
A dinuclear porphyrin-macrocycle as efficient catalyst for the hydrogen evolution reaction†
Julia
Jökel‡
a,
Fabian
Schwer‡
b,
Max
von Delius
 *b and
Ulf-Peter
Apfel
*ac
*b and
Ulf-Peter
Apfel
*ac
aInorganic Chemistry I, Ruhr-University Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: ulf.apfel@rub.de; ulf-peter.apfel@umsicht.fraunhofer.de
bInstitute of Organic Chemistry, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: max.vondelius@uni-ulm.de
cFraunhofer UMSICHT, Osterfelder Str. 3, 46047 Oberhausen, Germany
First published on 21st October 2020
Abstract
We report an unprecedented dinuclear catalyst for the electrochemical hydrogen evolution reaction (HER). A macrocyclic porphyrin complex comprising two nickel centres connected via redox mediating linker molecules gives rise to efficient catalysis, significantly outperforming a mononuclear reference catalyst.
Electrocatalytic water splitting using renewable energy is a promising process for the sustainable production of hydrogen.1 However, a shift from costly noble metal catalysts such as Pt to earth abundant elements is required to meet the growing global energy demand.2 While industrial catalysts are commonly of heterogeneous nature, molecular catalysts are well-defined and therefore provide insights into the fundamental mechanisms of catalysis.3 With naturally occurring enzymes enabling the interconversion of protons and H2 by [FeFe]- or [NiFe]-active sites, the potential of noble metal free bimetallic catalysts comprising for instance Fe or Ni seems evident.4
Although numerous structural models, inspired by the active site of both hydrogenases have been reported, most of them require high overpotentials and strong acids with low TON for HER.5 In contrast, functional analogues based on bimetallic electrocatalysts have a beneficial influence on HER and typically show greater efficiency than their mononuclear counterparts.4a,6
In the past decade, iron, cobalt and nickel complexes of porphyrins have gained increasing attention as HER catalysts4b,7 and the proton transfer ability and substrate access can be influenced by the substitution patterns on the porphyrin scaffold.8 For example, the monometallic Ni(II) hangman porphyrin was investigated towards HER activity and catalyzes the generation of H2 at −1.77 V vs. Fc/Fc+ with a faradaic efficiency of 92% (Fig. 1).8a,b Although bimetallic metalloporphyrins, such as Naruta's Pacman porphyrins (Fig. 1) were studied e.g. for the reduction of CO2,9 little is known about more rigid macrocyclic porphyrins. It therefore seems important to investigate bimetallic macrocyclic porphyrin scaffolds because of their redox properties and catalytic potential. During a recent study10 with a focus on the supramolecular chemistry of highly strained porphyrinylene nanohoops,11 we prepared an unstrained precursor, which features two cofacial porphyrins with a Ni–Ni distance of 7.4 Å (X-ray), but exhibits low solubility due to the choice of a 3,5-di-tert-butylphenyl residue (Ar1) (Fig. 1). When we compared this structure to literature-known bimetallic catalysts, such as Naruta's Pacman porphyrin,9 we hypothesized that this Ni–Ni distance§ could facilitate the activation/generation of small molecules such as CO2 or H2. Therefore, we decided to pursue the catalytic potential of this type of architecture and synthesize a novel, more soluble analogue based on a 2-butyloctyloxyphenyl residue (Ar2) (Fig. 2). To guarantee high stability of the macrocycle during catalysis (vide infra), we used the methoxy-substituted linker 2, which was reacted with the meso-brominated metalated porphyrin 1 in a Suzuki–Miyaura cross coupling (Fig. 2A). The crucial ring-closing step was carried out under template-assistance using pyrazine under highly diluted conditions (Fig. S5, ESI†). After chromatographic separation we were able to isolate pure 3 (50 mg, 8% yield) in sufficient quantity for this proof-of-principle study. In the case of the analogous Co(II) macrocycle, we were unable to isolate the product, presumably due to degradation during chromatography.
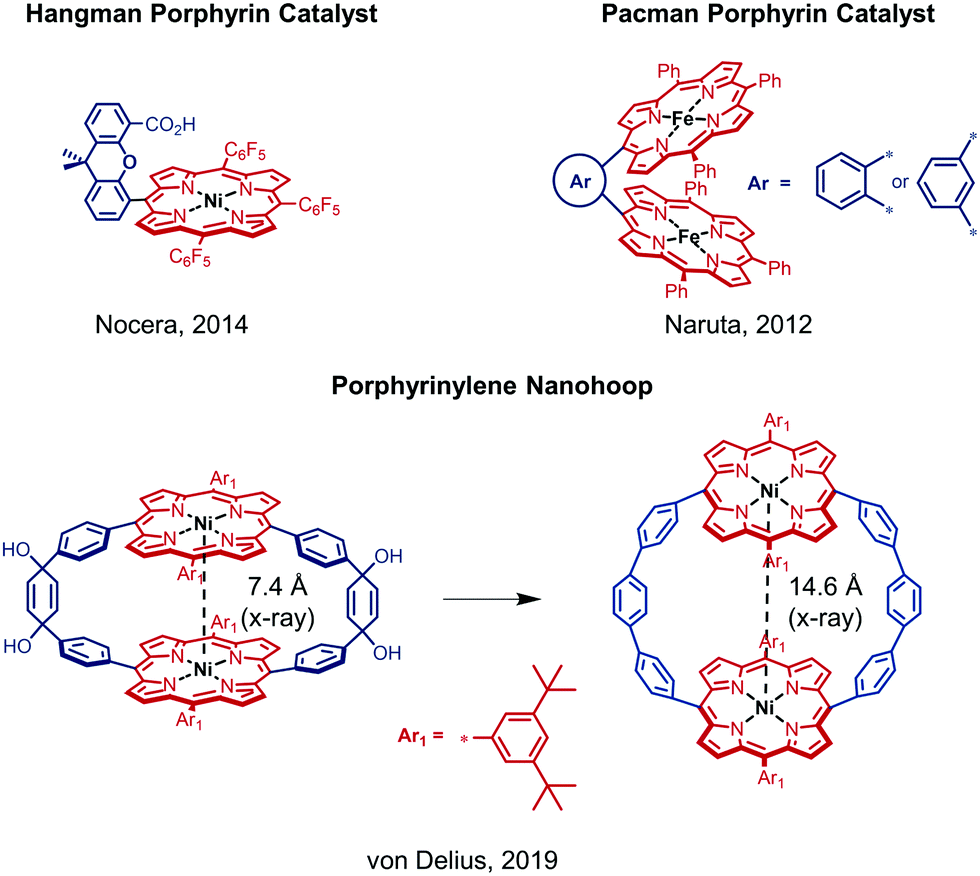 | ||
| Fig. 1 Schematic overview of relevant previous work on different metal porphyrin derivatives.8a,b,10 | ||
 | ||
| Fig. 2 (A) Synthesis of 3, Ni–Ni distance obtained by DFT (B3LYP/6-31G(d)). (B) UV/vis absorption spectrum (CH2Cl2) of 3 compared to the reference compound 4. (C) Cyclic voltammogram of 1 mM 3 and 1 mM complex 4 in CH2Cl2 with 0.1 M [nBu4N][PF6] at 100 mV s−1. | ||
The 1H NMR spectrum of compound 3 shows the typical shifts expected for the pyrrole as well as the olefinic protons. The signals corresponding to the aromatic protons in the phenylene bridges and the meso-aryl residues at the porphyrins are relatively broad, suggesting restricted C–C bond rotation within these moieties (for VT-NMR see Fig. S28, ESI†). To draw conclusions on the impact of the macrocyclic structure of 3 on its catalytic activity, we synthesized the monometallic reference compound 4 (Fig. 2B and Fig. S1, ESI†). Since the two porphyrins in 3 are not electronically coupled, there should be no difference in its photophysical, and electrochemical properties compared to 4. This assumption is confirmed by the superimposition of the recorded absorption spectra shown in Fig. 2B.
Compound 3 was furthermore characterized by cyclic voltammetry (CV) in dry dichloromethane with 0.1 M tetrabutylammonium hexafluorophosphate ([nBu4N][PF6]) as supporting electrolyte. The CV of 3 reveals one reduction and two oxidation waves at −1.30, 0.99 and 1.31 V vs. Fc/Fc+ (Fig. 2C). Previous studies on Ni-tetraphenyl porphyrins show reversible oxidation and reduction waves at similar half-wave potentials and allow for an assignment of redox states to the herein observed redox events.12 Hence, the reduction wave at −1.30 V vs. Fc/Fc+ is ascribed to the formation of [NiII(L)]−˙ (L = [2]CPT-OMe) while the oxidation waves at 0.99 and 1.31 V vs. Fc/Fc+ can be assigned to the formation of [NiII(L)]+˙ and [NiIII(L)]2+˙, respectively. Together with the superimposition of the CV of 3 and compound 4, these findings suggest that the macrocyclic structure has only minor influence on its electronic properties.
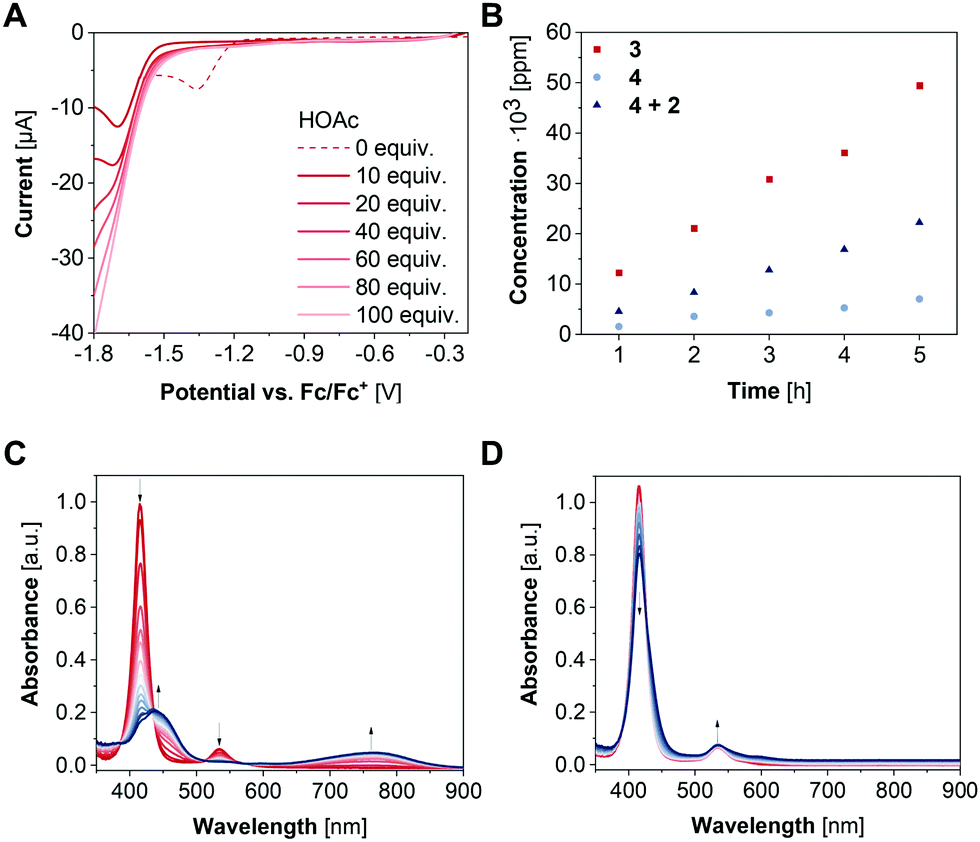 | ||
| Fig. 3 (A) Linear sweep voltammogram of 1 mM 3 with increasing equivalents of HOAc in CH2Cl2 with 0.1 M [nBu4N][PF6] at 100 mV s−1. (B) Quantification of the electrocatalytically generated H2 by GC-MS during CPC in CH2Cl2 with 0.1 M [nBu4N][PF6] and 0.1 M HOAc (3: 1 mM, CPC at −1.87 V vs. Fc/Fc+; 4: 2 mM, CPC at −1.90 V vs. Fc/Fc+). (C) SEC-UV/Vis spectrum of a 0.04 mM solution of 3 in CH2Cl2 with 0.1 M [nBu4N][PF6] at −2.0 V vs. Ag for 30 min (spectra recorded every 2 min) (d) SEC-UV/Vis spectrum of a 0.04 mM solution of 3 in CH2Cl2 with 0.1 M [nBu4N][PF6] and 4 mM acetic acid at −2.0 V vs. Ag for 30 min (spectra recorded every 2 min). | ||
To investigate the HER activity of 3, titration experiments were performed in the presence of varying amounts of acetic acid (HOAc) to a 1 mM solution of the complex in CH2Cl2. Upon addition of 10–100 equivalents acetic acid, the reduction wave at −1.36 V vs. Fc/Fc+ vanishes alongside with an ingrowing reduction wave at −1.71 V vs. Fc/Fc+ (Fig. 3A). These results suggest a transformation of the [NiII(L)]−˙ state into a species which is reduced at more anodic potential. Addition of an excess of acetic acid (>1000 equiv.) results in an onset potential of −1.67 V vs. Fc/Fc+ at −0.1 mA (Fig. S7, ESI†). Thus, 3 lowers the overpotential by 200 mV for proton reduction. Subsequently, controlled potential coulometry (CPC) experiments were performed in CH2Cl2 containing 0.1 M HOAc. The potential was held at −1.87 V vs. Fc/Fc+ for 5 h and the amount of the generated H2 gas was quantified hourly via GCMS (Fig. S8, ESI†). During the experiment, the faradaic efficiency ranged from 87 to 95% and a H2 production of 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 421 ppm after 5 h was obtained corresponding to 1.4 mmol g−1 h−1 (Fig. 3B).
421 ppm after 5 h was obtained corresponding to 1.4 mmol g−1 h−1 (Fig. 3B).
To verify the integrity of the catalyst during electrocatalysis, spectroelectrochemical (SEC) UV/vis spectroscopy of 3 was carried out in an electrochemical flow cell with a three-electrode setup (WE: glassy carbon electrode, RE: Ag wire, CE: Pt wire). For this purpose, CPC experiments were performed with a 0.04 mM solution of 3 in CH2Cl2 containing 0.1 M [nBu4N][PF6] as electrolyte for 30 min with UV/vis spectra being recorded every 2 min. The potential was held at −2.0 V to ensure the Nernst equilibrium being predominantly on the site of the reduced species. During CPC, the absorption bands at 416 nm and 534 nm decrease alongside with the growth of two new absorption bands at 437 nm and 771 nm (Fig. 3C). Notably, the altered SEC-UV/vis spectrum of 3 displays similar absorption bands as the porphyrinylene nanohoop reported in a previous work,11 suggesting a conversion of 3 into the fully or partially conjugated system as intermediate with the quinone-type motifs acting as electron reservoirs. Moreover, after application of −0.5 V to the newly generated species in a CPC experiment under otherwise same conditions, the SEC-UV/vis spectrum is identical to the initial one of 3, underlining the reversibility of the system. Similar SEC-UV/vis experiments were also performed under catalytic conditions in the presence of 4 mM acetic acid. Here, SEC-UV/vis spectra of 3 at −2.0 V differ significantly from the experiments without a proton source and reveal an isosbestic point at 426 nm (Fig. 3D). These findings indicate an interaction between the porphyrin complex and the proton source and are in line with previously reported remote ion-pair interactions which may play a pivotal role in the reactivity of the nickel centers of 3 towards HER.13
To examine whether the presence of two nickel centers has a beneficial influence on the catalytic performance, 4 was studied as catalyst under otherwise identical conditions. Interestingly, according to experiments in the presence of HOAc, complex 4 enables HER at −1.96 V vs. Fc/Fc+ at −0.1 mA (Fig. S7, ESI†), 290 mV more anodic compared to 3. During CPC with a 2 mM solution of 4 in CH2Cl2, which is twice as much as used in case of complex 3, in the presence of 0.1 M HOAc and a held potential of −1.90 V vs. Fc/Fc+ for 5 h, a smaller current was observed compared to 3 (Fig. S8, ESI†). Notably, an about 2-fold smaller H2 production with 0.64 mmol g−1 h−1 was determined with complex 4 as catalyst with a faradaic efficiency of 64–84% (Fig. 3B) and further supports the improved activity of macrocyclic complex 3. Since the linker unit of complex 3 was shown to participate as electron reservoir, its influence on the catalytic activity was investigated. Therefore, CPC experiments were also performed with a CH2Cl2 solution of 2 mM 2 and 4, respectively, with 0.1 M [nBu4N][PF6] and 0.1 M HOAc at −1.94 V vs. Fc/Fc+. After 5 h electrolysis, a H2 production of 0.99 mmol g−1 h−1 was obtained with a faradaic efficiency of 77–93% (Fig. S8, ESI†). Thus, the presence of the linker motif indeed has a major influence on the catalytic performance. Still, with the linker being covalently attached to the porphyrin units, the highest activity towards HER was observed. These findings might be attributed to redox mediating properties of the linker molecule within 3 resulting in cooperativity between the nickel centres and underline the beneficial effect of the macrocyclic structure.
In conclusion, a bimetallic macrocycle that features a relatively flexible arrangement of two cofacially linked Ni–porphyrins was successfully synthesized and characterized. Compared to the Ni(II) hangman porphyrin (Fig. 1),8a,b complex 3 achieves a higher faradaic efficiency with up to 95% and thus, shows the potential of dinuclear porphyrin complexes as HER catalysts. We also demonstrated that 3 exhibits the same photophysical and electrochemical properties as its mononuclear analogue 4. However, the bimetallic complex 3 showed increased activity towards HER compared to complex 4 as well as complex 4 together with 2. While 3 generates 49421 ppm H2 after 5 h electrolysis with a turnover frequency of 1.4 mmol g−1 h−1, complex 4 achieves only 7032 ppm H2 with 0.64 mmol g−1 h−1 and together with 2 22210 ppm with 0.99 mmol g−1 h−1. These results indeed show the positive impact of the linker molecule on HER activity in combination with 4. However, with 3 as catalyst, a 2-fold higher HER activity can be achieved highlighting the necessity of the macrocyclic scaffold. This behavior might be attributed to (i) the electron mediating function of the linker molecule which should be more efficient intramolecularly within the macrocyclic structure. Besides, (ii) the solvation within the cavity of 3 is different than for reference complex 4, leading to an enhanced catalytic activity towards HER. Further studies will have to be performed in the future to examine a third possibility, namely synergistic effects between the two nickel centers. Moreover, we plan to unravel the reaction mechanism, specifically whether HER proceeds via a homolytic pathway as already described for a monometallic Ni(II) porphyrin complex.14
Dr. Youzhi Xu is acknowledged for helpful discussions and providing precursor materials during the early stages of the project. This work was supported by the Fonds of the Chemical Industry (Liebig Grant to U.-P. A.) and the Deutsche Forschungsgemeinschaft (DFG, Emmy Noether Grant to U.-P. A., AP242/2-1 and AP242/5-1). This work was likewise supported by the Fraunhofer Internal Programs under Grant No. Attract 097-602175 and the DFG under Germany's Excellence Strategy – EXC-2033 – Projektnummer 390677874. M. v. D. acknowledges financial support by the DFG (182849149–SFB953 and 364549901–TRR 234). J. J. gratefully acknowledges financial support by the Deutsche Bundesstiftung Umwelt.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS; (b) A. Eftekhari, Int. J. Hydrogen Energy, 2017, 42, 11053–11077 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- (a) R. Francke, B. Schille and M. Römelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS; (b) K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1, 0039 CrossRef CAS; (c) J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- (a) F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, Chem. – Eur. J., 2018, 24, 1471–1493 CrossRef; (b) W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS; (c) J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. W. Adams, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1350–1369 CrossRef CAS; (d) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS.
- (a) J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC; (b) J. Dawson, C. Perotto, J. McMaster and M. Schröder, in Bioinspired Catalysis, ed. W. Weigand and P. Schollhammer, Wiley-VCH, Weinheim, 2014 Search PubMed; (c) U.-P. Apfel, F. Y. Pétillon, P. Schollhammer, J. Talarmin and W. Weigand, Bioinspired Catalysis, ed. W. Weigand and P. Schollhammer, Wiley-VCH, Weinheim, 2014 Search PubMed.
- (a) J. P. Collin, A. Jouaiti and J. P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS; (b) K. Hou, H. T. Poh and W. Y. Fan, Chem. Commun., 2014, 50, 6630–6632 RSC; (c) J.-P. Cao, T. Fang, Z.-Q. Wang, Y.-W. Ren and S. Zhan, J. Mol. Catal. A: Chem., 2014, 191–197 CrossRef CAS; (d) T. Fang, H.-X. Lu, J.-X. Zhao, S.-Z. Zhan and I. Qiying, J. Mol. Catal. A: Chem., 2015, 396, 304–309 CrossRef CAS.
- (a) J. P. Layfield and S. Hammes-Schiffer, Chem. Rev., 2014, 114, 3466–3494 CrossRef CAS; (b) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC; (c) M. O. Senge, S. A. MacGowan and J. M. O’Brien, Chem. Commun., 2015, 51, 17031–17063 RSC; (d) W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS; (e) Z.-Y. Wu, T. Wang, Y.-S. Meng, Y. Rao, B.-W. Wang, J. Zheng, S. Gao and J.-L. Zhang, Chem. Sci., 2017, 8, 5953–5961 RSC; (f) C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef CAS; (g) X. Guo, N. Wang, X. Li, Z. Zhang, J. Zhao, W. Ren, S. Ding, G. Xu, J. Li, U.-P. Apfel, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 8941–8946 CrossRef CAS; (h) L. Xie, J. Tian, Y. Ouyang, X. Guo, W. Zhang, U.-P. Apfel, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 15844–15848 CAS; (i) G. Xu, H. Lei, G. Zhou, C. Zhang, L. Xie, W. Zhang and R. Cao, Chem. Commun., 2019, 55, 12647–12650 RSC; (j) B. B. Beyene and C.-H. Hung, Coord. Chem. Rev., 2020, 410, 213234 CrossRef CAS.
- (a) D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001–15006 CrossRef CAS; (b) B. H. Solis, A. G. Maher, D. K. Dogutan, D. G. Nocera and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 485–492 CrossRef CAS; (c) D. J. Graham and D. G. Nocera, Organometallics, 2014, 33, 4994–5001 CrossRef CAS.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- Y. Xu, S. Gsänger, M. B. Minameyer, I. Imaz, D. Maspoch, O. Shyshov, F. Schwer, X. Ribas, T. Drewello, B. Meyer and M. von Delius, J. Am. Chem. Soc., 2019, 141, 18500–18507 CrossRef CAS.
- (a) H.-W. Jiang, T. Tanaka, H. Mori, K. H. Park, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 2219–2222 CrossRef CAS; (b) E. R. Darzi and R. Jasti, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC; (c) S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC; (d) E. J. Leonhardt and R. Jasti, Nat. Rev. Chem., 2019, 3, 672–686 CrossRef CAS.
- (a) E. C. Johnson, T. Niem and D. Doolphin, Can. J. Chem., 1978, 56, 1381–1388 CrossRef CAS; (b) D. Chang, T. Malinski, A. Ulman and K. M. Kadish, Inorg. Chem., 1984, 23, 817–824 CrossRef CAS.
- S. Kasemthaveechok, B. Fabre, G. Loget and R. Gramage-Doria, Catal. Sci. Technol., 2019, 9, 1301–1308 RSC.
- Y. Han, H. Fang, H. Jing, H. Sun, H. Lei, W. Lai and R. Cao, Angew. Chem., Int. Ed., 2016, 55, 5457–5462 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data, electrochemistry and HER results. See DOI: 10.1039/d0cc05229a |
| ‡ These authors contributed equally. |
| § The Ni–Ni distance in the nanohoop is 14.6 Å,10 which is too large to possibly give rise to synergistic catalysis. |
| This journal is © The Royal Society of Chemistry 2020 |