
Surface immobilized copper(I) diimine photosensitizers as molecular probes for elucidating the effects of confinement at interfaces for solar energy conversion†
Michael S.
Eberhart
 a,
Brian T.
Phelan
a,
Jens
Niklas
a,
Emily A.
Sprague-Klein
a,
David M.
Kaphan
a,
David J.
Gosztola
b,
Lin X.
Chen
ac,
David M.
Tiede
a,
Oleg G.
Poluektov
a and
Karen L.
Mulfort
*a
a,
Brian T.
Phelan
a,
Jens
Niklas
a,
Emily A.
Sprague-Klein
a,
David M.
Kaphan
a,
David J.
Gosztola
b,
Lin X.
Chen
ac,
David M.
Tiede
a,
Oleg G.
Poluektov
a and
Karen L.
Mulfort
*a
aDivision of Chemical Sciences and Engineering, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: mulfort@anl.gov
bCenter for Nanoscale Materials, Argonne National Laboratory, Lemont, IL 60439, USA
cDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA
First published on 14th September 2020
Abstract
Heteroleptic copper(I) bis(phenanthroline) complexes with surface anchoring carboxylate groups have been synthesized and immobilized on nanoporous metal oxide substrates. The species investigated are responsive to the external environment and this work provides a new strategy to control charge transfer processes for efficient solar energy conversion.
Copper(I) coordination complexes have attracted interest for integration in solar energy conversion schemes because of their broad absorption in the visible region and the relative earth abundance of copper compared to the ruthenium centre of the prototype molecular photosensitizer [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine).1–6 Recent work focused on ligand designs of Cu(I) photosensitizers has demonstrated the potential for these earth-abundant complexes to initiate light-driven H2 catalysis in multimolecular systems.7–9 Herein, we describe a fundamentally new strategy for controlling and directing photochemical processes through dimensional control of a metal oxide environment.
For Cu(I) bis(phen) (phen = 1,10-phenanthroline) complexes, substitution of the phen ligands and accompanied distortions to the ideal tetrahedral symmetry profoundly influence their ground- and excited-state properties.10–13 By comparing Cu(I)(dmp)2 and Cu(I)(dtbp)2 (where dmp = 2,9-dimethyl-1,10-phen, dtbp = 2,9-di-tert-butyl-1,10-phen),14 we know that increasing the steric bulk at the 2,9-phen position from methyl to tert-butyl results in a 29 nm blue shift of the absorbance feature assigned to MLCT from λmax = 454 nm to λmax = 425 nm and a 0.20 V increase in the Cu(II/I) oxidation potential.15 Both observations are explained by the bulkier substituents yielding a more idealized D2d symmetry in the Cu(I) state, increasing the Cu–N bond length, and restricting flattening in the Cu(II) oxidation state. The light-induced structural dynamics of these two complexes are well understood from investigations using multiple time-resolved optical and X-ray spectroscopy methods.16,17 In the non-coordinating solvent CH2Cl2, the 3MLCT decay for Cu(I)(dmp)2 is ∼90 ns, but increases to 1.9 μs for Cu(I)(dtbp)2 due to the substituents preventing deactivation pathways associated with structural distortion in the Cu(II) MLCT state and minimizing Cu(II)–solvent interaction. These general design principles are well-established for homoleptic Cu(I) bis(phen) complexes, and the same trends apply to heteroleptic Cu(I)(phenA)(phenB) complexes.18,19
Short excited-state lifetimes are problematic for diffusional reactions, but electrode surface immobilization provides a direct, well-defined pathway for interfacial electron transfer. Our group has shown that Cu(I) diimine complexes anchored to TiO2 substrates inject photogenerated electrons from the 1MLCT state on the sub-picosecond time scale while charge recombination occurs on the microsecond timescale, indicating a long lived charge separated state.5,20 Indeed, these favourable charge separation kinetics have led to an interest in deploying Cu(I) diimine complexes as the photosensitizer in dye-sensitized solar cells.1,21 In the pursuit of long-lived charge separated states from Cu(I) complexes, the immobilization approach provides more synthetic versatility than imposing the strict functionalization requirements for stable homoleptic complexes, and can also exploit what we have learned about the directional electron transfer in heteroleptic Cu(I) diimine complexes.6,22
The primary goal of this work is to understand whether the structural factors that dominate the solution phase photophysical properties of Cu(I) diimine complexes also apply in heterogeneous environments. To this end, we have synthesized three new heteroleptic Cu(I) bis(phen) (CuHETPHEN) complexes functionalized with carboxylic acid groups which are capable of binding to metal oxide surfaces (Chart 1). These complexes were designed to evaluate two different ligand factors that we anticipate will impact the photophysical properties, particularly when immobilized or confined. Complex 1 has a short anchor which is directly connected to the phen ligand. In contrast, the imidazole-containing anchoring ligand of complexes 2 and 3 is approximately 6.5 Å longer than that of 1 and should allow studies to investigate the effect of distance and packing efficiency on the ground- and excited-state properties. Complexes 2 and 3 differ only in the 2,9-phen substitution on the anchor ligand and provide a direct comparison for how the known solution trends apply to immobilized CuHETPHEN complexes.
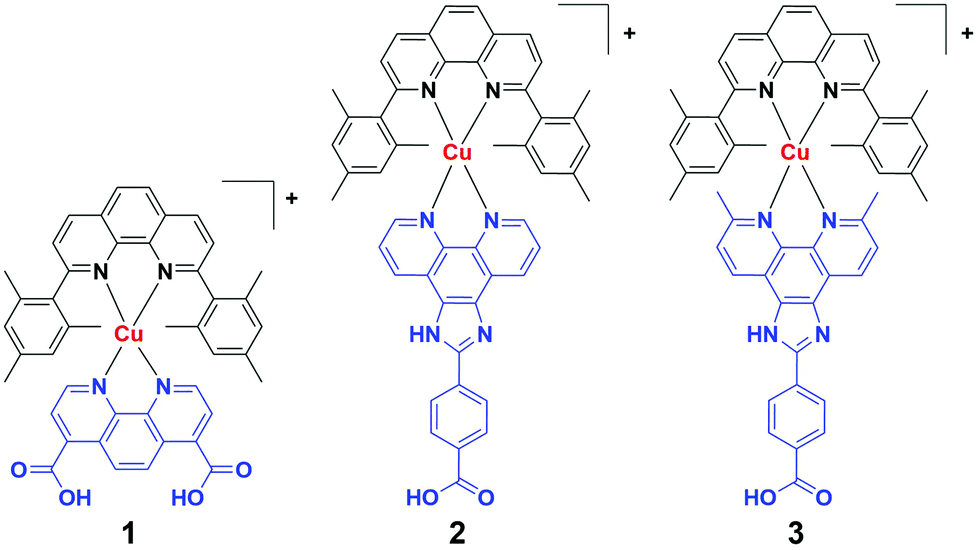 | ||
| Chart 1 CuHETPHEN complexes used in this study for surface immobilization. Blocking ligand indicated in black, surface anchoring ligand indicated in blue. | ||
Complexes 1, 2, and 3 were synthesized by the two-step one-pot HETPHEN approach that yields kinetically stable complexes with no formation of the homoleptic analogues.23–26 However, we had to refine the standard synthesis methods because of the poor solubility of the phenanthroline ligands functionalized with carboxylic acids. We dissolved Cu(CH3CN)4PF6 in de-aerated CH2Cl2 or THF depending on the target complex, then added a stoichiometric amount of 2,9-dimesityl-1,10-phenanthroline (L) followed by one equivalent of carboxylate-functionalized ligands L1, L2, or L3 to form complexes 1, 2, and 3. The synthesis of 1 was accomplished in neat CH2Cl2 but the somewhat more Lewis basic solvent THF was required for synthesis of 2 and 3.
To systematically investigate the ground- and excited-state properties of immobilized CuHETPHEN complexes, we selected nanoporous anodic aluminium oxide (AAO) wafers as the metal oxide platform. AAO wafers are commercially available in a variety of pore sizes ranging from 10 nm to 200 nm,27 and the controlled pore sizes offer a uniform chemical environment compared to the larger distribution of pore sizes that are present in more conventional metal oxide surfaces such as TiO2 nanoparticle films.28 Complexes 1, 2, and 3 were immobilized on AAO wafers by submerging the wafers in a CH2Cl2 or CH3OH solution of each complex overnight. The AAO wafers adopted the characteristic red-orange colour of the CuHETPHEN complexes and upon removal from solution the wafers were rinsed, soaked in pure solvent, and rinsed again to remove any residual weakly adsorbed complex.
UV-Visible absorbance spectra can be used to gather information on the coordination environment of Cu(I) diimine complexes.13,29,30 The AAO-immobilized MLCT band of 1 and 2 blue shift 10–30 nm as compared to each complex in CH2Cl2 solution (Fig. S3 and S4, ESI†), which is consistent with increased restriction to geometric changes in the complex.14,15 We observe only a minimal shift in the MLCT band for 3 between the immobilized complex and in solution (Fig. 1), which suggests that immobilization has less of an impact on the spectral response than the increased sterics of the phen ligand environment. Additionally, we were able to correlate the density of immobilized 1–3 on the surface of the AAO wafers from the UV-Vis absorbance spectra and the AAO pore sizes and densities (Tables S3 and S4, ESI†). We determined that 1–3 were adsorbed on the surfaces at a density of approximately 1–2 complexes per square nanometre which correlates to roughly monolayer coverage based on related immobilized complexes.31
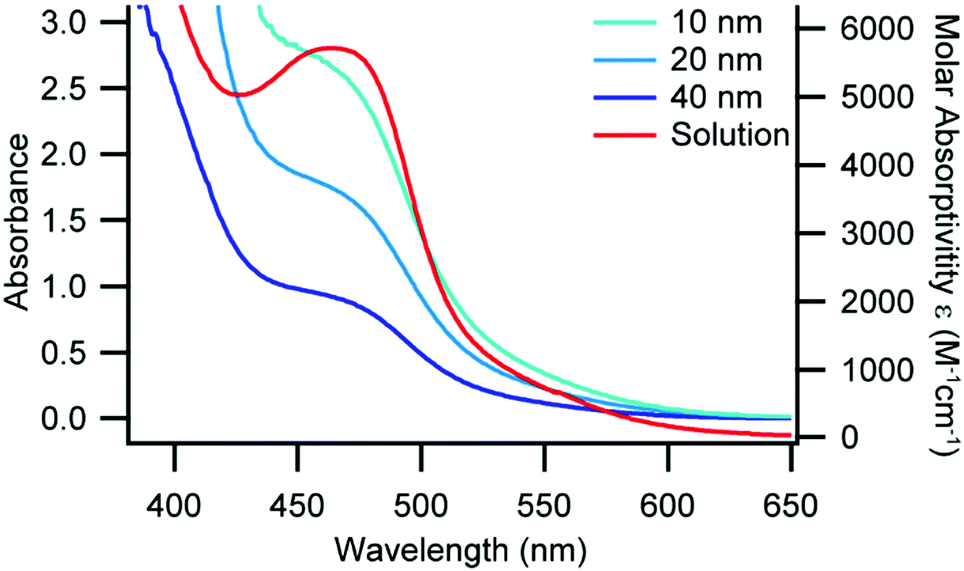 | ||
| Fig. 1 UV-Vis absorbance of 3 in CH2Cl2 (red, right axis) and immobilized in AAO pores of various pore size (blue traces, left axis), constant path length. | ||
To further probe the impact of pore immobilization on molecular and electronic structure, we utilized EPR spectroscopy due to its high sensitivity for paramagnetic centres and ability to elucidate structural and electronic information of the copper centre in the immobilized complexes. Since Cu(I) is diamagnetic and therefore EPR silent, we immobilized 3 on 10, 20, and 40 nm pore size AAO, oxidized the surface bound Cu(I) to the corresponding Cu(II) species using a solution of AgNO3 in CH3CN, and recorded X-band EPR spectra at 20 K (Fig. 2). For all of the AAO pore sizes, we observe spectra characteristic of Cu(II) 3d9 (S = 1/2),32,33 but with noticeable differences in the linewidth of the parallel, low field components as a function of AAO pore size. (We assign the sharp signal at 338 mT to an organic radical/defect, g ≈ 2.005, that we also observed in the bare AAO lacking any immobilized copper species, see Fig. S3, ESI†). The largest pore size of AAO investigated (40 nm) has the smallest linewidth (∼290 MHz), which increases as the pore size decreases from 20 nm (∼300 MHz) to 10 nm (∼550 MHz). Since there is no evidence of a chemical change to 3 in the different pore sizes, we interpret the broadening as a response to shorter neighbour–neighbour distances between Cu(II) ions in the smaller pores, which leads to stronger magnetic dipole–dipole interaction. Using the assumption that there is no substantial contribution to line width from spin–spin interactions in glassy frozen dilute solutions of Cu(II) complexes (see Fig. S7 (ESI†) and associated discussion), we estimate the distance between the immobilized Cu(II) neighbours to be ∼12 Å for the 40 and 20 nm pores, and ∼8.5 Å for 10 nm AAO pores.34 This difference in spin–spin interaction likely arises from decreasing distance between neighbouring chromophore tethering sites with changes in pore size and may also be related in part to increased curvature with decreasing pores size.
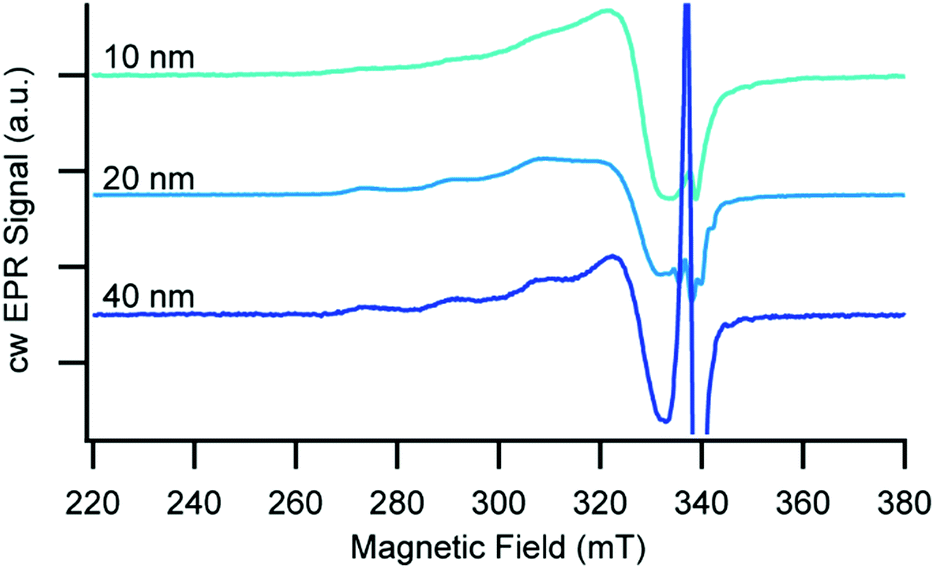 | ||
| Fig. 2 Continuous wave X-band EPR spectra of 3 in the Cu(II) oxidation state immobilized on 10, 20, and 40 nm AAO recorded at T = 20 K. | ||
Cyclic voltammetry of 1, 2, and 3 was performed in solution as well as immobilized on nanoITO which functions as both a substrate and working electrode since AAO is an insulator (Fig. 3 and Fig. S8, ESI†). The dominant factor in determining the Cu(II/I) oxidation potential is the substitution at the 2,9-phen position of the anchor ligand, consistent with previous observations.18 As expected, the Cu(II/I) potential for 3 is substantially more positive than for 1 and 2, both in solution and when immobilized (Fig. 3 and Fig. S8, Table S6, ESI†). Importantly, the potentials of 1 and 2 experience a more significant shift on immobilization than for 3 (230 mV for 1 and 2vs. 50 mV for 3) which suggests 3 is less influenced by the external environment, consistent with the observations based on UV-vis spectroscopy.
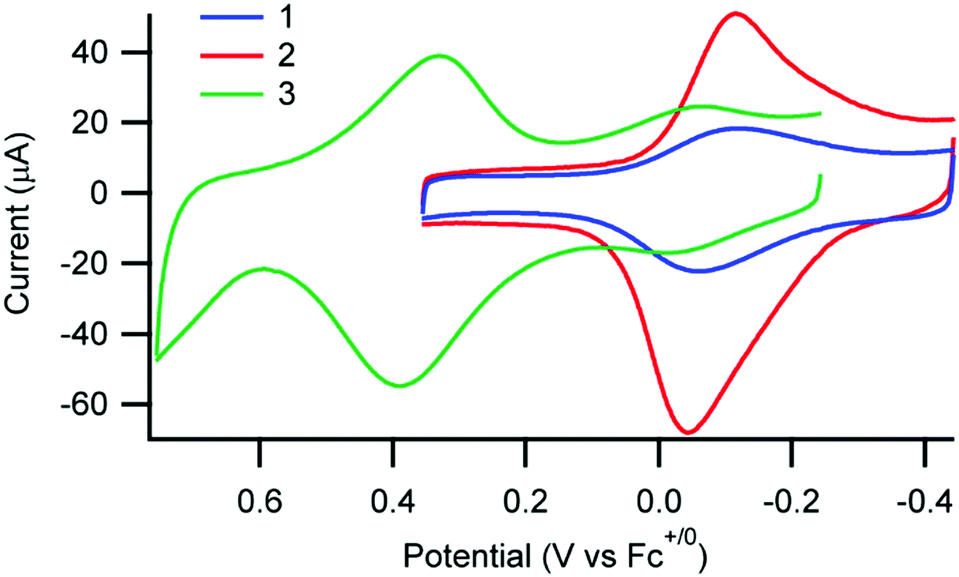 | ||
| Fig. 3 Cyclic voltammograms of 1, 2, and 3 immobilized on nanoITO substrates in 0.1 M nBu4PF6 in CH3CN. | ||
The excited-state dynamics of the AAO-immobilized complexes were measured by ultrafast and nanosecond transient optical spectroscopy. Because of the complicated spectral changes and multiple photophysical processes of Cu(I) diimine complexes,13,17,29 we used global analysis to fit the transient absorption data over a broad range of wavelengths with multi-exponential functions (complete fitting details are found in the ESI†). Similar to the well-documented photophysical behaviour for related complexes in solution, the transient spectra for the immobilized complexes are characterized by an initial broad featureless excited-state absorbance centred around 540 nm which evolves to a feature with two local λ-maxima at 530 and 570 nm within 30 ps (see for example 3 immobilized on 10 nm AAO in Fig. 4 and Fig. S8–S39, ESI†). As expected from literature precedent, methyl substitution in 3 results in a significant increase in excited-state lifetimes compared to 1 and 2, and the solution data are summarized in Table S7 (ESI†).
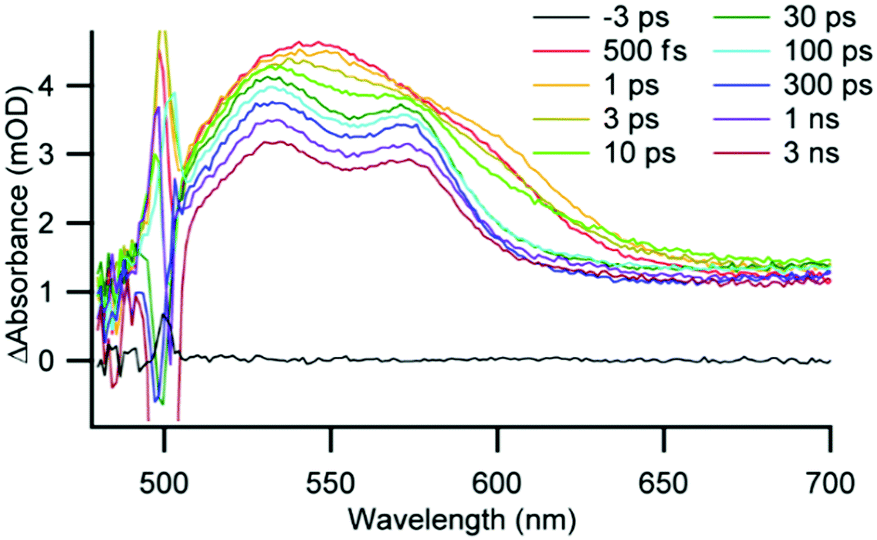 | ||
| Fig. 4 Ultrafast transient absorption spectra of 3 on 10 nm AAO in deaerated CH2Cl2. Excitation at 500 nm, time delay noted in legend. | ||
We immobilized complexes 1 and 3 on AAO and immersed the wafers in CH2Cl2 to investigate the impact of immobilization and pore size on the excited-state kinetics. Both 1 and 3 showed significantly longer excited-state lifetimes when immobilized on AAO than in solution, suggesting that immobilization restricts the ability of the complexes to undergo a flattening distortion and reduces exposure to solvent following photoexcitation. For complex 1, the 3MLCT lifetime is 1.0 ns in solution and increases to 1.7 ns when immobilized on 20 nm AAO. The average 3MLCT lifetime for 3 in CH2Cl2 is 49 ns, similar to that in 40 nm pores (51 ns), but the average 3MLCT lifetime increases to 69 ns for both 20 nm and 10 nm pores. This trend is similar to that previously observed for Cu(I)(dmp)2 dispersed into a polymer matrix where the 3MLCT lifetime increased from 90 ns in CH2Cl2 to 210 ns with increasing polymer viscosity.35 We interpret our results in a similar fashion: as 3 is immobilized in increasingly smaller pores and in closer proximity to other immobilized Cu centres (as measured by EPR), the typical flattening distortion is restricted which increases the lifetime of the excited state.
As a first step toward investigating the effect of nanoporous confinement on interfacial charge injection, we measured the electron transfer kinetics of 3 immobilized on mesoporous n-type semiconducting TiO2 films immersed in CH2Cl2 (Fig. S40 and S41, ESI†). Upon photoexcitation at 500 nm, we observe an average lifetime of 75 μs for the recovery of the ground-state bleach, which demonstrates that 3 does inject electrons into TiO2. The recombination lifetime is comparable to analogous cases of other Cu(I) species bound to TiO2.5,20
Immobilization of Cu(I) diimine complexes in nanoporous metal oxide materials presents an interesting opportunity to tune the photophysical properties of these complexes by external environmental factors. Here we have shown that decreasing the pore size of the AAO framework from 40 to 10 nm results in enhanced spin–spin interactions between Cu(II) centres, as well as a measurable increase in the 3MLCT lifetime, over twice that observed in solution. The increase in 3MLCT lifetime is consistent with inhibited structural flattening as pore size decreases and ongoing work is focused on restricting pore diameter even further to investigate the impact of extreme confinement on Cu(I) diimine excited-state dynamics. These results present a fundamentally new strategy for controlling and directing photochemical processes.
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, through Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Synthesis and electrochemistry were supported by the Early Career Research Program, EPR and TA facilities were supported by the Solar Photochemistry program. Transient optical spectroscopy was performed in part at the Center for Nanoscale Materials. The CNM is an Office of Science user facility supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386–8398 Search PubMed.
- M. S. Lazorski and F. N. Castellano, Polyhedron, 2014, 82, 57–70 Search PubMed.
- M. Sandroni, Y. Pellegrin and F. Odobel, C. R. Chim., 2016, 19, 79–93 Search PubMed.
- S.-P. Luo, E. Mejía, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419–423 Search PubMed.
- M. W. Mara, D. N. Bowman, O. Buyukcakir, M. L. Shelby, K. Haldrup, J. Huang, M. R. Harpham, A. B. Stickrath, X. Zhang, J. F. Stoddart, A. Coskun, E. Jakubikova and L. X. Chen, J. Am. Chem. Soc., 2015, 137, 9670–9684 Search PubMed.
- D. Hayes, L. Kohler, R. G. Hadt, X. Zhang, C. Liu, K. Mulfort and L. X. Chen, Chem. Sci., 2018, 9, 860–875 Search PubMed.
- R. S. Khnayzer, C. E. McCusker, B. S. Olaiya and F. N. Castellano, J. Am. Chem. Soc., 2013, 135, 14068–14070 Search PubMed.
- B. J. McCullough, B. J. Neyhouse, B. R. Schrage, D. T. Reed, A. J. Osinski, C. J. Ziegler and T. A. White, Inorg. Chem., 2018, 57, 2865–2875 Search PubMed.
- S. Saeedi, C. Xue, B. J. McCullough, S. E. Roe, B. J. Neyhouse and T. A. White, ACS Appl. Energy Mater., 2019, 2, 131–143 Search PubMed.
- M. K. Eggleston, D. R. McMillin, K. S. Koenig and A. J. Pallenberg, Inorg. Chem., 1997, 36, 172–176 Search PubMed.
- M. T. Miller, P. K. Gantzel and T. B. Karpishin, Inorg. Chem., 1998, 37, 2285–2290 Search PubMed.
- J. V. Lockard, S. Kabehie, J. I. Zink, G. Smolentsev, A. Soldatov and L. X. Chen, J. Phys. Chem. B, 2010, 114, 14521–14527 Search PubMed.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 Search PubMed.
- N. A. Gothard, M. W. Mara, J. Huang, J. M. Szarko, B. Rolczynski, J. V. Lockard and L. X. Chen, J. Phys. Chem. A, 2012, 116, 1984–1992 Search PubMed.
- O. Green, B. A. Gandhi and J. N. Burstyn, Inorg. Chem., 2009, 48, 5704–5714 Search PubMed.
- M. Iwamura, S. Takeuchi and T. Tahara, Phys. Chem. Chem. Phys., 2014, 16, 4143–4154 Search PubMed.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 Search PubMed.
- L. Kohler, D. Hayes, J. Hong, T. J. Carter, M. L. Shelby, K. A. Fransted, L. X. Chen and K. L. Mulfort, Dalton Trans., 2016, 45, 9871–9883 Search PubMed.
- L. Kohler, R. G. Hadt, D. Hayes, L. X. Chen and K. L. Mulfort, Dalton Trans., 2017, 46, 13088–13100 Search PubMed.
- J. Huang, O. Buyukcakir, M. W. Mara, A. Coskun, N. M. Dimitrijevic, G. Barin, O. Kokhan, A. B. Stickrath, R. Ruppert, D. M. Tiede, J. F. Stoddart, J.-P. Sauvage and L. X. Chen, Angew. Chem., Int. Ed., 2012, 51, 12711–12715 Search PubMed.
- M. Sandroni, M. Kayanuma, A. Planchat, N. Szuwarski, E. Blart, Y. Pellegrin, C. Daniel, M. Boujtita and F. Odobel, Dalton Trans., 2013, 42, 10818–10827 Search PubMed.
- D. Hayes, L. Kohler, L. X. Chen and K. L. Mulfort, J. Phys. Chem. Lett., 2018, 9, 2070–2076 Search PubMed.
- M. Schmittel and A. Ganz, Chem. Commun., 1997, 999–1000, 10.1039/A701509G.
- S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 Search PubMed.
- Y. Pellegrin, M. Sandroni, E. Blart, A. Planchat, M. Evain, N. C. Bera, M. Kayanuma, M. Sliwa, M. Rebarz, O. Poizat, C. Daniel and F. Odobel, Inorg. Chem., 2011, 50, 11309–11322 Search PubMed.
- M. Sandroni, A. Maufroy, M. Rebarz, Y. Pellegrin, E. Blart, C. Ruckebusch, O. Poizat, M. Sliwa and F. Odobel, J. Phys. Chem. C, 2014, 118, 28388–28400 Search PubMed.
- InRedox, http://www.inredox.com/product/aao-wafers/, accessed May 14, 2020.
- B. O'Regan, J. Moser, M. Anderson and M. Graetzel, J. Phys. Chem., 1990, 94, 8720–8726 Search PubMed.
- G. B. Shaw, C. D. Grant, H. Shirota, E. W. Castner, G. J. Meyer and L. X. Chen, J. Am. Chem. Soc., 2007, 129, 2147–2160 Search PubMed.
- M. W. Mara, N. E. Jackson, J. Huang, A. B. Stickrath, X. Zhang, N. A. Gothard, M. A. Ratner and L. X. Chen, J. Phys. Chem. B, 2013, 117, 1921–1931 Search PubMed.
- M. K. Brennaman, A. O. T. Patrocinio, W. Song, J. W. Jurss, J. J. Concepcion, P. G. Hoertz, M. C. Traub, N. Y. Murakami Iha and T. J. Meyer, ChemSusChem, 2011, 4, 216–227 Search PubMed.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–207 Search PubMed.
- J. R. Pilbrow, Transition Ion Electron Paramagnetic Resonance, Oxford University Press, 1991 Search PubMed.
- R. T. Ross, J. Chem. Phys., 1965, 42, 3919–3922 Search PubMed.
- A. O. Razgoniaev, C. E. McCusker, F. N. Castellano and A. D. Ostrowski, ACS Macro Lett., 2017, 6, 920–924 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods for molecular synthesis and immobilization, description of physical characterization methods, additional spectra, and details of kinetic analysis. See DOI: 10.1039/d0cc05972b |
| This journal is © The Royal Society of Chemistry 2020 |