
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhen sonochemistry meets heterogeneous photocatalysis: designing a sonophotoreactor towards sustainable selective oxidation†
Dimitrios A.
Giannakoudakis
 *,
Dariusz
Łomot
and
Juan Carlos
Colmenares
*
*,
Dariusz
Łomot
and
Juan Carlos
Colmenares
*
Institute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland. E-mail: DaGchem@gmail.com; dgiannakoudakis@ichf.edu.pl; jcarloscolmenares@ichf.edu.pl
First published on 3rd June 2020
Abstract
Exploration of the synergistic effect in catalysis upon simultaneous utilization of two sources of power, ultrasound and light, is barely explored and remains a challenging issue. A crucial reason behind this is the difficulty in designing and constructing a well-defined sonophotoreactor capable of taking advantage of the benefits of combining sonochemistry and photochemistry. Herein, we present our successful reactor and a detailed study regarding how the utilization of ultrasonication can act as a process intensification tool for selective and partial photooxidation of a biomass derived model compound, benzyl alcohol. The main outcome was the enhanced selectivity in the case of sonophotocatalysis compared to photocatalysis. This selectivity was ascribed to the effects derived from the cavitation phenomena, leading to phenomena such as hot spot formation and jetting. Our study can act as guidance towards understanding the unique effects of ultrasound irradiation as a hybrid process intensification method (HPIM) towards application in more complex chemical reactions and manipulation of the nanocatalysts’ photoreactivity in catalytic valorization applications.
Introduction
The utilization of natural and renewable source of power and feedstocks for the mass production of valuable compounds has become a hot technological and research challenge globally over the last few decades.1–3 Minimalization of the environmental footprint and “Green chemistry” approaches act as an oasis for synthetic and manufacturing processes in order to achieve a more sustainable way of obtaining a wide range of bio-products such as pharmaceuticals, polymers, antifungal agents, and organic conductors. The valorization of naturally available resources, like biomass, to obtain high-value chemicals by an economically and energetically feasible way is assumed to be a favorable strategy.2,4 In general, biomass valorization consists of two steps: the first one involves its conversion into crucial platform/building block chemicals and the second one their chemically conversion/upgrade to value-added chemicals. A characteristic paradigm for the latter conversion is the selective oxidation of biomass-derived alcohols, such as aromatics and furanics, to their corresponding carbonyl counterparts (aldehydes and carboxylic acids).4–8 A promising and advantageous method towards the above-mentioned direction is the exploitation of the most available natural power source, sunlight. The utilization of light for catalytic reactions led to a research field known as photocatalysis. Since the report of the photoelectrocatalytic water splitting using TiO2 by Fujishima and Honda9 in 1972, this semiconductor metal oxide has attracted worldwide research attention for use in light assisted processes. Additionally, heterogeneous photocatalysis is a success story for conventional organic synthetic chemistry.10–13However, a crucial drawback of photochemistry is that there are many cases, especially in aqueous media, wherein “nonselective” conversion cannot be avoided.3,14–17 Highly photoactive materials are widely studied for application in environmental remediation in order to decompose/degrade hazardous compounds, but in most of the cases the outcome is complete and non-selective mineralization.12,17–24The need for developing novel reactions for effective and selective valorization of organic compounds is in high demand, and two aspects can be assumed to be the most crucial: the development of a proper photoreactor and the use of a suitable photocatalyst for every specific photocatalytic reaction. Another strategy in order to elevate the photocatalytic reactions/transformations is to achieve process intensification by the application of alternative source of power. From the moment in 1919 when the Nobel Laureate in Chemistry, Friedrich Wilhelm Ostwald, introduced mechanochemistry as a separate sub-discipline of chemistry alongside photochemistry, electrochemistry, and thermochemistry,25 research efforts towards the manipulation and use of the effects derived from mechanochemical methods have increased. As a representative mechanochemical method, sonication (ultrasound irradiation) has been shown to be a promising tool for various applications, as well as a potential candidate in synthetic chemistry process intensification, and since 1980, when Nappiras introduced the term “sonochemistry”,26 this field has gained incremental volume of attention. The advantages of ultrasound irradiation are derived from the cavitation phenomena. The growth and implosion of acoustic cavitation can act as localized hotspots, since temperature and pressure can reach up to 5000 °C and 1000 bars, respectively.10,27 Mechanical effects, such as elevated mass transfer, de-passivation effect, and de-aggregating of the catalyst, and chemical effects, such as advance oxidation processes (AOP) by the formation of reactive oxygen species and radicals, are also vital.28–31It is feasible to control the extent of the chemical vs. physical effects by tuning the ultrasound wave frequency and power.30,32 Sonication can also replace the conventional mechanical stirring and, even more importantly, can be utilized in flow and micro-flow reactors.11,33–35 The latter method is a widely used approach for large scale synthesis in various modern industrial processes.
The combination of ultrasound and light irradiation in heterogeneous selective catalysis, known as sonophotocatalysis, is a very innovative approach that has attracted continuous attention over the last few years due to its cost-effective and environmentally friendly process intensification abilities. However, the majority of the successful applications of sonophotocatalysis have been focused on environmental remediation applications, especially for waste-water treatment against toxic organic compounds, through uncontrollable advanced oxidation processes (AOPs).36–40 The reported cases of successful combination of photo- and sonochemistry as a Hybrid Advanced Oxidation Process (HAOP) toward specific selective catalytic reactions and valorization of biomass are limited due to the complexity of manipulating the formed active oxygen species, developing an appropriate sonophotoreactor and of course an appropriate sonophotocatalyst.11 Regarding the latter issue, the ultimate goal is to avoid the usage of precious noble and/or rare-earth metal-based catalysts, addition of hazardous and non-reusable chemicals, and the need for harsh experimental conditions.
Our research efforts are focused on the utilization of sonophotocatalysis, herein referred to as SoPhoCatalysis, for the selective transformation of lignin-derived model molecules and on understanding of the features and phenomena that are important to achieve the goal. We found that coupling sonocatalysis with photocatalysis can lead to beneficial and specific effects leading to an improvement in conversion and selectivity. Our preliminary effort was to design and develop a reactor, a SonoPhotoReactor (SPR), in which we will be able to control a wide range of parameters, while allowing for the possibility of combining this reactor with a continuous flow system in the future.33 The commercially available TiO2 P25 (Evonik Degussa) is widely used as a reference/benchmark photocatalyst since it shows high photoreactivity for various reactions.41 For this reason, TiO2-P25, herein referred to as TiO2, was used as the main photocatalyst, while other commercial nanoparticles of metal oxides were tested for the sake of comparison (Fe2O3, Co3O4, or ZnO). The reaction to start with involved the selective partial oxidation of benzyl alcohol (C6H5CH2OH) to benzaldehyde (C6H5CHO). The former is referred to as BnOH and the latter as PhCHO.
Results and discussion
Our sonophotoreactor and its characterization
Various demo experimental setups and reactors were designed, constructed, and tested in order to establish a reactor capable of successfully utilizing simultaneously and controllable ultrasound (US) and light irradiation at a constant temperature. The main concept was to avoid the use of a probe inside the reaction vessel as the US source. By this, we eliminated the possibility of the probe acting as a catalytic spot, where part of the catalyst can stack on the surface, leading to side effects and collateral interactions. Additionally, a possible leaching from the tip/probe and/or possible corrosion were also avoided by this approach. But more importantly, the reactor should be capable with minimal modifications/additions of being adapted for continuous flow reactions. A schematic illustration of our first complete and functional sonophotoreactor, herein referred to a SoPhoReactror, can be seen in Fig. 1a, while photographs of the entire experimental setup are shown in Fig. S1.† The setup was built based on a cup horn sonicator (Qsonica sonicators, 20 kHz), in order to have the option to study the effect of ultrasound irradiation as an assist to photocatalytic reactions or to explore the sonochemical activity of the materials. US irradiation took place from the bottom, while light irradiation from the top. The ultrasound waves played the additional role to simultaneous mix the suspension. Simple commercial glass vials were used as the reaction vessels, while a homemade plastic holder and silicone rings held the reaction vessels stable at a specific distance from the upper surface of the horn. Technical drawings with the dimensions can be found in Fig. S2.† The light source was placed at the upper part of the reaction vessel using an optical fiber. A stainless-steel spiral was developed for the temperature stabilization of the water bath. By this setup, we were able to study and optimize a plethora of factors/parameters with the most critical to be: (i) acoustic frequency, power, and pulse protocol, (ii) wavelength and intensity of the irradiated light, (iii) solvent's nature and temperature, (iv) photocatalyst loading, and (v) mixture of solvents and pH of the solution.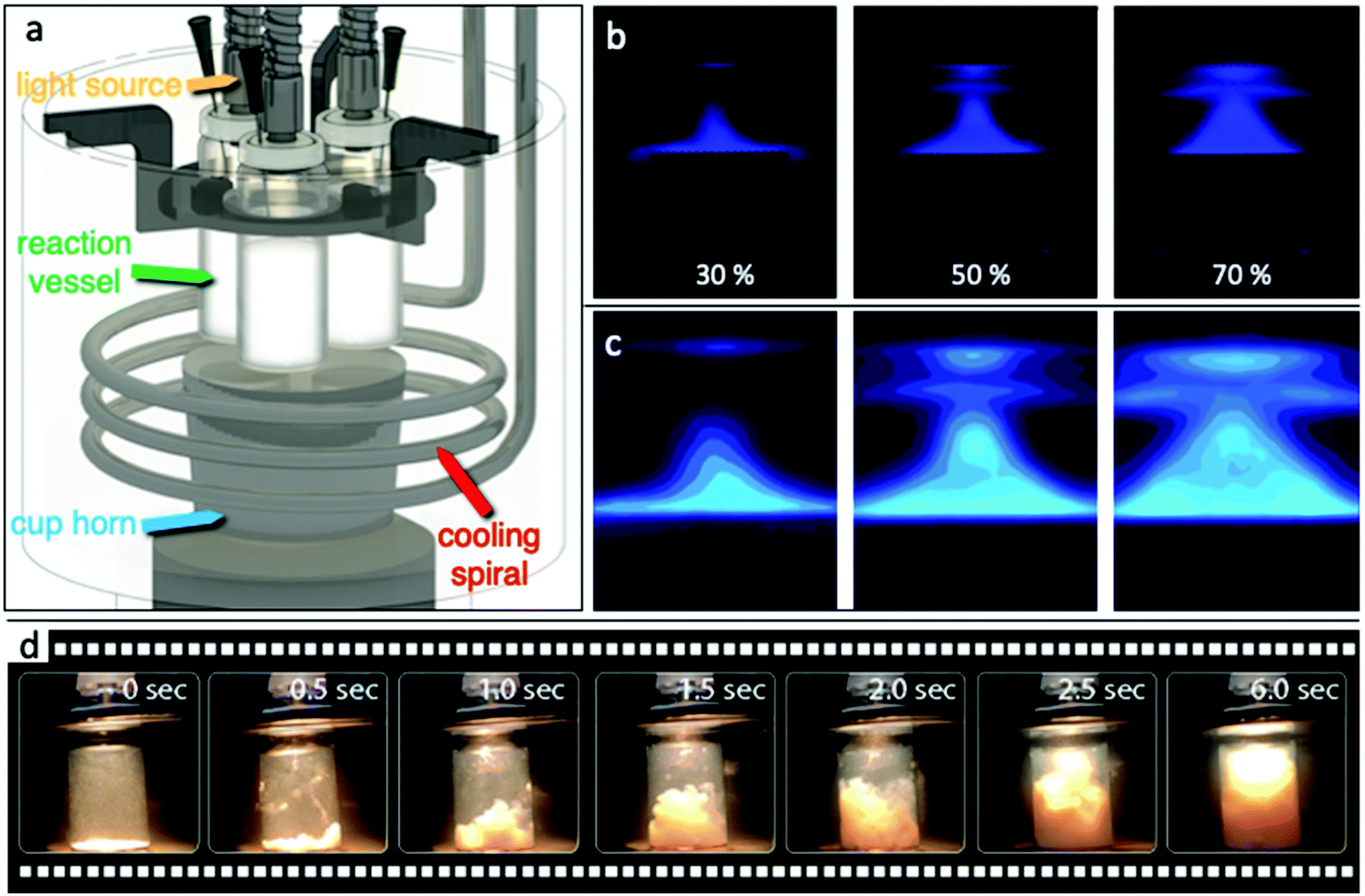 | ||
| Fig. 1 (a) Schematic illustration of the SonoPhotoReactor (SoPhoReactor, SPR); (b) real photographs during the luminol tests for different amplitudes/powers (power density per volume above the surface of the horn 0.16 ± 0.1, 0.33 ± 0.2, and 0.52 ± 0.2 W cm−3 for 30, 50, and 70% amplitude, respectively); (c) power mapping of sonochemiluminescence to highlight the most intense regions of ultrasound waves derived from luminescence tests as a result of OH radical formation due to “acoustic cavitation”; and (d) photographs of the reaction vessel after ultrasound irradiation (30% amplitude) loaded with 1 g L−1 of catalyst (captured with an iPhone X). | ||
A very important aspect when the discussion is around sono- as well as photochemistry is to determine the amount of energy entered into the system. This is a tricky part in order to compare different reports, since the irradiated power at every experimental setup is different, depending on plethora of parameters. Hence, the first task was to map the ultrasound waves in order to ensure their penetration inside the reaction vessel. The dispersion of the ultrasound waves inside the cup horn bath was analyzed by the luminol test.42–46 Luminol is photoluminating in aqueous solution upon reaction with free hydroxyl radicals, formed by acoustic cavitation phenomena. Real photographs of photoluminescence at three different amplitudes/power can be seen in Fig. 1b and sonochemiluminescence photographs of the US wave intensity can be seen in Fig. 1c. The increase of the US power has a positive impact on the radical formation density as well as on the volume in which they can be formed. The important outcome is that radical formation was observed at the entire volume above the cup horn surface.
The as reported power (W) from the instrument's indicator and the calculated power density per the surface area of the horn and per volume of water in the bath above the level of the cup horn surface are shown in Table S2.† The relationship between power and amplitude was found to have an almost perfect linear correlation. The power densities per volume above the surface of the horn were found to be 0.16 ± 0.1, 0.33 ± 0.2, and 0.52 ± 0.2 W cm−3. Although the most important factor is the real amount of the energy entering the reaction vessel, many factors like the shape and material of the reaction vessel can play a key role. For this reason, calorimetry tests were performed in order to calculate the actual ultrasonic power entering into the cup horn bath, as well as inside the reaction vessel, filled with water or acetonitrile.47–49 The temperature evolution upon ultrasound irradiation during the calorimetry tests is shown in Fig. S6† and the calculated powers are given in Table 1. The calculated powers based on calorimetry tests for the entire volume of the water bath of the setup are in good agreement with those reordered directly from the instrument and revealed a linear increment upon amplitude increase.
| Amplitude (%) | Power from calorimetry tests (W) | ||
|---|---|---|---|
| Cup horn water bath | Reaction vessel water | Reaction vessel acetonitrile | |
| 30 | 104.3 | 7.1 | 3.6 |
| 50 | 165.5 | 5.3 | 2.9 |
| 70 | 221.7 | 3.6 | 2.6 |
In contrast, the inserted power inside the reaction vessel, filled with 15 mL of water or acetonitrile, showed a decrease with the increase of the amplitude from 30 to 70%. This suggests that the shape, position, and material of the reaction vessel play a crucial role. The distribution of the ultrasound energy is also ultimately important and as can be seen in Fig. 1b, an increase in the amplitude leads to a higher dispersion of the power, and as a result less energy finally enters the reaction vessel. At an amplitude of 30%, around 7% of the ultrasound power is inserted in the reaction vessel (7.1 W) when the vial was filled with water. When the reaction vessel was filled with acetonitrile, the calorimetrically calculated power was found to be almost the half in comparison with water filling. This fact is due to the double heat capacity of water compared to acetonitrile. Interestingly, by applying a higher amplitude of US (70%), the inserted inside the reaction vessel power was decreased. This decrement of power upon the increase of the amplitude points out the key role played by the ultrasound power and the necessity to optimize it.
It was intended to perform the reaction under different but always stable temperature and/or US irradiation power in order to determine the optimum reaction parameters. Having in mind that irradiating with US will increase the temperature of the cup horn bath and as a result of the reaction vessel, it was crucial to be able to maintain stable the temperature. For this reason, a homemade stainless-steel spiral consisting of three windings was developed and a water circulator was used for cooling. This spiral was easy to be adjusted and be kept under the level of the cup horn upper surface. The temperature evolution of the system upon ultrasound and/or light irradiation was studied by temperature monitoring at different spots of the experimental setup (see ESI†). The spiral cooling system was found to be capable of overcoming the heat transfer effect upon US irradiation and of retaining the temperature stable. The cooling bath was tuned at two temperatures (21.5 and 26.5 °C) in order to evaluate the cooling capability of the spiral. The temperature evolution inside the bath above the horn (top) under four different ultrasound amplitudes and the two established cooling temperatures are shown in Fig. S5.† For cooling temperature of 21.5 °C, the stabilized temperature after around 40 min was 24.9 and 29.2 °C for 15 and 70% amplitudes, respectively. For cooling temperature of 26.5 °C, the temperatures were stabilized at 28.9 and 31.8 °C, respectively, for amplitudes of 15 and 70%. The temperature increase upon ultrasound irradiation is an in-direct effect which should always be considered, since the materials can act as thermocatalysts. Another crucial issue of the use of ultrasound waves is whether the mixing extent (mass transfer between the catalyst nanoparticles and the liquid phase of the reaction mixture) can be assumed to be sufficient. Based on Fig. 1d, it can be seen for 1 g L−1 of catalyst that even after less than six seconds, the catalyst is homogeneously dispersed to the entire volume of solvent. Analogue photographs for different amounts of catalyst can be seen in Fig. S3.†
Adsorption/reactivity in the dark
The first task prior to evaluating the catalytic behavior of the materials was to determine their potential adsorptive capability and the possibility of reactivity in the dark. For this reason, adsorption/removal tests in the absence of light were performed. The obtained results are shown in Table S1.† The removal of BnOH (the decrease in its concentration) can be due to adsorption phenomena and/or conversion to other products. The highest adsorption was observed for TiO2 (3.2%) and it can be assumed to be negligible. The adsorption extent was found to be the same in water or acetonitrile.Optimization of the sonophotocatalysis parameters
In order to determine the optimum US power, four different amplitudes (15, 30, 50, and 70%) and two different catalyst amounts (0.5 and 1 g L−1) were evaluated at two different temperatures of the cooling system (21.5 and 26.5 °C). In order to eliminate the formation of radicals and to study the mechanical effect of sonication rather than that of chemical effects, acetonitrile was used as a solvent and the concentration of BnOH was 1 mM.The conversion and selectivity were calculated based on the following formulas, where CBnOH,in and CBnOH,f are the initial and final concentrations of BnOH, respectively, and CPhCHO is the final concentration of PhCHO.
| BnOH conversion (%) = 100 × (CBnOH,in − CBnOH,f)/CBnOH,in |
| PhCHO selectivity (%) = 100 × CPhCHO/(CBnOH,in − CBnOH,f) |
The conversion extents for all the materials tested were negligible, except in the case of TiO2 P25, and so, our research effort was focused on this material. The obtained BnOH conversions and PhCHO selectivities are shown in Fig. 2. The most important outcome is that in all cases, 30% of amplitude led to the highest conversion extent, while the selectivity was also the maximum at this US power. In general, the temperature showed a negligible effect on the BnOH conversion, but the effect of temperature will be discussed in detail below. A low amplitude of 15% led to a very low conversion extent (23%). We link this to the fact that this amount of acoustic power is not sufficient in order to promote a sufficient level of mixing, mass transfer, and de-aggregation of catalyst particles, and as a result light penetration in the entire reaction volume. This can be confirmed by the comparison of the results with a lower amount of catalyst (0.5 g L−1). Only at this amplitude the lower amount of catalyst led to a higher conversion than that of 1 g L−1. At this point it is crucial to point out that negligible conversion or degradation of BnOH was observed without catalyst only under ultrasound irradiation (sonolysis) or under light irradiation (photolysis) for all the combinations of the tested parameters. No conversion was achieved under sonication in the presence of TiO2 alone. All these above facts confirm that the effect of US does not directly promote the oxidation of BnOH to PhCHO or to its decomposition/mineralization.
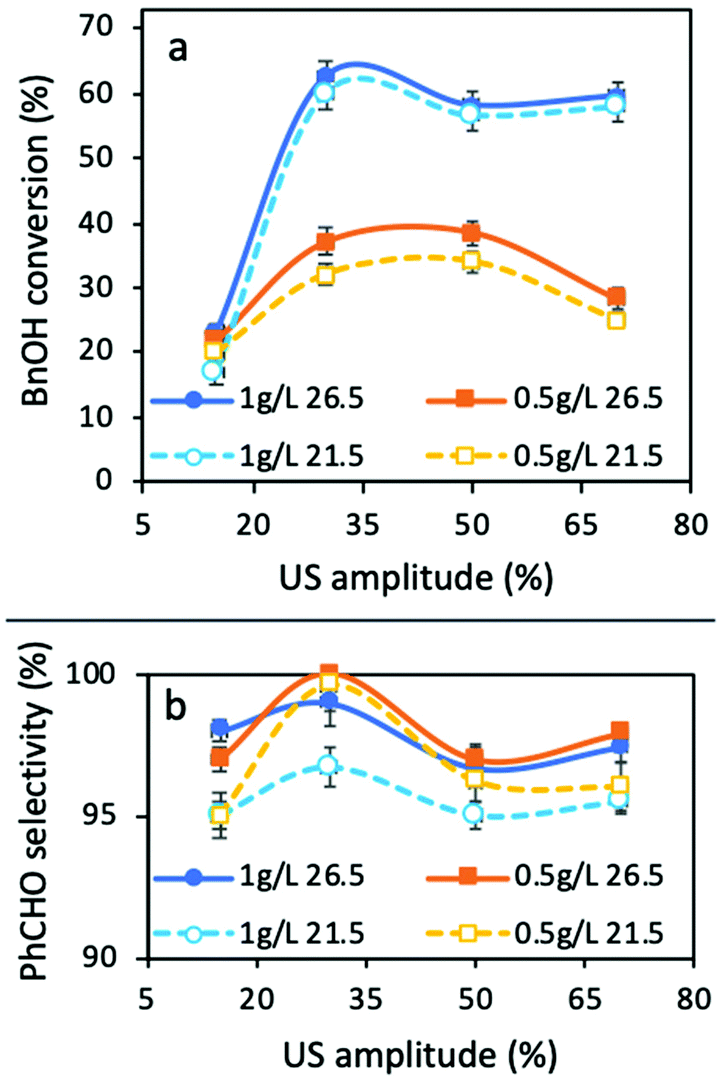 | ||
| Fig. 2 The BnOH conversion (a) and PhCHO selectivity (b) after 3 h under four different US amplitudes (15, 30, 50, and 70%), two different catalyst concentrations (0.5 and 1 g L−1), and two different temperature of the cooling system (21.5 and 26.5 °C) under irradiation of a solar light simulator (all the experiments were performed more than three times and the standard deviation/error bars were adopted). | ||
The maximum conversion of 62.6% was obtained for 1 g L−1 of catalyst at an amplitude of 30% and a reaction temperature of 29.3 °C (established temperature at the temperature control 26.5 °C), while the selectivity towards PhCHO was >99%. A 3.1 °C decrease of the reaction temperature (26.2 °C, established temperature at the temperature control of 21.5 °C) led to almost the same conversion of 60.1%, the fact that further supports the minimum effect of temperature. Increasing the amplitude to 50% resulted in lower conversions: 57 and 55% at 28.1 and 30.6 °C, respectively. Finally, the use of a high amplitude of 70% led to BnOH conversions of 58 and 60% at 29.2 and 31.8 °C, respectively. We can observe that the reaction temperature is the same (29.2 °C) in two cases with different amplitudes (30 and 70%), due to the different stabilization temperatures set. However, in the case of a 30% amplitude, the conversion as well as the selectivity are higher than that under a 70% amplitude, the fact that confirms the vital role of mixing during the reaction and that a high power of ultrasound power could not lead to optimum catalytic results. Based on all the above presented and discussed results, it can be concluded that for this experimental setup and reaction, the amplitude of 30% should be assumed to be the optimum one, and for this reason we focused on the optimization of the remaining parameters under a 30% amplitude.
Catalyst loading optimization
Even though two different loadings of catalyst were already discussed above, a more extended analysis of the catalyst amount was examined under ultrasound and light irradiation (sonophotocatalysis), as well as only under light irradiation (photocatalysis) using traditional magnetic stirring (MgS). The obtained results after the tests of the different amounts of catalyst, under solar light or UV light, are shown in Fig. 3. The main outcome is that the optimum catalyst loading from the point of view of performance versus cost is indeed 1 g L−1, both under UV irradiation or under solar light. A further increase in the catalyst can result in a slightly higher conversion, but with selectivity being sacrificed. This increment in conversion can also be linked to extended adsorption phenomena due to a higher amount of catalyst. The most important conclusions are that even though the conversion is slightly higher in the case of classic magnetic stirring, the selectivity is dramatically higher in the case of sonication, especially under UV irradiation. In the case of a solar light simulator, the application of US led to a conversion of ∼70% with an almost 98% of selectivity towards aldehyde (100% if we consider the around 2% of adsorption). In the case of magnetic stirring, the PhCHO selectivity follows a decreasing trend, suggesting further transformation to undesired products. The differences are more pronounced upon UV light irradiation, when in the case of magnetic stirring the PhCHO selectivity is found to be almost zero as a result of its decomposition. The reason behind this can also be assigned to the antenna effect.50 Due to the remaining aggregations, the formed e−/h+ pairs can travel through the network of particles and attack unselectively the BnOH molecules with a higher rate, especially inside the interstitial space. Additionally, the limited amount of adsorbed humidity on the nanoparticles’ surface adsorbed prior to the use can be concentrated inside this interstitial space, leading to harsh attack of the BnOH and/or PhCHO by formed radicals. This series of the results suggest a unique influence of US irradiation, which can be linked to the direct or indirect physical/mechanical effects and as a result to selectivity enhancement. In order to interpret these outcomes, we will discuss hereinafter the possible roles and effects upon ultrasound irradiation as a process intensification approach.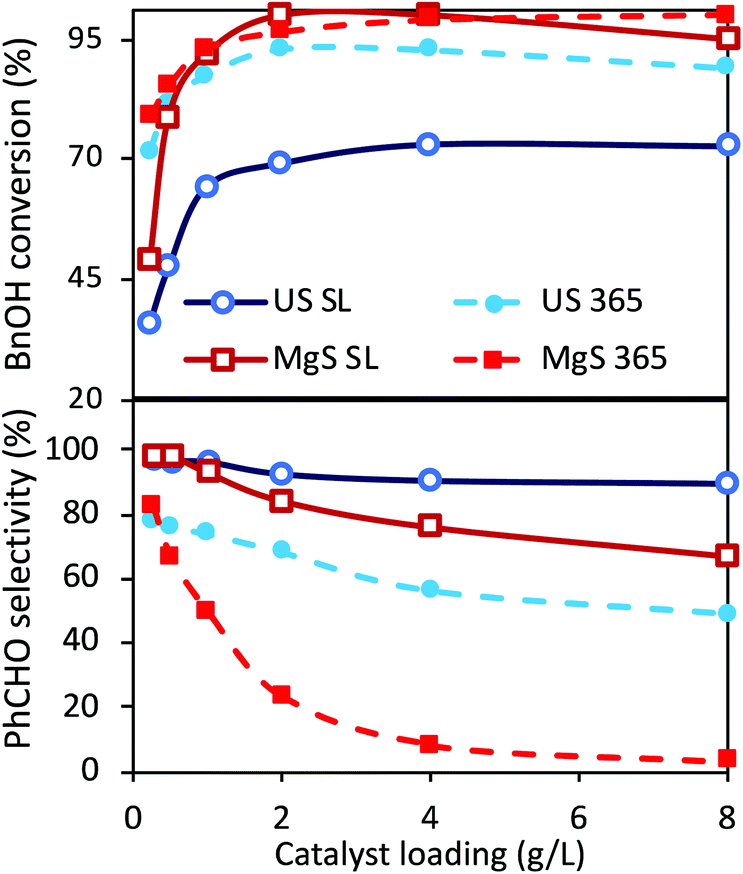 | ||
| Fig. 3 BnOH conversion (up) and PhCHO selectivity (down) after 3 h for different catalyst loadings (0.5, 1, 2, 4 and 8 g L−1) under a US amplitude of 30% (US) or magnetic stirring (MgS) irradiating with solar light (SL) or ultraviolet light of 365 nm wavelength (365); the reported values are the average of three experiments with the error to be less than 1.5%. | ||
Different light sources
Another ultimately important aspect in order to gain a better insight on the photoreactivity of P25 is to examine the behavior of the catalyst under the irradiation of light of different wavelengths. The optical band gap of the commercial P25, estimated from the DR-UV-Vis spectrum and by applying the Kubelka–Munk theory, was found to be equal to ∼3.2 eV (387 nm), a value on the border between ultraviolet and visible ranges of light. We also evaluated the catalytic performances under three specific wavelengths of light produced from LEDs (365, 455, and 510 nm) and under solar simulator light exposure. We also tested the solar light by applying a filter in order to cut the UV and IR regions. The spectra and the intensities of each light source are shown in Fig. 4 and Table S3,† while some photographs of the reaction vessel under different light source irradiation can be seen in Fig. S7.†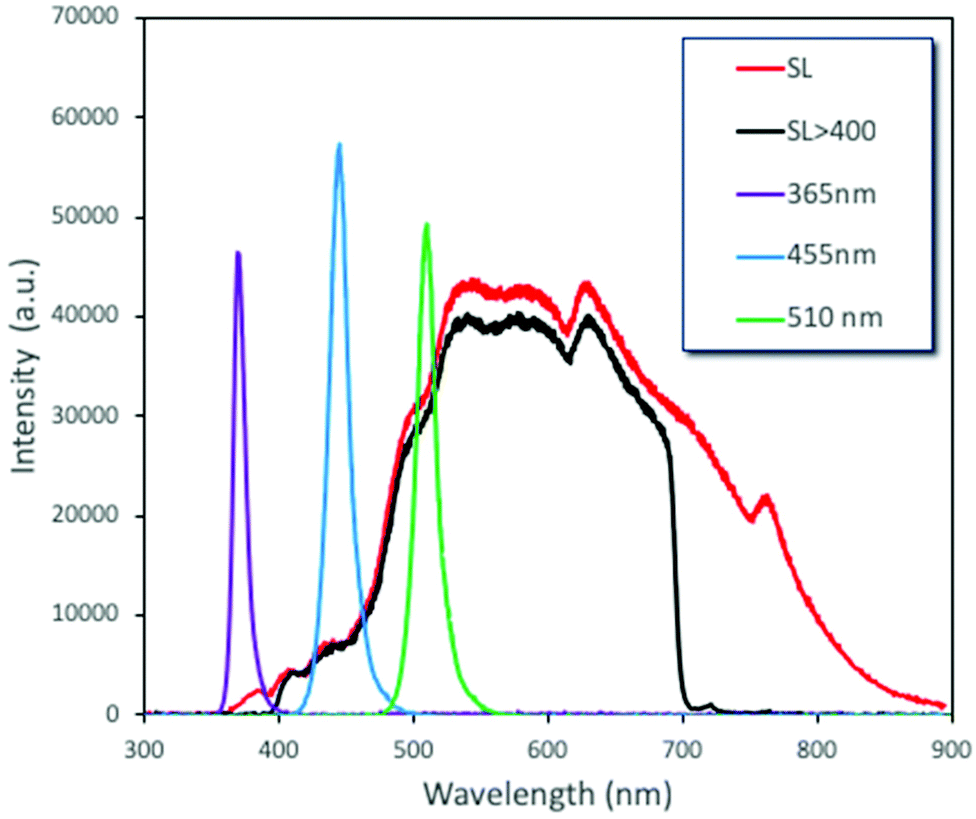 | ||
| Fig. 4 Emission spectra of the utilized light sources. | ||
The analysis of the results led to key conclusions. The first one is that P25 is photoactive even at 455 nm (Fig. 5a). This can be explained by the fact that the adsorbed molecules on the outer surface of the particles can act as antennas/photoscavengers and activate the TiO2 electron/hole formation.50 Secondly, the conversion in the case of ultrasonication (US) is lower compared to magnetic stirring (MgS) and this can be assigned to the higher extent of de-aggregation. However, the selectivity for all light sources, especially under 365 nm, was found to be higher for US than for MgS.
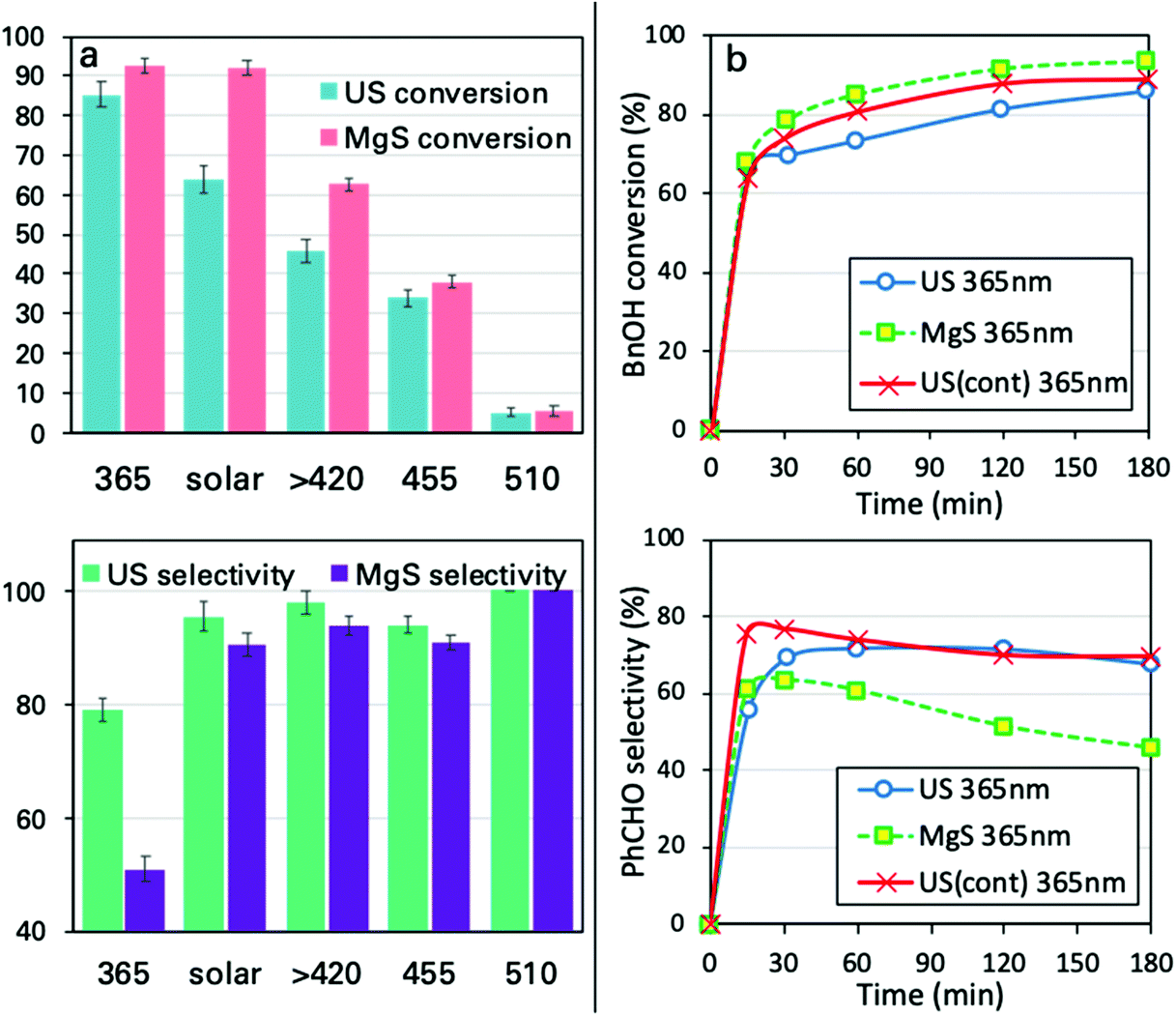 | ||
| Fig. 5 BnOH conversion (up) and PhCHO selectivity (down) for different light sources after 3 h (a), ultrasound irradiation in pulse mode, continuous mode and magnetic stirring (1 g L−1 catalyst, 1 mM BnOH, in acetonitrile at 21.6 °C) under UV light exposure (b). | ||
Pulse vs. continuous US irradiation mode
For all the reported experiments up to this point, the US irradiation protocol was in pulse mode (PM). We performed the sonophotocatalytic oxidation by continuous mode, US(cont), sonication (Fig. 5b). For continuous mode US irradiation, the conversion was found to be higher and the maximum conversion was achieved faster compared to PM irradiation and magnetic stirring. It is of uppermost importance that the selectivity reached a plateau in the case of the US irradiation, while it showed a declining trend for MgS. This can be associated with the possibility that US irradiation disturbs the specific equilibrium of the catalytic conversion. This can be explained based on the interactions/adsorption of benzyl aldehyde on the surface being weaker than that of BnOH51 and with the US irradiation getting desorbed prior to its further photocatalytic transformation.Effects of ultrasound irradiation
In order to elucidate the way by which the US positively affects the selective partial oxidation, it is vital to consider all possible effects upon ultrasound irradiation. The photoreactivity of titanium dioxide arises from the photon absorption which leads to the photoexcitation of electron/hole pairs which can further lead to the formation of different reactive oxygen species, depending on the conditions and the solvent used.52–56 The oxidation requires prior adsorption of the benzyl alcohol molecules on the surface and the formation of surface complexes.54,57,58 The presence of molecular oxygen is also an important aspect, since it can be involved in the oxidation process. The dissolved O2 is additionally removed due to cavitation formation, and this is an additional reason for the slightly slower partial oxidation kinetic/oxidation rate. On the other hand, the removal of the dissolved oxygen upon ultrasound irradiation is a positive aspect, avoiding further oxidation of the formed aldehyde. In order to verify the role of the dissolved air, tests were performed by simultaneous air purging, and the rate of benzyl aldehyde formation (expressed as yield, Fig. S8†) was found to be slightly faster compared with air purging. However, the conversion extent was lower. The continuous presence of dissolved air can also have an effect on the thermal effects and the dispersion of the nanoparticles upon US irradiation. Additionally, the oxygen purging can have an effect on the adsorbed amount of water molecules on the surface. The role of gas (oxygen, air, inert gas, etc.) is a complicated issue to study and is outside the scope of this work, since the effect of the dissolved gas, the rate of the purging, and the chemical nature of the gas can alter a variety of the ultrasound effects. Moreover, it is possible that the concentration of the diluted gas is not homogeneous, for instance closer to the ultrasound source the degassing is more pronounced.It is also possible for the water moieties, formed by side reactions or being initially adsorbed on the surface of the catalyst, to be transformed into hydroxyl radicals by the reaction with the photogenerated e−/h+ pairs.54,59 These species can lead to either overoxidation reactions or mineralization of the BnOH. However, the almost 100% aromatic balance and the selective partial oxidation to benzyl aldehyde suggests that if formed, the *OH acts as consuming agents of the released protons.53 The above results and others that follow can be linked to the harsh mechanical/physical effects of the cavitation formed upon ultrasound irradiation. In general, the nanoparticles are surrounded by a zone/phase of the solvent, in which the molecules of BnOH and moieties of water also exist (Fig. 6). Adsorbed BnOH on the particle surface can enhance the light adsorption due to the antenna light scavenger effect, especially under visible/blue light irradiation.50,51,60
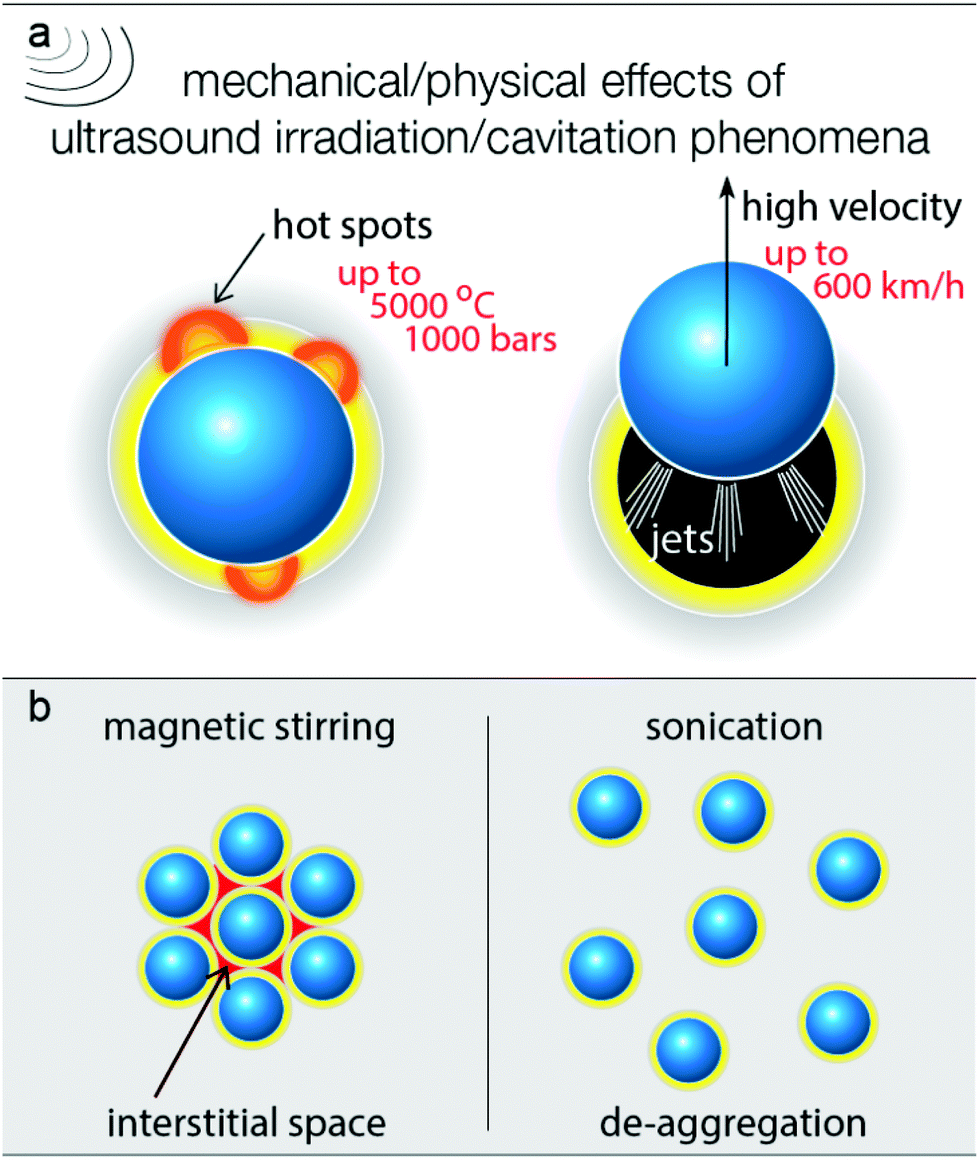 | ||
| Fig. 6 The main mechanical/physical effects derived from ultrasound irradiation due to the cavitation phenomena (a) and the difference in the extend of de-aggregation (b). | ||
In general, when a cavitation/bubble collapses near the surface of the solid, hot spots, microjets, microstreaming, turbulence, and agitation can be observed.29 This can result in a violent acceleration of the nanoparticles (jetting phenomena), as seen in Fig. 6a. It is also feasible for the latter to be distorted due to the mechanical effects of the bubbles collapse near the surface of the catalyst. It was reported that the liquid jet in the case of water can reach an instant velocity of up to 600 km h−1 (170 m s−1).26 This jetting acceleration is also sufficient to overcome the gravity and the solvent–nanoparticle interfacial interactions in order to achieve the desired mixing (Fig. 1d). Analogue photographs for various amounts of catalyst can be seen in Fig. S1,† in which it can be observed that US can lead to sufficient mixing within 20 s even for a high loading of 4 g L−1. Even in the case of pulse mode (ON![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OFF) US irradiation, the particles were moving from the bottom to the top inside the reaction vessel, while during the silent period, they were directed to settle toward the bottom of the reaction vessel. However, after some seconds of US irradiation, no particles were observable by the naked eye and the catalyst was homogeneously dispersed to the entire volume of solvent. Hence, the mixing extent by ultrasound can be assumed as sufficient and optimal.
:OFF) US irradiation, the particles were moving from the bottom to the top inside the reaction vessel, while during the silent period, they were directed to settle toward the bottom of the reaction vessel. However, after some seconds of US irradiation, no particles were observable by the naked eye and the catalyst was homogeneously dispersed to the entire volume of solvent. Hence, the mixing extent by ultrasound can be assumed as sufficient and optimal.
It is also possible that many nanoparticles remain aggregated during the magnetic stirring via polar interactions (Fig. 6b),61–63 and these aggregations move as one chunk during magnetic stirring. This leads to the creation of interstitial phases/spaces,64–66 where unselective reactions, like overoxidation, can take place, due to the presence of strongly adsorbed water moieties from the environmental humidity or/and retention of the diluted oxygen. So, ultrasound irradiation can also elevate de-aggregation (Fig. 6b),64,67 resulting in a positive impact on the reaction selectivity. De-aggregation also leads to an increase in the accessibility of the catalytic sites. Another important effect of the cavitation collapse close to the catalyst surface is the local increase in the temperature and pressure, forming hot spots and activating the surface reactivity. Additionally, ultrasonication results in the degassing of the solvent and of the entrapped gaseous molecules at the interface between the solid and the solvent. This aspect merits further investigation, considering the inserted in the reaction vessel ultrasonic power as well as the light scattering can be heterogeneous.
Water as solvent
To determine the role of the presence of water and to evaluate the potential synergistic effect of ultrasound irradiation in an aqueous environment, we also performed the experiments in water as the solvent (Fig. 7). The conversion was better and faster in the case of mechanochemical assisted processes, both under a solar light simulator (SL, without the UV cut-off) and UV irradiation. This can be linked to the removal of the water molecules from the surface of the catalyst that block/hinder the active reaction sites,6 due to the jetting phenomena or hot spot formation. The selectivity was also found to be improved almost by 45% for US under solar light irradiation, since the harsh movement of the nanoparticles affect the equilibrium of further oxidation/degradation of the formed PhCHO, by removing the adsorbed benzaldehyde molecules from the surface of the nanoparticles. In all cases, the decrease of the PhCHO was linked to decomposition/mineralization, since no benzoic acid or other organic compounds were detected by HPLC analysis.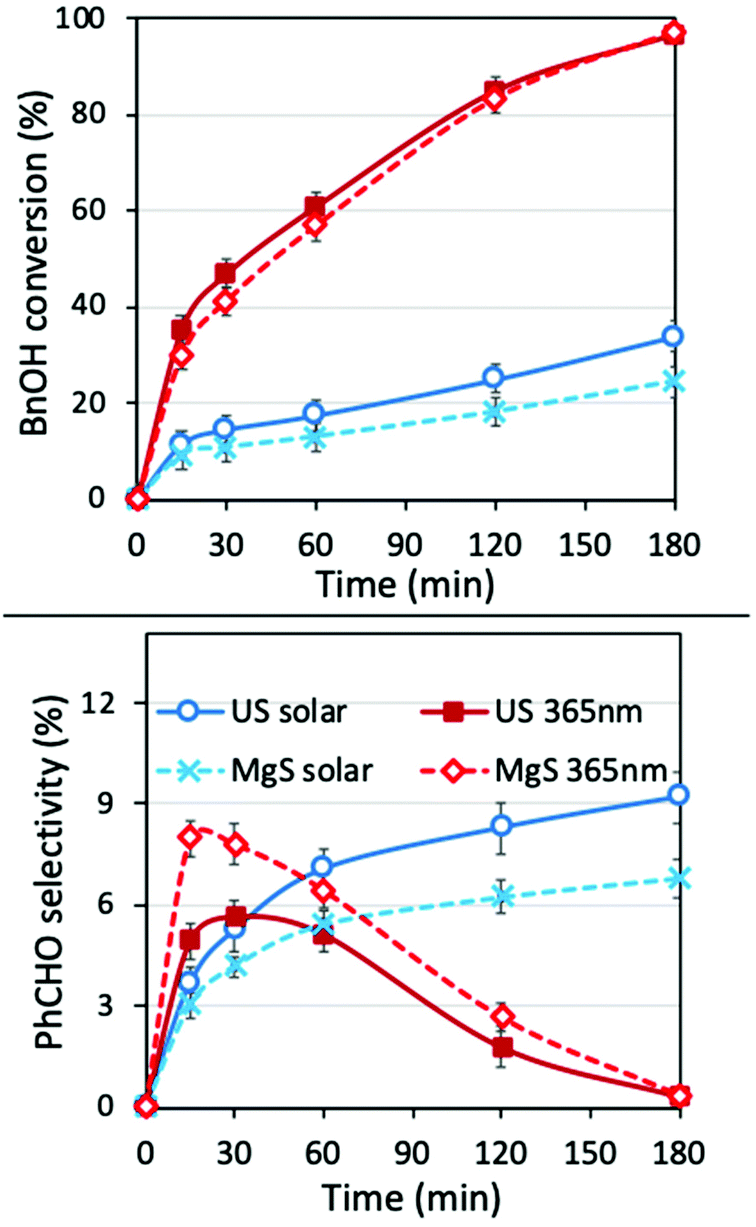 | ||
| Fig. 7 BnOH conversion (up) and PhCHO selectivity (down) in water as solvent for different light sources (solar: solar light simulator, UV: 365 nm), ultrasound irradiation in pulse mode of 30% amplitude and magnetic stirring (1 g L−1 catalyst, 1 mM BnOH, in acetonitrile at 21.6 °C). | ||
Conclusion
In conclusion, we presented for the first time the design and establishment of an easy-to-construct in the lab sono-photoreactor based on a cup horn sonicator and LED light sources. After the optimization of the most crucial parameters, we determined that the ultrasound irradiation can lead to equivalent to traditional magnetic stirring catalytic partial oxidative conversion of a biomass model compounds, benzyl alcohol. The important effect of the synergistic effect upon the simultaneous irradiation by two separate sources of power, light and ultrasound, was the higher selectivity of the partial oxidation to benzaldehyde. This can be due to the unique physical/mechanical effects arising from the acoustic cavitation phenomena, like hot spot and microjet formation, that play a key role in the controllable mass transfer and the elevated de-aggregation of the nanoparticles inside the liquid phase. This Hybrid Advance Oxidation Method (HAOM) have a great potential to be applied in the valorization/selective transformation of more complex biomass derived compounds, such as coniferyl, cinnamyl, veratyl, eugenol, etc., and our research effort is focused in this direction, since the photocatalytic capabilities of the catalyst can be manipulated by the control of the ultrasound power. Additionally, sonophotocatalysis can also be considered as a promising “green” approach for application in scaled-up reactors and more importantly in continuous flow reactors/microreactors, utilizing solar light, either for selective oxidation processes or for environmental remediation applications.Experimental
All the used chemicals, details regarding the sonophotoreactor and the ultrasound and light sources, and additional results are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful for the support from the National Science Centre in Poland within OPUS-13 project nr 2017/25/B/ST8/01592 (http://photo-catalysis.org). The help of Bartosz Zawadzki during the first steps of the research is also appreciated. Special acknowledgement should be expressed to Dr Vaishakh Nair for the fruitful discussion.References
- L. I. Granone, F. Sieland, N. Zheng, R. Dillert and D. W. Bahnemann, Green Chem., 2018, 20, 1169–1192 RSC.
- S. H. Li, S. Liu, J. C. Colmenares and Y. J. Xu, Green Chem., 2016, 18, 594–607 RSC.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef CAS.
- B. Sarmah, B. Satpati and R. Srivastava, ACS Omega, 2018, 3, 7944–7954 CrossRef CAS PubMed.
- D. A. Giannakoudakis, V. Nair, A. Khan, E. A. Deliyanni, J. C. Colmenares and K. S. Triantafyllidis, Appl. Catal., B, 2019, 256, 117803 CrossRef CAS.
- G. Fiorentino, M. Ripa and S. Ulgiati, Biofuels, Bioprod. Biorefin., 2017, 11, 195–214 CrossRef CAS.
- H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. C. Q. Colmenares, J. Nanosci. Nanotechnol., 2013, 13, 4787–4798 CrossRef PubMed.
- G. Chatel, S. Valange, R. Behling and J. C. Colmenares, ChemCatChem, 2017, 9, 2615–2621 CrossRef CAS.
- J. C. Colmenares, R. Luque, J. M. Campelo, F. Colmenares, Z. Karpiński and A. A. Romero, Materials, 2009, 2, 2228–2258 CrossRef CAS.
- Z. C. Kampouraki, D. A. Giannakoudakis, K. S. Triantafyllidis and E. A. Deliyanni, Green Chem., 2019, 21, 6685–6698 RSC.
- T. Zhou, X. Wu, J. Mao, Y. Zhang and T.-T. Lim, Appl. Catal., B, 2014, 160–161, 325–334 CrossRef CAS.
- H. Barndõk, D. Hermosilla, C. Han, D. D. Dionysiou, C. Negro and Á. Blanco, Appl. Catal., B, 2016, 180, 44–52 CrossRef.
- D. D. Dionysiou, M. T. Suidan, E. Bekou, I. Baudin and J. M. Laîné, Appl. Catal., B, 2000, 26, 153–171 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Appl. Catal., B, 2004, 54, 155–163 CrossRef CAS.
- D. A. Giannakoudakis, G. Chatel and J. C. Colmenares, Top. Curr. Chem., 2020, 378, 2 CrossRef CAS PubMed.
- A. Ibhadon and P. Fitzpatrick, Catalysts, 2013, 3, 189–218 CrossRef CAS.
- V. Nair, M. J. Muñoz-Batista, M. Fernández-García, R. Luque and J. C. Colmenares, ChemSusChem, 2019, 12, 2098–2116 CrossRef CAS PubMed.
- D. D. Dionysiou, M. T. Suidan, I. Baudin and J.-M. Laîné, Appl. Catal., B, 2004, 50, 259–269 CrossRef CAS.
- Y. Liu, X. He, X. Duan, Y. Fu and D. D. Dionysiou, Chem. Eng. J., 2015, 276, 113–121 CrossRef CAS.
- E. Evgenidou, K. Fytianos and I. Poulios, Appl. Catal., B, 2005, 59, 81–89 CrossRef CAS.
- E. Evgenidou, I. Konstantinou, K. Fytianos, I. Poulios and T. Albanis, Catal. Today, 2007, 124, 156–162 CrossRef CAS.
- W. Ostwald, Die chemische literatur und die organisation der wissenschaft, Akad. Verlag. Gesel., 1919 Search PubMed.
- E. A. Neppiras, Phys. Rep., 1980, 61, 159–251 CrossRef.
- J. C. Yu, J. Yu, W. Ho and L. Zhang, Chem. Commun., 2001, 1942–1943 RSC.
- A. Magdziarz and J. C. Colmenares, Molecules, 2017, 22(2), 216 CrossRef PubMed.
- G. Cravotto and P. Cintas, Chem. Sci., 2012, 3, 295–307 RSC.
- N. Pokhrel, P. K. Vabbina and N. Pala, Ultrason. Sonochem., 2016, 29, 104–128 CrossRef CAS PubMed.
- J. Berlan, F. Trabelsi, H. Delmas, A. M. Wilhelm and J. F. Petrignani, Ultrason. Sonochem., 1994, 1, S97–S102 CrossRef CAS.
- K. S. Suslick, D. A. Hammerton and R. E. Cline, J. Am. Chem. Soc., 1986, 108, 5641–5642 CrossRef CAS.
- S. R. Pradhan, V. Nair, D. A. Giannakoudakis, D. Lisovytskiy and J. C. Colmenares, Mol. Catal., 2020, 486, 110884 CrossRef CAS.
- S. Asgharzadehahmadi, A. A. Abdul Raman, R. Parthasarathy and B. Sajjadi, Renewable Sustainable Energy Rev., 2016, 63, 302–314 CrossRef CAS.
- R. J. Wood, J. Lee and M. J. Bussemaker, Ultrason. Sonochem., 2017, 38, 351–370 CrossRef CAS PubMed.
- N. Talebian, M. R. Nilforoushan and F. J. Mogaddas, Ceram. Int., 2013, 39, 4913–4921 CrossRef CAS.
- L. Liang, Y. Tursun, A. Nulahong, T. Dilinuer, A. Tunishaguli, G. Gao, A. Abulikemu and K. Okitsu, Ultrason. Sonochem., 2017, 39, 93–100 CrossRef CAS PubMed.
- V. Vinesh, A. R. M. Shaheer and B. Neppolian, Ultrason. Sonochem., 2018, 50, 302–310 CrossRef PubMed.
- S. Sunasee, K. T. Wong, G. Lee, S. Pichiah, S. Ibrahim, C. Park, N. C. Kim, Y. Yoon and M. Jang, Environ. Sci. Pollut. Res., 2017, 24, 15488–15499 CrossRef CAS PubMed.
- Z. Yan, L. Zhang, Z. Zhao, H. Qi, Y. Li and D. Cang, Ultrason. Sonochem., 2018, 47, 133–140 CrossRef CAS PubMed.
- B. Ohtani, O. O. Prieto-Mahaney, D. Li and R. Abe, J. Photochem. Photobiol., A, 2010, 216, 179–182 CrossRef CAS.
- K. S. Suslick, Science, 1990, 247, 1439–1445 CrossRef CAS PubMed.
- Y. Kojima, Y. Asakura, G. Sugiyama and S. Koda, Ultrason. Sonochem., 2010, 17, 978–984 CrossRef CAS PubMed.
- G. J. Price, N. K. Harris and A. J. Stewart, Ultrason. Sonochem., 2010, 17, 30–33 CrossRef CAS PubMed.
- T. J. Tiong, T. Chandesa and Y. H. Yap, Ultrason. Sonochem., 2017, 36, 78–87 CrossRef CAS PubMed.
- B. A. Corzo and M. F. Suárez-Herrera, Ultrason. Sonochem., 2018, 48, 281–286 CrossRef CAS PubMed.
- T. Kimura, T. Sakamoto, J. M. Leveque, H. Sohmiya, M. Fujita, S. Ikeda and T. Ando, Ultrason. Sonochem., 1996, 3, 0–4 CrossRef CAS.
- S. de La Rochebrochard d'Auzay, J. F. Blais and E. Naffrechoux, Ultrason. Sonochem., 2010, 17, 547–554 CrossRef PubMed.
- M. Toma, S. Fukutomi, Y. Asakura and S. Koda, Ultrason. Sonochem., 2011, 18, 197–208 CrossRef CAS PubMed.
- C. Y. Wang, R. Pagel, J. K. Dohrmann and D. W. Bahnemann, Mater. Sci. Forum, 2007, 544–545, 17–22 CAS.
- R. Li, H. Kobayashi, J. Guo and J. Fan, J. Phys. Chem. C, 2011, 115, 23408–23416 CrossRef CAS.
- C. Zheng, G. He, X. Xiao, M. Lu, H. Zhong, X. Zuo and J. Nan, Appl. Catal., B, 2017, 205, 201–210 CrossRef CAS.
- X. Xiao, C. Zheng, M. Lu, L. Zhang, F. Liu, X. Zuo and J. Nan, Appl. Catal., B, 2018, 228, 142–151 CrossRef CAS.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279–285 CrossRef CAS.
- L. Zhao, B. Zhang, X. Xiao, F. L. Gu and R. Q. Zhang, J. Mol. Catal. A: Chem., 2016, 420, 82–87 CrossRef CAS.
- C. J. Li, G. R. Xu, B. Zhang and J. R. Gong, Appl. Catal., B, 2012, 115–116, 201–208 CrossRef CAS.
- M. Zhang, Q. Wang, C. Chen, L. Zang, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2009, 48, 6081–6084 CrossRef CAS PubMed.
- X. Lang, W. Ma, C. Chen, H. Ji and J. Zhao, Acc. Chem. Res., 2014, 47, 355–363 CrossRef CAS PubMed.
- X. F. Zhang, Z. Wang, Y. Zhong, J. Qiu, X. Zhang, Y. Gao, X. Gu and J. Yao, J. Phys. Chem. Solids, 2019, 126, 27–32 CrossRef CAS.
- J. Cunningham and S. Srijaranai, J. Photochem. Photobiol., A, 1991, 58, 361–371 CrossRef CAS.
- D. A. Giannakoudakis, F. Pearsall, M. Florent, J. Lombardi, S. O'Brien and T. J. Bandosz, J. Colloid Interface Sci., 2018, 531, 233–244 CrossRef CAS PubMed.
- J. Lombardi, L. Yang, F. A. Pearsall, N. Farahmand, Z. Gai, S. J. L. Billinge and S. O'Brien, Chem. Mater., 2019, 31, 1318–1335 CrossRef CAS.
- R. Wallace, D. A. Giannakoudakis, M. Florent, C. Karwacki and T. J. Bandosz, J. Mater. Chem. A, 2017, 5, 4972–4981 RSC.
- D. A. Giannakoudakis, N. Farahmand, D. Łomot, K. Sobczak, T. J. Bandosz and J. C. Colmenares, Chem. Eng. J., 2020, 395, 125099 CrossRef CAS.
- D. A. Giannakoudakis, M. Jiang and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2016, 8, 31986–31994 CrossRef CAS PubMed.
- D. A. Giannakoudakis, J. Colón-Ortiz, J. Landers, S. Murali, M. Florent, A. V. Neimark and T. J. Bandosz, Appl. Surf. Sci., 2019, 467–468, 428–438 CrossRef CAS.
- D. A. Giannakoudakis, M. Barczak, F. Pearsall, S. O'Brien and T. J. Bandosz, Chem. Eng. J., 2020, 384, 123280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc00329h |
| This journal is © The Royal Society of Chemistry 2020 |