
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanocatalysis by noble metal nanoparticles: controlled synthesis for the optimization and understanding of activities
Thenner S.
Rodrigues
 a,
Anderson G. M.
da Silva
b and
Pedro H. C.
Camargo
*bc
a,
Anderson G. M.
da Silva
b and
Pedro H. C.
Camargo
*bc
aNanotechnology Engineering Program, Alberto Luiz Coimbra Institute for Graduate Studies and Research in Engineering, COPPE, Federal University of Rio de Janeiro, Av. Horácio Macedo, 2030, 21.941-972, Rio de Janeiro, Brazil
bDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, 05508-000, São Paulo, Brazil
cDepartment of Chemistry, University of Helsinki, A.I. Virtasen aukio 1, FI-00014, Helsinki, Finland. E-mail: pedro.camargo@helsinki.fi
First published on 19th February 2019
Abstract
Noble-metal nanoparticles have been widely employed in catalysis. As catalytic properties are dependent on their physical/chemical parameters, strategies for their controlled synthesis and the understanding of performance relationships have gained attention. In this review, we discuss developments on well-defined noble-metal nanoparticles focusing on relationships between performance and physical/chemical features. We begin with the control over shape, composition, and size. Then, we focus on nanoparticles with hollow interiors, which provide further possibilities for performance optimization. We provide a case-by-case discussion to illustrate how, in addition to the hollow interiors, the control over their composition, size, and surface morphology relate to catalytic performance.
 Thenner S. Rodrigues | Thenner S. Rodrigues received his B.Sc. and M.Sc. in Industrial Chemistry in 2010 and 2013, respectively, both from Federal University of Ouro Preto (Brazil). He obtained his Ph.D. in Chemistry from the University of São Paulo (Brazil) in 2017, under the supervision of Prof. Pedro Camargo. He was a visiting student in Prof. Younan Xia's group at the Georgia Institute of Technology (USA) from February 2015 to March 2016. In 2018, he was hired as an Assistant Professor at Federal University of Rio de Janeiro. His research interests are focused on the synthesis of metal nanostructures and metal oxides with controlled shapes for catalytic and photocatalytic applications. |
 Anderson G. M. da Silva | Anderson G. M. da Silva received his B.Sc. in Industrial Chemistry in 2010 and M.Sc. in Materials Engineering in 2013, both from the Federal University of Ouro Preto (Brazil). He pursued his Ph.D. at the University of São Paulo (Brazil) under the supervision of Dr Pedro Camargo. During his Ph.D., he spent one year in Prof. Younan Xia's group at the Georgia Institute of Technology (USA) as a visiting student. Currently, he is a postdoctoral researcher at the University of São Paulo. His research interest includes synthesis of nanomaterials displaying controlled shapes, compositions and architectures for applications in catalysis, plasmonic, and electrochemical applications. |
 Pedro H. C. Camargo | Pedro Camargo received my BS and MS in Chemistry in 2003 and 2005, respectively, both from the Federal University of Paraná in Brazil. In 2005, he was a recipient of a Fulbright/Capes Fellowship to pursue my Ph.D. in the USA and joined Professor Younan Xia's group at the University of Washington. In 2007, the Xia group relocated to Washington University in Saint Louis, where I obtained my Ph.D. in Biomedical Engineering in 2009. In 2011, he was hired as an Assistant Professor at the University of São Paulo. He was promoted to Associate and Full Professor in 2015 and 2018, respectively. In February of 2019, he relocated to a Professor position at the University of Helsinki, Department of Chemistry. His research interests include the synthesis of nanomaterials for nanocatalysis and plasmonic nanocatalysis. He also serves as an Editor of the Journal of Materials Science and as an Associate Editor of the Journal of the Brazilian Chemical Society. |
1. Introduction
Catalysis is a central discipline in science and industry.1–3 Catalysts are used in several chemical transformations (at least in one step of the productive process) encompassing the fields of petrochemistry, pharmaceuticals, transportation, environmental remediation, among others.1,4 The catalyst can be considered as the heart (and the brain) of catalysis, being responsible for the substrate conversion and the selectivity towards the desired products. Recently, due to the increasing demand for chemical processes with improved efficiencies, minimization on the generation of waste, less energy consumption, and use of green conditions, a lot of attention has been directed to optimizing the operating conditions (parameters of process engineering) and developing catalysts with improved activities and selectivity.In this context, the use of nanoparticles (NPs) as catalysts has emerged as a promising alternative towards a variety of chemical transformations.5 The recent interest in the utilization of metal NPs in catalysis is due to their high surface areas, which favor the interaction between the reactants and the surface of the catalysts. This, in turn, leads to high activities even under mild conditions. This attribute was originally associated with homogeneous catalysts. However, as nanoparticles are not soluble in the reaction mixture, we can say that they bridge the gap between homogeneous and heterogeneous catalysis, unifying the advantages of both approaches.2–4,6,7 Therefore, NPs can combine the maximum exposure of active sites as in the homogeneous catalysis (use of NPs with sizes progressively decreased until the single-atom limit) and the possibility of separation, purification, and reuse as in the heterogeneous catalysts.2–4,6,7 Interestingly, it has been shown that the catalytic properties of nanoparticle catalysts are strongly dependent, and therefore can be controlled and optimized, as a function of several physical and chemical parameters that include size, shape, composition, and structure (solid or hollow interiors). Consequently, the field of nanocatalysis has blossomed in the last decades, and important developments in terms of activities, selectivity, and durability have been reported.8–12 For instance, excellent and comprehensive reviews have been published on the control of size, shape, composition, and structure in NPs for catalytic applications.13–16
Among several nanocatalysts, those based on noble metal NPs deserve special attention due to their electronic, chemical, and even optical properties (in the case of transformations enhanced or mediated by plasmonic effects).17–19 Nevertheless, conventional catalysts based on noble metal NPs obtained by solution phase methods still display poor or little control over their properties (size, shape, composition, facet exposition, etc.) and, consequently, show limitations in terms of the understanding of stability, activity, and selectivity.20,21 As the catalytic performance in noble metal NPs is strongly dependent on their physicochemical properties, the precise control over these parameters has gained much importance and notoriety.22–28 For example, a large variety of noble metal-based nanomaterials have been reported as catalysts towards a wealth of transformations as shown in Table 1.
| Metal | Shape | Size (nm) | Molecular Transformation | Yield (%) | Ref. |
|---|---|---|---|---|---|
| Au | NPs | 1.0–1.9 | Homocoupling of phenylboronic acid | >99 | 106 |
| NPs | 1.0–3.0 | Oxidation of alcohols | >99 | 107 | |
| NPs | 10.5 ± 0.3 | Diboration of styrene | >99 | 108 | |
| NPs | 15–34 | Reduction of 4-nitrophenol | >99 | 24 | |
| NPs | 50 ± 9 | Hydrogen evolution | n.a. | 109 | |
| Pd | NPs | 5–10 | Hydrogenation of olefins | >99 | 110 |
| NPs | 7–10 | Heck coupling | 70–92 | 111 | |
| Suzuki coupling | 85–98 | ||||
| Sonogashira coupling | 90–95 | ||||
| Stille coupling | 88–98 | ||||
| NPs | 1.6 ± 0.3 | Heck coupling | >99 | 112 | |
| Suzuki coupling | >99 | ||||
| NPs | 1.4–1.8 | Hydrolysis of ammonia borane | n.a. | 113 | |
| NPs | 1.4 ± 0.3 | Dehydrogenation of formic acid | >99 | 114 | |
| Pt | NPs | 3.7 ± 0.7 | Oxidation of hydrazine | n.a. | 115 |
| Multioctahedra | 40 ± 4 | Reduction of oxygen | >99 | 116 | |
| Cubic | 12.3 ± 1.4 | Hydrogenation of benzene | n.a. | 117 | |
| Cuboctahedra | 13.5 ± 1.5 | n.a. | |||
| Tetrahedra | 4.8 ± 0.1 | Electron-transfer between hexacyanoferrate(III) and thiosulfate ions | n.a. | 66 | |
| Cubes | 7.1 ± 0.2 | n.a. | |||
| NPs | 4.9 ± 0.1 | n.a. | |||
| Nanoplates | 1.0 ± 0.2 | Oxidation of 2-propanol | 85 | 71 | |
| Rh | Nanoclusters | 0.4 ± 0.06 | Hydrogenation of cyclohexene | >99 | 118 |
| NPs | ∼1.3 | Hydrogenation of phenols | >99 | 119 | |
| NPs | ∼1.2–3.0 | Steam reforming of ethanol | >99 | 120 | |
| NPs | 1.9–4.9 | Hydrodechlorination of 4-chlorophenol | >99 | 121 | |
| Tetrahedra | 4.9 ± 0.4 | Hydrogenation of arenes | >99 | 122 | |
| NPs | 4.8 ± 0.4 | >99 | |||
| Ru | NPs | <3.0 | Hydrogenation of benzene | >99 | 123 |
| NPs | 5.3 ± 0.8 | Synthesis of ammonia | n.a. | 124 | |
| NPs | 2.3–10.2 | Conversion of synthesis gas | 68 | 125 | |
| NPs | 2.1–6.0 | Oxidation of carbon monoxide | n.a. | 126 | |
| NPs | 1.0–1.2 | Oxidation of D-glucose | >99 | 127 | |
| Ir | NPs | 1.0–3.0 | Hydrogenation of cyclohexene | >99 | 128 |
| NPs | 1.5–5.0 | Hydrogenation of 1-decene | >99 | 129 | |
| NPs | 2.0–3.0 | Hydrogenations of arenes and ketones | >99 | 130 | |
| NPs | ∼3.5 | Phenylborylation | 91 | 131 | |
| NPs | 3.3 ± 1.7 | Hydrogenation of cyclohexene and phenylacetylene | >99 | 132 |
The use of controlled metallic nanoparticles in catalysis has been recently reviewed by Schüth and Jia.19 The authors presented several strategies for the synthesis of colloidal metal NPs with controlled sizes and shapes as well as their applications in catalysis. The authors also discussed strategies to generate supported metal catalyst with pre-synthetized colloidal metal particles as a component and embedded metal catalysts, in which the colloidal NPs are isolated inside pores of the support material.19 Here, we aim at providing an update on the progress and examples involving the control over shape, composition and size of NPs for catalytic applications. We also include in our discussion catalysts in which the active phase is present as single atoms, which recently gave rise to the field of single-atom catalysis. Moreover, we discuss nanocatalysts based on hollow nanomaterials obtained by galvanic replacement. These materials allow one to tailor both electronic and geometric effects, and maneuver more than one physical/chemical parameter that define the nanocatalyst at a time, enabling unique possibilities for improved performances.29–33
As pointed by Cuenya and Behafarid,14 new catalytic properties encompassing significantly enhanced reactivities and selectivity have been reported for metal NPs. In particular, the authors discussed how, guided by the intuitive explanation based on surface-to-volume ratios, the progressive decrease in the size is a powerful tool to maximize surface area and thus enhance the catalytic performances.23–25,34,35 In addition, it was discussed how nanoparticle size and shape strongly influences the chemisorption of substrates, intermediates, and products, therefore influencing catalytic properties.14 For example, quantum size-effects and the presence of under-coordinated surface can play an important role over catalytic properties as the NPs size is decreased.14 The control over shape, on the other hand, enables the exposure of distinct atomic arrangements at the surface, which display different chemisorption features.14 While the authors reviewed some experimental approaches for the synthesis of controlled NPs and the role of size, shape, and metal–support interactions in nanocatalysis, we also aim to discuss herein how this knowledge can serve as the basis for the control and understanding in systems where more than one physical and chemical parameter can be controlled at a time, such as hollow nanoparticles obtained by galvanic replacement reaction.
In this context, in addition to the control over size, shape, and composition, the fabrication of metal NPs with hollow interiors have gained increased attention in nanocatalysis due to the possibility of creating active sites in the NPs.15,16,36–38 The control over the NPs structure by the introduction of hollow interiors allows for higher surface-to-volume ratios relative to solid counterparts. This enables one to reduce the loading of noble metals and avoid the presence of “wasted” atoms that do not come into contact with substrate molecules (atoms in the interior of a solid nanoparticle).39–45 Basically, a hollow nanomaterial is characterized by the formation of a hole or void in its interior, and the precise understanding on how this hole affects the catalytic performance remains challenging.46 With the creation of holes, an increase in the number of active sites is achieved due to larger surface areas and the fact that such structures can allow external reactants to access the interior of the nanostructures.15 Interestingly, it has been also established that hollow nanomaterials allow the formation of more reactive inner sites associated with the exposure of highly reactive sites including stacking faults, twin planes, and high index facets, which makes hollow nanomaterials promising candidates in the context of nanocatalysis.46–48 Because of these features, several well-defined hollow nanomaterials have been developed and employed in nanocatalysis as illustrated in Fig. 1. These include tubes, shells, cages, frames, dendrites, among others.47,49–63
 | ||
| Fig. 1 (A–X) SEM and TEM images showing examples of hollow nanomaterials employed as nanocatalysts towards a variety of molecular transformations. Adapted from ref. 27, 28, 47, 49–63 and 94 with permission. | ||
Lee et al.15 and Yu et al.16 recently reviewed important strategies for the synthesis of hollow nanomaterials. In addition to the syntheses, the authors discussed how the electrocatalytic performances were dependent on the materials physical and chemical features. While Yu et al.16 mainly focused on the synthesis of 1D hollow and alloyed nanotubes, Lee et al.15 also covered several NPs systems. In this case, the discussed experimental strategies for the synthesis of the hollow nanoparticles included heteroepitaxial growth followed by template removal, non-epitaxial growth followed by template removal, Kirkendall effect, and galvanic replacement reaction. In both these review articles, the authors targeted application of these controlled nanostructures in the field of electrocatalysis.
In this review, we focus on some important aspects concerning the physical and chemical control of noble metal NPs for applications in nanocatalysis. Our goal is to provide an update regarding the developments of the field and highlight the fundamentals of how their well-defined physical and chemical parameters affect their catalytic performances. We focus our discussion on representative studies that clearly demonstrate these concepts and relationships between performance and physical/chemical features. We begin with an account of the most traditionally employed strategies for optimizing catalytic performances in noble metal NPs: control over NPs shape, composition, and size. In these examples, only one of these properties is varied at a time, followed by the investigation of their influence on catalysis. We then focus on NPs with hollow interiors as an alternative approach to achieve high catalytic performances. In these systems, we will illustrate that we can maneuver more than one physical/chemical parameter that defines the NPs at a time, which opens further possibilities for the optimization of performances. Specifically, we provided a case-by-case discussion showing how, in addition to the hollow interiors, the control over their composition, size, and surface morphology relate to their catalytic properties and performance vs. physical/chemical features relationships.
2. Controlling shape, composition, and size in noble-metal NPs
Shape-controlled NPs have been widely employed as model systems to unravel facet-dependent catalytic performances, which are referred to as geometric effects. It has been demonstrated that there is a strong dependence between performance (both activity and selectivity) and shape, which have been demonstrated by both computational and experimental studies.64–67 There is a consensus that catalytic activity and selectivity is dependent on the atomic arrangement of the surface, which in turn can regulate adsorption energies and geometries of substrate molecules and intermediates during a molecular transformation.64–67 It is noteworthy that geometric effects that are important in nanocatalysis also include (in addition to the surface arrangement) the presence of steps, kinks, and atoms with low coordination numbers relative to atoms with high coordination numbers.68As a notable example for demonstrating how shape can affect the catalytic activity (geometric effects), we highlight the formic acid electrooxidation in the presence of Pd nanomaterials having different proportions of {100} and {111} facets exposed at their surfaces, as depicted in Fig. 2.69 In this case, a seed-mediated approach using preformed Pd nanocubes as the seeds for additional Pd deposition via the reduction of Na2PdCl4 in the presence of formaldehyde (considered a relatively mild reducing agent) was employed.70 By simply controlling the ratio between Na2PdCl4 and the Pd seeds, well-defined nanomaterials were obtained in high yields.70 This included cubes (Fig. 2A), truncated cubes (Fig. 2B), cuboctahedrons (Fig. 2C), truncated octahedrons (Fig. 2D), and octahedrons (Fig. 2E). Thus, as the obtained nanomaterials presented well-defined shapes and a systematic difference in the proportion of {111} to {100} facets, they could be employed as proof-of-concept systems to the precise understanding over the relationship between shape and the catalytic performance (Fig. 2F). The catalytic experiments showed that activity increased with the exposure of {100} surface facets (that are more active towards the formic acid electrooxidation compared to {111} facets).57 Specifically, a gradual increase in the maximum current density was observed when truncated cubes, cuboctahedrons, truncated octahedrons, and octahedrons were used as electrocatalysts. Here, following this sequence of shapes, a progressive increased in the exposure of {111} facets is observed, which justified the decrease in the catalytic performance due to the lower activity of the {111} facets compared to {100}.57
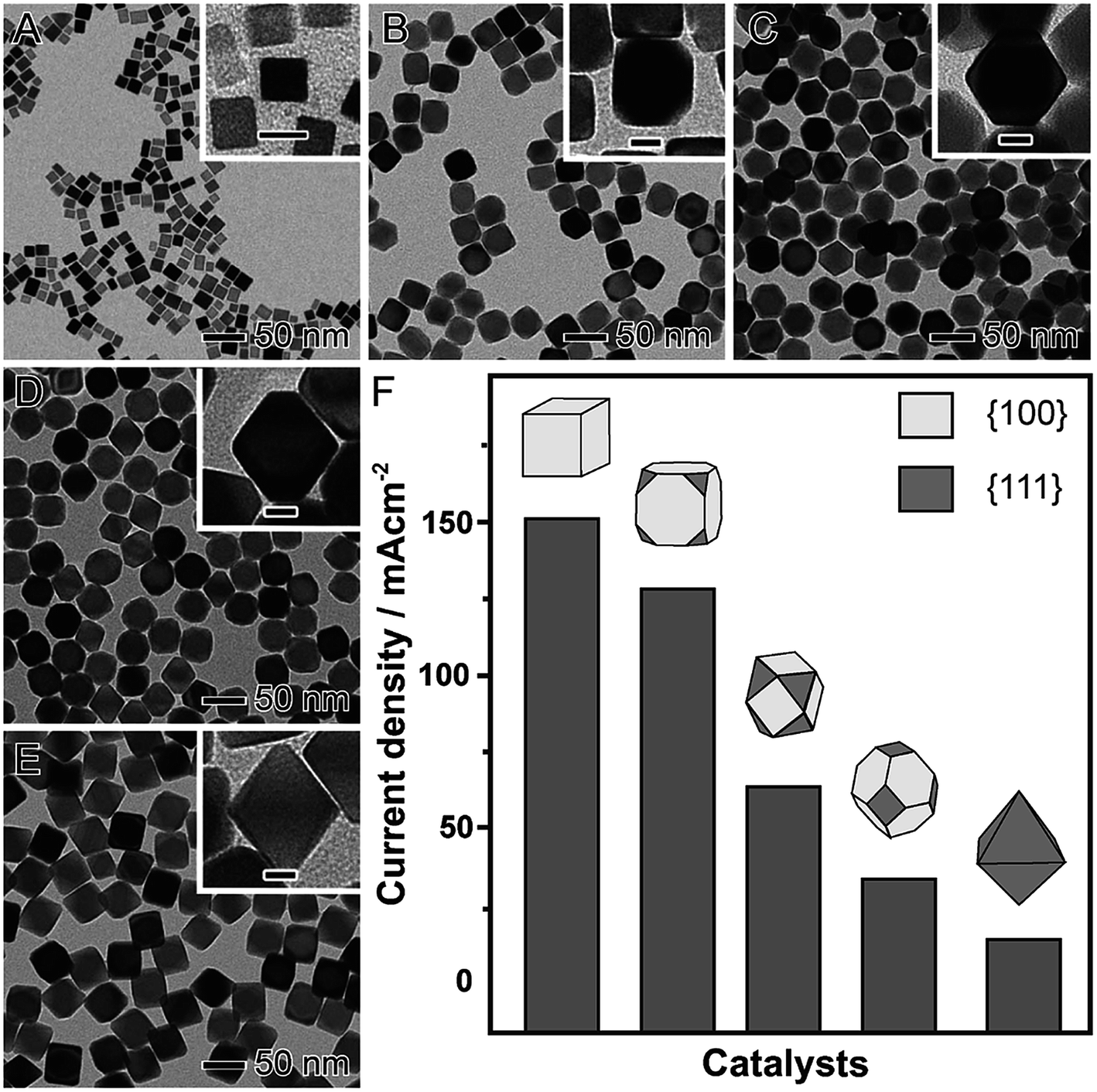 | ||
| Fig. 2 Effect of NPs shape over catalytic activity. TEM images of Pd nanomaterials having controlled shapes enabling a progressively increased exposure of {111} surface facets relative to {100}: cubes (A), truncated cubes (B), cuboctahedrons (C), truncated octahedrons (D), and octahedrons (E). The scale bars in the insets correspond to 10 nm. (E) Maximum current densities for formic acid oxidation employing Pd nanomaterials as electrocatalysts. Adapted from ref. 69 with permission. | ||
Furthermore, the control over nanoparticle composition (electronic effects) has also been effective towards the optimization of catalytic activities and selectivity.66,67,71,72 In this case, the electronic effects dictate the electronic status of active sites, which be adjusted by changing the local composition, for example.68 This is imperative for allowing the activation of the reactants while not enabling the binding of reaction intermediates or products too strongly to prevent poisoning of active sites (Sabatier principle).68,73 The electronic effect can, therefore, lead to a change in the electronic structure of an active site catalyst, changing the way that its d-band interacts with the molecules/adsorbates and therefore changing the binding strength.74,75 In this context, the synthesis of multimetallic nanoparticles represents an effective strategy to modify the electronic structure of metal nanoparticles and, consequently, enhance their catalytic properties.27,38 In the past few years, many studies have been reported on the correlation between geometric and electronic effects with catalytic activity. Traditionally, these effects are not easily decoupled from each other in dictating enhancement of catalytic activity of metal NPs.76 However, important contributions have been made recently regarding the understanding of the role played by electronic and geometric effects.76,77 In fact, this understanding represents an important challenge to modulate the performances of catalytic NPs. For example, Liang et al. recently reported on the synthesis of two Pt–Cu catalysts that served as model systems to investigate the contribution from each of these effects over the selective hydrogenation of p-chloronitrobenzene by Pt nanoparticles.77 It was found the electronic effect from Cu to Pt played a crucial role over the catalytic performance, while the geometric effect led to weak catalytic activity in this system.77
It is clear that composition strongly affects catalytic activities in noble-metal NPs.27,38,64,78–80 Considering the large applicability of catalysis, it can be stated that even slight changes in the composition of a catalyst (reducing the loading of a more expensive metal, for example) can have large economic and environmental impacts. In this context, the substitution of noble metals (Pt, Pd, Ru, Rh, and Ir) by less expensive ones (Cu, Fe, Ni, Co, etc.) or the enhancement on their properties by addition of other elements are highly desired. However, the origin of the improvement of catalytic activity coming from the difference of composition is very difficult to be unraveled (electronic effect). For example, in these systems, while the term “synergism” has been vastly explored, its origin remains unclear in several studies.
There are several examples in which multimetallic nanomaterials enabled one to enhance significantly catalytic performances as compared to the monometallic counterparts. It has been proposed that compositional variations in multimetallic nanomaterials directly affected the electronic state of their constituents and, consequently, their reactivity, adsorption energies, and geometries of substrate molecules, and thus catalytic behavior. However, compositional variations can also affect spatial arrangement and atomic ordering, which leads to significant changes in the properties as a consequence of surface segregation, faceting, diffusion, and others.81
In order to illustrate the effect of the composition on the catalytic activity, we selected two proof-of-concept examples. In the first, as shown in Fig. 3, PtxCu100−x nanocubes were prepared by a co-reduction approach, in which platinum(II) acetylacetonate ([Pt(acac)2]) and copper(II) acetylacetonate ([Cu(acac)2]) precursors were simultaneously reduced by oleylamine in the presence of 1-octadecene, 1,2-tetradecanediol, tetraoctylammonium bromide, and 1-dodecanethiol.82 In this approach, by adjusting the molar ratio between the metal precursors, nanocubes with different compositions were synthetized in the range of 20 up 46 at% for Cu (Fig. 3A–E). These systems were evaluated as electrocatalysts towards the formic acid oxidation (Fig. 3F). A decrease in the activity was observed when the amount of Cu was increased, probably due to the superior activity of Pt as compared to Cu. However, an interesting behavior was observed: the Pt80Cu20 nanocubes (20 at% of Cu) was the best electrocatalyst even comparing with pure Pt nanocubes with similar sizes. In fact, it showed showing superior activity, stability, and CO tolerance.82 The enhancement in the performance of this nanomaterial was associated with the formation of an alloyed structure displaying a lower tendency for the OH− chemisorption, which is a well-stablished poison for formic acid oxidation. This hypothesis was evidenced by a shift of the onset potentials of OH− electrosorption to more positive values.83
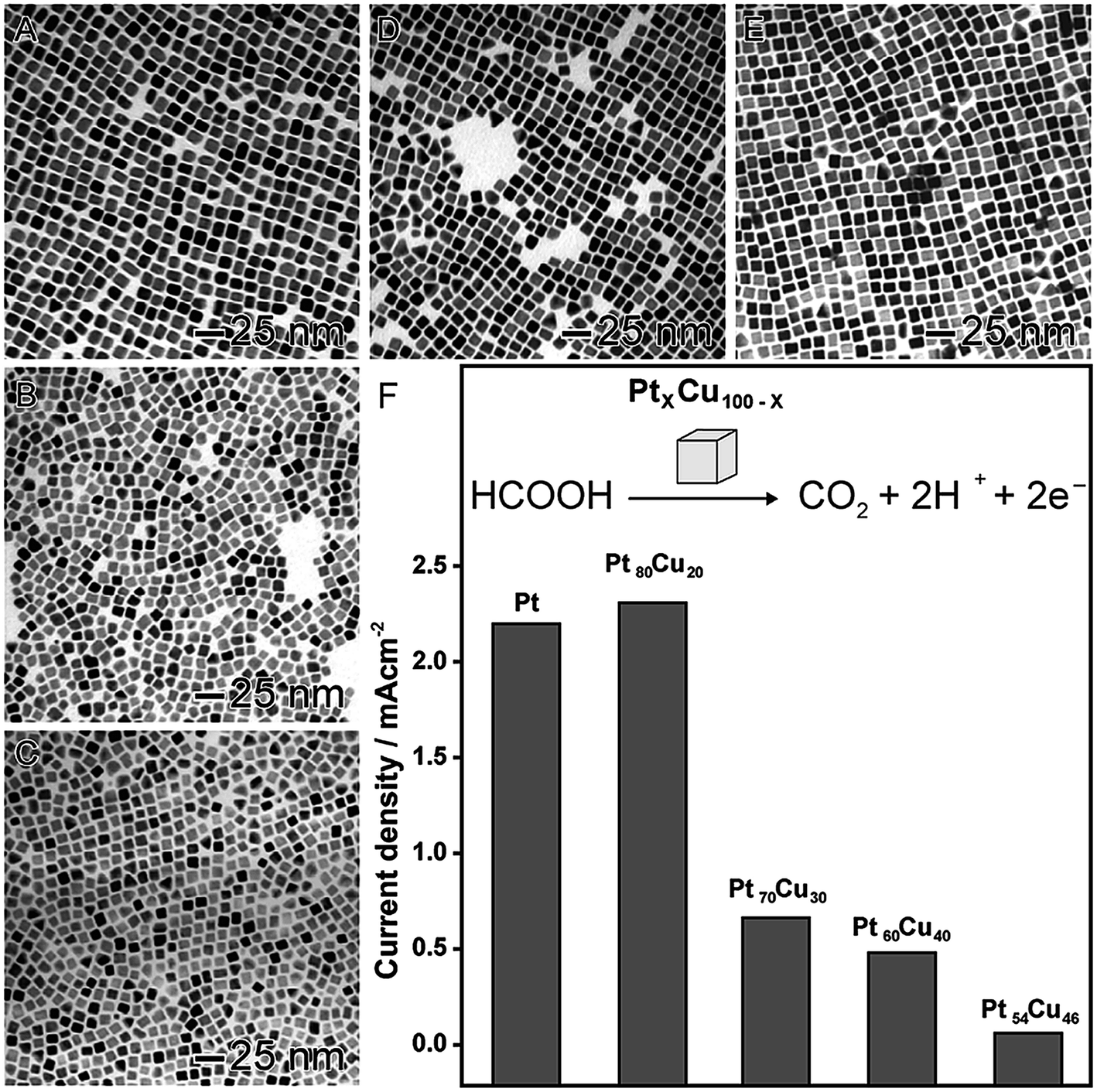 | ||
| Fig. 3 Effect of NPs composition over catalytic activity. TEM images of bimetallic PtxCu100−x nanocubes with controlled compositions: Pt100 (A), Pt80Cu20 (B), Pt70Cu30 (C), Pt60Cu40 (D), and Pt54Cu46 (E). (F) Maximum current densities for formic acid oxidation employing PtxCu100−x nanocubes as electrocatalysts. Adapted from ref. 82 with permission. | ||
In the second example (Fig. 4), AgxPd100−x nanomaterials displaying controlled compositions and narrow size distributions were also obtained by the co-reduction of the respective Ag and Pd precursors (AgNO3 and Pd(acac)2) using octadecylamine as both reducing agent and solvent in the absence of any other stabilizer or capping agent.84 This led to the formation of spherical nanoparticles with controlled compositions (Fig. 4A–E). The AgxPd100−x nanomaterials were employed as nanocatalysts towards hydrodechlorination of 4-chlorophenol under room temperature and atmospheric pressure, in which the effect of the nanomaterial composition showed a strong influence in the catalytic behavior. In terms of turnover frequency, a volcano-like profile was detected, in which the AgPd nanomaterial displaying 90 at% of Pd presented the best activity. This behavior was assigned considering the effect of each component together with a mechanism in which Ag atoms are responsible for the adsorption the chlorinated substrate and the cleavage of the C–Cl bond.66 Furthermore, Pd atoms aided the H2 dissociation in order to supply the H atoms to regenerate metallic Ag from AgCl.84
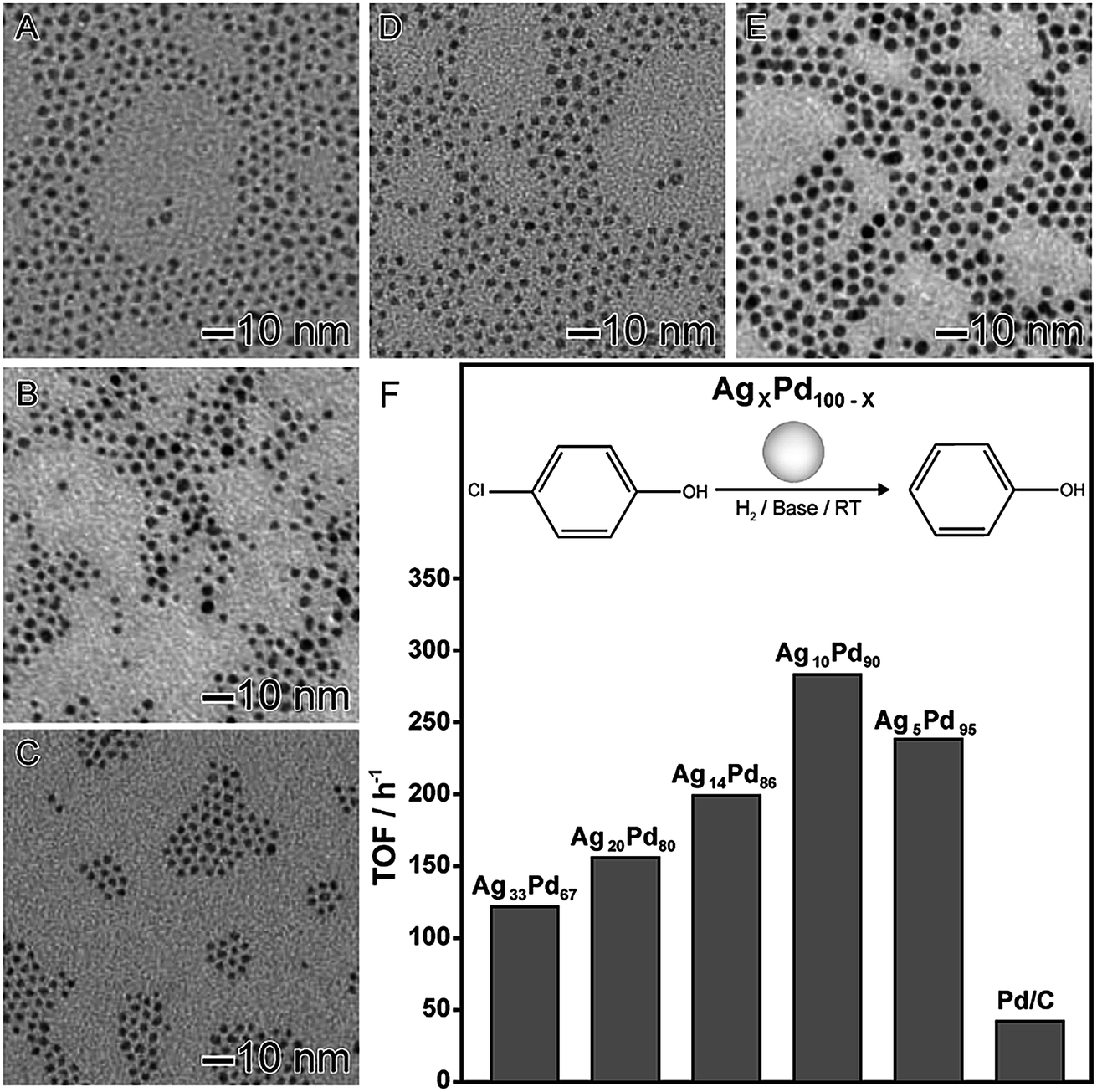 | ||
| Fig. 4 Effect of NPs composition over catalytic activity. TEM images of AgxPd100−x NPs with controlled compositions: Ag33Pd67 (A), Ag20Pd80 (B), Ag14Pd86 (C), Ag10Pd90 (D), and Ag5Pd95 (E). (F) Turnover frequencies (TOFs) in the first hour for the hydrodechlorination of 4-chlorophenol catalyzed by AgPd nanomaterials with controlled compositions. Adapted from ref. 84 with permission. | ||
The control over NPs size perhaps represents the most primitive and intuitive property in the context of synthesis, characterization, and application of nanomaterials in catalysis. This is because nanocatalysis is a surface phenomenon, in which molecules and active sites interact intimately as the reaction proceeds. Thus, in the context of the catalytic reaction itself, only surface atoms are considered active and, therefore, in addition to their reactivity, their availability relative to the atoms in the interior of the nanoparticle is crucial.24,34,58,85–87 Hence, it is not a surprise that the size-controlled synthesis of metal nanomaterials is an important strategy to improve the catalytic activity per mass unit of metal. In addition to increased surface areas, size can also enable the exposure of more reactive sites, such as surface defects, low-coordination, and unsaturated atoms.88,89
In order to illustrate these features, Rh nanomaterials displaying controlled sizes were quickly synthesized (15 minutes) by a microwave-assisted polyol method, in which different polyalcohols were employed as reducing agents under the same synthetic conditions, as shown in Fig. 5.90 More specifically, Rh3+ ions were reduced using ethanol (Fig. 5A), ethylene glycol (Fig. 5B), diethylene glycol (Fig. 5C), 2-ethoxyethanol (Fig. 5D), triethylene glycol (Fig. 5E), or 2-(2-ethoxyethoxy)ethanol (Fig. 5F) as reducing agents and PVP as the stabilizer.90 Polyalcohols with higher reduction potentials led to the formation of smaller Rh nanomaterials because of faster precursor reduction rate.17 After incorporation of the obtained Rh nanomaterials (1 wt% Rh basis) onto γ-Al2O3 support by a wet-impregnation method, their catalytic activities were investigated in the CO oxidation reaction as a function of size. Interestingly, no aggregation or sintering of Rh nanomaterials was observed after the wet-impregnation step and a good correlation between size and performance could be established.90Fig. 5G shows CO oxidation profiles as a function the temperature and each Rh nanocatalyst. As expected, the catalytic activity increased as NPs size decreased. These results were supported by CO chemisorption data, which showed a progressive increase in the number of Rh exposed atoms with the decrease in NPs size. As depicted in Fig. 5H, larger nanoparticles also showed higher temperatures for total CO conversion due to their lower catalytic activity.
 | ||
| Fig. 5 Effect of NPs size over catalytic activity. HAADF-STEM images of Rh nanomaterials with controlled sizes obtained using ethanol (A), ethylene glycol (B), diethylene glycol (C), 2-ethoxyethanol (D), triethylene glycol (E), and 2-(2-ethoxyethoxy)ethanol (F) as reducing agents. (G) Profiles of CO conversion as a function of the temperature employing Rh nanomaterials with controlled sizes as catalysts. (H) Bar graph showing the temperature in which 100% of CO conversion is achieved for each Rh nanomaterial. Adapted from ref. 90 with permission. | ||
Regarding the effect of size in nanocatalysis, it can be stated that the ultimate limit for size reduction is the use of single atoms as active sites. In fact, recent developments in synthesis, characterization, and modeling enabled the area of single-atom catalysis to emerge as a novel frontier in heterogeneous catalysis (and nanocatalysis).30–33 Single-atom nanocatalysts can be defined as systems that integrate spatially isolated metal atoms on appropriate host matrices.32,91 Therefore, these catalysts uniquely allow for the maximum utilization per metal atom while also being easily recovered and re-used (taking advantages of the most attractive features of both heterogeneous and homogeneous catalysis). Furthermore, they can present different reactivities and reaction pathways as compared to both nanoparticles and metal complexes analogs. In these systems, the support not only acts as a dispersing agent for the active phase atoms but also play an important role in tuning catalytic properties.29,30,33
One of the pioneering examples of single-atom catalysis is a system composed of Pt atoms dispersed in a FeOx matrix (denoted Pt1/FeOx).92 This catalyst was obtained by a co-precipitation method. In this case, low amounts of Pt precursor were added into an aqueous solution containing a Fe precursor. In a second step, both Pt and Fe were precipitated with Na2CO3 under controlled temperature and pH conditions.92 Here, two Pt/Fe atomic ratios were employed, which led to the formation of Pt1/FeOx catalysts with 0.17 and 2.5 wt% in Pt basis. More specifically, 0.17 wt% was achieved using a Pt/Fe atomic ratio of 1/1430 (sample A) and 2.5 wt% was achieved using a Pt/Fe atomic ratio of 1/95 (sample B). Fig. 6A, B and C, D show HAADF-STEM images of samples A and B, respectively, in which it can be observed that individual Pt atoms were uniformly incorporated in the FeOx structure (white circles). For the sample A (0.17 wt% in Pt basis), individual Pt atoms were uniformly incorporated in the FeOx structure, in which Pt atoms occupy the same positions of the Fe atoms. When the amount of Pt was increased to 2.5 wt% (sample B), besides isolated atoms (white circles), other Pt-based structures were also detected including rafts (black circles) and clusters (white squares). However, Pt nanomaterials with sizes above 2 nm were not identified, indicating the robustness of this reported approach.92 These Pt1/FeOx nanomaterials were evaluated towards both CO oxidation and preferential oxidation of CO in H2 (PROX) reactions. As depicted in Fig. 6E, both Pt1/FeOx samples showed extremely high catalytic activities for both CO oxidation and PROX reactions compared to the commercial Au/Fe2O3 catalyst (which was used as a standard to benchmark performance). Here, the authors proposed that the formation of Pt single-atoms are responsible for the increase in the number of vacant d orbitals due to the electron transfer from Pt atoms to the FeOx support, leading to the formation of positively charged Pt atoms, which are highly active and also contribute to the stabilization of the Pt atoms due to their strong binding with the FeOx support.92
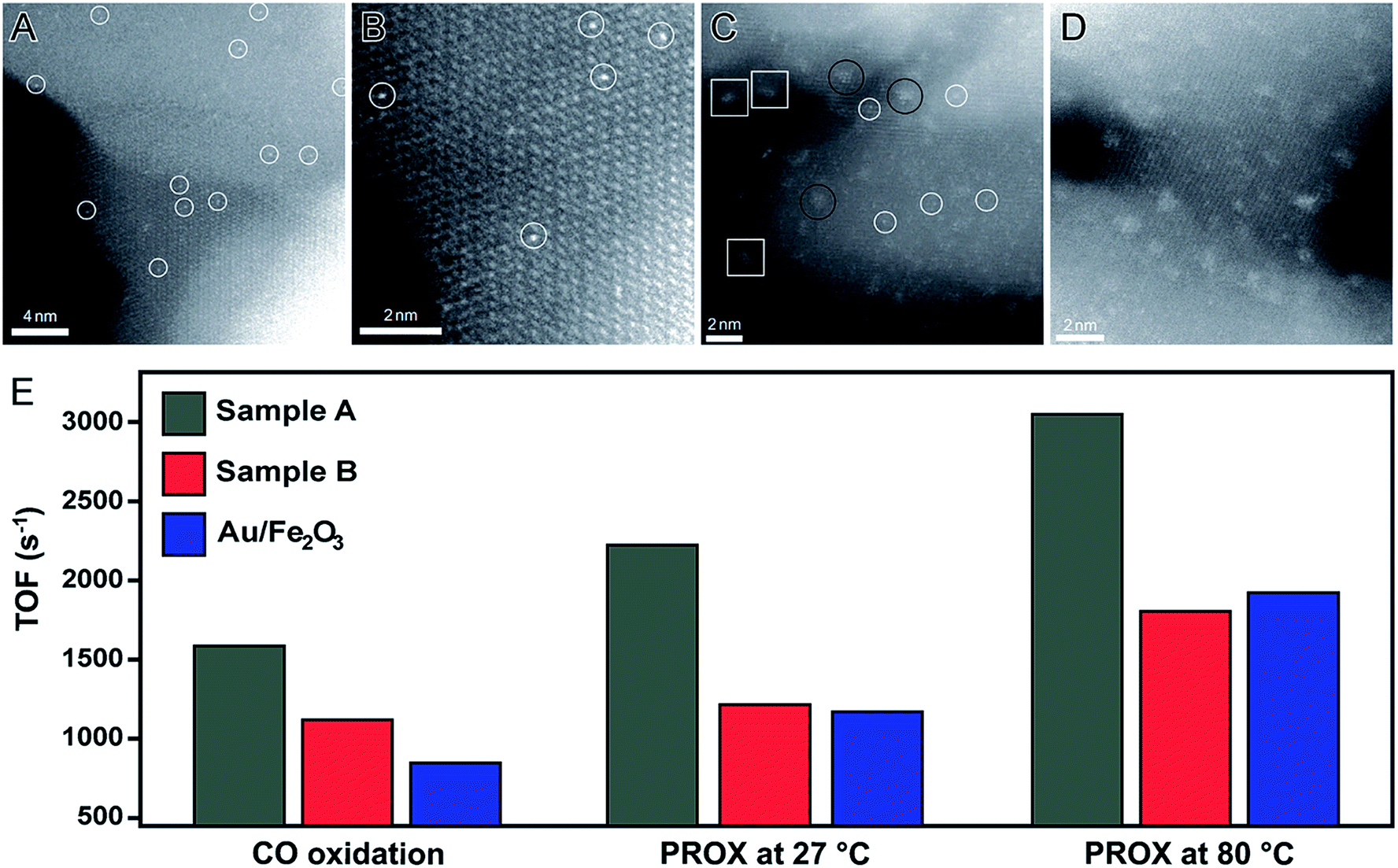 | ||
| Fig. 6 Effect of NPs size over catalytic activity. HAADF-STEM images of Pt single atoms (white circles) dispersed on FeOx support obtained using a Pt/Fe atomic ratio of 1/1430 (0.17 wt% of Pt, (A and B)) and 1/95 (2.5 wt% of Pt, (C and D)). (E) Bar graph showing the turnover frequencies towards CO oxidation and PROX reaction at 27 and 80 °C employing Pt1/FeOx as catalysts. Commercial Au/Fe2O3 was also included for comparison. Adapted from ref. 92 with permission. | ||
3. Hollow nanomaterials obtained by chemical templating: controlling more than one physical/chemical parameter at a time
In the previous sections, the development of controlled nanomaterials for the optimization of catalytic activities focused on the control over one parameter at a time, such as shape, composition, and size. However, it is plausible that major optimizations and understanding in nanocatalysis may require the manipulation of more than one physical/chemical parameter that defines a nanocatalyst at a time.6,58,93 The synthesis and utilization of hollow nanomaterials obtained by chemical templating may offer a good opportunity to move towards this goal. The galvanic replacement reaction, for example, represents a well-established and versatile method to the synthesis of hollow materials having bimetallic compositions and ultrathin walls in one step.38,41,94 It consists of a redox reaction between a less noble metal, employed as a sacrificial template, and metal ions in solution (more noble metal relative to the sacrificial template). Here, the differences in the electrical reduction potential drive the oxidation and dissolution of the sacrificial template together with the reduction of the metal ions in solution and its deposition at the surface of the template. This method is facile, relatively fast (reactions can take place under 15 min), and can be employed using water as a solvent and ambient conditions.28,41,95,96It is important to note that several hollow nanomaterials obtained by galvanic replacement have been reported and employed in nanocatalysis, in which better performances have been observed and assigned to the increased surface areas and bimetallic compositions.38,41,46,79 In one example depicted in Fig. 7, AgAu-based nanoboxes, nanocages, and their solid counterparts were investigated as catalysts towards the reduction of 4-nitrophenol by NaBH4.97 As depicted in Fig. 7A, these materials were synthesized by a galvanic replacement reaction between Ag nanocubes and AuCl4−. By adding progressively increased amounts of AuCl4− during the galvanic replacement reaction with Ag nanocubes, three different Au-based hollow nanostructures were prepared: partially hollow nanoboxes (Fig. 7B), nanoboxes (Fig. 7C), and nanocages (Fig. 7D). As expected, the catalytic activity increased with the degree of hollowing (Fig. 7E), which enabled the superior specific surface area compared to solid nanomaterials. They also presented ultrathin and porous walls, which can present a much higher activity due to the size effect and exposure of reactive sites.97
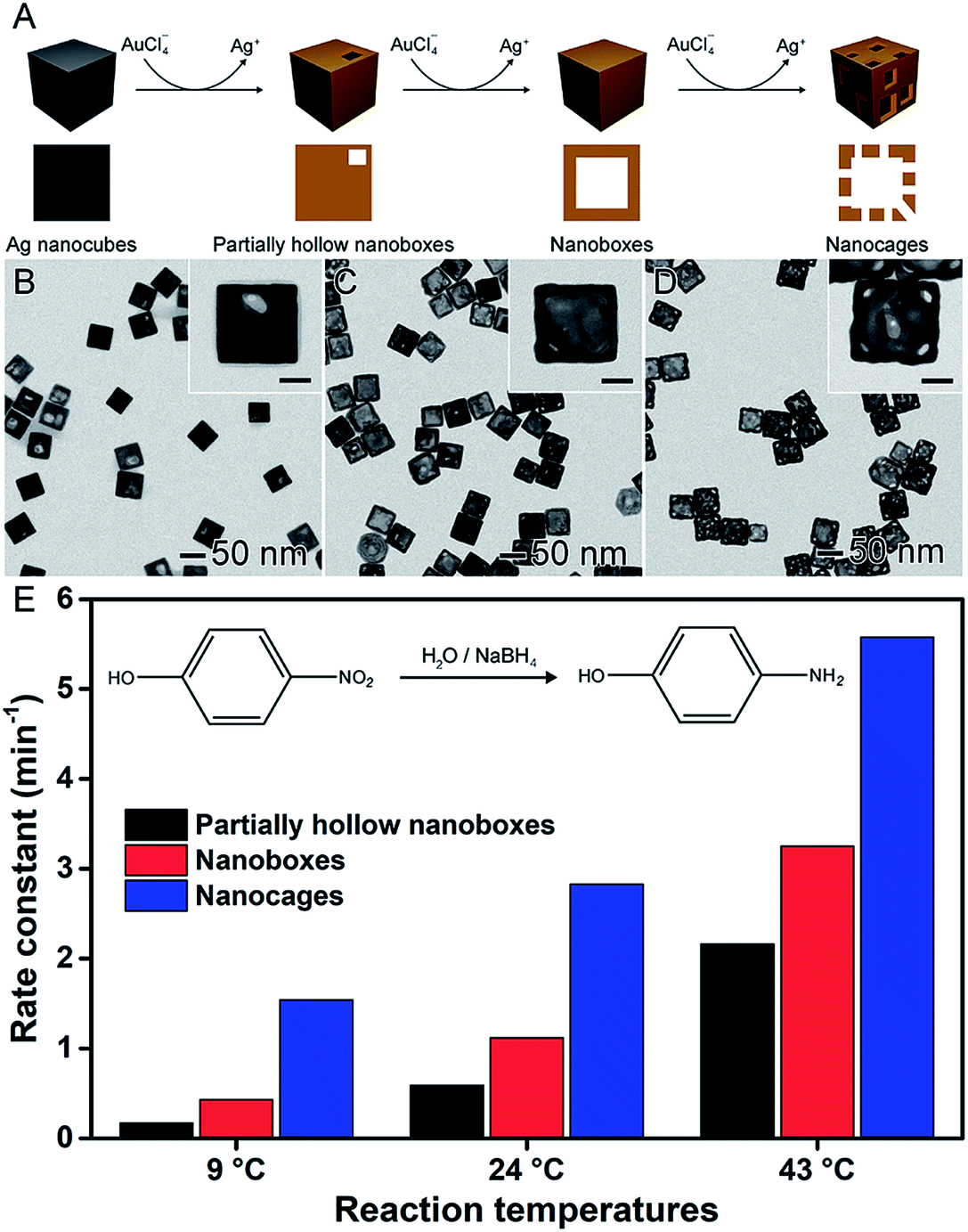 | ||
| Fig. 7 Effect of NPs structure (hollow interiors) over catalytic activity. (A) Scheme of the synthesis of Au-based hollow nanomaterials by the controlled galvanic replacement reaction between preformed Ag nanocubes and AuCl4− precursor. (B–D) TEM images of partially hollow nanoboxes (B), nanoboxes (C), and nanocages (D). The scale bars in the insets correspond to 20 nm. (E) Bar graph showing the rate constants towards the reduction of 4-nitrophenol using NaBH4 at 9, 24, and 43 °C as the reaction temperature employing Au-based hollow nanomaterials as catalysts. Adapted from ref. 97 with permission. | ||
In another example, the synthesis of AgAu nanorings by the galvanic replacement reaction between preformed Ag nanospheres and AuCl4(aq)− precursor was performed using water as solvent and PVP as a stabilizer, as depicted in Fig. 8A. Here, the AgAu nanorings could be formed via the precise control over the Ag/AuCl4(aq)− molar ratios (Fig. 8B).41,81,98,99 In order to investigate the catalytic activity of the produced AgAu nanorings, the 4-nitrophenol reduction was employed as a model reaction comparing to Au and Ag nanospheres with similar sizes as shown in Fig. 8C and D, respectively. In this case, probably due to their superior specific surface area, the AgAu nanorings displayed significantly enhanced performances relative to the both Ag and Au nanospheres (Fig. 8E–G). More specifically, considering the parameters obtained from the microscopic analyzes and the metal loading employed in the catalytic experiments, the calculated surface area of the nanostructures corresponded 1630, 1570, and 5100 m2 mol−1 for Ag nanospheres, Au nanospheres, and AgAu nanorings, respectively. Thus, the surface area for the nanorings was 3.1 and 3.2-fold higher relative to Ag and Au NPs, respectively, which reinforces the superior catalytic activity. Finally, the AgAu nanorings re-used for ten rounds without any loss of activity (Fig. 8H).
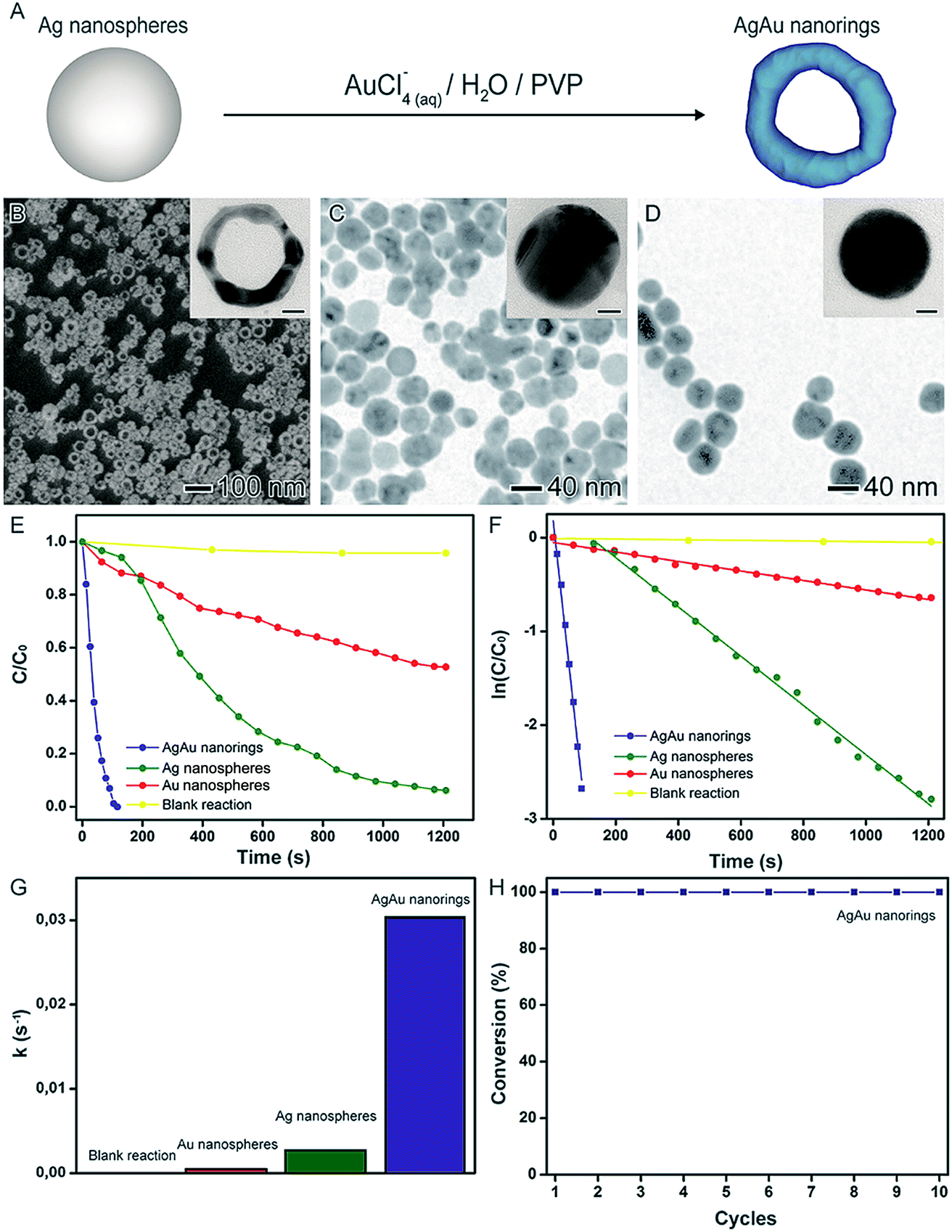 | ||
| Fig. 8 Effect of NPs structure (hollow interiors) over catalytic activity. (A) Scheme for the synthesis of AgAu nanorings by galvanic replacement reaction between Ag nanospheres and AuCl4(aq)−. SEM (B) and TEM (C and D) images of AgAu nanorings (B), Ag NPs (C), and Au NPs (D). The insets in the figures correspond to their respective HRTEM images (scale bars = 10 nm). (E) C/C0 and (F) ln(C/C0) profiles as a function of time employing of AgAu nanorings (blue trace), Ag NPs (green trace), and Au NPs (red trace) as catalysts. (G) Bar graph showing the pseudo-first-order rate constants calculated from (F). (H) Stability tests employing AgAu nanorings as catalysts. Adapted from ref. 94 with permission. | ||
In its simplest form, the galvanic replacement reaction allows us to control the degree of hollowing and thus surface area as well as surface composition (controlling the molar ratio between sacrificial templates and precursor metal ions). In this context, by using different metal ions as precursors, we can further control composition to enable the synthesis of trimetallic nanomaterials with hollow interiors. Besides, by controlling the shape of the metal employed as sacrificial templates, we can also control the shape of the final material (in addition to the degree of hollowing and composition). Finally, by coupling galvanic replacement with co-reduction, the surface morphology can be maneuvered in addition to the degree of hollowing, shape, and surface composition. In the next sections, we will highlight some examples in which the galvanic replacement reaction was employed to develop controlled nanomaterials maneuvering more than one physicochemical parameter at a time. This will be followed by their application as model systems in nanocatalysis targeting optimization of performances and the establishment of properties relationships.
3.1. Controlling composition in hollow NPs: from bimetallic to trimetallic systems
The synthesis of trimetallic nanomaterials represents an interesting strategy in nanocatalysis.27,42 Nevertheless, the exact role played by each metal component in the overall catalytic performances remains unclear. In most cases, in addition to different compositions, the reported catalysts have a large range of size distribution and poor control over shapes, which makes difficult to isolate the influence of the composition itself over the observed performances. In order to address this challenge, the galvanic replacement reaction represents an interesting approach. For instance, in the galvanic reaction, control over composition to generate trimetallic systems can be performed by using more than one metal ion precursor and adjusting the molar ratio among the precursors that will give rise to the desired nanomaterial.40,41,79Based on this strategy, the synthesis of trimetallic nanoshells has been recently reported by our group.27 We focused on trimetallic nanoshells having AgAuPd, AgAuPt, and AgPdPt compositions and compared their performances and their AgAu, AgPd, and AgPt bimetallic counterparts in order to gain insights on the role played by the addition of the third metal over the activities (Fig. 9).27 As depicted in Fig. 9A, bi- and trimetallic nanoshells with similar sizes and morphologies could be produced in a range of compositions by a sequential galvanic replacement approach, in which Ag nanospheres were employed as chemical/sacrificial templates in the presence of different amounts of the desired metal precursors (AuCl4(aq)−, PdCl4(aq)2−, and/or PtCl6(aq)2−). We selected three trimetallic nanoshells as model systems. In this case, the similar sizes and shapes allowed us to isolate the effect of the composition on the catalytic activity and evaluate the effect of the progressive addition of active components on a nanocatalyst. The following trimetallic nanoshells were selected: Ag56Au28Pd16 (Fig. 9B), Ag78Au9Pt13 (Fig. 9C), and Ag71Pd16Pt13 (Fig. 9D). For comparison, bimetallic nanoshells with the same atomic percentages of the active metals were also investigated (Ag72Au28 and Ag84Pd16; Ag91Au9 and Ag87Pt13; and Ag84Pd16 and Ag87Pt13). The 4-nitrophenol reduction was employed as a model reaction (Fig. 9E) and the positive effect of the composition, especially after the addition of the third metal, in the catalytic activities of all trimetallic nanoshells could be observed (Fig. 9F). Fig. 9F shows bar graphs representing the percentages of 4-nitrophenol reduction catalyzed by bi- and trimetallic nanoshells as a function of their atomic compositions: Ag, Au, and Pd (left); Ag, Au, and Pt (middle); and Ag, Pt, and Pd (right). In all cases, the trimetallic nanoshells showed superior catalytic activities compared to their bimetallic counterparts due to the addition of a third metal. The conversion percentages for the trimetallic nanoshells were superior to the sum of their bimetallic counterparts, illustrating the synergism of properties between the three metals relative to the bimetallic compositions.27 These results showed that the control over composition (moving from bi- to trimetallic) and structure (solid interiors) represents a promising strategy for boosting catalytic activities.
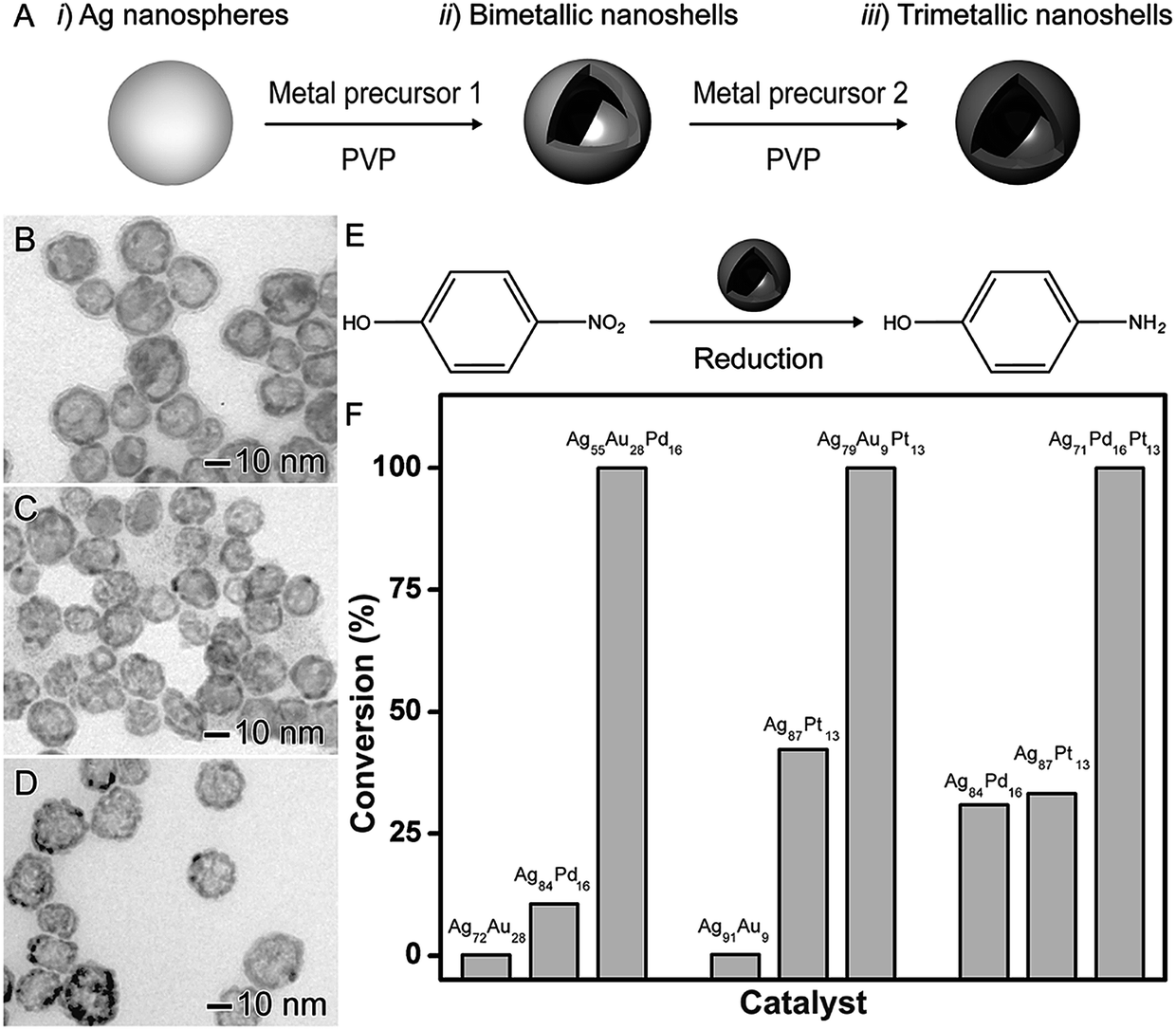 | ||
| Fig. 9 Effect of NPs structure (hollow interiors) and composition over catalytic activity. (A) Scheme for the synthesis of bimetallic and trimetallic nanoshells by sequential galvanic replacement reactions between Ag nanospheres and metal precursors. TEM images of Ag56Au28Pd16 (B), Ag78Au9Pt13 (C), and Ag71Pd16Pt13 (D) trimetallic nanoshells. (E) Scheme for the 4-nitrophenol reduction catalyzed by hollow metal nanomaterials. (F) Bar graphs showing the 4-nitrophenol conversion% as a function of the composition in bi- and trimetallic nanoshells having Ag, Au, and Pd (left), Ag, Au, and Pt (middle), and Ag, Pt, and Pd (right) compositions. Adapted from ref. 27 with permission. | ||
3.2. Controlling composition and surface morphology in hollow NPs: changing the size of Ag NPs as sacrificial templates
It is well-established that size plays an important role over catalytic activities of solid nanoparticles, in which activities usually increases with a decrease in size due to larger surface-to-volume ratios.24,34,100 In nanocatalysts comprised of hollow nanomaterials (such as nanoshells), these size variations may be more difficult to visualize. This is because, in addition to the outer diameter, shell thickness and surface morphology also influence the surface area. In order to better understand how outer diameter, shell thickness, and surface morphology collectively influence catalytic performances, the synthesis of AgPt nanoshells from the galvanic reaction with Ag NPs having different sizes was performed.58 They were prepared by a two-step synthesis. In the first step, Ag nanoparticles displaying controlled sizes were obtained by a seed-mediated synthesis. In the second step, the as-prepared Ag nanoparticles displaying controlled sizes are employed as templates in a galvanic replacement reaction using PtCl6(aq)2− precursor and hydroquinone as an auxiliary reducing agent. Here, the synthesis of AgPt hollow nanomaterials can be associated by the combination between the galvanic replacement process, which leads to the formation of hollow interiors due the Ag oxidation to Ag(aq)+ by PtCl6(aq)2−, and additional PtCl6(aq)2− reduction to Pt atoms by hydroquinone, which enables the Pt deposition at the surface of the Ag templates. By using this reported protocol, AgPt nanoshells with four different sizes were prepared: 95 ± 7, 105 ± 7, 133 ± 8, and 163 ± 11 nm, which were denoted AgPt 95 (Fig. 10A), AgPt 105 (Fig. 10B), AgPt 133 (Fig. 10C), and AgPt 163 nm (Fig. 10D), respectively. Interestingly, in addition to the outer diameters, by simply varying the sizes of Ag nanoparticles used as starting materials, we could also control other properties such as shell thickness and the surface morphology, which affected Pt surface area. Specifically, the surface area based on Pt (based on H2 chemisorption analyzes and HRTEM analyzes) increased with the size of the AgPt nanoshells because larger nanoshells were comprised of smaller Pt islands at their surface.58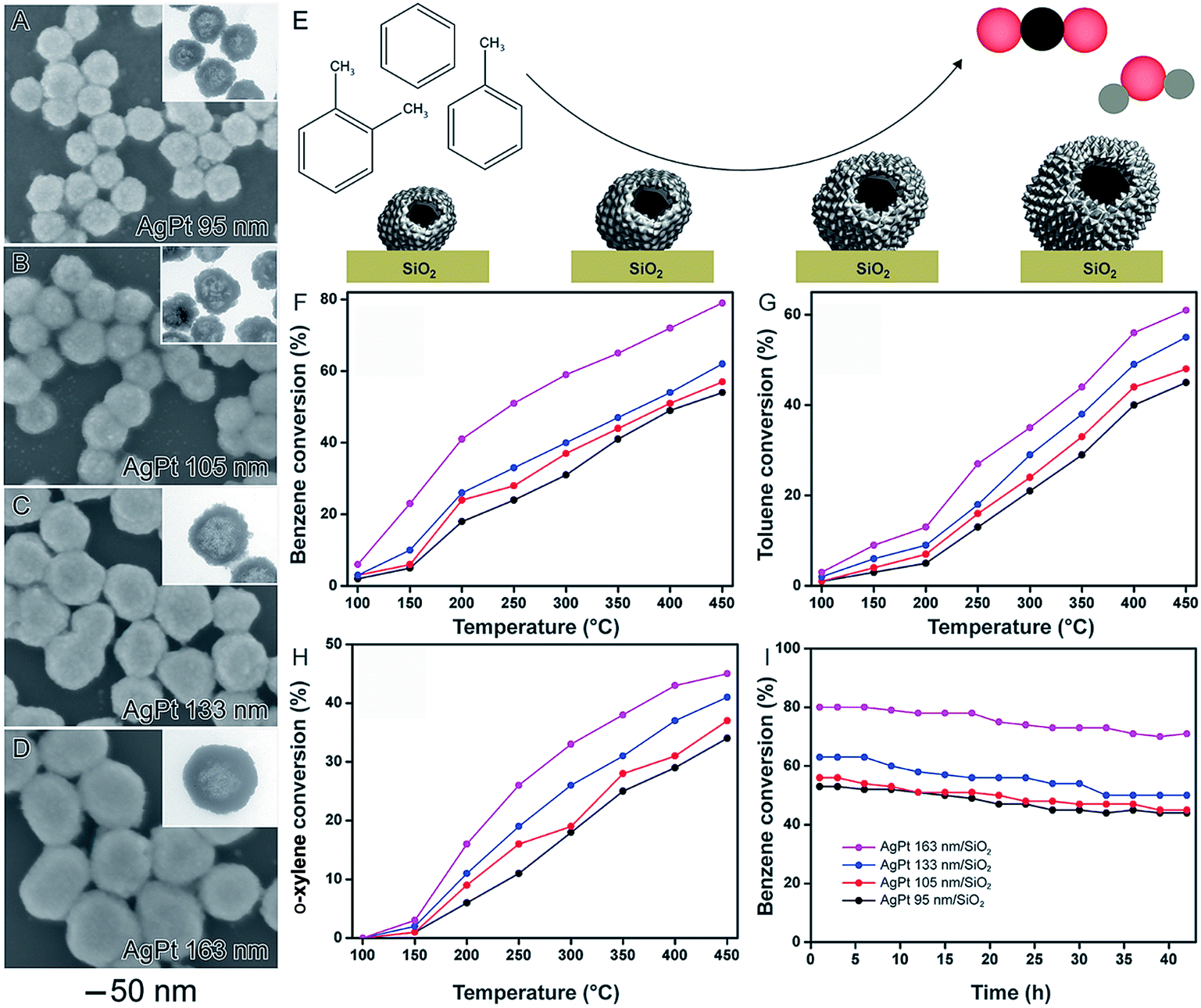 | ||
| Fig. 10 Effect of NPs structure (hollow interiors), size, and surface morphology over catalytic activity. SEM images for AgPt nanoshells with controlled sizes 95 nm (A), 105 nm (B), 133 nm (C), and 163 nm (D). The insets in the figures correspond to their respective TEM images. (E) Scheme showing the oxidation of benzene, toluene, and o-xylene catalyzed by AgPt nanoshells with controlled sizes. Conversion percentages of benzene (F), toluene (G), and o-xylene oxidation (H) as a function of temperature catalyzed by AgPt/SiO2. (I) Stability experiments for all AgPt/SiO2 catalysts over benzene oxidation at 450 °C. Adapted from ref. 58 with permission. | ||
After their incorporation onto the surface of commercial SiO2 support, the AgPt nanoshells were evaluated as a catalyst toward the gas-phase oxidation of benzene, toluene, and o-xylene as a function of their sizes (Fig. 10E–I). The best values of BTX oxidation were achieved for the bigger AgPt nanoshells. Specifically, the catalytic activities of the AgPt/SiO2 nanocatalysts decreased following the order: AgPt 163 nm/SiO2 > AgPt 133 nm/SiO2 > AgPt 105 nm/SiO2 > AgPt 95 nm/SiO2. This data indicates that the surface morphology played the dominant role relative to shell thickness and outer diameter to produce higher surface areas and therefore higher catalytic activities.58
3.3. Controlling composition, surface morphology, composition, and shape in hollow NPs: using Ag sacrificial templates having different shapes
In addition to the control over composition and degree of hollowing, the shape (and thus the nature of surface exposed facets) in hollow nanomaterials can be tuned by using NPs having different morphologies as sacrificial templates. Therefore, by combining the principles discussed in the previous sections with the use of nanocrystals with well-defined shapes as chemical templates, bimetallic nanomaterials with controlled surface morphologies, shapes, compositions, and hollow interiors can be obtained.In order to illustrate this concept, AgPt spherical nanoparticles displaying smooth or rough (assembled nanoparticles) surfaces and AgPt nanotubes displaying smooth and rough surfaces can be developed, as illustrated in Fig. 11A–D, respectively.28 For the synthesis of spherical AgPt hollow nanomaterials with controlled surface morphologies, Ag spheres were employed as templates in galvanic replacement reaction with PtCl6(aq)2− precursor. When the conventional galvanic reaction was performed at 100 °C in the presence of PVP, the formation of nanoshells with smooth surfaces was observed (Fig. 11A).41,81 However, after addition of hydroquinone as an auxiliary reducing agent to the galvanic reaction, AgPt assembled nanoparticles was formed (rough surfaces, Fig. 11B) due to the deposition of Pt islands at the surface provided by this additional reducing agent.101 In the synthesis of AgPt tubes, the temperature enabled the control over the surface morphology. In this case, the galvanic replacement reaction in the presence of PVP at 100 °C led to the formation of nanotubes with smooth surfaces (Fig. 11C) as a result of the epitaxial Pt deposition over the surface of the Ag nanowires templates, while nanotubes with rough surfaces were produced at room temperature (Fig. 11D).102
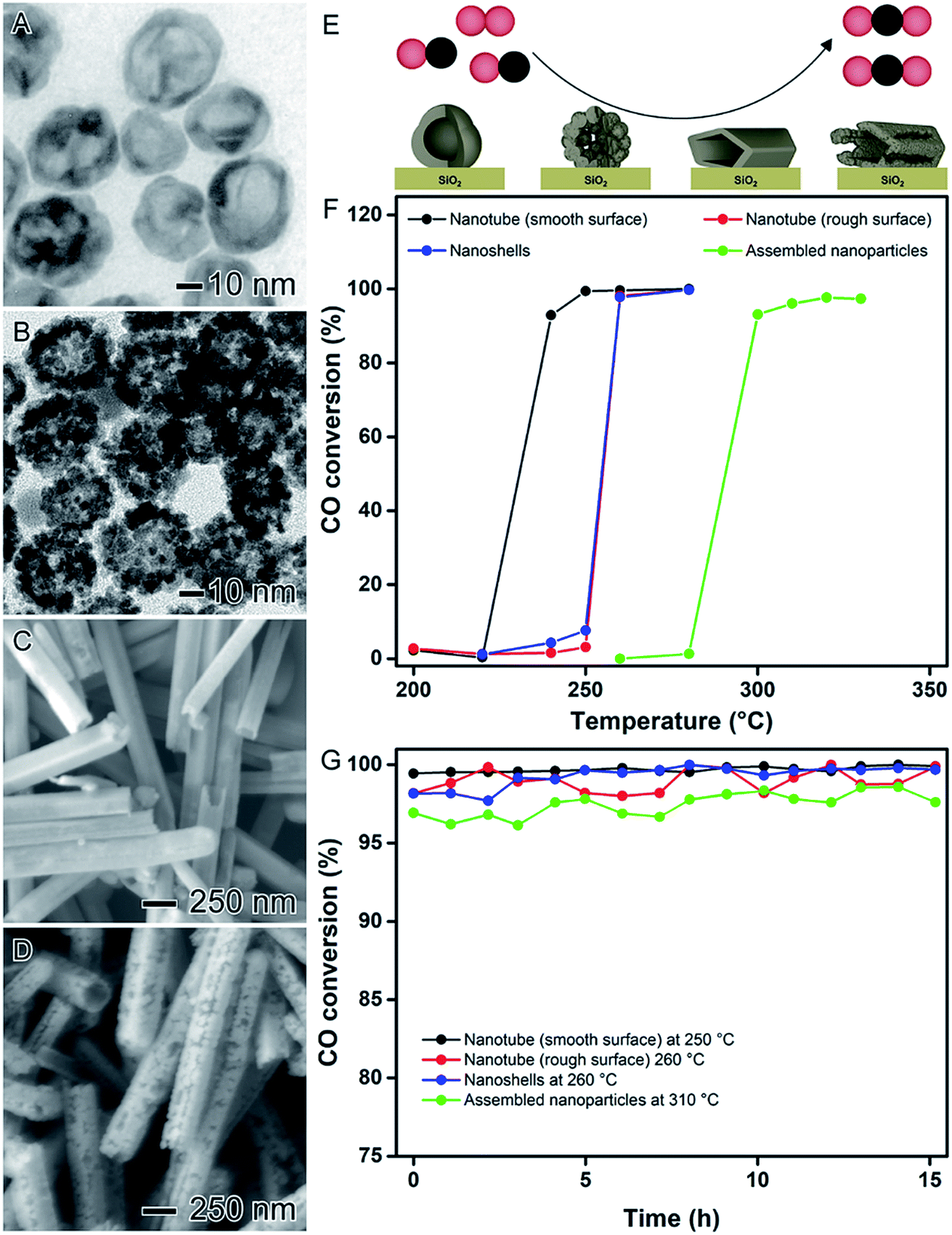 | ||
| Fig. 11 Effect of NPs structure (hollow interiors), shape, and surface morphology over catalytic activity. (A) Scheme showing the oxidation of CO catalyzed by AgPt hollow nanomaterials with controlled surface morphologies supported on SiO2. TEM images of AgPt nanoshells (B) and assembled nanoparticles (C). SEM images of AgPt nanotubes with smooth (D) and rough (E) surfaces. (F) CO conversion percentages as a function of temperature catalyzed by AgPt hollow nanomaterials with controlled surface morphologies supported on SiO2. (G) Stability tests employing the AgPt/SiO2 catalysts performed at their respective temperature of complete CO oxidation. Adapted from ref. 28 with permission. | ||
These characteristics led to significant differences in the surface properties of the obtained hollow nanomaterials such as crystallinity, preferential facet orientation, and available surface area. More specifically, AgPt nanoshells and assembled nanoparticles displayed polycrystalline surfaces with a mixture of {111} and {100} surface facets in agreement with the polycrystalline nature of the Ag spheres employed as templates. Here, the number of Pt surface atoms corresponded to 4.25 and 1.70 μmol per gram of catalysts for nanoshells and assembled nanoparticles, respectively. This difference can be associated with the thicker shell structure of the assembled nanoparticles. Regarding the AgPt nanotubes, the smooth nanotubes displayed a single-crystalline surface enclosed by {100} side facets while the rough nanotubes were polycrystalline enclosed by a mix of {111} and {100} surface facets. However, both nanotubes presented similar values of a number of Pt surface atoms (4.10, and 4.56 μmol per gram of catalysts for nanotubes with smooth and rough surfaces, respectively).
The catalytic activities of the AgPt hollow nanomaterials were evaluated towards the CO oxidation as a model reaction (Fig. 11E). To this end, all the syntheses were successfully scaled-up by 100 folds and all nanomaterials were incorporated onto commercial SiO2 support with Pt loading corresponding to 1 wt%. According to with the CO conversion percentages as a function of temperature (Fig. 11F), the catalytic performances of the AgPt/SiO2 materials decreased in the following order: nanotubes with smooth surfaces > nanotubes with rough = surfaces nanoshells > assembled nanoparticles, which could be clearly related to their surface properties. For the nanotubes, as both presented similar value of Pt surface atoms, the difference in their catalytic activities can be attributed to their surface facet exposition. In this case, it has been reported that the CO and formic acid oxidation catalyzed by noble metal nanoparticles display a facet-dependent behavior, in which {100} facets show enhanced catalytic activities towards the CO and formic acid oxidation.62,69,103–105 Thus, as the nanotubes with smooth surfaces were enclosed by {100} surface facets, is expected a superior catalytic performance. Interestingly, as depicted in Fig. 11G, all AgPt/SiO2 catalysts showed good stabilities under the employed experimental conditions without any significant loss of activity even after 15 hours of processing showing their applicability as catalysts in gas-phase transformations.
4. Concluding remarks and perspectives
We have discussed herein the importance of the control over several physical/chemical parameters in noble-metal nanomaterials for the optimization of the catalytic properties. We focused on the control over shape, size, composition, structure (solid or hollow interiors) and combinations thereof. We started by focusing on the control over one physical/chemical parameter at a time, such as shape, size, and composition, and then moved to experimental strategies that allow for the control over more than one physical/chemical parameter at a time in hollow nanomaterials. In this case, we focused on NPs prepared by galvanic replacement as a proof-of-concept system.By selecting some representative examples, we discussed how the control over these physical/chemical features was important for the understanding of catalytic performances. We believe that this precise understanding is crucial to enable the rational design of catalysts, in which the desired physical/chemical features can be planned and the nanomaterial prepared for achieving the target reactivity and selectivity towards a molecular transformation. This ambitious and exciting goal represents one of the bottlenecks in the field of nanocatalysis and requires progress in the context of NPs synthesis, catalytic assessment, the study of catalytic enhancement mechanisms, reaction pathways, modeling, and ex situ, in situ, and in operando characterizations.
Even though it was briefly discussed in this review, the ultimate control over size represented by single-atom catalysts has emerged as a promising new frontier in heterogeneous catalysts. These catalytic systems display an enormous potential and represent an exciting direction of the field as they possess the best advantages of both homogeneous and heterogeneous catalysis. This area is progressing rapidly, and developments in the areas of controlled synthesis, catalytic evaluation, and understanding of catalytic enhancements together with advanced characterization and modeling are of paramount importance towards the rational design of single-atom catalysts.
The combination of catalytic materials with semiconducting and plasmonic materials also represent interesting areas in which we can combine catalytic and optical properties, in which light can be employed as an additional and sustainable energy input to drive and control molecular transformations. This is attractive not only from the point of view of solar-to-chemical energy conversion but can also allow for the optimization of activities and control over reaction selectivity. For instance, in addition to classical photocatalysis, plasmonic photocatalysis has emerged as a unique strategy to combine plasmonic and catalytic properties at the nanoscale, and open new possibilities for driving molecular transformations under mild conditions and opening new avenues for controlling conversion percentages and product selectivity via selective activation of chemical bonds and enabling new reaction pathways.
Another important area comprises the reduction in the usage of noble metals and/or the replacement of expensive and noble-metals by more abundant and less noble metals while maintaining good catalytic activities and selectivities. The use of single-atom catalysis represents an important contribution towards this goal, as the utilization and efficiency of these noble metals can be maximized. Moreover, several noble-metal free nanomaterials having good activities have been reported. Some doped semiconducting materials and oxides, for example, have also been employed as alternative plasmonic materials to silver and gold. These topics deserve further attention and can play an important role in enabling future industrial applications.
Finally, the stability of nanocatalysts needed to be characterized and improved. Strategies to ensure recover and re-use without significant loss of activities and selectivities must be developed. This is particularly important for reactions that required high temperature and pressure, and transformations that generate products that can contaminate the catalytic surface. Therefore, the regulation over the surface interactions, metal leaching, metal–support interactions, and the nature of the catalytic process is imperative in order to address catalytic stability.
Nanocatalysis can play a central role in a sustainable future. We believe that the study and the optimization over the catalyst represents an important challenge in nanocatalysis (in addition to the overall process engineering). While we are still in the early stages with respect to a design-driven approach concerning the development of nanocatalysts towards target transformations, important progress has occurred in the last decade as a result of developments in synthesis, characterization, and modeling. Several emerging areas, such as single atom catalysis and plasmonic photocatalysis, represent novel directions of the field that can take nanocatalysis to a new level. It is clear that an interdisciplinary approach among precise synthesis, applications, benchmarking, understanding, modeling, and characterization is required. In this context, we believe that developments in the synthesis of catalytic systems, in which several physical/chemical parameters can be precisely tuned, is fundamental to enable understanding and allow us to move to the ultimate goal of a revolution in the chemical industry via nanocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by FAPESP (grant number 15/26308-7) and the Serrapilheira Institute (grant number Serra-1709-16900). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (CAPES) – Finance Code 001. PHCC thanks the CNPq for the research fellowships. AGMS thanks FAPESP for the fellowship.References
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–218 CrossRef CAS PubMed.
- A. Corma, Angew. Chem., Int. Ed., 2016, 55, 6112–6113 CrossRef CAS PubMed.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743 RSC.
- R. M. Mohamed, D. L. McKinney and W. M. Sigmund, Mater. Sci. Eng., R, 2012, 73, 1–13 CrossRef CAS.
- G. C. Bond, Heterogeneous catalysis, Oxford University Press, New York, NY, United States, 1987 Search PubMed.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2004, 108, 5726–5733 CrossRef CAS.
- M. A. Mahmoud, D. O'Neil and M. A. El-Sayed, Chem. Mater., 2014, 26, 44–58 CrossRef CAS.
- M. A. Mahmoud, R. Narayanan and M. A. El-Sayed, Acc. Chem. Res., 2013, 46, 1795–1805 CrossRef CAS PubMed.
- Y. Zhu, A. Chakrabarti and N. S. Hosmane, ed. D. Hnyk and M. McKee, Applications of Nanocatalysis in Boron Chemistry, Springer International Publishing, Cham, 2015, pp. 199–217 Search PubMed.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset, V. Polshettiwar, H. W. Zandbergen, F. M. F. de Groot, B. M. Weckhuysen, C. Morin, B. M. Weckhuysen and F. M. F. de Groot, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC.
- B. Roldan Cuenya, Acc. Chem. Res., 2013, 46, 1682–1691 CrossRef CAS PubMed.
- B. Roldan Cuenya and F. Behafarid, Surf. Sci. Rep., 2015, 70, 135–187 CrossRef CAS.
- J. Park, T. Kwon, J. Kim, H. Jin, H. Y. Y. Kim, B. Kim, S. H. H. Joo and K. Lee, Chem. Soc. Rev., 2018, 47, 8173–8202 RSC.
- H.-H. Li and S.-H. Yu, Adv. Mater., 2019, 1803503 CrossRef PubMed.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- J. L. Wang, R. A. Ando and P. H. C. Camargo, ACS Catal., 2014, 4, 3815–3819 CrossRef CAS.
- C.-J. Jia and F. Schüth, Phys. Chem. Chem. Phys., 2011, 13, 2457–2487 RSC.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- J. Otera and J. Nishikido, in Esterification, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 293–322 Search PubMed.
- K. Chanda, S. Rej and M. H. Huang, Chem.–Eur. J., 2013, 19, 16036–16043 CrossRef CAS PubMed.
- T. Shibata, B. A. Bunker, Z. Zhang, D. Meisel, C. F. Vardeman and J. D. Gezelter, J. Am. Chem. Soc., 2002, 124, 11989–11996 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, A. Macedo, R. T. P. da Silva and P. H. C. Camargo, Quim. Nova, 2014, 37, 1716–1720 CAS.
- Y. Watanabe, X. Wu, H. Hirata and N. Isomura, Catal. Sci. Technol., 2011, 1, 1490–1495 RSC.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- T. S. Rodrigues, A. G. M. Silva, A. Macedo, B. W. Farini, R. d. S. Alves and P. H. C. Camargo, J. Mater. Sci., 2015, 50, 5620–5629 CrossRef CAS.
- T. S. Rodrigues, A. H. M. da Silva, A. G. M. da Silva, D. G. Ceara, J. F. Gomes, J. M. Assaf and P. H. C. Camargo, Catal. Sci. Technol., 2016, 6, 2162–2170 RSC.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef.
- X.-F. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- S. Mitchell, E. Vorobyeva and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2018, 57, 15316–15329 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, L. S. K. Taguchi, H. V. Fajardo, R. Balzer, L. F. D. Probst and P. H. C. Camargo, J. Mater. Sci., 2016, 51, 603–614 CrossRef CAS.
- C. C. S. de Oliveira, R. A. Ando and P. H. C. Camargo, Phys. Chem. Chem. Phys., 2013, 15, 1887–1893 RSC.
- H. Zhang, M. S. Jin, J. G. Wang, W. Y. Li, P. H. C. Camargo, M. J. Kim, D. R. Yang, Z. X. Xie and Y. A. Xia, J. Am. Chem. Soc., 2011, 133, 6078–6089 CrossRef CAS PubMed.
- D. S. Wang and Y. D. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- A. G. M. Da Silva, T. S. Rodrigues, S. J. Haigh and P. H. C. Camargo, Chem. Commun., 2017, 53, 7135–7148 RSC.
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS.
- Y. Sun, B. Mayers and Y. Xia, Adv. Mater., 2003, 15, 641–646 CrossRef CAS.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- H.-L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21, 13705–13725 RSC.
- K. An and T. Hyeon, Nano Today, 2009, 4, 359–373 CrossRef CAS.
- X. W. Lou, L. A. Archer, Z. C. Yang, B. Xiong, W. David, L. A. Archer, X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- A. G. M. da Silva, M. L. de Souza, T. S. Rodrigues, R. S. Alves, M. L. A. Temperini and P. H. C. Camargo, Chem.–Eur. J., 2014, 20, 15040–15046 CrossRef CAS PubMed.
- M. A. Mahmoud, F. Saira and M. A. El-Sayed, Nano Lett., 2010, 10, 3764–3769 CrossRef CAS PubMed.
- X. Wang, L. Figueroa-Cosme, X. Yang, M. Luo, J. Liu, Z. Xie and Y. Xia, Nano Lett., 2016, 16, 1467–1471 CrossRef CAS PubMed.
- M. A. Mahmoud, B. Garlyyev and M. A. El-Sayed, J. Phys. Chem. Lett., 2014, 5, 4088–4094 CrossRef CAS PubMed.
- S. Guo, S. Dong and E. Wang, J. Phys. Chem. C, 2009, 113, 5485–5492 CrossRef CAS.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- W. Zhang, J. Yang and X. Lu, ACS Nano, 2012, 6, 7397–7405 CrossRef CAS PubMed.
- C. Xu, Y. Zhang, L. Wang, L. Xu, X. Bian, H. Ma and Y. Ding, Chem. Mater., 2009, 21, 3110–3116 CrossRef CAS.
- M. M. Shahjamali, M. Bosman, S. Cao, X. Huang, X. Cao, H. Zhang, S. S. Pramana and C. Xue, Small, 2013, 9, 2880–2886 CrossRef CAS PubMed.
- M. McEachran, D. Keogh, B. Pietrobon, N. Cathcart, I. Gourevich, N. Coombs and V. Kitaev, J. Am. Chem. Soc., 2011, 133, 8066–8069 CrossRef CAS PubMed.
- J. Li, X. Sun and D. Qin, ChemNanoMat, 2016, 2, 494–499 CrossRef CAS.
- J. Zhang, C. Qiu, H. Ma and X. Liu, J. Phys. Chem. C, 2008, 112, 13970–13975 CrossRef CAS.
- T. S. Rodrigues, A. G. M. da Silva, R. S. Alves, I. C. de Freitas, D. C. Oliveira and P. H. C. Camargo, Part. Part. Syst. Charact., 2018, 1700175 CrossRef.
- T. S. Rodrigues, A. G. M. Da Silva, M. C. Gonçalves, H. V. Fajardo, R. Balzer, L. F. D. Probst, A. H. M. Da Silva, J. M. Assaf and P. H. C. Camargo, Langmuir, 2016, 32, 9371–9379 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, V. G. Correia, T. V. Alves, R. S. Alves, R. A. Ando, F. R. Ornellas, J. Wang, L. H. Andrade and P. H. C. Camargo, Angew. Chem., Int. Ed., 2016, 55, 7111–7115 CrossRef CAS PubMed.
- M. Zhao, L. Figueroa-Cosme, A. O. Elnabawy, M. Vara, X. Yang, L. T. Roling, M. Chi, M. Mavrikakis and Y. Xia, Nano Lett., 2016, 16, 5310–5317 CrossRef CAS PubMed.
- J. W. Hong, S. W. Kang, B.-S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410–2419 CrossRef CAS PubMed.
- H. Zhang, M. Jin, H. Liu, J. Wang, M. J. Kim, D. Yang, Z. Xie, J. Liu and Y. Xia, ACS Nano, 2011, 5, 8212–8222 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- L. K. Yamada, A. G. M. da Silva, T. S. Rodrigues, S. J. Haigh and P. H. C. Camargo, ChemNanoMat, 2016, 2, 509–514 CrossRef CAS.
- C. Li, T. Liu, T. He, B. Ni, Q. Yuan and X. Wang, Nanoscale, 2018, 10, 4670–4674 RSC.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- J. Gu, Y.-W. Zhang and F. Tao, Chem. Soc. Rev., 2012, 41, 8050–8065 RSC.
- A. S. Bandarenka and M. T. M. Koper, J. Catal., 2013, 308, 11–24 CrossRef CAS.
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Energy Environ. Sci., 2012, 5, 6352–6357 RSC.
- T. S. Rodrigues, M. Zhao, T.-H. Yang, K. D. Gilroy, A. G. M. da Silva, P. H. C. Camargo and Y. Xia, Chem.–Eur. J., 2018, 24, 16944–16963 CrossRef CAS PubMed.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS PubMed.
- A. G. M. Da Silva, T. S. Rodrigues, J. Wang, L. K. Yamada, T. V. Alves, F. R. Ornellas, R. A. Ando and P. H. C. Camargo, Langmuir, 2015, 31, 10272–10278 CrossRef CAS PubMed.
- P. Sabatier, Ber. Dtsch. Chem. Ges., 1911, 44, 1984–2001 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed.
- H. Wang, X.-K. Gu, X. Zheng, H. Pan, J. Zhu, S. Chen, L. Cao, W.-X. Li and J. Lu, Sci. Adv., 2019, 5, eaat6413 CrossRef PubMed.
- X. Li, Y. Wang, L. Li, W. Huang, Z. Xiao, P. Wu, W. Zhao, W. Guo, P. Jiang and M. Liang, J. Mater. Chem. A, 2017, 5, 11294–11300 RSC.
- W. Yao, F.-L. Li, H.-X. Li and J.-P. Lang, J. Mater. Chem. A, 2015, 3, 4578–4585 RSC.
- L. Chen, J. M. Chabu and Y. Liu, RSC Adv., 2013, 3, 4391–4399 RSC.
- H. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- T. J. A. Slater, A. Macedo, S. L. M. Schroeder, M. G. Burke, P. O'Brien, P. H. C. Camargo and S. J. Haigh, Nano Lett., 2014, 14, 1921–1926 CrossRef CAS PubMed.
- D. Xu, S. Bliznakov, Z. Liu, J. Fang and N. Dimitrov, Angew. Chem., 2010, 122, 1304–1307 CrossRef.
- M. T. M. Koper, Fuel Cell Catalysis: A Surface Science Approach, 2008 Search PubMed.
- H. Rong, S. Cai, Z. Niu and Y. Li, ACS Catal., 2013, 3, 1560–1563 CrossRef CAS.
- M. Kourtelesis, P. Panagiotopoulou and X. E. Verykios, Catal. Today, 2015, 258, 247–255 CrossRef CAS.
- T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636 RSC.
- S.-I. Choi, S. R. Lee, C. Ma, B. Oliy, M. Luo, M. Chi and Y. Xia, ChemNanoMat, 2016, 2, 61–66 CrossRef CAS.
- A. G. M. da Silva, C. M. Kisukuri, T. S. Rodrigues, E. G. Candido, I. C. de Freitas, A. H. M. da Silva, J. M. Assaf, D. C. Oliveira, L. H. Andrade and P. H. C. Camargo, Appl. Catal., B, 2016, 184, 35–43 CrossRef CAS.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS PubMed.
- Y. Nishida, K. Sato, T. Yamamoto, D. Wu, K. Kusada, H. Kobayashi, S. Matsumura, H. Kitagawa and K. Nagaoka, Chem. Lett., 2017, 46, 1254–1257 CrossRef CAS.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- B. Wu and N. Zheng, Nano Today, 2013, 8, 168–197 CrossRef CAS.
- T. S. Rodrigues, A. G. M. da Silva, A. B. L. de Moura, I. G. Freitas and P. H. C. Camargo, RSC Adv., 2016, 6, 62286–62290 RSC.
- T. S. Rodrigues, A. G. M. Da Silva, A. B. L. De Moura, R. S. Geonmonond and P. H. C. Camargo, J. Braz. Chem. Soc., 2017, 28, 1630–1638 CAS.
- Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- J. Zeng, Q. Zhang, J. Y. Chen and Y. N. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
- Y. N. Xia, W. Y. Li, C. M. Cobley, J. Y. Chen, X. H. Xia, Q. Zhang, M. X. Yang, E. C. Cho and P. K. Brown, Acc. Chem. Res., 2011, 44, 914–924 CrossRef CAS PubMed.
- M. V. Petri, R. A. Ando and P. H. C. Camargo, Chem. Phys. Lett., 2012, 531, 188–192 CrossRef CAS.
- A. K. Samal, T. S. Sreeprasad and T. Pradeep, J. Nanopart. Res., 2010, 12, 1777–1786 CrossRef CAS.
- T. S. Rodrigues, A. G. M. da Silva, M. C. Gonçalves, H. V. Fajardo, R. Balzer, L. F. D. Probst and P. H. C. Camargo, ChemNanoMat, 2015, 1, 46–51 CrossRef CAS.
- Y. G. Sun and Y. N. Xia, Adv. Mater., 2004, 16, 264–268 CrossRef CAS.
- R. Wang, H. He, J. Wang, L. Liu and H. Dai, Catal. Today, 2013, 201, 68–78 CrossRef CAS.
- G. Chen, Y. Tan, B. Wu, G. Fu and N. Zheng, Chem. Commun., 2012, 48, 2758–2760 RSC.
- M. Jin, H. Liu, H. Zhang, Z. Xie, J. Liu and Y. Xia, Nano Res., 2011, 4, 83–91 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, N. Ichikuni, Y. Negishi and T. Tsukuda, Langmuir, 2004, 20, 11293–11296 CrossRef CAS PubMed.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS PubMed.
- J. Ramírez, M. Sanaú and E. Fernández, Angew. Chem., Int. Ed., 2008, 47, 5194–5197 CrossRef PubMed.
- G. Merga, N. Saucedo, L. C. Cass, J. Puthussery and D. Meisel, J. Phys. Chem. C, 2010, 114, 14811–14818 CrossRef CAS.
- H. Ohde, C. M. Wai, H. Kim, J. Kim and M. Ohde, J. Am. Chem. Soc., 2002, 124, 4540–4541 CrossRef CAS PubMed.
- S. Sawoo, D. Srimani, P. Dutta, R. Lahiri and A. Sarkar, Tetrahedron, 2009, 65, 4367–4374 CrossRef CAS.
- M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165–168 CrossRef CAS PubMed.
- H. Dai, J. Su, K. Hu, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 4947–4953 CrossRef CAS.
- Q.-L. Zhu, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2015, 137, 11743–11748 CrossRef CAS PubMed.
- C. Shin, T. E. Park, C. Park and S. J. Kwon, ChemPhysChem, 2016, 17, 1637–1641 CrossRef CAS PubMed.
- B. W. Lim, X. M. Lu, M. J. Jiang, P. H. C. Camargo, E. C. Cho, E. P. Lee and Y. N. Xia, Nano Lett., 2008, 8, 4043–4047 CrossRef CAS PubMed.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS PubMed.
- J. D. Aiken and R. G. Finke, Chem. Mater., 1999, 11, 1035–1047 CrossRef CAS.
- A. L. Maksimov, S. N. Kuklin, Y. S. Kardasheva and E. A. Karakhanov, Pet. Chem., 2013, 53, 157–163 CrossRef CAS.
- L. Coronel, J. F. Múnera, A. M. Tarditi, M. S. Moreno and L. M. Cornaglia, Appl. Catal., B, 2014, 160, 254–266 CrossRef.
- J. A. Baeza, L. Calvo, M. A. Gilarranz and J. J. Rodriguez, Chem. Eng. J., 2014, 240, 271–280 CrossRef CAS.
- K. H. Park, K. Jang, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2007, 46, 1152–1155 CrossRef CAS PubMed.
- S. Miao, Z. Liu, B. Han, J. Huang, Z. Sun, J. Zhang and T. Jiang, Angew. Chem., Int. Ed., 2006, 45, 266–269 CrossRef CAS PubMed.
- S. Guo, X. Pan, H. Gao, Z. Yang, J. Zhao and X. Bao, Chem.–Eur. J., 2010, 16, 5379–5384 CrossRef CAS PubMed.
- J. Kang, S. Zhang, Q. Zhang and Y. Wang, Angew. Chem., 2009, 121, 2603–2606 CrossRef.
- S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709–2713 CrossRef CAS PubMed.
- E. Sulman, V. Doluda, S. Dzwigaj, E. Marceau, L. Kustov, O. Tkachenko, A. Bykov, V. Matveeva, M. Sulman and N. Lakina, J. Mol. Catal. A: Chem., 2007, 278, 112–119 CrossRef CAS.
- E. Redel, J. Krämer, R. Thomann and C. Janiak, J. Organomet. Chem., 2009, 694, 1069–1075 CrossRef CAS.
- C. A. Stowell and B. A. Korgel, Nano Lett., 2005, 5, 1203–1207 CrossRef CAS PubMed.
- I. S. Park, M. S. Kwon, K. Y. Kang, J. S. Lee and J. Park, Adv. Synth. Catal., 2007, 349, 2039–2047 CrossRef CAS.
- Z. Yinghuai, K. Chenyan, A. T. Peng, A. Emi, W. Monalisa, L. Kui-Jin Louis, N. S. Hosmane and J. A. Maguire, Inorg. Chem., 2008, 47, 5756–5761 CrossRef PubMed.
- M. Zahmakiran, Dalton Trans., 2012, 41, 12690–12696 RSC.
| This journal is © The Royal Society of Chemistry 2019 |