
Single-atom catalysts for the oxygen evolution reaction: recent developments and future perspectives
Woong Hee
Lee†
 a,
Young-Jin
Ko†
b,
Jun-Yong
Kim
bc,
Byoung Koun
Min
ad,
Yun Jeong
Hwang
*aef and
Hyung-Suk
Oh
*aeg
a,
Young-Jin
Ko†
b,
Jun-Yong
Kim
bc,
Byoung Koun
Min
ad,
Yun Jeong
Hwang
*aef and
Hyung-Suk
Oh
*aeg
aClean Energy Research Center, Korea Institute of Science and Technology (KIST), Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: yjhwang@kist.re.kr; hyng-suk.oh@kist.re.kr; Tel: +82 (0)2 958 5227
bCenter for Electronic Materials, Korea Institute of Science and Technology (KIST), Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea
cDepartment of Materials Science and Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul, 02841, Republic of Korea
dGraduate School of Energy and Environmental, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
eDivision of Energy and Environmental Technology, KIST School, Korea University of Science and Technology, Seoul 02792, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, and Yonsei-KIST Convergence Research Institute, Yonsei University, Seoul 03722, Republic of Korea
gKHU-KIST Department of Conversing Science and Technology, Kyung Hee University, Seoul 02447, Republic of Korea
First published on 27th August 2020
Abstract
Single-atom catalysts (SACs) possess the potential to achieve unique catalytic properties and remarkable catalytic mass activity by utilizing low-coordination and unsaturated active sites. However, smaller particles tend to aggregate into clusters or particles owing to their high surface energy. In addition, support materials that have strong interactions with isolated metal atoms, extremely large surface areas, and electrochemical stability are required. Therefore, sufficient information about these factors is needed to synthesize and utilize SACs. Herein, we review the recent investigations and advances in SACs for the oxygen evolution reaction (OER). We present not only the structural characterization of SACs, but also in situ/operando spectroscopic techniques and computational research for SACs to understand the mechanism and reveal the origin of their excellent OER activity. Furthermore, the OER catalytic activity and stability of SACs are summarized to evaluate the current level of SACs. Currently, research on single-atoms as OER catalysts is in the infant stage for synthesis, characterization and mechanism studies. We discuss some challenges for understanding the fundamentals of SACs and enhancing the catalytic performance of SACs for industrial applications.
 Woong Hee Lee | Dr Woong Hee Lee received his BS (2010) and PhD (2016) degrees in Chemical Engineering from Yonsei University, Korea. From 2017 to 2018, he worked at the National Institute of Technology and Standards. He is currently a Postdoctoral Researcher in Korea Institute of Science and Technology (KIST). His research is focused on the electrochemical catalysts and devices for energy conversion and storage. |
 Young-Jin Ko | Dr Young-Jin Ko received a PhD degree in Advanced Materials Science and Engineering from Yonsei University, Korea, in 2017. He is currently a Postdoctoral Researcher in Korea Institute of Science and Technology (KIST). His research interests include in situ/operando analysis for electro-catalysts and nano-adsorbents for metal/organic pollutants from various solutions. |
 Jun-Yong Kim | Jun-Yong Kim is currently a PhD student at Korea University, and he is doing research at Center for Electronic Materials, Korea Institute of Science and Technology. He received a bachelor's degree from the Department of Materials Science and Engineering, Korea University. Recently, his research is focused on the synthesis of oxide catalysts, metal catalysts, oxide absorbents, and materials. |
 Byoung Koun Min | Dr Byoung Koun Min received his PhD degrees in 2004 in the Department of Chemistry at Texas A&M University. He moved to Harvard University as a postdoctoral fellow before joining the Korea Institute of Science and Technology (KIST) in 2006. He is now the Director of National Agenda Research Division in KIST and is also appointed as a KU-KIST professor in Green School at Korea University. His research interests have been centered around solar energy conversion techniques including thin film solar cells, solar-chemical conversion catalysts, etc. |
 Yun Jeong Hwang | Dr Yun Jeong Hwang received her BS (2003) and MS (2005) in Chemistry from Korea Advanced Institute of Science and Technology (KAIST), and then moved to the USA for her PhD study at Chemistry department, University of California, Berkeley where her research focused on photoelectron-chemical water splitting for a solar fuel application. In June 2012, she joined clean energy research center at Korea Institute of Science and Technology (KIST) and focused on the electrocatalyst development for electro-catalytic CO2 reduction and water oxidation for a sustainable carbon cycle technology and also explore new electrocatalytic reactions for valuable chemical synthesis. She is currently a principal researcher of KIST and an adjunct professor at department of chemical and biomolecular engineering in Yosei University. She is a recipient of KIST Young Fellowship, and currently serving as an Associate Editor of Journal of Materials Chemistry A (Royal Society of Chemistry). |
 Hyung-Suk Oh | Dr Hyung-Suk Oh received his BS (2006), MS (2008) and PhD (2012) degrees in Chemical Engineering from Yonsei University, Korea. From 2013 to 2017, he was a postdoctoral fellow at the Technical University Berlin (TUB) where his research focused on electrocatalysts and devices for water electrolysis. In February 2017, he joined clean energy research center at Korea Institute of Science and Technology (KIST) to study electrocatalysts and devices for energy conversion and storage. His research interests focus on electrocatalysts for oxygen evolution (OER), in-situ/operando analysis of various electrocatalysts, and optimization of electrolyzer operation. He is currently a Senior Researcher of KIST and an adjunct professor at KHU-KIST department of conversing science and technology in Kyung Hee University. |
Introduction
Catalysts are essential components in electrochemical industrial processes and enhance the chemical performance by increasing the rate of the chemical reaction. Precious metals have irreplaceable excellent catalytic activity in some reaction processes, but their high cost makes them less economically feasible for industrial applications.1–4 Therefore, the demand for developing catalysts with excellent mass activity has increased over recent decades. To meet this demand, tremendous efforts have been devoted to enhancing the catalytic mass activity by maximizing the surface composition and controlling the atomic morphology. Reducing the particle size of a catalyst or using a support material is a favorable and common strategy to increase the active sites.5–7 Furthermore, nanosized catalysts possess low-coordinated atoms at the surface, quantum size effects, and strong metal–support interactions; this makes their properties unique, unlike bulk materials.8,9 When these strategies are extremely applied, single atoms are monodispersed on the supporting material, referred to as single-atom catalysts (SACs), which have unique properties compared to the bulk catalysts.A prerequisite for unravelling the catalytic performance of SACs is to synthesize an atomically dispersed catalyst with an appropriate supporter material. The general challenge in the synthesis of atomically dispersed catalysts includes the following conditions: (i) support material with enough surface area; and (ii) enhancing the metal–support interactions. The proposed strategies are aimed at protecting isolated atoms from agglomeration and/or Ostwald ripening.
In general, most SACs are prepared by wet-synthesis methods, and a combination of the following sequential processes is required: (i) synthesis of metal ions on the support material by an ion-impregnation or -exchange, co-precipitation process; (ii) a drying process via evaporation or calcination; and (iii) a reduction process using photo/thermal energy. The advantage of a wet-approach is that it does not require specialized equipment compared to dry-processes. Furthermore, it is suitable for synthesizing SACs on individually separable support materials.
Recently, SACs have been extensively investigated and demonstrated to have effective heterogeneous catalysts for various electrochemical reactions.10–19 SACs can have special catalytic activity as a single metallic atom center coordinates with the surrounding atoms of the supporting matrix unlike traditional metal or metal oxide heterogeneous catalysts. The electronic states of the single atom are modified a lot depending on the supporting materials. For example, SACs of Pt supported on the nitride, carbide, and oxide exhibit superior activity and stability in the oxygen reduction reaction (ORR), hydrogen evolution reaction (HER), formic acid oxidation reaction, and methanol oxidation reaction.20–22 Ru single atoms distributed on N-doped carbon catalysts show superior electrocatalytic activity for NH3 production via electrochemical reduction of N2.23 Single transition metal–nitrogen moiety supported carbon (M–N–C) catalysts, which are the most widely studied SACs, exhibit remarkable catalytic activity and stability for the HER, ORR, and CO2 reduction reaction (CO2RR).24–29 However, not much emphasis has been placed on the use of SACs for the oxygen evolution reaction (OER).
The OER is a key reaction in electrochemical technology, as an oxidative half reaction of water electrolysis, electrochemical reductive synthesis and battery cells.30,31 However, the OER has sluggish kinetics and requires a large operation potential, increasing the needs for efficient and stable catalysts under the operational conditions. Noble metal catalysts such as Ir or Ru oxides show excellent OER catalytic activity in all potential ranges, and transition metal-based catalysts exhibit good performances in alkaline media.4,32 Nevertheless, developing OER catalysts with high mass activity and stability simultaneously remains a challenge. Here, we introduce the research of SACs for the OER including structural characterization, in situ/operando study, DFT calculations and electrochemical results for the OER. Finally, we discuss and propose new perspectives to develop SACs for the OER.
Characterization of SACs for the OER
Morphology and electronic structure of SACs
The coordination of a single atom in the supporting material is an ideal structure of a SAC whose morphology is difficult to obtain by conventional methods. To probe the SAC structure, experimental evidence of the synthesized catalyst is necessary. In typical morphology characterization such as X-ray diffraction (XRD), or high-resolution transmission electron microscopy (HR-TEM), the detection of single atoms is difficult owing to the limitations of their sensitivity. Rather, this limitation gives indirect evidence of a single atom structure. Gu et al. reported a high loading of Ir single atoms on NiO (Ir–NiO) for the OER.33 The XRD patterns of the Ir–NiO catalyst do not contain a peak associated with iridium but the diffractogram is consistent with that of pure NiO, suggesting that iridium is present in the atomic structure rather than the nanoparticles. The TEM images of Au single atoms on NiFe layered double hydroxide (LDH) (sAu/NiFe LDH) synthesized by Zhang et al. demonstrate the absence of Au nanoparticles, indicating the presence of Au at an atomic scale (Fig. 1a).34 However, these methods cannot clearly identify the morphology of a single atom. Two types of analysis are generally used to detect the single atoms microscopically and spectroscopically. One is the high-angle annular dark-field scanning TEM (HAADF-STEM) technique, which directly shows the electroscopic image of a single atom. The single Au atom of the sAu/NiFe LDH was observed by the HAADF-STEM micrograph in Fig. 1b not by its conventional TEM mode. Fig. 1c and d also show the high-angle annular dark-field (HAADF) element mapping and scanning transmission electron microscopy (STEM) micrographs of a single atom W-doped Ni(OH)2 synthesized by Ma et al., respectively.35 In the energy dispersive spectroscopy mapping, W was uniformly dispersed on the Ni(OH)2 structure. HAADF-STEM micrographs of the single atom W-doped Ni(OH)2 show single W atoms individually dispersed on the surface of Ni(OH)2. Transition-metal single atoms can also be observed by HAADF-STEM. Yao et al. reported atomically dispersed Ni catalysts on defective graphene (A-Ni@DG) and compared their properties with Ni nanoparticles on DG (Ni@DG).36 Abundant Ni single atoms in the A-Ni@DG catalyst were observed in the HAADF-STEM micrograph of the A-Ni@DG (Fig. 1e).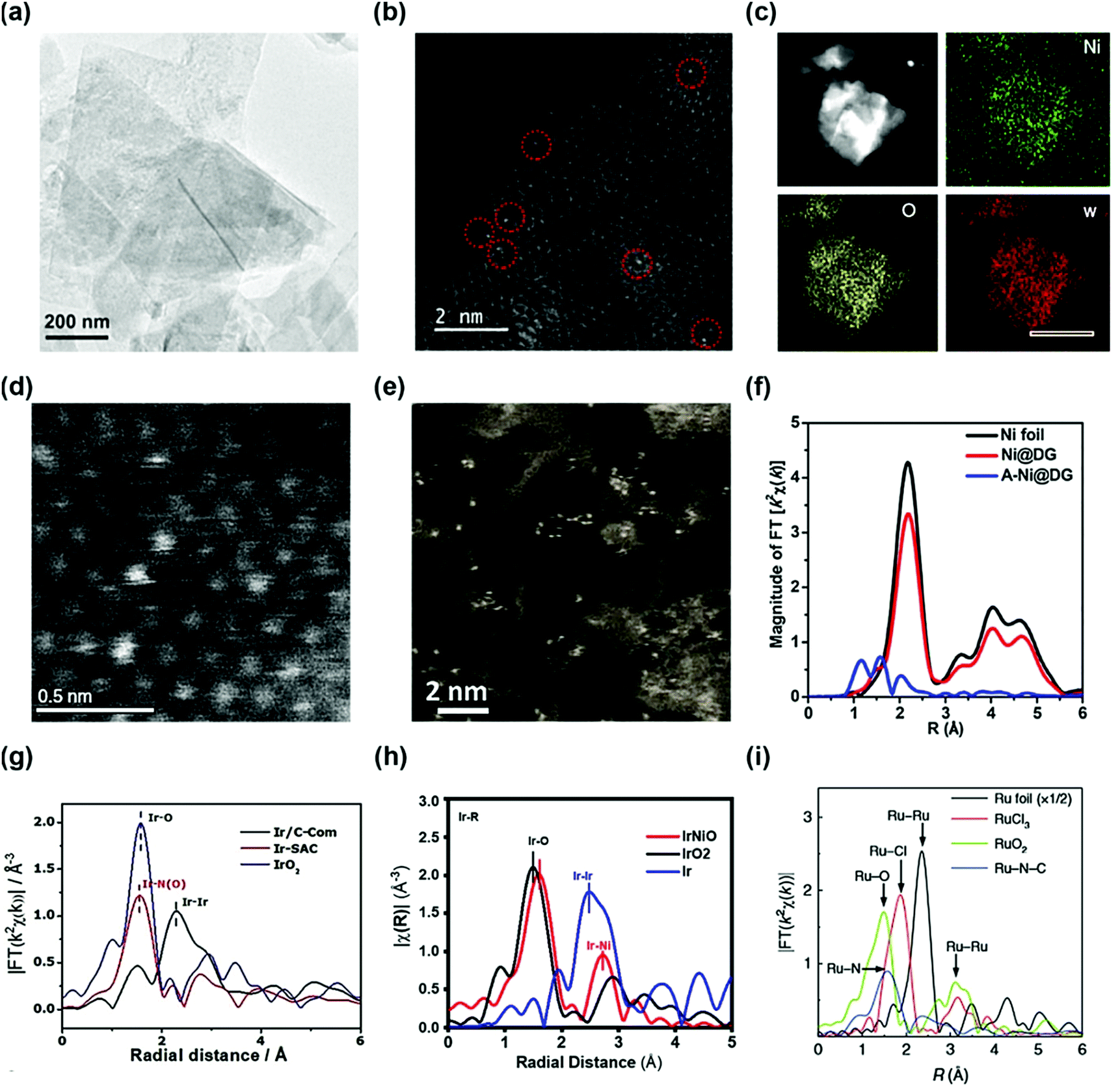 | ||
| Fig. 1 (a) TEM and (b) HAADF-STEM images of Au/NiFe LDH. (Reprinted with permission from ref. 34. Copyright 2018, American Chemical Society.) (c) The HAADF element mapping and (d) HADDF STEM images of single atom W-doped Ni(OH)2. (Reprinted with permission from ref. 35. Copyright 2019, Springer Nature.) (f) The k2-weighted Fourier transform spectra of Ni@DG, A-Ni@DG, and the Ni foil reference samples. (Reprinted with permission from ref. 36. Copyright 2018, Cell Press.) (g) The k3-weighted Fourier transform spectra of the Ir-based catalyst. (h) Fourier-transformed EXAFS spectra at the Ir L-edge of the IrNiO catalyst and the references. (Reprinted with permission from ref. 33. Copyright 2020, American Chemical Society.) (i) The Ru-k2-weighted Fourier transform spectra for Ru foil, RuCl3, RuO2, and Ru–N–C. (Reprinted with permission from ref. 37. Copyright 2019, Springer Nature.) | ||
However, the HAADF-STEM technique has a limitation of only showing the local information of the sample at a nanoscale which is not representative of the whole area of the sample. Meanwhile, the other widely used analytical method is the extended X-ray absorption fine structure (EXAFS) technique, which obtains representative coordination properties of a large area of the catalyst. Fourier-transformed (FT) EXAFS spectra provide information on interatomic distances and coordination numbers which can determine the single-atom morphology. The X-ray absorption near-edge spectroscopy (XANES) spectra and the FT EXAFS spectra at the Ni K-edge are used to verify the chemical state and coordination environment of the single-atom Ni in the A-Ni@DG catalyst. In the XANES spectra, the Ni@DG exhibits similar white line peaks with Ni foil, suggesting the presence of metallic Ni nanoparticles. Meanwhile, A-Ni@DG shows a higher white line peak than Ni@DG, indicating the oxidized electronic structure of the single-atom Ni owing to the presence of the Ni–C4 structure. In the EXAFS spectrum, Ni@DG had a prominent peak at 2.2 Å associated with Ni–Ni coordination (Fig. 1f). In contrast, the spectrum of A-Ni@DG showed the absence of Ni–Ni peaks and contains three peaks at 1,2, 1.6, and 2 Å, corresponding to different Ni–C coordination, which confirms the single-atom structure of Ni in the A-Ni@DG catalyst. Meanwhile, Ir SAC also showed a distinguishable Ir L3-edge Fourier transform spectrum (Fig. 1g), which supports that the Ir atom is strongly anchored on a nitrogen–carbon support. The main peak of Ir/C in the Ir L3-edge Fourier transform spectrum is at 2.3 Å, associated with Ir–Ir scattering, and the Ir L3-edge Fourier transform spectrum of IrO2 contains an Ir–O peak. The Ir L3-edge Fourier transform spectrum of Ir SAC shows no Ir–Ir scattering peak and only a single dominant peak at 1.5 Å which is assigned to the Ir–N scattering path, confirming that Ir coordinated with N atoms is atomically dispersed on the carbon support. In another study, the Ir L3-edge Fourier-transformed EXAFS spectra of the Ir–NiO catalyst exhibited Ir–O and Ir–Ni bonding instead of Ir–Ir, suggesting that Ir single atoms were doped on NiO without Ir particles (Fig. 1h). Yao et al. reported a Ru–N4 site anchored on a nitrogen–carbon support (Ru–N–C) and proved their single atom structure using EXAFS. The FT-EXAFS of Ru–N–C contained only one peak at 1.5 Å which represents the Ru–N bond, and the Ru–Ru peak was not observed in the spectrum (Fig. 1i).37 Furthermore, the EXAFS fitting results for Ru–N–C show four Ru–N coordination, suggesting formation of a Ru1–N4 site. These results show that EXAFS not only confirms the single atom structure but also provides a coordination environment for SAC.
In situ/operando analysis of SACs
Generally, single atoms are oxidized and strongly anchored onto the support materials. However, this property does not fully explain the catalytic activity and stability of single-atom active sites. Thus, various in situ/operando characterizations are essential to reveal the origin of the OER catalytic activity and stability of SACs. X-ray absorption fine structure (XAFS) is a powerful in situ/operando technique for observing the single-atom structure and electronic structure during the OER. For instance, Sun et al. reported a single-atom Ru catalyst anchored on a NiFe LDH (Ru/CoFe-LDH) catalyst and obtained the in situ/operando Ru XANES spectra of Ru/CoFe-LDH.38 When a potential of 1.6 V is applied, the oxidation state of Ru is increased during the OER but still lower than that of RuO2 (Fig. 2a and b). This indicates that a single atomic Ru was not transformed into an unstable phase of Ru during the OER. Under OCV after the OER, the oxidation state of Ru was returned, showing the reversible property of a single Ru atom. In contrast, Co and Fe were highly oxidized during the OER and they maintained their oxidation state under OCV conditions after the OER (Fig. 2c and d). Operando X-ray absorption spectroscopy (XAS) results show that a low oxidation state under the OER conditions and the reversible properties of the single-atom Ru led to the high stability of Ru/CoFe-LDH.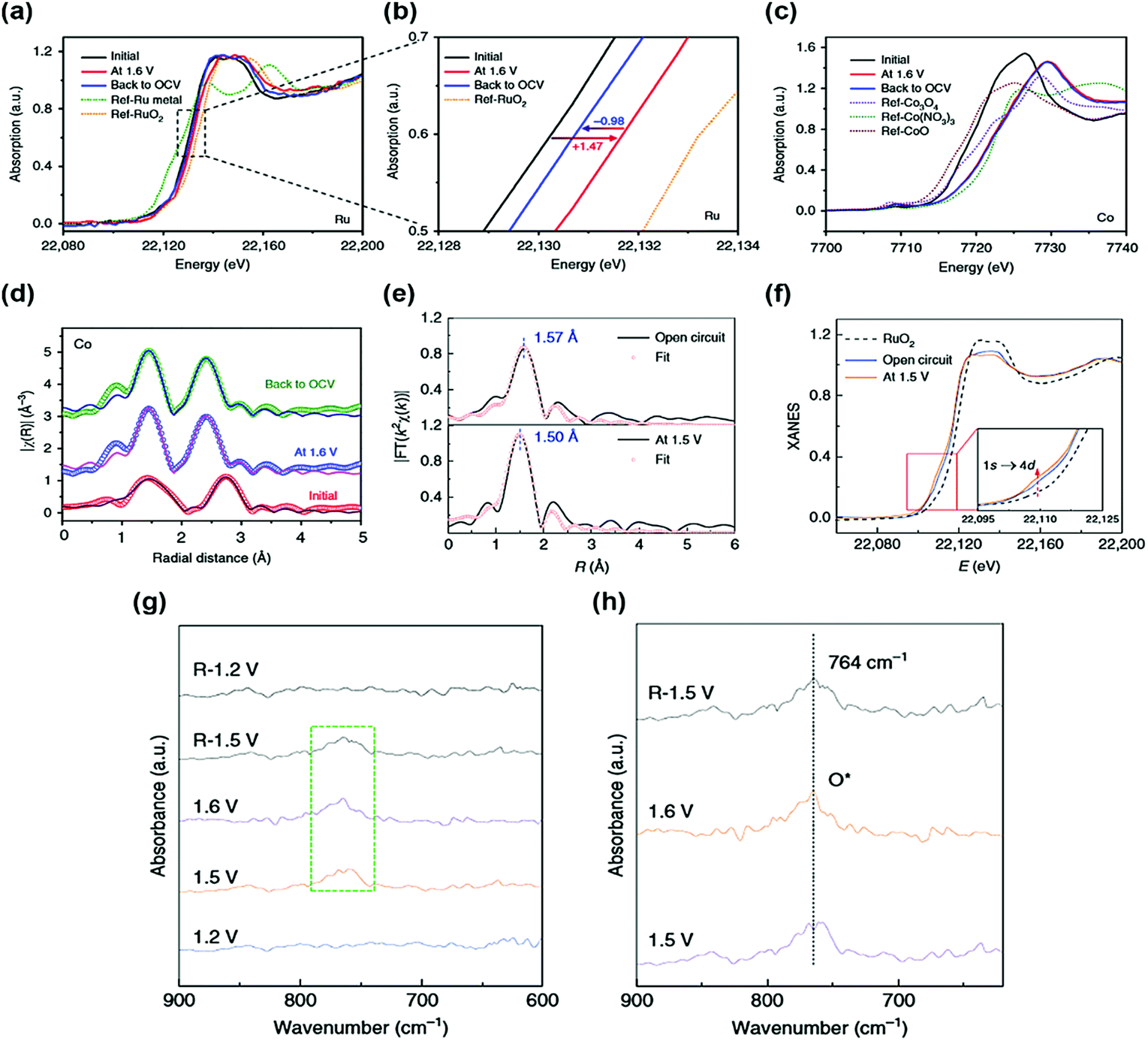 | ||
| Fig. 2 In situ/operando XAS measurement of Ru/CoFe-LDHs. In situ/operando XANES under the electrochemical conditions of (a and b) Ru K-edge and (c) Co K-edge. R-Space fitting curves of Co (d) EXAFS at the reaction potential of 1.6 V, indicating that the valence of Ru is always kept <4+ even under high overpotential. (Reprinted with permission from ref. 38. Copyright 2019, Springer Nature.) (e) Operando EXAFS spectra and first-shell fitting curves for Ru–N–C at different applied voltages from the open circuit condition to 1.5 V during the OER. (f) Operando XANES spectra for Ru–N–C during the OER. Inset: Magnified pre-edge XANES region. (g) Operando SR-FTIR spectroscopy measurements for Ru–N–C during the acid OER. (h) Enlarged infrared signal at ∼764 cm−1. All potentials are normalized to RHE. (Reprinted with permission from ref. 37. Copyright 2019, Springer Nature.) | ||
In another study, operando XAS measurements were conducted for the Ru–N–C catalyst.37 Based on the FT-EXAFS curve of Ru–N–C under the OER conditions, the first coordination peak decreased by 0.07 Å, suggesting strong interaction and hybridization for Ru-intermediate coordination (Fig. 2e). The oxidation state of Ru gradually increases during the OER, but it remained lower than that of RuO2 (Fig. 2f). These results indicate a stable Ru–N–C catalyst under OER conditions. The in situ/operando Fourier transform infrared (FT-IR) spectroscopy of the Ru–N–C catalyst was also carried out to monitor the OER intermediates. At a potential of 1.2 V vs. reversible hydrogen electrode (RHE), there was no obvious peak over the low vibration frequency region between 600 and 900 cm−1 (Fig. 2e). When the potential was increased to 1.6 V vs. RHE, a new peak was observed at 764 cm−1, suggesting the presence of single oxygen adsorption (O*) intermediates (Fig. 2f), showing the O1–Ru1–N4 configuration of the Ru–N–C catalyst during the OER. In situ Raman spectroscopy was also carried out to observe the phase transition of the SAC support.34 At a potential of 1.12 to 1.32 V, the sAu/NiFe LDH had two peaks located at 462 and 535 cm−1, these peaks are associated with the NiFe LDH phase. Under the OER conditions, the NiFe LDH structure was irreversibly transformed into the NiFe oxyhydroxide phase. The authors expected that the sAu/NiFeOOH–NiFe LDH structure would act as a real catalytic mechanism for the OER.
Only a few studies use the in situ/operando technique to identify intermediates and the real electronic structures of SACs during the OER. The nature of SACs under OER conditions is still unresolved and remains the key to enhancing the activity of SACs. Thus, it is essential to utilize various in situ/operando characterizations, such as infrared adsorption spectroscopy, synchrotron-based X-ray absorption spectroscopy, and Raman spectroscopy. These characterization methods help to understand the fundamentals of SACs and provide an insight into the development of SACs for the OER.
DFT calculations of SACs
Advanced computational methods such as density functional theory (DFT) calculations have been used to confirm the structural stability of single-atom catalysts, and it provides new perspectives for determining the detailed structure of the catalyst. In particular, it provides complementary data to forecast mutual interactions between isolated metals and their supports, while also providing specific catalytic reaction mechanisms on the supported single-atoms. The DFT calculations are significantly simplified compared to those of metal nanoparticles and clusters; this is because only one metal atom is present in the structure (Fig. 3). The structure of the SAC to which the DFT calculations is applied can be roughly classified into two: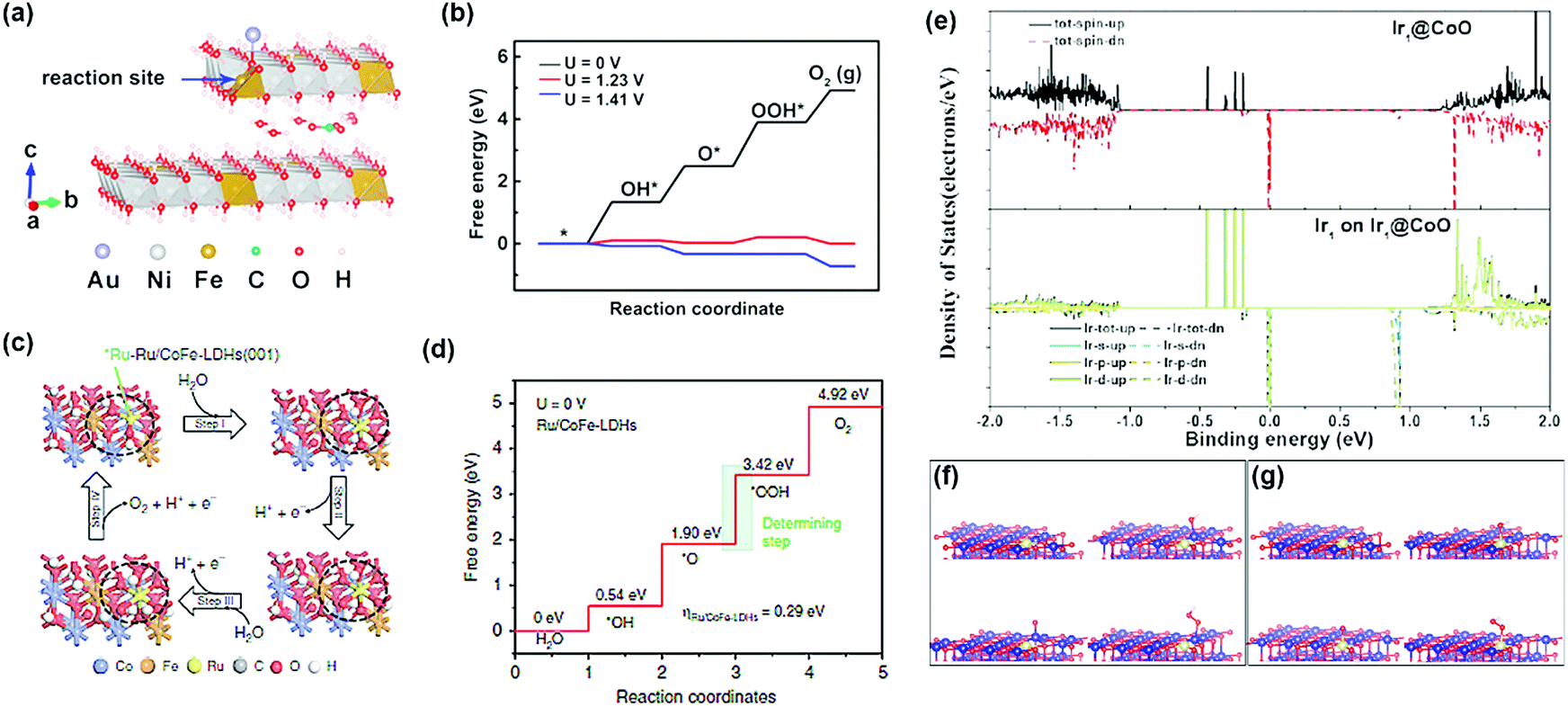 | ||
| Fig. 3 (a) Two-layer slab model for the CO32−/H2O molecule-intercalated Au/NiFe LDH catalyst, and (b) its Gibbs free energy diagram (a and b were adapted with permission from ref. 19. Copyright 2018, American Chemical Society). (c) Proposed OER for Ru/CoFe-LDH catalysts, and (d) the Gibbs free energy diagram for OER on Ru/CoFe-LDH catalysts (c and d were adapted with permission from ref. 39. Copyright 2019, Springer Nature.) (e) Density states of Ir@Co, and Ir@CoO, respectively. (f and g) Optimized adsorption structures of OH, O, and OOH on (f) Ir1@CoO (Co), and (g) Ir1@CoO (Ir) (e–g were adapted with permission from ref. 40. Copyright 2019, Wiley-VCH). | ||
(1) Precious metal SACs: there are recent DFT calculation-based reports on the origin of activity boosting when precious metal SAC is supported on an oxide, LDH, and metal alloys, which are well known to have OER activity in alkaline media. Zhang et al. investigated the origin of the OER activity of an LDH system and significant activity enhancement by Au-SA decoration through DFT+U + vdW calculations.19 They provided evidence for the activity enhancement that each of the Au-SA and NiFe oxyhydroxides transformed from the LDH structure with interlayer CO32− anions contributed to the local Au interface active site. Li et al. studied the improvement of the intrinsic activity and stability of Ru-SA anchored with CoFe LDH using DFT+U calculations.39 Since the strong synergetic electron coupling between Ru-SA and the LDH structure led to the optimal adsorption free energy of *OOH, the OER activity can be significantly boosted. Furthermore, avoiding the formation of Ru with a high oxidation state led to enhanced stability. Lai et al. showed the OER performance of four possible catalytic sites and calculated the free energies of their intermediates at pH = 14.40 Since the Ir SA produces OOH* and O2 without further decrease in free energy, the structure consisting of Ir SA on Co nanoparticles was the most active catalytic site for the OER.40 Computational calculations also confirmed that the Ir SA can efficiently improve the d electron dominance near the Fermi level and promote electron transfer to the conduction band. This leads to higher conductivity of the catalyst and enhanced catalytic activity.
(2) Other transition metal (TM) SACs: the transition metal nanoparticles are physically or chemically unstable. Therefore, these nanoparticles are very vulnerable to agglomeration and sintering when energy is applied. In particular, a single-atom can be considered as the most unstable structure. To overcome the physical/chemical instability of a single-atom, a single-atom bonded with nitrogen or carbon in the support structure has been used as a catalyst for the OER, and the origin of the activity has also been studied through DFT calculations. Deng et al. studied the OER activity of the SA-N4 bond (Fe-, Mn-, Ni-) in a tetracyanoquinodimethane (TCNQ) monolayer.41 They predicted that the adsorption strength of the reaction intermediates on transition metal active sites can be further optimized by adopting grafting axial ligands and external strain (Fig. 4a and b). Fei et al. studied a series of monodispersed transition metals (Fe-, Co-, Ni-) embedded in nitrogen-doped graphene with an SA-N4C4 bond.42 The unambiguous structure determination allows density functional theoretical prediction of SA-N4C4 moieties with a di-vacancy in the graphene lattice as an efficient oxygen evolution catalyst with activities following the trend Ni > Co > Fe (Fig. 4c and d).42
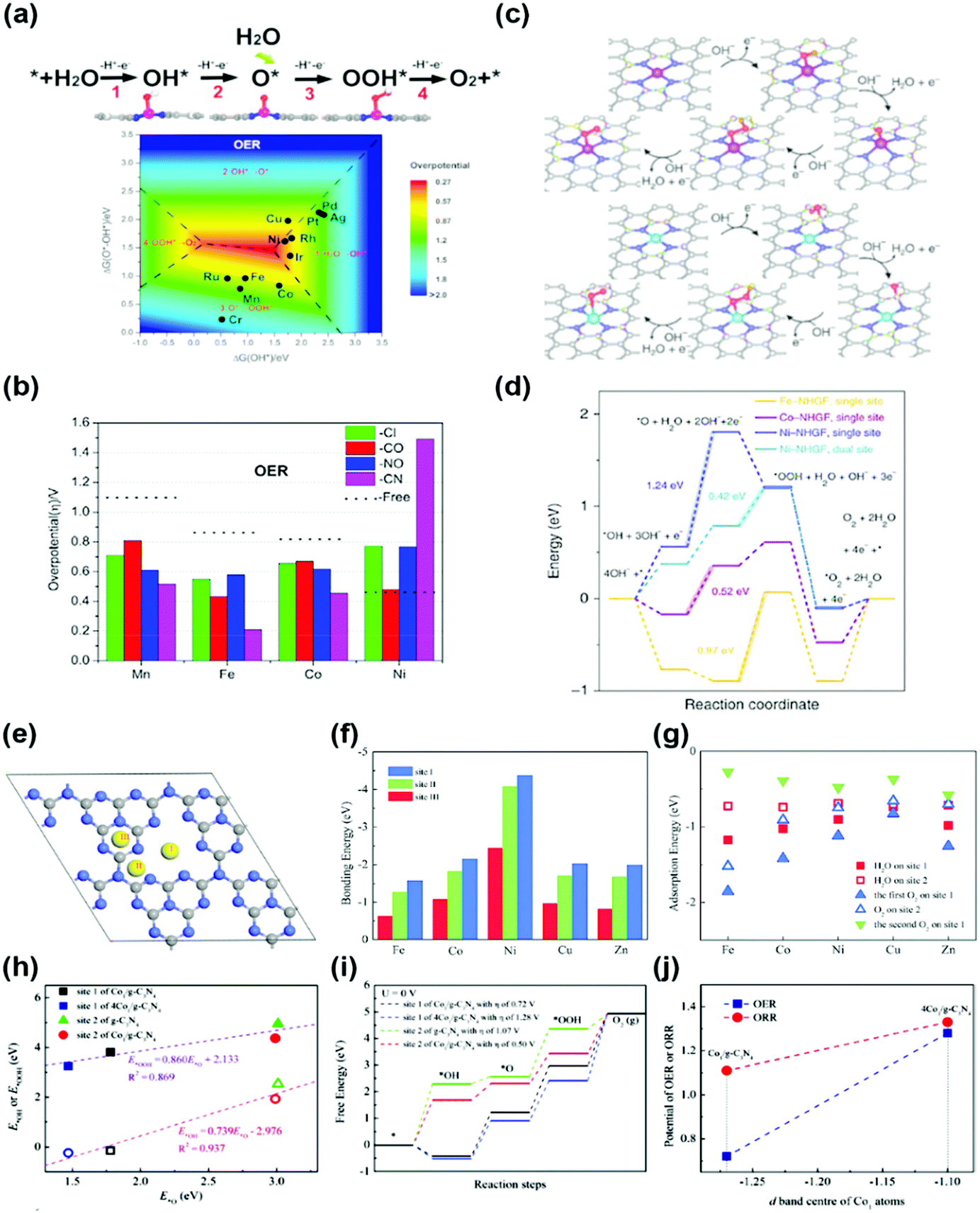 | ||
| Fig. 4 (a) Four-electron-transfer OER pathway through the intermediates OH*, O* and OOH*, and the contour map of the OER limiting overpotential (η) as a function of Gibbs free energies of O*–OH* and OH*. (b) Limiting overpotentials (η) of M–TCNQ (M = Mn, Fe, Co) toward the OER by coordinating Cl, CO, NO and CN as axial the ligands as well as the ligand free monolayers for comparison (a and b were adapted with permission from ref. 41. Copyright 2019, Elsevier Inc.). (c) Proposed for OER with the intermediates on the single-site and dual-site of a metal–NHGF catalyst. (d) The Gibbs free energy diagrams @ 1.23 V for the OER on the single-site of Fe–NHGF, Co–NHGF and Ni–NHGF catalysts (c and d were adapted with permission from ref. 42. Copyright 2018, Springer Nature.) (e) Three different bonding sites of SA on g-C3N4. (f) The bonding energies of SA at three different sites, and (g) the adsorption energies of H2O and O2 molecules on the (site 1) M1/g-C3N4 and (site 2) M1/g-C3N4. (h) Transition states of OH, O and OOH groups from site 2 to site 1 of Co1/g-C3N4. (i) The linear relationships of the adsorption energies at site 1 of Co1/g-C3N4 and 4Co1/g-C3N4 plus site 2 of g-C3N4 and Co1/g-C3N4. (j) The Gibbs free energy changes during the OER of Co1/g-C3N4 and 4Co1/g-C3N4 specimens (e–j were adapted with permission from ref. 43. Copyright 2019, Royal Society of Chemistry). | ||
Wu et al. and Li et al. predicted the OER activity of SA-N bonds embedded in 2D structured graphitic carbon nitride (g-CN) (Fig. 4e–j).43,44 They proposed two conditions for enhancement of the OER activity. First, the redox reaction of the transition metal SA plays a critical role in determining the activity of the SAC. Second, based on the stability of the reactants and intermediates on the surface, adjacent host atoms of SA can be considered as the secondary active sites. Although the probability of a reaction on the host atoms is significantly smaller than that on the SA active sites, the host atoms play a salient role in reducing the overpotentials for the OER. All information obtained served as a foundation for further optimization of the reaction system, including the catalyst design and rationalization of the reaction conditions. Since the OER application of SAC is still in its infancy, the calculation studies have not been diversified. Therefore, it will be possible to predict OER activity for various SA structures in the future.
Investigation of the electrochemical properties for the OER
The OER catalytic activity of SACs
Precious metals with high OER catalytic activity, such as Ru and Ir, have been intensively studied as promising candidates for OER SACs. For example, Ru–N–C shows high OER catalytic activity with low overpotential of 267 mV and 340 mV in 0.5 M H2SO4 solution to achieve a current density of 10 and 100 mA cm−2, respectively (Fig. 5a).37 Furthermore, the mass activity of the Ru–N–C is 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 284 A gmetal−1 at an overpotential of 300 mV, which is 410 times higher than that of the RuO2/C catalyst. Pristine N–C catalysts have poor OER activity, indicating that atomic Ru coordinated with N is a highly active site for the OER. Sun et al. reported a single atom ruthenium catalyst anchored on NiFe LDH (Ru/CoFe-LDH), which requires an overpotential of 237 mV for a current density of 10 mA cm−2 in 1 M KOH electrolyte (Fig. 5b).38 NiFe LDH shows overpotentials of 263 mV at a current density of 10 mA cm−2, indicating that single atom Ru improves the OER kinetic rate. Zeng's research group synthesized Ir single-atoms on Co0.8Fe0.2Se2 (A-Ir1/Co0.8Fe0.2Se2) by anodic deposition.45 The overpotential of A-Ir1/Co0.8Fe0.2Se2 was 230 mV to reach 10 mA cm−2 in 1 M KOH media, which is 135 mV lower than IrO2 (Fig. 5c). Noble metals with low OER activity are also used as SACs for the OER. Yu et al. developed a Pt single-atom grafted Fe–N4 center (Pt1@Fe–N–C) catalyst with OER catalytic activity as well as HER and ORR catalytic activity.46 In alkaline media, Pt1@Fe–N–C exhibited an onset potential of 1.33 V and an overpotential of 310 mV at 10 mA cm−2 (Fig. 5d). The results of Pt1@Fe–N–C are superior to those of the benchmark RuO2 catalyst. They expected that the synergistic effect of the heteroatoms and low-coordinated dangling Pt4+ atoms may promote the OER catalytic activity.
284 A gmetal−1 at an overpotential of 300 mV, which is 410 times higher than that of the RuO2/C catalyst. Pristine N–C catalysts have poor OER activity, indicating that atomic Ru coordinated with N is a highly active site for the OER. Sun et al. reported a single atom ruthenium catalyst anchored on NiFe LDH (Ru/CoFe-LDH), which requires an overpotential of 237 mV for a current density of 10 mA cm−2 in 1 M KOH electrolyte (Fig. 5b).38 NiFe LDH shows overpotentials of 263 mV at a current density of 10 mA cm−2, indicating that single atom Ru improves the OER kinetic rate. Zeng's research group synthesized Ir single-atoms on Co0.8Fe0.2Se2 (A-Ir1/Co0.8Fe0.2Se2) by anodic deposition.45 The overpotential of A-Ir1/Co0.8Fe0.2Se2 was 230 mV to reach 10 mA cm−2 in 1 M KOH media, which is 135 mV lower than IrO2 (Fig. 5c). Noble metals with low OER activity are also used as SACs for the OER. Yu et al. developed a Pt single-atom grafted Fe–N4 center (Pt1@Fe–N–C) catalyst with OER catalytic activity as well as HER and ORR catalytic activity.46 In alkaline media, Pt1@Fe–N–C exhibited an onset potential of 1.33 V and an overpotential of 310 mV at 10 mA cm−2 (Fig. 5d). The results of Pt1@Fe–N–C are superior to those of the benchmark RuO2 catalyst. They expected that the synergistic effect of the heteroatoms and low-coordinated dangling Pt4+ atoms may promote the OER catalytic activity.
Recently, some researchers have reported that transition metal SACs are used as OER catalysts. In alkaline media, similar to precious metals, transition metals are highly stable and possess good catalytic properties for the OER. This suggests that transition metals are promising candidates for OER SACs. Wu et al. reported atomically dispersed Fe–Nx species on an N and S co-decorated hierarchical carbon (S,N–Fe/N/C-CNT) catalyst for the ORR and OER under alkaline media.47 In Fig. 5e, the overpotential of the S,N–Fe/N/C-CNT catalyst was 370 mV at 10 mA cm−2 with a Tafel slope of 82 mV dec−1, indicating the potential of transition SACs for the OER. Qiao et al. represent that Co–C3N4 catalyst OER exhibits a potential of 1.61 V to achieve a current density of 10 mA cm−2 in KOH solution.48 A recently reported Ni SAC coordinated to oxygen sites on a graphene-like carbon (Ni–O–G-SAC) catalyst possesses an overpotential of 224 mV for 10 mA cm−2 and Tafel slope of 42 mV dec−1 for the OER in alkaline solution because of a high valence state of Ni.49 As shown in Fig. 5f, the A-Ni@DG catalyst exhibits a low overpotential of 270 mV and achieved a current density of 10 mA cm−2, which outperformed DG (340 mV), Ni@DG (310 mV), and Ir/C catalyst (320 mV).36 This suggested that atomic Ni coordinated with carbon is a superior active site compared to metallic Ni nanoparticles for the OER.
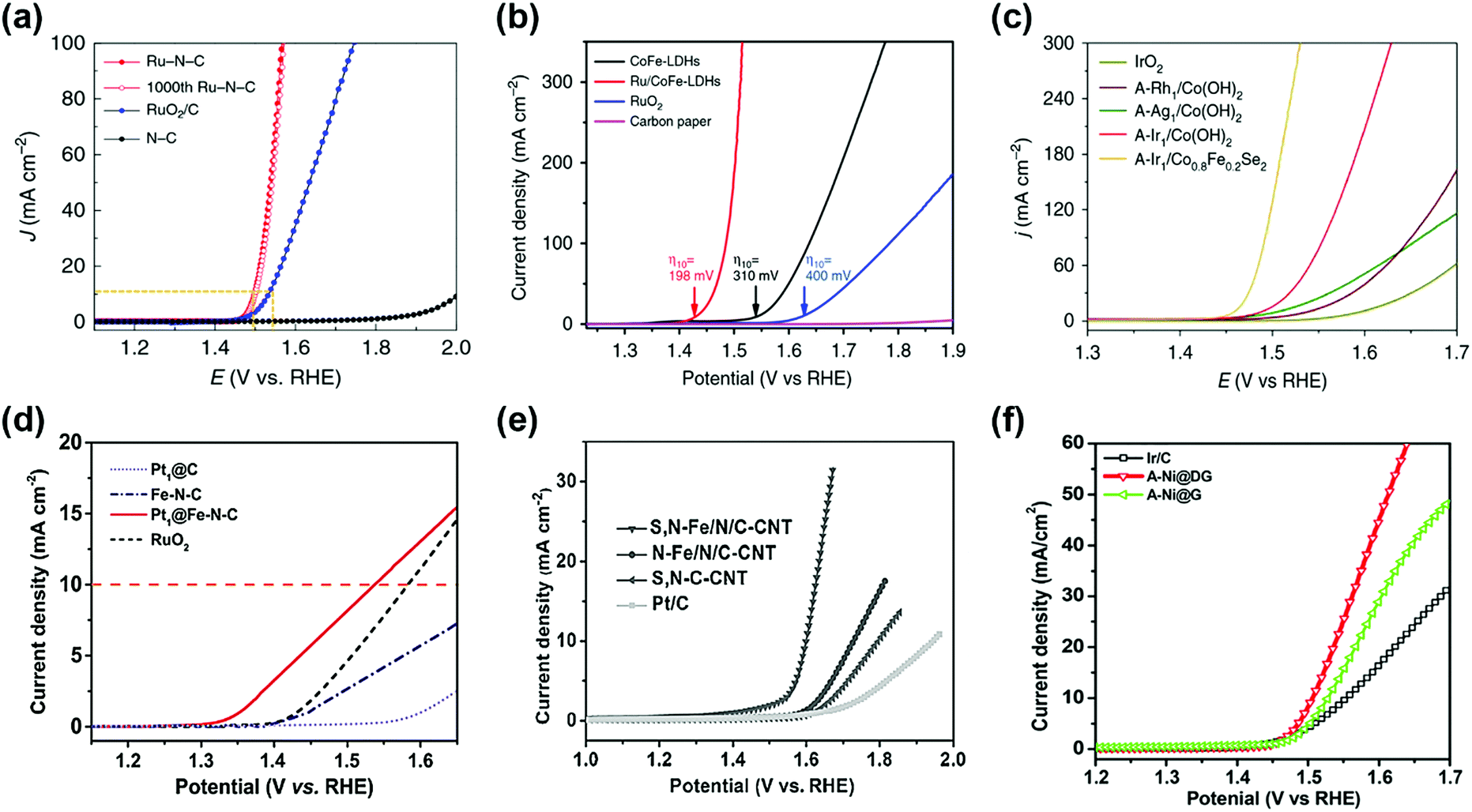 | ||
| Fig. 5 (a) Electrocatalytic OER performances of the Ru–N–C and commercial RuO2/C in a 0.5 M H2SO4 electrolyte. (Reprinted with permission from ref. 37. Copyright 2019, Springer Nature.) (b) Comparison of iR compensated polarization curves of Ru/CoFe-LDHs with CoFe-LDHs, carbon paper and commercial RuO2 catalyst. The η10 stands for the overpotential with current density of 10 mA cm−2. (Reprinted with permission from ref. 38. Copyright 2019, Springer Nature.) (c) Polarization curves of anodically deposited SACs for the OER. All measurements were conducted in 1 M KOH. (Reprinted with permission from ref. 45. Copyright 2020, Springer Nature.) (d) OER polarization curves of single atom catalysts and the reference catalysts measured in 0.1 M KOH. (Reprinted with permission from ref. 46. Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinhei.) (e) iR-corrected OER polarization curves of S,N-Fe/N/C-CNT, N-Fe/N/C-CNT, and S,N-C-CNT samples, and commercial Pt/C, in 0.1 M KOH solution. (Reprinted with permission from ref. 47. Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinhei.) (f) OER polarization curves of A-Ni@DG, A-Ni@G, and Pt/C performed in 1 M KOH electrolyte. (Reprinted with permission from ref. 36. Copyright 2018, Cell Press.) | ||
The stability of SACs during the OER
The stability of SACs with high activity was of concern due to the low coordination of single atoms and low stability of bulk materials such as Ru. Thus, the stability of SACs with high OER catalytic activity was also evaluated. The stability of the Ru–N–C catalyst was measured by chronoamperometry at 1.5 V (Fig. 6a).37 Only slight degradation (5%) was observed for Ru–N–C over the 30 h operation. During 30 h of operation, the Ru dissolution ratio of Ru–N–C was 5%, suggesting excellent structural stability of Ru coordinated with N site. The stability of Ru/CoFe-LDH was evaluated by CV and constant potential test.38 The stable performance of Ru/CoFe-LDH was observed after 1000 CV cycles between 1.35 and 1.5 V vs. RHE. Under chronovoltametry tests in Fig. 6b, Ru/CoFe-LDH exhibits a stable current density after 24 h (99% maintained), which is better than that of CoFe-LDH (90% after 12 h) and RuO2 (64% after 10 h). The stability of the A-Ir1/Co0.8Fe0.2Se2 catalyst was tested in a two-electrode system using a nickel foam substrate (Fig. 6c).45 At a current density of 500 mA cm−2, the cell voltage was only 1.62 V and maintained for over 100 h. This suggests high stability of an Ir single atom for the OER. The stability of transition metal SACs was also evaluated in alkaline media. Tavakkoli et al. reported Ni, Co, Mo single atom doped graphene nanoflake–carbon nanotube hybrid material (N–Co–M–GF/CNT) for the ORR and OER under alkaline conditions. N–Co–M–GF/CNT exhibits stable performance during 24 h at 20 mA cm−2.51 A-Ni@G showed stable potential at a current density of 5 mA cm−2, demonstrating stable single-atom Ni on the graphene substrate (Fig. 6d).36 The high activity and stability of SACs suggests their potential for real applications of water splitting. The performance and stability summary of SACs for the OER is tabulated in Table 1.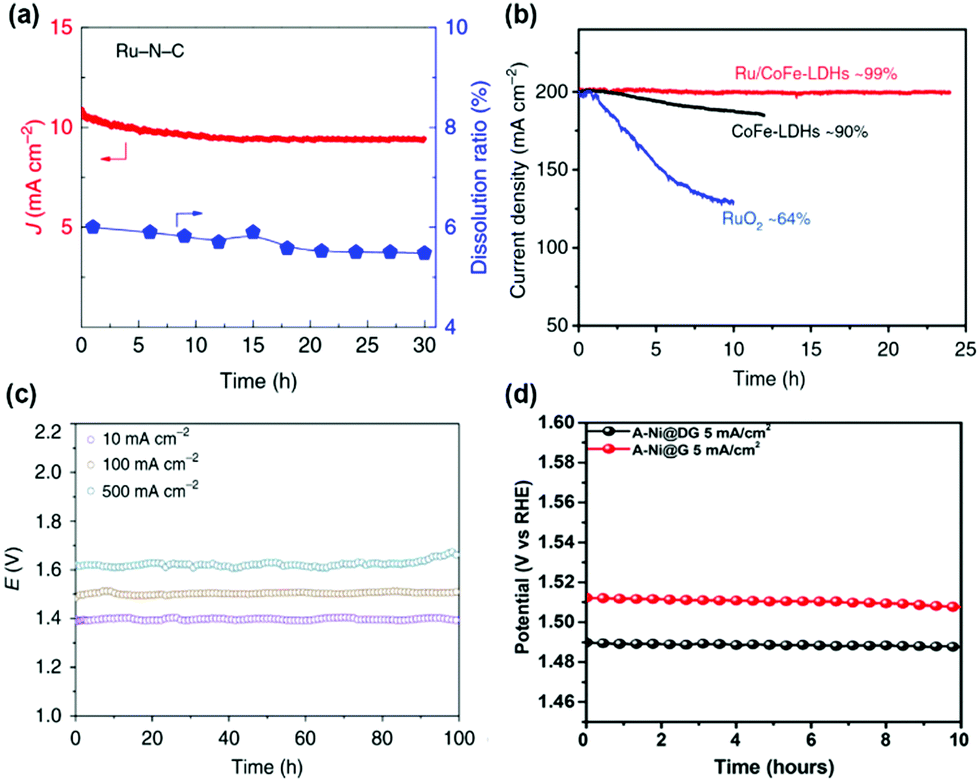 | ||
| Fig. 6 (a) Plot of current density and Ru dissolved mass ratio versus time for Ru–N–C at a constant anode voltage of 1.49 V versus RHE in a 0.5 M H2SO4 electrolyte. (Reprinted with permission from ref. 37. Copyright 2019, Springer Nature.) (b) The potentiostatic curves of Ru/CoFe-LDHs with CoFe-LDHs under a certain overpotential for initial current density of 200 mA cm−2. (Reprinted with permission from ref. 38. Copyright 2019, Springer Nature.) (c) Chronopotentiometric curves of Ir1/Co0.8Fe0.2Se2@Ni foam for overall water splitting at 10, 100, and 500 mA cm−2 for 100 h. (Reprinted with permission from ref. 45. Copyright 2020, Springer Nature.) (d) The chronopotentiometry curves of A-Ni@DG and A-Ni@G at an anodic current density of 5 mA cm−2 in a 1 M KOH electrolyte. (Reprinted with permission from ref. 36. Copyright 2018, Cell Press.) | ||
| Catalyst | Electrolyte | Overpotential @ 10 mA cm−2 (mV) | Tafel slope (mV dec−1) | Durability | Ref. |
|---|---|---|---|---|---|
| Ru–N–C | 0.5 M H2SO4 | 267 | 52.6 | 30 h @ 10 mA cm−2 | 37 |
| A-Ir1/Co0.8Fe0.2Se2 | 1 M KOH | 230 | 100 h @ 500 mA cm−2 | 45 | |
| Ir–NiO | 1 M KOH | 215 | 38 | 10 h @ 10 mA cm−2 | 33 |
| sAu/NiFe LDH | 1 M KOH | 237 | 36 | 20 h @ 100 mA cm−2 | 34 |
| A-Ni@DG | 1 M KOH | 270 | 47 | 10 h @ 5 mA cm−2 | 36 |
| Ru/CoFe-LDH | 1 M KOH | 198 | 39 | 25 h @ 200 mA cm−2 | 38 |
| Pt1@Fe–N–C | 0.1 M KOH | 310 | 62 | — | 46 |
| S,N–Fe/N/C-CNT | 0.1 M KOH | 370 | 82 | — | 47 |
| W-Ni(OH)2 | 1 M KOH | 237 | — | 3.5 h @ 100 mA cm−2 | 35 |
| Ir/Co | 1 M KOH | 235 | 70.2 | 10 h @ 10 mA cm−2 | 50 |
| Ni-CN-200 | 1 M KOH | — | 86 | — | 48 |
Conclusions and perspectives
SACs have received great attention because of their potential as a new generation catalyst with remarkable mass activity. SACs have constituted a bridge connecting heterogeneous and homogeneous catalysts by providing isolated active sites on the support surface. Furthermore, because of the zero-dimension (quantum) size effect and low-coordination status, the SACs show unique physical/chemical properties such as density of states, chemical potential, high electrochemical activity, and reaction selectivity. Researchers have recently begun to understand the applications of SACs for the OER, and it is encouraging that research on catalyst synthesis, characterization of the physical/chemical properties, performance evaluation, and investigation of the reaction mechanisms has been performed. Precious metal-based SACs exhibit excellent OER activity in alkaline as well as acidic media. Transition metal-based SACs have enhanced OER performance under alkaline conditions.Single-atom catalysis for OERs has shown attractive performance and unique properties; however, there are still many challenges to be solved. (1) SACs should guarantee sufficient loading content for practical applications while maintaining the single-atom reaction sites under catalytic conditions of the OER. The general challenge in the synthesis of the high content SA supported catalysts includes the following conditions: (a) support material with extremely high surface area and (b) high number of bonding sites to coordinate isolated single-atoms with strong interaction, such as nitrogen and sulfur. The activity is enhanced when the loading density of the SA is sufficient.
(2) The durability of a single-atom should be guaranteed. The single-atom should not agglomerate and dissolved during the OER process. Moreover, the support materials should also be stable at the OER potential. Carbon materials, which are the most commonly used support for the ORR and HER, are relatively easy to synthesize and they have large surface areas. In addition, carbon materials can be modified to increase the strong bonding sites via the doping process. However, carbon materials have a corrosion reaction that produces carbon dioxide over 0.207 V. The kinetics of the corrosion reaction is slow, but in OER conditions, a very high potential is applied. In addition, the supported metal acts as a catalyst for the corrosion reaction.52 As a result, the kinetics of the corrosion reaction is extremely increased in OER conditions. Therefore, it is necessary to study the synthesis of support materials that can replace carbon materials.
Here, we present examples of infant steps taken in this research area. Currently, researchers in this research area are struggling to comprehend the electronic structures of the active site, which is a fundamental component for designing SACs. Furthermore, advanced characterization techniques are necessary to understand SACs, and it can further provide advanced information about the reaction mechanism, such as the role of light elements (e.g., hydrogen, nitrogen, and sulfur) bonded with single-atoms. The use of theoretical methods is also expected to unravel the possible reaction pathway. Thus, innovative synthesis techniques, investigation of the reaction mechanisms through advanced characterization technologies, and theoretical calculations can bring about great strides in understanding the fundamentals of SACs (Fig. 7).
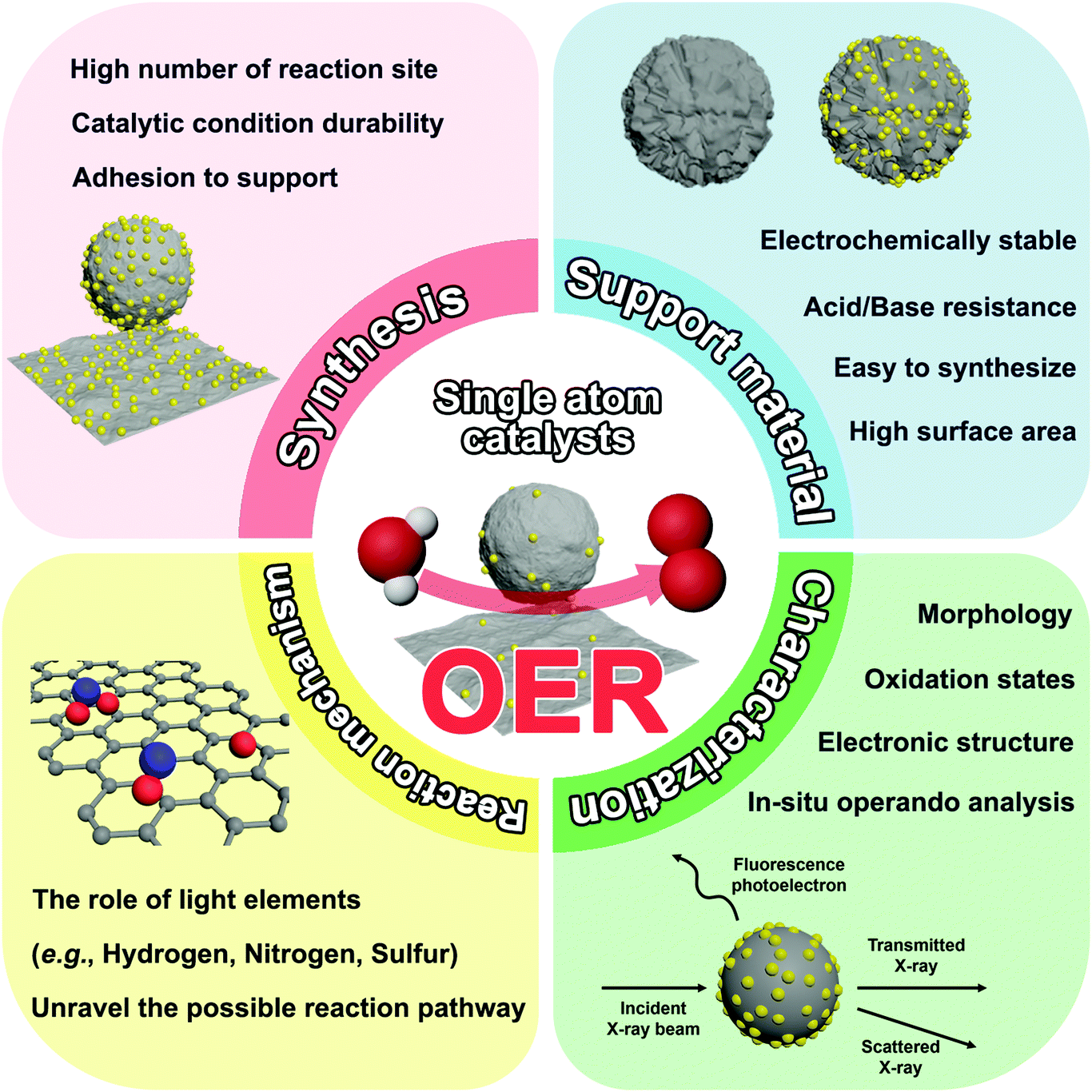 | ||
| Fig. 7 Issues to be improved in the future for single-atom OER catalysts and related challenges. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Science and Technology (KIST) institutional program and supported by “Carbon to X Project” (Project No. 2020M3H7A1098229) through the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Republic of Korea.Notes and references
- K. Neyerlin, R. Srivastava, C. Yu and P. Strasser, J. Power Sources, 2009, 186, 261–267 Search PubMed.
- A. Holewinski, J.-C. Idrobo and S. Linic, Nat. Chem., 2014, 6, 828 Search PubMed.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. r. Selve, A. Bergmann, H. N. Nong and R. Schlögl, J. Am. Chem. Soc., 2015, 137, 13031–13040 Search PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 Search PubMed.
- K. Bergamaski, A. L. Pinheiro, E. Teixeira-Neto and F. C. Nart, J. Phys. Chem. B, 2006, 110, 19271–19279 Search PubMed.
- M. Tian, G. Wu and A. Chen, ACS Catal., 2012, 2, 425–432 Search PubMed.
- F. Raimondi, G. G. Scherer, R. Kötz and A. Wokaun, Angew. Chem., Int. Ed., 2005, 44, 2190–2209 Search PubMed.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- H. Yano, J. Inukai, H. Uchida, M. Watanabe, P. K. Babu, T. Kobayashi, J. H. Chung, E. Oldfield and A. Wieckowski, Phys. Chem. Chem. Phys., 2006, 8, 4932–4939 Search PubMed.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 Search PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 Search PubMed.
- C. Zhu, Q. Shi, S. Feng, D. Du and Y. Lin, ACS Energy Lett., 2018, 3, 1713–1721 Search PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 Search PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 Search PubMed.
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1–10 Search PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-j. Liu and L. Zhuang, Nat. Commun., 2017, 8, 1–10 Search PubMed.
- S. Shin, J. Kim, S. Park, H.-E. Kim, Y.-E. Sung and H. Lee, Chem. Commun., 2019, 55, 6389–6392 Search PubMed.
- M. Jia, S. Hong, T.-S. Wu, X. Li, Y.-L. Soo and Z. Sun, Chem. Commun., 2019, 55, 12024–12027 Search PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 Search PubMed.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 Search PubMed.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 Search PubMed.
- J. Kim, C. W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2018, 8, 1701476 Search PubMed.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 Search PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai and R. Chen, Nat. Energy, 2018, 3, 140–147 Search PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang and J. Lu, Angew. Chem., 2019, 131, 15013–15018 Search PubMed.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 Search PubMed.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie and A. S. Jalilov, Adv. Energy Mater., 2018, 8, 1703487 Search PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. R. Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 1–9 Search PubMed.
- H. Ju, G. Kaur, A. P. Kulkarni and S. Giddey, J. CO2 Util., 2019, 32, 178–186 Search PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 Search PubMed.
- Y. Mo, S. P. Ong and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205446 Search PubMed.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 Search PubMed.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 Search PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 Search PubMed.
- J. Yan, L. Kong, Y. Ji, J. White, Y. Li, J. Zhang, P. An, S. Liu, S.-T. Lee and T. Ma, Nat. Commun., 2019, 10, 1–10 Search PubMed.
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang and A. Du, Chem, 2018, 4, 285–297 Search PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang and W. Liu, Nat. Commun., 2019, 10, 1–9 Search PubMed.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma and Z. Feng, Nat. Commun., 2019, 10, 1–11 Search PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 1–6 Search PubMed.
- W. H. Lai, L. F. Zhang, W. B. Hua, S. Indris, Z. C. Yan, Z. Hu, B. W. Zhang, Y. N. Liu, L. Wang, M. Liu, R. Liu, Y. X. Wang, J. Z. Wang, Z. P. Hu, H. K. Liu, S. L. Chou and S. X. Dou, Angew. Chem., Int. Ed., 2019, 58, 11868–11873 Search PubMed.
- Q. M. Deng, J. Zhao, T. T. Wu, G. B. Chen, H. A. Hansen and T. Vegge, J. Catal., 2019, 370, 378–384 Search PubMed.
- H. L. Fei, J. C. Dong, Y. X. Feng, C. S. Allen, C. Z. Wan, B. Volosskiy, M. F. Li, Z. P. Zhao, Y. L. Wang, H. T. Sun, P. F. An, W. X. Chen, Z. Y. Guo, C. Lee, D. L. Chen, I. Shakir, M. J. Liu, T. D. Hu, Y. D. Li, A. I. Kirkland, X. F. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 Search PubMed.
- Y. Wu, C. Li, W. Liu, H. H. Li, Y. Y. Gong, L. Y. Niu, X. J. Liu, C. Q. Sun and S. Q. Xu, Nanoscale, 2019, 11, 5064–5071 Search PubMed.
- X. Y. Li, P. Cui, W. H. Zhong, J. Li, X. J. Wang, Z. W. Wang and J. Jiang, Chem. Commun., 2016, 52, 13233–13236 Search PubMed.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si and S. Zhou, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 Search PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu and C. Wu, Angew. Chem., Int. Ed., 2017, 56, 610–614 Search PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 Search PubMed.
- Y. Li, Z. S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 Search PubMed.
- Y. Zhang, C. Wu, H. Jiang, Y. Lin, H. Liu, Q. He, S. Chen, T. Duan and L. Song, Adv. Mater., 2018, 30, 1707522 Search PubMed.
- M. Tavakkoli, E. Flahaut, P. Peljo, J. Sainio, F. Davodi, E. V. Lobiak, K. Mustonen and E. I. Kauppinen, ACS Catal., 2020, 10, 4647–4658 Search PubMed.
- S. Moller, S. Barwe, J. Masa, D. Wintrich, S. Seisel, H. Baltruschat and W. Schuhmann, Angew. Chem., Int. Ed., 2020, 59, 1585–1589 Search PubMed.
Footnote |
| † The authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |