
Conjugated polymers for visible-light-driven photocatalysis
Chunhui
Dai
 and
Bin
Liu
*
and
Bin
Liu
*
Department of Chemical & Biomolecular Engineering, National University of Singapore, 117585, Singapore. E-mail: cheliub@nus.edu.sg
First published on 1st October 2019
Abstract
Conjugated polymers have recently been under active investigation as promising alternatives to traditional inorganic semiconductors for photocatalysis. This is due to their unique advantages of low cost, high chemical stability, and molecularly tunable optoelectronic properties. This critical review summarizes the recent advancements in π-conjugated polymers for visible-light-driven photocatalytic applications including water splitting, CO2 reduction, organic transformation and degradation of organic dyes. Special emphasis is placed on how the changes in the polymer structure could influence their physicochemical properties and photocatalytic activities. This structure–activity relationship analysis should guide rational molecular design of conjugated polymers for improved photocatalytic activity.
 Chunhui Dai | Chunhui Dai obtained his PhD degree (2016) in organic chemistry from the School of Chemistry and Chemical Engineering at Nanjing University, China. He is now a Research Fellow in Prof. Bin Liu's research group at the Department of Chemical and Biomolecular Engineering, National University of Singapore. His research interest is centred on the design and synthesis of conjugated polymers for photocatalytic conversions. |
 Bin Liu | Bin Liu is currently Provost's Chair Professor in the Department of Chemical and Biomolecular Engineering at the National University of Singapore (NUS), where she received her PhD degree in 2001. After postdoctoral work at the University of California, Santa Barbara, she joined NUS in late 2005. Her research focuses on the development of conjugated polymers and organic nanomaterials and exploration of their applications in sensing, imaging and solar energy conversion. |
Broader contextThe exploitation of efficient photocatalysts to directly convert the radiant sunlight into chemical energy is of great importance to address the current energy and environmental challenges. Conjugated polymers (CPs) consisting of photoactive π-systems represent an attractive platform for solar energy utilization. They have been intensively studied for a variety of photocatalytic applications and many exciting performances were reported through facile molecular design. A comprehensive review is thus timely to summarize the progress of the field. This review article systematically presents the recent advances in conjugated polymers for visible-light-driven photocatalysis, including water splitting, CO2 reduction, organic transformation and degradation of organic dyes. The synthesis and design principles of conjugated polymers in these photocatalytic applications are illustrated, with an emphasis on the correlation between polymer structures and their photocatalytic activities. We expect that the systematic discussion in this review will not only provide a general overview of the field, but also promote the development of conjugated polymers with fascinating properties for photocatalysis. |
1. Introduction
The sun is known as a super “energy warehouse” to irradiate solar light continuously to the Earth. On average, the solar energy reaching the Earth's surface per hour is enough to meet the current human energy demand in a whole year.1,2 Despite the advantages of inexpensive, abundant and clean features of sunlight, it is difficult to utilize solar energy very efficiently because of its low energy density as well as diffuse and intermittent nature. Therefore, it is desirable to convert sunlight into chemical energy, which represents an appealing solution to address several challenges facing the world today. For example, clean hydrogen fuel production by photocatalytic water splitting holds great promise to alleviate the global energy crisis, which promises to be clean, renewable and eco-friendly.3–9 Enabling photocatalytic technology will also allow the reduction of CO2 into value-added chemicals (CO, CH4, and CH3OH, etc.), thereby creating huge benefits by simultaneously solving the current energy shortage and environmental issues caused by overloaded CO2 release.10–14 Moreover, light-driven organocatalysis is considered as an attractive strategy for direct chemical bond functionalization with a broad substrate scope under mild conditions, which is a vitally important contribution to organic synthetic methodology.15–20 Last but not least, photocatalytic degradation of organic dyes in wastewater is a powerful technology for environmental remediation.21–24In a photocatalytic process, the photon energy required for photoexcitation is dependent on the optical gap of the photocatalyst. As compared to the UV region that comprises only 5% of solar light, the visible light (400–800 nm) accounts for about 53% of the incident solar energy (Fig. 1).4,25,26 Therefore, it is crucial to develop photocatalysts with optical gap below 3.0 eV to increase the absorption of solar light with a focus on the visible light region.
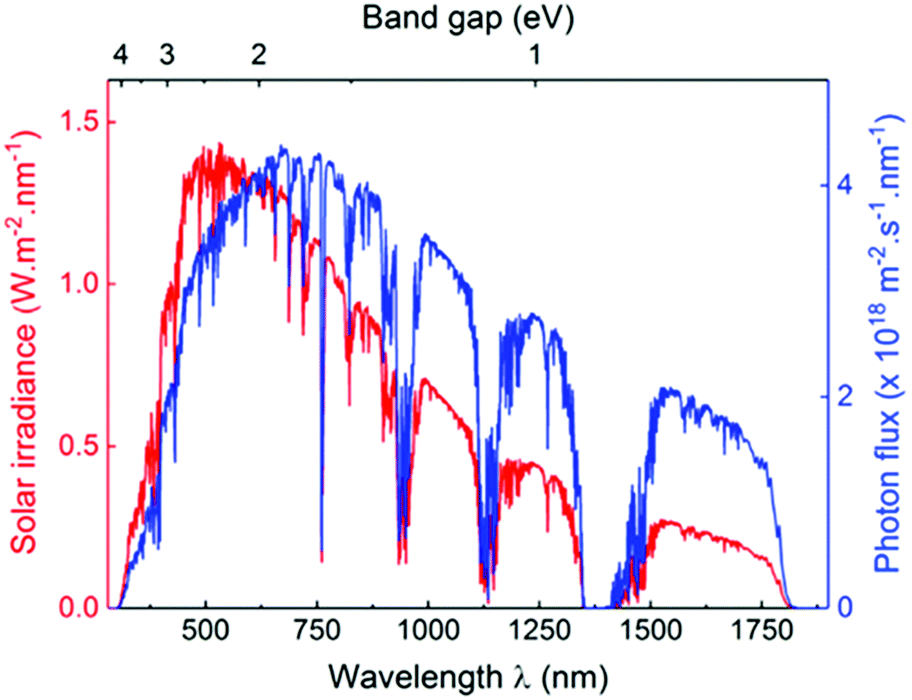 | ||
| Fig. 1 AM1.5 solar light radiation spectrum (red) and photon flux curve (blue) as functions of light wavelength and material band gap. Reprinted with permission from ref. 25. Copyright 2017 Wiley-VCH. | ||
Over the past few decades, research efforts have been intensively devoted to pursuing suitable photocatalysts for practical applications. Among them, organic π-conjugated polymers have recently garnered significant attention as promising candidates for various photocatalytic applications. Many interesting features allow π-conjugated polymers to stand out in the field of photocatalysis over inorganic semiconductors. Firstly, they are environmentally friendly semiconductor materials, consisting of earth-abundant elements, and they possess tunable energy levels for oxidation and reduction reactions. Secondly, they could be facilely prepared under mild conditions and have excellent chemical stability against photobleaching. Thirdly, their molecular structures can be fine-tuned to better utilize visible light, while many traditional inorganic photocatalysts are only UV-active. Last but not least, the π-conjugation along the polymer backbone enables them with unique photogenerated charge carrier separation and transport properties, which is critical to trigger photoredox reactions.
Upon diverse modification of conjugated polymers (CPs) at the molecular level, many exciting achievements in photocatalysis have recently been reported, which clearly justify a comprehensive review on this research topic. So far, most CP based photocatalysis reviews have been based on the family of graphitic carbon nitride (g-C3N4) materials with a focus on one or two photocatalytic applications.18,27–35 There is an urgent need to provide a review focused on π-conjugated polymers in photocatalysis from a broad perspective to present an overview of the recent developments on this research topic.
This review presents the recent advances of CPs in applications of photocatalytic water splitting, CO2 photoreduction, light-driven organic transformation and photocatalytic degradation of organic dyes (Fig. 2). We focus on an in-depth understanding of how the CP structure modification could influence the optical properties, photogenerated charge separation and transport, and photocatalytic activity. The review starts from a brief discussion of general photocatalytic processes and the synthesis of CP photocatalysts. This is followed by the illustration of CPs for each photocatalytic application. Finally, perspectives toward exploring new CPs for photocatalytic applications are proposed, which is expected to stimulate the future development of more exciting CPs for photocatalysis.
 | ||
| Fig. 2 Overview of π-conjugated polymers for various photocatalytic applications, which are summarized in this review. | ||
2. Basic aspects of π-conjugated polymers for photocatalytic applications
2.1 Fundamentals of heterogeneous photocatalysis
The main processes of polymeric semiconductors in specific light-driven redox reactions are described in many reviews3,11,14,27,28,36–39 and the general pathway is briefly illustrated in Fig. 3a. Upon light irradiation, the semiconductor harvests incident photons and charges (electron–hole pairs) are formed inside the photocatalyst. The photogenerated charges subsequently separate and migrate to the semiconductor surface where redox reactions take place. On the other hand, most of the photogenerated electrons and holes could undergo bulk or surface recombination, which are competitive processes to the surface reactions. | ||
| Fig. 3 (a) The main process of polymeric semiconductors for photocatalysis (D: electron donor; A: electron acceptor) and (b) critical factors affecting their photocatalytic performances. | ||
According to the proposed mechanism of photocatalytic redox reactions, an efficient CP photocatalyst should at least have the following properties: (a) a strong and broad absorption in the visible region to effectively capture solar light; (b) high charge separation and migration efficiency for surface redox reactions; (c) appropriate energy level alignments to enable efficient charge separation as well as the reduction or oxidation half reaction. Therefore, light harvesting, energy levels as well as photogenerated charge separation and migration are three key factors affecting the photocatalytic activities of these CPs (Fig. 3b). This requires rational design and synthesis of CPs for effective photocatalysis.
2.2 Synthesis of conjugated polymers for heterogeneous photocatalysis
The extensively available conjugated organic monomers and diverse synthetic methods enable facile synthesis of a broad range of CPs. Moreover, the polymer structures could be precisely controlled for specific photocatalytic applications. Among these methods, Pd-catalyzed Suzuki–Miyaura coupling polymerization between halogenated aromatics and diboronyl ester has been widely used to prepare linear copolymers and conjugated porous polymers (Fig. 4). The reaction is typically carried out in the presence of a base (always K2CO3) under a N2 or Ar atmosphere. Another commonly used method for preparing CPs by C–C coupling is the Sonogashira coupling reactions of aromatic halides and terminal alkynes, in which Pd(PPh3)2Cl2 was employed as the catalyst with an amine as the base, and CuI as the cocatalyst. Furthermore, Ni(0)-promoted Yamamoto dehalogenation coupling reaction is a convenient pathway to synthesize CPs from dibromoaromatic monomers. In this reaction, Ni(COD)2 and 2,2′-bipyridyl are used as the catalyst and promoter for polymerization, respectively. Oxidative coupling reactions are exploited as a versatile methodology for performing self-polymerization of arylethynylene monomers, in which CuBr is used as the catalyst and a base is used as the binding agent. | ||
| Fig. 4 Typical polymerization methods used to synthesize conjugated polymers for photocatalysis. | ||
Interestingly, conjugated polymer nanomaterials could be readily prepared under appropriate reaction conditions. For example, conjugated microporous polymer nanosheets could be obtained by one-step oxidative coupling of aromatic alkynes40 or a condensation reaction.41,42 Conjugated microporous polymer nanoparticles could be directly prepared by polymerization in high internal phase emulsion, which is concentrated emulsion with a dispersed phase volume fraction over 74.05%.43,44 The morphologies of nanoparticles can be varied by introducing monomers with different electron-donating/withdrawing properties.45
Additionally, the condensation reaction between amino and aldehyde groups based on Schiff-chemistry is widely performed to prepare crystalline covalent organic frameworks (COFs) for photocatalysis.51 The reaction is typically performed under harsh experimental conditions with carefully controlled temperature (usually 120 °C), pressure and reaction time (usually 3–5 days). Acetic acid is employed as the catalyst in the reaction. It is believed that an error-correction process occurs during the conversion of amorphous intermediates to crystalline products. The reaction solvent is crucial for the assembly of framework units. In a typical condensation reaction, mesytilene/dioxane and n-butanol/o-dichlorobenzene are commonly used reaction solvents. Besides, a suitable building block is also important to obtain a highly crystalline COF. Rigid and planar π-conjugated units are preferred to prepare 2D COFs.
Covalent triazine frameworks (CTFs) could be prepared by a trimerization reaction of aromatic nitriles in the presence of a Brønsted or Lewis acid catalyst.53 In a typical ionothermal preparation, ZnCl2 is chosen as the catalyst and high temperature (>400 °C) is always required for the reversible reaction. By contrast, a much lower temperature (<100 °C) could be employed when CF3SO3H is used as the catalyst for the condensation reaction. Alternatively, CTFs could be synthesized by condensation of an aldehyde and an amidine dihydrochloride, which involves the generation of a Schiff base followed by Michael addition. The reaction is carried out in DMSO with Cs2CO3 as a base.
Graphitic carbon nitride (g-C3N4) consisting of s-triazine or tri-s-triazine (heptazine) units has been synthesized by thermal polycondensation of nitrogen-rich precursors, such as melamine, thiourea, urea, dicyanadiamide and cyanamide, etc.18,29,31 For more detailed preparation of the above-mentioned CPs, please refer to other reviews46–53 and books.54,55
3. Conjugated polymers for photocatalytic water splitting
Photocatalytic water splitting is of particular interest as the technology provides an effective pathway for clean hydrogen production using water and sunlight, both of which are abundant in nature.In a light-driven water splitting process, photogenerated electrons and holes were used for the reduction and oxidation of water, respectively. The photocatalytic reaction is a thermodynamically uphill reaction and a Gibbs free energy (ΔG0) of +237.2 kJ mol−1 is required.3 In principle, to drive the redox half reaction, the energy level alignments of the polymer photocatalyst and the redox potential of water must be well matched. This means that the lowest unoccupied molecular orbital (LUMO) of the polymer photocatalyst should be more negative than the reduction potential of H+/H2 (0 eV vs. normal hydrogen electrode (NHE), pH = 0), while its highest occupied molecular orbital (HOMO) must be more positive than the oxidation potential of O2/H2O (1.23 eV vs. NHE, pH = 0). Therefore, the band gap (Eg) of the polymer should be over 1.23 eV to trigger water splitting. In this part, we summarize the state-of-the-art advancements in photocatalytic H2 evolution using various types of organic CPs, and their applications in photocatalytic O2 evolution and overall water splitting are also briefly described.
3.1 Photocatalytic H2 evolution
Hydrogen is an ideal fuel with gravimetric energy density up to 142 MJ kg−1, which is almost three times larger than that of gasoline.56 More importantly, H2 combustion produces water with no pollution to the environment. Photocatalytic hydrogen evolution using semiconductors is a sustainable and economical technology to convert solar energy into hydrogen energy. Since Honda and Fujishima reported water splitting on TiO2 photoelectrodes in 1972, a huge number of semiconductors have been studied for hydrogen production.3,34,57–64As promising photocatalysts, CPs have been employed for hydrogen production in the early 1980s. Unfortunately, they haven’t attracted much attention until three-dimensional (3D) poly(azomethine) networks were demonstrated to be efficient photocatalysts for hydrogen production from water in 2010.65 Since then, various types of organic CPs have been developed to yield greatly improved H2-production efficiency. This was achieved through the optimization of CP photophysical properties, mainly light harvesting in the visible region, band gap alignment, as well as photogenerated charge generation and transport, etc.
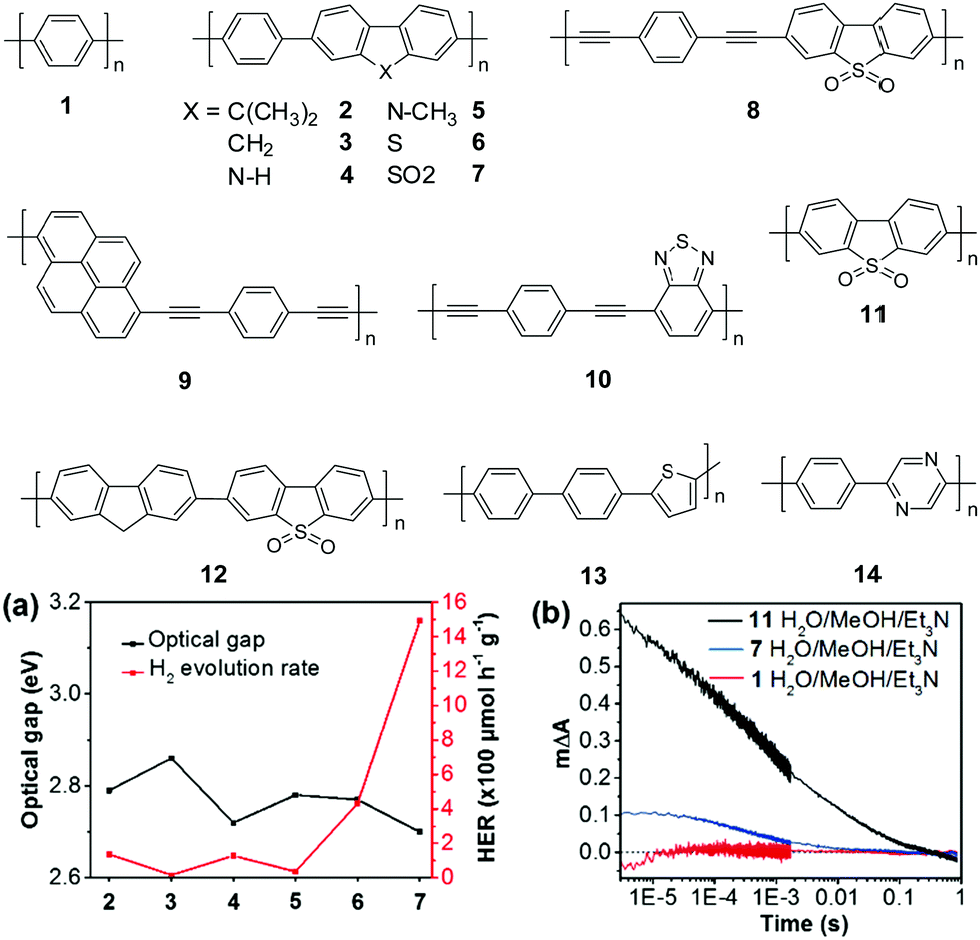 | ||
| Fig. 5 Chemical structures of polymers 1–14, (a) the correlation between the photocatalytic H2 evolution and their optical gaps of polymers 2–7 (>420 nm); (b) transient kinetics probed at 630 nm for polymers 1, 7, and 11 in the Et3N/CH3OH/H2O mixture (excited at 355 nm). Reprinted with permission from ref. 72. Copyright 2018 Nature Publishing Group. | ||
In 2016, Cooper and co-workers demonstrated the new breakthrough in H2 evolution by incorporating extended planarized units into the main chain of 1, which is expected to facilitate the photogenerated charge dissociation and thus promote the proton reduction reaction.70 Polymers 2–7 show band gaps ranging from 2.70 to 2.86 eV (Fig. 5a). Among them, polymers 2 and 4 exhibited an almost equal HER of ∼128 μmol h−1 g−1, which was 3-fold improvement over 1. More excitingly, polymer 7 with the lowest band gap of 2.70 eV exhibited an impressive HER of 1492 μmol h−1 g−1, which was 46-fold higher than that of 1 (Table 1).
| Polymer | Optical gap (eV) | S BET (m2 g−1) | Cocatalyst | Sacrificial agent | Light sourcea | HER (μmol h−1 g−1) | AQYb (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Xe: xenon lamp. b At 420 nm. NA: no activity; AQY: apparent quantum yield; HER: hydrogen evolution rate; DEA: diethylamine; TEA: trimethylamine; TEOA: triethanolamine; AA: ascorbic acid. | ||||||||
| 1 | 2.9 | — | — | DEA | >400 nm (Xe) | Trace | — | 66 |
| 2 | 2.79 | — | — | TEA | >420 nm (Xe) | 136 | — | 70 |
| 3 | 2.86 | — | — | TEA | >420 nm (Xe) | 16 | — | 70 |
| 4 | 2.72 | — | — | TEA | >420 nm (Xe) | 128 | — | 70 |
| 5 | 2.78 | — | — | TEA | >420 nm (Xe) | 36 | — | 70 |
| 6 | 2.77 | — | — | TEA | >420 nm (Xe) | 432 | 1.1 | 70 |
| 7 | 2.70 | — | — | TEA | >420 nm (Xe) | 1492 | 2.3 | 70 |
| 8 | 1.89 | — | — | TEOA | >420 nm (Xe) | 6023 | 4.2 | 71 |
| 9 | 1.64 | — | — | TEOA | >420 nm (Xe) | 4270 | 0.85 | 71 |
| 10 | 1.94 | — | — | TEOA | >420 nm (Xe) | 3187 | — | 71 |
| 11 | 2.62 | — | — | TEA | >420 nm (Xe) | 3260 | 11.6 | 72 |
| 12 | 2.07 | — | — | TEA | >420 nm (Xe) | 5040 | 2.13 | 74 |
| 13 | 2.42 | 17 | — | TEA | >420 nm (Xe) | 420 | — | 76 |
| 14 | 2.45 | 69 | — | TEA | >420 nm (Xe) | 960 | — | 77 |
| 15 | 2.38 | — | — | AA | >420 nm (LED) | 8300 | ∼0.73 | 82 |
| 16 | 1.98 | — | — | AA | >420 nm (LED) | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
∼0.88 | 83 |
| 17 | 2.46 | — | — | AA | >420 nm (LED) | NA | — | 83 |
| 18 | 2.03 | — | — | DEA | >420 nm (LED) | 12700 |
— | 84 |
| 19 | 2.11 | — | — | TEA | >420 nm (Xe) | 512 | — | 89 |
| 20 | — | — | Pt | AA | >420 nm (Xe) | 12800 |
0.13 | 90 |
| 21 | 2.33 | 995 | — | DEA | >420 nm (Xe) | 174 | — | 96 |
| 22a | 1.81 | 39 | — | TEA | >400 nm (Xe) | 17 | — | 105 |
| 22b | 1.95 | 59 | — | TEA | >400 nm (Xe) | 140 | — | 105 |
| 23a | 2.89 | 228 | — | TEA | >400 nm (Xe) | 397 | — | 105 |
| 23b | 2.45 | 279 | — | TEA | >400 nm (Xe) | 1460 | — | 105 |
| 24a | 2.31 | 121 | — | TEA | >400 nm (Xe) | 260 | — | 105 |
| 24b | 2.19 | 99 | — | TEA | >400 nm (Xe) | 786 | — | 105 |
| 25a | 2.33 | 80 | — | TEA | >400 nm (Xe) | 414 | — | 105 |
| 25b | 2.06 | 104 | — | TEA | >400 nm (Xe) | 1900 | — | 105 |
| 26 | 1.88 | — | — | TEA | >400 nm (Xe) | 2590 | — | 106 |
| 27 | 2.44 | 280 | Pt | TEOA | >420 nm (Xe) | 400 | — | 107 |
| 28 | 2.18 | 58 | — | TEOA | ≥420 nm (Xe) | 9600 | 1.8 | 110 |
| 29 | 2.19 | 499 | Pt | TEOA | >420 nm (Xe) | 30 | — | 111 |
| 30 | 2.21 | 669 | Pt | TEOA | >420 nm (Xe) | 134 | — | 112 |
| 31 | 2.28 | 750 | Pt | TEOA | >420 nm (Xe) | 598 | — | 112 |
| 32 | 2.14 | 564 | Pt | TEOA | >420 nm (Xe) | 908 | 2.0 | 112 |
| 33 | 2.37 | 834 | Pt | TEOA | >420 nm (Xe) | 620 | — | 112 |
| 34 | 2.53 | 811 | — | TEOA | >420 nm (Xe) | 2460 | — | 113 |
| 35 | 2.62 | 915 | — | TEOA | >420 nm (Xe) | 188 | — | 113 |
| 36 | 2.64 | 447 | — | TEOA | >420 nm (Xe) | 116 | — | 113 |
| 37 | 2.84 | 895 | — | TEA | >420 nm (Xe) | 120 | — | 114 |
| 38 | 2.56 | 431 | — | TEA | >420 nm (Xe) | 3106 | 13.2 | 115 |
| 39 | 2.37 | 723 | — | TEOA | >420 nm (Xe) | 5697 | — | 116 |
| 40 | 2.11 | 692.5 | Pt | TEOA | >420 nm (Xe) | 2103.2 | 6.4 | 117 |
| 41 | 2.8 | 1603 | Pt | TEOA | >420 nm (Xe) | 1970 | — | 119 |
| 42 | 2.67 | 702 | Pt | TEOA | 420–900 nm (Xe) | 22.5 | — | 120 |
| 43 | 2.68 | 326 | Pt | TEOA | 420–900 nm (Xe) | 90 | 0.08 | 120 |
| 44 | 2.62 | 1537 | Pt | TEOA | 420–900 nm (Xe) | 438 | 0.19 | 120 |
| 45 | 2.65 | 1046 | Pt | TEOA | 420–900 nm (Xe) | 1703 | 0.44 | 120 |
| 46 | 2.10 | 985 | Pt | AA | >420 nm (Xe) | 4440 | — | 121 |
| 47 | 1.85 | 1288 | Pt | AA | >420 nm (Xe) | 10100 |
— | 121 |
| 48 | 2.28 | 919 | Pt | AA | >420 nm (Xe) | 1600 | — | 121 |
| 49 | 2.34 | 523 | Pt | TEOA | ≥395 nm (Xe) | 30 | — | 122 |
| 50 | 2.31 | 758 | Pt | TEOA | ≥395 nm (Xe) | 324 | 1.8 | 122 |
| 51 | 2.09 | 541 | Pt | TEOA | >420 nm (Xe) | 8300 | — | 123 |
| 52 | 2.06 | 348 | Pt | TEOA | >420 nm (Xe) | 1560 | — | 123 |
| 53 | 1.92 | 162 | Pt | TEOA | >420 nm (Xe) | 220 | — | 123 |
| 54 | 2.03 | 602 | Pt | TEOA | >420 nm (Xe) | 1360 | — | 124 |
| 55 | 2.94 | 19 | Pt | TEOA | ≥420 nm (Xe) | 200 | — | 128 |
| 56 | 2.38 | 52 | Pt | TEOA | >420 nm (Xe) | 6600 | 7.3 | 130 |
| 57 | 2.22 | 807 | Pt | TEOA | >420 nm (Xe) | 1238 | — | 132 |
| 58 | 2.13 | 764 | Pt | TEOA | >420 nm (Xe) | 1582 | — | 132 |
| 59 | 2.33 | 663 | Pt | TEOA | >420 nm (Xe) | 1460 | — | 132 |
| 60 | 2.42 | 757 | Pt | TEOA | >420 nm (Xe) | 2647 | — | 132 |
| 61 | 2.17 | 569 | Pt | TEOA | >420 nm (Xe) | 10760 |
4.07 | 133 |
| 62 | 2.47 | 533 | Pt | TEOA | >420 nm (Xe) | 5320 | 4.11 | 133 |
| 63 | 2.67 | 650 | Pt | TEOA | >420 nm (Xe) | 1440 | 2.10 | 133 |
| 64 | — | 599 | Pt | TEOA | >420 nm (Xe) | 5100 | — | 138 |
| 65 | — | 520 | Pt | TEOA | >420 nm (Xe) | 2400 | — | 138 |
| 66 | — | 598 | Pt | TEOA | >420 nm (Xe) | 650 | — | 138 |
| CN–F | 2.63 | — | Pt | TEOA | >420 nm (Xe) | ∼130 | — | 142 |
| CN–N | 2.65 | 9.21 | Pt | TEOA | >400 nm (Xe) | 553.5 | — | 143 |
| CN–C/P | 2.98 | 141.1 | Pt | TEOA | >420 nm (Xe) | 1493.3 | 2.14 | 144 |
| CN–BA | 2.57 | 22 | Pt | TEOA | >420 nm (Xe) | 294 | — | 145 |
| CN–P15 | — | — | — | TEOA | >420 nm (Xe) | 722.3 | — | 152 |
| CN–P17 | 2.24 | — | — | TEOA | >400 nm (Xe) | 929.3 | 5.7 | 153 |
| CN–PyP | 2.46 | 120 | — | TEOA | ≥420 nm (Xe) | 600 | — | 154 |
| CN–COF | — | 85.2 | Pt | TEOA | >420 nm (Xe) | 10100 |
— | 155 |
| CN–CTF | — | — | Pt | TEOA | ≥420 nm (Xe) | 850 | — | 156 |
Encouraged by the good activity of polymer 7, Chen et al. further modified this polymer by introducing an ethynyl group into the main chain, which led to a reduced band gap and accelerated charge separation of the resulting polymers. Polymer 8 has a band gap of 1.89 eV and showed a HER of up to 6023 μmol h−1 g−1 when triethanolamine (TEOA) was used as the sacrificial agent (>420 nm).71 Similarly, compared to their counterparts without ethynyl insertion, polymers 9 and 10 showed large red shifts of 167 and 154 nm in their light absorption edges, respectively. The HER was enhanced to 4270 μmol h−1 g−1 for 9 and 3187 μmol h−1 g−1 for 10.
Notably, dibenzo[b,d]thiophene sulfone has been recognized as one of the most attractive building blocks for the synthesis of H2-production polymer photocatalysts. As shown above, many polymers with incorporated sulfone units show outstanding photocatalytic performances. To study the critical role of the sulfone unit in H2 evolution, Cooper and co-workers investigated the photophysical properties of polymers 1, 7 and 11.72 Compared with 1 and 7, the homopolymer of sulfone 11 showed a much higher HER of 3260 μmol h−1 g−1. The decay kinetics at 630 nm exhibited a significantly larger signal for 11 than 7, while almost no signal was observed for 1 (Fig. 5b). This result indicated a higher polaron concentration of 11 and 7, which is in line with the H2 evolution activities of the three polymers. Recently, they further prepared nanoparticles of about 160 nm using polymer 11 by mini-emulsion polymerization.73 With a polymer concentration of 0.1 mg mL−1, the nanoparticle suspension yielded a HER of 14.52 mmol h−1 g−1, while a significantly lower HER of 6.13 mmol h−1 g−1 was measured for the bulk material of 11, evidently suggesting a size-dependent H2 evolution of the polymer. Moreover, when the polymer concentration was deceased to 13 μg mL−1, the suspension gave a superior average HER of 60.6 mmol h−1 g−1 over 5 h, which is the highest value among the reported organic polymers so far.
To further extend the conjugation degree of polymer 7, we introduced large planar units (fluorene and pyrene) into the main chain.74 Polymer 12 exhibited a smaller band gap of 2.07 eV in comparison with 7 (2.70 eV) and a good HER of 5040 μmol h−1 g−1 was achieved. Very recently, Wang et al. reported an analogue of polymer 12 by replacing the fluorene in the main chain with electron-rich dibenzothiophene to construct a local D–A heterojunction, which led to a reduced exciton binding energy from 91 to 88 meV and 1.3-fold HER as compared to 12 in the presence of TEOA.75
Another modification of PPP (1) is the copolymerization of 2,5-phenylene with heterocycles, which results in optimized band gaps and energy levels of the as-prepared polymers. A maximum HER enhancement of 6.5-fold compared to PPP was achieved by polymer 13 with 33% thiophene fraction and a band gap of 2.42 eV.76 Polymer 14 bearing a 2,5-pyrazine attained a 15-fold higher HER than PPP.77
Compared to those insoluble bulk polymers, the development of soluble CPs provides a good choice for easy processing of polymers into a desirable pattern for photocatalytic H2 production. By virtue of its inherent hydrophobicity, CP could be self-assembled into polymer dots (Pdots) in water.78–80 Pdots have much smaller size relative to the bulk materials; hence, the distance covered by photogenerated charges migrating to the surface of photocatalyst particles becomes short, and the recombination probability of electrons and holes decreases to improve the photocatalytic efficiency.81 In 2016, Tian et al. prepared a Pdot suspension by the nano-precipitation method using polymer 15 and a matrix polymer PS–PEG–COOH.82 In the preparation, the mixture of polymer and matrix in THF was injected into pure water under sonication, and a clear Pdot suspension was obtained after THF evaporation (Fig. 6). The as-prepared Pdots displayed an impressive initial rate of 8300 μmol h−1 g−1. Compared to polymer 15, Pdots of polymer 16 with extended absorption renders an excellent initial HER of up to 50 mmol h−1 g−1.83 The theoretical calculations suggested that N atoms in the 2,1,3-benzothiadiazole (BT) units provided the reactive sites in the formation of hydrogen, polymer 17 without a BT unit did not show any activity although its optical gap and reduction potential are appropriate for proton reduction. Using the nano-precipitation method, Chou et al. prepared cycloplatinated Pdots using polymer 18, which provided an impressive HER of 12.7 mmol h−1 g−1, while only 1.3 and 5.46 mmol h−1 g−1 were obtained for Pdots without a Pt complex and their Pt-complex-blended counterpart under identical conditions, respectively.84 This was likely attributed to the improved charge transfer after grafting the Pt complex into the polymer backbone.
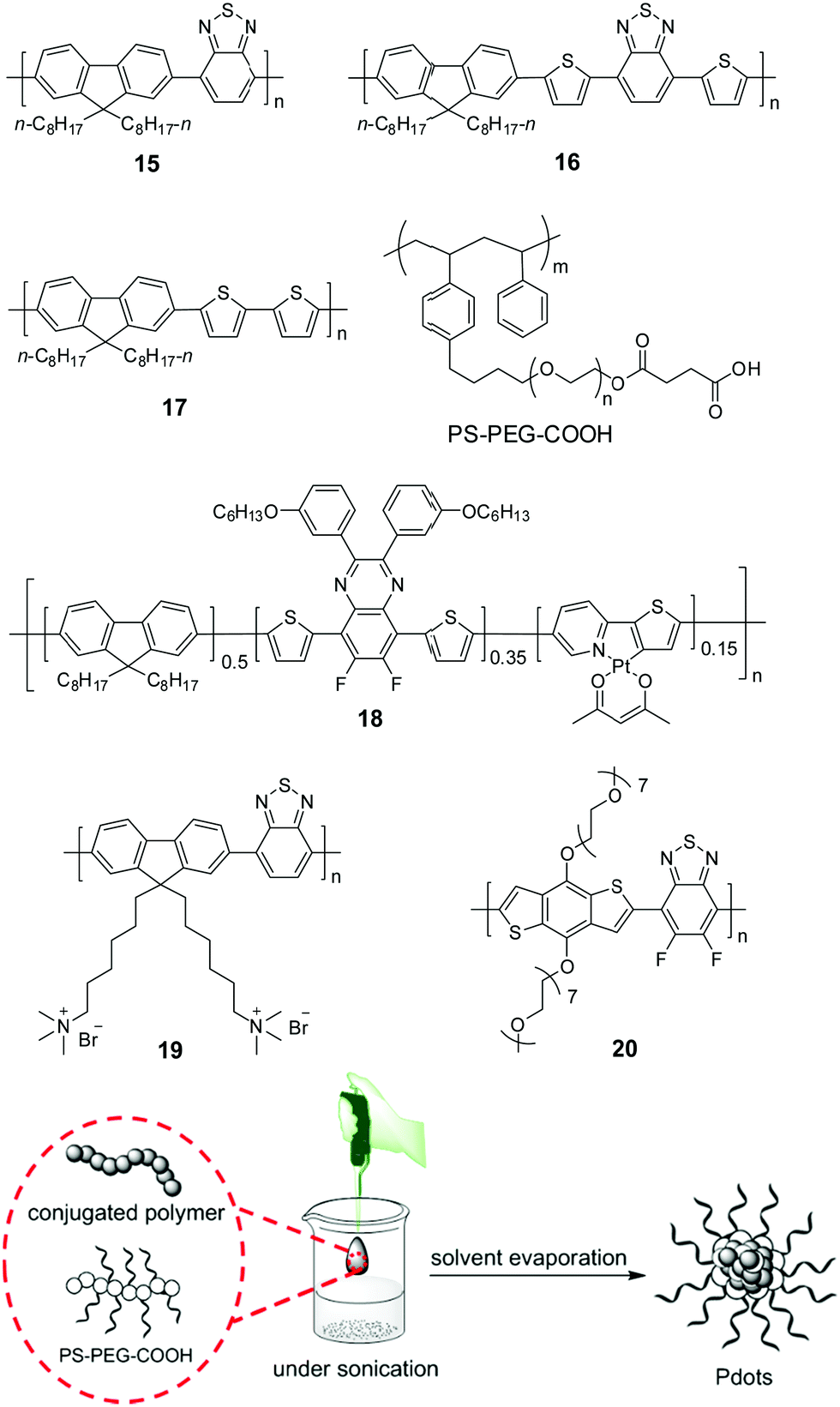 | ||
| Fig. 6 Chemical structures of polymers 15–20 and preparation of polymer dots (Pdots) by the nano-precipitation method for photocatalytic H2 production. The scheme is adapted with permission from ref. 82, Copyright 2016 Wiley-VCH. | ||
Conjugated polyelectrolytes (CPEs) are a subclass of interesting water-soluble CPs with wide applications in photovoltaic devices, biomedical imaging and therapy, as well as biosensing, etc.85–88 Their rigid conjugated structures endow them with excellent optoelectronic properties, while the hydrophilic side chains with charged groups allow for unique dispersibility of CPEs in polar solvents. We prepared cationic CPE 19 by one-step quaternization of the precursor polymer attaching a hexyl bromide side chain.89 In the mixture of Et3N/CH3OH/H2O, polymer 19 could self-assemble into homogeneous nanoparticles of ∼180 nm. The suspension gives a HER of 512 μmol h−1 g−1, which is 3-fold that of its counterpart without the side chain. The enhanced HER was attributed to the improved light absorption and charge separation of the nanoparticles.
Different from the cationic CPEs, attaching oligoethylene glycol (OEG) side chains endows polymer 20 with more interesting functions.90 The O atom in the OEG side chain favours the interface contact of the polymer backbone with water, which resulted in a much smaller water contact angle of 20 (27.5°) than that of an alkyl-functionalized polymer (101.2°). Moreover, the side chains could interact with Pt, resulting in more efficient charge transport from the polymers to Pt. Polymer 20 loaded with 3 wt% Pt gave a HER of 12.8 mmol h−1 g−1, which is much higher than its alkyl-attached counterpart.
Besides, soluble CPs could also be processed into films for photocatalytic H2 production. By drop-casting, Cooper's group prepared a film of polymer 4 attaching a 2-ethylhexyl side chain. The film demonstrated a HER of 72 μmol h−1 g−1 when immersed in the Et3N/CH3OH/H2O mixture (>420 nm).91
The statistical copolymerization of aromatic monomers was reported as an important strategy to enhance the catalytic activity of CPPs by improving their physical properties such as band gap, porosity and charge mobility, etc. For instance, Cooper and co-workers synthesized fifteen conjugated microporous polymers (CMPs) by adjusting the feed ratio of the four comonomers shown in Fig. 7.96 The CMPs have high surface areas of 597–1710 m2 g−1 and their band gaps gradually decrease from 2.95 to 1.94 eV with the increase of pyrene content in the polymer. Without adding a metal cocatalyst, the polymers showed gradually enhanced HER when the band gap decreased from 2.95 to 2.33 eV. With the ratio of 1,4-benzene diboronic acid and 1,2,4,5-tetrabromobenzene of 2:1, polymer 21 exhibited the highest HER of 174 μmol h−1 g−1. However, further narrowing the band gap of polymer led to a decrease in the H2 production. This was likely ascribed to the increased nonradiative recombination of electron and hole in the pyrene-rich polymers.
 | ||
| Fig. 7 Synthesis of conjugated microporous polymers with varying ratios of benzene and pyrene as well as the correlation of optical gap (red line) and hydrogen production (black line) as a function of increasing pyrene content in the polymer backbone. | ||
Similarly, Jiang et al. synthesized D–π–A CPPs using pyrene as a donor, benzothiadiazole as an acceptor, and benzene or biphenyl as a π-crosslinker unit.97 Compared to D–A polymers (without π unit), the extended π-conjugation endows the D–π–A polymers with more efficient charge migration to the surface of polymer particles. In addition, the electron push–pull interaction in the D–π–A polymer backbone could facilitate charge separation in the photocatalytic process. Thereby, D–π–A polymers exhibited better hydrogen production than the D–A and D–π–D polymers (without acceptor). When the ratio of pyrene to benzothiadiazole was 9:2, a maximum HER of 296 μmol h−1 g−1 was obtained (>420 nm). Xiang et al. developed a series of CPPs by copolymerizing 1,3,6,8-tetrabromopyrene and 3,8-dibromophenanthroline. A remarkable HER of 4200 μmol h−1 g−1 was achieved by the polymer having a 3:1 ratio for 1,3,6,8-tetrabromopyrene and 3,8-dibromophenanthroline.98 In addition, Wang et al. further modified polymer 21 by gradually replacing the benzene units in the polymer network with dibenzo[b,d]thiophene sulfone units.99 They claimed that the sulfone unit could act as an electron-output “tentacle” to trap photogenerated electrons from polymers, which is beneficial for the proton reduction reaction. The HERs of the resulting polymers were significantly enhanced with increasing content of sulfone unit in the polymer. This molecular modification provided a maximum HER of 400 μmol h−1.
The donor–acceptor combination approach is one of the most prevailing methods used in polymer solar cells to enhance the power conversion efficiency.100–104 The alternating electron-rich donor (D) and electron-deficient acceptor (A) in the main chain could induce intramolecular charge transfer (ICT) interactions between D and A units, resulting in a narrowing of the band gap and molecular energy level modulation. Owing to this, the D–A approach has attracted a great deal of interest in designing CPs for photocatalytic H2 evolution.
In 2016, Yu et al. reported CPPs by the combination of various chromophores (M1–M4) with biphenyl and bipyridyl, respectively.105 With the chromophores from strong acceptors (M1) to strong donors (M4), the band gaps could be fine-tuned from 1.81 to 2.89 eV for 22a–25a. The replacement of biphenyl (weak donor) with bipyridyl (weak acceptor) led to lower band gaps of 2.06–2.45 eV for 23b–25b (Fig. 8a). The overall H2 evolution of bipyridyl containing polymers is better than that of biphenyl containing polymers (Fig. 8b). Polymer 22b gave the highest HER of 1900 μmol h−1 g−1 (>400 nm) as a result of the improved light absorption, wettability and charge separation. Later, a higher HER of 2590 μmol h−1 g−1 was obtained by polymer 26 through further optimizing nitrogen-containing acceptors in M4 based polymers.106
 | ||
| Fig. 8 Chemical structures of D–A type CPPs and (a) the optical gaps as well as (b) photocatalytic H2 production of polymers 22–25 (P-a: 22a–25a; P-b: 22b–25b). Reprinted with permission from ref. 105. Copyright 2016 American Chemical Society. | ||
2,1,3-Benzothiadiazole (BT) is a strong acceptor unit used in designing D–A copolymers. Wang and co-workers demonstrated that the substitution position of BT on the central phenyl unit has a profound effect on the porosity and morphology of the resulting polymers.107 CPPs with 1,2,4,5 and 1,2,4-substitution exhibited a fused particle-like shape and a fibre shape was observed for their counterpart with 1,3,5-substitution, while the BET surface areas of the three polymers ranged from 40 to 280 m2 g−1 with optical gaps of 2.25–2.44 eV. Consequently, polymer 27 loaded with 3 wt% Pt yielded a HER of 400 μmol h−1 g−1, while 8 μmol h−1 g−1 and 26 μmol h−1 g−1 were observed for the polymers with BT at the 1,2,4 and 1,3,5-positions of the central phenyl ring, respectively. Interestingly, compared to those 3D CPPs, a 1D linear polymer consisting of alternating phenyl and benzothiadiazole units showed a much higher HER of 2320 μmol h−1 g−1, which was ascribed to its narrowest band gap (2.17 eV) and improved charge transport in the polymer.
Fluorine (F) substitution on the CP backbones is an effective way to engineer their optoelectronic properties.108 The van der Waals radii of F atom is only 1.35 Å, which is close to that of the hydrogen atom (1.2 Å).109 Besides, with a Pauling electronegativity value of up to 4.0, the F atom tends to impair the electron density in the D–A type CPs. As a consequence, the introduction of F does not cause a significant steric hindrance, but has a profound influence on the molecular orbits of the polymers.
Zhu and co-workers studied the F substitution effect in the case of BT incorporated linear CPs and conjugated porous polymers on their photocatalytic activities.110 It was found that F substitution had a weak influence on the optical gaps of polymers but the charge transfer efficiency was improved significantly. With the increase of F atom number in BT from 0 to 2, the HOMO and LUMO levels of polymers decrease simultaneously. Polymer 28 incorporating CH3O and F substituted BT units showed a HER of 9.6 mmol h−1 g−1, which was 28.8-fold as large as that of its counterpart without fluorination. The F substitution was also examined by Wang and co-workers.111 They found that the F substitution on BT led to a more negative LUMO level, which could increase the driving force for proton reduction. In this work, polymer 29 showed a 5-fold enhancement of HER compared to its non-fluorinated counterpart.
In general, extending the π-conjugation along the skeleton had multiple impacts on the morphologies, porous properties and photophysical properties of the resulting polymers, which have a synergistic effect on the photocatalytic performances of the CPs. For example, Zhang and co-workers examined the π-extended conjugation effect of polymers 30–33 on their H2 production performances.112 The spacer change led to a profound effect on the porosity of the polymers. Polymer 30 with a small para-phenylene spacer has micropores, while both micro- and mesopores were found for 31 and 33 with a longer spacer. By contrast, polymer 32 showed a much broader pore size distribution, which was due to the longest polyphenylene spacer in the polymer network. As a result, the BET surface areas were 669, 750, 564, and 834 m2 g−1 for 31, 32, 33, and 34, respectively. In addition, the four polymers showed different energy level alignments due to the donor (arene units) and acceptor (tricyanomesitylene) interactions (Fig. 9a). The HERs were found to be 134, 598, 908, and 620 μmol h−1 g−1 for 30, 31, 32, and 33, respectively. Despite the similar band gap, the highest photocatalytic activity of 33 as compared to 30 and 31 was ascribed to its nanoparticle morphology and the largest surface area.
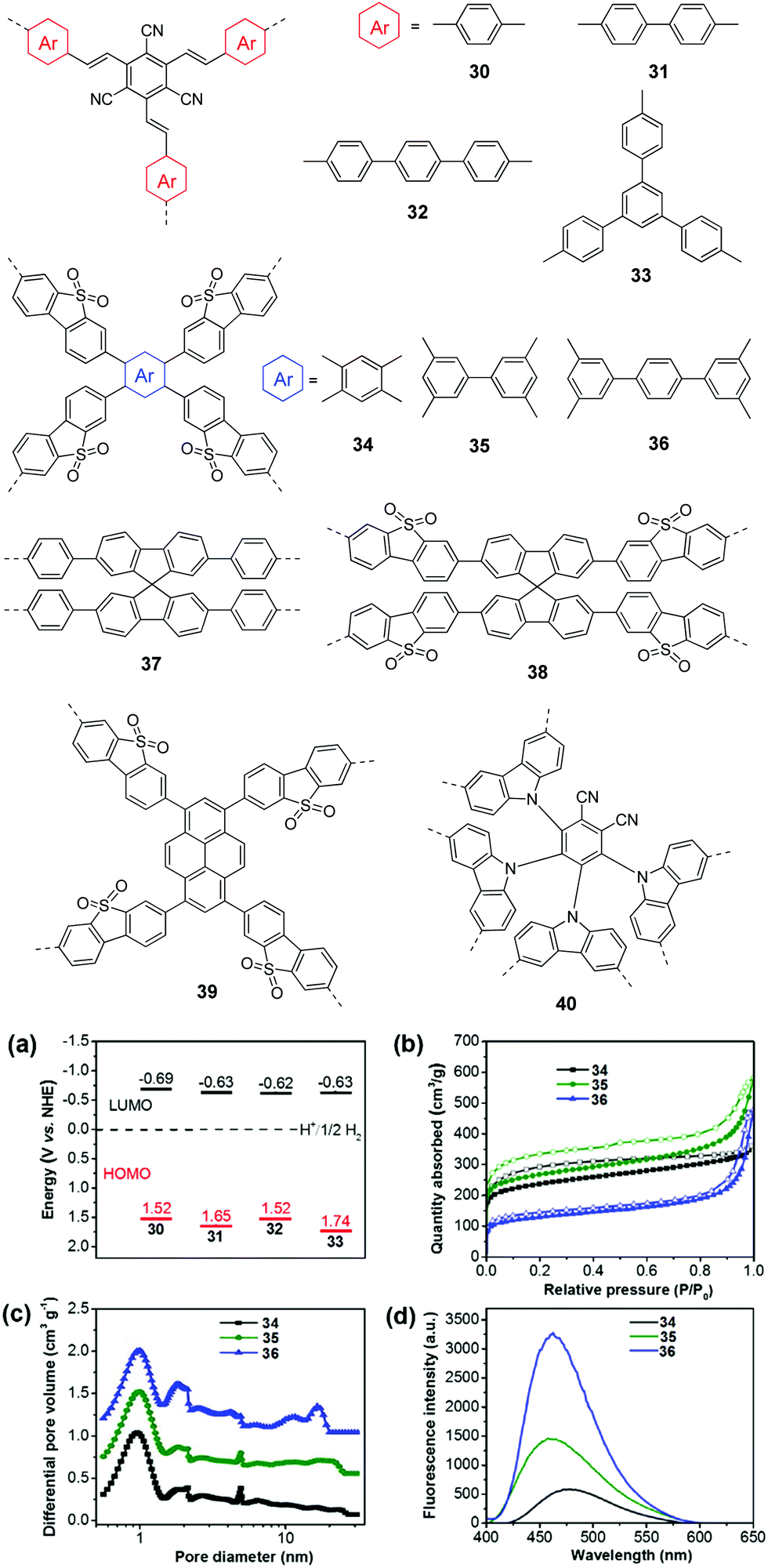 | ||
| Fig. 9 Chemical structures of polymers 30–40. (a) HOMO and LUMO band positions of polymers 30–33. (b) Nitrogen sorption isotherms, (c) photoluminescence spectra, and (d) hydrogen evolution of polymers 34–36. Reprinted with permission from ref. 112 and 113. Copyright 2017 Wiley-VCH and 2016 American Chemical Society. | ||
Similarly, Jiang et al. developed polymers 34–36 by changing the central π-linker from benzene to p-terphenyl.113 Because of the higher steric hindrance of the biphenyl than phenyl, 34 showed a smaller BET surface area of 811 m2 g−1 than 35 (915 m2 g−1), while 36 with the longest linker showed the lowest surface area of 447 m2 g−1 (Fig. 9b). Moreover, the introduction of the extended linker seemed to favour the formation of larger pores. The three polymers showed a similar pore distribution but more mesopores or macropores were found for 35 and 36 (Fig. 9c). In addition, polymer 34 has a smaller band gap of 2.53 eV than 35 (2.62 eV) and 36 (2.64 eV). Polymer 34 exhibits better charge separation efficiency as evidenced by a much lower emission than that of 35 and 36 (Fig. 9d). Accordingly, polymer 34 yielded the highest HER of 2460 μmol h−1 g−1, while HERs of 188 and 116 μmol h−1 g−1 were obtained by 35 and 36, respectively.
Cooper et al. found that the optical gaps of the CMPs could be tuned by changing the monomer linker length and type of linkage, which led to different H2 evolution activities.114 With spirobifluorene as the core, polymer 37 exhibited a 15-fold HER as compared to that of the analogue with phenyl as the core. In addition, by extending the central core from phenyl to spirobifluorene, polymer 38 was reported with an improved HER of 3106 μmol h−1 g−1.115
Jiang et al. demonstrated that the substitution position of dibenzo[b,d]thiophene sulfone had a significant influence on the photocatalytic performances.116 They prepared two CMPs by linking pyrene at the 3,7 and 2,8 positions of sulfone, respectively. Polymer 39 with the 3,7-linking pattern showed a narrower optical gap and lower emission intensity than the other polymer, which indicated better photogenerated charge migration and thus led to a much higher HER of 8523 μmol h−1 g−1 than that of the 2,8-linked polymer (2650 μmol h−1 g−1). Similarly, Su and co-workers found that substitution positions of CN groups and carbazole on the central phenyl ring could affect spectral absorption and charge separation of the resulting polymers. With an optimized structure, polymer 40 showed an excellent HER of 2103.2 μmol h−1 g−1 and 6.4% apparent quantum yield at 420 nm.117
In an early contribution, Lotsch et al. synthesized COF 41 (TFPT-COF) by a condensation reaction of 2,5-diethoxy-terephthalohydrazide and 1,3,5-tris(4-formylphenyl)triazine in a 1,4-dioxane/mesitylene mixture.119 COF 41 showed a honeycomb-type layered structure with mesopores of 3.8 nm and its BET surface area was up to 1603 m2 g−1. Pt-loaded COF 41 could produce H2 from water at a constant rate of 1970 μmol h−1 g−1. Subsequently, they reported a series of azine-linked COFs 42–45 (Nx-COFs, x = 0–3) with different N numbers in the central aryl ring (Fig. 10).120 The molecular design is based on the varied dihedral angles between the central aryl ring and peripheral phenyl rings by replacing the H atom (green dots) with a N atom (Fig. 11a), which led to different planarity of the platform and thus varied crystallinity and porosity of the resulting COFs. As displayed in Fig. 11b, compared to 42 (N0-COF) and 43 (N1-COF), 44 (N2-COF) and 45 (N4-COF) have better crystallinity as revealed by their sharper PXRD peaks, which is beneficial for charge migration in the photocatalytic reaction. Besides, the four COFs showed close band gaps of 2.6–2.7 eV, but different BET areas of 326 to 1536 m2 g−1. The HERs of Pt-modified 42, 43, 44 and 45 were 23, 90, 438 and 1703 μmol h−1 g−1, respectively. The progressively enhanced HER was likely attributed to the increased surface area and improved charge migration when the number of N atoms was increased in the frameworks.
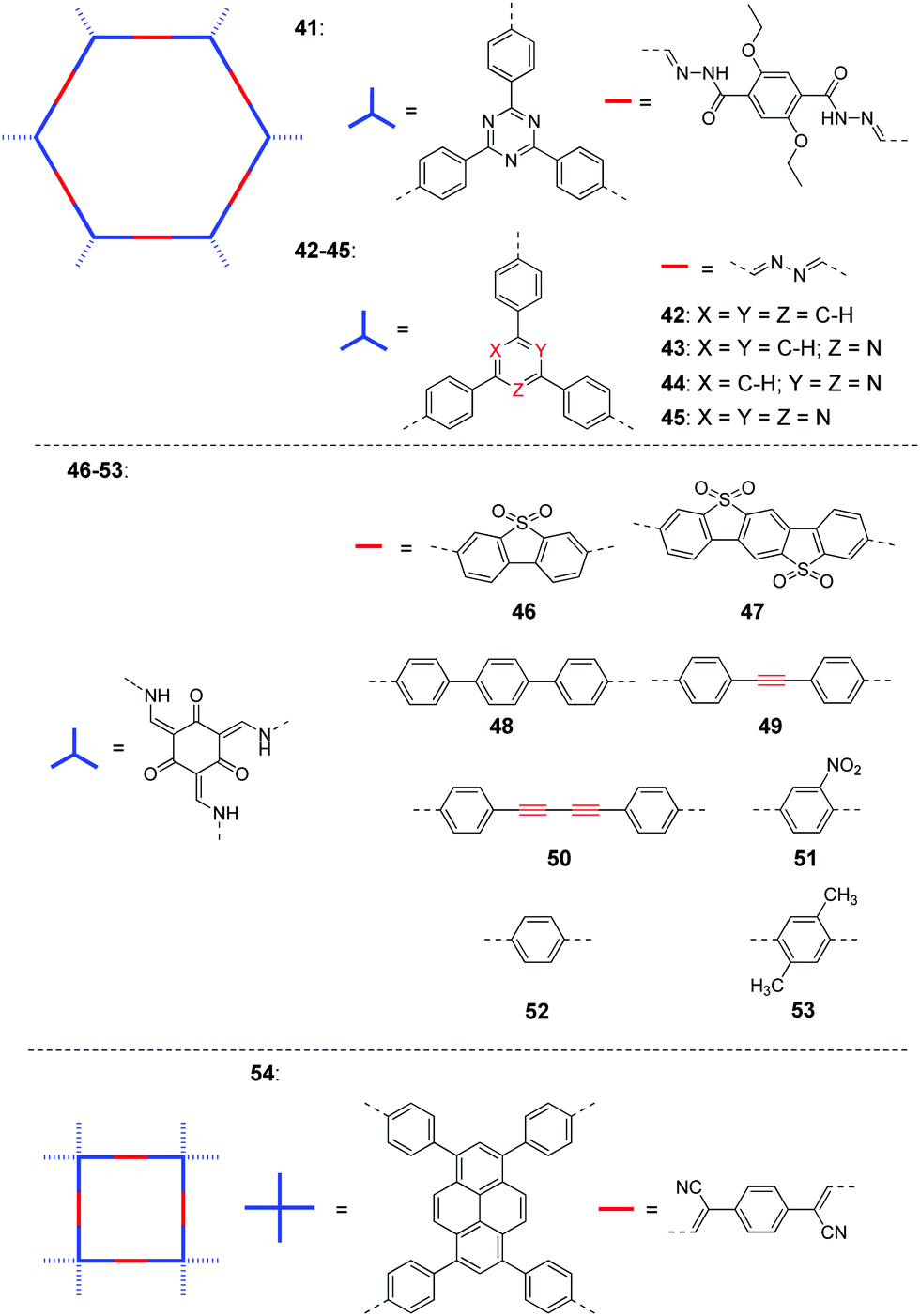 | ||
| Fig. 10 Chemical structures of COFs 41–54 for photocatalytic H2 production under visible light. | ||
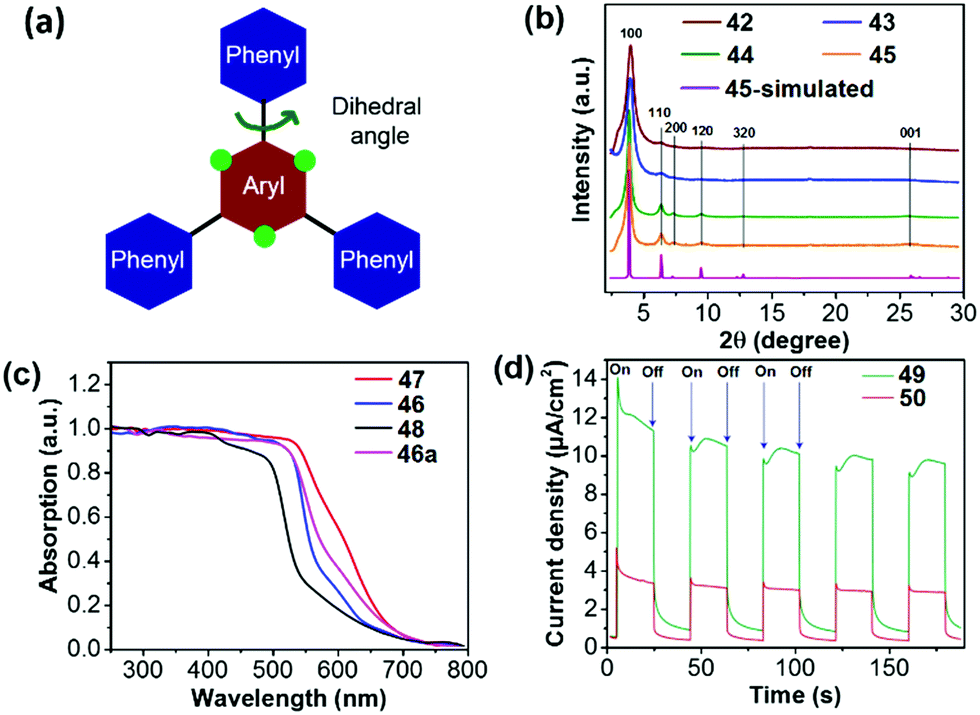 | ||
| Fig. 11 (a) Design principle and (b) PXRD patterns of COFs 42–45. (c) Diffuse reflectance spectra of COFs 46–48 (46a is an amorphous analogue of 46). (d) Transient photocurrent for COFs 49 and 50 measured in 0.5 M KCl solution under visible light irradiation. Reprinted with permission from ref. 120–122. Copyright 2015 Nature Publishing Group, 2018 Nature Publishing Group and 2018 American Chemical Society. | ||
The facile choice of building blocks did permit a good modulation of the porosity, wettability and optoelectronic properties of β-ketoenamine linked COFs, which led to enhanced photocatalytic performance. When different building blocks were introduced, the corresponding band gaps were 2.10, 1.85 and 2.28 eV for 46 (F-COF), 47 (FS-COF) and 48 (TP-COF), respectively (Fig. 11c).121 Moreover, 46 and 47 containing a sulfone unit exhibited better wettability than 48 as revealed by the water contact angle measurement, which facilitates dispersion of COF particles in water and the interaction between COF and water in the photocatalytic reaction. Loaded with 3 wt% Pt, the HER of 46 was up to 4.44 mmol h−1 g−1, while 10.1 and 1.6 mmol h−1 g−1 were reached for 47 and 48, respectively. The best H2 evolution of 47 was ascribed to its lowest band gap, good wettability, highest BET area (985 m2 g−1 for 46, 1288 m2 g−1 for 47 and 919 m2 g−1 for 48).
In addition, the photocatalytic activity of COFs is susceptible to linking bond length between comonomers. Thomas and co-workers reported that the catalytic activity of the diacetylene-based COF 49 (TP-EDDA) was much better than the acetylene-based COF 50 (TP-BDDA).122 Despite the almost identical band gaps (2.34 eV for 49 and 2.31 eV for 50), COF 50 has an HER of 324 μmol h−1 g−1, whereas a relatively low HER of 30 μmol h−1 g−1 was achieved for 49. The significantly enhanced HER was attributed to the higher charge carrier generation of 50 under visible light, which could be revealed by the larger photocurrent response of 50 as shown in Fig. 11d.
Substituent modification in the COF could also bring good photocatalytic performances. Sun et al. prepared three ketoenamine-based COFs (51–53) and studied the effect of different functional groups on their photocatalytic activities.123 The photocurrent density of COF 53 (TpPa-COF-(CH3)2) is about 2.3-fold and 6.5-fold higher than that of 52 (TpPa-COF-H) and 51 (TpPa-COF-NO2), respectively. Moreover, the electron transfer resistance value decreased gradually when the electron-donating ability of the substituent groups became stronger (from NO2 to H, then CH3). These results indicated that the charge separation of COFs was improved by increasing the electron-donating properties of the substituents. With an optical gap of 2.06 eV and the best charge separation efficiency, COF 52 showed a HER of 8.33 mmol h−1 g−1, which was much higher than that of 51 (1.56 mmol h−1 g−1) and 53 (0.22 mmol h−1 g−1).
In comparison with those reported COFs based on imine-, hydrazone-, or azine linkers, fully π-conjugated COFs are expected to allow for more efficient exciton separation and migration over the framework, which is good for H2 production. Very recently, Jiang and co-workers reported a 2D sp2 carbon-COF 54 (sp2c-COF) as the H2-producing photocatalyst, into which electron-withdrawing cyano groups were incorporated to impart a conjugated electron donor–acceptor structure and the phenylenevinylene further improved π conjugation.124 The BET surface area of COF 54 was 602 m2 g−1 and its optical gap was 2.05 eV. A stable HER of 1360 μmol h−1 g−1 was obtained under visible light. The HER could be enhanced to 2120 μmol h−1 g−1 by attaching 3-ethylrhodanine as an end-capping group to strengthen the push–pull effect.
In 2015, Wu and co-workers prepared CTF 55 (CTF-T1) by a trimerization reaction of terephthalonitrile using CF3SO3H as the catalyst at room temperature (Fig. 12).128 The CTF exhibits a band gap of 2.94 eV and a low HER of 200 μmol h−1 g−1 after loading 3 wt% Pt.
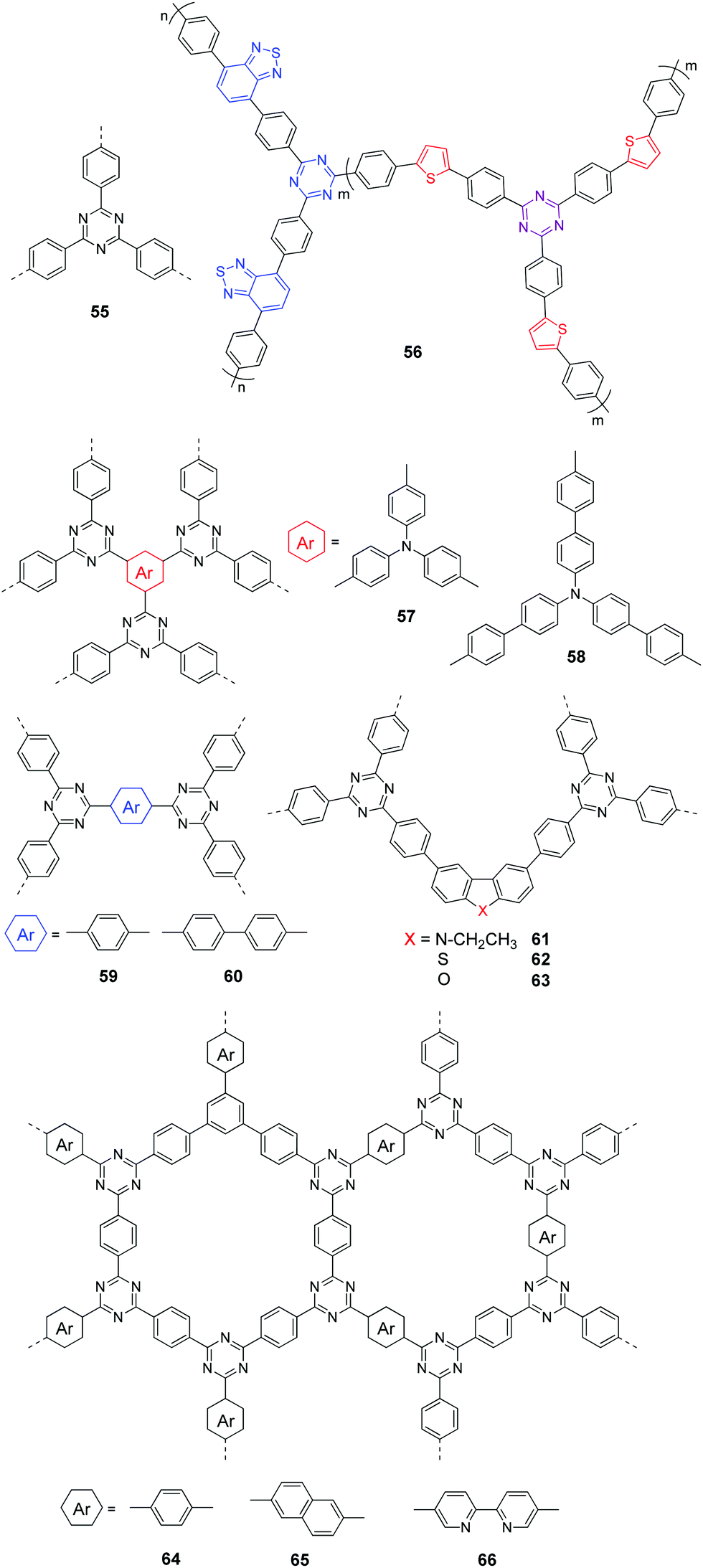 | ||
| Fig. 12 Chemical structures of polymers 55–66. | ||
Afterward, Thomas et al. showed that a CTF with a low HER could be converted into a more efficient photocatalyst by further mixing the pre-synthesized CTF in CHCl3/CF3SO3H with ZnCl2 salt for 10 minutes. The HER could increase from 186 to 1072 μmol h−1 g−1 due to the decreased optical gap (2.42 eV) and improved crystallinity.129 Alternatively, Wang et al. further processed the pre-synthesized CTF 55 through thermal annealing at 300 °C, which induced structural reorganization of 55 and yielded new conjugated heterocycles in the polymer. With improved light absorption and charge mobility, 3 times enhancement of HER was achieved under visible light as a result of the thermal treatment of 55.130
The fast recombination of photogenerated electrons and holes of CTFs is another major disadvantage preventing them from achieving higher efficiencies. In this context, Li et al. developed a molecular heterostructure 56 (CTF-Th) via a two-step synthetic strategy.131 CTF 56 exhibited higher photocurrent and charge carrier density than its single-component counterparts, which suggested a largely improved charge separation efficiency. As a result, a HER of 6.6 mmol h−1 g−1 was obtained for 56 under visible light.
Compared to the commonly used ionothermal method (usually at 400 °C), polycondensation under relatively gentle conditions is desirable for the preparation of CTFs with tunable functions and geometries. Tan and co-workers developed CTFs 57–60 by condensation between 1,4-benzenedicarboximidamide and different aldehydes in the presence of Cs2CO3.132 The structural variations have an obvious impact on the porosity and light absorption of the resulting CTFs. The BET surfaces areas are 807, 764, 663, and 757 m2 g−1 for 57 (CTF-HUST-3), 58 (CTF-HUST-3), 59 (CTF-HUST-1) and 60 (CTF-HUST-2), respectively (Fig. 13a). CTFs 57 and 58 showed broader absorption compared to 59 and 60 (Fig. 13b). Under visible light, CTF 60 showed the highest HER of 2647 μmol h−1 g−1, while the HERs were 1238, 1582 and 1460 μmol h−1 g−1 for 57, 58 and 59, respectively. Using a similar synthetic method, Tan et al. prepared donor–acceptor CTFs 61–63.133 Upon incorporating a strong electron-donating carbazole unit, 61 (CTF-N) showed the lowest band gap of 2.17 eV compared to 62 (CTF-S, 2.47 eV) and 63 (CTF-O, 2.67 eV) (Fig. 13c). Meanwhile, the enhanced charge separation/transfer efficiency of 61 was studied by employing fluorescence and photoelectrochemical measurements (Fig. 13d). Therefore, CTF 61 decorated with 3 wt% Pt exhibits the best HER of 10760 μmol h−1 g−1.
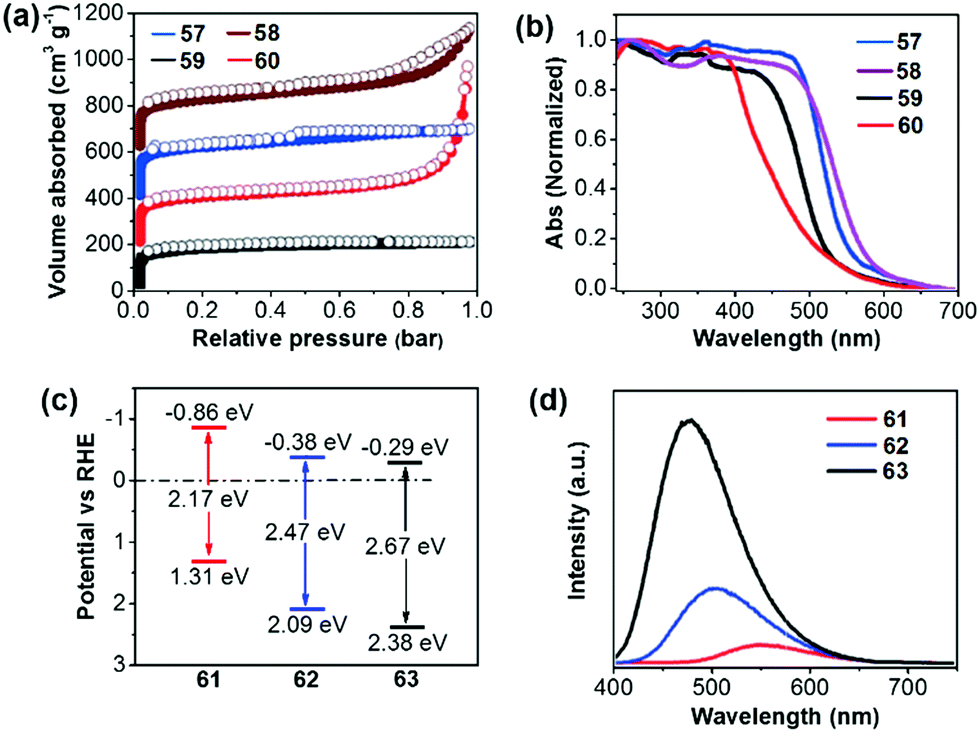 | ||
| Fig. 13 (a) Nitrogen sorption curves and (b) UV-vis absorption spectra of CTFs 57–60. (c) Energy diagram and (d) fluorescence spectra of CTFs 61–63. Reproduced with permission from ref. 132 and 133. Copyright 2017 Wiley-VCH and 2018 Royal Society of Chemistry. | ||
CTFs could also be fabricated by a Pd-catalyzed coupling reaction. In 2017, Cooper et al. synthesized two series of CTFs with similar structures by either Suzuki coupling of 2,4,6-tris(4-bromophenyl)-1,3,5-triazine and aromatic monomers or trimerization of dicyanobenzene derivatives.134 In this case, Pt loaded CTFs from trimerization exhibited higher activity than their counterparts from Suzuki coupling. Moreover, the length of the para-phenylene spacer has a profound effect on the band gaps and porosity for both series of CTFs and thus affects their photocatalytic activities. Based on the D–A interaction between thiophene derivatives (donor) and triazine units (acceptor), Bojdys et al. designed eight CTFs with fine-tuned band gaps (2.06–2.70 eV) and charge transfer in the polymer.135 As a result, a maximum HER of 3158 μmol h−1 g−1 was achieved by the CTF consisting of benzotrithiophene and triazine.
Instead of introducing different building blocks into CTFs, heteroatom doping is another efficient method to boost the HERs of CTFs. Su et al. prepared S-doped CTF samples (CTFS10) by annealing treatment of CTFs with sulfur.136 The S concentration in the framework was 0.5–0.52 atom% when the CTF was mixed with different amounts of S (5–30 wt%). The characteristic peak of C–S at 286.5 eV revealed by XPS analysis showed that the S atom is covalently bound to the CTF backbone. After depositing 3 wt% Pt, the CTFs mixed with 10 wt% S showed the best HER of 2000 μmol h−1 g−1, which is 5-fold enhanced compared to that of the pristine CTFs. This is due to the sharply decreased band gap (from 2.87 eV to 1.87 eV) and improved charge transport after S doping. Alternatively, the photocatalytic activity of CTFs could be enhanced by phosphorus doping due to the narrower band gap and more efficient charge transfer.137
Besides the above-mentioned amorphous CTFs, Tan and co-workers reported the successful preparation of crystalline CTFs 64–66 by in situ oxidizing alcohols into aldehyde monomers and studied their H2 production activities.138 It was claimed that high crystallinity of CTFs could be achieved by slowing down their nucleation process through controlling aldehyde monomer generation in the polycondensation reaction. All CTFs showed good crystallinity with BET surface areas of 520–599 m2 g−1. The HERs of 64 (CTF-HUST-C1), 65 (CTF-HUST-C5) and 66 (CTF-HUST-C6) were 5100, 2400 and 650 μmol h−1 g−1, respectively. In addition, Zou and co-workers prepared crystalline CTFs by condensation of melamine (MA) and pyromellitic dianhydride (PMDA) under thermal conditions. The obtained CTF displayed a band gap of 2.7 eV and HER of 7 μmol h−1.139
The elemental doping strategy has been intensively developed to boost the photocatalytic performance of g-C3N4 by tuning its band structure, light absorption and charge carrier transport.27–32 Various elements such as F, Br, I, N, P, O, S and B have been doped in the skeleton of tri-s-triazines based g-C3N4, as shown in Fig. 14.141 For instance, Wang et al. prepared F doped g-C3N4 (CN–F) by adding NH4F in the polymerization of dicyanamide. The formation of C–F bonds in the polymer led to a slightly red-shifted optical absorption and 2.7 times higher HER (∼130 μmol h−1 g−1) than that of the unmodified sample (>420 nm).142 Nitrogen-doped g-C3N4 (CN–N) could be synthesized using pre-treated melamine with hydrazine hydrate.143 This modification gave rise to improved charge transfer and a slightly narrowed band gap from 2.72 eV to 2.65 eV, as a result, CN–N showed a 1.8-fold higher HER of 553.5 μmol h−1 g−1 than that of pristine g-C3N4 (>400 nm). On the other hand, the doping of multiple elements in g-C3N4 provided more variable properties.141 Dai et al. reported carbon and phosphorus co-doped g-C3N4 (CN–C/P), which showed a significantly larger surface area of 141.1 m2 g−1 than pristine g-C3N4 (24.9 m2 g−1).144 Moreover, this modulation also led to extended optical absorption and promoted charge transfer in the polymer. Benefiting from this, CN–C/P exhibited a HER of 1493.3 μmol h−1 g−1, which was 9.7-fold as compared to that of pristine g-C3N4.
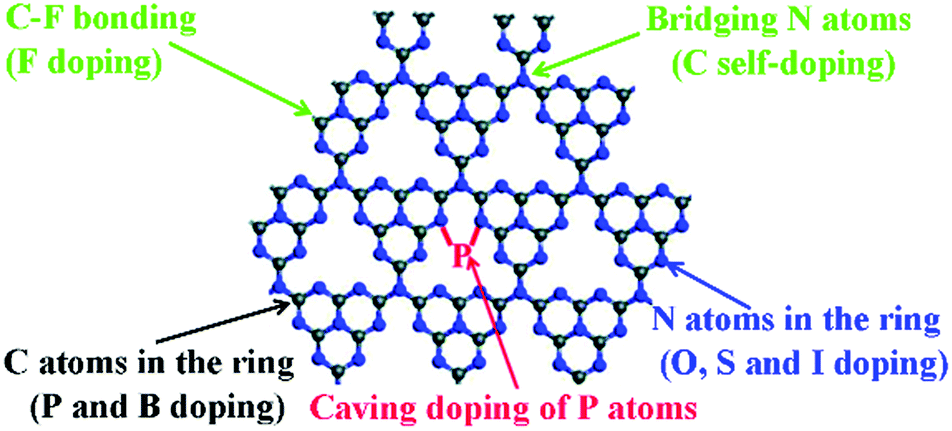 | ||
| Fig. 14 Possible elemental doping sites in the single layer of g-C3N4. Reproduced with permission from ref. 141. Copyright 2017 Royal Society of Chemistry. | ||
Organic small molecules are also capable of doping into g-C3N4 by copolymerization to adjust its π-electronic system and band structure. In 2010, Wang and co-workers prepared modified g-C3N4 (CN–BA) using dicyandiamide and barbituric acid as the precursor and dopant, respectively.145 With an optimization of amount of dopants, CN–BA exhibited a maximum HER of 294 μmol h−1 g−1 mainly due to the extended light absorption. After that, many other small molecules such as pyrimidine,146 quinoline,147 and thiophene148 were incorporated into g-C3N4 by several groups.
Structure defect engineering is another efficient approach for improving the photocatalytic performance of g-C3N4. The introduction of defects into the g-C3N4 backbone can not only modify the band structure by generating mid-gap states to extend the light absorption, but also act as trapping sites for charge carriers to inhibit the recombination of electrons and holes. In the presence of KOH, Zhang et al. introduced nitrogen vacancies and cyano groups into g-C3N4, which resulted in a reduced band gap from 2.68 to 2.36 eV and enhanced charge carrier separation.149 Therefore, the defect-rich g-C3N4 showed a HER of 6.9 mmol h−1 g−1, which was nearly twice that of pristine g-C3N4. In a very recent study, Wang et al. introduced defects into g-C3N4 by NaBH4 reduction of a crystalline pristine polymer under an inert atmosphere, which achieved nearly 8 times higher HER than the pristine polymer.150 Apart from those modification strategies, the development of analogous polymers of g-C3N4 such as C3N3S3151 offered new opportunities for exciting findings in photocatalytic H2 production.
In some cases, the construction of polymer heterojunctions (PHJs) can efficiently modulate the band structure and charge transport of g-C3N4 through inter/intramolecular π–π interactions between g-C3N4 and the aromatic structures. Shen et al. prepared three PHJs by coating different polyfluorene polymers on the surface of g-C3N4, which resulted in extended optical absorption and promoted charge transfer within the PHJs.152 Under visible light, all three pure CPs have no photocatalytic activity and pure g-C3N4 showed a very low HER of 87.4 μmol h−1 g−1. Among the three PHJs, 5% polymer 15 coated g-C3N4 (CN–P15) exhibited a maximum HER of 722.2 μmol h−1 g−1. Using a similar method, Chen et al. deposited Pdots on the surface of g-C3N4 nanosheets, which yielded an enhanced HER via improved charge transfer from Pdots to g-C3N4 nanosheets.153 In this case, 5 wt% polymer 17 deposited PHJ (CN–P17) exhibited a HER of 929.3 μmol h−1 g−1, which is higher than that of its counterpart coated with polymer 15 (439.6 μmol h−1 g−1). Besides, Wang et al. fabricated type II heterojunctions by anchoring pyrene-based polymers onto CN to improve light absorption and interface charge transfer.154 By optimizing the amount and molecular structures of polymers, CN–PyP hybrids attained a maximum HER of 600 μmol h−1 g−1 without Pt deposition. PHJs based on g-C3N4 could also be prepared by linking another component through a chemical bond. Yan et al. prepared heterojunctions (CN–COF) by in situ formation of a COF on the surface of pre-synthesized g-C3N4 through condensation of amino group in g-C3N4 and aldehyde group in the COF.155 CN–COF attained a dramatically enhanced HER of 10.1 mmol h−1 g−1, which is much higher than those of the pure COF (0.16 mmol h−1 g−1), g-C3N4 (1.05 mmol h−1 g−1) and their mixture (0.93 mmol h−1 g−1). Zou and co-workers fabricated heterostructured photocatalysts (CN–CTF) by linking g-C3N4 and CTFs with amide bonds, which demonstrated a good HER of 850 μmol h−1 g−1 and high stability compared to their counterpart (CN and COFs connected via van der Waals forces).156 These investigations offer more opportunities for the design of H2-production polymer photocatalysts. The photocatalytic activity and stability of these materials remain a great concern.
Among the commonly used metal cocatalysts such as Ru, Pd, Rh, Au, and Ag, Pt is a versatile cocatalyst mainly because of its largest work function for trapping photogenerated electrons and lowest overpotential for photocatalytic H2 evolution.158 In general, Pt is deposited on CPs by photoreduction of H2PtCl6 in the reaction system. Under light illumination, Pt(VI) is reduced by the photoinduced electrons of the polymer to produce Pt(0) nanoparticles with sizes of 3–5 nm in situ on the polymer surface. Compared to the pristine polymer, the Pt–polymer composite exhibited greatly enhanced H2 evolution. H2PtCl6 with a Pt mass concentration of 3 wt% was commonly used in the photodeposition experiment, because too low Pt concentration provided only a few active sites for proton reduction, which resulted in a low enhancement of catalytic activity. Excessive amount of Pt deposited on the surface of the polymer photocatalyst could prevent light absorption by shielding the incident light and stimulate the aggregation of Pt nanoparticles, which is detrimental to the activity of Pt.
Pd-catalyzed cross-coupling polymerization reactions are widely used to synthesize CPs for photocatalytic water splitting. Residual Pd inevitably existed in the polymer and may play a vital role in its photocatalytic process.160 Yu et al. have shown that the Pd content in the porous polymer greatly affected the H2 evolution.105 By changing the initial feeding ratio of Pd(PPh3)4 and monomer from 0.005 to 0.40, the Pd content was increased from 0.04% to 1.88% as measured by inductively coupled plasma mass spectrometry (ICP-MS). As shown in Fig. 15a, the HER of polymer 25b was sharply improved with increasing Pd content from 0.04% to 0.46%, which became saturated at a higher residual Pd content. In addition, upon loading with 2 wt% Pt, a ∼5-fold enhancement of the AQY at 350 nm was measured for 25b (Fig. 15b), which verified the importance of the Pt cocatalyst in the enhancement of photoactivity.
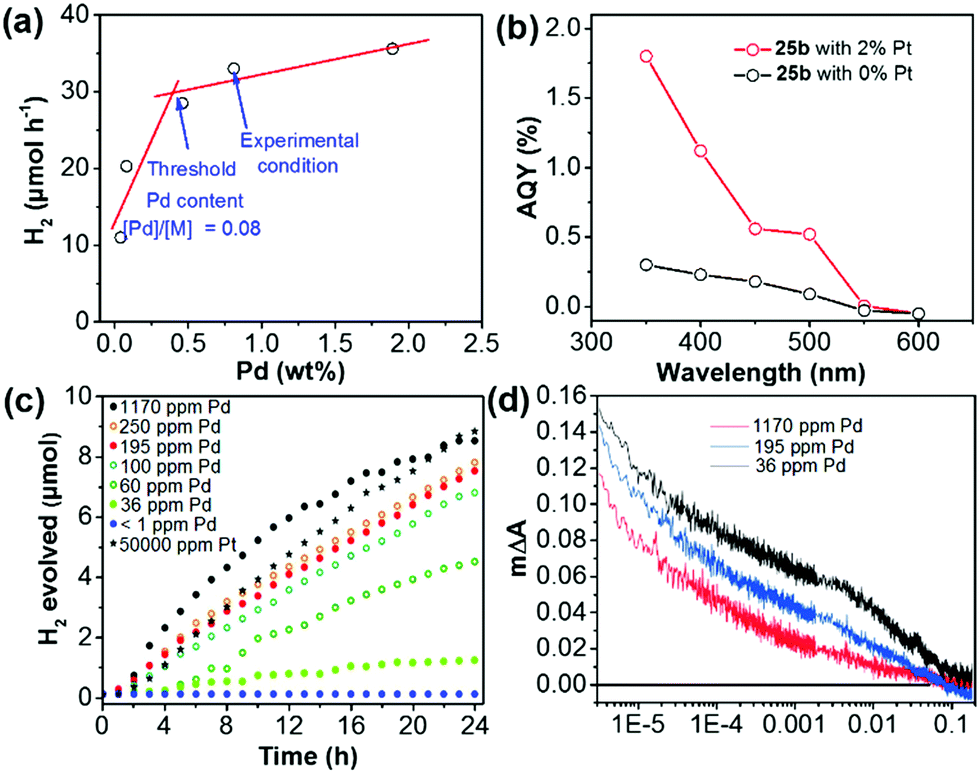 | ||
| Fig. 15 (a) H2 evolution of polymer 25b with different residual Pd contents. (b) Apparent quantum yield (AQY) of polymer 25b with 2% and without Pt loading at different wavelengths. (c) H2 evolution of nanoparticles of polymer 15 with different amounts of Pd or Pt. (d) Transient kinetics of nanoparticles of polymer 15 with different Pd concentrations in a diethylamine/water mixture. Reprinted with permission from ref. 105 and 161. Copyright 2016 American Chemical Society and 2018 Wiley-VCH. | ||
To study the role of Pd residues in determining the H2 evolution activity, Jan Kosco and co-workers prepared colloidal polymer 15 nanoparticles with different Pd contents.161 In the sample preparation, they first purified the as-prepared polymer 15 by gel permeation chromatography (GPC), and then washed the polymer with diethyldithiocarbamate. After the treatment, the Pd content in polymer 15 was below 1 ppm and the content could be gradually adjusted to 1170 ppm by adding Pd2(dba)3 solution. Fig. 15c displays that the H2 evolution of nanoparticles of polymer 15 is gradually enhanced with increasing Pd content from <1 ppm to ∼100 ppm, at which point the evolved H2 begins to saturate. Transient absorption spectroscopy (TAS) suggested that Pd could mediate electron transfer from the nanoparticles of polymer 15 to protons in the reaction media (Fig. 15d). It was found that photogenerated electrons were increasingly accumulated with decreasing Pd content in nanoparticles of polymer 15. This indicates that the Pd cocatalyst could effectively suppress the accumulation of long-lived electrons and thereby accelerate photocatalytic H2 generation of polymer 15.
3.2 Conjugated polymers for photocatalytic O2 evolution
The photocatalytic O2 evolution is a critical step in water splitting because the four-electron transfer is an uphill process with large overpotentials and slow kinetics, which makes it very challenging to develop robust photocatalysts for water oxidation.162,163To date, most of the O2 evolution photocatalysts have been based on nitrogen-rich CPs. In 2017, Xu et al. reported polymer 67, which has a nanosheet morphology and a very low band gap of ∼1.22 eV.41 With AgNO3 as the electron scavenger, 67 exhibits an oxygen evolution rate (OER) of 1.0 μmol h−1, which could be enhanced to 14.3 μmol h−1 after loading Co(OH)2 as the cocatalyst. More interestingly, polymer 67 can catalyze water oxidation under near-infrared light (>800 nm) and the OER is ∼0.4 μmol h−1 while g-C3N4 is not active.
Inspired by the facile tunability of CTFs, Wang et al. synthesized polymers 68–70 with different numbers of phenyl units in the polymer backbone.164 The band gaps of 68–70 decreased from 2.98 eV to 2.36 eV with increasing number of phenyl units (Fig. 16a). Importantly, their HOMO/LUMO levels straddle the potential of water splitting, suggesting sufficient driving force for both photocatalytic water reduction and oxidation. Furthermore, both the HOMO and LUMO levels decreased with the increase of phenyl number in the polymer. After loading with 3 wt% Co(NO3)2 as a cocatalyst, polymer 69 showed the highest OER of 3 μmol h−1 (>300 nm) compared to 68 (∼1 μmol h−1) and 70 (∼0.4 μmol h−1) in the presence of AgNO3, which was attributed to the synergistic effect of improved light absorption and charge transfer. In addition, the catalytic activity of 69 is also much better than that of C3N4 (Fig. 16b) due to the lower LUMO position of 69. Notably, polymer 69 could produce 7.2 μmol of O2 after 8 h visible-light irradiation. Considering that high crystallinity of CTF materials is desirable for exciton separation and thus enhancing the photocatalytic activity, Tang and co-workers reported highly ordered CTFs made by microwave-assisted polymerization.165 The crystallinity of the resulting CTFs became higher with the increase of microwave power from 20 to 100 W. However, a further increase in microwave power can destroy the hexagonal units. At a power of 100 W, polymer 71 has a well-defined planar structure as evidenced by a G+ peak at 1613 cm−1 from the Raman spectra analysis of CTFs. In addition, polymer 71 has conduction band (CB)/valence band (VB) levels of −0.74/+2.18 eV (vs. NHE, pH = 0), which is suitable for the photocatalytic water oxidation and reduction reaction. Polymer 71 gave an O2 production rate of 140 μmol h−1 g−1 upon loading with 3 wt% RuOx in the presence of AgNO3, which is higher than those of its counterparts prepared under different powers.
 | ||
| Fig. 16 Chemical structures of polymers 67–71; (a) band structure diagram and (b) time course of the O2 production for polymer 69 and C3N4. Reprinted with permission from ref. 164. Copyright 2018 Wiley-VCH. | ||
3.3 Conjugated polymers for photocatalytic overall water splitting
In comparison with the aforementioned photocatalytic water reduction or water oxidation, stoichiometric splitting of pure water into hydrogen and oxygen is cost-saving because both photogenerated electrons and holes are utilized without using a sacrificial agent to suppress one of the half-reactions.8,166–168 Until now, the reports of efficient CPs for photocatalytic overall water splitting remains rare.In 2016, Wang et al. demonstrated that Pt modified g-C3N4 was an efficient photocatalyst for overall water splitting and the morphology of g-C3N4 significantly affected the catalytic activity.169 Among the three types of g-C3N4, the urea-derived g-C3N4 exhibits a 2D nanosheet structure and compact layer-like morphology, which showed better charge mobility as compared to the other two samples made from dicyandiamide and ammonium thiocyanate as the precursor. All three types of pure g-C3N4 have no photocatalytic activity in the absence of Pt. After depositing Pt on the three samples, g-C3N4 nanosheets exhibited the best activity with H2 and O2 evolution rates of 1.2 and 0.6 μmol h−1, respectively (>420 nm). Very recently, Zhang and co-workers prepared 3D g-C3N4 with high crystallinity, displayed a larger surface area (130 m2 g−1) than those of bulk g-C3N4 (10.83 m2 g−1) and g-C3N4 nanosheets (93.84 m2 g−1).170 Surprisingly, after loading 1 wt% Pt and 3 wt% IrO2, 3D g-C3N4 exhibited H2 and O2 generation rates of 101.4 and 49.1 μmol h−1 g−1, respectively, which are about 11.8 and 5.1-fold higher than those of bulk g-C3N4 and g-C3N4 nanosheets. These examples demonstrated the critical role of the cocatalyst and surface morphology control of g-C3N4 in photocatalytic overall water splitting performance.
In 2017, Xu and co-workers prepared 1,3-diyne-linked CMPs 72–73 by Glaser oxidative coupling of terminal alkynes.40 TEM measurements revealed that both polymers have sheet-like structures (Fig. 17a and b), which favours the separation and transport of photogenerated excitons to the surface of polymer nanosheets for water redox reactions. The CB/VB levels were determined to be −3.55/−6.5 eV for 72 and −2.9/−5.77 eV for 73, indicating that both polymers have appropriate electronic band structures for overall water splitting under the given conditions. As a result, both H2 and O2 are evolved for 73 with expected 2:1 stoichiometry upon light irradiation (Fig. 17c). The HER is 218 μmol h−1 g−1 for 73, which is higher than that of 72 (102 μmol h−1 g−1). This could be attributed to the lower band gap of 73 (2.85 eV) compared to 72 (2.94 eV). This work demonstrates the great potential of polymer nanosheets as unique photocatalysts for overall water splitting.
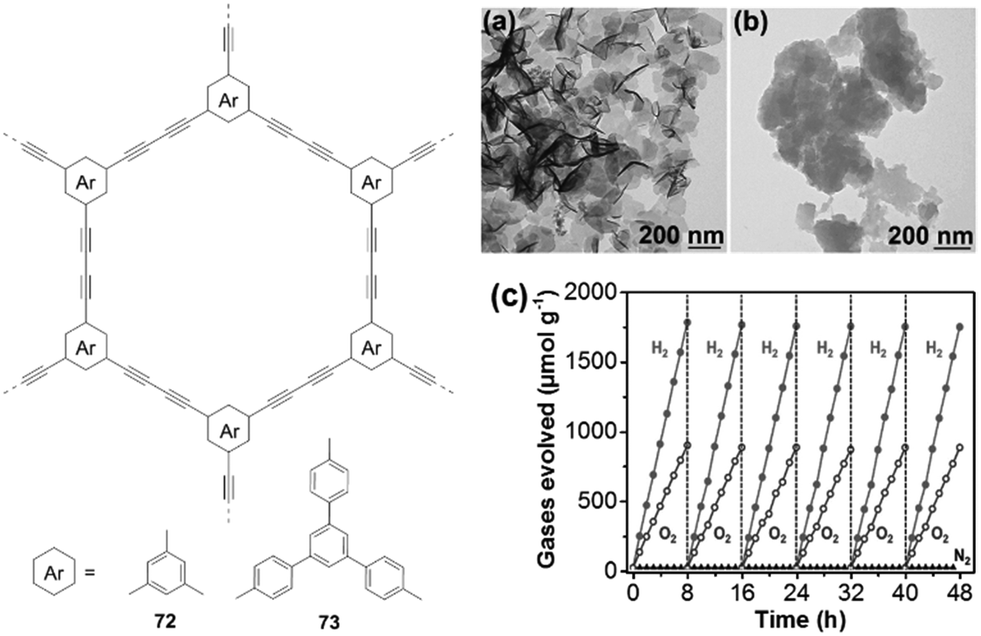 | ||
| Fig. 17 Chemical structures of polymers 72 and 73; TEM pictures of (a) 72 and (b) 73 as well as (c) time course of H2 and O2 production of polymer 73. Reprinted with permission from ref. 40. Copyright 2017 Wiley-VCH. | ||
4. Conjugated polymers for photocatalytic CO2 reduction
4.1 Photocatalytic CO2 reduction
The massive consumption of fossil fuels has caused the CO2 levels in the atmosphere to increase over the years, which has raised serious environmental and safety concerns. One of the best solutions to address this challenge is to convert CO2 into hydrocarbon fuels using solar light as an energy source.10,11,13,171–174 The technology could create huge economic and environmental benefits by simultaneously reducing the greenhouse effect and solving the energy shortage issue. However, CO2 is a very stable linear molecule. Compared with that of C–C (336 kJ mol−1), C–H (411 kJ mol−1), and C–O (327 kJ mol−1), an energy input of ∼750 kJ mol−1 is required for the cleavage of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond,13 which makes the conversion of CO2 very challenging. In a typical CO2 photoreduction process, water is required to provide a hydrogen source to achieve CO2 transformation. The adsorption and activation of CO2 by photocatalysts was a prerequisite for this transformation. Importantly, the redox potential alignment of the photocatalyst must be sufficient to drive the reaction. The potentials of CO2 reduction in water are given in Table 2 (pH = 7).13 Reduction of CO2 by one-electron transfer to generate CO2˙− is not feasible due to the large negative reduction potential of −1.85 V (vs. NHE). However, relatively lower reduction potentials of −0.61, −0.53, −0.48, −0.38 and −0.24 V are required for the generation of HCOOH, CO, HCHO, CH3OH, and CH4, respectively. A favourable potential of −0.42 V is required for water reduction to generate H2, which is a strong competitive reaction during the CO2 photoreduction process. Therefore, depending on the reaction pathways taken and the number of photogenerated electrons transferred, many different products are possible for CO2 photoreduction, making it difficult to achieve both good selectivity and high efficiency by a given photocatalyst. The selectivity (Φ) of the reacted electrons for CO2 reduction could be calculated using the following equation:
O bond,13 which makes the conversion of CO2 very challenging. In a typical CO2 photoreduction process, water is required to provide a hydrogen source to achieve CO2 transformation. The adsorption and activation of CO2 by photocatalysts was a prerequisite for this transformation. Importantly, the redox potential alignment of the photocatalyst must be sufficient to drive the reaction. The potentials of CO2 reduction in water are given in Table 2 (pH = 7).13 Reduction of CO2 by one-electron transfer to generate CO2˙− is not feasible due to the large negative reduction potential of −1.85 V (vs. NHE). However, relatively lower reduction potentials of −0.61, −0.53, −0.48, −0.38 and −0.24 V are required for the generation of HCOOH, CO, HCHO, CH3OH, and CH4, respectively. A favourable potential of −0.42 V is required for water reduction to generate H2, which is a strong competitive reaction during the CO2 photoreduction process. Therefore, depending on the reaction pathways taken and the number of photogenerated electrons transferred, many different products are possible for CO2 photoreduction, making it difficult to achieve both good selectivity and high efficiency by a given photocatalyst. The selectivity (Φ) of the reacted electrons for CO2 reduction could be calculated using the following equation:Heterogeneous CO2 photoreduction over porous polymers was performed in either liquid–solid or gas–solid systems. In a liquid–solid system, the polymer particles were suspended in the CO2-saturated aqueous solution. Under light irradiation, the CO2 reduction reactions take place at the interface of the liquid–solid phase. Organic sacrificial agents such as triethanolamine are needed to consume undesirable holes. In contrast, CO2 photoreduction in a gas–solid system is much simpler and only water vapour is used as the electron donor. In this case, photocatalysts were uniformly dispersed at the bottom of the reactor. Water vapour and CO2 were chemisorbed and activated by a porous polymer followed by the reduction of CO2 under light irradiation.
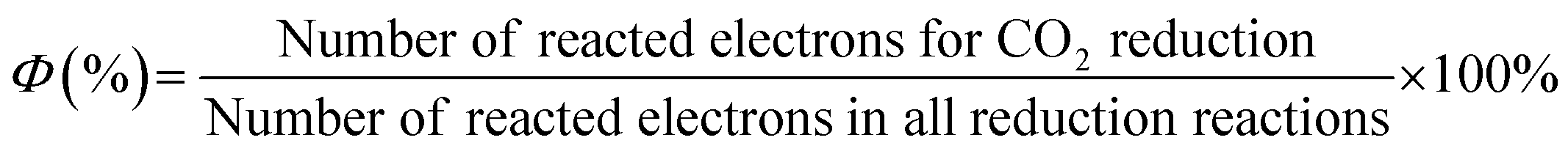
| Entry | Reaction | E 0 vs. NHE (V) |
|---|---|---|
| 1 | CO2 + e− → CO2˙− | −1.85 |
| 2 | CO2 + 2H+ + 2e− → HCOOH | −0.61 |
| 3 | CO2 + 2H+ + 2e− → CO + H2O | −0.53 |
| 4 | CO2 + 2H+ + 4e− → HCHO + H2O | −0.48 |
| 5 | CO2 + 6H+ + 6e− → CH3OH + 6H2O | −0.38 |
| 6 | CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
| 7 | 2H+ + 2e− → H2 | −0.42 |
Most of the photocatalysts for CO2 reduction are inorganic compounds such as metal oxides or sulphides, e.g. TiO2, CdS, BiVO4, etc.163,171 Organic polymers have been relatively less explored so far. For the development of highly active porous CPs for CO2 photoreduction, some critical factors such as band gaps, surface area, CO2 absorption, energy level alignments and photogenerated charge separation of the polymers are needed to be taken into account for pursuing good photocatalytic activities and selectivity. In addition, the external environment and operating conditions, such as catalyst quantity, solution pH, CO2 pressure, temperature, etc. will also likely influence the activity and selectivity of the reactions. For the liquid–solid system, the very low CO2 solubility in the reaction media is one major hurdle to achieve high CO2 reduction efficiency for the photocatalyst. To address this issue to some extent, Liu et al. used ionic liquids to capture more CO2 from air for the reduction reaction.175 It was estimated that ∼33 mg of CO2 was captured by 1 g of ionic liquid. By introducing different functional monomers, the CBs of polymers 74–78 (Fig. 18) are varied from −0.78 and −1.52 eV, indicating sufficient driving force for the reduction of CO2 to CO. The CO production rate follows the order: 78 > 76 > 77 > 75 > 74. The result was largely determined by the CB values of the CPs. Polymer 78 shows the highest CO production rate of 47.37 μmol h−1 g−1 with an excellent selectivity of up to 98.3%, which is attributed to its most negative CB value and the largest photocurrent.
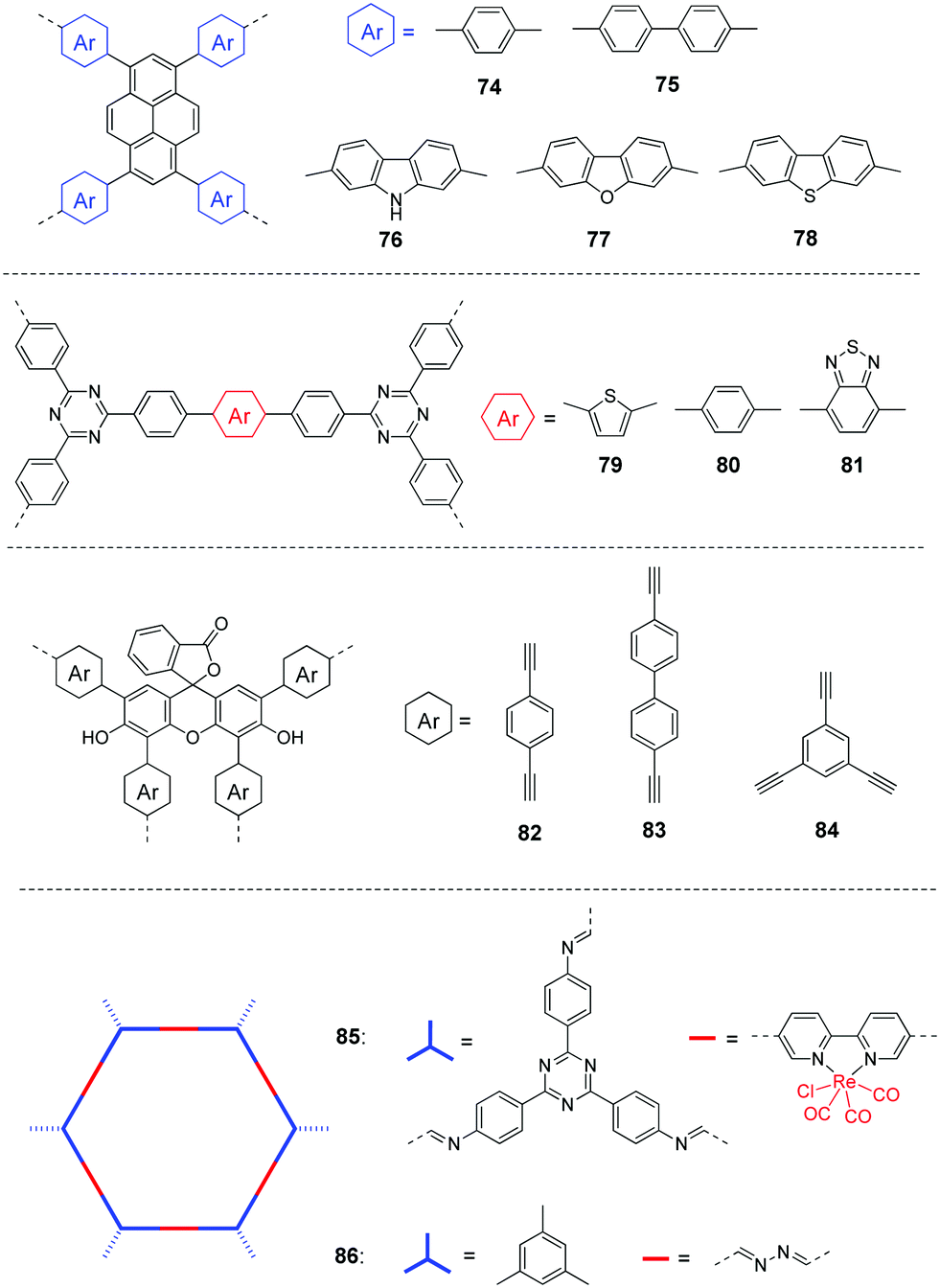 | ||
| Fig. 18 Chemical structures of polymers 74–86 for photocatalytic CO2 reduction. | ||
g-C3N4 also has a favourable CB position to drive the reduction of CO2 into hydrocarbons, which makes them potential candidates in this field.30,176 For example, Huang et al. reported that fluorine doped boron carbon nitride (BCN–F) could efficiently catalyse the reduction of CO2 into CO in the CH3CN/H2O/TEOA mixture, which gave 3-times higher CO generation rate (7.75 μmol h−1) than pristine BCN.177 Wang et al. synthesized barbituric acid modified g-C3N4 nanosheets (CN–BAN) through copolymerization between urea and barbituric acid (BA).178 With an optimal amount of BA, CN–BAN could produce CO and H2 with a rate of ∼14 μmol h−1 and ∼3.1 μmol h−1, respectively. In addition, Guo and co-workers prepared g-C3N4 nanosheets with nitrogen vacancies (CN–NV) by polymerization of dicyandiamide in the presence of tartaric acid.179 It was found that the introduction of defects could not only prolong the lifetimes of charge carriers, but also improve the visible light absorption and charge transfer; thereby, CN–NV exhibited a CO formation rate of 56.9 μmol h−1 g−1, which is about 8 times higher than that of pristine g-C3N4.
Apart from g-C3N4, CTFs with high nitrogen content are expected to have good CO2-philic ability and thus facilitate the reduction of CO2. Wang and co-workers developed triazine-based conjugated polymers 79–81,180 which showed tunable band gaps of 2.24–2.93 eV due to the D–A interaction between the triazine unit and comonomers. The BET surface areas were 409, 52, and 37 m2 g−1 for 79, 80, and 81, respectively. After loading CoCl2 and dipyridyl as cocatalysts, polymer 81 gave the highest CO generation rate of 18.2 μmol h−1 with 81.6% selectivity, which is due to the enhanced charge separation by incorporating electron-withdrawing BT units into the polymer.
Considering the advantages of relatively simple and inexpensive gas–solid systems, Liu et al. reported polymers 82–84 by introducing different arylethynylenes.181 The BET surface areas were 445, 131, and 610 m2 g−1 for 82, 83, and 84, respectively. The highest BET surface area of polymer 84 has thus led to a CO2 adsorption capacity of 62 mg g−1 at 1.0 bar, while lower capacities of 39 and 24 mg g−1 were measured for 82 and 83, respectively. In addition, the band gaps are 2.42–2.53 eV for the three polymers. The combination of electrochemical impedance spectroscopy (EIS) and transient photocurrent response measurements demonstrated that 82 had the best charge transfer efficiency. Therefore, polymer 82 exhibited the highest CO production rate of 33 μmol g−1 h−1 with 92% selectivity (Table 3).
| Polymer | Optical gap (eV) | HOMO/LUMO | S BET (m2 g−1) | Cocatalyst | Sacrificial agent | Main products and highest yield | Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Reaction conditions:a 50 mg of polymer, 10 g of CO2-saturated ionic liquid, 1 g of TEOA, 220 W xenon lamp (>420 nm).b Polymer (15 mg), CoCl2 (1 mmol), dipyridyl (5 mmol), TEOA (1 mL), solvent (5 mL, acetonitrile/water = 4:1), CO2 (1 atm), white light (>420 nm), 30 °C.c 10 mg of polymer powder, 300 W Xe lamp (>420 nm), CO2 and H2O vapour.d 0.9 mg of polymer, 3 mL of CH3CN, and 0.2 mL of TEOA, 225 W Xe lamp (>420 nm), irradiated for 10 h.e 10 mg of polymer, 500 W Xe lamp (800 nm ≥ λ ≥ 420 nm), CO2 (0.4 MPa) and H2O vapour, the reactor temperature was kept at 80 °C.f 50 mg of catalyst, dipyridine (20 mg), CoCl2·6H2O (1 mmol), solvent (5 mL, acetonitrile:H2O = 3:2), TEOA (1 mL), CO2 (1 atm), 300 W xenon lamp (≥420 nm).g 30 mg of catalyst, 1 μmol CoCl2, 15 mg of bipyridine, 5 mL of solvent containing 1 mL of TEOA, CO2 (1 atm), 300 W xenon lamp (>420 nm).h 5 mg of catalyst, 4 mL of CH3CN, 1 mL of TEOA, bipyridine (10 mmol L−1), as well as 25 μL of 20 mmol L−1 CoCl2 aqueous solution, 300 W xenon lamp (>400 nm).i 50 mg of sample, CO2 and H2O vapour, 300 W xenon lamp (>420 nm).j 30 mg of polymer, CO2 and H2O vapour, 300 W xenon lamp (≥420 nm). |
||||||||
| 74 | 2.48 | 1.70/−0.78 (eV) | 27.6 | — | TEOA | 14.53 (CO)/5.90 (H2) (μmol h−1 g−1) | 71.1 | 175 |
| 75 | 2.17 | 1.26/−0.91 (eV) | 14.0 | — | TEOA | 27.45 (CO)/1.16 (H2) (μmol h−1 g−1) | 95.9 | 175 |
| 76 | 2.86 | 1.40/−1.46 (eV) | 442.8 | — | TEOA | 41.44 (CO)/1.20 (H2) (μmol h−1 g−1) | 97.2 | 175 |
| 77 | 2.76 | 1.61/−1.15 (eV) | 4.5 | — | TEOA | 31.76 (CO)/1.16 (H2) (μmol h−1 g−1) | 96.5 | 175 |
| 78 | 2.66 | 1.14/−1.52 (eV) | 23.9 | — | TEOA | 47.37 (CO)/0.81 (H2) (μmol h−1 g−1) | 98.3 | 175 |
| 79 | 2.24 | 1.55/−0.69 (V vs. NHE) | 52 | CoCl2 and dipyridyl | TEOA | 4 (CO)/2 (H2) (μmol h−1) | 66.7 | 180 |
| 80 | 2.93 | 1.66/−0.77 (V vs. NHE) | 409 | CoCl2 and dipyridyl | TEOA | 10 (CO)/3 (H2) (μmol h−1) | 76.9 | 180 |
| 81 | 2.43 | 2.08/−0.85 (V vs. NHE) | 37 | CoCl2 and dipyridyl | TEOA | 18.2 (CO)/4.1 (H2) (μmol h−1) | 81.6 | 180 |
| 82 | 2.45 | 1.50/−0.95 (eV) | 445 | — | — | 33 (CO)/2.8 (H2) (μmol h−1) | 92 | 181 |
| 83 | 2.53 | 1.57/−0.96 (eV) | 131 | — | — | ∼26 (CO)/∼3.0 (H2) (μmol h−1) | 90 | 181 |
| 84 | 2.42 | 1.48/−0.94 (eV) | 610 | — | — | ∼12 (CO)/∼29 (H2) (μmol h−1) | 29 | 181 |
| 85 | — | — | — | — | TEOA | ∼900 (CO)/∼18 (H2) (μmol h−1 g−1) | 98 | 182 |
| 44 | 2.57 | — | 1412 | — | — | 37 (CH3OH) (μmol h−1 g−1) | ∼100 | 185 |
| 86 | 2.69 | — | 1053 | — | — | 57 (CH3OH) (μmol h−1 g−1) | ∼100 | 185 |
| BCN–F | 2.81 | 1.93/−0.88 (eV) (V vs. NHE) | — | CoCl2 and dipyridyl | TEOA | 7.75 (CO) (μmol h−1) | ∼100 | 177 |
| CN–BAN | — | — | 54 | CoCl2 and dipyridyl | TEOA | ∼14 (CO)/∼3.1 (H2) μmol h−1 | 81.9 | 178 |
| CN–NV | 2.49 | 1.66/−0.83 (V vs. NHE) | 29.5 | CoCl2 and dipyridyl | TEOA | 56.9 (CO)/10.3 (H2) μmol h−1 | 84.7 | 179 |
| CN–O | 2.61 | 1.73/−0.88 (V vs. Ag/AgCl) | 36 | — | — | 0.88 (CH3OH) μmol h−1 g−1 | 100 | 183 |
| CN–CV | 2.79 | 1.60/−1.19 (V vs. NHE) | 135.66 | — | — | 4.18 (CO) μmol h−1 g−1 | 100 | 184 |
COFs have inherent features of crystalline porous structures, which facilitate more efficient charge separation. Thus, this kind of material is used for CO2 photoreduction. Huang and co-workers developed COF 85, in which the photoactive COF served as the photosensitizer and Re complex (Re(bpy)(CO)3Cl) was incorporated as a CO2 reduction catalyst.182 Transient absorption spectroscopy showed a lifetime of 171 ps for the excited state of 85, which was much longer than that of the pristine COF, indicating that the incorporation of the Re moiety inhibits charge recombination in 85. COF 85 could produce ∼15 mmol CO/g with 98% selectivity in the TEOA/water mixture after visible light illumination for 22 h.
g-C3N4 could also be modified for CO2 photoreduction in the gas phase. For instance, Yu et al. prepared O-doped g-C3N4 (CN–O) with a tube-like morphology by a two-step process, which involves thermal exfoliation of the bulk g-C3N4 into a nanosheet and curling-condensation of the nanosheet into a nanotube.183 Compared with bulk g-C3N4, the as-prepared hierarchical nanotube exhibited a narrower band gap, higher CO2 uptake and better charge separation. Therefore, CN–O showed a methanol production rate of 0.88 μmol h−1 g−1, which is 5 times larger than that of bulk g-C3N4. Wang and co-workers prepared modified g-C3N4 with carbon vacancies (CN–CV) by heat treatment of pre-synthesized g-C3N4 under an NH3 atmosphere.184 This modification improves light absorption and CO2 adsorption/activation for CN–CV, as compared to pristine g-C3N4. Moreover, the charge generation and separation of g-C3N4 are also enhanced. As a result, CN–CV displayed a CO production rate of 4.18 μmol h−1 g−1, nearly 3.3 times higher than that of pristine g-C3N4.
COFs are also efficient in a gas–solid system; azine-linked COFs 45 and 86 were reported for the photoconversion of CO2 with gaseous H2O into methanol under visible light.185 COF 45 showed a higher BET surface area of 1412 m2 g−1, much higher than 86 (1053 m2 g−1), which is expected to absorb more CO2 for the reduction reaction. With a smaller optical gap of 2.57 eV than 86 (2.67 eV) and better photogenerated charge transfer, COF 45 produced 13.7 μmol g−1 of CH3OH after 24 h. The amount is much higher than that of 86 (8.6 μmol g−1) and C3N4 (4.8 μmol g−1) under the same conditions.
5. Conjugated polymers for photocatalytic organic transformation
Light-initiated organic transformation is a fascinating tool for the synthesis of many important compounds under mild conditions.15–17 In recent years, CPs have been intensively studied for organic transformation and different types of photochemical reactions were carried out in high yields with good selectivity under visible light (Fig. 19).18,19,157–160 As compared to those homogeneous systems for organic transformation, heterogeneous polymer photocatalysts have distinct advantages in terms of structural robustness, non-toxicity, low cost and good recyclability. In general, a photocatalytic organic transformation is initiated by excitation of the polymer photocatalyst to generate electrons and holes, which react with organic substrates to form radical intermediates. These active intermediate species are further converted into final products through rearrangements or reactions with other species at the surface or in bulk solution.18–20 | ||
| Fig. 19 Typical organic transformation reactions using CPs as the photocatalysts. | ||
5.1 Photocatalytic oxidative coupling of amine
Under light irradiation, polymer semiconductors could generate singlet oxygen (1O2), which promotes amine oxidative coupling with a broad range of substrates. In 2012, Lin et al. synthesized phosphorescent porous polymer 87 with incorporated [Ru(bpy)3]2+ by oxidative homocoupling (Fig. 20).186 Polymer 87 possesses [Ru(bpy)3]2+ loadings of up to 91 wt%, which not only improves the light absorption but also favours energy migration of the triplet excited states in the polymer network to promote oxidative reactions. Under visible light and at 60 °C, stoichiometric conversions (99%) were achieved for all three substituted benzylamines. Subsequently, Zhang et al. developed three CMPs 88–90 by changing the heteroatoms in the benzochalcogenadiazole unit from oxygen to sulfur to selenium.187 All three polymers can catalyze the oxidative coupling of benzylamine derivatives at room temperature and polymer 89 exhibits the best activity compared to 88 and 90, which was rationalized by the synergistic effect of the highest BET area, optimized VB and CB band levels and the highest efficiency of electron–hole pair generation. With polymer 89 as the photocatalyst, the reaction illustrates a broad substrate scope with conversions of 5–74%. In parallel, they showed that substitution positions of the benzoxadiazole unit at the centred phenyl unit has a significant impact on the CB/VB of the polymers and their photocatalytic activities. Compared to the other two 3D polymers connecting benzoxadiazole on the 1,2,4- and 1,2,4,5-positions of the phenyl unit, polymer 88 with the 1,3,5-substitution position of the centred phenyl ring demonstrated superior photocatalytic activity.188 With a high specific surface area of 1137 m2 g−1 and a broad light absorption in the visible region, the excited polymer 91 could reduce the molecular O2 to generate superoxide radicals (Ered = −0.86 V vs. SCE), which can oxidize the primary amine to imine products with 94–98% yields.189 Directly using natural sunlight, polymer 92 exhibited good conversion ranging from 77 to 99% for oxidative homocoupling of benzylamine, which provided a cost-effective way for large-scale photochemical applications in the future.190 | ||
| Fig. 20 Typical polymerization methods used to synthesize conjugated polymers for photocatalysis. | ||
5.2 Photocatalytic selective sulfide oxidation
In recent years, significant progress has been made by applying CPPs as photocatalysts for selective sulfide oxidation.191–193 In 2014, Zhang et al. presented the photooxidation of thioanisole using polymer 93 with high porosity.194 By introducing a BT unit as the strong acceptor into the conjugated backbone, polymer 93 shows a lower band gap of 2.47 eV compared to its counterpart polymer incorporating a carbazole unit. A number of thioanisole derivatives could be photooxidized by polymer 93 with 33–98% conversion and selectivity up to 88–99% after 24 h irradiation of visible light. Upon incorporation of BODIPY dyes into the polymer network, polymer 94 exhibited a bright red emission in the solid state with efficient singlet oxygen generation, which contributed to trigger the oxidation of sulfides with excellent conversion and selectivity.195 With superior surface area and good activity to reduce O2 into the superoxide radical, polymer 91 could oxidize thioanisole to the corresponding sulfoxide with excellent selectivity (>93%) and 90–99% yields.189 Notably, the reactions were completed in a short reaction time of 4.5–12 h, which was strongly influenced by the electron-donating/withdrawing substituents on various substrates.5.3 Hydroxylation of arylboronic acid
As versatile intermediates for the synthesis of important natural products and pharmaceutical compounds, phenols could be directly synthesized by the hydroxylation of arylboronic acids in air under light illumination. Zhang et al. designed polymer 95 using biphenyl as the core and the polymer had a BET area of up to 2065 m2 g−1.196 Moreover, polymer 95 exhibited a half wave potential at +1.38 V (vs. SEC), indicating its strong oxidation ability. With Et3N as a sacrificial reductant, this polymer can catalyze the hydroxylation of arylboronic acids bearing various substituents with excellent yields (71–97%). Subsequently, Wang et al. reported the conversion of arylboronic acid derivatives to the corresponding phenols using benzoxazole-based COF 103 as the metal-free photocatalyst.197 Prepared by reversible/irreversible cascade reactions, COF 103 exhibited super stability against light, strong acids and bases. The COF has an optical gap of 2.02 eV with a BET area of up to 1035 m2 g−1. Under air, polymer 103 displayed 55–99% yields for a range of substrates and the polymer could be recycled for at least 20 catalytic runs.5.4 Aza-Henry reaction
The aerobic cross dehydrogenative coupling reaction offers a powerful method for the conversion of C–H bonds adjacent to nitrogen atoms into C–C bonds under mild conditions. This reaction plays an increasingly critical role in obtaining natural products and biologically active molecules. Over the last decade, there has been increasing interest in developing CP photocatalysts for this reaction. As a commonly used organic dye, rose bengal was incorporated into polymer 96, which shows a broad absorption from 350 to 700 nm and a BET surface area of 833 m2 g−1.198 23 substrates were transformed into desirable products with 80–97% isolated yields using a common fluorescent lamp as the light source at room temperature. With good crystallinity and stability, 2D COF 104 exhibited excellent performance for the C–C/C–P coupling reaction with 45–92% yields and a reaction time of 6–11 h without loading any cocatalysts.199 In this case, the interaction between the donor (2,5-dimethoxybenzene) and acceptor (2,4,6-triphenyl-1,3,5-triazine) in the polymer was expected to improve the photoelectric properties of the resulting COF. Based on the donor–acceptor strategy, they recently synthesized three CMPs by alternating the central core with different electron-deficient units.200 Compared to the CMPs with incorporated benzene (2.385 eV) and 4,7-diphenyl-2,1,3-benzothiadiazole (2.199 eV), polymer 97 exhibited the smallest optical gap of 1.994 eV. Under visible light irradiation, polymer 97 could catalyse the conversion of 1,2,3,4-tetrahydroisoquinoline derivatives to the desired products with 82–94% yields in 1 h.5.5 Reductive dehalogenation of haloketones
Besides photo-oxidation reactions, CPs are also very efficient in catalysing some reduction reactions. Photocatalytic reductive dehalogenation of haloketones was one of the most widely investigated reactions. In 2014, Zhang et al. reported that polymer 98, prepared by Sonogashira-coupling in a high internal phase emulsion, exhibited excellent activity for dehalogenation of haloketones with 15–97% yields in 4–5 h at room temperature.43 The incorporation of benzobisthiadiazole (BBT) unit as a strong electron acceptor into the polymer backbone led to a much lower band gap of 1.5 eV and significantly higher electron paramagnetic resonance (EPR) signals for polymer 98 as compared to its counterpart without the BBT unit. With promising photoredox properties and high porosity, polymer 95 can catalyze the transformation of phenacyl bromide derivatives into acetophenones in 79–98% yields after 24 h light irradiation.201 Polymer 102 exhibits good crystallinity with a strong diffraction peak at 2.67° and forms irregular block nanocrystals with a size of 60–80 nm.202 With a BET area of 945 m2 g−1 and a band gap of 2.08 eV, polymer 102 could catalyse many α-bromoacetophenones to the corresponding acetophenone compounds in 69–88 yields.5.6 C–C coupling reactions
Cross-coupling reactions, such as Pd(0)-catalyzed Suzuki or Stille coupling reactions, are one of the most important methods for the construction of C–C bonds in organic synthesis.203,204 In general, these coupling reactions are carried out in homogeneous systems under thermal conditions. However, the Pd(0) catalysts are not stable enough and tend to undergo rapid deactivation during the reaction, which greatly affects the reaction efficiency. Therefore, the synthesis of these targets by stable and robust photocatalysts has attracted increasing attention.205,206 Zhang et al. reported Pd decorated CMP 88 (Pd@P88) for C–C coupling reactions at ambient temperature.207 TEM images demonstrated that Pd NPs with sizes of 5–10 nm were distributed in the polymer network after treating the polymer with Pd(OAc)2 at 90 °C for 1 h. The Pd content in Pd@P88 was determined to be 3% by ICP measurements. After immobilization with Pd NPs, the band gap of polymer 88 was slightly decreased from 2.43 eV to 2.38 eV. Pd@P88 has a BET surface area of 176 m2 g−1 and VB/CB of +1.43/−0.95 eV (vs. SCE). 75–98% conversion was achieved for the coupling between aryl halides and arylboronic acid with a range of substrates. CPs could also be used for the photocatalytic Stille coupling reaction. Zhang et al. disclosed that an azulene-containing CMP 99 could catalyse the Stille coupling reaction between aryl iodides and aryl stannanes under visible light at room temperature.208 Polymer 99 exhibits a band gap of 2.03 eV with HOMO/LUMO of 1.14/−1.10 V (vs. SCE). Notably, only electron-withdrawing-group substituted aryl iodides could be coupled with aryl stannanes due to their lower activated energies than the LUMO of 99 (−1.10 V vs. SCE), while electron-donating-group substituted or unsubstituted phenyl iodides are not applicable.Besides those critical advances in photoredox organic transformations, several new reactions were recently exploited using CPs as the photocatalysts. For instance, polymer 89 was used for photocatalytic C–C double bond cleavage in aqueous media.209 Various styrene derivatives could be cleaved to afford aldehydes with 27–65% conversion and 92–99% selectivity. Yu and co-workers reported three conjugated microporous polymers for C-3 functionalization of indoles;210 morphology and oxidation of polymers played an important role in determining the catalytic efficiency.
6. Conjugated polymers for photocatalytic degradation of organic dyes
As one of the most common contaminants in wastewater from textile industries, commercial organic dyes such as methylene blue (MB), rhodamine B (RhB), methyl orange (MO) are chemically stable and could not be removed by microbial processes.211 Other techniques such as adsorption, ozonation and flocculation have been proven to be insufficient for treatment of these effluents. Photocatalytic degradation of organic dyes into less hazardous compounds offers an attractive method for environmental remediation.22,23,36,39,212,213 The degradation rate of organic dyes could be facilely monitored using UV-vis absorption spectroscopy.The degradation process of organic dyes using semiconductors is complicated and the detailed mechanism is not well revealed in the literature. A combination of electron paramagnetic resonance (ESR) analyses and radical species trapping experiments revealed that singlet oxygen (1O2), superoxide radical (˙O2−), photogenerated hole (h+), and HO˙ are the main reactive species in the process, and the generation of these species could be proposed as follows:
| e− + O2 → O2˙− | (1) |
| O2˙− + H+ → HOO˙ | (2) |
| HOO˙ → H2O2 + O2 | (3) |
| H2O2 + O2˙− → HO˙ + O2 + HO− | (4) |
| H2O2 + h+ → 2HO˙ | (5) |
| H2O2 + hν → 2HO˙ | (6) |
Linear CPs with unique nanostructures show great promise for organic dye degradation. Remita et al. prepared nanofibers of poly(diphenylbutadiyne) (105) by photopolymerization using a soft templating approach (Fig. 21).214 The nanofibers are a few micrometres long with a diameter of about 19 nm. Without additional sacrificial reagents or cocatalysts, 75% MO were decomposed in water after 240 min irradiation, whereas only 17% MO degradation was achieved by Ag-modified TiO2. The photocatalytic performance of the nanofibers was almost unchanged even after 15 repeated cycles. Using the same method, they further prepared polymer 106 with vesicle and spindle morphologies, respectively.215 The spindles are 40 nm thick and the length is several hundred nanometres, while the vesicles are spherical hollow capsules with a diameter of around 1 μm and the thickness of the wall is around 40 nm. A clear shape-dependent photocatalytic activity was demonstrated as shown in Fig. 22a. The nanospindles displayed 100% degradation of MO after 180 min visible light irradiation; by contrast, the vesicles remained totally inactive under identical conditions. Besides, the photocatalytic activity of the polymer 107 nanospindles is much better than that of the polymer 106 nanofibers.
 | ||
| Fig. 21 Representative conjugated polymers for photocatalytic degradation of organic dyes. | ||
With good photoactivity and excellent water dispersibility, conjugated polyelectrolytes (CPEs) were reported by Zhang and co-workers for organic dye degradation.216 They prepared polymer 108 by attaching 1-alkyl-3-vinylimidazolium bromide onto the side chains of the poly(benzothiadiazolylfluorene) backbone, which was conducted through a self-initiated radical polymerization process under visible light. With a band gap of 2.11 eV, polymer 108 could almost quantitatively decompose MB and RhB after 70 and 90 min, respectively (Fig. 22b). The good singlet oxygen generation ability of polymer 107 under light excitation is likely responsible for the organic dye photodegradation. In addition, RhB was degraded almost quantitatively in repeated 10 runs, demonstrating excellent stability and reusability of 108.
 | ||
| Fig. 22 (a) Comparative photocatalytic degradation of MO of commercial TiO2, Ag-TiO2, polymer nanofibers of 105, vesicles and nanospindles of polymer 106 under visible light (P25-TiO2 is TiO2 with a diameter of 25 nm). (b) Photocatalytic degradation of RhB and MB using polymer 107. (c) The adsorption isotherms of RhB for polymers 108a, 108b and 109 at 25 °C. (d) Removal of RhB with polymers 108a, 108b and 109 in the supernatant. Reprinted with permission from ref. 215–217. Copyright 2015 Nature Publishing Group, 2015 Wiley-VCH and 2015 Royal Society of Chemistry. | ||
 | ||
| Fig. 23 (a) The adsorption isotherms for RhB on polymers 110–112. (b) Photocatalytic degradation kinetics of RhB using polymer 110. (c) UV/vis absorption spectra of NPs of polymers 114–116 in a water dispersion. (d) Photocatalytic degradation of RhB in the presence of NPs of polymers 114–116 in water. Reprinted with permission from ref. 218–220. Copyright 2018 American Chemical Society, 2019 American Chemical Society and 2015 Royal Society of Chemistry. | ||
Recently, Ouyang and co-workers synthesized three polymers with similar backbone structures and studied the effect of structural variation on their photocatalytic activities.217 Among them, polymer 109 had a 3D network structure, while the linear polymers 108a and 108b were obtained by varying the concentrations of the feeding monomers.
The BET areas were 1204, 571, 1265 m2 g−1 for 109a, 109b and 109, respectively. As shown in Fig. 22c, with a high surface area, the adsorption capacities of 110 (661 mg g−1) and 108a (688 mg g−1) toward RhB were much higher than that of 108b (189 mg g−1). Polymers 108a and 108b exhibited a very close band gap (1.95 eV for 108a and 1.97 eV for 108b), which was narrower than that of 109 (2.28 eV). By the synergistic adsorption-photocatalysis, RhB was totally decomposed in 30 min for both 109 and 108a, while 120 min was needed for 108b to achieve the same performance (Fig. 22d). These results implied that the BET surface area of the polymer played an important role in photocatalytic activity.
The adsorption-photocatalysis synergy was also reported by Chen et al.218 They prepared 9,9′-bifluorenylidene-based porous polymers 110–112 by introducing different comonomers such as pyrene, tetraphenylethene and triphenyl. Polymer 110 with incorporated pyrene units exhibited the lowest band gap of 1.55 eV and the largest BET surface area of 1306 m2 g−1, while lower surface areas of 777 and 590 m2 g−1 were determined for 111 and 112, respectively. All three polymers demonstrated fast uptake of RhB in an aqueous solution. After 45 min, the absorbed percentages of RhB were 97%, 96% and 89% for 110, 111 and 112, respectively. As shown in Fig. 23a, polymer 110 exhibited a remarkably high adsorption capacity of 1905 mg g−1 of RhB, which is much higher than those of 111 (1024 mg g−1) and 112 (926 mg g−1). As shown in Fig. 23b, more than 81% of RhB was decomposed in the presence of polymer 110 after 30 min visible light irradiation (>450 nm).
Very recently, Jin and co-workers prepared three CMPs with thiazolo[5,4-d] thiazole as the linker for photodegradation of organic dyes.219 Interestingly, the polymers showed nanosheet morphologies and their porous and photophysical properties could be fine-tuned by introducing diphenyl, triphenylbenzene and tetraphenylbenzene. With the highest surface area of 673 m2 g−1 among the three polymers, polymer 113 could absorb 57% of RhB, while the amount of RhB absorbed by the other two polymers is lower than 15%. Polymer 113 can completely degrade RhB in 30 min and 60 min is needed for the degradation of 80% MO.
Conjugated microporous polymer (CMP) nanoparticles offer unique advantages of high porosity and excellent solution dispersibility compared to bulk catalysts, which is desirable for organic dye photodegradation. Inspired by this, Zhang et al. reported the direct preparation of CMP nanoparticles via palladium-catalysed polycondensation reactions in a miniemulsion.220 Interestingly, the difference in the comonomers and linkages has a clear impact on the morphologies and optical properties of the resulting polymers. CMP nanoparticles obtained via Sonogashira-coupling are larger than the ones obtained via Suzuki-coupling. For BT-containing polymers, polymer 116a showed a rod-like shape, while polymer 116b formed nanorings in the reaction media. As shown in Fig. 23c, the introduction of BT into the backbone has led to a narrower band gap of the resulting polymers. Over 80% of RhB was degraded in the presence of 116b after 25 min, while only about 50% of RhB was decomposed for 116a (band gap: 1.96 eV) after the same period (Fig. 23d). This result is in accordance with the lowest optical band gap of 116b (1.76 eV).
7. Summary and outlook
In this Review, we have summarized the recent advances in CPs for various photocatalytic applications, including water splitting, CO2 reduction, organic transformation and organic dye degradation. Since g-C3N4 was first demonstrated for water splitting under visible light in 2009,140 CPs have drawn intensive interest and represent a new platform in the field of photocatalysis on account of their inherent advantages of earth-abundant nature, tunable band structure and high stability in the photocatalytic process. We discussed all classes of CPs including g-C3N4, linear CPs, porous CPs, water-soluble conjugated polyelectrolytes (CPEs), and organic framework materials (COFs and CTFs) in this review article and presented how polymer design could contribute to different photocatalytic reactions. Structurally, linear CPs consist of alternative conjugated units, while CPPs have 3D porous structures, which are expected to facilitate charge transfer in multiple dimensions. Compared to CPPs and linear CPs, crystalline COFs possess a stacked 2D structure, which is beneficial for photogenerated charge transfer, thus boosting the photocatalytic activity. On the other hand, to date, COFs used for heterogeneous photocatalysis still suffer from limited structural variations. Moreover, a Pt cocatalyst is commonly needed for COFs in photocatalytic H2 production while CPs prepared by Pd-catalyzed coupling reactions could be directly used without adding any cocatalysts. Besides those noble metal cocatalysts (Pt and Pd), the development of earth-abundant and low-cost cocatalysts has recently attracted attention. For instance, nickel-thiolate cluster was found to be a very efficient H2 production cocatalyst on a COF photoabsorber.221 This system demonstrated long-term hydrogen evolution activity with a turnover number of up to 103 and the maximum HER reached 941 μmol h−1 g−1 in the TEOA/H2O mixture. To improve the water-wettability of the polymers, a family of polymer photocatalysts enriched with CPEs with conjugated backbones and charged side-chains have also been developed, which offered new potential to this field.The molecular design has undoubtedly played a vital role in achieving the preferred properties of the resulting polymers, such as strong and wide visible light absorption, appropriate band structure, high efficiency of charge separation and transport, as well as good wettability and surface reaction efficiency. These synergy improvements in principle could result in remarkably enhanced catalytic activity. Among the reported strategies, elemental and molecular doping and the introduction of structural defects into the skeleton of g-C3N4 have been well developed for achieving enhanced photocatalytic water splitting and CO2 reduction. In addition, copolymerization of well-designed donors and acceptors has been widely employed to design CPs for photocatalysis, which have brought extended light absorption and improved charge mobility of polymers. The statistical copolymerization for the synthesis of CPPs offered additional opportunity to adjust the band gaps and porosity of polymers.
Despite the great progress made so far, the investigations in this field are in its infancy. The reported CPs suffer from low photocatalytic efficiency, which are far from the practical requirements. Many issues need to be resolved which may include the following:
(1) The development of photocatalytic H2 production using CPs has evolved rapidly in the past few years. A recorded H2 generation rate of 60.6 mmol h−1 g−1 has been achieved, which is very competitive to inorganic counterparts. It is highly expected that more exciting performance could be achieved in the future by designing new polymers, and the combination of computation and experiment appears to be a promising approach for high-throughput screening of polymer libraries. In a very recent report, Cooper and co-workers computationally studied photocatalytic H2 production using 6354 potential alternating co-polymers, among which more than 170 co-polymers have been synthesized and examined for their H2 evolution activities.222 This work well explored the structure–property-performance relationships of candidate co-polymers and successfully discovered new polymers with HERs greater than 6 mmol h−1 g−1. Apart from photocatalytic water reduction or oxidation, photocatalytic overall water splitting has been reported by conjugated microporous polymer nanosheets.40 However, the fundamentals of the photocatalytic process and structure–activity relationship of the polymer are not well understood. Besides, loading metals as cocatalyst could considerably increase the photocatalytic H2 production activity, but knowledge about the interaction mechanism between the polymer and metal is still not clear.
(2) Until now, relatively few CPs were exploited for photocatalytic CO2 reduction and most of them suffer from low activities. Likewise, knowledge about reaction pathways and fundamental enhancements for CO2 reduction is scarce. In order to increase the catalytic performance of the reaction, more work is in urgent demand to optimize the photoactivity and porosity of polymers.
(3) The use of CPs as heterogeneous photocatalysts is rapidly expanding the toolbox of chemical transformations. They have demonstrated good catalytic activities and recyclability for a variety of photochemical reactions. In particular, some reactions were performed directly under natural sunlight. However, the applied reaction types and substrate scope are still limited as compared to those of traditional transition-metal complexes. It is highly desirable to explore new organic molecule transformations, such as more challenging C-heteroatom or C–C functionalization, for heterogeneous polymer photocatalysts.
(4) Photodegradation of organic dyes using CPs presented an effective technology for environmental remediation. Interestingly, nanostructured CPs with good water dispersion have demonstrated excellent degradation ability. Another important finding is that CPPs could decompose organic dyes very efficiently by the adsorption-photocatalysis synergy. In order to achieve better catalytic activity, advanced research should be devoted to optimize the photophysical properties of conjugated polymers to improve the reactive species generation efficiency.
As CPs have the inherent advantages of diverse molecular structures and readily tunable physicochemical properties, there are unlimited opportunities and challenges in developing high-efficiency CPs for various photocatalytic applications. We expect that this review will be a good reference for the next decade of research in this exciting field. With more advances in polymer chemistry and materials science, it is anticipated that critical innovations in photocatalytic technology using CPs could play an increasingly important role in practical applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Singapore National Research Foundation (grant no. R279-000-444-281 and R279-000-483-281) and the National University of Singapore (grant no. R279-000-482-133).References
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- A. J. Carrillo, J. González-Aguilar, M. Romero and J. M. Coronado, Chem. Rev., 2019, 119, 4777–4816 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- T. Wang and J. Gong, Angew. Chem., Int. Ed., 2015, 54, 10718–10732 CrossRef CAS PubMed.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed.
- R. Shi, G. I. N. Waterhouse and T. Zhang, Sol. RRL, 2017, 1, 1700126 CrossRef.
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610–7627 CrossRef CAS.
- C. Wang, Z. Sun, Y. Zheng and Y. H. Hu, J. Mater. Chem. A, 2019, 7, 865–887 RSC.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- J. W. Beatty and C. R. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735–4750 CrossRef CAS.
- A. Savateev and M. Antonietti, ACS Catal., 2018, 8, 9790–9808 CrossRef CAS.
- U. G. Akpan and B. H. Hameed, J. Hazard. Mater., 2009, 170, 520–529 CrossRef CAS.
- L. G. Devi and R. Kavitha, Appl. Catal., B, 2013, 140–141, 559–587 CrossRef CAS.
- C.-C. Wang, J.-R. Li, X.-L. Lv, Y.-Q. Zhang and G. Guo, Energy Environ. Sci., 2014, 7, 2831–2867 RSC.
- L. Nie and Q. Zhang, Inorg. Chem. Front., 2017, 4, 1953–1962 RSC.
- L. Bucher, N. Desbois, P. D. Harvey, G. D. Sharma and C. P. Gros, Sol. RRL, 2017, 1, 1700127 CrossRef.
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- V. S. Vyas, V. W.-h. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS PubMed.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- M. Marszewski, S. Cao, J. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261–278 RSC.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS.
- L. V. Bora and R. K. Mewada, Renewable Sustainable Energy Rev., 2017, 76, 1393–1421 CrossRef CAS.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed.
- L. Wang, Y. Wan, Y. Ding, Y. Niu, Y. Xiong, X. Wu and H. Xu, Nanoscale, 2017, 9, 4090–4096 RSC.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2014, 2, 18720–18724 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. I. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- J. L. Segura, M. J. Mancheno and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS.
- M. Liu, L. Guo, S. Jin and B. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC.
- M. Leclerc and J.-F. Morin, Design and synthesis of conjugated polymers, John Wiley & Sons, 2010 Search PubMed.
- Y. Chujo, Conjugated polymer synthesis: methods and reactions, John Wiley & Sons, 2011 Search PubMed.
- M. Karayilan, W. P. Brezinski, K. E. Clary, D. L. Lichtenberger, R. S. Glass and J. Pyun, Angew. Chem., Int. Ed., 2019, 58, 7537–7550 CrossRef CAS.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS.
- L.-Z. Wu, B. Chen, Z.-J. Li and C.-H. Tung, Acc. Chem. Res., 2014, 47, 2177–2185 CrossRef CAS.
- S. Kaufhold, L. Petermann, R. Staehle and S. Rau, Coord. Chem. Rev., 2015, 304, 73–87 CrossRef.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- M. Wen, K. Mori, Y. Kuwahara, T. An and H. Yamashita, Appl. Catal., B, 2017, 218, 555–569 CrossRef CAS.
- Y. Huang and B. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14804–14806 CrossRef CAS.
- Y.-J. Yuan, D. Chen, Z.-T. Yu and Z.-G. Zou, J. Mater. Chem. A, 2018, 6, 11606–11630 RSC.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 8, 474–475 RSC.
- T. Shibata, A. Kabumoto, T. Shiragami, O. Ishitani, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 2068–2076 CrossRef CAS.
- S. Matsuoka, T. Kohzuki, A. Nakamura, C. Pac and S. Yanagida, J. Chem. Soc., Chem. Commun., 1991, 8, 580–581 RSC.
- S. Matsuoka, H. Fujii, T. Yamada, C. Pac, A. Ishida, S. Takamuku, M. Kusaba, N. Nakashima and S. Yanagida, J. Phys. Chem., 1991, 95, 5802–5808 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2018, 57, 2520 CrossRef CAS.
- X.-H. Zhang, X.-P. Wang, J. Xiao, S.-Y. Wang, D.-K. Huang, X. Ding, Y.-G. Xiang and H. Chen, J. Catal., 2017, 350, 64–71 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef.
- C. M. Aitchison, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2019, 7, 2490–2496 RSC.
- C. Dai, S. Xu, W. Liu, X. Gong, M. Panahandeh-Fard, Z. Liu, D. Zhang, C. Xue, K. P. Loh and B. Liu, Small, 2018, 14, 1801839 CrossRef.
- Z.-A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Chem. Mater., 2018, 30, 5733–5742 CrossRef CAS.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS.
- K. Li and B. Liu, J. Mater. Chem., 2012, 22, 1257–1264 RSC.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- S. Kundu and A. Patra, Chem. Rev., 2016, 117, 712–757 CrossRef PubMed.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- P.-J. Tseng, C.-L. Chang, Y.-H. Chan, L.-Y. Ting, P.-Y. Chen, C.-H. Liao, M.-L. Tsai and H.-H. Chou, ACS Catal., 2018, 8, 7766–7772 CrossRef CAS.
- C. V. Hoven, A. Garcia, G. C. Bazan and T. Q. Nguyen, Adv. Mater., 2008, 20, 3793–3810 CrossRef CAS.
- H. Jiang, P. Taranekar, J. R. Reynolds and K. S. Schanze, Angew. Chem., Int. Ed., 2009, 48, 4300–4316 CrossRef CAS.
- A. Duarte, K.-Y. Pu, B. Liu and G. C. Bazan, Chem. Mater., 2010, 23, 501–515 CrossRef.
- Q. Cui and G. C. Bazan, Acc. Chem. Res., 2017, 51, 202–211 CrossRef PubMed.
- C. Dai, M. Panahandeh-Fard, X. Gong, C. Xue and B. Liu, Sol. RRL, 2019, 3, 1800255 CrossRef.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef CAS PubMed.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 CrossRef.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- J. Zhu, C. Yang, C. Lu, F. Zhang, Z. Yuan and X. Zhuang, Acc. Chem. Res., 2018, 51, 3191–3202 CrossRef CAS PubMed.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819–7832 RSC.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- Y. Xu, N. Mao, C. Zhang, X. Wang, J. Zeng, Y. Chen, F. Wang and J.-X. Jiang, Appl. Catal., B, 2018, 228, 1–9 CrossRef.
- Y. Liu, Z. Liao, X. Ma and Z. Xiang, ACS Appl. Mater. Interfaces, 2018, 10, 30698–30705 CrossRef CAS PubMed.
- Z.-A. Lan, W. Ren, X. Chen, Y. Zhang and X. Wang, Appl. Catal., B, 2019, 245, 596–603 CrossRef CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- C. Gao, L. Wang, X. Li and H. Wang, Polym. Chem., 2014, 5, 5200–5210 RSC.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS.
- L. Li, W.-y. Lo, Z. Cai, N. Zhang and L. Yu, Macromolecules, 2016, 49, 6903–6909 CrossRef CAS.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS.
- Q. Zhang, M. A. Kelly, N. Bauer and W. You, Acc. Chem. Res., 2017, 50, 2401–2409 CrossRef CAS.
- W. Zhong, J. Liang, S. Hu, X.-F. Jiang, L. Ying, F. Huang, W. Yang and Y. Cao, Macromolecules, 2016, 49, 5806–5816 CrossRef CAS.
- Y. Xiang, X. Wang, L. Rao, P. Wang, D. Huang, X. Ding, X. Zhang, S. Wang, H. Chen and Y. Zhu, ACS Energy Lett., 2018, 3, 2544–2549 CrossRef CAS.
- C. Cheng, X. Wang, Y. Lin, L. He, J.-X. Jiang, Y. Xu and F. Wang, Polym. Chem., 2018, 9, 4468–4475 RSC.
- S. Bi, Z. A. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang and E. Brunner, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef.
- Z. Wang, X. Yang, T. Yang, Y. Zhao, F. Wang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J.-X. Jiang, ACS Catal., 2018, 8, 8590–8596 CrossRef CAS.
- R. S. Sprick, B. Bonillo, M. Sachs, R. Clowes, J. R. Durrant, D. J. Adams and A. I. Cooper, Chem. Commun., 2016, 52, 10008–10011 RSC.
- R. S. Sprick, Y. Bai, A. A. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- Y. Zhao, W. Ma, Y. Xu, C. Zhang, Q. Wang, T. Yang, X. Gao, F. Wang, C. Yan and J.-X. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- G. Zhang, W. Ou, J. Wang, Y. Xu, D. Xu, T. Sun, S. Xiao, M. Wang, H. Li, W. Chen and C. Su, Appl. Catal., B, 2019, 245, 114–121 CrossRef CAS.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg and R. S. Sprick, Nat. Chem., 2018, 10, 1180 CrossRef CAS.
- P. Pachfule, A. Acharjya, J. r. m. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- J. L. Sheng, H. Dong, X. B. Meng, H. L. Tang, Y. H. Yao, D. Q. Liu, L. L. Bai, F. M. Zhang, J. Z. Wei and X. J. Sun, ChemCatChem, 2019, 11, 2313–2319 CrossRef CAS.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS.
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600 CrossRef CAS.
- P. Puthiaraj, Y.-R. Lee, S. Zhang and W.-S. Ahn, J. Mater. Chem. A, 2016, 4, 16288–16311 RSC.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- S. Kuecken, A. Acharjya, L. Zhi, M. Schwarze, R. Schomäcker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC.
- Z.-A. Lan, Y. Fang, X. Chen and X. Wang, Chem. Commun., 2019, 55, 7756–7759 RSC.
- W. Huang, Q. He, Y. Hu and Y. Li, Angew. Chem., Int. Ed., 2019, 131, 1–6 Search PubMed.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. I. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS PubMed.
- L. Guo, Y. Niu, H. Xu, Q. Li, S. Razzaque, Q. Huang, S. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J.-S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 CrossRef CAS PubMed.
- L. Li, W. Fang, P. Zhang, J. Bi, Y. He, J. Wang and W. Su, J. Mater. Chem. A, 2016, 4, 12402–12406 RSC.
- Z. Cheng, W. Fang, T. Zhao, S. Fang, J. Bi, S. Liang, L. Li, Y. Yu and L. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 41415–41421 CrossRef CAS PubMed.
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., 2018, 130, 12144–12148 CrossRef.
- S. Chu, Y. Wang, Y. Guo, P. Zhou, H. Yu, L. Luo, F. Kong and Z. Zou, J. Mater. Chem., 2012, 22, 15519–15521 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406–23433 RSC.
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS.
- J. Fang, H. Fan, M. Li and C. Long, J. Mater. Chem. A, 2015, 3, 13819–13826 RSC.
- H. Wang, B. Wang, Y. Bian and L. Dai, ACS Appl. Mater. Interfaces, 2017, 9, 21730–21737 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- X. Fan, L. Zhang, R. Cheng, M. Wang, M. Li, Y. Zhou and J. Shi, ACS Catal., 2015, 5, 5008–5015 CrossRef CAS.
- J. Zhang, M. Zhang, S. Lin, X. Fu and X. Wang, J. Catal., 2014, 310, 24–30 CrossRef CAS.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- W. Ren, J. Cheng, H. Ou, C. Huang, M. M. Titirici and X. Wang, ChemSusChem, 2019, 12, 3257–3262 CrossRef CAS.
- Z. Zhang, J. Long, L. Yang, W. Chen, W. Dai, X. Fu and X. Wang, Chem. Sci., 2011, 2, 1826–1830 RSC.
- J. Chen, C. L. Dong, D. Zhao, Y. C. Huang, X. Wang, L. Samad, L. Dang, M. Shearer, S. Shen and L. Guo, Adv. Mater., 2017, 29, 1606198 CrossRef PubMed.
- W. Zhou, T. Jia, H. Shi, D. Yu, W. Hong and X. Chen, J. Mater. Chem. A, 2019, 7, 303–311 RSC.
- S. Zang, G. Zhang, P. Yang, D. Zheng and X. Wang, Chem. – Eur. J., 2019, 25, 6102–6107 CrossRef CAS PubMed.
- M. Luo, Q. Yang, K. Liu, H. Cao and H. Yan, Chem. Commun., 2019, 55, 5829–5832 RSC.
- G. Zhou, L.-L. Zheng, D. Wang, Q.-J. Xing, F. Li, P. Ye, X. Xiao, Y. Li and J.-P. Zou, Chem. Commun., 2019, 55, 4150–4153 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- J. Kosco and I. McCulloch, ACS Energy Lett., 2018, 3, 2846–2850 CrossRef CAS.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- D. Kong, Y. Zheng, M. Kobielusz, Y. Wang, Z. Bai, W. Macyk, X. Wang and J. Tang, Mater. Today, 2018, 21, 897–924 CrossRef CAS.
- Z. A. Lan, Y. Fang, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 470–474 CrossRef CAS.
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261–5274 RSC.
- L. Wang, Y. Zhang, L. Chen, H. Xu and Y. Xiong, Adv. Mater., 2018, 30, 1801955 CrossRef PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- X. Chen, R. Shi, Q. Chen, Z. Zhang, W. Jiang, Y. Zhu and T. Zhang, Nano Energy, 2019, 59, 644–650 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 RSC.
- J. Mao, K. Li and T. Peng, Catal. Sci. Technol., 2013, 3, 2481–2498 RSC.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- Y. Chen, G. Ji, S. Guo, B. Yu, Y. Zhao, Y. Wu, H. Zhang, Z. Liu, B. Han and Z. Liu, Green Chem., 2017, 19, 5777–5781 RSC.
- Y. Fang and X. Wang, Chem. Commun., 2018, 54, 5674–5687 RSC.
- F. Xing, Q. Liu, M. Song and C. Huang, ChemCatChem, 2018, 10, 5270–5279 CrossRef CAS.
- J. Qin, S. Wang, H. Ren, Y. Hou and X. Wang, Appl. Catal., B, 2015, 179, 1–8 CrossRef CAS.
- H. Shi, S. Long, J. Hou, L. Ye, Y. Sun, W. Ni, C. Song, K. Li, G. G. Gurzadyan and X. Guo, Chem. – Eur. J., 2019, 25, 5028–5035 CrossRef CAS PubMed.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem. – Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS PubMed.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- J. Fu, B. Zhu, C. Jiang, B. Cheng, W. You and J. Yu, Small, 2017, 13, 1603938 CrossRef PubMed.
- M. Shen, L. Zhang, M. Wang, J. Tian, X. Jin, L. Guo, L. Wang and J. Shi, J. Mater. Chem. A, 2019, 7, 1556–1563 RSC.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- J.-L. Wang, C. Wang, K. E. deKrafft and W. Lin, ACS Catal., 2012, 2, 417–424 CrossRef CAS.
- Z. J. Wang, K. Garth, S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemSusChem, 2015, 8, 3459–3464 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Mater., 2015, 27, 6265–6270 CrossRef CAS PubMed.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- V. R. Battula, H. Singh, S. Kumar, I. Bala, S. K. Pal and K. Kailasam, ACS Catal., 2018, 8, 6751–6759 CrossRef CAS.
- C. Yang, Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. Deng, Green Chem., 2009, 11, 1401–1405 RSC.
- R. D. Chakravarthy, V. Ramkumar and D. K. Chand, Green Chem., 2014, 16, 2190–2196 RSC.
- B. Zhang, J. Li, B. Zhang, R. Chong, R. Li, B. Yuan, S.-M. Lu and C. Li, J. Catal., 2015, 332, 95–100 CrossRef CAS.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- M. Liras, M. Iglesias and F. L. Sánchez, Macromolecules, 2016, 49, 1666–1673 CrossRef CAS.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS.
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- J.-X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779–8783 CrossRef CAS.
- Y. Zhi, Z. Li, X. Feng, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, J. Mater. Chem. A, 2017, 5, 22933–22938 RSC.
- Y. Zhi, S. Ma, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, Appl. Catal., B, 2019, 244, 36–44 CrossRef CAS.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS.
- Z. Lia, Y. Zhi, P. Shao, H. Xia, G. Li, X. Feng and X. Chen, Appl. Catal., B, 2019, 245, 334–342 CrossRef.
- V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS PubMed.
- I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137–173 CrossRef CAS.
- Q. Xiao, S. Sarina, A. Bo, J. Jia, H. Liu, D. P. Arnold, Y. Huang, H. Wu and H. Zhu, ACS Catal., 2014, 4, 1725–1734 CrossRef CAS.
- F. Wang, C. Li, H. Chen, R. Jiang, L.-D. Sun, Q. Li, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Mater., 2015, 27, 1921–1924 CrossRef CAS.
- S. Ghasimi, S. A. Bretschneider, W. Huang, K. Landfester and K. A. I. Zhang, Adv. Sci., 2017, 4, 1700101 CrossRef PubMed.
- C. Ayed, L. C. da Silva, D. Wang and K. A. I. Zhang, J. Mater. Chem. A, 2018, 6, 22145–22151 RSC.
- W. Zhang, J. Tang, W. Yu, Q. Huang, Y. Fu, G. Kuang, C. Pan and G. Yu, ACS Catal., 2018, 8, 8084–8091 CrossRef CAS.
- T. Robinson, G. McMullan, R. Marchant and P. Nigam, Bioresour. Technol., 2001, 77, 247–255 CrossRef CAS PubMed.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouame, S. Remita, L. Ramos, F. Goubard, P.-H. Aubert, A. Dazzi, A. Deniset-Besseau and H. Remita, Sci. Rep., 2015, 5, 18002 CrossRef CAS PubMed.
- S. Ghasimi, S. Prescher, Z. J. Wang, K. Landfester, J. Yuan and K. A. I. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14549–14553 CrossRef CAS PubMed.
- J. Wang, H. Yang, L. Jiang, S. Liu, Z. Hao, J. Cheng and G. Ouyang, Catal. Sci. Technol., 2018, 8, 5024–5033 RSC.
- B. Wang, Z. Xie, Y. Li, Z. Yang and L. Chen, Macromolecules, 2018, 51, 3443–3449 CrossRef CAS.
- Q. Huang, L. Guo, N. Wang, X. Zhu, S. Jin and B. Tan, ACS Appl. Mater. Interfaces, 2019, 11, 15861–15868 CrossRef CAS PubMed.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- B. P. Biswal, H. A. Vignolo-González, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |