
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ambidentate bonding and electrochemical implications of pincer-type pyridylidene amide ligands in complexes of nickel, cobalt and zinc†
Philipp
Melle
,
Nathalie
Ségaud
 and
Martin
Albrecht
*
and
Martin
Albrecht
*
Department für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: martin.albrecht@dcb.unibe.ch
First published on 21st August 2020
Abstract
Pincer-type tridentate pyridyl bis(pyridylidene amide) (pyPYA2) ligand systems were coordinated to the Earth-abundant first row transition metals nickel, cobalt and zinc. A one-pot synthesis in water/ethanol afforded octahedral homoleptic bis-PYA complexes, [M(pyPYA2)2](PF6)2, whereas five-coordinate mono-PYA dichloride complexes, [M(pyPYA2)Cl2], were obtained upon slow addition of the ligand to the metal chlorides in DMF. Electrochemical measurements further revealed a facile oxidation of the metal centers from Ni2+ to Ni4+ and Co2+ to Co3+, respectively, while the Zn2+ system was redox inactive. These experiments further allowed for quantification of the much stronger electron donor properties of neutral N,N,N-tridentate pyPYA2 pincer ligands as compared to terpy. Remarkably, ortho-PYA pincer ligands feature amide coordination to the metal center via oxygen or nitrogen. This ambidentate ligand binding constitutes another mode of donor flexibility of the PYA ligand system, complementing the resonance structure dynamics established previously. NMR spectroscopic and MS analysis reveal that the meta-PYA ligand undergoes selective deuteration when coordinated to cobalt. This reactivity suggests the potential of this ligand as a transient proton reservoir for HX bond activation and, moreover, indicates the relevance of several resonance structures and therefore supports the notion that meta-PYA ligands are mesoionic.
Introduction
The large-scale production of bulk chemicals requires the utilization of inexpensive materials, and many industrially relevant processes rely on heterogeneous catalysts based on Earth-abundant metals. Prominent examples are the iron oxide catalysts used in the Haber–Bosch process,1–3 nickel catalysts in steam reforming for the production of syngas4–6 or RANEY® nickel catalysts utilized in hydrogenation reactions.7,8 In contrast, the development of homogeneous catalysts has long focused on complexes based on platinum group and coinage metals.9 These noble metals feature excellent reactivity and stability properties, though their high price10 and scarcity11,12 as well as the high toxicity13 demand for more sustainable solutions and have led to extensive research activities aiming for the replacement of noble metals by first row transition metals.14 As a consequence, iron catalysis has made major progress over the last years,15 including for example the development of highly efficient hydrogenation catalysts that are competitive with the best ruthenium catalysts.16 Likewise manganese(I) pincer complexes have been explored as attractive alternative to established ruthenium- and iridium-based catalysts in (de)hydrogenation as well as in C–C and C–X bond-forming reactions.17 Also nickel complexes have gained much attention recently, e.g. as low-cost replacement for palladium in cross-coupling reactions.18We have recently developed a new pincer platform based on pyridylidene amide (PYA) ligands (A, Fig. 1), which imparted high catalytic activity to ruthenium centers in transfer hydrogenation catalysis.19,20 Based on the relatively hard yet formally neutral amide donor site of these PYA ligands, we sought to expand the application of this ligand system towards first row transition metals in view of providing a more sustainable alternative to noble metals. PYA ligands have been shown to induce attractive catalytic properties to iridium, ruthenium21–26 and palladium27 complexes, in parts by stabilizing high-valent oxidation states and in other parts due to their unique electron donor flexibility.28–30 Despite these attractive features, no first row transition metal complexes with PYA ligands have been reported, but analogous pyridylidene amine (PYE) ligands have been coordinated to nickel centers by the groups of Johnson31 and Douthwaite32 (B and C, Fig. 1). Here we report nickel, cobalt, and zinc complexes containing the pyridyl bis(PYA) (pyPYA2) pincer ligand and disclose some unique reactivity properties, including the linkage isomerism between N- to O-coordination of the PYA ligand as well as isotope exchange reactions at the meta-PYA unit that support a mesoionic notion of this ligand.
 | ||
| Fig. 1 Pincer pyridylidene amide (PYA) ruthenium complex A with two limiting PYA resonance structures, and pyridylidene amine (PYE) nickel complexes B and C reported earlier. | ||
Results and discussion
Synthesis and characterization of bis(PYA-pincer) complexes
The known para- and meta-pyPYA2 ligand precursor salts 1a and 1b were prepared according to standard procedures.19,33 Similarly, the ortho-pyPYA2 ligand precursor 1c was prepared from 2-aminopyridine and 2,6-pyridinedicarbonyl dichloride followed by selective methylation of both aminopyridine sites and anion metathesis. Notably, methylation required an excess of MeOTf (5 eq.) to be quantitative, while the use of MeI, which was sufficient for methylation of the para and meta analogues, resulted in only partial methylation. Complexation to cobalt and nickel was accomplished by reacting the dicationic ligand salts 1a–c with the corresponding chloride salt (CoCl2, NiCl2) in the presence of Na2CO3 as a base (Scheme 1). Complexation under these conditions consistently afforded the octahedral homoleptic complexes [M(pyPYA2)2](PF6)2 (M = Co, 2a–c; M = Ni, 3a–c) even when a large excess of the metal salt was used. This reactivity pattern contrasts the analogous ruthenation, which exclusively afforded mono-ligated [Ru(pyPYA2)(MeCN)3]2+ complexes. Complexes 2 and 3 are paramagnetic and NMR silent, their quantitative elemental composition, structure and purity was therefore confirmed by CHN microanalysis, single crystal X-ray diffraction, and HR ESI-MS. The latter showed the monocationic complex fragments [M − PF6]+, e.g. at 898.1733 amu for 2a (calcd 898.1733).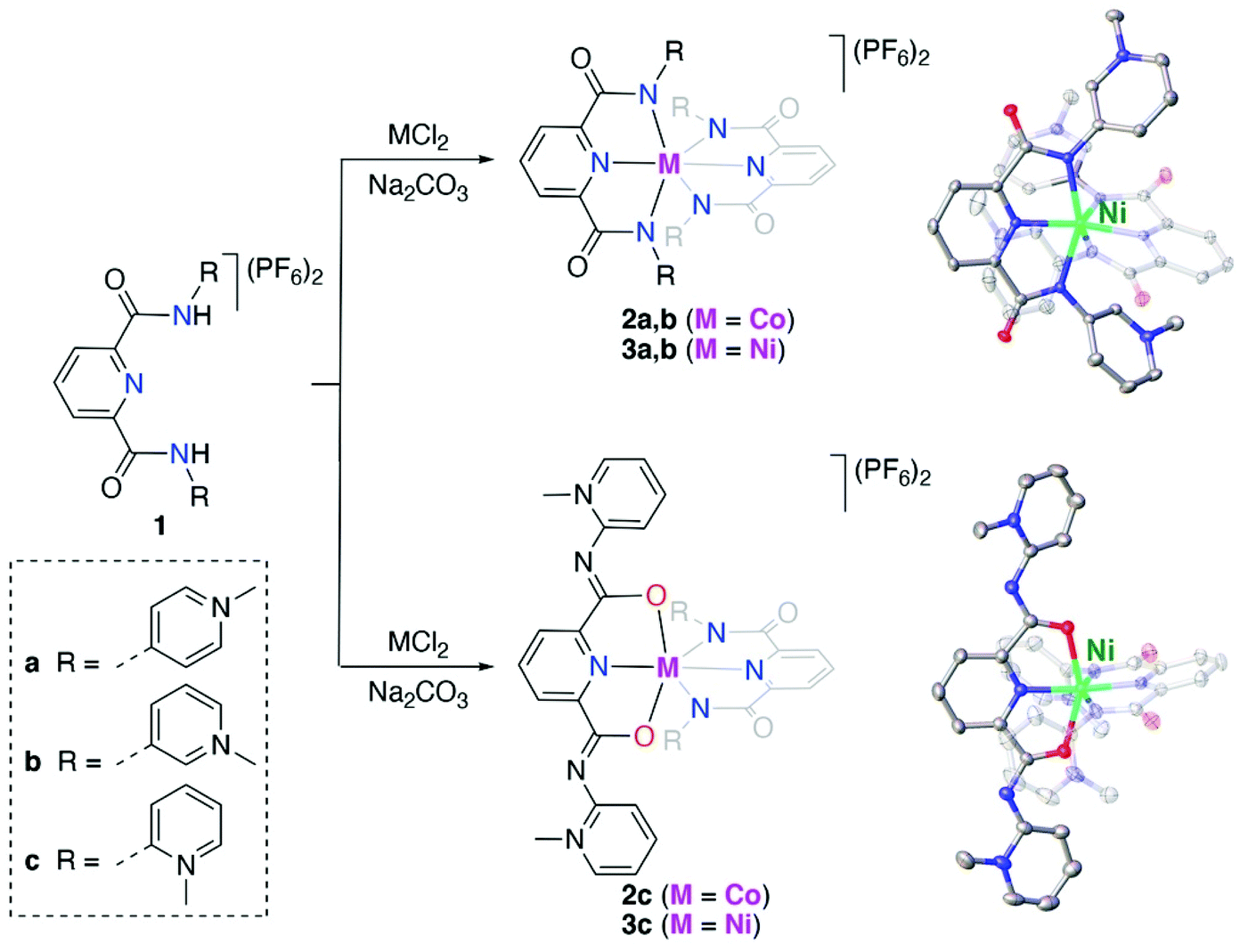 | ||
Scheme 1 Synthesis of Co(II) and Ni(II) bis-pyPYA2 pincer complexes 2a–c and 3a–c in H2O/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). Crystal structures of complex cations 3b and 3c; all ellipsoids at 50% probability level (hydrogen atoms and co-crystallized solvent molecules omitted for clarity). :1 v/v). Crystal structures of complex cations 3b and 3c; all ellipsoids at 50% probability level (hydrogen atoms and co-crystallized solvent molecules omitted for clarity). | ||
Crystallographic analysis revealed ambidentate coordination of the ortho-pyPYA2 ligand in 2c and 3c, as one of the ligands is bound to the metal center in the typical κ3-N,N,N bonding mode, while the other ligand binds with the amide through oxygen in a κ3-O,N,O coordination mode (cf.Scheme 1, Table S1, Fig. S1†). Both meta-pyPYA2 ligands in complexes 2b and 3b show the expected κ3-N,N,N coordination through the amide nitrogens and the central pyridine as observed for related dianionic pyridine dicarboxamidate systems.34–36 Such a linkage isomerism of PYA ligands has been proposed,37 yet never established so far38 and presents a further type of flexibility of the PYA system. Notably, the same ortho-pyPYA2 ligand displayed the classic κ3-N,N,N-coordination mode when bound to ruthenium(II) in [Ru(ortho-pyPYA2)(MeCN)3](PF6)2 (Fig. S2†). Likewise, pyridine dicarboxamide systems with ortho-dimethylated aryl substituents show exclusively κ3-N,N,N-bonding.35,39 The κ3-O,N,O coordination mode is unprecedented and presumably a consequence of the Lewis acidity of the first row transition metal centers and their ensuing oxophilicity, combined with the unique donor flexibility of the formally neutral and sterically constrained ortho-PYA donor site, and distinguishes these ligands from the more commonly used pyridine carboxamide systems.
Synthesis and characterization of mono(PYA-pincer) complexes
The synthesis of cobalt and nickel mono-PYA complexes, [M(pyPYA2)Cl2] complexes 4a–c (M = Co) and 5a–c (M = Ni; Scheme 2), was accomplished by slow addition of a filtered DMF solution containing the ligand and Na2CO3 to a DMF solution containing the metal chloride. The slow addition of ligand as well as the choice of solvent played a crucial role. Initial attempts to control the reaction of the PYA ligands 1a–c with MCl2 by quenching the reaction at early reaction times or by using a large excess of the metal salt (up to 10 eq.) invariably resulted in the formation of the homoleptic [M(pyPYA2)2]2+ compounds 2 and 3. | ||
| Scheme 2 Synthesis of Ni(II) and Co(II) [M(pyPYA2)Cl2] complexes 4a–c and 5a–c by slow addition of deprotonated ligand at room temperature. | ||
Formation of complexes 4 and 5 was established by CHN microanalysis as well as HR ESI-MS, which showed the monocationic complex fragments [M − Cl]+, e.g. at 441.0409 amu for 4a (calcd 441.0403). The set of cobalt complexes 4a–c was furthermore characterized by single crystal X-ray diffraction analysis (Fig. 2). The complexes feature a strongly distorted trigonal bipyramidal arrangement (τ5 = 0.45–0.49),40 which is in good agreement with the reported analogous structure of [Co(terpy)Cl2] (terpy = terpyridine).41 The para- and meta-PYA pincer ligands in complexes 4a,b bind to the metal center in a κ3-N,N,N coordination mode whereas the ortho-PYA pincer ligand in 4c is again κ3-O,N,O coordinated as observed for the bis-PYA pincer complexes. Notably, steric constraints should be less relevant when the PYA pincer ligand is bound to the CoCl2 fragment than in bis-PYA pincer complexes, indicating an electronic stabilization of the PYA coordination through oxygen.
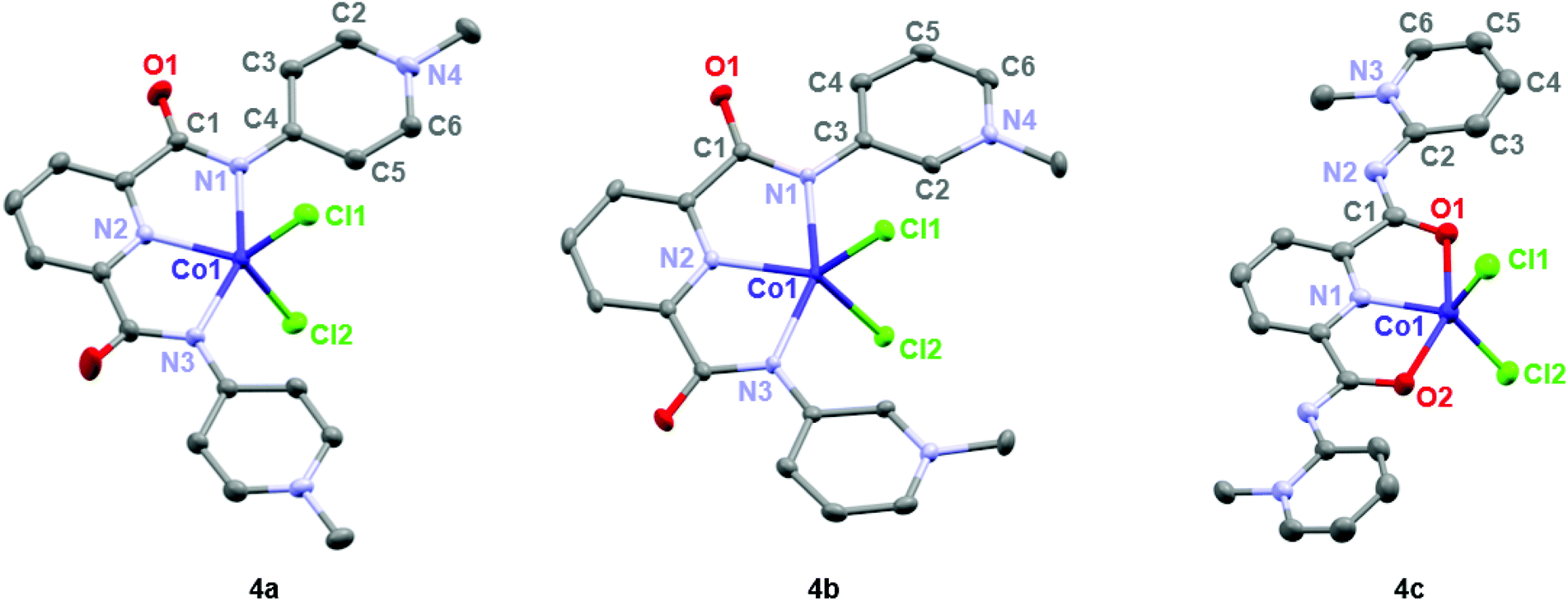 | ||
| Fig. 2 ORTEP representations of mono-pyPYA2 cobalt complexes 4a–c; all ellipsoids at 50% probability level (hydrogen atoms and co-crystallized solvent molecules omitted for clarity). | ||
The para- and meta-PYA pincer ligands in complexes 4a,b are almost planar, i.e. the dihedral angle between the PYA heterocycles and central pyridine ring are only 8.5°, 9.8° in 4a and 10.4° in 4b respectively (Table 1), which is distinctly different when compared to the correspondent homoleptic complex variation [Co(meta-pyPYA2)2](PF6)2 which show a 55° twist between the PYA and central pyridyl planes. This high planarity may hint to a significant π-contribution of the Namide–CPYA bonds (N1–C3/4) in both complexes. In such a model, the amide nitrogen is acting predominantly as neutral L-type N-donor ligand, presumably as a consequence of the higher electron density at the formally neutral metal center in complexes 4 and 5 compared to the dicationic center in the bis(pincer) complexes 2 and 3. In agreement with a more pronounced L-type and π-acidic binding mode of the amide nitrogen, the para-PYA heterocycles of complex 4a reveal a pronounced diene-imine resonance form with localized double and single bonds (average C2–C3 and C5–C6 1.365(4) Å, average C3–C4 and C4–C5 bonds (1.415(7) Å; Table 1). This bond alteration implies considerable contribution of the imine resonance structure of the PYA unit (cf. bottom structure of A in Fig. 1).
| 4a | 4b | 4c | ||
|---|---|---|---|---|
| a Average of two bonds. b Dihedral angle θ between the PYA heterocycles and the pyridyl-amide plane. c See ref. 40. | ||||
| Co1–N1 | 2.227(2) | 2.185(1) | Co1–O1 | 2.217(2) |
| Co1–N2 | 2.020(2) | 2.005(1) | Co1–N1 | 2.018(1) |
| Co1–N3 | 2.217(2) | 2.185(1) | Co1–O2 | 2.225(2) |
| Co1–Cl1 | 2.315(1) | 2.330(1) | Co1–Cl1 | 2.249(1) |
| Co1–Cl2 | 2.296(1) | 2.330(1) | Co1–Cl2 | 2.262(1) |
| N1–C1a | 1.360(3) | 1.347(2) | N2–C1a | 1.331(3) |
| C1–O1a | 1.231(2) | 1.244(2) | C1–O1a | 1.254(2) |
| N1–C3/4a | 1.373(2) | 1.391(2) | N2–C2a | 1.353(3) |
| C3–C4a | 1.412(3) | 1.406(2) | C2–C3a | 1.405(3) |
| C4–C5a | 1.419(3) | 1.388(2) | C3–C4a | 1.368(4) |
| C5–C6a | 1.364(3) | 1.366(2) | C4–C5a | 1.393(4) |
| C6–N4a | 1.345(3) | 1.349(2) | C5–C6a | 1.353(3) |
| N4–C2a | 1.353(2) | 1.342(2) | C6–N3a | 1.356(3) |
| C2–C3a | 1.365(3) | 1.399(2) | N3–C2a | 1.375(3) |
| N1–Co1–N3 | 153.1(1) | 154.1(1) | O1–Co1–O2 | 152.0(2) |
| θ PYA-pyrb | 8.5 & 9.8 | 10.4 & 10.4 | θ PYA-pyrb | 11.3 & 25.8 |
| τ 5 | 0.46 | 0.49 | τ 5 | 0.45 |
The κ3-O,N,O coordination mode of the ortho-PYA pincer ligand in mono-pyPYA2 complex 4c suggests this complex to be an intermediate en route to the formation of the heteroleptic bis-pyPYA2 pincer complex 2c. The stronger coordination of the O,N,O donor set may then accommodate a second PYA pincer ligand in the more typical κ3-N,N,N mode.
Dynamic linkage isomerism of the ortho-PYA-pincer ligand system
For better spectroscopic analysis in solution, the analogous zinc(II) complexes [Zn(pyPYA2)2](PF6)26a–c were prepared by reacting ligands 1a–c with 0.5 eq. ZnCl2 under mildly basic conditions (Scheme 3). In contrast to the paramagnetic and NMR-silent octahedral Co(II) and Ni(II) complexes 2 and 3, complexes 6a–c are diamagnetic and allow for NMR spectroscopic characterization. Ligand coordination to the ZnII center resulted in the loss of the NH amide proton resonances and in a marked upfield shift of the aromatic HPYA proton resonances, e.g. from δH = 9.76 (H6PYA) in meta-PYA ligand 1b to δ = 8.43 in complex 6b.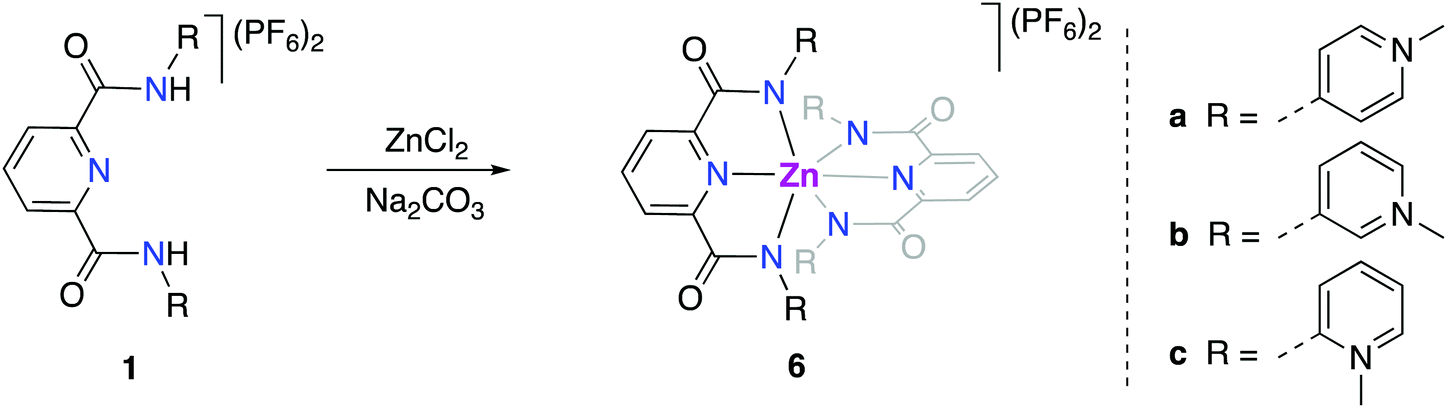 | ||
| Scheme 3 Synthesis of [Zn(pyPYA2)2](PF6)2 complexes 6a–c. (a) 0.5 eq. ZnCl2; 3 eq. Na2CO3 in MeCN. | ||
All zinc complexes 6a–c feature one distinct set of PYA ligand proton resonances, including N-CH3 (δH around 4), pyridyl (δH 8.25–8.70) and pyridylidene (δH 7.08–8.43) signals. This symmetric arrangement indicates that both pyPYA2 ligands in each zinc complex are coordinated in the same mode, most presumably the typical κ3-N,N,N-coordination based on the structures of the cobalt and nickel analogues and previously described structures.19,42–45
The symmetric ligand bonding in 6c containing ortho-pyPYA2 ligands contrasts the solid state bonding established for the analogous nickel and cobalt complexes 2c and 3c comprised of a mixture of κ3-N,N,N and κ3-O,N,O pincer coordination. NMR spectroscopic analysis of complex 6c at low temperature indicated decoalescence of the signals into two sets in a 9:1 integral ratio, indicating the presence of two species (Fig. 3). Based on the six inequivalent aromatic resonances, the major species was assigned to the homoleptic bis-κ3-N,N,N-coordinated pincer isomer. The minor isomer features at least ten inequivalent proton signals, which were therefore attributed to mixed κ3-N,N,N/κ3-O,N,O-coordination isomer (cf. structures of 2c, 3c). These measurements therefore suggest that at room temperature, complex 6c undergoes rapid κN-/κO-amide isomerization.
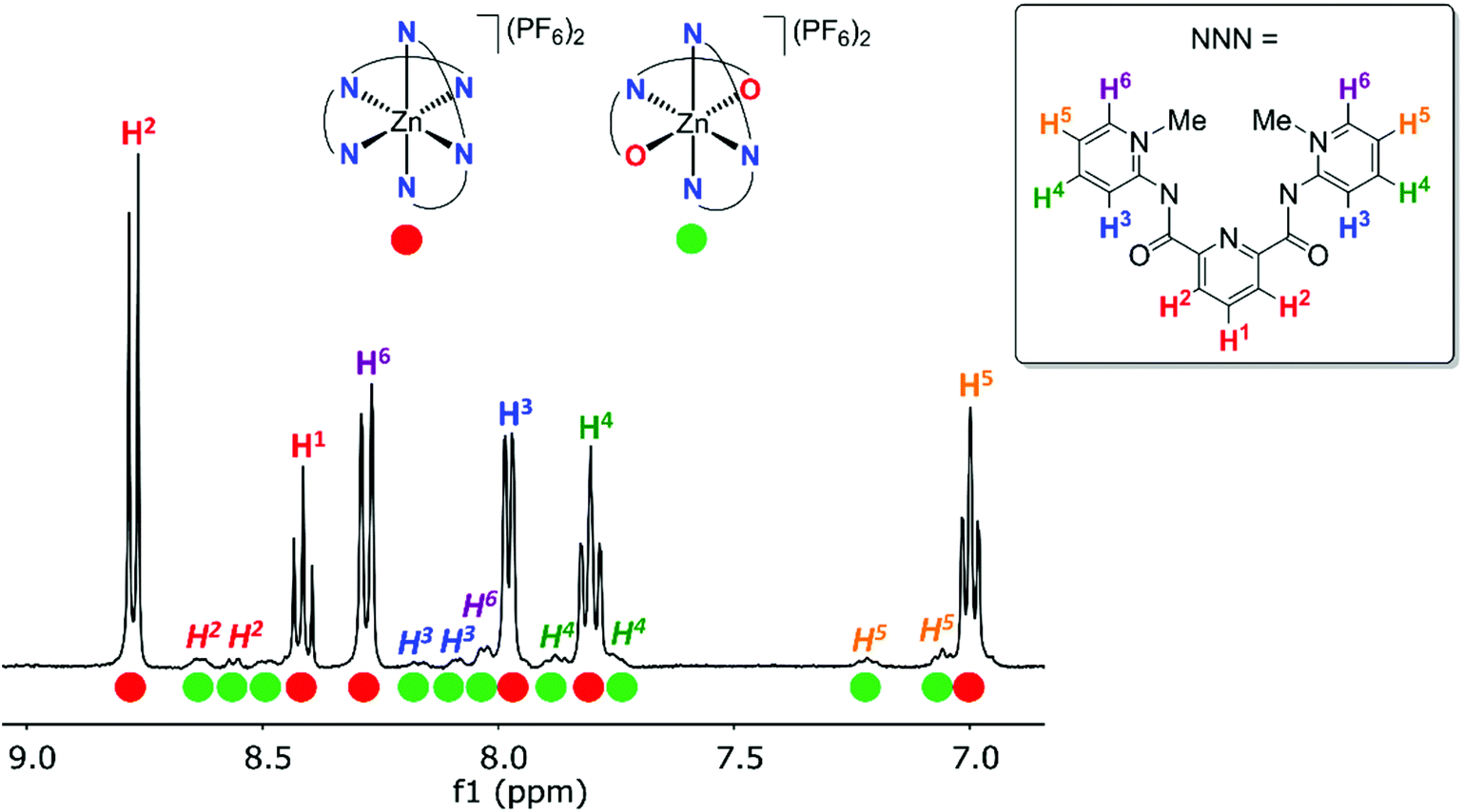 | ||
| Fig. 3
1H NMR spectrum of [Zn(ortho-pyPYA2)2](PF6)26c in CD2Cl2 at −80 °C, revealing two different complexes in 9:1 ratio. The major species was assigned to the bis-(κ3-N,N,N)-coordination isomer (red circles), the minor species to the mixed κ3-N,N,N/κ3-O,N,O-coordination isomer (green circles). | ||
The dynamic process of this complex was investigated by variable temperature 1H NMR spectroscopic experiments in CD2Cl2 in the +20 to −80 °C temperature range (Fig. S3†). Decoalescence of most of the pyridyl and PYA proton resonances was observed around −40 °C, apart from the PYA H6 signal, which undergoes the largest shift and features a coalescence temperature Tc around −20 °C. Based on these values and applying the Eyring equation, an average activation energy ΔG‡ = 11.4 ± 0.3 kcal mol−1 was calculated for this PYA linkage isomerism (Table S2†).46,47 A similar behavior was noted in CD3CN (Fig. S4†), though due to the higher melting point of this solvent, the decoalescence was not resolved. Nonetheless, these experiments suggest that the dynamic bonding is not significantly depending on the solvent.
The bonding mode in the bis-(pyPYA2) complexes was further analyzed by solid state infrared (IR) spectroscopy of the pure compounds. The ligand precursor 1a features three characteristic absorption bands for the amide carbonyl and PYA-imine units in the 1580–1700 cm−1 region (Table 2, Fig. S5–S11†).48 Upon metalation, the two lower bands around 1590 and 1645 cm−1 remain essentially unchanged, while the high energy band at 1706 cm−1 shifts characteristically to 1617(±1) cm−1, presumably due to metal coordination to the amide unit (Fig. 4a). Likewise, the strong absorption above 1700 cm−1 for ligand precursors 1b and 1c shifts to lower energy upon metalation, though assignments are more complicated as all bands shift considerably (Fig. 4b and Fig. S5†). It is worth noting that the shift is independent of the metal center and that the IR spectra are essentially identical for Co(II), Ni(II), and Zn(II) complexes for the para- and meta-pyPYA2 ligand even in the low frequency area. This high similarity suggests an identical coordination mode for all three metal complexes. For the ortho-pyPYA2 series, however, subtle differences are noted in the 1550–1650 cm−1 range which distinguish the Zn from the Ni and Co systems (Fig. 4c). These differences may point to a different ligand behavior, which may be associated with the κ3-N,N,N vs. κ3-O,N,O linkage isomerism established for the [Zn(ortho-pyPYA2)2](PF6)2 complex 6c by NMR spectroscopy. IR spectroscopy therefore does not conclusively indicate whether this linkage isomerism is exclusively occurring at Zn2+, or whether it is more general for ortho-pyPYA2 ligands and also takes place at Co2+ and Ni2+. The exclusive κ3-O,N,O bonding in the solid state of the latter two complexes (cf. structures of 2c–5c) might be induced by lower solubility or a preferential crystal packing of the mixed coordination isomer, leading to a dynamic kinetic resolution of the two isomers through crystallization.
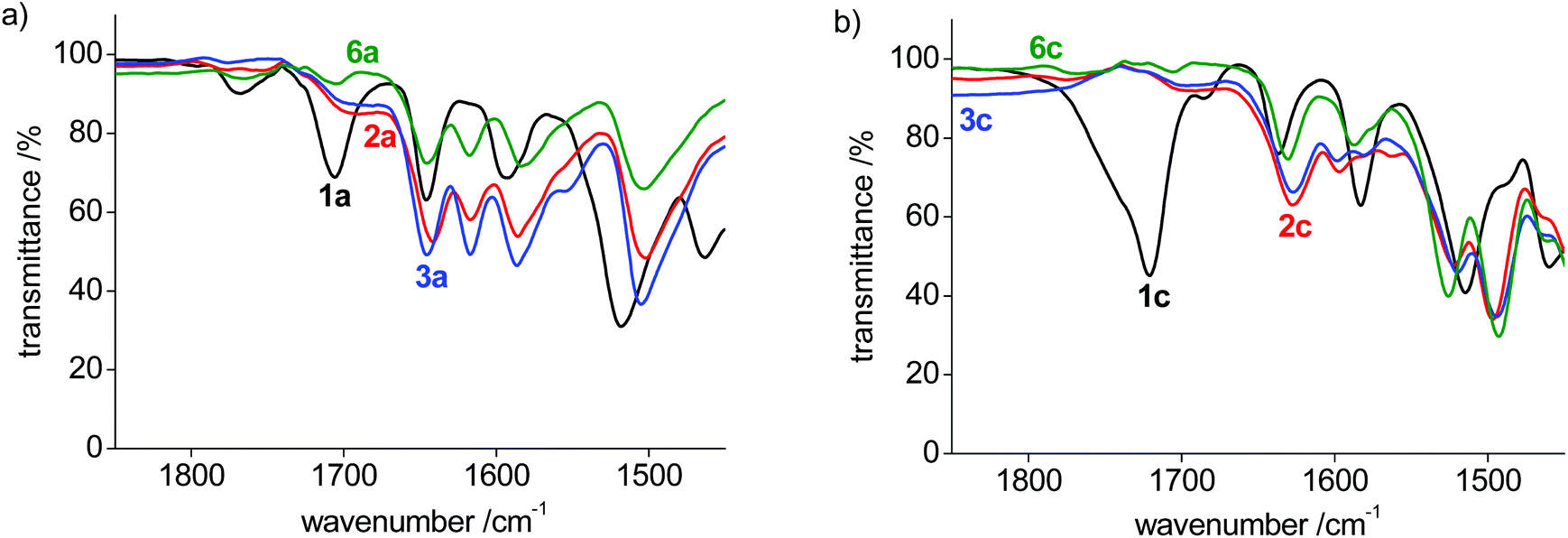 | ||
| Fig. 4 IR spectra of (a) para-pyPYA2 pincer complex series; (b) ortho-pyPYA2 pincer complex series. | ||
| Entry | Ligand motif | Bonding partner |
ν
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CN [cm−1] O/CN [cm−1] |
E 1/2 (MII/III)a [V] | E 1/2 (MIII/IV)a [V] | |
|---|---|---|---|---|---|---|
| a Potentials in MeCN vs. SSCE at 100 mV s−1 scan rate using the Fc+/Fc couple as standard; E1/2 = +0.43 V and (nBu4N)PF6 as electrolyte, 0–2.2 V sweep range; ΔEp = |Epc − Epa| [mV] in parenthesis. | ||||||
| 1 | para-PYA | H | 1a | 1593, 1646, 1706 | — | — |
| 2 | para-PYA | Co | 2a | 1586, 1617, 1642 | 0.20 (90) | — |
| 3 | para-PYA | Ni | 3a | 1587, 1617, 1645 | 1.20 (80) | 1.72 (150) |
| 4 | para-PYA | Zn | 6a | 1584, 1618, 1646 | — | — |
| 5 | meta-PYA | H | 1b | 1695 | — | — |
| 6 | meta-PYA | Co | 2b | 1567 | 0.01 (80) | — |
| 7 | meta-PYA | Ni | 3b | 1564 | 1.03 (80) | 1.54 (90) |
| 8 | meta-PYA | Zn | 6b | 1566 | — | — |
| 9 | ortho-PYA | H | 1c | 1583, 1637, 1721 | — | — |
| 10 | ortho-PYA | Co | 2c | 1597, 1628 | 0.40 (100) | — |
| 11 | ortho-PYA | Ni | 3c | 1598, 1627 | 1.34 (100) | — |
| 12 | ortho-PYA | Zn | 6c | 1587, 1630 | — | — |
Redox properties of homoleptic bis-PYA pincer complexes
The orange meta-pyPYA2 cobalt(II) complex 2b oxidized to the green Co(III) analogue 2b+ under aerobic conditions in a saturated aqueous solution (Scheme 4). The oxidation state of the cobalt center in 2b+ was unambiguously identified by crystallographic analysis due to the 0.2 Å shorter Co–N bond distances compared to 2b as well as the presence of three PF6− counter ions. This oxidation under mild conditions indicates strong donor properties of the PYA pincer ligand in these cobalt and nickel complexes. Therefore, the electrochemical properties of the complexes were investigated by cyclic voltammetry. | ||
| Scheme 4 Oxidation of [Co(meta-pyPYA2)2](PF6)22b in H2O under aerobic conditions, to the Co(III) complex 2b+. Crystal structure of complex cation 2b+; all ellipsoids at 50% probability level (hydrogen atoms and co-crystallized solvent molecules omitted for clarity). | ||
Cobalt complexes 2a–c show reversible redox processes in the 0 to +0.5 V potential range that were assigned to metal-centered Co2+ to Co3+ oxidations (all potentials vs. Ag/AgCl; Fig. 5). The octahedral complex [Co(terpy)2](PF6)249 was also included to compare the donor strength of the formally neutral tridentate PYA pincer ligands with a well-studied reference compound. The reversibility of the MII/III oxidation processes in complexes 2 and 3 is supported by the linear relationship of anodic (Ipa) and cathodic peak currents (Ipc) vs. the square root of the scan rate (Fig. S12†). This linearity together with the small peak potential difference ΔEp < 105 mV (Table 2) indicates a diffusion limited electron transfer process.50–52 The oxidation potential is dependent on the PYA substitution pattern and is lowest for cobalt coordinated to the meta-PYA pincer ligand (E1/2 = 0.01 V in 2b) and increases with para-PYA (E1/2 = 0.20 V in 2a)53 and even more with ortho-PYA pincer ligands (E1/2 = 0.40 V in 2c). It is noteworthy that [Co(ortho-pyPYA2)2](PF6)2 complex 2c features two consecutive redox processes in the potential range of 0.3–0.6 V, which might be caused by possible linkage isomerism in solution and the presence of two coordination isomers (exclusively κ3-N,N,N-coordinated ligand and mixed coordination mode, cf. NMR studies of analogous ortho-PYA zinc complex above). These half wave potentials indicate an increase of ligand donor strength along the series ortho-PYA < para-PYA < meta-PYA, a trend that has been previously observed for [Ru(pyPYA2)L3]2+ and [IrCp*(phenyl-PYA)L]+ complexes.19,54 This trend points to a strongly π-basic character of the meta-PYA donor, while the donor properties of the ortho-PYA unit are probably attenuated by the vicinity of the formally positively charged pyridinium nitrogen. When compared to the redox properties of [Co(terpy)2]2+ (E1/2 = 0.32 V)55 the ortho-PYA pincer ligand is less donating than terpy, while the para and even more so the meta analogue are stronger donors.
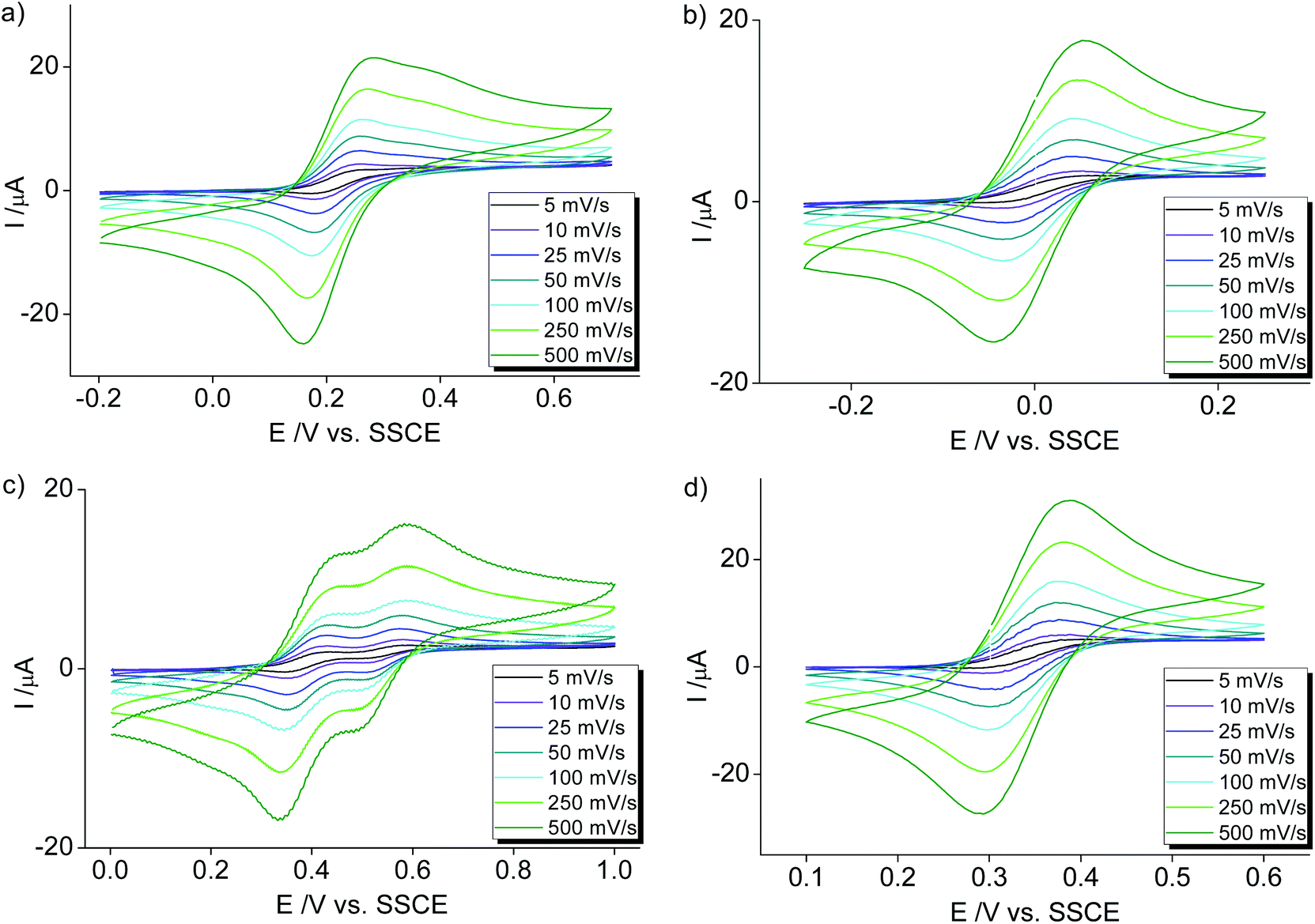 | ||
| Fig. 5 Cyclic voltammetry measurements of homoleptic [Co(pyPYA2)2](PF6)2 complexes in MeCN: (a) 2a [Co(para-pyPYA2)2](PF6)2; (b) 2b [Co(meta-pyPYA2)2](PF6)2; (c) 2c [Co(ortho-pyPYA2)2](PF6)2; (d) [Co(terpy)2](PF6)2. Conditions: 10 mL solvent, 1 mM sample and 100 mM (nBu4N)PF6 as supporting electrolyte at 5–500 mV s−1 scan rate (potentials vs. Ag/AgCl using the Fc+/Fc couple as standard; E1/2 = +0.4319). | ||
The metal-centered nature of the oxidation process was confirmed by redox analyses of the Zn analogues 6a–c. These complexes do not show any redox activity in the 0–2.2 V potential window (Fig. S14†), indicating that the coordinated PYA pincer ligands are redox inactive within this range. Likewise, the three pyridinium salt ligand precursors 1a–c do not show any oxidation process up to 2 V and feature an irreversible reduction processes around −1 V (Fig. S13†). This reduction is dependent on the PYA substitution pattern and easiest for ortho-PYA system 1c (Epc = −0.98 V), intermediate for meta-PYA 1b (Epc = −1.22 V) and at lowest potential for the para analogue 1a (Epc = −1.33 V).
Electrochemical characterization of the nickel complexes supports the trend in donor strength of the PYA pincer ligands as observed for the Co series. Thus, the Ni3+ oxidation state was stabilized better by the meta-PYA pincer ligand (E1/2 = 1.03 V in 3b) than the para analogue (E1/2 = 1.20 V in 3a) or the ortho-pyPYA2 system (E1/2 = 1.34 V in 3c; Fig. 6a–c). In addition, the stronger donor sets also provided access to NiIV complexes via a second reversible oxidation at higher potential (E1/2 = 1.72 V and 1.54 V for complexes 3a and 3b, respectively). Notably, [Ni(terpy)2](PF6)2 as reference compound is oxidized at considerably higher potential (E1/2 = 1.65 V),55 suggesting weaker donation of the terpy ligand than even the least basic ortho-pyPYA2 ligand.
 | ||
| Fig. 6 Cyclic voltammetry measurements of homoleptic [Ni(pyPYA2)2](PF6)2 complexes in MeCN: (a) 3a [Ni(para-pyPYA2)2](PF6)2; (b) 3b [Ni(meta-pyPYA2)2](PF6)2; (c) 3c [Ni(ortho-pyPYA2)2](PF6)2. Conditions: 10 mL solvent, 1 mM sample and 100 mM (nBu4N)PF6 as supporting electrolyte at 5–500 mV s−1 scan rate (potentials vs. SSCE using the Fc+/Fc couple as standard; E1/2 = +0.4319). | ||
H/D exchange in meta-PYA sites
A closer investigation of the homoleptic cobalt complex 2b with meta-PYA pincer ligands revealed intriguing ligand-based reactivity. Crystallization of this complex in D2O under aerobic conditions resulted in a mixture of orange and green crystals. The latter are a product of spontaneous oxidation of a the metal center and furthermore revealed selective deuterium incorporation into the PYA heterocycle, as demonstrated by MS and NMR analyses. Material crystallized from D2O showed the m/z signals for the mono-, di-, and tri-cationic species at 8, 4 and 2.66 amu higher, respectively, than samples of 2b+ crystallized from H2O (Table S3†). Formation of 2b+-d8 was further supported by the absence of the PYA resonances at δH 8.11 and 8.25 in the 1H NMR spectrum of the complex generated in D2O (Fig. S15†). Based on NOE spectroscopy as well as the pertinent coupling constants, these resonances were assigned to H2PYA and H6PYA, respectively, i.e. the positions ortho to the PYA nitrogen. These data indicate a highly selective H/D exchange. Interestingly, the isomeric complexes 2a and 2c containing para- and ortho-PYA pincer ligands, respectively do not undergo any detectable deuteration or oxidation (MS, NMR) when exposed to D2O, presumably because of the higher oxidation potential of these complexes (vide supra). The selective deuterium incorporation in the PYA 2- and 6-position of 2b may be rationalized by considering resonance forms B and C of the meta-PYA ligand system, which imply a formally neutral imine donor site and opposite charges within the pyridylidene heterocycle (Scheme 5). Reversible deuteration/protonation at the partially negatively charged positions would account for the observed selectivity and indicates that resonance forms B and C are contributing in addition to resonane form A. This contribution of several resonance structures therefore validates meta-PYA systems as mesoionic compounds.56 | ||
| Scheme 5 Limiting resonance structures of the mesoionic meta-PYA ligand site. The negative charge is localized on the amide nitrogen and in 2-, 4- and 6-position of the PYA heterocycle. | ||
Conclusions
Here, we have shown that PYA pincer ligands form stable complexes with nickel, cobalt and zinc, which expands the application potential of this ligand beyond platinum group metals to first row metals. The substitution pattern of the PYA heterocycle influences the binding mode of the pincer ligand to these Earth-abundant transition metals. meta- and para-pyPYA2 ligand systems are exclusively κ3-N,N,N-coordinated, whereas the ortho-methylated PYA pincer ligand forms either κ3-O,N,O-coordinated mono-pyPYA2 compounds or bis-pyPYA2 complexes with a mixed κ3-O,N,O/κ3-N,N,N coordination mode. According to electrochemical analyses, the donor strength of the PYA ligand systems decrease in the order meta-PYA > para-PYA > ortho-PYA. The mesoionic meta-PYA pincer cobalt complex displays unique reactivity, since the 2- and 6-positions of the pyridinium heterocycle are selectively deuterated in D2O under mild conditions. This deuteration suggests partial delocalization of the negative charge and therefore mesoionic properties of the meta-PYA unit. Moreover, this reactivity may become potentially useful for metal–ligand cooperative HX bond activation catalysis.Experimental
General
All reagents were commercially available and used as received. Ligands 1a and 1b have been prepared according to previously described methods.19 Unless specified otherwise, 1H NMR spectra were recorded at 25 °C on Bruker spectrometers operating at 300 MHz. Chemical shifts (δ in ppm, coupling constants J in Hz) were referenced to residual solvent signals. Assignments are based on homo- and heteronuclear shift correlation spectroscopy. Purity of bulk samples of the complexes has been established by NMR spectroscopy, and when possible by elemental analysis. Elemental analyses were performed at DCB Microanalytic Laboratory using a Thermo Scientific Flash 2000 CHNS-O elemental analyzer. High-resolution ESI mass spectrometry was carried out with a Thermo Scientific LTQ Orbitrap XL (ESI-TOF). Cyclic voltammograms were recorded using an Autolab PGSTAT101 from Metrohm in MeCN solutions: 10 mL solvent, 1 mM sample and 100 mM tetrabutylammonium hexafluorophosphate (nBu4N)PF6 as supporting electrolyte. Solutions were deaerated with argon gas for 10 min prior to each run. The scan rate was 100 mV s−1. Redox potentials were measured using a Pt-button working electrode, an Ag/AgCl reference electrode and a Pt-wire auxiliary electrode and are tabulated versus a ferrocene internal standard. The Fc+/Fc couple is 0.43 V vs. Ag/AgCl in 0.1 M (nBu4N)PF6 MeCN solutions.57 ATR-FTIR spectra were recorded with a Jasco FT/IR-4700 spectrometer with a Jasco ATR Pro One unit containing a diamond crystal.Synthetic procedures
:2). The product crystallized upon slow cooling to room temperature, was filtered and washed with small portions of cold water (3 × 20 mL) and dried under vacuum. 1c was obtained as white crystalline material (0.73 g; 72%). 1H NMR (300 MHz, MeCN-d3) δ 10.45 (s, 2H, NH), 8.64–8.51 (m, 6H, HPYA, Hpyr), 8.46–8.36 (m, 3H, HPYA, Hpyr), 7.87–7.79 (m, 2H, HPYA), 4.29 (s, 6H, N-CH3). 13C NMR (75 MHz, MeCN-d3) δ 163.1 (CO), 148.1 (Cpyr), 148.0 (CPYA), 147.9 (CHPYA), 146.5 (CHPYA), 141.9 (CHpyr), 128.8 (CHpyr), 125.1 (CHPYA), 124.7 (CHPYA), 45.4 (N-CH3). HR ESI-MS: m/z calculated for C19H19O2N5F6P [M − PF6]+ = 494.1151; found: 494.12. Elemental analysis: anal. calculated for C19H19F12N5O2P2: C: 35.70; H: 3.00; N: 10.95. Found: C: 36.28; H: 2.95; N: 11.03.
General procedure for compounds 2/3a–c
The respective amounts of the ligands 1a–c (150 mg, 0.235 mmol, 1 eq.), Na2CO3 (75 mg, 0.705 mmol, 3 eq.) and the MCl2·6H2O salts (28 mg, 0.117 mmol, 0.5 eq.) were dissolved in a H2O/EtOH (1:1) mixture (10 mL) and stirred at r.t. for 30 min. The solvents were evaporated under reduced pressure. The crude product was redissolved in MeCN, cooled down to −18 °C and filtered over a pad of Celite to remove sodium salt residues. The final products were obtained upon precipitation/crystallization from Et2O.
2b+: ESI-MS: m/z calculated for [M − PF6]+ = 1043.1375; found: 1043.1393. 1H NMR (300 MHz, MeCN-d3): δ 8.39 (t, 3JHH = 7.7 Hz, 2H, Hpyr), 8.27–8.22 (m, 4H, H6PYA), 8.15 (d, 3JHH = 7.7 Hz, 4H, Hpyr), 8.11 (s, 4H, H2PYA), 7.62–7.56 (m, 8H, H4/5PYA), 4.09 (s, 12H, N-CH3) ppm.
2b+-d8: ESI-MS: m/z calculated for [M − PF6]+ = 1051.1877; found: 1051.1883. 1H NMR (300 MHz, MeCN-d3): δ 8.39 (t, 3JHH = 7.7 Hz, 2H, Hpyr), 8.15 (d, 3JHH = 7.7 Hz, 4H, Hpyr), 7.61–7.56 (m, 8H, H4/5PYA), 4.10 (s, 12H, N-CH3) ppm.
General procedure for compounds 4/5a–c
The respective amounts of the ligands 1a–c (102 mg, 0.16 mmol, 1 eq.) and Na2CO3 (45 mg, 0.42 mmol, 3 eq.) were dissolved in DMF (4 mL) and stirred for 20 min at r.t. In a second vial the MCl2·6H2O salts (38 mg, 0.16 mmol, 1 eq.) were dissolved in DMF (4 mL). The ligand solution was filtered and slowly added to the metal solution. The combined reaction phase was stirred for another 2 h at r.t. and the final products were obtained upon precipitation/crystallization from Et2O and MeCN/Et2O.General procedure for compounds 6a–c
The respective amounts of ligands 1a–c (100 mg, 0.16 mmol, 1 eq.) were reacted with Na2CO3 (85 mg, 0.80 mmol, 5 eq.) in MeCN (5 mL) for 1 h at room temperature in a vial. Then a 0.2 M ZnCl2 solution in MeCN (470 μL, 0.09 mmol, 0.55 eq.) was added to the ligand solution. The vial was sealed and heated up to 50 °C for 16 h under constant stirring. The reaction mixture was cooled to r.t. and filtered through Celite. A white solid was precipitated from Et2O (20 mL). The solid was collected by filtration over a fritted funnel, rinsed with Et2O (3 × 5 mL) and dried.Crystal-structure determination
Suitable single crystals of 2b–c, 3b–c, and 4a–c were mounted in air at ambient conditions and measured on an Oxford Diffraction SuperNova area-detector diffractometer58 using mirror optics monochromated Mo Kα radiation (λ = 0.71073 Å) and Al filtered.59 The unit cell constants and an orientation matrix for data collection were obtained from a least-squares refinement of the setting angles of reflections. Data reduction was performed using the CrysAlisPro58 program. The intensities were corrected for Lorentz and polarization effects, and a numerical absorption correction based on Gaussian integration over a multifaceted crystal model was applied. The structure was solved by direct methods using SHELXT,60 which revealed the positions of all non-hydrogen atoms of the title compounds. The non-hydrogen atoms were refined anisotropically. All H-atoms were placed in geometrically calculated positions and refined using a riding model where each H-atom was assigned a fixed isotropic displacement parameter with a value equal to 1.2Ueq of its parent atom (1.5Ueq for methyl groups and water). Refinement of the structure was carried out on F2 using full-matrix least-squares procedures, which minimized the function ∑w(Fo2 − Fc2)2. The weighting scheme was based on counting statistics and included a factor to downweight the intense reflections. All calculations were performed using the SHELXL-2014/761 program in OLEX2.62 The PF6− anions in the crystals of complex 2b, 2c, and 3b are disordered as indicated by the high residual electron density peak near its P-atom. No conclusive disorder model was found for 2b and 3b, while for 2c, disorders about two sites were restrained by SHELX SIMU and RIGU instructions. Further details are given in Tables S4–S6.† Crystallographic data for all structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication numbers 2015875 (2b), 2015876 (2b+), 2015877 (2c), 2015878 (3b), 2015879 (3c), 2015880 (4a), 2015881 (4b), 2015882 (4c), and 2015883 ([Ru(ortho-pyPYA2)(MeCN)3](PF6)2).†
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge generous financial support from the European Research Council (CoG 615653) and from the Swiss National Science Foundation (200021_162868 and 200020_182663, R'equip projects 206021_128724 and 206021_170755). We thank the group of Chemical Crystallography of the University of Bern for the X-ray analysis of all reported structures.References
- F. Haber and G. van Oordt, Z. Anorg. Allg. Chem., 1905, 44, 341–378 CrossRef.
- M. Appl, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2011 Search PubMed.
- Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- G. F. Froment, J. Mol. Catal. A: Chem., 2000, 163, 147–156 CrossRef CAS.
- L. Wang, D. Li, M. Koike, H. Watanabe, Y. Xu, Y. Nakagawa and K. Tomishige, Fuel, 2013, 112, 654–661 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 2000, 63, 159–164 CrossRef CAS.
- L. W. Covert and H. Adkins, J. Am. Chem. Soc., 1932, 54, 4116–4117 CrossRef CAS.
- H. Adkins and H. R. Billica, J. Am. Chem. Soc., 1948, 70, 695–698 CrossRef CAS.
- M. Albrecht and G. Van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- B. Su, Z. C. Cao and Z. J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS.
- H. C. Urey, Rev. Mod. Phys., 1952, 28, 53–74 Search PubMed.
- J. W. Morgan and E. Anders, Proc. Natl. Acad. Sci. U. S. A., 1969, 77, 6973–6977 CrossRef.
- P. E. Brubaker, J. P. Moran, K. Bridbord and F. G. Hueter, Environ. Health Perspect., 1975, 10, 39–56 CrossRef CAS.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS.
- W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1084 CrossRef CAS.
- M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344–4362 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS.
- P. Melle, Y. Manoharan and M. Albrecht, Inorg. Chem., 2018, 57, 11761–11774 CrossRef CAS.
- P. Melle, J. Thiede, D. A. Hey and M. Albrecht, Chem. – Eur. J., 2020 DOI:10.1002/chem.202001145.
- P. Melle and M. Albrecht, Chimia, 2019, 73, 299–303 CrossRef CAS.
- K. F. Donnelly, C. Segarra, L.-X. Shao, R. Suen, H. Müller-Bunz and M. Albrecht, Organometallics, 2015, 34, 4076–4084 CrossRef CAS.
- V. Leigh, D. J. Carleton, J. Olguin, H. Mueller-Bunz, L. J. Wright and M. Albrecht, Inorg. Chem., 2014, 53, 8054–8060 CrossRef CAS.
- K. Salzmann, C. Segarra and M. Albrecht, Angew. Chem., 2020, 59, 8932–8936 CrossRef CAS.
- A. Weilhard, K. Salzmann, M. Navarro, J. Dupont, M. Albrecht and V. Sans, J. Catal., 2020, 385, 1–9 CrossRef CAS.
- M. Navarro, C. Segarra, P. Tim and M. Albrecht, Organometallics, 2020, 39, 2383–2391 CrossRef CAS.
- M. Navarro, V. Rosar, T. Montini, B. Milani and M. Albrecht, Organometallics, 2018, 37, 3619–3630 CrossRef CAS.
- M. Navarro, M. Li, H. Müller-Bunz, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2016, 22, 6740–6745 CrossRef CAS.
- M. Navarro, M. Li, S. Bernhard and M. Albrecht, Dalton Trans., 2017, 47, 659–662 RSC.
- M. Navarro, C. A. Smith, M. Li, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2018, 24, 6386–6398 CrossRef CAS.
- M. E. Doster and S. A. Johnson, Angew. Chem., Int. Ed., 2009, 48, 2185–2187 CrossRef CAS.
- Q. Shi, R. J. Thatcher, J. Slattery, P. S. Sauari, A. C. Whitwood, P. C. McGowan and R. E. Douthwaite, Chem. – Eur. J., 2009, 15, 11346–11360 CrossRef CAS.
- A. Dorazco-González, H. Höpfl, F. Medrano and A. K. Yatsimirsky, J. Org. Chem., 2010, 75, 2259–2273 CrossRef.
- A. K. Patra and R. Mukherjee, Inorg. Chem., 1999, 38, 1388–1393 CrossRef CAS.
- D. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4701 CrossRef CAS.
- P. Pirovano, E. R. Farquhar, M. Swart and A. R. McDonald, J. Am. Chem. Soc., 2016, 138, 14362–14370 CrossRef CAS.
- C. M. A. Muller, M. V. Babak, M. Kubanik, M. Hanif, S. M. F. Jamieson, C. G. Hartinger and L. J. Wright, Inorg. Chim. Acta, 2016, 450, 124–130 CrossRef CAS.
- With iron and cobalt observed in pyridine dicarboxamide systems, see: (a) G. L. Guillet, J. B. Gordon, G. N. Di Francesco, M. W. Calkins, E. Čižmár, K. A. Abboud, M. W. Meisel, R. García-Serres and L. J. Murray, Inorg. Chem., 2015, 54, 2691–2704 CrossRef CAS; (b) T. C. Harrop, L. A. Tyler, M. M. Olmstead and P. K. Mascharak, Eur. J. Inorg. Chem., 2003, 475–481 CrossRef CAS.
- P. Pirovano, B. Twamley and A. R. McDonald, Chem. – Eur. J., 2018, 24, 5238–5245 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- E. Goldschmied and N. C. Stephenson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 1867–1875 CrossRef CAS.
- X. Zhang, D. Huang, Y. S. Chen and R. H. Holm, Inorg. Chem., 2012, 51, 11017–11029 CrossRef CAS.
- D. Bansal, G. Kumar, G. Hundal and R. Gupta, Dalton Trans., 2014, 43, 14865–14875 RSC.
- D. Bansal and R. Gupta, Dalton Trans., 2019, 48, 14737–14747 RSC.
- For examples of κ3-O,N,O coordination due to grid restrains, see: (a) L. K. Thompson, L. Zhao, Z. Xu, D. O. Miller and W. M. Reiff, Inorg. Chem., 2003, 42, 128–139 CrossRef CAS; (b) L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Inorg. Chem., 2009, 48, 3323–3341 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- The TC were obtained upon comparing 1H NMR spectra of 6c recorded in CD2Cl2 in the +20 to −80 °C temperature range. The difference of the resonance frequencies (δν) was calculated from chemical shift differences for protons of the major species, i.e. bis-(κ3-N,N,N-pincer), to the average chemical shift
of the corresponding protons in the minor species (κ3-N,N,N/κ3-O,N,O-isomer). The energy barrier (ΔG‡) between the two isomers was estimated by using the following equation: ΔG‡ = 4.57·Tc·(9.97 + logTc/δν) according to:
H. Günther, NMR-Spektroskopie, Thieme, Stuttgart, 1973 Search PubMed.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Thieme, Stuttgart, Germany, 8th edn, 2012 Search PubMed.
- C. M. Harris, T. N. Lockyer, R. L. Martin, R. L. Patil, E. Sinn and I. M. Stewart, Aust. J. Chem., 1969, 22, 2105–2126 CrossRef CAS.
- M. Navarro, Doctoral dissertation, University of Bern, 2017.
- C. Arana, S. Yan, M. Keshavarz-K, K. T. Potts and H. D. Abruña, Inorg. Chem., 1992, 31, 3680–3682 CrossRef CAS.
- Ideal value one electron process is 56.5 mV according to Butler–Volmer equation, experimental values are known to be slightly higher; T. Erdey-Grúz and M. Volmer, Z. Phys. Chem., 1930, 150, 203–213 Search PubMed.
- We note that the CV of complex 2a indicates a second oxidation process at slightly higher potential. This process might be rationalized by a linkage isomerism also in this compound, though we do not have any experimental support for dynamic O- vs. N-coordination of para-pyPYA2 ligands.
- J.-M. Savéant, Elements of Molecular and Biomolecular Electrochemistry, John Wiley & Sons, Hoboken, United States, 2006 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentaland Applications, John Wiley & Sons, Hoboken, United States, 2nd edn, 2001 Search PubMed.
- Mesoionic compounds are defined by IUPAC as “dipolar five- (possibly six-) membered heterocyclic compounds in which both the negative and the positive charge are delocalized, for which a totally covalent structure cannot be written, and which cannot be represented satisfactorily by any one polar structure. The formal positive charge is associated with the ring atoms, and the formal negative charge is associated with ring atoms or an exocyclic nitrogen or chalcogen atom”. IUPAC. Compendium of Chemical Terminology, 2nd Ed. Gold Book Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 1997. XML online corrected version: http://goldbook.iupac.org (2006) created by M. Nic, J. Jirat, and B. Kosata updates compiled by A. Jenkins. ISBN 0-9678550-9-8.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- Oxford Diffraction Ltd., CrysAlisPro, Yarnton, Oxfordshire, U.K., Version 1., 2010 Search PubMed.
- P. Macchi, H.-B. Bürgi, A. S. Chimpri, J. Hauser and Z. Gál, J. Appl. Crystallogr., 2011, 44, 763–771 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details, IR and NMR spectra, electrochemical analyses, synthetic details for the Ru complex. CCDC 2015875–2015883. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02482a |
| This journal is © The Royal Society of Chemistry 2020 |