
Modifying the luminescent properties of a Cu(I) diphosphine complex using ligand-centered reactions in single crystals†
Kyounghoon
Lee
a,
Po-Ni
Lai
 b,
Riffat
Parveen
c,
Courtney M.
Donahue
a,
Mikayla M.
Wymore
a,
Blake A.
Massman
a,
Bess
Vlaisavljevich
c,
Thomas S.
Teets
b and
Scott R.
Daly
*a
b,
Riffat
Parveen
c,
Courtney M.
Donahue
a,
Mikayla M.
Wymore
a,
Blake A.
Massman
a,
Bess
Vlaisavljevich
c,
Thomas S.
Teets
b and
Scott R.
Daly
*a
aDepartment of Chemistry, The University of Iowa, E331 Chemistry Building, Iowa City, IA 52242, USA. E-mail: scott-daly@uiowa.edu
bDepartment of Chemistry, University of Houston, 3585 Cullen Boulevard, Room 112, Houston, TX 77204, USA
cDepartment of Chemistry, The University of South Dakota, 414 E. Clark Street, Vermillion, SD 57069, USA
First published on 30th June 2020
Abstract
Here we report how reactions at a chemically reactive diphosphine shift the long-lived luminescent colour of a crystalline three-coordinate Cu(I) complex from green to blue. The results demonstrate how vapochromism and single-crystal-to-single-crystal transformations can be achieved using ligand-centered reactions.
Luminescent Cu(I) complexes are highly sought after as potential alternatives to those containing iridium, platinum, and ruthenium due to the higher abundance and lower cost of copper. Cu(I) complexes typically exhibit photoluminescence via thermally-activated delayed fluorescence (TADF), which occurs when the energy gap between the S1 and T1 excited states (ΔEST) is thermally accessible, and/or by phosphorescence induced by appreciable spin–orbit coupling.1 This gives rise to widely tuneable emission lifetimes needed for use in diverse applications such as OLEDs,2 chemical sensing,3 photosensitizers,4 and photocatalysis.5
A challenge that has limited the use of luminescent Cu(I) complexes is their propensity to adopt tetrahedral coordination geometries that undergo large excited-state geometric distortions that increase non-radiative decay processes and decrease quantum efficiencies.1a,3a,4c,6 It has been shown, however, that non-radiative decay processes can be suppressed by using ligands with sterically-bulky substituents to form two- or three-coordinate Cu(I) complexes.6b,7 Another challenge is developing stable and efficient blue emitters, which has proven difficult with noble metal complexes because of energetically low-lying metal-centered (3MC) d–d states that provide an alternate pathway of non-radiative decay.8 In contrast, d10 complexes have no 3MC states, and several efficient Cu(I) blue emitters have been reported.7e,9
We recently described a new class of triaminoborane-bridged diphosphine ligands derived from 1,8,10,9-triazaboradecalin (TBD) called TBDPhos.10 When bound to Ni(II) and Pd(II), phenyl-substituted TBDPhos (PhTBDPhos) can undergo cooperative ligand-centered reactions with water and alcohols to form trans N–H and B–OH or B–OR bonds on the TBD backbone (Scheme 1).10a Given that numerous examples of luminescent Cu(I) diphosphine complexes are known,1a,2d,4c,11 and some form highly emissive three-coordinate complexes,6b,7d,e we postulated that ligand-centered reactions in Cu(I) TBDPhos complexes could be used to modify their photophysical properties. Here we report the first such examples using single crystals of (PhTBDPhos)CuCl (1).
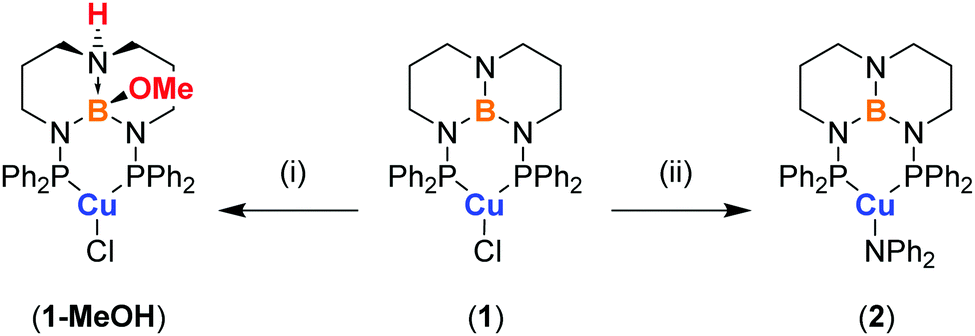 | ||
| Scheme 1 Structure of (PhTBDPhos)CuCl (1) and synthesis of 1-MeOH and 2. (i) Excess MeOH at RT. (ii) Addition of 1 eq. of HNPh2 and KN(SiMe3)2 in toluene at −78 °C. | ||
Complex 1 was prepared by treating CuCl with PhTBDPhos in CH2Cl2 (Scheme 1). The reaction was monitored by NMR spectroscopy, and new signals supporting the formation of 1 were observed at δ 24.8 ppm and δ 40.9 ppm in the 11B and 31P NMR spectra, respectively. Light greenish-yellow crystals were grown from CH2Cl2 solution by vapor diffusion with Et2O. As expected, the crystals were highly luminescent, appearing green when exposed to UV light. X-ray diffraction data collected on the crystals revealed the structure to be three-coordinate monomeric 1 (Fig. 1). The geometry around Cu and B are best described as trigonal planar with the sum of the bond angles being 359.81(4)° and 360.0(3)°, respectively, though the three angles around Cu are less congruent due to the PhTBDPhos bite angle (P–Cu–P) of 101.52(2)°. The Cu–P bond distances of 2.1952(6) and 2.1953(6) Å are 0.04–0.06 Å shorter compared to other CuCl complexes with aryl-substituted diphosphines.6b,12
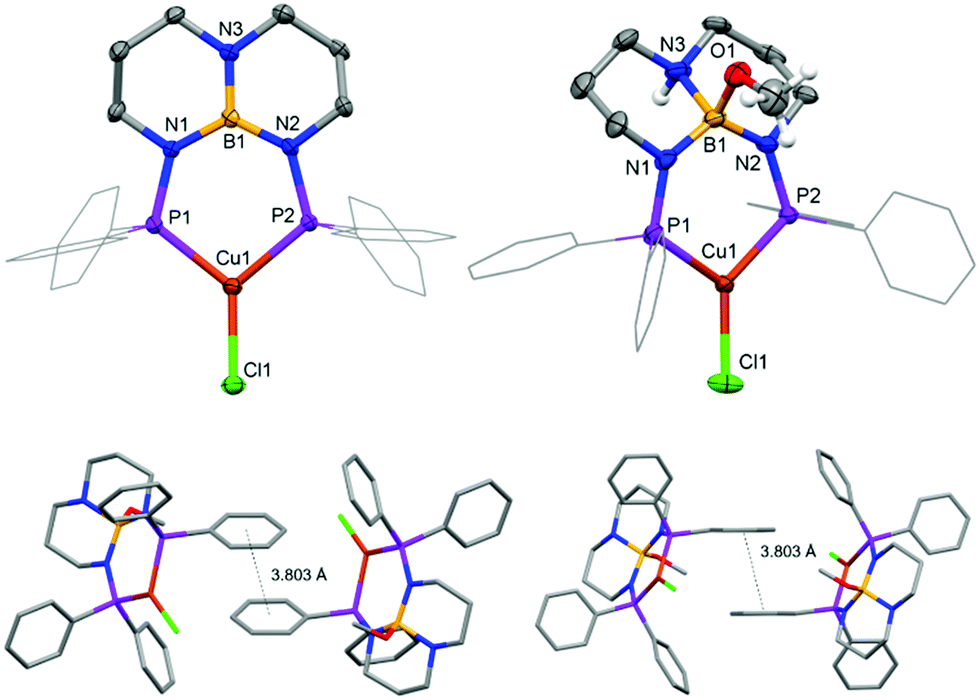 | ||
| Fig. 1 Top: Molecular structures of 1 (left) and 1-MeOH (right) with thermal ellipsoids at the 50% probability level. Phenyl groups are depicted as wire frames, and hydrogen atoms except for NH and OCH3 were omitted from the figures. Bottom: Intermolecular π–π stacking in the extended XRD structure of 1-MeOH. | ||
To give insight into how different ancillary ligands affect the photophysical properties of Cu(I) PhTBDPhos complexes, we replaced the chloride in 1 with diphenylamide, another ligand known to yield luminescent Cu(I) complexes with diphosphines.7e The synthesis of (PhTBDPhos)Cu(NPh2) (2) was performed by treating a mixture of 1 and HNPh2 in toluene with KN(SiMe3)2 at −78 °C, which formed an intense yellow solution (Scheme 1). XRD studies on single crystals obtained by diffusion of pentane into toluene confirmed the three-coordinate complex and revealed similar PhTBDPhos bond distances and angles as 1 (Fig. S2; ESI†). Crystals of 2 exhibit similarly green luminescence like 1 despite the change in ancillary ligand.
Although 1 is highly luminescent in the solid-state, its luminescence is dramatically attenuated in solution, which is attributed in part to dynamic changes in its composition and structure when dissolved. NMR analysis of isolated crystals of 1 in CD2Cl2 revealed small, broad resonances in the baseline that became more resolved upon cooling (Fig. S6 and S7; ESI†). The 31P NMR spectrum collected at −80 °C for example revealed three resonances, suggesting that monomeric 1 dimerizes to some extent in solution and/or undergoes ligand exchange to form [Cu(PhTBDPhos)2][CuCl2], as has been reported for other CuCl diphosphine complexes.13
Fortunately, the attenuated solution luminescence did not prevent our investigation of ligand-centered reactivity on the photophysical properties of 1. We discovered that crystals of 1 are not appreciably soluble in MeOH, but soaking them for a few hours to overnight depending on their size changes their photoluminescence from green to blue. To ensure complete conversion, the crystals were soaked for two days and then analyzed by single-crystal XRD. The crystals had the same monoclinic space group as 1 with similar cell parameters, although the cell setting changed from P21/n to P21/c and the unit cell volume increased from 2850.3(5) to 3017.4(5) Å3 (Table S1; ESI†). Modeling the crystal data revealed these changes to be due to trans addition of MeOH to the TBD backbone to yield (PhTBDPhos-MeOH)CuCl (1-MeOH; Fig. 1). The Cu–P and Cu–Cl distances in 1 and 1-MeOH are effectively identical (Table S2; ESI†), but the P–Cu–P angle increased from 101.52(2)° in 1 to 105.13(4)° in 1-MeOH. As expected, the biggest change occurred at the diphosphine. The N–B bond distances of 1.427(3), 1.464(3), and 1.468 Å in 1 increase to 1.628(5), 1.529(5), and 1.544(5) Å in 1-MeOH.
One of the most remarkable features of the ligand-centered reactivity with 1 is that it also occurs when crystals are exposed to MeOH vapor. Crystal changes that occur in response to vapor are called solvent-induced single-crystal-to-single-crystal (SCSC) transformations.14 Not surprisingly, the SCSC transformation was slower than that observed when soaking the crystals in MeOH, as followed over the course of several days by the photoluminescent colour change from green to blue. Vapochromism with luminescent Cu(I) complexes is known,2d,3e,f,15 and it is typically initiated by solvent induced rearrangement of ligands and metal coordination geometry,16 solvent intercalation in the crystal lattice,17 or solvent binding at the metal.18 Complex 1 is unique because it relies on a ligand-centered reaction to induce the SCSC and the vapochromic response with MeOH. The vapochromic reaction with 1 did not appear to be reversible; the blue luminescence persisted when placing 1-MeOH under dynamic vacuum at ca. 10−2 Torr overnight. Moreover, the crystals appeared to decompose when heated above 60 °C under vacuum, as indicated by their quenched luminescence.
In contrast to 1, attempts to test the ligand-centered reactivity of 2 were unsuccessful. Exposing 2 to MeOH, for example, quenched its photoluminescence. Given that Cu–NPh2 bonds in 2 are highly susceptible to protonolysis, as described previously for similar Cu(I) amido complexes,7e and because emission of 2 is quenched in the solid state when exposed to MeOH, we did not pursue further investigations into its reactivity.
Room-temperature excitation of solid samples of 1 at 360 nm yielded an emission peak at 502 nm consistent with its green luminescence (Fig. 2), and triplicate measurements revealed the quantum yield (ΦPL) to be 67(1)%. The emission spectrum for 2 is similar to 1 with λmax = 505 nm and a slight shoulder at 454 nm, but the quantum yield decreased to ΦPL = 33(1)%. The MeOH-bound complex 1-MeOH showed the most significant change, as expected based on the change in photoluminescence colour; excitation at 310 nm yielded a blue-shifted λmax of 466 nm and ΦPL = 9.2(6)%.
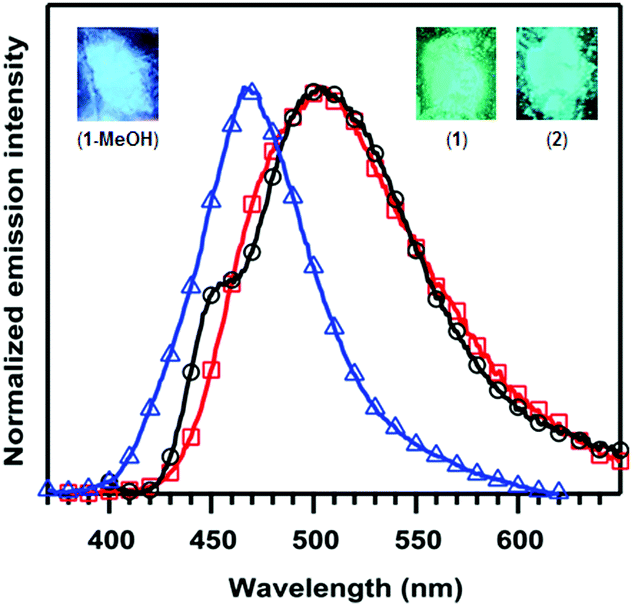 | ||
| Fig. 2 Emission spectra for 1 (red; □), 2 (black; ○) and 1-MeOH (blue; △) in the solid-state. Photoluminescence colours for 1, 2, and 1-MeOH are provided at the upper right or left corners of the figure. Data were collected every 1 nm, and the symbols are present to help distinguish the overlaid plots. | ||
Photoemission decay plots for 1, 2, and 1-MeOH showed bi-exponential curves with weighted-average lifetimes of 670, 550, and 770 μs, respectively. The average radiative decay rates (kr) decreased across the series in the order 1 > 2 > 1-MeOH from 1.1 × 103 to 1.3 × 102 s−1, whereas the non-radiative decay rates (knr) increased from 5.0 × 102 s−1 in 1 to 1.2 × 103 s−1 in 2 and 1-MeOH (Table 1). Such long lifetimes suggest that phosphorescence is the dominant photoemission processes,7g and the lifetimes are remarkable when combined with their relatively high quantum yields.7g
| Complex | λ em/nm | Φ PL | τ/μs | k r/102 s−1 | k nr/102 s−1 |
|---|---|---|---|---|---|
| 1 | 502 | 0.67(1) | 670 | 11 | 5 |
| 2 | 454(sh), 505 | 0.33(1) | 550 | 6.4 | 12 |
| 1-MeOH | 466 | 0.092(6) | 770 | 1.3 | 12 |
DFT and TDDFT calculations were used to investigate the photophysical differences in 1, 1-MeOH, and 2. The calculations were performed in the gas phase and using SMD solvation models with toluene and CH2Cl2 for comparison. Consistent with our solution UV-vis data,19 analysis of the TDDFT calculations and associated Kohn–Sham orbitals confirm that UV absorptions in 1 are best assigned as MLCT transitions between the HOMO (Cu–Cl π* and Cu–P σ*) and unoccupied phenyl-derived π orbitals localized on the PhTBDPhos ligand. Despite addition of MeOH to the TBD backbone, the calculated absorption transitions for 1-MeOH are effectively the same as 1.
Evaluating the emissive properties of the Cu(I) complexes required determining the structures of their T1 excited states. The optimized structures revealed that 1 undergoes a distortion from trigonal planar in the ground state to T-shaped in the T1 state, similar to that reported for other emissive three-coordinate complexes (Fig. 3).6b,20 The cis and trans P–Cu–Cl angles were 113.2° and 150.7° and the P–Cu–P angle was 96.1° (Σ = 360.0°). A similar, albeit less dramatic bending distortion was calculated for the T1 structure of 2, but also with rotation of the NPh2 phenyl groups around the Cu–N bond. In contrast to 1 and 2, 1-MeOH undergoes a different Jahn–Teller distortion in the excited state. Instead of trigonal planar to T-shaped, the T1 coordination geometry in 1-MeOH is trigonal pyramidal with more congruent P–Cu–Cl angles of 123.4° and 119.4° and a P–Cu–P angle of 86.7° (Σ = 329.5°). Given that structural excited-state distortions are known to influence the availability of non-radiative decay modes, it is likely that the different excited-state Jahn–Teller distortions for 1-MeOH compared to 1 contributes to its decreased quantum yield.
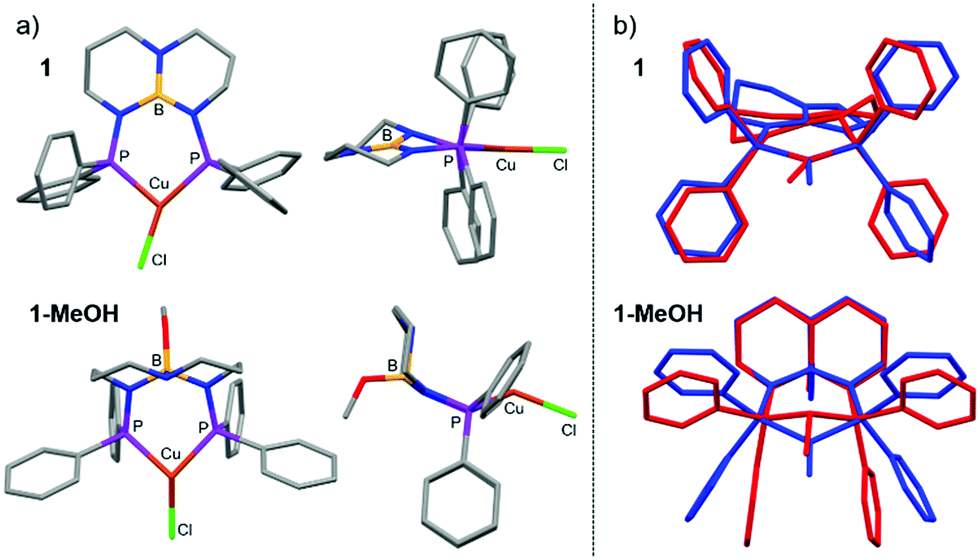 | ||
| Fig. 3 (a) Top and side views of optimized gas-phase DFT structures for the triplet state (T1) of 1 (top) and 1-MeOH (bottom). (b) Stack plot of calculated structures for 1 and 1-MeOH: gas-phase singlet ground state (S0; red) and triplet excited state (T1; blue). | ||
Calculations performed on discrete complexes of 1 and 1-MeOH did not offer a clear reason for the colorimetric shift between the two, which suggests that the shift is tied to differences in their extended solid-state structures. The Kohn–Sham orbitals involved in the transitions do not have appreciable boron or nitrogen character in the optimized ground- and excited-state structures, which appears to rule out that the colour change is associated with chemical engagement of boron and nitrogen orbitals on the ligand (Fig. S3 and S4; ESI†). Analysis of the calculated T1 → S0 emission energies was also inconclusive because they varied in magnitude and sign depending on the conditions selected (gas-phase vs. solvation model vs. solvent selection; Table S4, ESI†). As has been described for luminescent complexes containing aryl-substituted ligands,17,21 we suspect that the change in luminescence colour can be attributed to solid-state differences in intermolecular π–π stacking between adjacent aryl groups in response to the SCSC transformation. Evidence in support of this hypothesis is afforded by analysis of the XRD data. The crystal structure of 1 shows no intermolecular π–π stacking, whereas the extended structure of 1-MeOH has adjacent complexes with overlapping, parallel-offset phenyl groups with Ph⋯Ph centroid distances of 3.803 Å (Fig. 1).
We briefly investigated the scope of the solid-state ligand-centered reactivity of 1 with other Brønsted acids. As with MeOH, the green photoluminescence of crystalline 1 slowly turns blue when exposed to vapor from water and aqueous HCl solutions (Fig. 4). It appears that PhTBDPhos in crystals of 1 undergoes solid-state ligand-centered reactions in the same way as it does with MeOH under these conditions, and this observation is consistent with solution reactivity reported previously with (PhTBDPhos)NiCl2 and (PhTBDPhos)PdCl2.10a,b
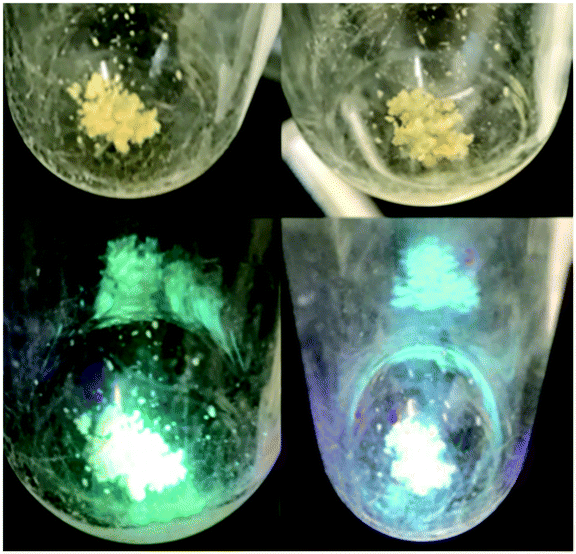 | ||
| Fig. 4 Single crystals of 1 before (left) and after (right) exposure to vapor from an aqueous HCl solution for 12 h under normal light (top) and commercial violet laser pointer (bottom). | ||
In summary, we have described how three-coordinate Cu(I) complexes with a reactive diphosphine ligand called PhTBDPhos exhibit green photoemission, relatively high quantum yields, and long luminescent lifetimes. Exposing crystals of 1 to MeOH solution or vapor turns the photoemission blue by way of ligand-centered reactions at the TBD backbone that cause a single-crystal-to-single-crystal transformation. Similar ligand-centered reactivity appears to be operative when 1 is exposed to vapor from water and aqueous HCl solutions. Collectively, these results demonstrate how ligand-centered reactions can be used to modify the luminescent properties of crystalline Cu(I) complexes, which may be useful for the development of new materials for optical sensing and luminescent devices.
This work was generously supported by the University of Iowa Center for Health Effects of Environmental Contamination (CHEEC) and the National Science Foundation (1650894). We thank Dale Swenson for collecting the single-crystal XRD data. T. S. T. acknowledges the Welch Foundation (Grant no. E-1887) for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3648 CrossRef CAS PubMed; (b) H. Yersin, Highly efficient OLEDs with phosphorescent materials/edited by Hartmut Yersin, Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim, Germany, 2008 Search PubMed; (c) M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann and S. Bräse, Chem. – Eur. J., 2014, 20, 6578–6590 CrossRef CAS PubMed; (d) W. Liu, Y. Fang and J. Li, Adv. Funct. Mater., 2018, 28, 1705593 CrossRef; (e) L. P. Ravaro, K. P. S. Zanoni and A. S. S. de Camargo, Energy Rep., 2020, 6, 37–45 CrossRef.
- (a) M. J. Leitl, D. M. Zink, A. Schinabeck, T. Baumann, D. Volz and H. Yersin, Top. Curr. Chem., 2016, 374, 25 CrossRef PubMed; (b) H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS; (c) F. Dumur, Org. Electron., 2015, 21, 27–39 CrossRef CAS; (d) E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef CAS.
- (a) D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201–1220 CrossRef PubMed; (b) M. H. Keefe, K. D. Benkstein and J. T. Hupp, Coord. Chem. Rev., 2000, 205, 201–228 CrossRef CAS; (c) E. J. O’Neil and B. D. Smith, Coord. Chem. Rev., 2006, 250, 3068–3080 CrossRef; (d) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC; (e) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed; (f) A. Kobayashi and M. Kato, Chem. Lett., 2017, 46, 154–162 CrossRef CAS.
- (a) N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC; (b) S.-P. Luo, E. Mejía, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2012, 52, 419–423 CrossRef PubMed; (c) Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- (a) Chemical Photocatalysis, ed. B. König, de Gruyter, 2013 Search PubMed; (b) S. Paria and O. Reiser, ChemCatChem, 2014, 6, 2477–2483 CrossRef CAS; (c) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647 CrossRef CAS PubMed; (d) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681 CrossRef CAS PubMed.
- (a) N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69–115 CrossRef CAS; (b) M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133, 10348–10351 CrossRef CAS PubMed; (c) M. Iwamura, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2007, 129, 5248–5256 CrossRef CAS PubMed.
- (a) V. A. Krylova, P. I. Djurovich, M. T. Whited and M. E. Thompson, Chem. Commun., 2010, 46, 6696–6698 RSC; (b) R. Hamze, R. Jazzar, M. Soleilhavoup, P. I. Djurovich, G. Bertrand and M. E. Thompson, Chem. Commun., 2017, 53, 9008–9011 RSC; (c) S. Shi, P. I. Djurovich and M. E. Thompson, Inorg. Chim. Acta, 2018, 482, 246–251 CrossRef CAS; (d) M. Osawa, Chem. Commun., 2014, 50, 1801–1803 RSC; (e) K. J. Lotito and J. C. Peters, Chem. Commun., 2010, 46, 3690–3692 RSC; (f) S. Shi, L. R. Collins, M. F. Mahon, P. I. Djurovich, M. E. Thompson and M. K. Whittlesey, Dalton Trans., 2017, 46, 745–752 RSC; (g) R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed; (h) S. Shi, M. C. Jung, C. Coburn, A. Tadle, M. R. Daniel Sylvinson, P. I. Djurovich, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 3576–3588 CrossRef CAS PubMed.
- (a) H. Na and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 6353–6360 CrossRef CAS PubMed; (b) T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed; (c) K. P. S. Zanoni, R. L. Coppo, R. C. Amaral and N. Y. Murakami Iha, Dalton Trans., 2015, 44, 14559–14573 RSC.
- (a) M. J. Leitl, F.-R. Küchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117, 11823–11836 CrossRef CAS PubMed; (b) T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed; (c) T. Gneuß, M. J. Leitl, L. H. Finger, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44, 20045–20055 RSC; (d) A. Schinabeck, N. Rau, M. Klein, J. Sundermeyer and H. Yersin, Dalton Trans., 2018, 47, 17067–17076 RSC.
- (a) K. Lee, C. M. Donahue and S. R. Daly, Dalton Trans., 2017, 46, 9394–9406 RSC; (b) K. Lee, C. Kirkvold, B. Vlaisavljevich and S. R. Daly, Inorg. Chem., 2018, 57, 13188–13200 CrossRef CAS PubMed; (c) K. Lee, C. W. Kim, J. L. Buckley, B. Vlaisavljevich and S. R. Daly, Dalton Trans., 2019, 48, 3777–3785 RSC.
- (a) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (b) R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS; (c) K. Tsuge, Y. Chishina, H. Hashiguchi, Y. Sasaki, M. Kato, S. Ishizaka and N. Kitamura, Coord. Chem. Rev., 2016, 306, 636–651 CrossRef CAS; (d) C. Kutal, Coord. Chem. Rev., 1990, 99, 213–252 CrossRef CAS.
- S. Daly, M. F. Haddow, A. G. Orpen, G. T. A. Rolls, D. F. Wass and R. L. Wingad, Organometallics, 2008, 27, 3196–3202 CrossRef CAS.
- (a) K. Saito, S. Saijo, K. Kotera and T. Date, Chem. Pharm. Bull., 1985, 33, 1342–1350 CrossRef CAS; (b) P. C. Healy, J. C. McMurtrie and J. Bouzaid, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2010, 66, m493–m494 CrossRef CAS PubMed.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Cryst. Growth Des., 2017, 17, 2893–2910 CrossRef CAS.
- (a) X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427–11441 RSC; (b) E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
- (a) E. Cariati, X. Bu and P. C. Ford, Chem. Mater., 2000, 12, 3385–3391 CrossRef CAS; (b) E. Cariati and J. Bourassa, Chem. Commun., 1998, 1623–1624 RSC.
- X.-W. Chen, H.-L. Yuan, L.-H. He, J.-L. Chen, S.-J. Liu, H.-R. Wen, G. Zhou, J.-Y. Wang and W.-Y. Wong, Inorg. Chem., 2019, 58, 14478–14489 CrossRef CAS PubMed.
- T. Hasegawa, A. Kobayashi, H. Ohara, M. Yoshida and M. Kato, Inorg. Chem., 2017, 56, 4928–4936 CrossRef CAS PubMed.
- See ESI† for details.
- K. A. Barakat, T. R. Cundari and M. A. Omary, J. Am. Chem. Soc., 2003, 125, 14228–14229 CrossRef CAS PubMed.
- (a) K.-H. Wong, K.-K. Cheung, M. C.-W. Chan and C.-M. Che, Organometallics, 1998, 17, 3505–3511 CrossRef CAS; (b) W. Lu, M. C. W. Chan, K.-K. Cheung and C.-M. Che, Organometallics, 2001, 20, 2477–2486 CrossRef CAS; (c) E. A. Mikhalyova, A. V. Yakovenko, M. Zeller, M. A. Kiskin, Y. V. Kolomzarov, I. L. Eremenko, A. W. Addison and V. V. Pavlishchuk, Inorg. Chem., 2015, 54, 3125–3133 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data, NMR spectra, molecular structure of 2, and XYZ coordinates of the calculated structures. CCDC 2003378–2003380. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03427d |
| This journal is © The Royal Society of Chemistry 2020 |