
Hydrogen evolution reaction mediated by an all-sulfur trinuclear nickel complex†
Cyril
Pieri
a,
Anirban
Bhattacharjee
b,
Alexandre
Barrozo
 a,
Bruno
Faure
a,
Michel
Giorgi
c,
Jennifer
Fize
d,
Marius
Réglier
a,
Martin
Field
*d,
Maylis
Orio
*a,
Vincent
Artero
d and
Renaud
Hardré
*a
a,
Bruno
Faure
a,
Michel
Giorgi
c,
Jennifer
Fize
d,
Marius
Réglier
a,
Martin
Field
*d,
Maylis
Orio
*a,
Vincent
Artero
d and
Renaud
Hardré
*a
aAix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: renaud.hadre@univ-amu.fr; maylis.orio@univ-amu.fr
bSocivolta Inc., 3 Place Ville Marie, suite 400, Montreal, QC H3B 2E3, Canada
cAix Marseille Univ, CNRS, Centrale Marseille, Spectropole FR1739, Marseille, France
dUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 17 rue des Martyrs, 38000 Grenoble, France. E-mail: martin.field@cea.fr
First published on 5th August 2020
Abstract
We report the synthesis and the characterization of a trinuclear nickel complex. Solid state and solution studies using X-ray diffraction, NMR and UV-vis spectroscopy highlight the square planar geometries around the metal centers and an all-sulfur coordination sphere. It exhibits significant electrocatalytic activity for hydrogen evolution in DMF using Et3NHCl as the proton source. DFT studies suggest that sulfur atoms act as proton relay, as proposed in [NiFe] hydrogenases.
The limited sources of fossil fuels, together with their environmental impact, render them unviable in the long run. This is aggravated by an increasing energy demand worldwide, resulting in higher prices, as well as with the lack of renewability of fossil energy at the human time scale.1 To the contrary, dihydrogen seems to be an ideal substitute, since it is based on the most abundant atom in the universe, and its combustion yields high energy, with water as the only waste.2 The main issue remains with its production. While water electrolysis appears as the most efficient way to produce hydrogen, the flexible proton exchange membrane technology requires the use of platinum, a rare and expensive metal with limited stocks.3 In the search for an economically competitive way of producing dihydrogen with non-noble metal-based catalysts, Nature can guide us and inspiration can be found with the [NiFe] hydrogenases. This family of enzymes is a very attractive example as it is the most abundant hydrogenases among living organisms. Their active sites are composed of two metal ions, iron and nickel, the latter lying in a rare all-sulfur and distorted tetrahedral coordination geometry.4–6 Many bio-inspired complexes mimicking this active site can be found in literature7–14 These models generally show some degree of variation from the enzyme active site: metals, coordinating atoms, bridging and exogenous ligands, e.g. CO and CN−.15 To reproduce the features of the [NiFe] hydrogenase active site, an all-sulfur nickel complex was obtained by Yamamura et al. in 1985, long before the crystal structure of the enzyme was resolved.16 Sellman et al. also explored a certain number of all-sulfur nickel complexes.17 Once the structure of the [NiFe] hydrogenase was known, many heterobimetallic complexes were then developed.18 Unlike the enzyme, all of these complexes suffered from a lack of nickel centred reactivity6 and the situation only changed recently.11,19–23 Moreover, the hydrogen producing catalytic performances of these complexes with respect to overpotential requirement and turnover frequency (TOF) towards hydrogen production were far below those of [NiFe] hydrogenases, operating at the thermodynamic equilibrium with a TOF around 1000 s−1.24,25 The high activity of hydrogenase depends on the presence of proton relays at the vicinity of the active site. In [NiFe] hydrogenases, terminal cysteinates have been assigned this role.6 While there are several examples of FeFe hydrogenase models with terminal thiolate ligands prone to protonation,26,27 there is only one known example of a NiFe complex, and this was only observed under strongly acidic conditions, not pertinent for catalysis.28 In this context, we have developed a bio-inspired nickel complex bearing an all-sulfur and constrained ligand that resembles the Ni-center of the enzyme active site with its cysteine-thiolate ligands implicated as bases in proton transfer reactions.
Inspired by the seminal work of Bosnich and Pickett,29–31 our strategy to prepare the trithiol 3 was to start the synthesis from the thiol 1 easily accessible according to the Bosnich procedure.29,32 Compound 2 was then obtained by S-alkylation of 1 with 2-bromoethyl-trityl-sulfide. Removal of the trityl substituent in dry acidic condition (CF3COOH) and the reduction of the disulfide moiety by triethyltin hydride led to the trithiol 3. This overall process provided access to the trithiol 3 which was used for the synthesis of Ni(II) complexes. Thus, the reaction of the ligand 3 with 1 eq. of [Ni(CH3CN)6](BF4)2 in acetonitrile with K2CO3 as base led to the formation of a soluble complex that rapidly evolved by the formation of a brown precipitate. Filtration followed by liquid chromatography with CH2Cl2 as eluant allowed us to get the pure Ni(II) complex 4 in 45% yield (Scheme 1).
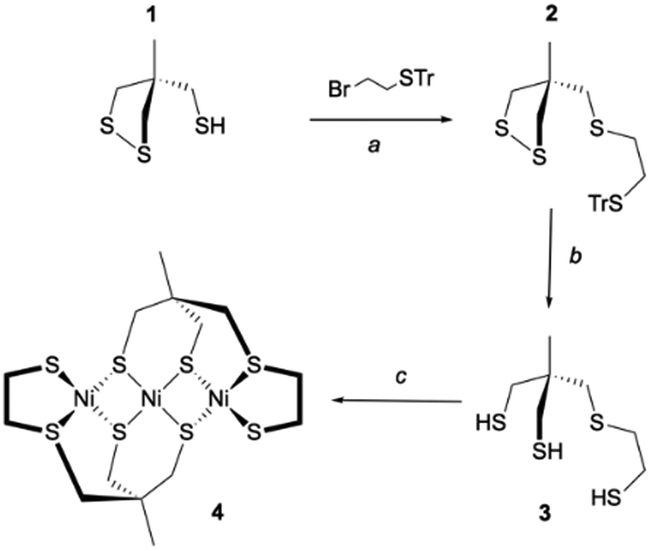 | ||
| Scheme 1 Synthetic route for the nickel complex 4. Conditions: (a) NaH, THF, 0 °C to RT, overnight, 66%; (b) CF3COOH (excess), Et3SiH (3 eq.), CH2Cl2, RT overnight, 81%; (c) [Ni(CH3CN)6](BF4)2, K2CO3 (3 eq.), CH3CN, RT, 2 h. | ||
Suitable crystals were obtained from a saturated solution of the complex in CH3CN. The complex crystallizes as a symmetric trimer with the central Ni-atom lying on the crystallographic two-fold axis (Fig. 1).
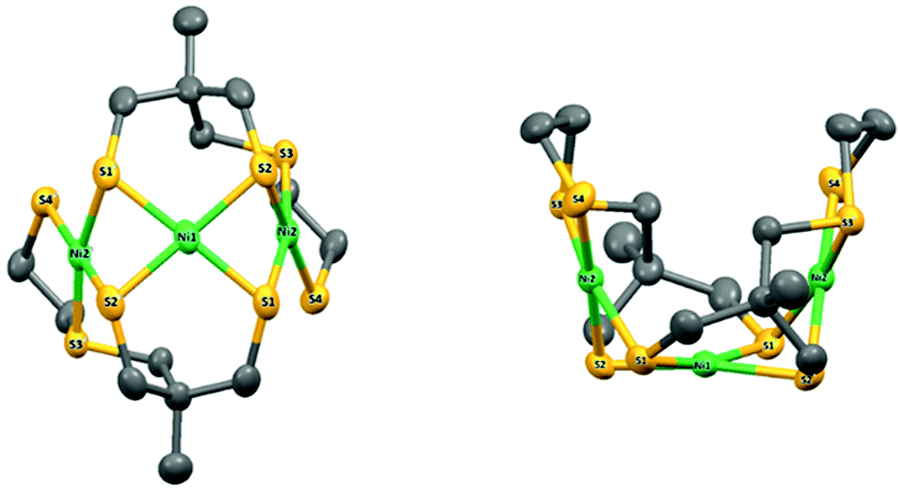 | ||
| Fig. 1 X-ray molecular structure of 4 with atom labelling scheme and ellipsoid parameters at 50% probability level. Hydrogen atoms and carbon labels are omitted for clarity. | ||
The asymmetric unit is thus composed of half the complex. The coordination geometries of the metals are square planar with small deviations from planar (defined by the Ni and the four coordinated S atoms, Fig. S6 and Table S1, ESI†): the maximum out-of-plane distances are close to 0.15 Å and 0.19 Å (S1 in plane P1 including Ni2 and S1 in plane P2 including Ni1, respectively). The dihedral angle between P1 and P2 is equal to 73.0697(4)°, while the dihedral angle between P1 and the symmetric plane P1′ (symmetry code: 1 − x, y, 3/2 − z) is equal to 33.8471(8). The structure of compound 4 is therefore very compact and folded with a bowl shape and a small accessible cavity toward the central Ni-atom (mean width and depth equal to 3.4 Å and 4.9 Å, respectively, Table S2, ESI†).
To check that the structure of 4 corresponds to the one found in solution, we performed the UV-vis titration of the ligand 3 by nickel perchlorate in the presence of triethylamine in DMF. Upon addition of [Ni(H2O)6](ClO4), the UV-vis spectrum displays three absorption bands at 290 nm, 410 nm and 540 nm (Fig. S3, ESI†).
Plotting the molecular extinction coefficient as a function of the number of Ni(II) equivalents for each absorption band allowed us to determine the stoichiometry of the complex (Fig. S4, ESI†). Analysis of the titration curves for both the 410 and 540 nm bands support the formation of a trinuclear species like 4 with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 nickel to ligand ratio, e.g. 2 ligands for 3 metal ions.
:3 nickel to ligand ratio, e.g. 2 ligands for 3 metal ions.
To support the above assignment, we used time-dependent DFT to calculate the theoretical UV-vis spectrum of 4 (Fig. S9 and Table S5, ESI†) in the trimer form revealed by X-ray diffraction. Our computations support that its UV-vis spectrum is dominated by three main absorption bands of different intensity (Fig. S10, ESI†). The low-energy absorption is assigned to a d–d transition while the two other spectral features are attributed to charge transfers from the ligand to metal (Fig. S11 and Table S4, ESI†). Both the shape and relative intensities of the calculated spectrum are in pretty fair agreement with the spectrum measured in solution.
The electrochemical study of 4 was performed by cyclic voltammetry between +0.5 and −2.5 V vs. Fc+/0 (Fig. 2). In the cathodic region, two one-electron redox systems were observed: one quasi-reversible at −1.65 V (1/2(Epc* + Epa*)) and another irreversible at −2.09 V vs. Fc+/0 (Epc**). The plot of the peak current versus the square root of the scan rates for the reversible cathodic wave was found to be linear, indicating that it is a diffusion-controlled process (Fig. S7, ESI†). In the anodic direction, the CV revealed one oxidative process with an irreversible wave observed at +0.22 V vs. Fc+/0.
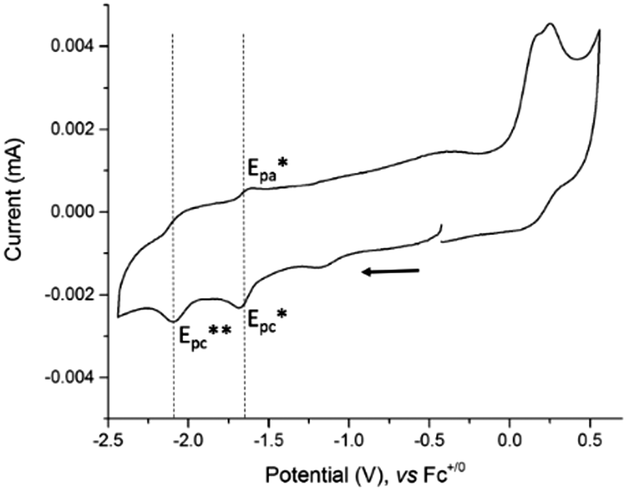 | ||
| Fig. 2 Cyclic voltammogram of 4, concentration 0.3 mM at a stationary glassy carbon electrode (3 mm diameter) in DMF + 0.1 M NEt4ClO4. Scan rate 100 mV s−1. | ||
In an effort to gain insight into the nature of the species observed during cyclic voltammetry experiments, we calculated the structures of the putative species resulting from one and two-electron reduction of 4 (Fig. S12–S14 and Tables S6–S8, ESI†), and their electronic properties. Upon reduction, our results indicate that most of the spin density is distributed at one nickel center (Fig. S15, ESI†). The first reduction process of 4 occurs at the central metal site leading to the formation of a Ni(II)–Ni(I)–Ni(II) complex. The two-electron reduction of 4 results in a triplet ground spin state S = 1 with a stabilization energy of 24 kJ mol−1 over the singlet state S = 0. This result supports a metal-based reduction occurring on another nickel center (Fig. S16, ESI†) and leading to the formation of a Ni(I)–Ni(II)–Ni(I) complex. We can notice a subsequent electronic rearrangement between the first and second reduction, which explains the irreversibility observed experimentally for the second redox system.
Redox potential calculations were conducted to fully assign the electrochemical events (Table S3, ESI†). The computed values for the one- and two-electron reduction processes are in good agreement with experiment which indicates that the two cathodic systems correspond to a monoelectronic processes and support the above assignments of the reduction loci in 4.
To examine the capability of complex 4 to mediate proton reduction catalysis, its electrochemical behaviour was studied by cyclic voltammetry in the presence of Et3NHCl (pKa = 9.2 in DMF)33,34 as proton source.
Cyclic voltammograms of 4 were recorded under argon in DMF solutions with increased amounts of Et3NHCl (Fig. 3) and show a large irreversible catalytic wave with a mid-wave potential E0cat of −1.70 ± 0.03 V vs. Fc+/0, i.e. close to the reduction potential of 4. The current response of this peak-shaped wave is directly proportional to the concentration of protons in the electrolyte solution (Fig. S8, ESI†). A value of 590 mV was calculated for the overpotential requirement of 4 for proton reduction in the presence of Et3NHCl in DMF.34
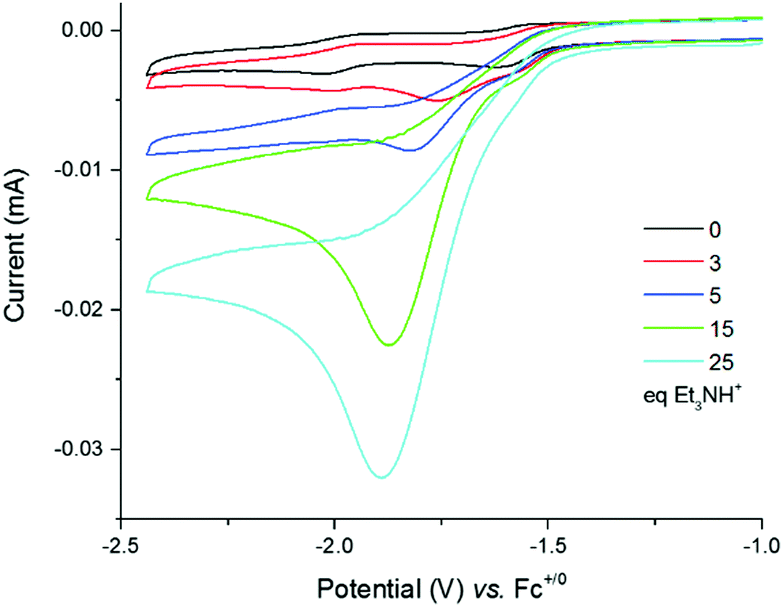 | ||
| Fig. 3 Cyclic voltammograms of a 0.25 mM solution of 4 in DMF (0.1 M NEt4ClO4) with increasing amounts of Et3NHCl (0–55 equiv.) recorded at a glassy carbon electrode (3 mm diameter) and 100 mV s−1. | ||
To confirm that the observed catalytic waves correspond to the electrocatalytic reduction of protons into hydrogen, we performed a bulk electrolysis experiment of a DMF solution of Et3NHCl in excess (200 mM) and 4 (1 mM). Gas products were monitored by continuous in-line GC analysis. A potential of −1.60 V versus Fc+/0 was applied to the mercury-pool working electrode resulting in the immediate formation of hydrogen. This experiment rules out the possibility that proton reduction occurs as the result of deposition of nickel nanoparticles on the electrode surface since they would amalgamate within the mercury pool electrode. Consequently, under these conditions catalysis mediated by 4 is expected to be homogeneous in nature. During the 15 h experiment, a turnover number of 5 was achieved with a moderate faradaic yield of 30%; indicating some degradation of the complex during turn-over.
DFT calculations were finally conducted to decipher the reaction mechanism of hydrogen evolution by 4 (Scheme 2). We propose the catalytic cycle for H2 evolution presented in Scheme 2 and based on the ECEC sequence, classically found for cobalt and nickel complexes.35–37 UV/vis measurements performed in the absence and presence of Et3NHCl (Fig. S5, ESI†) display almost identical features, indicating that the structure of 4 is unaffected by the addition of a proton source of such strength. From the cyclic voltammetry measurements in the presence of acid, it can be inferred that the reaction proceeds from the one-electron reduced species with a midwave electrocatalytic potential equal to that of the first reduction in 4 denoted [Ni3′S4′2]. The first reduction at the Ni site was calculated at potentials of −1.84 and −2.02 V vs. Fc+/0 when using the B86 and B3LYP functionals, respectively (Table S3, ESI†). We then investigated the protonation of the reduced complex [Ni′3S4′2]− with Et3NH+ yielding [HNi3′S4′2]. Two distinct protonation sites could be found. The most stable isomer (by 28 and 61 kJ mol−1 with BP86/B3LYP, respectively) is protonated on a terminal thiolate ligand coordinated on a peripheral Ni ion that remains in a square-planar environment (Fig. S17 and Table S9, ESI†). This reaction is calculated to be almost isergonic. Another isomer, with higher energy, is obtained when protonation occurs at the Ni site thus yielding a hydride derivative with a distorted square-based pyramidal geometry (Fig. S18 and Table S10, ESI†). Reduction of the two isomers of [HNi3′S4′2] yielding [HNi3′S4′2]− is calculated to occur at potentials close to the one calculated for the first reduction of [Ni3′S4′2]. We note that the stability order of the two isomers is inverted after this reduction step: in that case the sulfur-protonated species (Fig. S19 and Table S11, ESI†) is less stable (by 76 and 24 kJ mol−1 with BP86/B3LY, respectively) than the Ni-hydride square-planar isomer (Fig. S20 and Table S12, ESI†) in which one thioether ligand has been eliminated from the Ni coordination sphere and replaced by the hydride ligand.
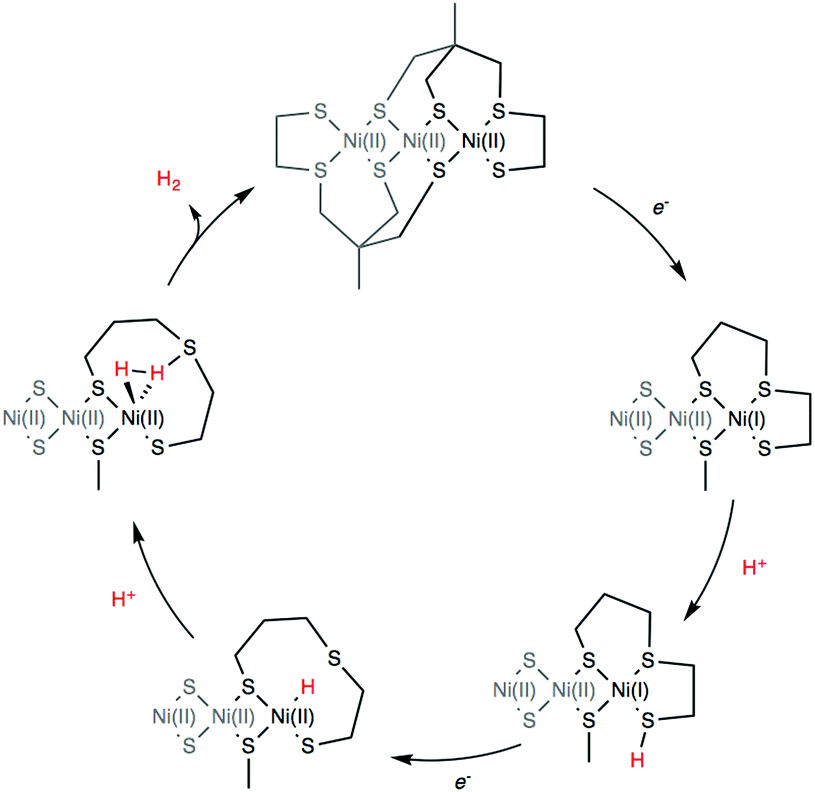 | ||
| Scheme 2 Proposed DFT computed reaction mechanism for hydrogen evolution mediated by 4. | ||
Finally, protonation of the reduced hydride species [HNi3′S4′2]− leads to formation of a dihydrogen complex [(H2)Ni3′S4′2] (Fig. S21 and Table S13, ESI†). This reaction is calculated to be thermodynamically favorable. The structure is reminiscent of a dihydrogen bond between nickel and sulfur that has been proposed as a key intermediate in the route to H2 evolution at the active site of [NiFe] hydrogenases (Ni-R in Scheme 3).38,39 From this dihydrogen complex, liberation of H2 is strongly and entropically favored. Removal of H2 leads to the original structure of [Ni3′S4′2] thus closing the catalytic cycle.
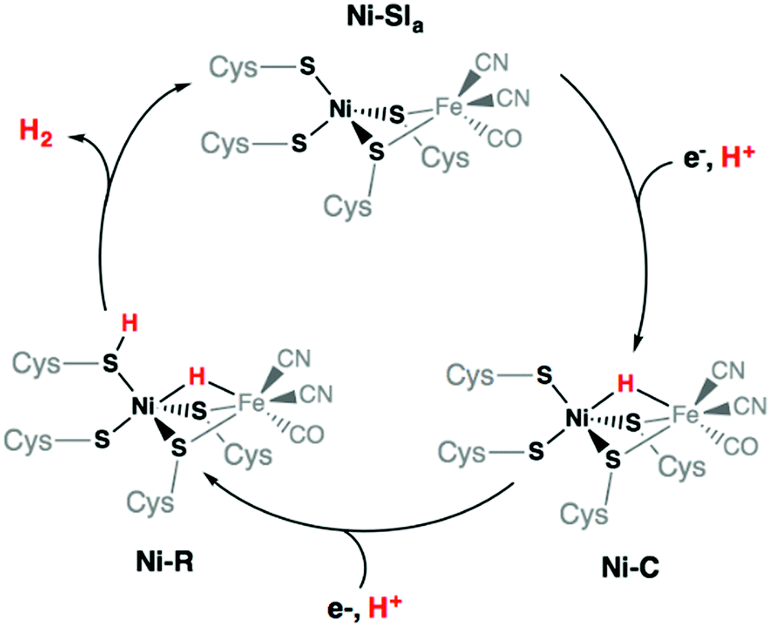 | ||
| Scheme 3 Catalytic cycle of the [NiFe] hydrogenases involving the Ni-SIa, Ni-R, and Ni-C states. | ||
In this work, we have reported the experimental and theoretical study of an original trinuclear nickel complex that displays an all-sulfur coordination sphere and exhibits a significant catalytic activity with a moderate overpotential requirement for hydrogen production in DMF using Et3NHCl as the proton source. Detailed analysis of the electrocatalytic data combined with DFT calculations allowed us to propose an ECEC catalytic pathway involving Ni-bound terminal thiolate ligand as proton relay mimicking the role of terminal cysteinate ligand in the active site of [NiFe] hydrogenases.
Further work is currently in progress in our group to develop new bio-inspired mimics of the [NiFe] hydrogenase and to design original polynuclear metal complexes with thiolate ligands capable of mediating efficient hydrogen evolution.
The authors gratefully acknowledge research support of this work by the French National Research Agency (CODEC, ANR-19-CE05_0030_01; CatH2, ANR-07-BLAN-0298; NiFe-Cat, ANR-2010-BLAN-711-1; NiFemim, ANR-DFG-2016-NLE; Labex ARCANE; Graduate school of Chemistry, Biology and Health of Univ. Grenoble Alpes, CBH-EUR-GS, ANR-17-EURE-0003).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. A. King, Science, 2004, 303, 176 CrossRef CAS PubMed.
- P. P. Edwards, V. L. Kuznetsov, W. I. F. David and N. P. Brandon, Energy Policy, 2008, 36, 4356 CrossRef.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865 RSC.
- A. Volbeda, M.-H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J.-C. Fontecilla-Camps, Nature, 1995, 373, 580 CrossRef CAS PubMed.
- A. Volbeda, A. E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J.-C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS PubMed.
- D. Sirbu, T. Straistari and A. C. Benniston, Hydrogenases: From Biomimetic to Bioinspired Models, Bioinspired Chemistry, From Enzymes to Synthetic Models, 2019, vol. 5, p. 89 Search PubMed.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107 CrossRef CAS PubMed.
- M. Y. Darensbourg, E. J. Lyon and J. J. Smee, Coord. Chem. Rev., 2000, 206, 533 CrossRef.
- Z. Li, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2005, 127, 8950 CrossRef CAS PubMed.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pecaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054 CrossRef CAS PubMed.
- S. Canaguier, M. Field, Y. Oudart, J. Pécaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876 RSC.
- D. Basu, T. Spencer Bailey, N. Lalaoui, C. P. Richers, T. J. Woods, T. B. Rauchfuss, F. Arrigoni and G. Zampella, Inorg. Chem., 2019, 58(4), 2430 CrossRef CAS PubMed.
- N. Coutard, N. Kaeffer and V. Artero, Chem. Commun., 2016, 52, 13728 RSC.
- T. R. Simmons, G. Berggrena, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127 CrossRef CAS.
- T. Yamamura, H. Miyamae, Y. Katayama and Y. Sasaki, Chem. Lett., 1985, 269 CrossRef CAS.
- D. Sellmann, S. Fünfgelder, G. Pohlmann, F. Knoch and M. Moll, Inorg. Chem., 1990, 29, 4772 CrossRef CAS.
- S. Kaur-Ghumaan and M. Stein, Dalton Trans., 2014, 43, 9392 RSC.
- H. Tang and M. B. Hall, J. Am. Chem. Soc., 2017, 139, 18065 CrossRef CAS PubMed.
- D. Brazzolotto, L. Wang, H. Tang, M. Gennari, N. Queyriaux, C. Philouze, S. Demeshko, F. Meyer, M. Orio, V. Artero, M. B. Hall and C. Duboc, ACS Catal., 2018, 8, 10658 CrossRef CAS.
- G. M. Chambers, M. T. Huynh, Y. Li, S. Hammes-Schiffer, T. B. Rauchfuss, E. Reijerse and W. Lubitz, Inorg. Chem., 2016, 55, 419 CrossRef CAS PubMed.
- S. Ding, P. Ghosh, A. M. Lunsford, N. Wang, N. Bhuvanesh, M. B. Hall and M. B. Y. Darensbourg, J. Am. Chem. Soc., 2016, 138, 12920 CrossRef CAS PubMed.
- S. Ding, P. Ghosh, M. Y. Darensbourg and M. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9775 CrossRef CAS PubMed.
- M. E. Ahmed and A. Dey, Curr. Opin. Electrochem., 2019, 15, 155 CrossRef CAS.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, et al. , Chem. Rev., 2016, 116, 8693 CrossRef CAS PubMed.
- U.-P. Apfel, D. Troegel, Y. Halpin, S. Tschierlei, U. Uhlemann, H. Görls, M. Schmitt, J. Popp, P. Dunne, M. Venkatesan, M. Coey, M. Rudolph, J. G. Vos, R. Tacke and W. Weigand, Inorg. Chem., 2010, 49, 10117 CrossRef CAS PubMed.
- R. Zaffaroni, T. B. Rauchfuss, D. L. Gray, L. De Gioia and G. Zampella, J. Am. Chem. Soc., 2012, 134, 19260 CrossRef CAS PubMed.
- K. Weber, T. Krämer, H. S. Shafaat, T. Weyhermüller, E. Bill, M. van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 20745 CrossRef CAS PubMed.
- C. Kolomyjec, J. Whelan and B. Bosnich, Inorg. Chem., 1983, 22, 2343 CrossRef CAS.
- M. Razavet, S. C. Davies, D. L. Hughes and C. J. Pickett, Chem. Commun., 2001, 847 RSC.
- M. Razavet, S. C. Davies, D. L. Hughes, J. E. Barclay, D. J. Evans, S. A. Fairhurst, X. Liu and C. J. Pickett, Dalton Trans., 2003, 586 RSC.
- R. A. Valiulin and A. G. Kutateladze, J. Org. Chem., 2008, 73, 335 CrossRef CAS PubMed.
- K. Izutsu, Acid-Base Dissociation Constants, Dipolar Aprotic Solvents, Blackwell Scientific, Oxford, UK, 1990 Search PubMed.
- V. Fourmond, P. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338 CrossRef CAS PubMed.
- V. Artero and J.-M. Saveant, Energy Environ. Sci., 2014, 7, 3808 RSC.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286 CrossRef CAS PubMed.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304–305, 3 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414 CrossRef CAS PubMed.
- G. Dong and U. Ryde, JBIC, J. Biol. Inorg. Chem., 2016, 21, 383 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 939713. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04174b |
| This journal is © The Royal Society of Chemistry 2020 |