
Recent advances in sulfonylation reactions using potassium/sodium metabisulfite
Shengqing
Ye
a,
Min
Yang
 b and
Jie
Wu
*ac
b and
Jie
Wu
*ac
aSchool of Pharmaceutical and Materials Engineering & Institute for Advanced Studies, Taizhou University, 1139 Shifu Avenue, Taizhou 318000, China. E-mail: jie_wu@fudan.edu.cn
bSchool of Basic Medicine, Gannan Medical University, 1 Yixueyuan Road, Ganzhou 341000, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 24th March 2020
Abstract
Recently, sulfonylation reactions using potassium/sodium metabisulfite as the sulfur dioxide surrogate have been developed rapidly. In most cases, the transformations go through radical processes with the insertion of sulfur dioxide under mild conditions. Additionally, transition metal catalysis is applied in the reactions for the synthesis of sulfonyl-containing compounds. Among the approaches, photoinduced conversions under visible light or ultraviolet irradiation are also involved. In this updated report, the insertion of sulfur dioxide from potassium metabisulfite or sodium metabisulfite is summarized.
 Shengqing Ye | Shengqing Ye received his BSc in chemistry from Fudan University in 2008. He pursued his PhD studies at Fudan University under the guidance of Prof. Jie Wu (2008–2013). After obtaining his PhD degree in 2013, he joined Prof. Jin-Quan Yu's group as a postdoctoral fellow at The Scripps Research Institute (2013–2015). In the year of 2015, he worked as a Process Chemist in Suzhou Novartis Pharma Technology Co., Ltd. (2015–2019). In 2019, he joined the School of Pharmaceutical and Materials Engineering at Taizhou University as an associate professor. He got the Thieme Chemistry Journals Award in 2020. Currently, his research interests mainly focus on the development of new methods for the synthesis of bioactive compounds. |
 Min Yang | Min Yang received his BSc in the College of Chemistry and Chemical Engineering from Jiangxi Normal University in 2010. He pursued his PhD studies at East China Normal University under the guidance of Prof. Jie Tang (2011–2014). After obtaining his PhD degree in 2014, he joined Pharmaron Inc. as an organic synthetic chemist and senior team leader in drug research and development (2014–2018). In 2019, he joined the School of Basic Medicine at Gannan Medical University as a lecturer. Currently, his research interests mainly focus on the synthesis of drug-related compounds and organic fluorescence probes for toxicity analysis. |
 Jie Wu | Jie Wu received his BSc in chemistry from Jiangxi Normal University in 1995 and he pursued his PhD studies at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the supervision of Prof. Xue-Long Hou (1995–2000). After conducting research as a postdoctoral fellow at Harvard University (2000–2001), he worked as a visiting scientist at the Aaron Diamond AIDS Research Centre (2001–2002) and a staff scientist at VivoQuest, Inc (2002–2004). In 2004, he moved to the Department of Chemistry at Fudan University and held the Professor rank two years later. In 2019, he joined the School of Pharmaceutical and Materials Engineering at Taizhou University. He got the Thieme Chemistry Journals Award in 2010 and Dow Innovation Challenge Award in 2013. He serves as a member of the Editorial Advisory Board of ACS Combinatorial Science. |
1. Introduction
It is known that sulfonyl-containing compounds are important in the field of pharmaceuticals and materials science.1Fig. 1 presents some examples of sulfonyl-containing drug molecules on the market.2 Thus, continuous efforts have been made in the synthesis of sulfonyl compounds including sulfones and sulfonamides. So far, there are many conventional methods for the generation of sulfonyl compounds.3 For example, sulfonated indolo[1,2-a]quinolines with excellent fluorescence properties could be synthesized from arylsulfonyl hydrazides and 1-(2-(arylethynyl)phenyl)indoles in the presence of TBAI/TBHP.3f Recently, the focus has been centered on the insertion of sulfur dioxide.4 In the past decade, several sulfur dioxide surrogates have been used as the source of sulfur dioxide in organic transformations. For instance, the bench-stable reagent of 1,4-diazabicyclo[2.2.2]octane-sulfur dioxide (DABCO·(SO2)2), as initially disclosed by Santos and Mello in 1988,5 has been used broadly as the source of sulfur dioxide6 although the preparation of DABCO·(SO2)2 suffers from harsh reaction conditions from gaseous sulfur dioxide at −78 °C.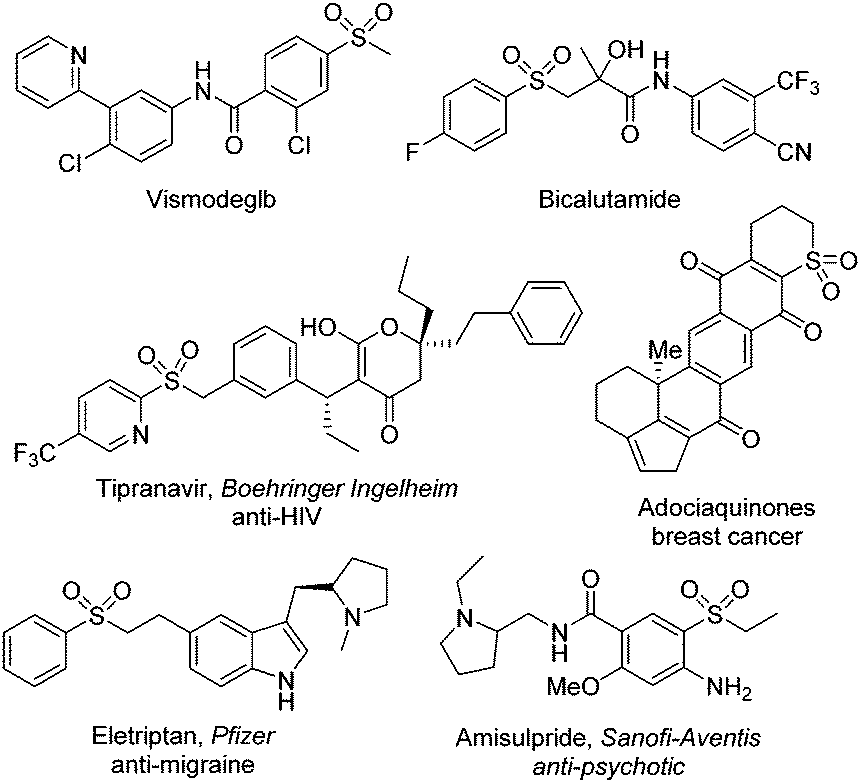 | ||
| Fig. 1 Marketed sulfonyl-containing drugs. | ||
Inorganic sulfites as another sulfur dioxide surrogate have attracted continuous interest, because inorganic sulfites are easily available, cheap and abundant in nature. Since the first example of using potassium metabisulfite as the source of sulfur dioxide in the palladium-catalyzed sulfonylation of aryl halides with hydrazines as described by Wu and co-workers in 2012,7 significant progress for the synthesis of sulfonyl-containing compounds by using potassium/sodium metabisulfite has been witnessed. Among the inorganic sulfites screened, potassium/sodium metabisulfite was demonstrated as the most efficient one. At the end of 2018, the insertion of sulfur dioxide from potassium metabisulfite or sodium metabisulfite was summarized,4j since inorganic sulfites would be ideal as the source of sulfur dioxide in organic transformations. From 2019, methods developed using potassium/sodium metabisulfite in sulfonylation reactions boomed. In most cases, the transformations went through radical processes with the insertion of sulfur dioxide under mild conditions. Additionally, transition metal catalysis was applied in the reactions for the synthesis of sulfonyl-containing compounds. Among the approaches, photoinduced conversions under visible light or ultraviolet irradiation were also involved. In this updated report, we herein report the recent advances in sulfonylation reactions using potassium/sodium metabisulfite as the source of sulfur dioxide from Dec. 2018.
2. Sulfonylation from aryldiazonium tetrafluoroborates and potassium/sodium metabisulfite
The combination of DABCO·(SO2)2 with aryldiazonium tetrafluoroborates was demonstrated efficiently for the generation of the arylsulfonyl radical and tertiary amine (DABCO) radical cation under mild conditions via single electron transfer.8 Potassium/sodium metabisulfite was applied as well in the reaction of aryldiazonium tetrafluoroborates as a replacement for DABCO·(SO2)2. It is known that the scaffold of the furan-2(5H)-one core broadly exists in biologically active natural products, pharmaceuticals, and pesticides.9 Additionally, furan-2(5H)-ones are useful building blocks in organic synthesis for the construction of complex molecules.10 Starting from sodium metabisulfite, 4-sulfonylated furan-2(5H)-ones could be easily prepared from a copper(II)-catalyzed multicomponent reaction of 2,3-allenoic acids and aryldiazonium tetrafluoroborates under mild conditions (Scheme 1).11 This method is quite effective under mild conditions, giving rise to a range of 4-sulfonyl furan-2(5H)-ones in moderate to good yields. It was found that various functional groups including fluoro, chloro, bromo, trifluoromethyl, methoxy, nitro, and ester were all compatible with this transformation. The plausible mechanism showed that the aryl radical generated in situ through single electron transfer of aryldiazonium tetrafluoroborate with Cu(I) would react with sodium metabisulfite to form the arylsulfonyl radical, which would undergo addition to the central-C position of 2,3-allenoic acid leading to an allylic radical intermediate. Subsequently, oxidative single electron transfer would occur with the assistance of the copper catalyst to produce an allylic cation intermediate, which would go through further intramolecular nucleophilic attack by the carboxylate in the presence of a base to provide the corresponding 4-sulfonylated furan-2(5H)-one. | ||
| Scheme 1 A copper(II)-catalyzed multicomponent reaction of 2,3-allenoic acids, sodium metabisulfite and aryldiazonium tetrafluoroborates. | ||
As important synthetic building blocks, thiosulfonates have been broadly applied in organic synthesis due to their advantages in reactivity and stability.12 Sodium metabisulfite was found to be effective for the synthesis of S-aryl thiosulfonates as well. Wu and co-workers described a photoinduced three-component reaction of aryldiazonium tetrafluoroborates, sodium metabisulfite, and thiourea, leading to S-aryl thiosulfonates in good yields (Scheme 2).13 A range of aryldiazonium tetrafluoroborates were applied in this transformation. However, the limitation was obvious, since only S-aryl thiosulfonates could be accessed. This method was not suitable for S-alkyl thiosulfonates because of the stability issue of alkyldiazonium tetrafluoroborates. Moreover, the reaction was not workable for 2-substituted aryldiazonium tetrafluoroborates. The steric hinderance might hamper the reaction. For the mechanism, a radical coupling pathway was proposed in the presence of a photocatalyst under visible light irradiation. It was postulated that thiophenolate anions, sulfur dioxide, and urea would be formed from aryldiazonium tetrafluoroborate, sodium metabisulfite and thiourea via a salt intermediate. In the meantime, the aryl radical generated in situ from aryldiazonium tetrafluoroborate assisted by the excited state of the photocatalyst via single electron transfer (SET) would react with sulfur dioxide leading to the arylsulfonyl radical. Subsequently, the thiophenolate anion would afford the sulfur radical in the presence of the photocatalyst, which would combine with the arylsulfonyl radical, giving rise to the corresponding S-aryl thiosulfonate.
 | ||
| Scheme 2 Photoinduced three-component reaction of aryldiazonium tetrafluoroborates, sodium metabisulfite and thiourea. | ||
In the above transformation, thiourea could be replaced by thiols. Ji and co-workers reported the synthesis of thiosulfonates through a TFA-promoted multi-component reaction of aryldiazonium tetrafluoroborates, sodium metabisulfite and thiols (Scheme 3).14 In this reaction, not only S-aryl thiosulfonates but also S-alkyl thiosulfonates could be easily accessed. Thiosulfonates bearing various functional groups were all tolerated. Mechanistic studies showed that the generated arylsulfonyl radical would react with the thiol anion to afford a radical anion intermediate, which would undergo single electron transfer with aryldiazonium tetrafluoroborate, giving rise to the corresponding thiosulfonate.
 | ||
| Scheme 3 TFA-promoted multi-component reaction of aryldiazonium tetrafluoroborates, sodium metabisulfite and thiols. | ||
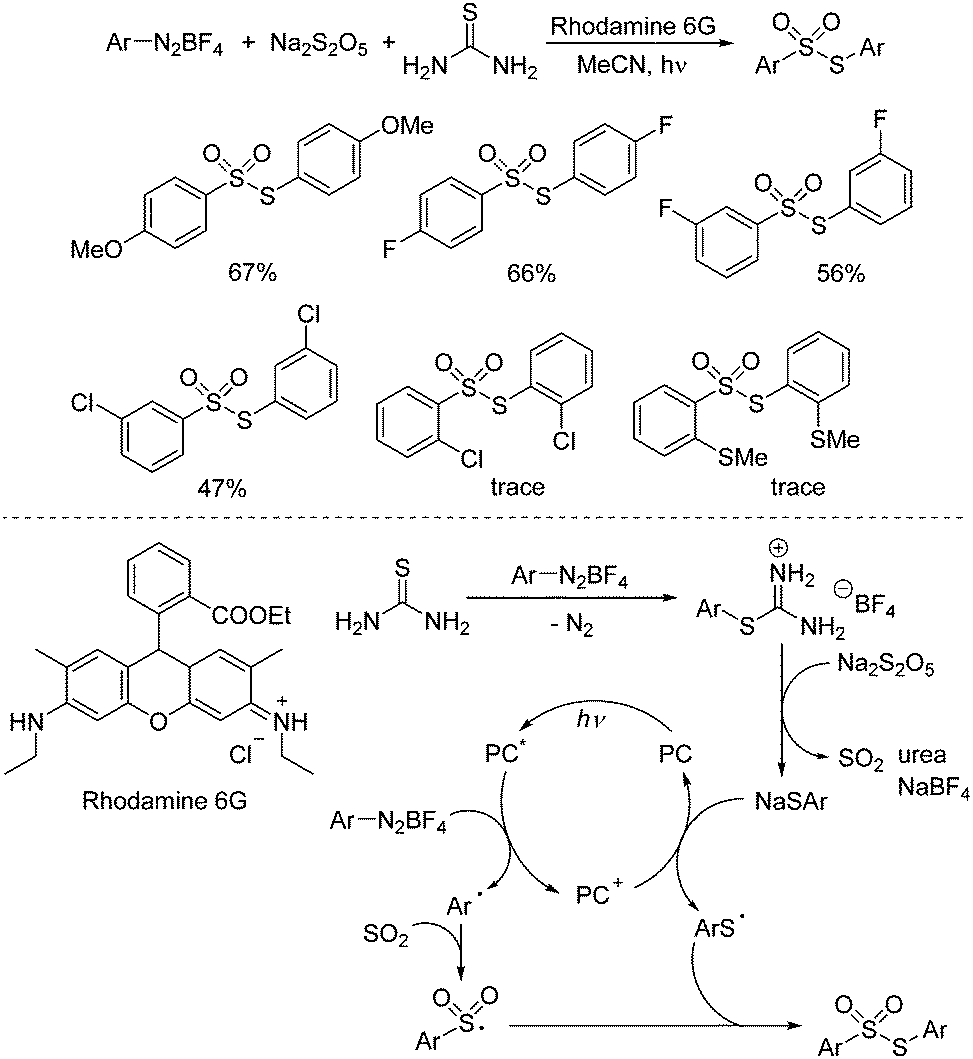
Construction of 2-sulfonyl-substituted 9H-pyrrolo [1,2-a]indoles was accomplished through a photoinduced reaction of aryldiazonium tetrafluoroborates, potassium metabisulfite and N-propargylindoles under visible light irradiation (Scheme 4).15 This cascade sulfonylation/cyclization with the insertion of sulfur dioxide was firstly disclosed by Wu's group, starting from aryldiazonium tetrafluoroborates and 1,4-diazabicyclo[2.2.2]octane-sulfur dioxide (DABCO·(SO2)2).16 In this transformation, a photocatalyst was involved. The mechanism indicated that the aryl radical would be generated from aryldiazonium tetrafluoroborate with the assistance of Ru(bpy)32+* under irradiation with visible light via single electron transfer. Then aryl radical would react with potassium metabisulfite, leading to the arylsulfonyl radical, which would attack the triple bond of N-propargylindole, giving rise to a vinyl radical intermediate. Intramolecular cyclization would subsequently occur. Following oxidative single electron transfer, the cation intermediate would be formed, which would undergo deprotonation and isomerization to produce the cyclic product.
 | ||
| Scheme 4 Photoinduced reaction of aryldiazonium tetrafluoroborates, potassium metabisulfite and N-propargylindoles. | ||
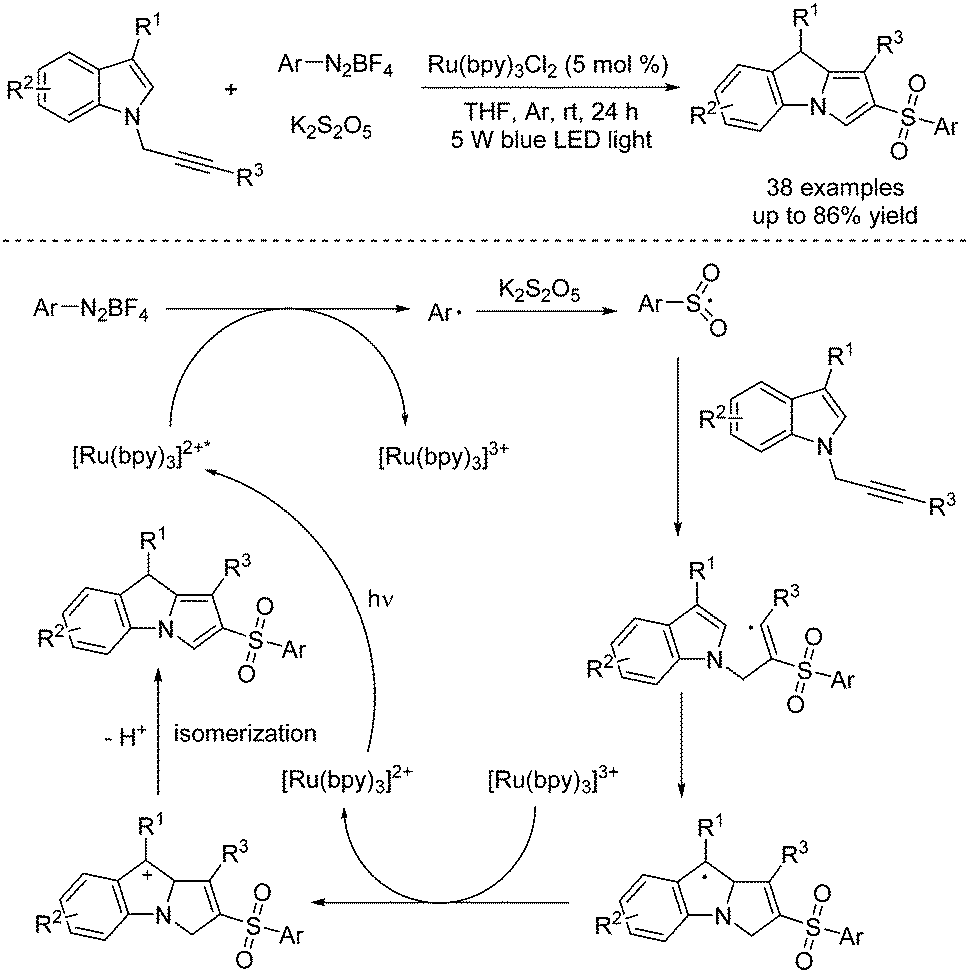
Singh and co-workers described a multicomponent reaction for the synthesis of β-keto sulfones through a reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and styrenes or alkynes (Scheme 5).17 Potassium metabisulfite or DABCO·(SO2)2 was used as the source of sulfone. Mechanistic studies from controlled liquid chromatography-mass spectrometry and 18O-labelling experiments revealed that the source of the incoming oxygen atom of the keto group in β-keto sulfones was from the air.
 | ||
| Scheme 5 Synthesis of β-keto sulfones through a reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and styrenes or alkynes. | ||
Xia and co-workers developed a metal-free three-component reaction of quinoline N-oxides, sodium metabisulfite and aryldiazonium tetrafluoroborates via a radical process (Scheme 6).18 2-Sulfonyl quinolines or isoquinolines were obtained in moderate to good yields under mild conditions. On the basis of control experiments and literature reports, it was reasoned that aryldiazonium tetrafluoroborate would go through a decomposition process leading to the aryl radical, which would subsequently react with sodium metabisulfite to provide the arylsulfonyl radical. This arylsulfonyl radical would attack the quinoline N-oxide via a Minisci-like radical transformation. The resulting O-radical would capture another arylsulfonyl radical, with the following release of arylsulfonic acid to furnish the corresponding 2-sulfonylquinolines.
 | ||
| Scheme 6 Metal-free three-component reaction of quinoline N-oxides, sodium metabisulfite and aryldiazonium tetrafluoroborates. | ||
3. Sulfonylation from organometallic reagents and potassium/sodium metabisulfite
As basic raw chemical materials, nitroarenes are widely applied in the pharmaceutical, pesticide, and dye industries.19 So far, applications of nitroarenes have been extensively explored in synthetic chemistry. It was found that nitroarenes could be used as the coupling partner with sulfur dioxide for the preparation of sulfonamides. Synthesis of sulfonamides through a copper-catalyzed reaction of triarylbismuthines, sodium metabisulfite, and nitro compounds was developed by using a deep eutectic solvent as a reaction medium, as reported by Guillena and J. Ramón (Scheme 7).20 In this transformation, triarylbismuthines were used as the substrates for the incorporation of sulfur dioxide into organic motifs, and the utilization of a deep eutectic solvent as a reaction medium was crucial for the conversion. A plausible mechanism was proposed, indicating that the insertion of sulfur dioxide from sodium metabisulfite into triarylbismuthine would be the first step. Subsequently, arylsulfinate would be formed with the release of BiCl3. In the presence of a copper catalyst, arylsulfinate would react with nitroarene. Following double reduction with NaHSO3, the corresponding sulfonamide would be furnished (Scheme 3).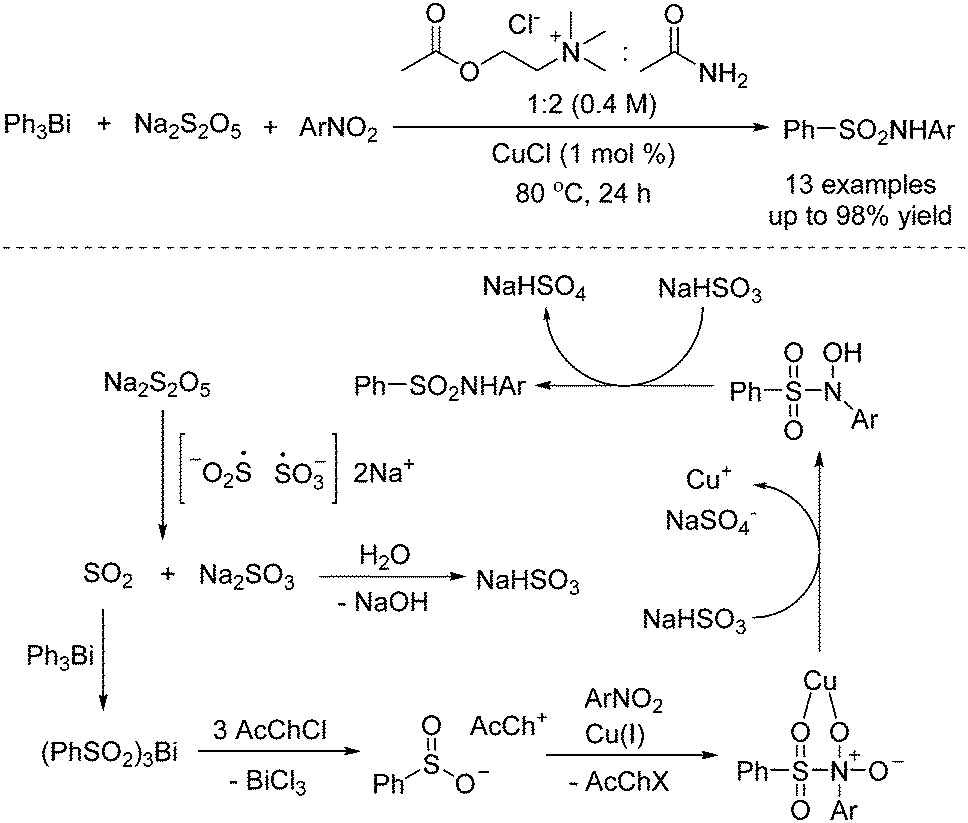 | ||
| Scheme 7 Copper-catalyzed reaction of triarylbismuthines, sodium metabisulfite, and nitroarenes in a deep eutectic solvent. | ||
Later, Wu and co-workers described a copper-catalyzed reaction of arylboronic acids, nitroarenes, and potassium metabisulfite (Scheme 8).21 A range of sulfonamides was afforded in good to excellent yields. Broad substrate scope was demonstrated, and various functional groups including hydroxyl, cyano, amino, and carbonyl were all compatible. Mechanistic studies showed that arylsulfinate was the intermediate, which was generated from the copper-catalyzed transmetallation of arylboronic acid and subsequent insertion of sulfur dioxide. The copper-assisted interaction of nitroarene and arylsulfinate was also the key for success. In this transformation, better results were obtained when isopropanol was used as the reductant. The late-stage modification of a currently marketed drug (Flutamide) was performed as well.
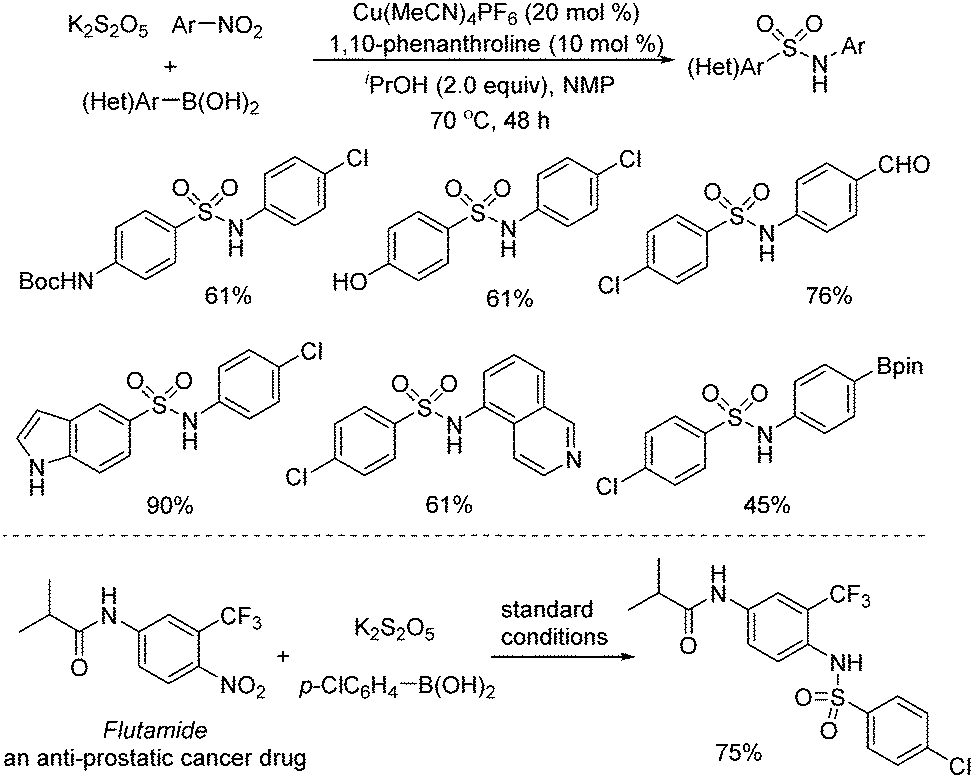 | ||
| Scheme 8 Copper-catalyzed reaction of arylboronic acids, nitroarenes, and potassium metabisulfite. | ||
In the meantime, Chen, Wu, and co-workers described a one-pot three-component reaction of nitroarenes, (hetero)arylboronic acids, and potassium metabisulfite for the synthesis of sulfonamides (Scheme 9).22 Interestingly, this transformation went through sequential C–S and S–N coupling under metal catalyst-free conditions, although a high temperature was required. A broad reaction scope was present, and a range of substrates was examined under the conditions. It was proposed that at the outset, this reaction proceeded through nucleophilic addition of arylboronic acid to sulfur dioxide from potassium metabisulfite in the presence of a base, leading to arylsulfinate. Then arylsulfinate would react with nitroarene, followed by reduction of sulfur dioxide and double deoxygenation to produce the corresponding sulfonamide. This method was also successfully applied in the late-stage modification of currently marketed drugs (Scheme 9).
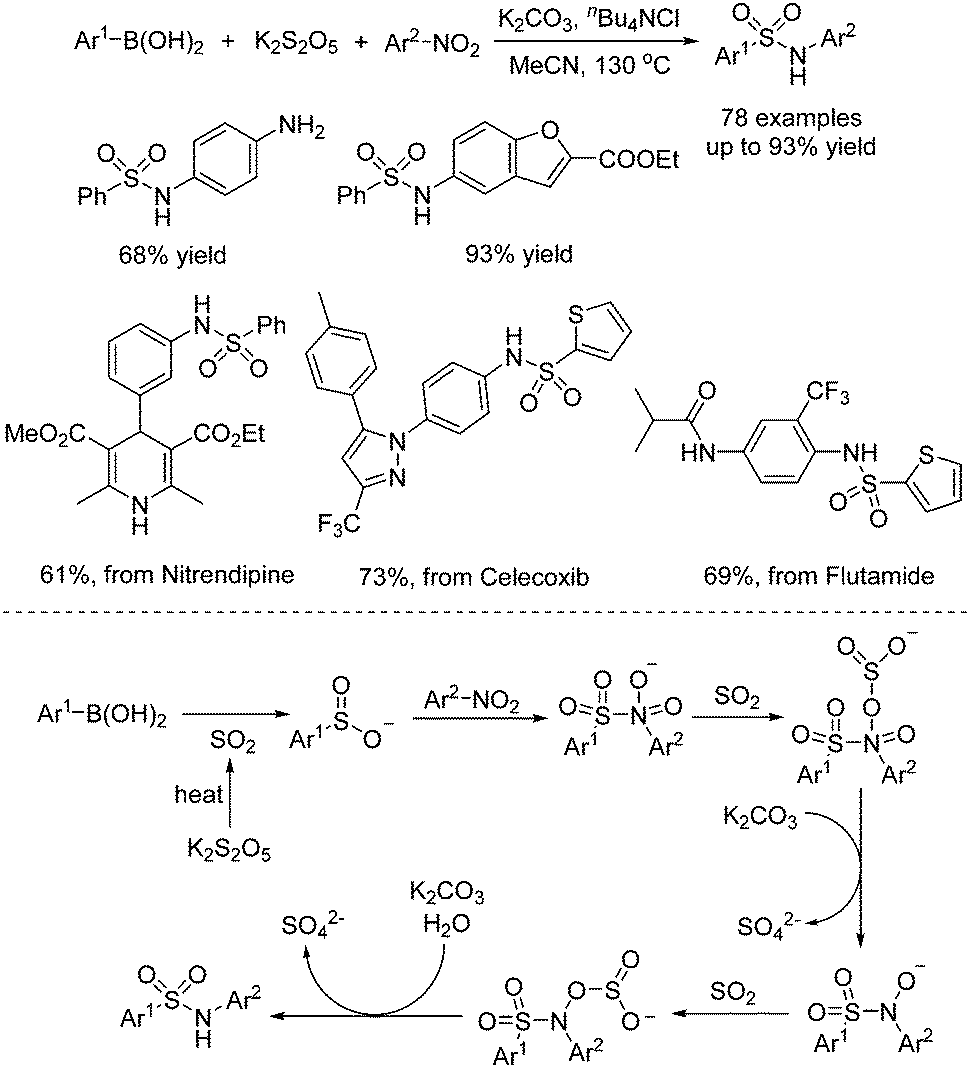 | ||
| Scheme 9 One-pot reaction of nitroarenes, (hetero)arylboronic acids, and potassium metabisulfite. | ||
Due to the broad applications of methylsulfonyl-containing compounds in pharmaceuticals and bioactive molecules, methylsulfonylation for the generation of methylsulfonyl-containing compounds is one of the important transformations in organic synthesis. Jiang and co-workers reported another approach for the generation of methylsulfonyl-containing compounds. Diverse methyl sulfones could be prepared through a palladium-catalyzed three-component reaction of arylboronic acids, sodium metabisulfite, and dimethyl carbonate (Scheme 10).23 Interestingly, dimethyl carbonate was used as the methyl reagent in this transformation. Additionally, the late-stage modification of pharmaceuticals and the synthesis of Firocoxib were efficiently established by using this strategy. Experimental evidence revealed that the radical pathway was excluded. It was postulated that the transmetallation of arylboronic acid with the palladium catalyst would occur firstly. Subsequently, sulfur dioxide insertion into the palladium complex would take place with the assistance of the electron-rich ligand. Finally, alkylation with dimethyl carbonate would produce the corresponding methyl sulfone product with the regeneration of the palladium catalyst.
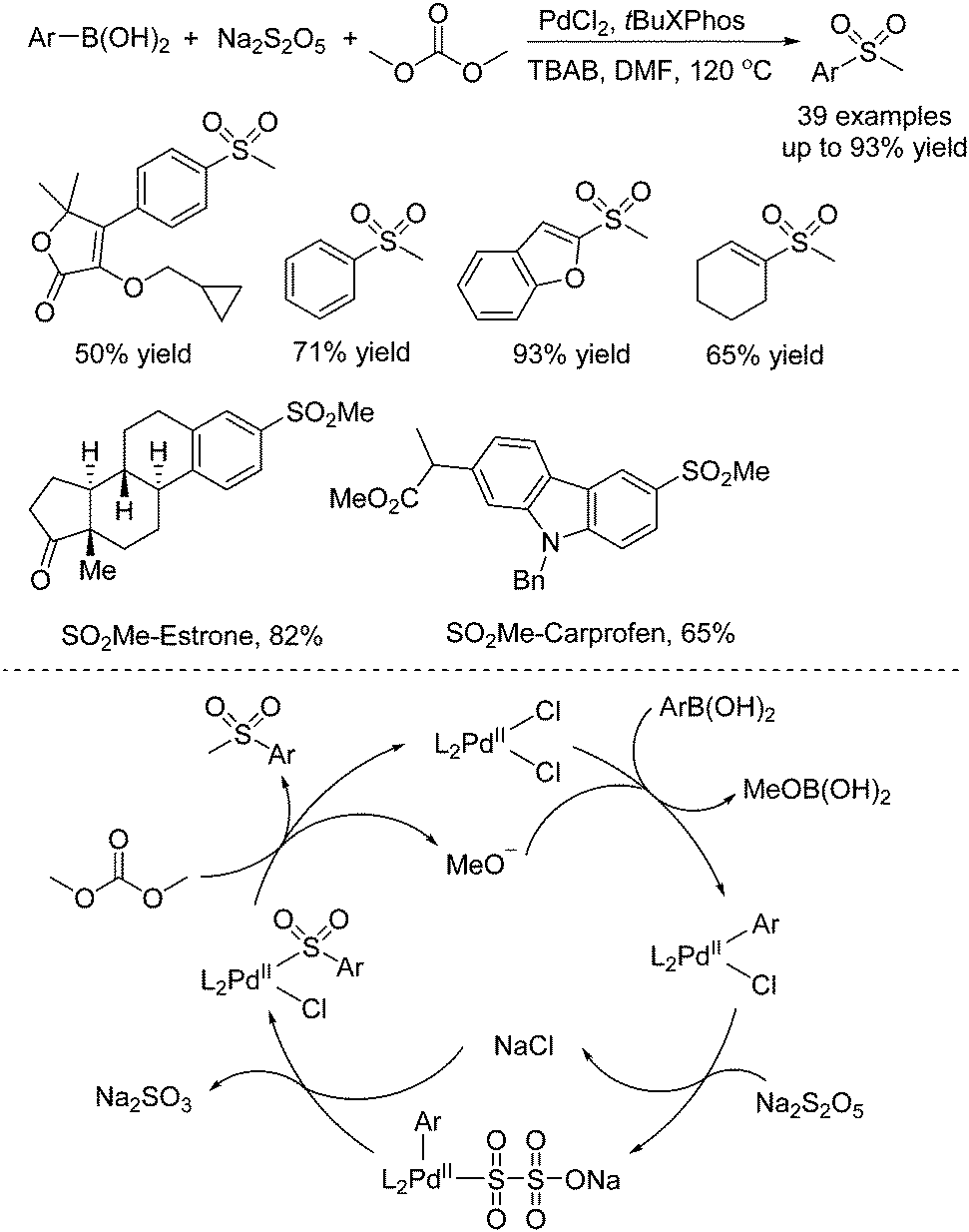 | ||
| Scheme 10 Palladium-catalyzed three-component reaction of arylboronic acids, sodium metabisulfite, and dimethyl carbonate. | ||
Synthesis of ortho-substituted diarylsulfones through a reaction of arylboronic acids, potassium metabisulfite, and diaryliodonium salts was established by using an acenaphthoimidazolylidene gold complex as the catalyst (Scheme 11).24 This sulfonylation process allowed the sterically hindered aryl groups in diaryliodonium salts to be preferentially transferred over less bulky ones. The chemoselectivity might be attributed to the more stable bulky Ar+ formed by the diaryliodonium salt. Diverse (poly-)ortho-substituted diarylsulfones could be generated. The proposed mechanism showed that transmetallation of NHC–Au(I) species with arylboronic acid would occur to provide NHC–Au–Ar species, as reported by Toste.25 Then sulfur dioxide insertion would take place to afford sulfonyl Au(I) complex NHC–Au–SO2–Ar, which would further furnish an arylsulfinate intermediate.25 Meanwhile, the more stable bulky Ar+ from the diaryliodonium salt would be captured by arylsulfinate, giving rise to a sterically hindered diarylsulfone.
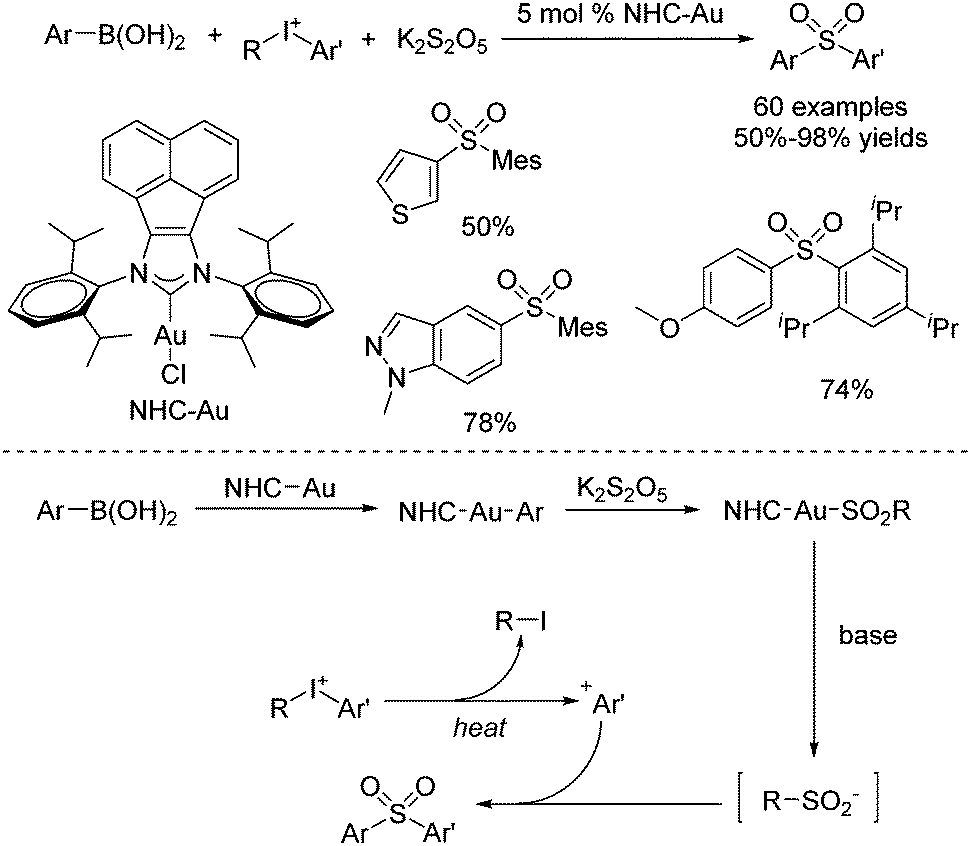 | ||
| Scheme 11 Synthesis of ortho-substituted diarylsulfones through a gold-catalyzed reaction of arylboronic acids, potassium metabisulfite, and diaryliodonium salts. | ||
Recently, radical processes initiated by the treatment of organotrifluoroborates under photoredox catalysis have developed rapidly.26 In general, carbon radical species would be formed from organotrifluoroborates in the presence of a photocatalyst under visible light irradiation. Compared with aryldiazonium tetrafluoroborates, alkyl radicals would be produced from potassium alkyltrifluoroborates. Thus, a photoinduced reaction of potassium alkyltrifluoroborates, sodium metabisulfite, and alkynyl bromides under visible light irradiation at room temperature under photocatalysis was designed and developed (Scheme 12).27 A broad substrate scope was presented, and diverse alkylalkynyl sulfones were generated in moderate to good yields. A similar mechanism was proposed, which showed that the alkyl radical was formed initially from potassium alkyltrifluoroborate in the presence of the photocatalyst under visible light irradiation. Subsequent sulfonylation and addition to alkynyl bromide would provide the final outcome. During the reaction, the presence of ammonium fluoride would enhance the reaction efficiency.
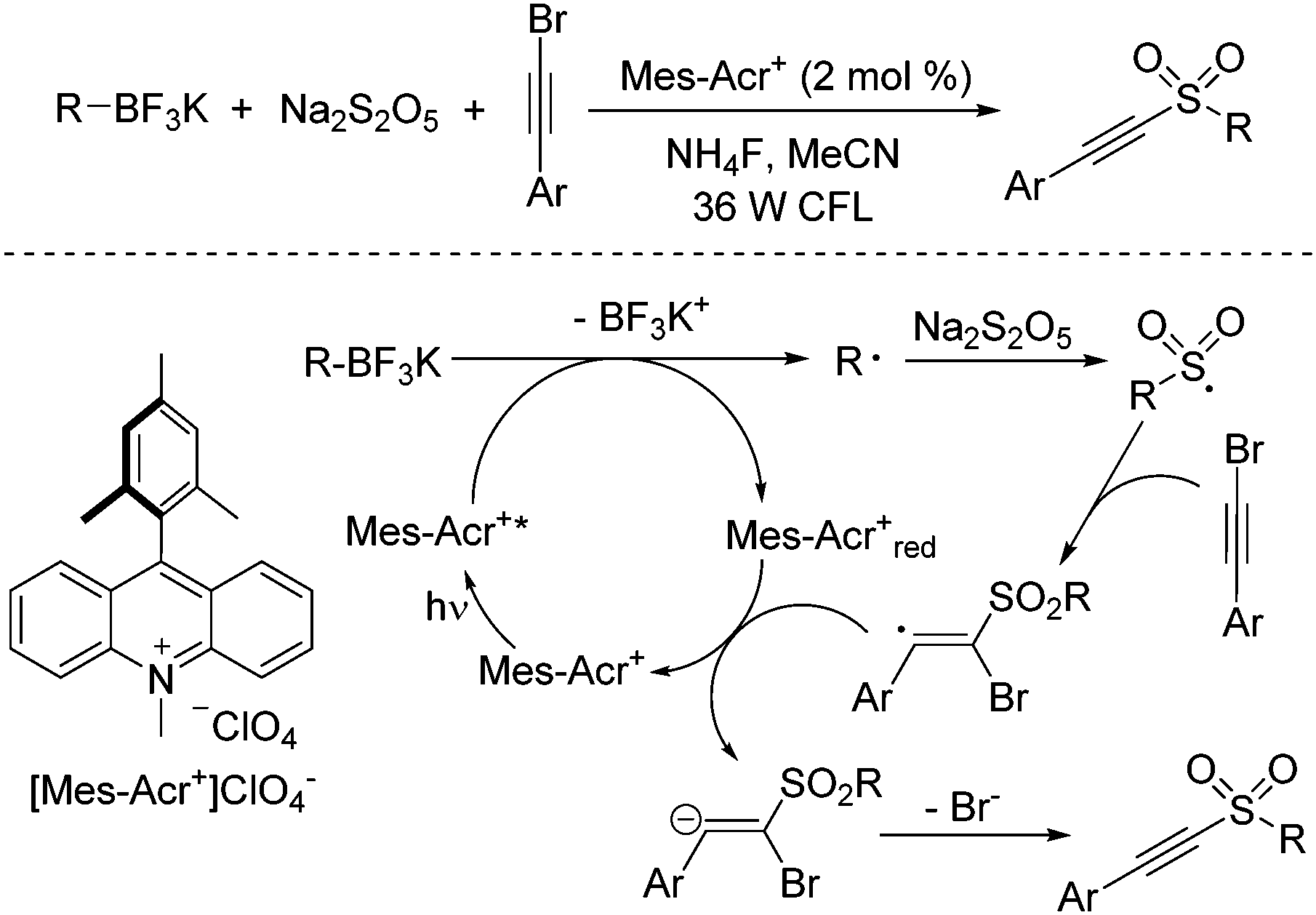 | ||
| Scheme 12 Photoinduced reaction of potassium alkyltrifluoroborates, sodium metabisulfite, and alkynyl bromides. | ||
4. Others
As mentioned above, methylsulfonylation for the generation of methylsulfonyl-containing compounds is one of important transformations in organic synthesis, especially for C–H bond methylsulfonylation. Recently, the synthesis of (E)-2-methyl styrenyl sulfones via direct C–H methyl sulfonylation of alkenes with sodium metabisulfite was developed (Scheme 13).28 During the reaction process, di-tert-butyl peroxide (DTBP) was employed as the methyl source, and a range of (E)-2-methyl styrenyl sulfones were produced in good yields. In this transformation, 1.0 equiv. of iron(III) chloride and a high temperature had to be used for successful conversion. It was postulated that the homolytic cleavage of DTBP would afford a tert-butyl radical, which would be further converted to a methyl radical and acetone. Then, the methyl radical would react with sodium metabisulfite, leading to a methylsulfonyl radical intermediate, which would attack the double bond of the alkene to produce a carbon radical species. With the assistance of iron(III), this carbon radical would be oxidized to a cation intermediate via single electron transfer (SET), which would subsequently undergo deprotonation to provide the desired (E)-2-methyl styrenyl sulfone.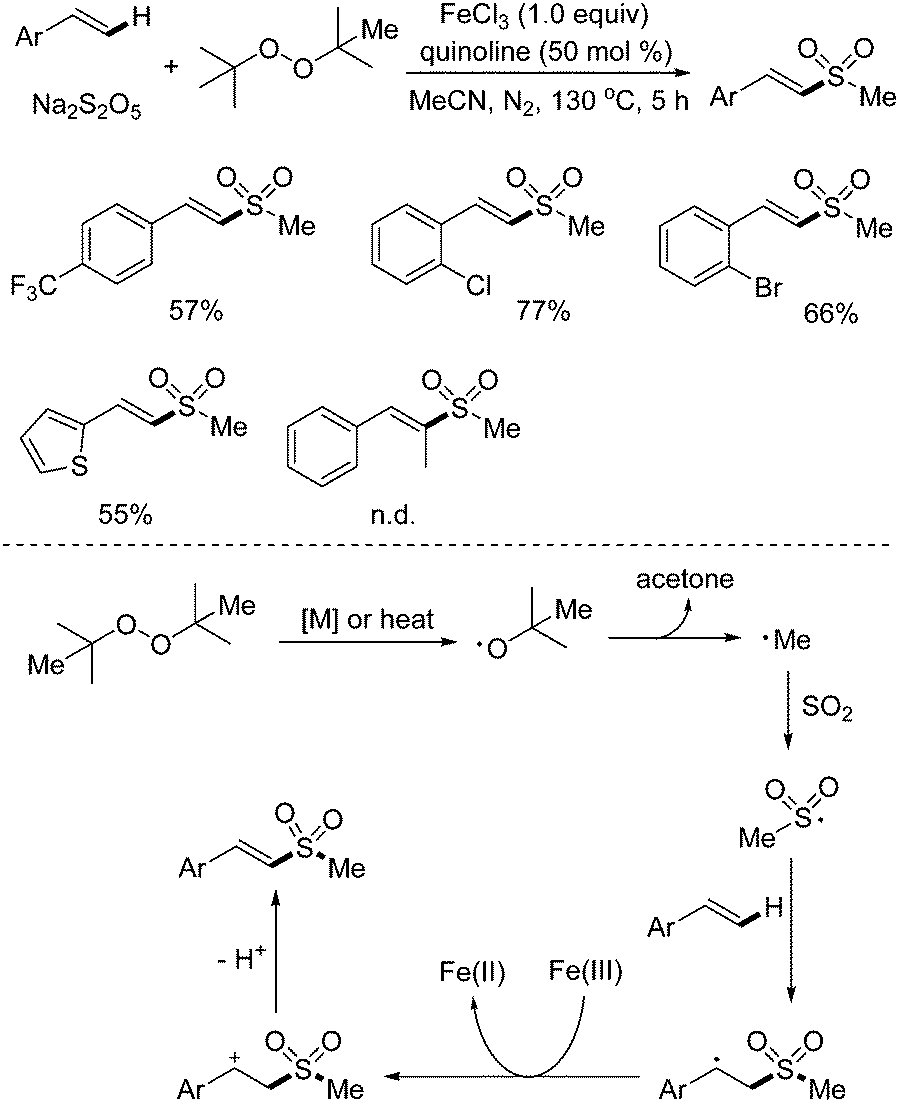 | ||
| Scheme 13 Synthesis of (E)-2-methyl styrenyl sulfones via direct C–H methyl sulfonylation of alkenes with sodium metabisulfite. | ||
A radical relay strategy for the generation of 3-(methylsulfonyl)benzo[b]thiophenes was developed. This transformation starting from methyl(2-alkynylphenyl)sulfanes and sodium metabisulfite proceeded smoothly in the presence of a photocatalyst under visible light irradiation (Scheme 14).29 Interestingly, a catalytic amount of sodium methylsulfinate was used as an initiator in this photoinduced sulfonylation process under mild conditions. The generality of this radical relay reaction was examined, and the corresponding 3-(methylsulfonyl)benzo[b]thiophenes were obtained in moderate to good yields. Several sensitive functional groups including chloro, bromo, fluoro, cyano, ester, and aldehyde were all compatible under the conditions. The R2 group attached on the triple bond of the substrate was crucial for the successful transformation. It was found that only an aryl group was effective, and no desired product was obtained when the group at the R2 position of the substrate was changed to 2-pyridinyl, tert-butyl, trimethylsilyl, or ester. On the basis of experimental evidence, the proposed mechanism showed that the methylsulfonyl radical generated in situ from methylsulfinate via single electron transfer in the presence of the excited state of the photocatalyst would initiate the reaction, which would subsequently attack the triple bond of methyl(2-alkynylphenyl)sulfane to produce a methyl radical. This released methyl radical went through a radical relay with sulfur dioxide from sodium metabisulfite, giving rise to the corresponding 3-(methylsulfonyl)benzo[b]thiophene through a vinyl radical intermediate. During the reaction process, it was assumed that the aryl group at the R2 position would stabilize the vinyl radical intermediate. The reaction of sodium metabisulfite under the standard conditions was further extended to the substrates of 1-methoxy-2-(phenylethynyl)benzene and N,N-dimethyl-2-alkynylaniline as a replacement for methyl(2-alkynylphenyl)sulfanes. However, the desired 3-(methylsulfonyl)benzo[b]furans and 3-(methylsulfonyl)indoles could not be formed.
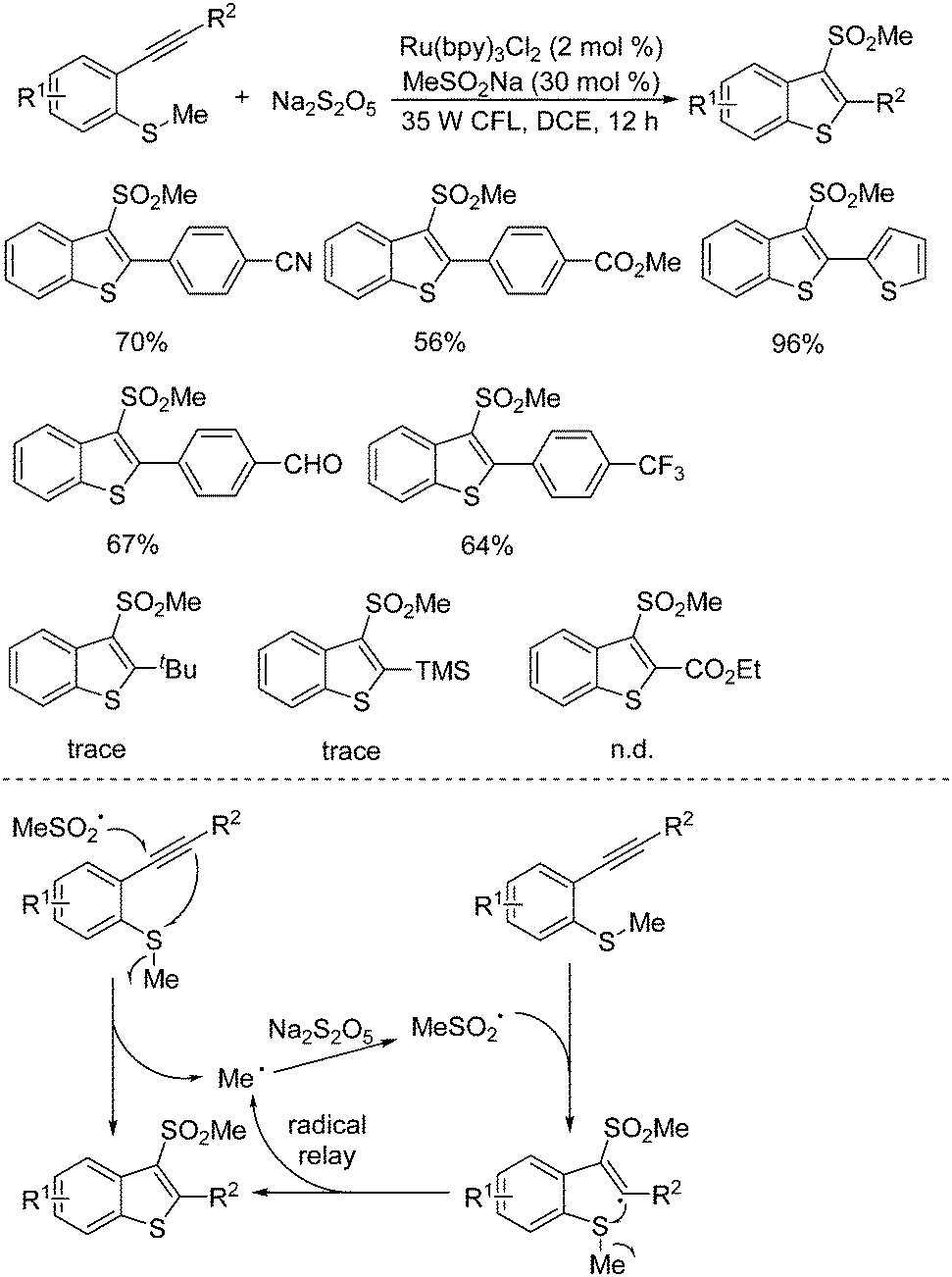 | ||
| Scheme 14 Generation of 3-(methylsulfonyl)benzo[b]thiophenes from methyl(2-alkynylphenyl)sulfanes and sodium metabisulfite. | ||
Recently, N-functionalized pyridinium salts such as Katritzky salts have been used as effective alkyl precursors under photoredox catalysis in various transformations.30 The combination of sulfur dioxide and Katritzky salts was reported as well. Wu and co-workers developed the first example by using Katritzky salts as alkyl radical precursors in the reaction of potassium metabisulfite and silyl enol ethers under photoredox catalysis, leading to diverse dialkyl sulfones (Scheme 15).31 A broad reaction scope was demonstrated and various functional groups were all tolerated including amino, cyano, hydroxy, and trifluoromethyl groups. This transformation proceeds efficiently under mild conditions in the presence of a photocatalyst and visible light irradiation, giving rise to the corresponding dialkyl sulfones in good yields. A photoinduced radical pathway under visible-light conditions was proposed. The experimental results showed that an alkyl radical generated in situ from the Katritzky salt via reductive single electron transfer by visible-light excited Ir(III) species would initiate the reaction. After combination of sulfur dioxide from potassium metabisulfite, the alkylsulfonyl radical would be produced, which would be trapped by silyl enol ether, leading to a carbon radical intermediate. In the presence of Ir(IV), this carbon radical intermediate would be oxidized to carbocation species, which would then undergo desilylation assisted by a base to afford the corresponding β-keto sulfone.
 | ||
| Scheme 15 Reactions of Katritzky salts, potassium metabisulfite and silyl enol ethers under photoredox catalysis. | ||
4-Substituted Hantzsch esters as alkyl radical reservoirs have been demonstrated, and the alkyl units from 4-substituted Hantzsch esters could be easily introduced into various small molecules.32 In the presence of a photoredox catalyst under visible light irradiation, alkyl radicals would be generated from 4-alkyl Hantzsch esters through single electron transfer (SET). Thus, synthesis of alkynyl sulfones through a reaction of 4-alkyl Hantzsch esters, sodium metabisulfite, and alkynyl bromides under metal-free photoinduced conditions was accomplished (Scheme 16).33 This transformation proceeds smoothly under visible light irradiation at room temperature, giving rise to the corresponding alkylalkynyl sulfones in moderate to good yields. The control experiments showed that the alkyl radical generated in situ from 4-alkyl Hantzsch esters in the presence of the photocatalyst would initiate the reaction. After capture of sulfur dioxide from sodium metabisulfite, the alkylsulfonyl radical would be formed, which would attack the triple bond of alkynyl bromide leading to a vinyl radical intermediate. Single electron transfer would then occur with the assistance of the excited photocatalyst to produce a vinyl anion, which would afford the corresponding alkylalkynyl sulfone with the release of a bromide anion.
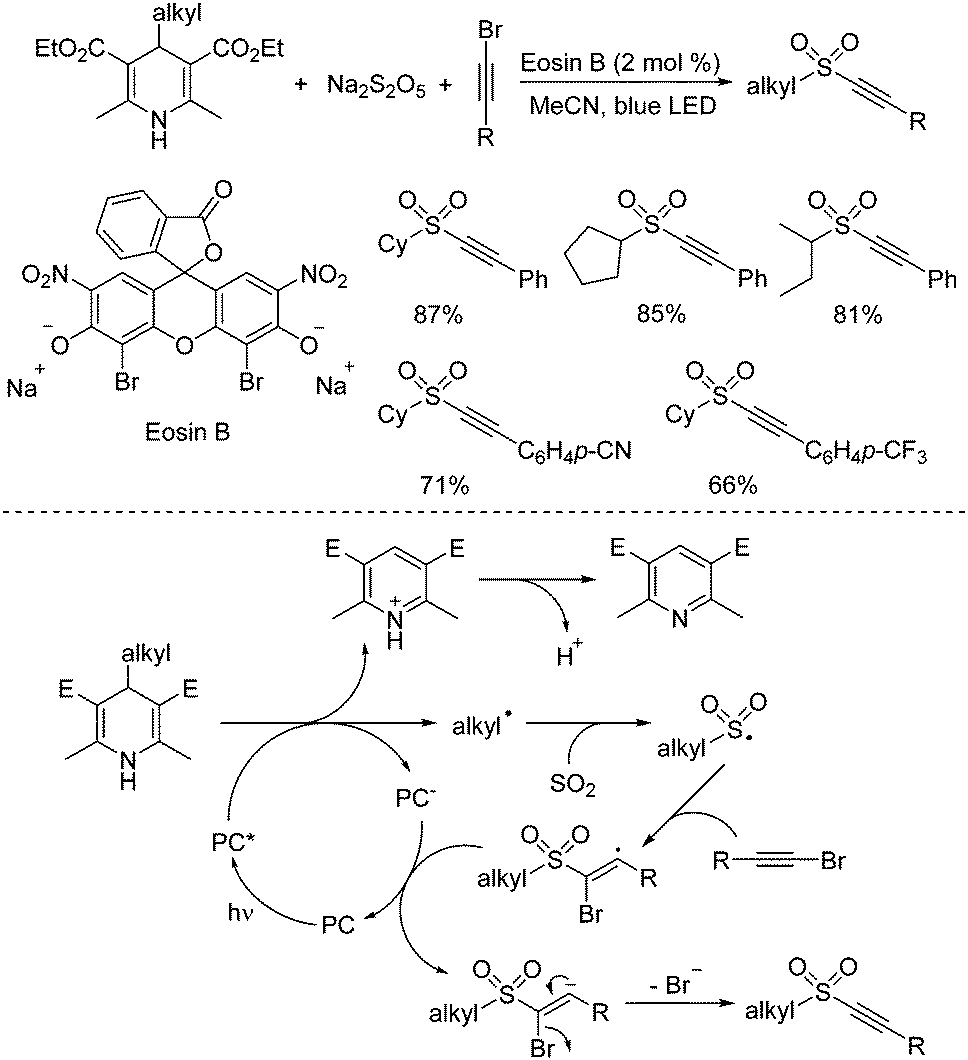 | ||
| Scheme 16 Synthesis of alkynyl sulfones through a reaction of 4-alkyl Hantzsch esters, sodium metabisulfite, and alkynyl bromides under metal-free photoinduced conditions. | ||
So far, much focus has been centered on the reactions of iminyl radicals. In some cases, iminyl radicals could be easily formed via a photoreductive strategy from O-acyl oximes under visible light irradiation.34 A photoredox-catalyzed multicomponent reaction of O-acyl oximes, potassium metabisulfite, alkenes, and nucleophiles under visible light irradiation is developed (Scheme 17).35 This transformation proceeded through sulfonylation of O-acyl oximes via iminyl radicals with the insertion of sulfur dioxide. The nucleophiles included alcohols and water. A range of β-alkoxy sulfones and β-hydroxyl sulfones was obtained in moderate to good yields with good functional group compatibility. Primary alcohols were effective in this transformation. However, the reactions of secondary alcohols and tertiary alcohols gave inferior results. Reaction in methan-d3-ol-d instead of methanol was workable as well. β-Hydroxy sulfones could be prepared when the reactions took place in a mixture of water and MeCN. Additionally, this photoredox-catalyzed multicomponent reaction showed excellent chemoselectivity during the reaction process. Mechanistic studies were performed, which showed that the reaction was initiated by the iminyl radical formed in situ from O-acyl oxime via N–O bond dissociation under visible light irradiation in the presence of the photocatalyst. Then, the intramolecular C–C bond cleavage of the iminyl radical would occur to provide a carbon radical, which would react with sulfur dioxide from potassium metabisulfite, leading to a sulfonyl radical intermediate. Subsequently, the sulfonyl radical would attack the double bond of alkene giving rise to another carbon radical intermediate, which would then undergo oxidative single electron transfer via the oxidized form of the photocatalyst to produce the corresponding cation intermediate. With the assistance of a base, the alcohol or water would act as the nucleophile to attack the cation intermediate, affording the expected β-alkoxy sulfone and β-hydroxyl sulfone.
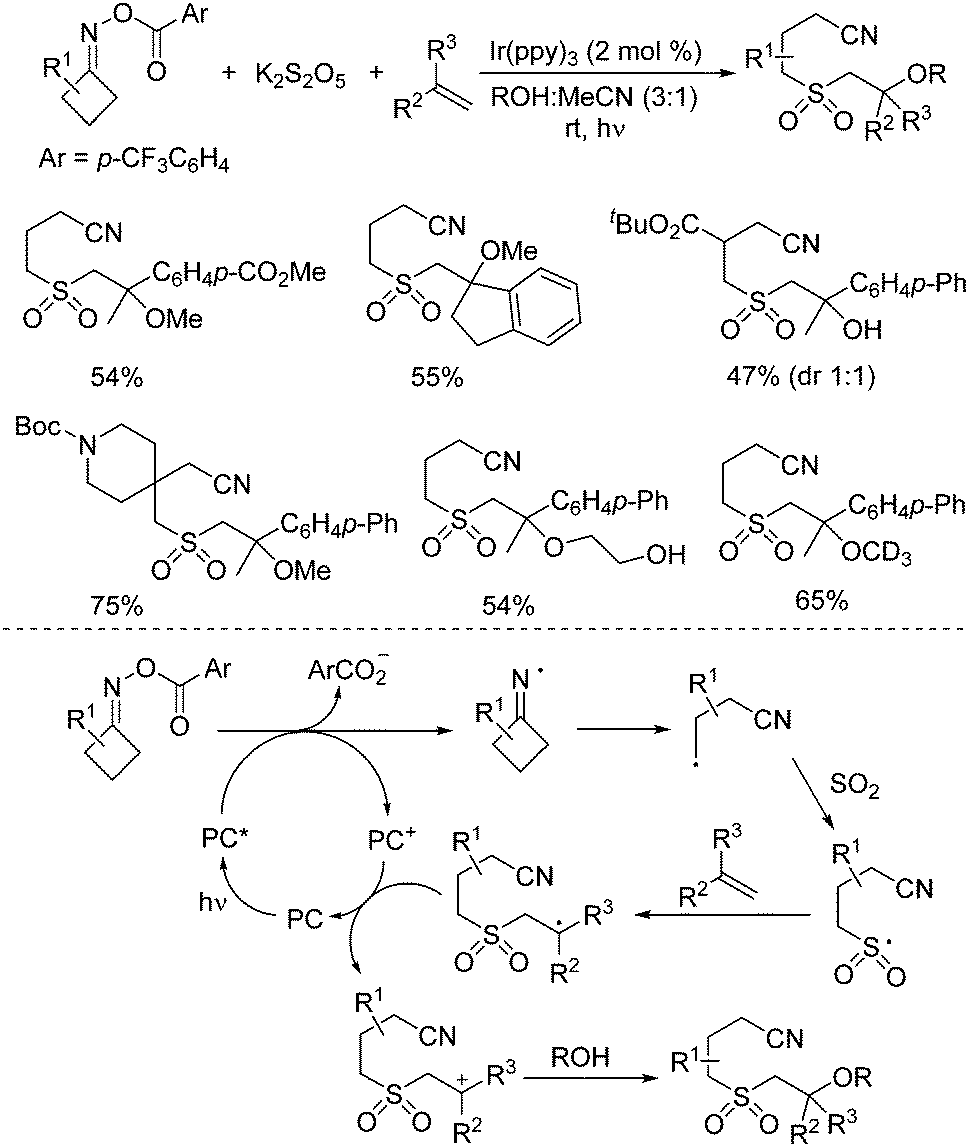 | ||
| Scheme 17 Photoredox-catalyzed reaction of O-acyl oximes, potassium metabisulfite, alkenes, and nucleophiles under visible light irradiation. | ||
Tang and co-workers described the synthesis of 2-cyanoalkylsulfonated 3,4-dihydro-naphthalenes via visible-light photoredox-catalyzed dual carbon–carbon bond cleavage of methylenecyclopropanes, potassium metabisulfite and cycloketone oximes (Scheme 18).36 Compared with the above method as reported by Wu,35 methylenecyclopropanes were used as a replacement for alkenes. For the mechanism, similar to Wu's report, the iminyl radical formed in situ from O-acyl oxime via N–O bond dissociation under visible light irradiation in the presence of the photocatalyst would initiate the reaction. Then, the intramolecular C–C bond cleavage of the iminyl radical would take place to provide a carbon radical, which would react with sulfur dioxide from potassium metabisulfite, giving rise to a sulfonyl radical intermediate. This sulfonyl radical intermediate would attack the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of methylenecyclopropane, leading to a carbon radical intermediate. Subsequently, ring-opening of cyclopropane would occur with another carbon–carbon bond cleavage. Following intramolecular cyclization and oxidative single electron transfer assisted by the photocatalyst, the final product would be obtained.
C double bond of methylenecyclopropane, leading to a carbon radical intermediate. Subsequently, ring-opening of cyclopropane would occur with another carbon–carbon bond cleavage. Following intramolecular cyclization and oxidative single electron transfer assisted by the photocatalyst, the final product would be obtained.
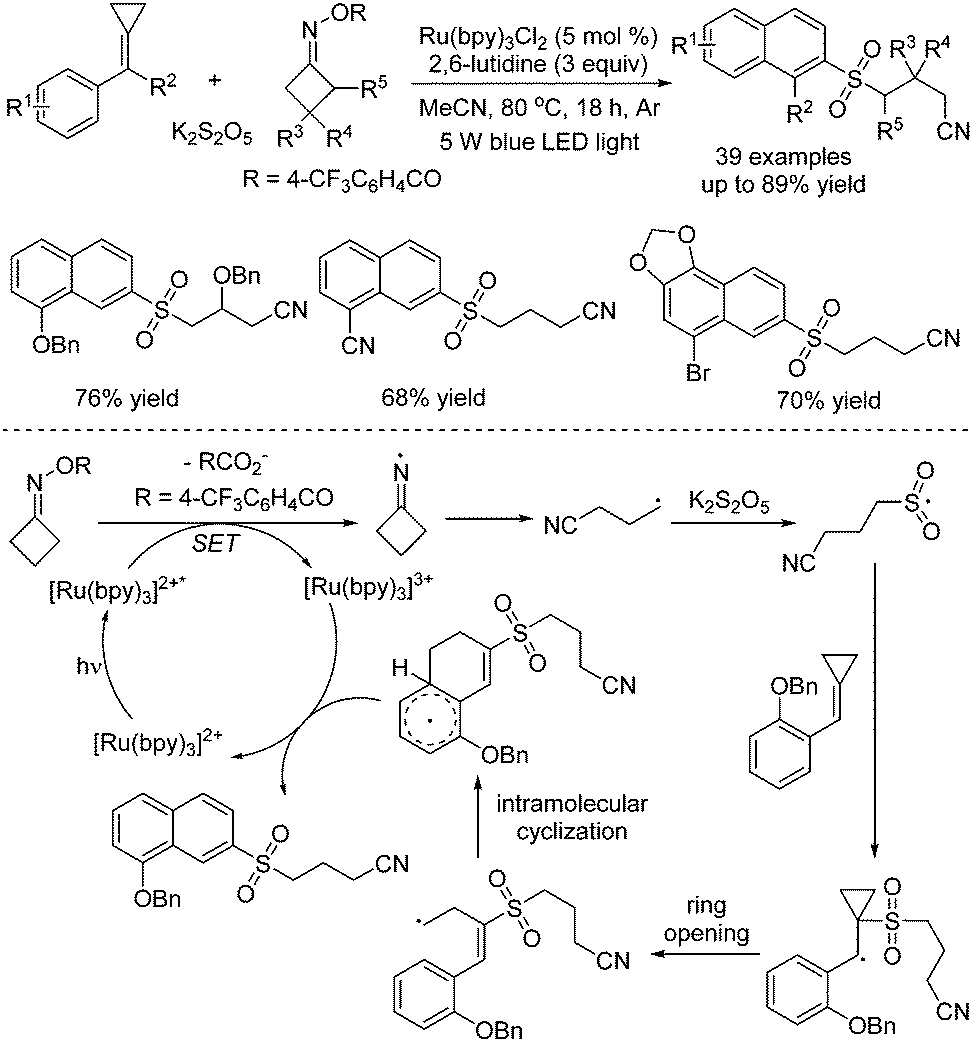 | ||
| Scheme 18 Photoredox-catalyzed reaction of potassium metabisulfite, methylenecyclopropanes and cycloketone oximes. | ||
A metal-free three-component reaction of aryl/alkyl iodides, sulfur dioxide and 3-azido-2-methylbut-3-en-2-ol under ultraviolet irradiation at room temperature is developed, leading to 2-(arylsulfonyl)acetonitriles in moderate to good yields.37 Various functional groups are compatible including amino, ester, halo, and trifluoromethyl groups. Although DABCO·(SO2)2 was used as the source of sulfur dioxide, the reaction using potassium metabisulfite or sodium metabisulfite was also effective with lower yields. The aryl radical generated in situ from aryl iodide under ultraviolet irradiation initiated the reaction, which underwent sulfonylation with sulfur dioxide, leading to an arylsulfonyl radical intermediate.
Jiang and co-workers described the synthesis of sulfones through a palladium-catalyzed three-component reductive cross-coupling reaction of sodium metabisulfite, aryl iodides, and alkyl halides (Scheme 19).38 Although this transformation proceeded in the presence of tin at high temperature, some biomolecules including steroids, saccharides, and amino acids were all compatible under the conditions. Intramolecular cyclic sulfones were prepared as well from five- to twelve-membered rings. Additionally, four drug molecules bearing functional groups were obtained via late-stage sulfur dioxide insertion. From mechanistic studies, it was shown that an alkyl radical would be formed from the combination of alkyl bromide with tin via a single-electron transfer process. This alkyl radical would be trapped by sodium metabisulfite, leading to an alkylsulfonyl radical intermediate, which would be reduced by tin, affording the alkylsulfonyl anion. In the meantime, the oxidative addition of aryl iodide to Pd(0) would provide Pd(II) species, which would react with the alkylsulfonyl anion leading to another Pd(I) complex. Further reductive elimination from this Pd(II) complex would produce the desired sulfone with the release of the Pd(0) catalyst.
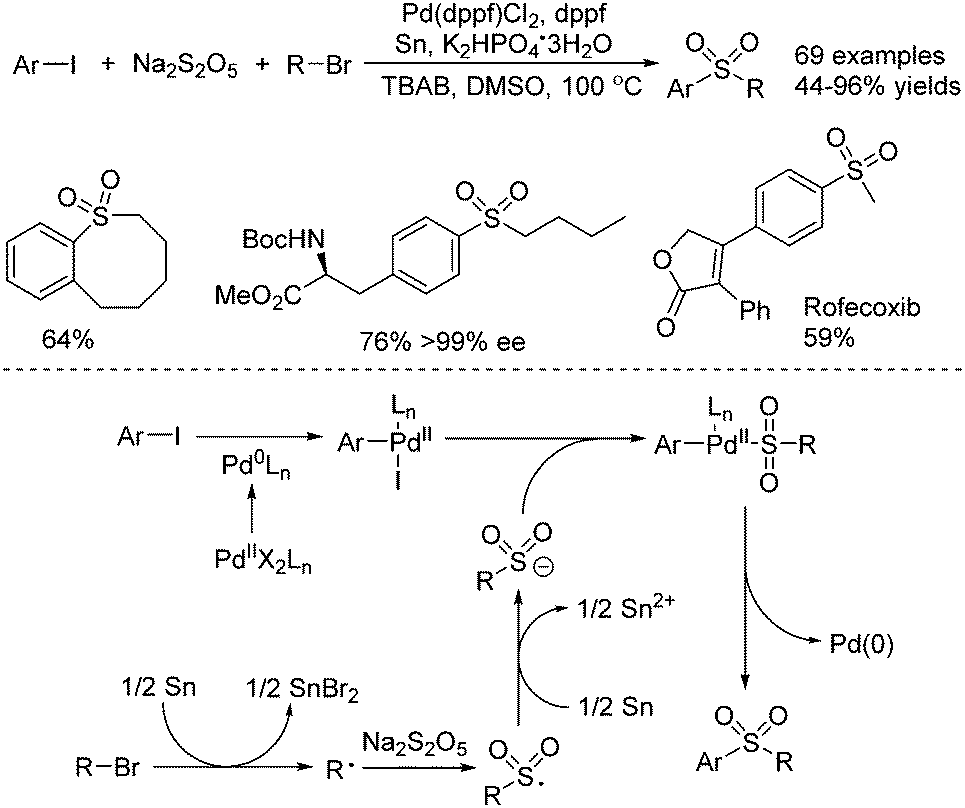 | ||
| Scheme 19 A palladium-catalyzed three-component reductive cross-coupling reaction of sodium metabisulfite, aryl iodides, and alkyl halides. | ||
5. Conclusions and outlook
In this updated report, the recent advances in sulfonylation reactions using potassium/sodium metabisulfite as the source of sulfur dioxide from Dec. 2018 are summarized. In this field, method development by using potassium/sodium metabisulfite in sulfonylation reactions boomed last year. As mentioned above, these approaches are attractive and promising, since potassium/sodium metabisulfite is abundant, easily available and cheap. Diverse sulfonyl compounds including sulfones and sulfonamides can be accessed easily and conveniently. In most cases, the transformations go through radical processes with the insertion of sulfur dioxide under mild conditions. Additionally, transition metal catalysis is applied in the reactions for the synthesis of sulfonyl-containing compounds. Among the approaches, photoinduced conversions under visible light or ultraviolet irradiation are also involved.However, some challenges are still to be explored. Since many marketed drugs are chiral molecules, asymmetric synthesis of sulfonyl-containing compounds through enantioselective reactions of potassium/sodium metabisulfite will be the focus in the near future, which has not been disclosed yet. Additionally, examples are rare of using potassium/sodium metabisulfite as the source of sulfur dioxide for the preparation of sulfonyl-containing drugs, although the developed methods have been employed successfully for the late-stage modification of currently marketed drugs. Thus, more efficient methods using potassium/sodium metabisulfite in organic synthesis will continuously appear, and the application of these methods for the synthesis of drugs, especially chiral sulfonyl-containing drugs, is anticipated.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from National Natural Science Foundation of China (No. 21532001 and 21871053) and the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2019R01005) is gratefully acknowledged.Notes and references
- (a) J. Drews, Science, 2000, 287, 1960 CrossRef CAS PubMed; (b) N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, New York, 1993 Search PubMed; (c) A. EI-Awa, M. N. Noshi, X. M. du Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315 CrossRef PubMed.
- (a) M. Bartholow, Top 200 Drugs of 2011, Pharmacy Times, http://www.pharmacytimes.com/publications/issue/2012/July2012/Top-200-Drugs-of-2011, accessed on Jan 9, 2013; (b) For a list of top drugs by year, see: http://cbc.arizona.edu/njardarson/group/toppharmaceuticals-poster, accessed on Jan 9, 2013.
- For selected examples, see: (a) Z. Chen, S. Liu, W. Hao, G. Xu, S. Wu, J. Miao, B. Jiang, S. Wang, S. Tu and G. Li, Chem. Sci., 2015, 6, 6654 RSC; (b) V. Khakyzadeh, Y. Wang and B. Breit, Chem. Commun., 2017, 53, 4966 RSC; (c) L. Zheng, Z. Zhou, Y. He, L. Li, J. Ma, Y. Qiu, P. Zhou, X. Liu, P. Xu and Y. Liang, J. Org. Chem., 2016, 81, 66 CrossRef CAS PubMed; (d) X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205 CrossRef CAS PubMed; (e) G. Zhang, L. Zhang, H. Yi, Y. Luo, X. Qi, C. Tung, L. Wu and A. Lei, Chem. Commun., 2016, 52, 10407 RSC; (f) K. Sun, X.-L. Chen, Y.-L. Zhang, K. Li, X.-Q. Huang, Y.-Y. Peng, L.-B. Qua and B. Yu, Chem. Commun., 2019, 55, 12615 RSC.
- For reviews, see: (a) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691 RSC; (b) K. Hofman, N. Liu and G. Manolikakes, Chem. – Eur. J., 2018, 24, 11852 CrossRef CAS PubMed; (c) D. Zheng and J. Wu, Sulfur Dioxide Insertion Reactions for Organic Synthesis, Nature Springer, Berlin, 2017 CrossRef; (d) G. Liu, C. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592 RSC; (e) E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602 CrossRef CAS; (f) A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 2701 Search PubMed; (g) P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393 RSC; (h) G. Qiu, L. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405 RSC; (i) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561 RSC; (j) S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013 RSC.
- P. S. Santos and M. T. S. Mello, J. Mol. Struct., 1988, 178, 121 CrossRef CAS.
- For recent selected examples, see: (a) Z. Chen, N.-W. Liu, M. Bolte, H. Ren and G. Manolikakes, Green Chem., 2018, 20, 3059 RSC; (b) Q. Lin, Y. Liu, Z. Xiao, L. Zheng, X. Zhou, Y. Guo, Q.-Y. Chen, C. Zheng and C. Liu, Org. Chem. Front., 2019, 6, 447 RSC; (c) X. Y. Qin, L. He, J. Li, W. J. Hao, S. J. Tu and B. Jiang, Chem. Commun., 2019, 55, 3227 RSC; (d) R. J. Reddy, A. H. Kumari, J. J. Kumar and J. B. Nanubolu, Adv. Synth. Catal., 2019, 361, 1587 CrossRef CAS; (e) L. Lu, C. Luo, H. Peng, H. Jiang, M. Lei and B. Yin, Org. Lett., 2019, 21, 2602 CrossRef CAS PubMed; (f) S. Ye, D. Zheng, J. Wu and G. Qiu, Chem. Commun., 2019, 55, 2214 RSC; (g) J. Zhang, W. Xie, S. Ye and J. Wu, Org. Chem. Front., 2019, 6, 2254 RSC; (h) Y. Chen, P. R. D. Murray, A. T. Davies and M. C. Willis, J. Am. Chem. Soc., 2018, 140, 8781 CrossRef CAS PubMed; (i) B. Ni, B. Zhang, J. Han, B. Peng, Y. Shan and T. Niu, Org. Lett., 2020, 22, 670 CrossRef CAS PubMed.
- S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037 RSC.
- (a) D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451 CrossRef CAS PubMed; (b) D. Zheng, J. Yu and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 11925 CrossRef CAS PubMed.
- For selected examples, see: (a) B. C. M. Potts and D. J. Faulkner, J. Nat. Prod., 1992, 55, 1701 CrossRef CAS PubMed; (b) A. Evidente and L. Sparapano, J. Nat. Prod., 1994, 57, 1720 CrossRef CAS; (c) T. Ishikawa, K. Nishigaya, H. Uchikoshi and I.-S. Chen, J. Nat. Prod., 1998, 61, 534 CrossRef CAS PubMed; (d) J. K. Son, D. H. Kim and M. H. Woo, J. Nat. Prod., 2003, 66, 1369 CrossRef CAS PubMed; (e) E. E. Shults, J. Velder, H.-G. Schmalz, S. V. Chernov, T. V. Rubalava, Y. V. Gatilov, G. Henze, G. A. Tolstikov and A. Prokop, Bioorg. Med. Chem. Lett., 2006, 16, 4228 CrossRef CAS PubMed; (f) R. Muddala, J. A. M. Acosta, L. C. A. Barbosa and J. Boukouvalas, J. Nat. Prod., 2017, 80, 2166 CrossRef CAS PubMed; (g) B. Mao, M. Fanãnás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502 CrossRef CAS PubMed.
- For selected examples, see: (a) L. G. Monovich, Y. Le Huérou, M. Roön and G. A. Molander, J. Am. Chem. Soc., 2000, 122, 52 CrossRef CAS; (b) D. F. Taber, K. Nakajima, M. Xu and A. L. Rheingold, J. Org. Chem., 2002, 67, 4501 CrossRef CAS PubMed; (c) T. Yoshimitsu, T. Makino and H. Nagaoka, J. Org. Chem., 2004, 69, 1993 CrossRef CAS PubMed; (d) S. Gao, Q. Wang and C. Chen, J. Am. Chem. Soc., 2009, 131, 1410 CrossRef CAS PubMed.
- K. Zhou, J. Zhang, G. Qiu and J. Wu, Org. Lett., 2019, 21, 275 CrossRef CAS PubMed.
- (a) Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059 CrossRef CAS PubMed; (b) R. J. Reddy, M. P. Ball-Jones and P. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 13310 CrossRef CAS PubMed.
- X. Gong, X. Li, W. Xie, J. Wu and S. Ye, Org. Chem. Front., 2019, 6, 1863 RSC.
- C.-M. Huang, J. Li, S.-Y. Wang and S.-J. Ji, Chin. Chem. Lett., 2020 DOI:10.1016/j.cclet.2019.12.032.
- Y. Liu, Q.-L. Wang, Z. Chen, P. Chen, K.-W. Tang, Q. Zhou and J. Xie, Org. Biomol. Chem., 2019, 17, 10020 RSC.
- H. Chen, M. Liu, Q. Qiu and J. Wu, Adv. Synth. Catal., 2019, 361, 146 CrossRef CAS.
- M. Kumar, R. Ahmed, M. Singh, S. Sharma, T. Thatikonda and P. P. Singh, J. Org. Chem., 2020, 85, 716 CrossRef CAS PubMed.
- G. You, D. Xi, J. Sun, L. Hao and C. Xia, Org. Biomol. Chem., 2019, 17, 9479 RSC.
- N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 1st edn, 2001 Search PubMed.
- X. Marset, J. Torregrosa-Crespo, R. M. Martinez-Espinosa, G. Guillena and D. J. Ramón, Green Chem., 2019, 21, 4127 RSC.
- X. Wang, M. Yang, K. Kuang, J.-B. Liu, X. Fan and J. Wu, Chem. Commun., 2020, 56, 3437 RSC.
- K. Chen, W. Chen, B. Han, W. Chen, M. Liu and H. Wu, Org. Lett., 2020, 22, 1841 CrossRef CAS PubMed.
- M. Wang, J. Zhao and X. Jiang, ChemSusChem, 2019, 12, 3064 CrossRef CAS PubMed.
- H. Zhu, Y. Shen, D. Wen, Z.-G. Le and T. Tu, Org. Lett., 2019, 21, 974 CrossRef CAS.
- M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404 CrossRef CAS.
- For reviews: (a) G. Duret, R. Quinlan, P. Bisseret and N. Blanchard, Chem. Sci., 2015, 6, 5366 RSC; (b) G. A. Molander, J. Org. Chem., 2015, 80, 7837 CrossRef CAS; (c) S. Roslin and L. R. Odell, Eur. J. Org. Chem., 2017, 1993 CrossRef CAS.
- X. Gong, M. Yang, J.-B. Liu, F.-S. He and J. Wu, Org. Chem. Front., 2020, 7, 938 RSC.
- F.-S. He, X. Gong, P. Rojsitthisak and J. Wu, J. Org. Chem., 2019, 84, 13159 CrossRef CAS PubMed.
- X. Gong, M. Wang, S. Ye and J. Wu, Org. Lett., 2019, 21, 1156 CrossRef CAS.
- F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943 CrossRef CAS.
- X. Wang, Y. Kuang, S. Ye and J. Wu, Chem. Commun., 2019, 55, 14962 RSC.
- For review, see: (a) W. Huang and X. Cheng, Synlett, 2017, 148 CAS; (b) S. Ye and J. Wu, Acta Chim. Sin., 2019, 77, 814 CrossRef.
- X. Gong, M. Yang, J.-B. Liu, F.-S. He, X. Fan and J. Wu, Green Chem., 2020, 22, 1906 RSC.
- For reviews: see: (a) W. Yin and X. Wang, New J. Chem., 2019, 43, 3254 RSC; (b) X. Wu and C. Zhu, Chin. J. Chem., 2019, 37, 171 CAS; (c) M. M. Jackman, Y. Cai and S. L. Castle, Synthesis, 2017, 1785 CAS; (d) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem. – Eur. J., 2018, 24, 12154 CrossRef CAS PubMed.
- J. Zhang, X. Li, W. Xie, J. Wu and S. Ye, Org. Lett., 2019, 21, 4950 CrossRef CAS PubMed.
- Y. Liu, Q.-L. Wang, Z. Chen, H. Li, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, Chem. Commun., 2020, 56, 3011 RSC.
- K. Zhou, J.-B. Liu, W. Xie, S. Ye and J. Wu, Chem. Commun., 2020, 56, 2554 RSC.
- Y. Meng, M. Wang and X. Jiang, Angew. Chem., Int. Ed., 2020, 59, 1346 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |