Metal-oxide-modified carbons are the characteristic surface-redox pseudocapacitive materials for symmetric and asymmetric CSs, since high capacitances could be achieved through the electrochemical reactions of electrolyte ions onto/near the active surface.6,141–145 Shi's group prepared a free-standing MnO2/carbon sphere/graphene electrode film by a simple vacuum filtration process and subsequent solution infiltration.146 After modifying with high-capacitive MnO2, the composite film delivered higher volumetric capacitance (277.8 F cm−3) than that of a carbon sphere/graphene film, and correspondingly higher energy density of 25.6 W h L−1 coupled with capacitance retention of 94.1% over 5000 cycles in a Na2SO4-based symmetric supercapacitor. Yushin's group demonstrated the integration of N-doped carbon nanowires@metal oxide (MnO2 and Fe2O3) composites into conductive fabrics for asymmetric supercapacitors (Fig. 10a).147 N-doped carbon nanowires functioned as a stable conductive substrate, while the shortened diffusion channels within the metal oxide particles largely facilitated ion transportation, giving rise to high areal capacitance (72 mF cm−2) for the aqueous-gel-based asymmetric device operated at 1.6 V. Metal-oxide-modified carbons can also be employed as electrode materials in ionic-liquid-based supercapacitors.148–150 Oschatz's group reported the facile deposition of MnO2 nanosplotches on N/S-doped mesoporous carbon matrix, and the as-obtained composite electrode exhibited outstanding electrochemical properties in an ionic liquid (EMIMBF4)-based symmetric supercapacitor, i.e., large capacitance (200 F g−1 at 2 mV s−1) at 3.5 V and ultrastable cycling feature within a wide operating temperature.148 Further theoretical calculations revealed that the high capacitance of the composite electrode could be attributed to the enhanced electrolyte adsorption and restructuring at the electrolyte/metal oxide@carbon interface, which could promote further improvements in high energy storage and broader operation applicability.
 | ||
| Fig. 10 Schematic of N-doped carbon nanowires@MnO2 and N-doped carbon nanowires@Fe2O3 (a). (Reproduced with permission.147 Copyright 2018, Wiley-VCH.) Schematic for the potassium storage reaction mechanism (b). (Reproduced with permission.157 Copyright 2019, Wiley-VCH.) | ||
Battery–capacitor hybrid devices combine capacitive carbon and battery-type electrodes, exhibiting energy storage close to those of batteries and power output approximately that of supercapacitors.7,151–154 Nevertheless, battery-type materials generally exhibit limited kinetics during redox reactions rooted from the sluggish phase transformation rate.8 Wang's group synthesized and employed hierarchical porous carbon nanosheets and MnO@C nanocomposites as the positive and negative electrodes for Li-ion hybrid capacitors with kapok fiber as the precursor.155 Nanoscale diffusion paths in the MnO@C nanocomposite facilitated Li+ insertion/extraction and electron transfer, resulting in the improved electrochemical kinetics to balance energy delivery (100 W h kg−1 at 83 W kg−1) and power density (20 kW kg−1 at 30 W h kg−1) for a high-potential (4 V) device. Lee's group developed the simple fabrication of Nb2O5@C/rGO nanocomposite in a water-in-oil microemulsion assembly system for the high-power Na-ion hybrid capacitor anode.156 The hybrid device using the Nb2O5@C/rGO anode and an activated carbon cathode showed high energy density of 76 W h kg−1 in the voltage window of 1.0–4.3 V as well as exceptional power density (20.8 kW kg−1), and the stable cycling life could be attributed to the uniform Nb2O5@C particle deposition between the rGO layers to prevent layer restacking. Lu's group employed a simple solvothermal approach to prepare 3D N-doped carbon-coated FeSe2 clusters as the anode for K-ion hybrid capacitors.157 Carbon coating and unique 3D architecture effectively offered abundant active sites, improved conductivity, and charge transfer shortcuts, and therefore, the assembled hybrid device delivered an energy density of 230 W h kg−1 due to the transformation from FeSe2 to Fe3Se4 in electrochemical reactions (Fig. 10b). The long lifetime and high power density (920 W kg−1) further demonstrated the role of FeSe2-modified carbons as emerging hybrid capacitor anodes.
3. Electrolytes
3.1. Traditional aqueous electrolytes
The electrolyte constituent and its stable operation potential play a key role in the double-layer formation and faradaic redox reaction to finally determine the energy storage and other parameters related to the electrochemical performance (Table 3). CSs using aqueous electrolytes (e.g., KOH and H2SO4) exhibit high specific capacitance and rate performance due to their small ion diameter and excellent conductivity (one order of magnitude higher than those of nonaqueous media).5,6 Unfortunately, the inherent obstacle in traditional aqueous electrolytes is their narrow operation potential window (below 1.23 V) impeded by water hydrolysis. Hydrogen/oxygen evolution occurs at the anode/cathode potential of around 0/1.23 V, respectively, which consequently threatens device configuration and electrochemical performance. One solution is utilizing the high overpotential of neutral electrolytes (e.g., Na2SO4, Li2SO4, etc.) to reduce the H+/OH− availability for hydrogen/oxygen evolution reactions.158–161 Frackowiak's group investigated neutral Li2SO4, Na2SO4, and Na2SO4 electrolytes in a wide concentration range (0.1–2.5 mol L−1) for electrochemical capacitors, and the strong ion solvation enabled a working voltage of 2.2 V in 1 mol L−1 Li2SO4 with low capacitance decay (from 140 to 120 F g−1) after 15000 charge/discharge cycles (Fig. 11).162 Another strategy is to introduce supplementary pseudocapacitance with redox-active pairs (e.g., I−/I3−, Fe(CN)63−/Fe(CN)64−, MV2+/MV+, and hydroquinone/quinone) as the electrolyte additives.163–167 Fischer's group introduced KI as a redox additive into a traditional H2SO4 electrolyte, and the significant increase in the capacitance from 74 to 521 F g−1 could be ascribed to the redox processes between the active couples of 3I−/I3−, 2I3−/3I2, 2I−/I2, and I2/IO3.43 Additionally, the hybrid energy storage system extended the operational voltage (1.6 V) by altering the oxygen evolution potential, which afforded a novel strategy to achieve battery-grade energy and capacitor-grade power density for CSs.
| Electrolyte | Potential (V) | Electrode | Energy density (W h kg−1) | Power density (W kg−1) | Ref. |
|---|---|---|---|---|---|
| 6 M KOH | 1.0 | N-doped porous carbon nanosheets | 11.77 | 34.11 | 29 |
| 1 M H2SO4 | 1.0 | O/N codoped porous carbons | 18.2 | 300 | 134 |
| 1 M Na2SO4 | 1.8 | B/N-codoped carbon nanosheets | 30.1 | 225.1 | 124 |
| 1 M Li2SO4 | 2.2 | Activated carbon | — | — | 162 |
| 1 M KBr | 1.9 | Activated carbon | 12.0 | — | 163 |
| 1 M Na2SO4 + K3Fe(CN)6/K4Fe(CN)6 | 2.0 | Activated carbon | 18.9 | 11500 |
164 |
| 1 M H2SO4 + hydroquinone | 1.0 | Activated carbon | 31.3 | — | 165 |
| 1 M KBr + MVCl2 | 1.4 | Activated carbon | ∼14 | — | 166 |
| 1 M H2SO4 + KI | 1.6 | 2D nanoporous carbon sheets | 89.73 | 533 | 43 |
| 5 M LiTFSI | 2.4 | Hierarchical porous carbon monoliths | 24 | 480 | 169 |
| 8 M NaClO4 | 2.5 | Activated carbon | — | — | 176 |
| 8 M LiOAc + 32 M KOAc | 2.85 | Activated carbon | 77.9 | 149.1 | 181 |
| 1 M TEABF4/ACN | 2.7 | Porous interconnected carbon nanosheets | ∼30 | 13000 |
72 |
| 1.5 M SBPBF4/PC + DMC | 2.7 | Activated carbon | 29.6 | 12500 |
184 |
| EMIMBF4 | 4.0 | Cross-coupled macro–mesoporous carbon | 92 | 1000 | 104 |
| EMIMFSI | 3.5 | Activated carbon | 42 | — | 198 |
| TMABF4 + EMIMBF4 | 3.5 | Mesoporous carbon materials | — | 7500 | 203 |
| PI13FSI + PY14FSI | 3.4 | Activated carbon | 42.5 | — | 205 |
| c-P4VPh-EMIMTFSI | 4.0 | Porous carbon | 72 | — | 212 |
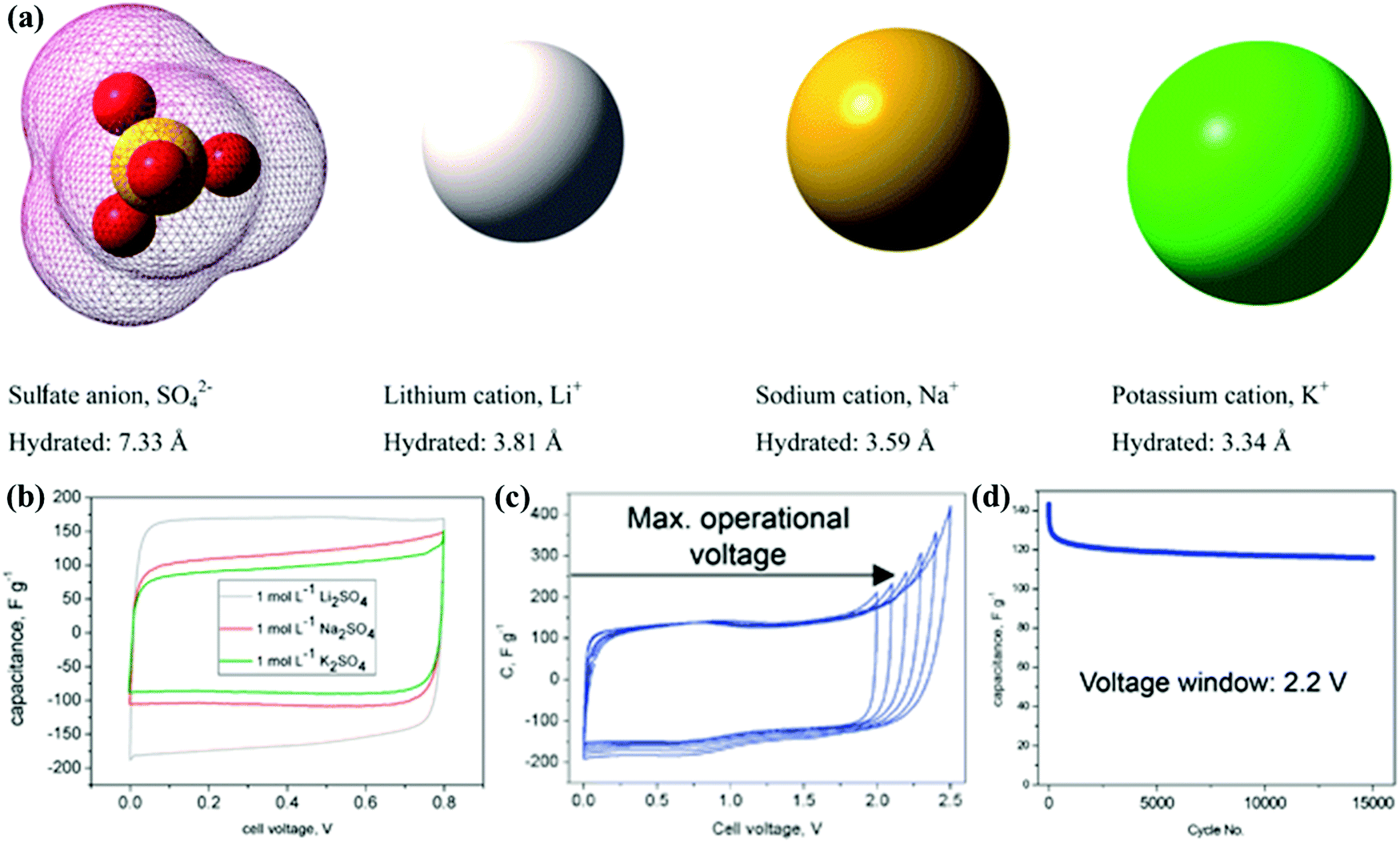 | ||
| Fig. 11 Ion shapes/dimensions in aqueous solutions (a). Cyclic voltammograms in 1 mol L−1 Li2SO4/Na2SO4/K2SO4 at 1 mV s−1 (b) and 1 mol L−1 Li2SO4 at 10 mV s−1 (c). Cyclability in 1 mol L−1 Li2SO4 under 2.2 V (d). (Reproduced with permission.162 Copyright 2012, The Royal Society of Chemistry.) | ||
3.2. WIS electrolytes
Highly concentrated aqueous electrolytes have also been used to effectively widen the operational potential window by relieving the electrolyte/carbon interphase from water availability. WIS electrolytes refer to super-concentrated salt solutions, and they have been frequently applied in energy-related applications of high-potential batteries and supercapacitors.116,168–172 Such an exceptional high-potential feature can be attributed to two aspects:168,173 (i) the super-concentrated salt layer creates an anion-involved Li+ solvation shell strongly bonded by the coordination of Li+ and water molecules outside, suppressing the possibility of water electrolysis; (ii) the dominant reduction process leads to dense LiF distribution as a solid–electrolyte interphase, kinetically avoiding endless decomposition of both water and anions. Nakanishi's group explored the performance of a WIS-electrolyte-based supercapacitor using activated carbon electrodes, and the assembled device containing 5 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) achieved high operation potential of 2.4 V with prominent energy/power performances of 24 W h kg−1 at 0.48 kW kg−1 and 10 W h kg−1 at 7.6 kW kg−1.169 The low ionic conductivity is generally considered to be the main disadvantage of WIS electrolytes due to the strong coordination between Li+ and water molecules, leading to unsatisfactory rate/power capabilities of CSs.174–177 More recently, introducing extra ions with weak/inert interactions into the WIS electrolytes has further boosted the salt/water ratio to an unprecedented level, and these water-in-bisalt electrolytes with upgraded operational potential still maintain competitive conductivities and viscosities in contrast to typical organic electrolytes (Fig. 12).178–182 | ||
| Fig. 12 Visualization of WIS and water-in-bisalt systems. (Reproduced with permission.178 Copyright 2018, The Royal Society of Chemistry.) | ||
3.3. Organic electrolytes
Currently, most commercial CSs employ organic electrolytes (TEABF4 in ACN/PC and tetraethylammonium tetrafluoroborate in acetonitrile/propylene carbonate) with stable operational potential of 2.5–2.8 V.5,183–185 As mentioned above, nonaqueous media—free from the limitation of water hydrolysis—facilitate the fabrication of high-potential CSs. When compared with aqueous CSs, organic-electrolyte-based devices generally provide higher energy storage, even though their lower micropore accessibility and ionic conductivity lead to lower capacitances.186–188 Sevilla's group synthesized highly microporous carbon nanosheets via the one-step carbonization of potassium citrate for supercapacitor electrodes, and the as-obtained electrode material exhibited capacitances of 150/120 F g−1 at 1/200 mV s−1 in 1 M TEABF4/ACN-based organic device with energy/power densities of ∼30 W h kg−1 at 13 kW kg−1 operated at 2.7 V.72 The enhancement in specific capacitances can be realized when solvated ions undergo desolvation/distortion under certain potential and they are endowed with easy access to the size-matched pores.9,69 Another disadvantage of organic media is the lower ionic conductivity (1 M TEABF4/ACN: 0.06 S cm−1 at 25 °C) when compared with aqueous solutions (1 M H2SO4:0.8 S cm−1), which dramatically reduces the power performances owing to the sluggish mass diffusion.186 The imposed confinement of electrolyte ions within the solvation shells notably decreases the diffusion coefficients of bulk TEA+/BF4− and pure ACN solvent molecules by 1–2 orders of magnitude.189 In addition, problems such as high cost, complicated cell assembly, and rising concerns of toxicity and flammability of organic solvents remain to be resolved.190
3.4. Ionic liquid electrolytes
Ionic liquids as solvent-free electrolytes have recently aroused significant interest due to their wide operation potential window (>3 V), broad temperature applicability, negligible volatility, and low flammability.190,191 Ionic liquids are the organic salt matrix of large organic cations and inorganic/organic anions, and the highly asymmetric conjugation leads to their liquid form at room temperature or lower temperatures, consequently getting rid of additional solvent shells for charge transfer. Generally, ionic liquids explored for CS applications consist of imidazolium/pyrrolidinium/pyridinium/piperidinium/tetraalkylammonium cations coupled with anions such as Cl−, tetrafluoroborate (BF4−), bis(fluorosulfonyl)imide (FSI−), and bis(trifluoromethanesulfonyl)imide (TFSI−). The physicochemical properties of ionic liquid electrolytes are strongly dependent on the choice of cation/anion couples.192–196 Ma's group assembled ionic-liquid-based CSs with an operational potential window of 4.0 V by pairing 1-ethyl-3-methylimidazolium (EMIM+) and BF4−/TFSI−; notably, the energy/power densities in EMIMBF4 and EMIMTFSI were 106.4 and 135.6 W h kg−1, respectively, at a power output of 10 kW kg−1.197 Chang's group achieved optimized capacitive performances of 120/60 F g−1 at 0.5/10 A g−1 in EMIMFSI because of its low viscosity (17.9 MPa s) and rapid ion transfer, and replacing EMIM+ with PMP+ enhanced the device potential of the activated carbon-assembled supercapacitor to 3.5 V and energy delivery to 42 W h kg−1 at the expense of long cycling lifetime.198 Ionic liquids can work well in a broad temperature range since their negligible and stable volatility endows them with extended fluid scope and combustion resistance.199–201 Oschatz's group demonstrated ultrastable cycling performance (over 90% capacitance retention after 5000 cycles) of the EMIMBF4-based supercapacitor operated in a broad temperature range of −20 to 60 °C, wherein the low resistance could be attributed to the low viscosity and high conductivity of the unelectrolyzed EMIMBF4 at higher temperatures and its low glass transition temperature (−91 °C).148 Grinstaff's group described the physicochemical features of a class of alkyl-ether-containing piperidinium-based ionic liquid electrolytes associated with LiTFSI for use in hybrid supercapacitors. When compared with a standard LiTFSI/PC electrolyte, the piperidinium-based electrolyte showed advantageous high conductivity and it could be effectively operated for over 10000 cycles at 100 °C, where the LiTFSI/PC-assembled device failed after 3200 charge/discharge cycles.202 Moreover, there is still plenty of room for the modification of ionic liquid electrolytes for use in high-performance CSs. Gogotsi's group demonstrated the in-cell charging selectivity in the TMABF4/EMIMBF4 ionic mixture electrolyte for simultaneously improved power/energy densities.203 Two cations (TMA+/EMIM+) with strong cationic interactions could be arranged in denser distribution at the electrode/electrolyte interface to increase the capacitance, while the selective sieving effect of TMA+ with weaker interactions allowed fast ion dynamics in micropores without reducing the power output (Fig. 13). The modified features of ionic mixture electrolytes invite additional experimental/theoretical exploitation for comprehensive enhancements in CS performances.204–207
 | ||
| Fig. 13 Schematic of the selective charging behavior of TMABF4/EMIMBF4 ionic mixture electrolyte (a). Ragone plots (b). (Reproduced with permission.203 Copyright 2017, American Chemical Society.) | ||
3.5. (Quasi-)solid-state electrolytes
With the growing demand for flexible and portable electronics, (quasi-)solid-state electrolytes have become popular for use in CSs as both ion-conducting media and permeable separators, and they can be divided into gel polymer electrolyte, dry polymer electrolyte, and polyelectrolyte.6,138 Among them, liquid-phase gel polymer electrolytes are also considered to be quasi-solid-state electrolytes, and they currently dominate solid-state CS studies because of their highest ionic conductivity.160,186,208–212 Gel polymer electrolytes consist of a polymer matrix (e.g., polyvinyl alcohol (PVA)) and liquid-state electrolyte (aqueous electrolyte, organic medium, ionic liquid, etc.) or conducting salts dispersed in an aqueous/organic solvent. Wu's group developed a zwitterionic poly(propylsulfonate dimethylammonium propylmethacrylamide) (PPDP)–LiCl gel electrolyte by the free-radical polymerization of propylsulfonate dimethylammonium propylmethacrylamide (PDP) and physically mixing with LiCl.213 This PPDP–LiCl hydrogel electrolyte afforded synergy between liquid preservation capacity and ion transfer pathways; subsequently, the PPDP–LiCl-based solid-state supercapacitor exhibited a higher energy density (41.78 W h L−1) than that of a PVA–LiCl-based one (26.89 W h L−1) (Fig. 14). MacFarlane's group proposed a novel preparation approach for self-supported ionic liquid gel membranes by incorporating two ionogels [EMIM][NTf2]/[C4mpyr][eFAP] into the poly(vinylidene fluoride-co-hexafluoropropylene) polymer matrix.214 The [EMIM][NTf2]-ionogel-membrane-based symmetric supercapacitor could achieve capacitances of 153/101 F g−1 at 0.1/10 A g−1, and the flexible CS still retained specific capacitance of 119 F g−1 at 1 A g−1. Dry polymer electrolytes consist of a polymeric network (e.g., PEO) and conducting salt (LiCl, LiClO4, etc.) without any aqueous/organic solvents.186,215,216 As an example of polyelectrolytes, Zhi's group fabricated a self-healable and stretchable polyelectrolyte via the dual crosslinking of polyacrylic acid by H-bond and vinyl hybrid SiO2, and the corresponding polyelectrolyte-based CS delivered superior functions when compared with that offered by PVA-based acidic devices.217 (Quasi-)solid-state electrolytes can simplify the assembling/packaging manufacture of CSs, and they can also prevent the potential leakage of liquid-state electrolytes.3 Nevertheless, issues related to ionic conductivity (10−8–10−5 S cm−1), mechanical strength, electrochemical stability, and interfacial contact in CS applications still remain to be fully addressed for flexible and portable devices.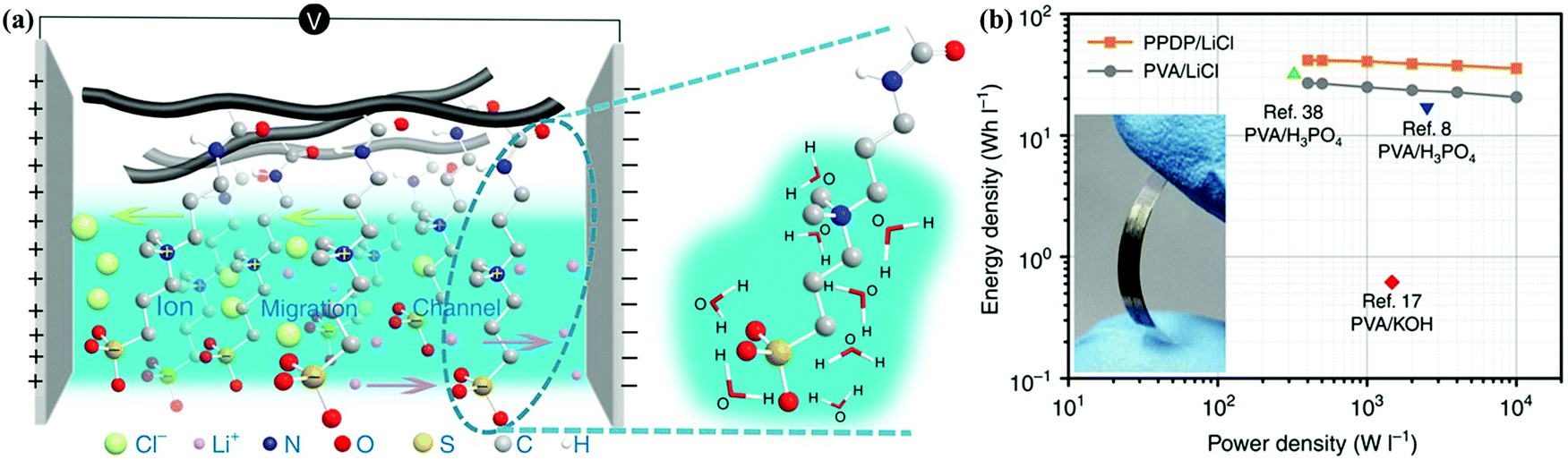 | ||
| Fig. 14 Zwitterionic PPDP under an external electric field (a). Ragone plots (b). (Reproduced with permission.213 Copyright 2016, Springer Nature.) | ||
4. Summary and outlook
CSs are promising large-power systems that have dominated the commercial market and they offer fascinating features such as fast pulses of energy and millions of charge/discharge cycles. Porous carbons have long been the most popular electrode material due to their versatile syntheses as well as physicochemical stability and electronic properties. To meet the growing demand for energy storage, significant efforts have been devoted toward exploring large-capacitance carbon-based electrodes and high-potential electrolytes for advanced CSs. In this review, recent advances made in both the core components of CSs have been summarized. For the role of carbon-based electrodes, novel synthesis strategies of large-surface-area carbon materials with morphology control, pore structure regulation, heteroatom functionalization, and metal composite, have been highlighted in pursuit of large-capacitance electrodes. Furthermore, with regard to exploiting electrolytes, WIS and ionic-liquid-based electrolytes with wider potential windows, broader temperature applicability, or higher electrochemical inertness and operation safety, have emerged as the new trend to settle the concern regarding water hydrolysis in traditional media. Despite the significant progresses made in the development of advanced CSs in recent years, some in-depth efforts are still underway and future research directions are suggested to focus on the following:(1) One strategy is maximizing the capacitances of carbon-based electrodes by a facile fabrication strategy to achieve synergy among the surface area, geometrical morphology, pore structure, and pseudocapacitive activity. Instead of external templating or random functionalization, self-templating and doping syntheses based on the direct pyrolysis of organic salt/polymer precursors (e.g., metal complexes, conjugated polymer networks, ionic liquids, and poly(ionic liquid)s) have become in considerable demand as straightforward and cost-effective strategies for fabricating pseudo-active hierarchical porous carbons. Through the judicious assembly of molecular building blocks, pseudo-active functionalities can be doped in situ into the precursor skeletons; meanwhile, the multifunctional reactants serve as the self-porogen that may undergo a series of stages such as framework decomposition, filler etching/expansion, and intermediate foaming during annealing, affording micro-, meso-, and macroporous structures. Besides, in-depth correlation between the precursor chemistry and carbon architecture should be established for the task-specific fabrication of carbon-based electrodes at the molecular level by quantifying the morphological growth with solvent/precursor solubility parameters, tracking the network polymerization/crosslinking degree, speculating the possible pyrolysis mechanism, etc. Moreover, molecular dynamics studies and simulations help to clarify the energy storage mechanisms in carbon architectures and consequently provide the guidelines for maximizing the electrode capacitance in a certain electrolyte.
(2) Another strategy is modifying high-potential electrolytes in terms of water-in-bisalt media and ionic mixtures. The exploitation of high-potential electrolytes is a long-sought-after target shared with battery applications. When compared with traditional media, emerging WIS and ionic-liquid-based electrolytes possess overwhelming superiorities such as wider potential windows, applicability at distinct temperatures, higher electrochemical inertness, and operational safety. The physicochemical properties of electrolytes are strongly dependent on the cation/anion couples and ion concentrations of conducting salts as well as those of water or organic solvents. Introducing additional conducting salts into such super-ion-concentrated aqueous/nonaqueous systems can further elevate the energy density to a higher level, even realizing comprehensive enhancements in energy/power performances. Apart from salt mixtures, solvent mixtures have also been recently developed to modify salt solvent systems for improved ion dynamics and reduced electrolyte viscosity for comprehensively enhanced energy/power densities. Besides, ionic liquids or poly(ionic liquid)s trapped in polymer matrixes appear to resolve the leakage crisis of existing energy storage cells, and they hold a position as potential gel polymer electrolytes for flexible and portable supercapacitors.
(3) Lastly, the carbon/electrolyte compatibility can be optimized for ideal device configurations. The fundamental energy storage mechanism of CSs can be derived from fast and reversible ion adsorption at the carbon/electrolyte interfaces. Future studies should attempt to enhance interface accessibility, particularly in high-potential electrolytes by optimizing the carbon/electrolyte compatibility, including tailoring the micropore diameter/shape to accommodate larger electrolyte ions, introducing mesopore/macropore paths to transfer viscous media, and doping pseudo-active sites to enhance the interfacial interactions. With regard to upgraded device configurations, the carbon/electrolyte compatibility still remains to be optimized for relieving the limited ion kinetics/interfacial contact of redox reactions and phase transformation for hybrid/solid-state CSs. Despite these challenges in maximizing the carbon/electrolyte synergy, we believe that high-energy, large-power, and long-running CSs can be realized in the near future for ongoing commercial applications of electronics and other multipurpose fields.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21875165, 51772216 and 21905207), the Science and Technology Commission of Shanghai Municipality, China (14DZ2261100), the Fundamental Research Funds for the Central Universities, and the Recruitment Program of Global Experts of China.Notes and references
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, eaan8285 CrossRef CAS PubMed.
- X. Zeng, Y. Xu, Y. Yin, X. Wu, J. Yue and Y. Guo, Mater. Today Nano, 2019, 8, 100057 CrossRef.
- T. Lv, M. Liu, D. Zhu, L. Gan and T. Chen, Adv. Mater., 2018, 30, 1705489 CrossRef PubMed.
- X. Chen, R. Paul and L. Dai, Nat. Sci. Rev., 2017, 4, 453–489 CrossRef CAS.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2012, 46, 1094–1103 CrossRef PubMed.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- D. Tie, S. Huang, J. Wang, J. Ma, J. Zhang and Y. Zhao, Energy Storage Mater., 2019, 21, 22–40 CrossRef.
- M. Lukatskaya, B. Dunn and Y. Gogotsi, Nat. Commun., 2016, 7, 12647 CrossRef PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P.-L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- P. Wu, J. Huang, V. Meunier, B. Sumpter and R. Qiao, ACS Nano, 2011, 5, 9044–9051 CrossRef CAS PubMed.
- M. Benzigar, S. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- W. Tian, H. Zhang, X. Duan, H. Sun, G. Shao and S. Wang, Adv. Funct. Mater., 2020, 30, 1909265 CrossRef CAS.
- S. Dutta, A. Bhaumik and C. W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- B. Bhadra, A. Vinu, C. Serre and S. Jhung, Mater. Today, 2019, 25, 88–111 CrossRef CAS.
- F. Xu, D. Wu, R. Fu and B. Wei, Mater. Today, 2017, 20, 629–656 CrossRef CAS.
- T. P. Fellinger, A. Thomas, J. Yuan and M. Antonietti, Adv. Mater., 2013, 25, 5838–5854 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- P. Qiu, B. Ma, C. Hung, W. Li and D. Zhao, Acc. Chem. Res., 2019, 52, 2928–2938 CrossRef CAS PubMed.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- R. Yan, M. Antonietti and M. Oschatz, Adv. Energy Mater., 2018, 8, 1800026 CrossRef.
- B. Qiu, C. Pan, W. Qian, Y. Peng, L. Qiu and F. Yan, J. Mater. Chem. A, 2013, 1, 6373–6378 RSC.
- M. Yang and Z. Zhou, Adv. Sci., 2017, 4, 1600408 CrossRef PubMed.
- C. Cui, Y. Gao, J. Li, C. Yang, M. Liu, H. Jin, Z. Xia, L. Dai, Y. Lei, J. Wang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2–8 CrossRef.
- D. Puthusseri, V. Aravindan, S. Madhavi and S. Ogale, Energy Environ. Sci., 2014, 7, 728–735 RSC.
- J. Chen, Y. Han, X. Kong, X. Deng, H. J. Park, Y. Guo, S. Jin, Z. Qi, Z. Lee and Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 13822–13827 CrossRef CAS PubMed.
- K. Sing, D. Everett, R. Haul, L. Moscou, R. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS PubMed.
- Q. Niu, K. Gao, Q. Tang, L. Wang, L. Han, H. Fang, Y. Zhang, S. Wang and L. Wang, Carbon, 2017, 123, 290–298 CrossRef CAS.
- H. Xu, C. Wu, X. Wei and S. Gao, J. Mater. Chem. A, 2018, 6, 15340–15347 RSC.
- O. Barbieri, M. Hahn, A. Herzog and R. Kotz, Carbon, 2005, 43, 1303–1310 CrossRef CAS.
- J. Liu, N. Wickramaratne, S. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef CAS PubMed.
- L. Peng, C. Hung, S. Wang, X. Zhang, X. Zhu, Z. Zhao, C. Wang, Y. Tang, W. Li and D. Zhao, J. Am. Chem. Soc., 2019, 141, 7073–7080 CrossRef CAS PubMed.
- H. Zhang, O. Noonan, X. Huang, Y. Yang, C. Xu, L. Zhou and C. Yu, ACS Nano, 2016, 10, 4579–4586 CrossRef CAS PubMed.
- J. Wang, H. Liu, X. Zhang, M. Shao and B. Wei, J. Mater. Chem. A, 2018, 6, 17653–17661 RSC.
- T. Wang, Y. Sun, L. Zhang, K. Li, Y. Yi, S. Song, M. Li, Z. Qiao and S. Dai, Adv. Mater., 2019, 31, 1807876 CrossRef PubMed.
- Y. He, X. Zhuang, C. Lei, L. Lei, Y. Hou, Y. Mai and X. Feng, Nano Today, 2019, 24, 103–119 CrossRef CAS.
- J. Gong, H. Lin, M. Antonietti and J. Yuan, J. Mater. Chem. A, 2016, 4, 7313–7321 RSC.
- H. Lyu, J. Zhu, B. Zhou, H. Cao, J. Duan, L. Chen, W. Jin and Q. Xu, Carbon, 2018, 139, 740–749 CrossRef CAS.
- L. Yao, Q. Wu, P. Zhang, J. Zhang, D. Wang, Y. Li, X. Ren, H. Mi, L. Deng and Z. Zheng, Adv. Mater., 2018, 30, 1706054 CrossRef PubMed.
- L. Miao, D. Zhu, M. Liu, H. Duan, Z. Wang, Y. Lv, W. Xiong, Q. Zhu, L. Li and X. Chai, Chem. Eng. J., 2018, 347, 233–242 CrossRef CAS.
- G. Yuan, Y. Liang, H. Hu, H. Li, Y. Xiao, H. Dong, Y. Liu and M. Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 26946–26955 CrossRef CAS PubMed.
- K. Jayaramulu, D. P. Dubal, B. Nagar, V. Ranc, O. Tomanec, M. Petr, K. K. R. Datta, R. Zboril, P. Gómez-Romero and R. A. Fischer, Adv. Mater., 2018, 30, 1705789 CrossRef PubMed.
- B. Ding, J. Wang, Y. Wang, Z. Chang, G. Pang, H. Dou and X. Zhang, Nanoscale, 2016, 8, 11136–11142 RSC.
- W. Tian, Q. Gao, L. Zhang, C. Yang, Z. Li, Y. Tan, W. Qian and Z. Hang, J. Mater. Chem. A, 2016, 4, 8690–8699 RSC.
- J. Yu, C. Yu, W. Guo, Z. Wang, S. Li, J. Chang, X. Tan, Y. Ding, M. Zhang and L. Yang, Nano Energy, 2019, 64, 103921 CrossRef CAS.
- G. Zhou, L. Xu, G. Hu, L. Mai and Y. Cui, Chem. Rev., 2019, 119, 11042–11109 CrossRef CAS PubMed.
- S. Li, X. Song, X. Wang, C. Xu, Y. Cao, Z. Xiao, C. Qi, M. Wu, Z. Yang, L. Fu, X. Ma and J. Gao, Carbon, 2020, 160, 176–187 CrossRef CAS.
- P. Shang, J. Zhang, W. Tang, Q. Xu and S. Guo, Adv. Funct. Mater., 2016, 26, 7766–7774 CrossRef CAS.
- X. Wang, Z. Na, D. Yin, C. Wang, G. Huang and L. Wang, Energy Storage Mater., 2018, 12, 103–109 CrossRef.
- L. Miao, D. Zhu, M. Liu, H. Duan, Z. Wang, Y. Lv, W. Xiong, Q. Zhu, L. Li and X. Chai, Electrochim. Acta, 2018, 274, 378–388 CrossRef CAS.
- D. Zhu, K. Cheng, Y. Wang, D. Sun, L. Gan, T. Chen, J. Jiang and M. Liu, Electrochim. Acta, 2017, 224, 17–24 CrossRef CAS.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS PubMed.
- D. Bo, G. Xiang, Y. Xu, F. Yan, H. Liang, J. Zhou and L. Zhang, Nano Energy, 2016, 27, 482–491 CrossRef.
- S. He, C. Zhang, C. Du, C. Cheng and W. Chen, J. Power Sources, 2019, 434, 226701 CrossRef CAS.
- L. Wang, T. Wei, L. Sheng, L. Jiang, X. Wu, Q. Zhou, B. Yuan, J. Yue, Z. Liu and Z. Fan, Nano Energy, 2016, 30, 84–92 CrossRef CAS.
- L. Jiang, J. Wang, X. Mao, X. Xu, B. Zhang, J. Yang, Y. Wang, J. Zhu and S. Hou, Carbon, 2017, 111, 207–214 CrossRef CAS.
- C. Liu, X. Huang, J. Wang, H. Song, Y. Yang, Y. Liu, J. Li, L. Wang and C. Yu, Adv. Funct. Mater., 2018, 28, 1705253 CrossRef.
- Z. Chen, S. Ye, S. Evans, Y. Ge, Z. Zhu, Y. Tu and X. Yang, Small, 2018, 14, 1704015 CrossRef PubMed.
- S. Chen, D. M. Koshy, Y. Tsao, R. Pfattner, X. Yan, D. Feng and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10297–10304 CrossRef PubMed.
- M. Liu, J. Qian, Y. Zhao, D. Zhu, L. Gan and L. Chen, J. Mater. Chem. A, 2015, 3, 11517–11526 RSC.
- L. Miao, D. Zhu, Y. Zhao, M. Liu, H. Duan, W. Xiong, Q. Zhu, L. Li, Y. Lv and L. Gan, Microporous Mesoporous Mater., 2017, 253, 1–9 CrossRef CAS.
- D. Qiu, J. Guan, M. Li, C. Kang, J. Wei, Y. Li, Z. Xie, F. Wang and R. Yang, Adv. Funct. Mater., 2019, 29, 1903496 CrossRef.
- X. Qian, L. Miao, J. Jiang, G. Ping, W. Xiong, Y. Lv, Y. Liu, L. Gan, D. Zhu and M. Liu, Chem. Eng. J., 2020, 388, 124208 CrossRef CAS.
- Z. Xu, X. Zhuang, C. Yang, J. Cao, Z. Yao, Y. Tang, J. Jiang, D. Wu and X. Feng, Adv. Mater., 2016, 28, 1981–1987 CrossRef CAS PubMed.
- W. Song, Y. Zhang, A. Varyambath and I. Kim, ACS Nano, 2019, 13, 11753–11769 CrossRef CAS PubMed.
- H. Itoi, H. Nishihara, T. Kogure and T. Kyotani, J. Am. Chem. Soc., 2011, 133, 1165–1167 CrossRef CAS PubMed.
- W. Lu, M. Liu, L. Miao, D. Zhu, X. Wang, H. Duan, Z. Wang, L. Li, Z. Xu and L. Gan, Electrochim. Acta, 2016, 205, 132–141 CrossRef CAS.
- N. Jäckel, P. Simon, Y. Gogotsi and V. Presser, ACS Energy Lett., 2016, 1, 1262–1265 CrossRef.
- K. Kim, T. Lee, Y. Kwon, Y. Seo, J. Song, J. K. Park, H. Lee, J. Y. Park, H. Ihee, S. J. Cho and R. Ryoo, Nature, 2016, 535, 131–135 CrossRef CAS PubMed.
- M. Oschatz, S. Boukhalfa, W. Nickel, J. P. Hofmann, C. Fischer, G. Yushin and S. Kaskel, Carbon, 2017, 113, 283–291 CrossRef CAS.
- M. Sevilla and A. Fuertes, ACS Nano, 2014, 8, 5069–5078 CrossRef CAS PubMed.
- W. Li, S. Hu, X. Luo, Z. Li, X. Sun, M. Li, F. Liu and Y. Yu, Adv. Mater., 2017, 29, 1605820 CrossRef PubMed.
- L. Miao, H. Duan, Z. Wang, Y. Lv, W. Xiong, D. Zhu, L. Gan, L. Li and M. Liu, Chem. Eng. J., 2020, 382, 122945 CrossRef.
- J. Zhou, Z. Li, W. Xing, H. Shen, X. Bi, T. Zhu, Z. Qiu and S. Zhuo, Adv. Funct. Mater., 2016, 26, 7955–7964 CrossRef CAS.
- L. Borchardt, Q.-L. Zhu, M. E. Casco, R. Berger, X. Zhuang, S. Kaskel, X. Feng and Q. Xu, Mater. Today, 2017, 20, 592–610 CrossRef CAS.
- M. Kim, P. Puthiaraj, Y. Qian, Y. Kim, S. Jang, S. Hwang, E. Na, W.-S. Ahn and S. E. Shim, Electrochim. Acta, 2018, 284, 98–107 CrossRef CAS.
- Y. Lu, J. Liang, S. Deng, Q. He, S. Deng, Y. Hu and D. Wang, Nano Energy, 2019, 65, 103993 CrossRef CAS.
- T. Kyotani, N. Sonobe and A. Tomita, Nature, 1988, 331, 331–333 CrossRef.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- Y. Fang, Y. Lv, F. Gong, Z. Wu, X. Li, H. Zhu, L. Zhou, C. Yao, F. Zhang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 2808–2811 CrossRef CAS PubMed.
- Z. Song, L. Li, D. Zhu, L. Miao, H. Duan, Z. Wang, W. Xiong, Y. Lv, M. Liu and L. Gan, J. Mater. Chem. A, 2019, 7, 816–826 RSC.
- Y. Bu, T. Sun, Y. Cai, L. Du, O. Zhuo, L. Yang, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2017, 29, 1700470 CrossRef PubMed.
- L. Estevez, V. Prabhakaran, A. L. Garcia, Y. Shin, J. Tao, A. M. Schwarz, J. Darsell, P. Bhattacharya, V. Shutthanandan and J.-G. Zhang, ACS Nano, 2017, 11, 11047–11055 CrossRef CAS PubMed.
- J. Du, L. Liu, Z. Hu, Y. Yu, Y. Qin and A. Chen, Adv. Funct. Mater., 2018, 28, 1802332 CrossRef.
- Y. Pei, G. Choi, S. Asahina, J. Yang, A. Vinu and J. Choy, Chem. Commun., 2019, 55, 3266–3269 RSC.
- C. Liang, K. Hong, G. Guiochon, J. Mays and S. Dai, Angew. Chem. Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- Y. Wan, Y. Shi and D. Zhao, Chem. Mater., 2008, 20, 932–945 CrossRef CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2015, 54, 588–593 CAS.
- J. Chen, J. Xu, S. Zhou, N. Zhao and C. Wong, Nano Energy, 2016, 25, 193–202 CrossRef CAS.
- Y. Liu, Z. Wang, W. Teng, H. Zhu, J. Wang, A. Elzatahry, D. Al-Dahyan, W. Li, Y. Deng and D. Zhao, J. Mater. Chem. A, 2018, 6, 3162–3170 RSC.
- T. Liu and G. Liu, J. Mater. Chem. A, 2019, 7, 23476–23488 RSC.
- H. Peng, B. Yao, X. Wei, T. Liu, T. Kou, P. Xiao, Y. Zhang and Y. Li, Adv. Energy Mater., 2019, 9, 1803665 CrossRef.
- J. Wang, J. Tang, B. Ding, V. Malgras, Z. Chang, X. Hao, Y. Wang, H. Dou, X. Zhang and Y. Yamauchi, Nat. Commun., 2017, 8, 15717 CrossRef CAS PubMed.
- W. Li, B. Li, M. Shen, Q. Gao and J. Hou, Chem. Eng. J., 2020, 384, 123309 CrossRef CAS.
- Y. Song, J. Yang, K. Wang, S. Haller, Y. Wang, C. Wang and Y. Xia, Carbon, 2016, 96, 955–964 CrossRef CAS.
- Z. Qiang, Y. Xia, X. Xia and B. D. Vogt, Chem. Mater., 2017, 29, 10178–10186 CrossRef CAS.
- D. Liu, G. Cheng, H. Zhao, C. Zeng, D. Qu, L. Xiao, H. Tang, Z. Deng, Y. Li and B. Su, Nano Energy, 2016, 22, 255–268 CrossRef CAS.
- T. Liu, F. Zhang, Y. Song and Y. Li, J. Mater. Chem. A, 2017, 5, 17705–17733 RSC.
- Y. Kim, J. Park and J. Lee, Carbon, 2018, 126, 215–224 CrossRef CAS.
- B. Zhang, C. Song, C. Liu, J. Min, J. Azadmanjiri, Y. Ni, R. Niu, J. Gong, Q. Zhao and T. Tang, J. Mater. Chem. A, 2019, 7, 22912–22923 RSC.
- D. Xue, D. Zhu, M. Liu, H. Duan, L. Li, X. Chai, Z. Wang, Y. Lv, W. Xiong and L. Gan, ACS Appl. Nano Mater., 2018, 1, 4998–5007 CrossRef CAS.
- N. Fechler, T. P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75–79 CrossRef CAS PubMed.
- J. Li, N. Wang, J. Tian, W. Qian and W. Chu, Adv. Funct. Mater., 2018, 28, 1806153 CrossRef.
- D. Xue, D. Zhu, H. Duan, Z. Wang, Y. Lv, W. Xiong, L. Li, M. Liu and L. Gan, Chem. Commun., 2019, 55, 11219–11222 RSC.
- J. M. Serrano, T. Liu, A. U. Khan, B. Botset, B. J. Stovall, Z. Xu, D. Guo, K. Cao, X. Hao, S. Cheng and G. Liu, Chem. Mater., 2019, 31, 8898–8907 CrossRef CAS.
- K. Yan, L. Kong, Y. Dai, M. Shi, K. Shen, B. Hu, Y. Luo and L. Kang, J. Mater. Chem. A, 2015, 3, 22781–22793 RSC.
- J. Yang, G. Li, M. Cai, P. Pan, Z. Li, Y. Bao and Z. Chen, Chem. Commun., 2017, 53, 5028–5031 RSC.
- Y. Tong, X. Li, L. Xie, F. Su, J. Li, G. Sun, Y. Gao, N. Zhang, Q. Wei and C. Chen, Energy Storage Mater., 2016, 3, 140–148 CrossRef.
- L. Miao, X. Qian, D. Zhu, T. Chen, G. Ping, Y. Lv, W. Xiong, Y. Liu, L. Gan and M. Liu, Chin. Chem. Lett., 2019, 30, 1445–1449 CrossRef CAS.
- D. Zhu, Y. Wang, W. Lu, H. Zhang, Z. Song, D. Luo, L. Gan, M. Liu and D. Sun, Carbon, 2018, 111, 667–674 CrossRef.
- J. Zhang, X. Zhong, H. Chen, Y. Gao and H. Li, Electrochim. Acta, 2014, 148, 203–210 CrossRef CAS.
- B. Hu, W. B. Zhang, K. Yan, T. Zhang, K. Li, X. W. Chen, L. Kang and L. B. Kong, RSC Adv., 2018, 8, 35083–35093 RSC.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Small Methods, 2020, 4, 1900853 CrossRef CAS.
- Y. Zhao, M. Lu, P. Tao, Y. Zhang, X. Gong, Z. Yang, G. Zhang and H. Li, J. Power Sources, 2016, 307, 391–400 CrossRef CAS.
- Z. Song, H. Duan, D. Zhu, Y. Lv, W. Xiong, T. Cao, L. Li, M. Liu and L. Gan, J. Mater. Chem. A, 2019, 7, 15801–15811 RSC.
- D. Xue, D. Zhu, W. Xiong, T. Cao, Z. Wang, Y. Lv, L. Li, M. Liu and L. Gan, ACS Sustainable Chem. Eng., 2019, 7, 7024–7034 CrossRef CAS.
- G. Zhang, L. Wang, Y. Hao, X. Jin, Y. Xu, Y. Kuang, L. Dai and X. Sun, Adv. Funct. Mater., 2016, 26, 3340–3348 CrossRef CAS.
- X. Wu, L. Jiang, C. Long and Z. Fan, Nano Energy, 2015, 13, 527–536 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- T. Lin, I.-W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS PubMed.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 RSC.
- M. Perovic, Q. Qin and M. Oschatz, Adv. Funct. Mater., 2020, 1908371 CrossRef.
- L. Yang, D. Wu, T. Wang and D. Jia, ACS Appl. Mater. Interfaces, 2020, 12, 18692–18704 CrossRef CAS PubMed.
- P. Zhang, J. Yuan, T. P. Fellinger, M. Antonietti, H. Li and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 6028–6032 CrossRef CAS PubMed.
- Y. Zhang, L. Liu, P. Zhang, J. Wang, M. Xu, Q. Deng, Z. Zeng and S. Deng, Chem. Eng. J., 2019, 355, 309–319 CrossRef CAS.
- B. Yang, J. Chen, L. Liu, P. Ma, B. Liu, J. Lang, Y. Tang and X. Yan, Energy Storage Mater., 2019, 23, 522–529 CrossRef.
- J. Hwang, R. Yan, M. Oschatz and B. V. K. J. Schmidt, J. Mater. Chem. A, 2018, 6, 23521–23530 RSC.
- H. Li, J. Li, A. Thomas and Y. Liao, Adv. Funct. Mater., 2019, 29, 1904785 CrossRef.
- D. Zhu, J. Jiang, D. Sun, X. Qian, Y. Wang, L. Li, Z. Wang, X. Chai, L. Gan and M. Liu, J. Mater. Chem. A, 2018, 6, 12334–12343 RSC.
- Z. Zhou, X. Zhang, L. Xing, J. Liu, A. Kong and Y. Shan, Electrochim. Acta, 2019, 298, 210–218 CrossRef CAS.
- Y. He, Z. Cheng, H. Zuo, C. Yan and Y. Liao, ChemElectroChem, 2020, 7, 959–966 CrossRef CAS.
- H. Jin, X. Feng, J. Li, M. Li, Y. Xia, Y. Yuan, C. Yang, B. Dai, Z. Lin and J. Wang, Angew. Chem. Int. Ed., 2019, 58, 2397–2401 CrossRef CAS PubMed.
- Z. Song, L. Miao, L. Li, D. Zhu, Y. Lv, W. Xiong, H. Duan, Z. Wang, L. Gan and M. Liu, J. Mater. Chem. A, 2020, 8, 3717–3725 RSC.
- S. Zhang, K. Dokko and M. Watanabe, Mater. Horiz., 2015, 2, 168–197 RSC.
- H. Li, C. J. Zhang, Y. Yan, K. Hu, X. Q. Shi, N. Wang, H. J. Lin, K. Rui, J. X. Zhu and W. Huang, J. Power Sources, 2020, 449, 22750 Search PubMed.
- L. Miao, H. Duan, M. Liu, W. Lu, D. Zhu, T. Chen, L. Li and L. Gan, Chem. Eng. J., 2017, 317, 651–659 CrossRef CAS.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- W. Zhang, S. Wei, Y. Wu, Y.-L. Wang, M. Zhang, D. Roy, H. Wang, J. Yuan and Q. Zhao, ACS Nano, 2019, 13, 10261–10271 CrossRef CAS PubMed.
- J. Gong, M. Antonietti and J. Yuan, Angew. Chem., Int. Ed., 2017, 56, 7557–7563 CrossRef CAS PubMed.
- T. Liu, Z. Zhou, Y. Guo, D. Guo and G. Liu, Nat. Commun., 2019, 10, 675 CrossRef CAS PubMed.
- G. Li, J. Zhang, W. Li, K. Fan and C. Xu, Nanoscale, 2018, 10, 9252–9260 RSC.
- K. Qin, E. Liu, J. Li, J. Kang, C. Shi, C. He, F. He and N. Zhao, Adv. Energy Mater., 2016, 6, 1600755 CrossRef.
- M. Liu, M. Shi, W. Lu, D. Zhu, L. Li and L. Gan, Chem. Eng. J., 2017, 313, 518–526 CrossRef CAS.
- M. Liu, X. Wang, D. Zhu, L. Li, H. Duan, Z. Xu, Z. Wang and L. Gan, Chem. Eng. J., 2016, 308, 240–247 CrossRef.
- Y. Liu, X. Cai, B. Luo, M. Yan, J. Jiang and W. Shi, Carbon, 2016, 107, 426–432 CrossRef CAS.
- W. Fu, E. Zhao, X. Ren, A. Magasinski and G. Yushin, Adv. Energy Mater., 2018, 8, 1703454 CrossRef.
- F. Lai, J. Feng, R. Yan, G. C. Wang, M. Antonietti and M. Oschatz, Adv. Funct. Mater., 2018, 28, 1801298 CrossRef.
- F. Lai, J. Feng, T. Heil, G. Wang, P. Adler, M. Antonietti and M. Oschatz, Energy Storage Mater., 2019, 20, 188–195 CrossRef.
- H. Cheng, J. Meng, G. Wu and S. Chen, Angew. Chem., Int. Ed., 2019, 58, 17465–17473 CrossRef CAS PubMed.
- Q. Tan, X. Chen, H. Wan, B. Zhang, X. Liu, L. Li, C. Wang, Y. Gan, P. Liang, Y. Wang, J. Zhang, H. Wang, L. Miao, J. Jiang, P. A. van Aken and H. Wang, J. Power Sources, 2020, 448, 227403 CrossRef CAS.
- R. Wang, P. Liu, J. Lang, L. Zhang and X. Yan, Energy Storage Mater., 2017, 6, 53–60 CrossRef.
- S. Li, W. He, B. Liu, J. Cui, X. Wang, D. Peng, B. Liu and B. Qu, Energy Storage Mater., 2020, 25, 636–643 CrossRef.
- J. Zhao, G. Wang, R. Hu, K. Zhu, K. Cheng, K. Ye, D. Cao and Z. Fan, J. Mater. Chem. A, 2019, 7, 4047–4054 RSC.
- Y. Zhao, Y. Cui, J. Shi, W. Liu, Z. Shi, S. Chen, X. Wang and H. Wang, J. Mater. Chem. A, 2017, 5, 15243–15252 RSC.
- E. Lim, C. Jo, M. S. Kim, M.-H. Kim, J. Chun, H. Kim, J. Park, K. C. Roh, K. Kang, S. Yoon and J. Lee, Adv. Funct. Mater., 2016, 26, 3711–3719 CrossRef CAS.
- J. Ge, B. Wang, J. Wang, Q. Zhang and B. Lu, Adv. Energy Mater., 2020, 10, 1903277 CrossRef CAS.
- M. Yu, D. Lin, H. Feng, Y. Zeng, Y. Tong and X. Lu, Angew. Chem., Int. Ed., 2017, 56, 5454–5459 CrossRef CAS PubMed.
- J. C. Icaza and R. K. Guduru, J. Power Sources, 2016, 336, 360–366 CrossRef CAS.
- N. Batisse and E. Raymundo-Piñero, J. Power Sources, 2017, 348, 168–174 CrossRef CAS.
- Z. Zhou, L. Miao, H. Duan, Z. Wang, Y. Lv, W. Xiong, D. Zhu, L. Li, M. Liu and L. Gan, Chin. Chem. Lett., 2020, 31, 1226–1230 CrossRef CAS.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 RSC.
- Q. Li, M. Haque, V. Kuzmenko, N. Ramani, P. Lundgren, A. D. Smith and P. Enoksson, J. Power Sources, 2017, 348, 219–228 CrossRef CAS.
- J. Y. Hwang, M. Li, M. F. El-Kady and R. B. Kaner, Adv. Funct. Mater., 2017, 27, 1605745 CrossRef.
- S. Roldán, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Angew. Chem., Int. Ed., 2011, 50, 1699–1701 CrossRef PubMed.
- S. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- J. Y. Hwang, M. F. El-Kady, M. Li, C. Lin, M. Kowal, X. Han and R. B. Kaner, Nano Today, 2017, 15, 15–25 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- G. Hasegawa, K. Kanamori, T. Kiyomura, H. Kurata, T. Abe and K. Nakanishi, Chem. Mater., 2016, 28, 3944–3950 CrossRef CAS.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- Q. Dou, S. Lei, D.-W. Wang, Q. Zhang, D. Xiao, H. Guo, A. Wang, H. Yang, Y. Li and S. Shi, Energy Environ. Sci., 2018, 11, 3212–3219 RSC.
- M. Ma, Z. Shi, Y. Li, Y. Yang, Y. Zhang, Y. Wu, H. Zhao and E. Xie, J. Mater. Chem. A, 2020, 8, 4827–4835 RSC.
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschamps and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC.
- H. Avireddy, B. W. Byles, D. Pinto, J. M. Delgado Galindo, J. J. Biendicho, X. Wang, C. Flox, O. Crosnier, T. Brousse, E. Pomerantseva, J. R. Morante and Y. Gogotsi, Nano Energy, 2019, 64, 103961 CrossRef CAS.
- D. Xiao, Q. Dou, L. Zhang, Y. Ma, S. Shi, S. Lei, H. Yu and X. Yan, Adv. Funct. Mater., 2019, 29, 1904136 CrossRef CAS.
- Q. Dou, Y. Lu, L. Su, X. Zhang, S. Lei, X. Bu, L. Liu, D. Xiao, J. Chen, S. Shi and X. Yan, Energy Storage Mater., 2019, 23, 603–609 CrossRef.
- J. Yan, D. Zhu, Y. Lv, W. Xiong, M. Liu and L. Gan, Chin. Chem. Lett., 2020, 31, 579–582 CrossRef CAS.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS PubMed.
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lü, C.-J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569, 245–250 CrossRef CAS PubMed.
- W. Deng, X. Wang, C. Liu, C. Li, J. Chen, N. Zhu, R. Li and M. Xue, Energy Storage Mater., 2019, 20, 373–379 CrossRef.
- L. Chen, J. Zhang, Q. Li, J. Vatamanu, X. Ji, T. P. Pollard, C. Cui, S. Hou, J. Chen, C. Yang, L. Ma, M. S. Ding, M. Garaga, S. Greenbaum, H.-S. Lee, O. Borodin, K. Xu and C. Wang, ACS Energy Lett., 2020, 5, 968–974 CrossRef CAS.
- N. Jung, S. Kwon, D. Lee, D. Yoon, Y. Park, A. Benayad, J. Choi and J. Park, Adv. Mater., 2013, 25, 6854–6858 CrossRef CAS PubMed.
- Z. Shi, X. Yu, J. Wang, H. Hu and C. Wu, Electrochim. Acta, 2015, 174, 215–220 CrossRef CAS.
- X. Yu, D. Ruan, C. Wu, J. Wang and Z. Shi, J. Power Sources, 2014, 265, 309–316 CrossRef CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CrossRef CAS PubMed.
- K. Nomura, H. Nishihara, N. Kobayashi, T. Asada and T. Kyotani, Energy Environ. Sci., 2019, 12, 1542–1549 RSC.
- L. Borchardt, D. Leistenschneider, J. Haase and M. Dvoyashkin, Adv. Energy Mater., 2018, 8, 1800892 CrossRef.
- A. Eftekhari, Energy Storage Mater., 2017, 9, 47–69 CrossRef.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- D. Wang, Y. Wang, H. Liu, W. Xu and L. Xu, Chem. Eng. J., 2018, 342, 474–483 CrossRef CAS.
- J. Ewert, D. Weingarth, C. Denner, M. Friedrich, M. Zeiger, A. Schreiber, N. Jäckel, V. Presser and R. Kempe, J. Mater. Chem. A, 2015, 3, 18906–18912 RSC.
- P. Iamprasertkun, A. Krittayavathananon and M. Sawangphruk, Carbon, 2016, 102, 455–461 CrossRef CAS.
- C. Kong, W. Qian, C. Zheng and W. Fei, J. Mater. Chem. A, 2015, 3, 15858–15862 RSC.
- Z. Song, H. Duan, L. Li, D. Zhu, T. Cao, Y. Lv, W. Xiong, Z. Wang, M. Liu and L. Gan, Chem. Eng. J., 2019, 372, 1216–1225 CrossRef CAS.
- C. Li, X. Zhang, K. Wang, X. Sun, G. Liu, J. Li, H. Tian, J. Li and Y. Ma, Adv. Mater., 2017, 29, 1604690 CrossRef PubMed.
- Q. D. Nguyen, J. Patra, C. Hsieh, J. Li, Q. Dong and J. K. Chang, ChemSusChem, 2019, 12, 449–456 CAS.
- R. Thangavel, A. G. Kannan, R. Ponraj, V. Thangavel, D.-W. Kim and Y.-S. Lee, J. Power Sources, 2018, 383, 102–109 CrossRef CAS.
- J. Xu, N. Yuan, J. Razal, Y. Zheng, X. Zhou, J. Ding, K. Cho, S. H. Ge, R. Zhang, Y. Gogotsi and R. Baughman, Energy Storage Mater., 2019, 22, 323–329 CrossRef.
- L. Zhang, K. Tsay, C. Bock and J. Zhang, J. Power Sources, 2016, 324, 615–624 CrossRef CAS.
- J. Varela, K. Sankar, A. Hino, X. Lin, W. Chang, D. Coker and M. Grinstaff, Chem. Commun., 2018, 54, 5590–5593 RSC.
- X. Wang, A. Y. Mehandzhiyski, B. Arstad, K. L. Van Aken, T. S. Mathis, A. Gallegos, Z. Tian, D. Ren, E. Sheridan and B. A. Grimes, J. Am. Chem. Soc., 2017, 139, 18681–18687 CrossRef CAS PubMed.
- C. Lian, K. Liu, K. L. Van Aken, Y. Gogotsi, D. J. Wesolowski, H. L. Liu, D. E. Jiang and J. Z. Wu, ACS Energy Lett., 2016, 1, 21–26 CrossRef CAS.
- A. Borenstein, R. Attias, O. Hanna, S. Luski, R. B. Kaner and D. Aurbach, ChemElectroChem, 2017, 4, 2660–2668 CrossRef CAS.
- K. L. Van Aken, M. Beidaghi and Y. Gogotsi, Angew. Chem., Int. Ed., 2015, 54, 4806–4809 CrossRef CAS PubMed.
- L. Dagousset, G. Pognon, G. Nguyen, F. Vidal, S. Jus and P. H. Aubert, J. Power Sources, 2017, 359, 242–249 CrossRef CAS.
- Y. Cho, C. Hwang, D. Cheong, Y. Kim and H. Song, Adv. Mater., 2019, 31, 1804909 CrossRef PubMed.
- Z. Song, D. Zhu, L. Li, T. Chen, H. Duan, Z. Wang, Y. Lv, W. Xiong, M. Liu and L. Gan, J. Mater. Chem. A, 2019, 7, 1177–1186 RSC.
- Y. Gong, D. Li, C. Luo, Q. Fu and C. Pan, Green Chem., 2017, 19, 4132–4140 RSC.
- Z. Song, D. Zhu, D. Xue, J. Yan, X. Chai, W. Xiong, Z. Wang, Y. Lv, T. Cao and M. Liu, ACS Appl. Energy Mater., 2018, 1, 4293–4303 CrossRef CAS.
- Y. Ahn, B. Kim, J. Ko, D. You, Z. Yin, H. Kim, D. Shin, S. Cho, J. Yoo and Y. S. Kim, J. Mater. Chem. A, 2016, 4, 4386–4391 RSC.
- X. Peng, H. Liu, Q. Yin, J. Wu, P. Chen, G. Zhang, G. Liu, C. Wu and Y. Xie, Nat. Commun., 2016, 7, 11782 CrossRef CAS PubMed.
- X. Zhang, M. Kar, T. Mendes, Y. Wu and D. MacFarlane, Adv. Energy Mater., 2018, 8, 1702702 CrossRef.
- X. Chen, X. Liu, A. Pandian, K. Lou, F. Delnick and N. Dudney, ACS Energy Lett., 2019, 4, 1080–1085 CrossRef CAS.
- V. Vijayakumar, B. Anothumakkool, A. T. A. Torris, S. B. Nair, M. V. Badiger and S. Kurungot, J. Mater. Chem. A, 2017, 5, 8461–8476 RSC.
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6, 10310 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |