
Chemoenzymatic, biomimetic total synthesis of (−)-rugulosin B, C and rugulin analogues and their biosynthetic implications†
Amit
Mondal
 ,
Shailesh Kumar
Singh
,
Tanaya
Manna
and
Syed Masood
Husain
*
,
Shailesh Kumar
Singh
,
Tanaya
Manna
and
Syed Masood
Husain
*
Molecular Synthesis and Drug Discovery Unit, Centre for Biomedical Research, Sanjay Gandhi Postgraduate Institute of Medical Sciences Campus, Raebareli Road, Lucknow 226014, India. E-mail: smhusain@cbmr.res.in; smhusain.cbmr@gmail.com
First published on 13th February 2020
Abstract
Herein, we report the chemoenzymatic synthesis of a heterodimeric (−)-rugulosin B, homodimeric (−)-rugulosin C, and several rugulin analogues in three to four steps starting from anthraquinones. This work supports dimerization between variously substituted putative monomeric intermediates during the biosynthesis of naturally occurring (+)-rugulosin B and C.
Nature produces a large number of homo- and hetero-dimeric natural products with fascinating structures which display a variety of useful biological activities. Among these, modified bisanthraquinones (1–5 and 7–9) having five to eight chiral centres isolated mainly from fungi constitute a class of secondary metabolites consisting of two chiral monomeric units bonded together with two to four bonds (Fig. 1). The most abundant is homodimeric (+)-rugulosin (1)1,2 (Fig. 1A) isolated initially from Penicillium rugulosum Thom2a and later from a dozen of other fungal strains,2b,c and is known to play a role as a bioinsecticide3 along with many other prominent biological activities.2a,4 The other related but less abundant homodimeric bisanthraquinones such as (+)-rugulosin C (2),5 (+)-2,2′-epi-cytoskyrin A (3),6 (−)-flavoskyrin (4)1,7 and (−)-rugulosin (5)1,8 and an exceptional four bonded cage-like compound, rugulin (7),9 have also been isolated from various fungal and lichen species (Fig. 1A). However, more interesting is the existence of heterodimeric bisanthraquinones such as (+)-rugulosin B (8) isolated from Penicillium radicum FKI-3765-2 which shows antimicrobial activity against methicillin-resistant Staphylococcus aureus5 and (−)-deoxyluteoskyrin (9) isolated from Penicillium islandicum (Fig. 1B).1 All these natural products appear to be biosynthesized by a unifying strategy that involves the dimerization of chiral monomeric anthraquinone units, as proposed by Shibata and others.10–12 Although the total synthesis of (+)-rugulosin (1) and (+)-2,2′-epi-cytoskyrin A (3) was successfully demonstrated by the group of Nicolaou,13a a much simpler chemoenzymatic, biomimetic, and protecting group free synthesis of their enantiomers, (−)-rugulosin (5) and (−)-2,2′-epi-cytoskyrin A (6), has been reported by us (Fig. 1A).14
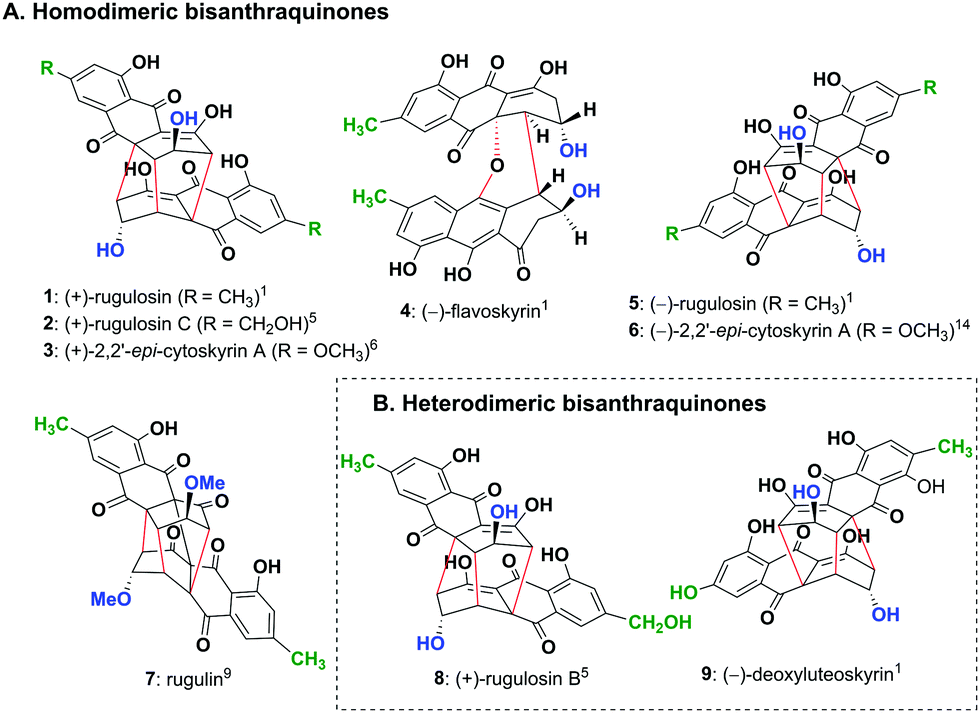 | ||
| Fig. 1 (A) Homodimeric bisanthraquinones 1–5 and 7 isolated from various fungal species and not yet isolated analogue 6 and (B) heterodimeric bisanthraquinones 8 and 9 isolated from fungi. | ||
In the current work, we envisaged a biomimetic, chemoenzymatic strategy for the preparation of (−)-rugulosin C (ent-2), (−)-rugulosin B (ent-8) and rugulin analogues, in just three and four steps, respectively. In order to simplify our strategy, at first, a retro(bio)synthetic route to rugulin type compounds has been proposed (Scheme 1). The rugulin analogue 10 with four C–C bonds might be formed by the oxidative coupling of a dimeric rugulosin type molecule 11. Based on the earlier proposals by Shibata, 11 can be formed using flavoskyrin type compound 12 following a rearrangement through a number of dimeric intermediates.10,11 The formation of 12 will require dimerization of monomeric intermediate 13b, a dienone tautomer of 13avia a hetero-Diels–Alder reaction, whereas 13a/13b appeared to be formed by the enzymatic reduction of anthraquinones (Scheme 1). For the proposed synthesis of ent-2 and ent-8, we planned to use an anthrol reductase from Talaromyces islandicus (ARti) identified recently for the chemoenzymatic reduction of emodin (15) and citreorosein (19).15
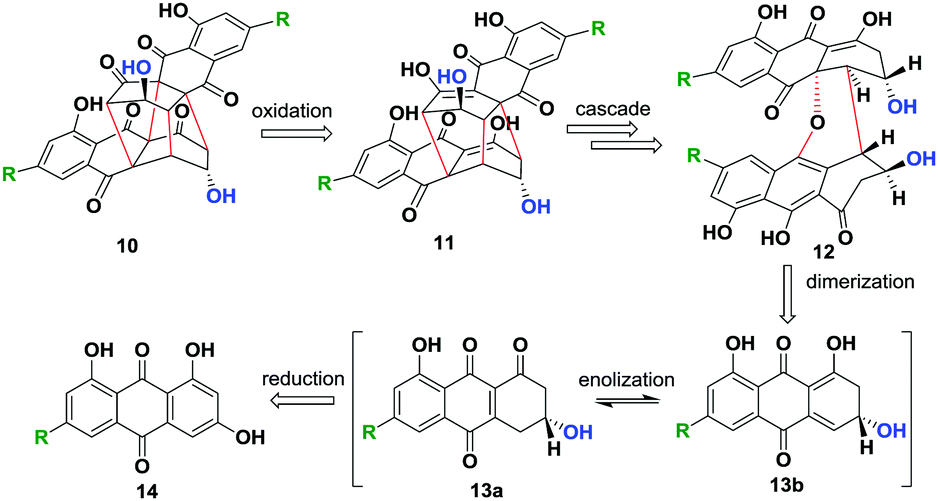 | ||
| Scheme 1 Proposed retro(bio)synthesis of modified bisanthraquinones. | ||
For this purpose, the pET-19b plasmid containing the gene for ARti_his was transformed into E. coli BL21 (DE3) cells and the protein was expressed and purified using Ni-NTA affinity chromatography. The anthraquinone, emodin (15) or citreorosein (19) dissolved in dimethyl sulfoxide taken into potassium phosphate buffer (50 mM, 1 mM EDTA, 1 mm DTT, pH 7) along with NADPH (regenerated through glucose/glucose dehydrogenase system) in the presence of Na2S2O4 is incubated with ARti_his for 14 h. ARti catalyses the stereoselective reduction of anthrols/hydroanthraquinones 16a/16b or 20a/20b (formed in situ) to dihydroanthracen-1(2H)-ones, 17 and 21, respectively. These transformations also resulted in the formation of a small amount of chrysophanol (18) and aloe-emodin (22) as side products formed by the non-enzymatic processes that took place during extraction. Nevertheless, the formation of the deoxy-side products is avoided through optimization of the extraction and purification procedure giving (R)-17 and (R)-21 in 74% and 79% yield, respectively (Scheme 2). Both 17 and 21 reportedly show >99% of enantiomeric excess as analysed through chiral HPLC using racemic 17 and 21 (see the ESI†).15
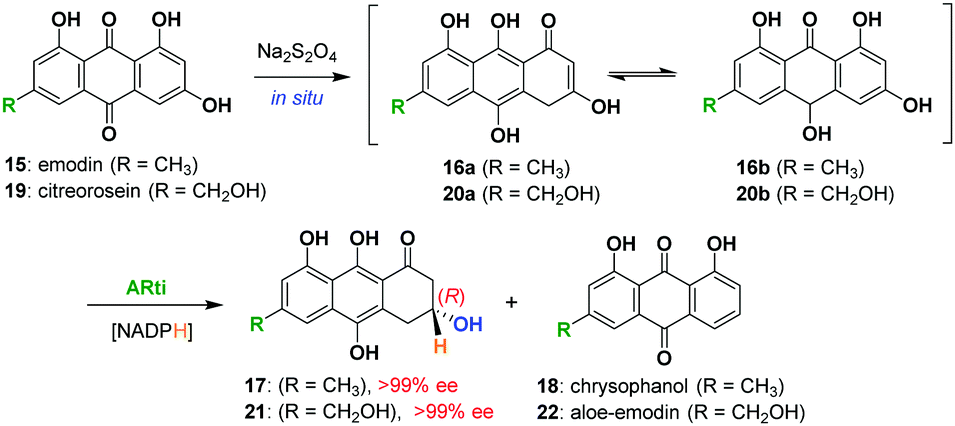 | ||
| Scheme 2 Chemoenzymatic reduction of emodin (15) and citreorosein (19) by ARti using NADPH (regenerated by glucose/glucose dehydrogenase). | ||
Next, we focused on to obtain flavoskyrin type dimers, desired for the synthesis of proposed bisanthraquinones. At first, we oxidized 21 using Pb(OAc)4 in AcOH, which resulted in the formation of (R)-3,4-dihydrocitreorosein 23a and its tautomer 23b. The dienol tautomer 23b undergoes dimerization through the hetero-Diels–Alder reaction to give (−)-flavoskyrin C (24) in 48% yield after purification with oxalic acid impregnated silica gel (Scheme 3A). Then, we oxidized the mixture of dihydroanthracenones 17 and 21 taken in ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 using Pb(OAc)4 in AcOH to maximize the formation of the desired heterodimeric flavoskyrins (Scheme 3B).
:1 using Pb(OAc)4 in AcOH to maximize the formation of the desired heterodimeric flavoskyrins (Scheme 3B).
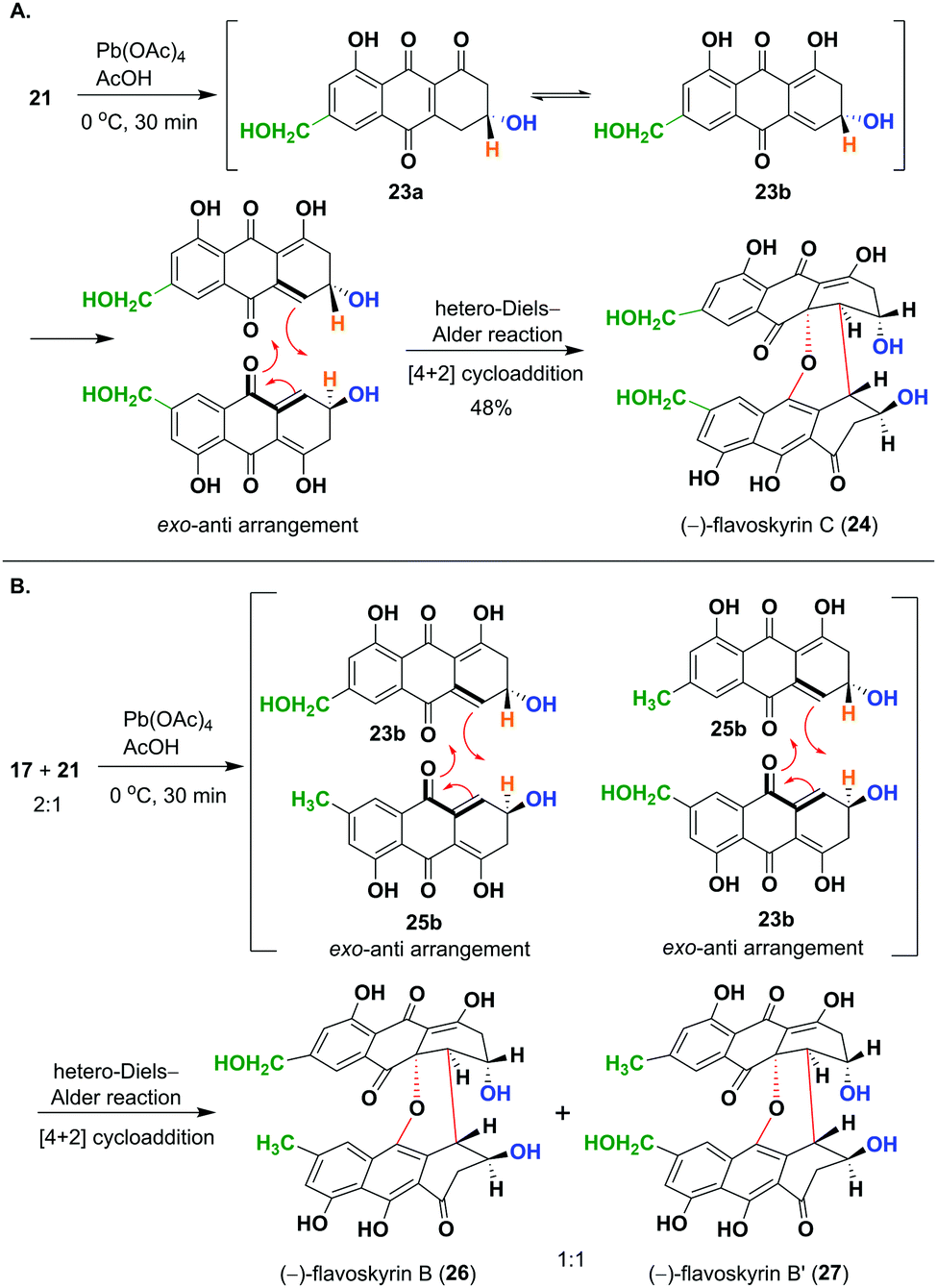 | ||
| Scheme 3 (A) Oxidation of 21 by Pb(OAc)4 forms (−)-flavoskyrin C (24) through the hetero Diels–Alder reaction. (B) Oxidation of a mixture of 17 and 21 (2:1) by Pb(OAc)4 yielded flavoskyrin B (26) and flavoskyrin B′ (27) as a major product. | ||
This has resulted in the formation of an inseparable mixture of diastereomers, (−)-flavoskyrin B (26) and (−)-flavoskyrin B′ (27) in 55% yield (Scheme 3B). This was expected as the dienol tautomer 23b of (R)-3,4-dihydrocitreorosein 23a and dienol tautomer 25b of (R)-3,4-dihydroemodin 25a acted as diene as well dienophile during the hetero-Diels–Alder reaction. In addition to 26/27, we also isolated a small amount of (−)-flavoskyrin (4), and (−)-flavoskyrin C (24), giving us the possibility to obtain all the isomers in just a single step. This also shows that the heterodimerization between two differently substituted monomeric units is also feasible, which would have significant implications on the biosynthesis of many related heterodimeric natural products.
In the next step, we aim to convert these flavoskyrin analogues to modified bisanthraquinones in the presence of a base, following a cascade.13,14 For this purpose, we incubated 24 in pyridine under an oxygen atmosphere. This resulted in the formation of (−)-rugulosin C (ent-2) in 50% yield and (−)-dianhydrorugulosin C (31) in 23% yield (Scheme 4A). The conversion would have taken place via a cascade involving several putative intermediates 28–30. By performing the cascade under an argon atmosphere, we prevented the oxidation of 29 to 30. This way we isolated and characterized intermediate 29 whereas intermediates 28 and 30 could not be observed. However, when we perform the cascade in pyridine-d5, we observed the formation of both 29 and 30 through 1H NMR.
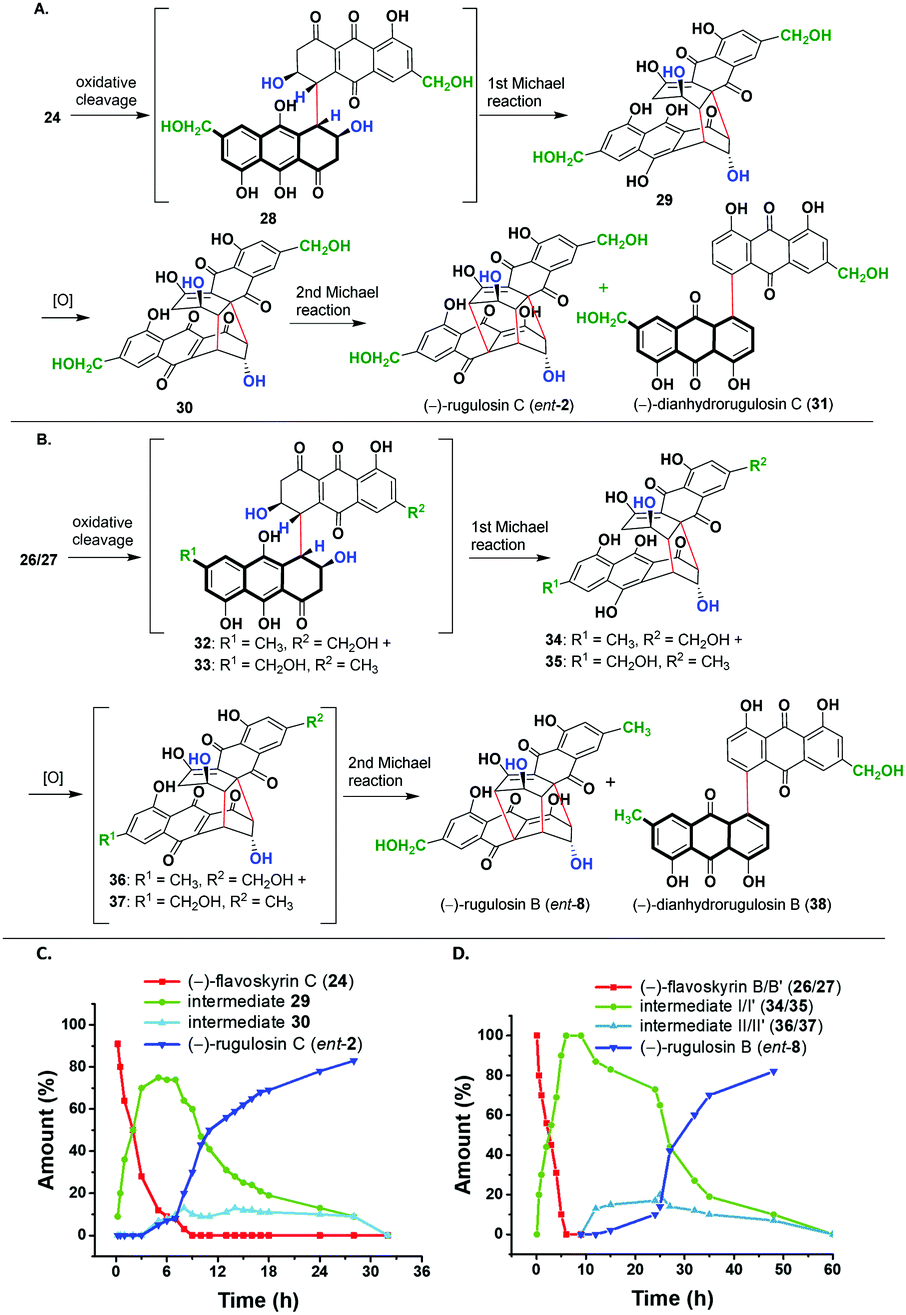 | ||
| Scheme 4 (A) Cascade conversion of (−)-flavoskyrin C (24) to (−)-rugulosin C (ent-2). (B) Cascade conversion of (−)-flavoskyrin B/B′ (26/27) to (−)-rugulosin B (ent-8). (C) Plot of percentage conversion of the products formed with time (h) for the cascade in (A). (D) Plot of percentage conversion of the products formed with time (h) for the cascade in (B). | ||
The plot of % conversion to products (determined through 1H NMR spectra) versus time shows an initial conversion of (−)-flavoskyrin C (24) into intermediate I (29) in six hours and only a small amount of (−)-rugulosin C (ent-2) was formed. This is due to the small amount of dissolved oxygen present in pyridine-d5. However, when the sample was bubbled with molecular oxygen, intermediate 29 got converted mostly to (−)-rugulosin C (ent-2) within 30 h via intermediate II (30), which was only observed in small amounts during the course of the reaction.
Likewise, we treated the mixture of two diastereomers, namely (−)-flavoskyrin B (26) and (−)-flavoskyrin B′ (27), with pyridine for 24 h in the presence of oxygen. The reaction resulted in the formation of only single enantiomers that is (−)-rugulosin B (ent-8) and (−)-dianhydrorugulosin B (38) in 60% and 18% yields, respectively. For a detailed study of the cascade, we incubated the mixture of 26/27 in pyridine-d5 and observed the reaction course through 1H NMR after regular intervals (see the ESI,† Scheme S3). Although the proposed putative intermediates 32/33 were not detected during the cascade, we could observe the formation of a mixture of diastereomeric intermediate I/I′ (34/35) through 1H NMR as a major product. It is only when we bubbled oxygen through the reaction mixture after 10 h that the formation of a trace amount of the oxidized intermediate II/II′ (36/37) was observed. In 50 h, most of the diastereomeric mixture 34/35 got converted into (−)-rugulosin B (ent-8). This has been further verified by the isolation of the diastereomeric mixture of intermediate I/I′ (34/35), which is then treated with pyridine-d5 and again observed through 1H NMR spectroscopy (see the ESI,† Fig S1). No spectral changes were observed until oxygen is introduced into the system. After bubbling of molecular oxygen into the mixture of 34/35, we observed the formation of (−)-rugulosin B (ent-8) as the only product (see the ESI,† Fig. S1D). This is because of symmetry considerations, where the formation of a third C–C bond from either intermediate II (36) or II′ (37) will lead to (−)-rugulosin B (ent-8) (see the ESI,† Scheme S3). The comparison of CD spectra and optical rotation for (−)-rugulosin B (ent-8) and (−)-rugulosin C (ent-2) with the reported data of (+)-rugulosin B (8) and (+)-rugulosin C (2),5 respectively, further confirms the enantiomeric nature and the optical purity of the synthesized compounds.
In order to further extend the limit of our approach, we planned to convert all the dimeric bisanthraquinones synthesised herein to their rugulin analogues through oxidation in a single step. This is in contrast to the 17-step procedure reportedly used for the synthesis of the enantiomers of rugulin analogues.13a For this, we used (−)-rugulosin (5) as a model compound for optimization, which was synthesized using emodin (15) following a chemoenzymatic procedure reported earlier.14 Inspired by our retro-biosynthetic analysis, we proposed that the oxidation of rugulosin to rugulin analogues must involve the conversion of enol to keto followed by oxidative coupling. To achieve this, at first, we incubated 5 in tris buffer (50 mM, pH 9) by bubbling molecular oxygen for 6 h (entry 1) (Table 1). However, this does not result in any change in the starting material as observed by TLC. Next, we employed Pb(OAc)4 in AcOH for oxidation at room temperature (entry 2). The reaction led to the formation of expected rugulin analogue 39 (<20%) along with several inseparable side products. Likewise, when the reaction was performed for 10 h, it resulted in the formation of 39 with 32% conversion (entry 3). When the reaction was performed at 0 °C and then warmed up to room temperature (rt), it resulted in the formation of a complex mixture of compounds and hence the conversion to the product was not determined (entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | Reagent (equiv.) | Solventb | Time (h) | Conv.a (%) |
| a Determined from the 1H NMR spectra. b 0 °C to rt. c Dry solvent. | ||||
| 1 | O2 | Tris buffer (pH 9) | 6 | — |
| 2 | Pb(OAc)4 (1.1) | AcOH | 2 | <20 |
| 3 | Pb(OAc)4 (1.1) | AcOH | 10 | 32 |
| 4b | Pb(OAc)4 (2) | AcOH | 10 | n.d. |
| 5 | MnO2 (10) | Dry DCM | 10 | 18 |
| 6c | MnO2 (10) | DCM:Et2O (5:1) |
3 | 47 |
| 7 | MnO2 (10) | Dry THF | 4 | 64 |
| 8c | MnO2 (10) | DCM:ACN (5:1) |
4 | 83 |
| 9 | CAN (2) |
ACN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :H
2
O (1:1) :H
2
O (1:1)
|
2 | 95 |
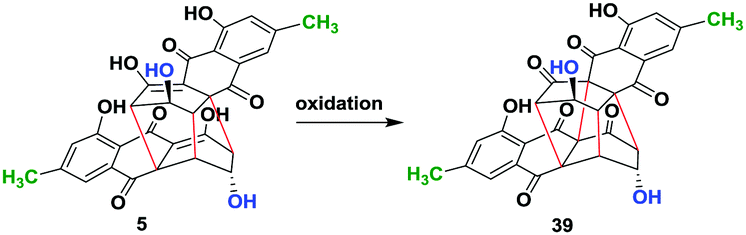
We then employed MnO2 as an oxidizing agent. However, when 5 was stirred in dry dichloromethane (DCM) with 10 equivalent of MnO2 for 4 h, only 18% conversion to the product was observed (entry 5). This was due to the very low solubility of 5 in DCM. Therefore, we used dry Et2O to dissolve 5 and added to dry DCM containing 10 equiv. of MnO2. This resulted in the formation of 39 with 47% conversion (entry 6). Furthermore, improvement in conversion up to 64% was observed by using dry THF as a solvent for the reaction (entry 7). However, when we used acetonitrile (ACN) as a co-solvent to dissolve 5 and added it to dry DCM containing MnO2, we observed 83% conversion to 39 (entry 8). Hence, the solubility of the starting material during oxidation is a key factor for high conversion. Therefore, we employed ceric ammonium nitrate (CAN), which is water-soluble and is a strong oxidizing agent. (−)-Rugulosin (5) was dissolved in acetonitrile and added to the solution of CAN in water. This shows the best conversion of 95% (entry 9). Finally, product 39 was purified using oxalic acid impregnated silica gel in 84% yield. The optical rotation was found to be opposite to that of its enantiomer which was reportedly synthesized in nearly 17 steps (see the ESI†).
To further generalize the optimized procedure, we oxidized (−)-2,2′-epi-cytoskyrin A (6) obtained using a procedure reported earlier,14 (−)-rugulosin B (ent-8) and (−)-rugulosin C (ent-2) to their respective rugulin analogues using CAN in water (Fig. 2). Although 6 resulted in the formation of the expected rugulin analogue 40 in 57% yield, ent-8 on oxidation with CAN gave 41 in 52% yield, formed by the oxidation of alcohol to aldehyde. Despite several attempts, the rugulin analogue of (−)-rugulosin C (ent-2) could not be obtained, which may be due to over oxidation resulting in an unstable product. Nevertheless, we have shown that rugulin analogues can be synthesized in a biomimetic fashion in just four steps starting from anthraquinones.
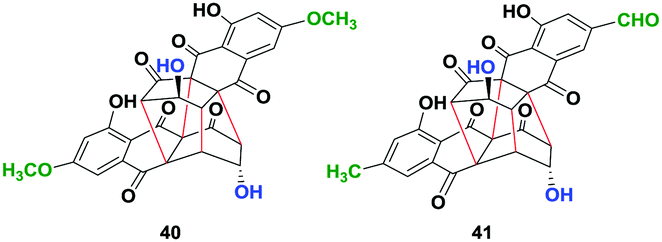 | ||
| Fig. 2 Rugulin analogues (40 & 41) synthesized by the oxidation of rugulosin analogues using ceric ammonium nitrate (CAN) in water. | ||
In conclusion, a chemoenzymatic, biomimetic, protecting group free strategy for the synthesis of a homodimeric (−)-rugulosin C (ent-2) and a heterodimeric (−)-rugulosin B (ent-8) and several rugulin analogues (39–41) is developed. This provides very first evidence in support of the homo/hetero dimerization step during the biosynthesis of (+)-rugulosin (5) and (+)-rugulosin B (8) and C (2) in P. radicum FKI-3765-2. A detailed investigation of a cascade conversion of a diastereomeric mixture of flavoskyrins B/B′ (26/27) to (−)-rugulosin B (ent-8) using NMR spectroscopy supports the formation of a single enantiomer at the end during hetero dimerization. Furthermore, the synthesis of rugulin analogues (39, 40, and 41) through oxidation of 5, 6 and ent-8 using CAN in water further highlights the feasibility of the current procedure to synthesize complex dimeric bisanthraquinones in fewer steps without the need for any protecting groups. The procedure developed here will serve as a model for (bio)synthetic studies in future and may give access to other more complex homo- and hetero-dimeric bisanthraquinones in a biomimetic fashion.
We are grateful to the Council of Scientific and Industrial Research (02(0258)/16/EMR-II), Science and Engineering Board, New Delhi (CRG/2018/002682), for funding this research, the Department of Biotechnology for fellowship to S. K. S, the CSIR for fellowship to A. M and T. M, Nirmal Saha for his skillful help, and the Director, Centre of Biomedical Research for research facilities.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Takeda, S. Seo, Y. Ogihara, U. Sankawa, I. Iitaka, I. Kitagawa and S. Shibata, Tetrahedron, 1973, 29, 3703 CrossRef CAS.
- (a) J. Breen, J. C. Dacre, H. Raistrick and G. Smith, Biochem. J., 1955, 60, 618 CrossRef CAS PubMed; (b) Y. Yamamoto, A. Hamaguchi, I. Yamamoto and S. Imai, Yakugaku Zasshi, 1956, 76, 1428 CrossRef CAS; (c) S. Shibata and T. Ikekawa, Chem. Pharm. Bull., 1963, 11, 368 CrossRef CAS.
- (a) J. Dobias, V. Betina and P. Nemec, Biol. Czechoslov., 1980, 36, 431 Search PubMed; (b) P. Watts, P. Kittakoop, S. Veeranondha, S. Wanasith, R. Thongwichian, P. Saisaha, S. Intamas and N. L. Hywel-Jones, Mycol. Res., 2003, 107, 581 CrossRef CAS PubMed; (c) J. D. Miller, M. W. Sumarah and G. W. Adams, J. Chem. Ecol., 2008, 34, 362 CrossRef CAS PubMed.
- Y. Ueno, N. Sato, T. Ito, I. Ueno, M. Enomoto and H. Tsunoda, J. Toxicol. Sci., 1980, 5, 295 CrossRef CAS PubMed.
- H. Yamazaki, N. Koyama, S. Ōmura and H. Tomoda, Org. Lett., 2010, 12, 1572 CrossRef CAS PubMed.
- A. Agusta, K. Ohashi and H. Shibuya, Chem. Pharm. Bull., 2006, 54, 579 CrossRef CAS PubMed.
- (a) B. H. Howard and H. Raistrick, Biochem. J., 1954, 56, 56 CrossRef CAS PubMed; (b) K. Kawai, Y. Nozawa, H. Mori and Y. Ogihara, Toxicol. Lett., 1986, 30, 105 CrossRef CAS PubMed.
- (a) D.-M. Yang, U. Sankawa, Y. Ebizuka and S. Shibata, Tetrahedron, 1976, 32, 333 CrossRef CAS; (b) S. Nakamura, F. Nii, M. Shimizu and I. Watanabe, Jpn. J. Microbiol., 1971, 15, 113 CrossRef CAS PubMed; (c) S. Nakamura, F. Nii, S. Inoue, I. Nakanishi and M. Shimizu, Jpn. J. Microbiol., 1974, 18, 1 CrossRef CAS PubMed.
- (a) P. Sedmera, M. Podojil, J. Vokoun, V. Betina and P. Nemec, Folia Microbiol., 1978, 23, 64 CrossRef CAS PubMed; (b) V. Betina and P. Nemec, Spôsob prípravy antibiotika rugulínu a metabolitov skyrínu a rugulozínu z mikroorganizmu Penicillium rugulosum, Czechoslovakian patent No. 187049, 1978.
- S. Seo, U. Sankawa, Y. Ogihara, Y. Iitaka and S. Shibata, Tetrahedron, 1973, 29, 3721 CrossRef CAS.
- U. Sankawa, in The Biosynthesis of Mycotoxins, ed. P. S. Steyn, Academic Press, New York, 1980, pp. 357–394 Search PubMed.
- E. Gravel and E. Poupon, Eur. J. Org. Chem., 2008, 27 CrossRef.
- (a) K. C. Nicolaou, Y. H. Lim, J. L. Piper and C. D. Papageorgiou, J. Am. Chem. Soc., 2007, 129, 4001 CrossRef CAS PubMed; (b) B. B. Snider and X. Gao, J. Org. Chem., 2005, 70, 6863 CrossRef CAS PubMed.
- N. Saha, A. Mondal, K. Witte, S. K. Singh, M. Müller and S. M. Husain, Chem. – Eur. J., 2018, 24, 1283 CrossRef CAS PubMed.
- S. K. Singh, A. Mondal, N. Saha and S. M. Husain, Green Chem., 2019, 21, 6594 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and supplementary figures and tables. See DOI: 10.1039/d0cc00406e |
| This journal is © The Royal Society of Chemistry 2020 |