
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA light-responsive, self-immolative linker for controlled drug delivery via peptide- and protein-drug conjugates†‡
Chuanlong
Zang
a,
Huawei
Wang
a,
Tiantian
Li
b,
Yingqian
Zhang
a,
Jiahui
Li
a,
Mengdi
Shang
a,
Juanjuan
Du
b,
Zhen
Xi
a and
Chuanzheng
Zhou
 *a
*a
aState Key Laboratory of Elemento-Organic Chemistry, Department of Chemical Biology, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: chuanzheng.zhou@nankai.edu.cn
bSchool of Pharmaceutical Sciences, Tsinghua University, 30 Shuangqing Rd., Beijing 100084, China
First published on 19th August 2019
Abstract
When designing prodrugs, choosing an appropriate linker is the key to achieving efficient, controlled drug delivery. Herein, we report the use of a photocaged C4′-oxidized abasic site (PC4AP) as a light-responsive, self-immolative linker. Any amine- or hydroxyl-bearing drug can be loaded onto the linker via a carbamate or carbonate bond, and the linker is then conjugated to a carrier peptide or protein via an alkyl chain. The PC4AP linker is stable under physiologically relevant conditions. However, photodecaging of the linker generates an active intermediate that reacts intramolecularly with a primary amine (the ε-amine of a lysine residue and the N-terminal amine) on the carrier, leading to rapid and efficient release of the drug via an addition–elimination cascade, without generating any toxic side products. We demonstrated that the use of this self-immolative linker to conjugate the anticancer drug doxorubicin to a cell-penetrating peptide or an antibody enabled targeted, controlled delivery of the drug to cells. Our results suggest that the linker can be used with a broad range of carriers, such as cell-penetrating peptides, proteins, antibodies, and amine-functionalized polymers, and thus will find a wide range of practical applications.
Introduction
In drug design and development, prodrug strategies are widely used to improve the pharmacokinetic properties of drugs, especially targeted delivery.1,2 A prodrug is generally constructed by conjugation of a drug molecule to a carrier via a linker containing a trigger moiety. After delivery of the prodrug to the target cells or tissues, the active drug is released via cleavage of the linker, either by an endogenous stimulus such as a pH change,3,4 a redox reaction,5,6 or an enzyme,7,8 or by an exogenous stimulus such as light9–11 or a small-molecule trigger.2,12 The key to achieving efficient, controlled drug release is to choose an appropriate linker.13,14In some cases, the close proximity of the drug and the carrier impairs linker cleavage by the stimulus. This problem can be overcome by introducing an additional linker, referred to as a self-immolative linker, between the trigger and the drug.15,16 Removal or cleavage of the trigger by an appropriate stimulus induces a cascade of disassembly reactions that ultimately lead to drug release. So far, only two types of self-immolative linkers have gained wide acceptance, and both types undergo self-immolative elimination, cyclization, or both to release the conjugated drug.16,17 However, disassembly of these self-immolative linkers can generate toxic side products such as quinone methides, which can have unwanted side effects.18 Therefore, the development of biocompatible self-immolative linkers has attracted considerable attention.11,19,20
We and others have shown that primary amines can catalyze DNA cleavage at C4′-oxidized abasic sites (C4APs, which are formed by abstraction of the C4′–H from 2′-deoxyribose; Fig. 1A).21–26 Specifically, addition of a primary amine to the C1 of a C4AP via Schiff base formation leads to sequential elimination of the C3 and C5 phosphates, generating the 5-methylene pyrrolone (5MP) derivative of the primary amine.23 On the basis of these addition–elimination cascade reactions, we designed a photocaged C4AP (PC4AP, Fig. 1B) as a novel light-responsive, self-immolative linker for controlled drug delivery via peptide- and protein-drug conjugates.
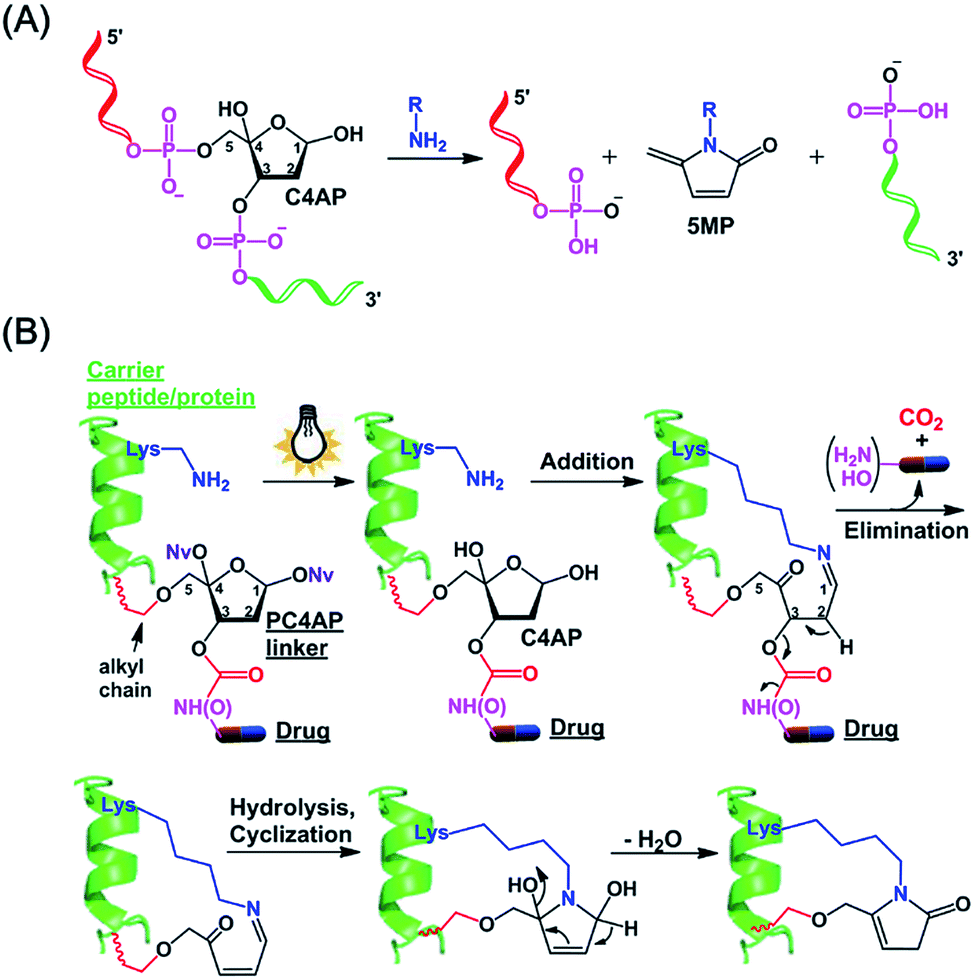 | ||
| Fig. 1 Design of a photocaged C4′-oxidized abasic site (PC4AP) as a light-responsive, self-immolative linker for controlled drug delivery via peptide- and protein-drug conjugates. (A) Primary-amine-catalyzed DNA cleavage at the C4′-oxidized abasic site (C4AP). (B) Principle of PC4AP-based drug delivery via peptide- and protein-drug conjugates. | ||
Results and discussion
Design of the PC4AP linker
The PC4AP linker is constructed by protection of the C1–OH and C4–OH of the C4AP with photolabile O-nitrobenzyl groups (–ONv),27,28 which make the system light-responsive.22,29 The C3–OH and C5–OH are used to load the drug and the carrier, respectively (Fig. 1B). Specifically, an amine- or hydroxyl-bearing drug payload is attached to the C3–OH via a carbamate or carbonate bond, and the C5–OH of the PC4AP linker is conjugated to a carrier peptide or protein via an alkyl chain. Upon photodecaging, the C4AP moiety undergoes an intramolecular addition reaction with any nearby amine on the carrier, and a subsequent elimination reaction leads to cleavage of the carbamate or carbonate bond and concomitant release of the active drug, along with nontoxic CO2. Because C5–OH is linked to the carrier via a stable O–C bond, cleavage of the linker via departure of C5–O− is disfavored. Therefore, the linker remains attached to the carrier, and a cyclic species is generated.Synthesis of a doxorubicin prodrug based on the PC4AP linker
Doxorubicin (DOX) is a cytotoxic anthracycline antibiotic and anticancer drug. Because of its natural fluorescence, DOX is widely used as a model cytotoxin for cellular delivery studies.30–32 Here, in a proof-of-principle experiment, DOX was employed to examine the efficacy of controlled drug delivery by means of the PC4AP linker. To this end, we designed and synthesized Mal-PC4AP-DOX (9, Scheme 1), in which DOX and a maleimide (Mal) moiety are bridged by the PC4AP linker. The purpose of the maleimide moiety was to enable site-specific bioconjugation of 9 to a Cys residue in carrier peptides and proteins. | ||
| Scheme 1 Synthesis of Mal-PC4AP-DOX (9). | ||
The synthesis of 9 started from acetal 1, in which the C5–OH is protected with a benzoyl group. After protection of the C3–OH as a t-butyldiphenylsilyl ether, the benzoyl group was selectively removed by treatment with NaOMe, and the C5–OH was then allowed to react with propargyl bromide to afford alkyne 3, which was transformed into dithiane 4 in 85% yield. After oxidation of the C4–OH to a ketone, the dithiane moiety of 5 was oxidatively cleaved in the presence of 4,5-dimethoxy-2-nitrobenzyl alcohol (NvOH) to afford protected PC4AP 6 as a mixture of four stereoisomers.29 The C3–OH was then deprotected by treatment with tetra-n-butylammonium fluoride, and three isomers of 7 were isolated and characterized by NMR spectroscopy. The predominant isomer, 7a, was subjected to further transformations. Specifically, treatment of 7a with triphosgene converted the C3–OH to the corresponding carbonochloridate (8′), which, without purification, was allowed to react selectively with the amino group of DOX to give 8 in 65% yield. Finally, target product 9 was obtained by coupling 8 with Mal-PEG3-N3via click chemistry. It is worth noting that various functional groups can be introduced into the PC4AP linker by means of click reactions between 8 and azide-containing molecules. The linker can thereby be site-specifically attached to a broad range of residues in carrier peptides and proteins.
Although the carbonochloridate intermediate reacts readily with amines, it can also react with alcohols and thiols.33–35 For example, when 8′ was treated in situ with 1-pyrenemethanol (PyrCH2OH), carbonate 10 was obtained in 50% yield.
Analysis of PC4AP linker stability and disassembly
To evaluate the stability of the PC4AP linker under physiologically relevant conditions, we incubated 8 in pH 7.5 and 5.2 buffers, which simulate intercellular and lysosomal environments, respectively. Ultra performance liquid chromatography (UPLC) revealed that under both conditions, 8 was completely stable after 24 h of incubation (Fig. S1‡).Next, we examined the kinetics and mechanism of the photodecaging process by using model compounds 8 and 9. Analysis of the UV-vis absorption spectra of 9 as a function of irradiation time (365 nm, 7.0 mw cm−2) revealed a rapid decrease in absorbance at 350 nm along with a concomitant increase at 270 nm,31 and irradiation for 3 min was sufficient to completely remove the photolabile O-nitrobenzyl groups (Fig. S2‡). Similarly, UPLC-MS analysis showed that irradiation of 8 under the same conditions furnished the decaged intermediate 11 quantitatively (Fig. 2 and S3A‡). Compound 11 was stable in a neutral buffer, but upon addition of 20 equiv. of benzyl amine (BnNH2), 11 decomposed to DOX and 12, the structure of which was determined unambiguously by means of NMR spectroscopy (Fig. S3A and S25–S28‡). The release of DOX was complete in 5 min, and the yield was essentially quantitative. In contrast, no reaction occurred upon incubation of 8 and BnNH2 in the absence of light (Fig. 2C). These results indicate that the PC4AP was a stable linker and that both light and a primary amine were required to trigger release of the drug.
 | ||
| Fig. 2 Mechanism of primary-amine-catalyzed disassembly of the PC4AP linker. (A) Proposed mechanism of disassembly of the PC4AP linker in 8 by irradiation and subsequent reaction with BnNH2. (B) Disassembly of the PC4AP linker in 10 by irradiation and subsequent reaction with BnNH2. (C) Ultra performance liquid chromatography-mass spectrometry (UPLC-MS) analysis of light-triggered decomposition of 8 in the presence of BnNH2. (D) UPLC-MS analysis of light-triggered decomposition of 8 in the presence of BnNH2 and NaBH3CN. DOX was reduced by NaBH3CN to give DOXR, which contains two more hydrogen atoms than DOX. (E) UPLC-MS analysis of decomposition of 10 by irradiation and subsequent reaction with BnNH2. | ||
We propose the following mechanism for the primary-amine-catalyzed release of DOX (Fig. 2A). Nucleophilic attack at the anomeric carbon (C1) of 11 by the primary amine produces Schiff base 13. The presence of the C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond increases the acidity of the C2–H, which in turn promotes the departure of the C3-carbamate via β-elimination to furnish 14. Concomitant cleavage of the carbamate bond releases CO2 and DOX. Subsequently, hydrolysis of the C1N bond of 14 regenerates a nucleophilic amine, which intramolecularly attacks the C4-ketone, generating cyclic intermediate 15. Finally, elimination of a H2O molecule followed by isomerization produces 12.
N bond increases the acidity of the C2–H, which in turn promotes the departure of the C3-carbamate via β-elimination to furnish 14. Concomitant cleavage of the carbamate bond releases CO2 and DOX. Subsequently, hydrolysis of the C1N bond of 14 regenerates a nucleophilic amine, which intramolecularly attacks the C4-ketone, generating cyclic intermediate 15. Finally, elimination of a H2O molecule followed by isomerization produces 12.
To confirm this proposed mechanism, we carried out photodecaging of 8 in the presence of both BnNH2 and NaBH3CN. After 0.5 h, a single product with a [MH]+ value of 817.3194 was obtained (Fig. 2D and S3B‡). We assumed that this product was compound 16§ and that it had been generated via intermediates 17 and 18 (Fig. 2A).25 It is known that NaBH3CN is unreactive toward aldehydes and ketones but can reduce CN to C–N. Therefore, NaBH3CN would reduce intermediate 13 to give 17, the C2–H of which is not acidic enough for rapid elimination of the C3-carbamate. Instead, 17 would undergo rapid cyclization followed by another reduction to afford observed product 16.
Intermediates 13–15, 17, and 18 were not observed either in the presence or in the absence of NaBH3CN, suggesting that once the CN bond was formed by the reaction of the C4AP with a primary amine, the drug was rapidly released via intramolecular reactions.
Similarly, photoirradiation of 10 generated intermediate 19, which decomposed to release PyrCH2OH quantitatively upon treatment with BnNH2 for 5 min (Fig. 2B, E and S4‡). This result indicates that a payload attached to the PC4AP linker via a carbonate bond could be released as efficiently as a payload attached via a carbamate bond.
Requirement of either a lysine residue or an N-terminal amine for drug release from peptide-PC4AP-drug conjugates
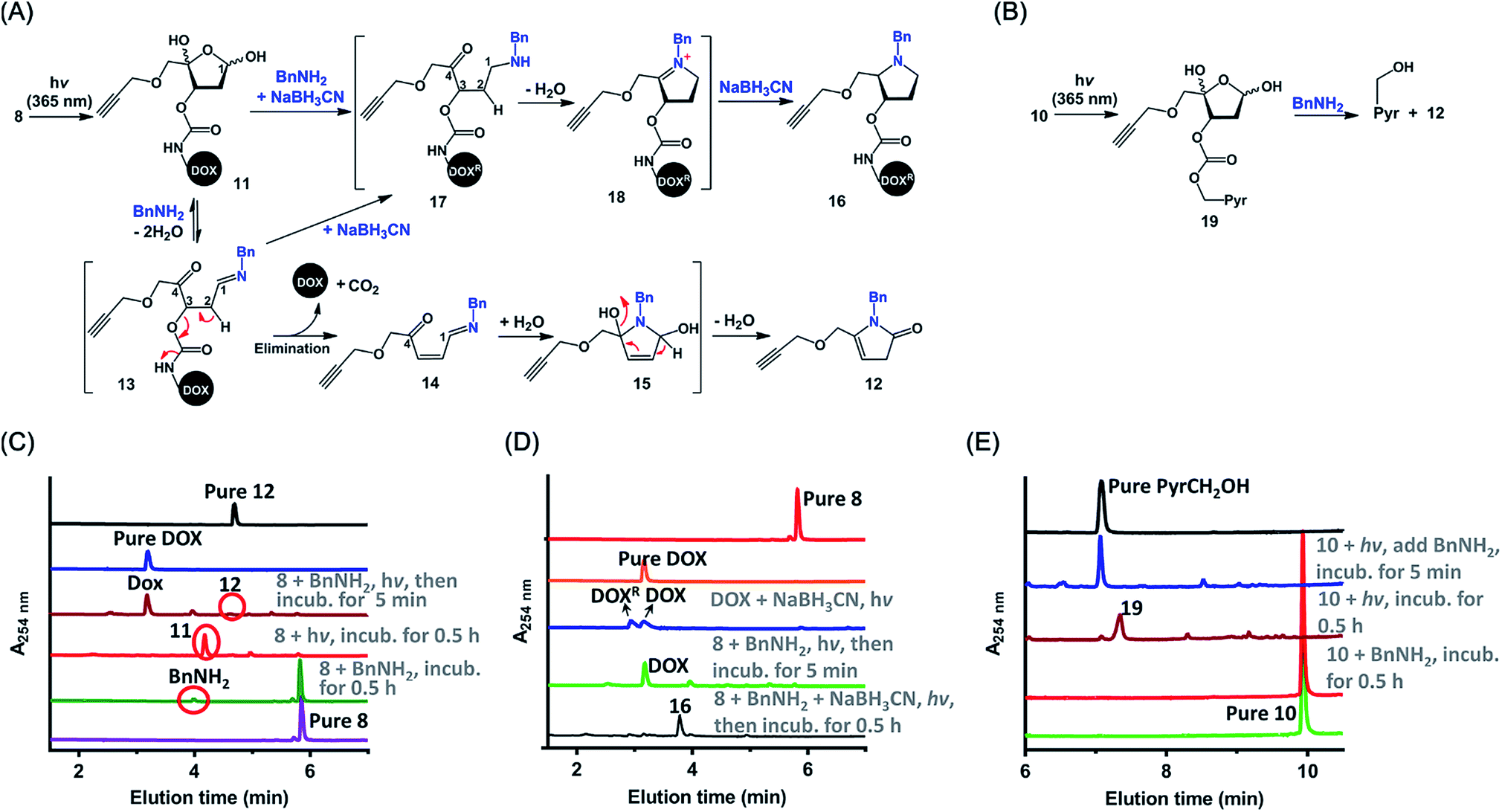
The above-described results confirmed that the intermolecular reaction with BnNH2 could trigger rapid disassembly of the PC4AP linker. To determine whether a primary amine in a peptide, such as the ε-amine of a Lys residue or the N-terminal amine, could induce efficient drug release from a peptide-PC4AP-drug conjugate via an intramolecular process, we prepared peptide-PC4AP-DOX conjugates 25–29 by attaching 9 to the Cys residues of peptides 20–24 (Fig. 3A). Irradiation of 25, the Cys residue of which is flanked by the N-terminal Gly and a Lys residue, followed by incubation in phosphate buffer (pH 7.5) at 37 °C for 5 min led to the formation of intermediate 30 and release of approximately 50% of the DOX (Fig. 3B). The amount of intermediate 30 decreased with increasing incubation time, and complete release of DOX was achieved after incubation for 1 h. Simultaneously, the peptide moiety was transformed into a cyclic species (Fig. S5A‡). In contrast, irradiation of conjugate 26, which lacks the N-terminal amine and has no Lys residues, followed by incubation in phosphate buffer for 0.5 h produced intermediate 31 quantitatively, and none of the DOX was released (Fig. 3C). We also found that photoirradiation of conjugates 27 and 28, which contain either an N-terminal amine or a Lys residue, respectively (but not both), resulted in efficient release of DOX (Fig. S5C and D‡). Taken together, these results confirm that only a primary amine in the peptides (i.e., the ε-amine of Lys and the N-terminal amine) could intramolecularly induce drug release from the peptide-PC4AP-drug conjugates.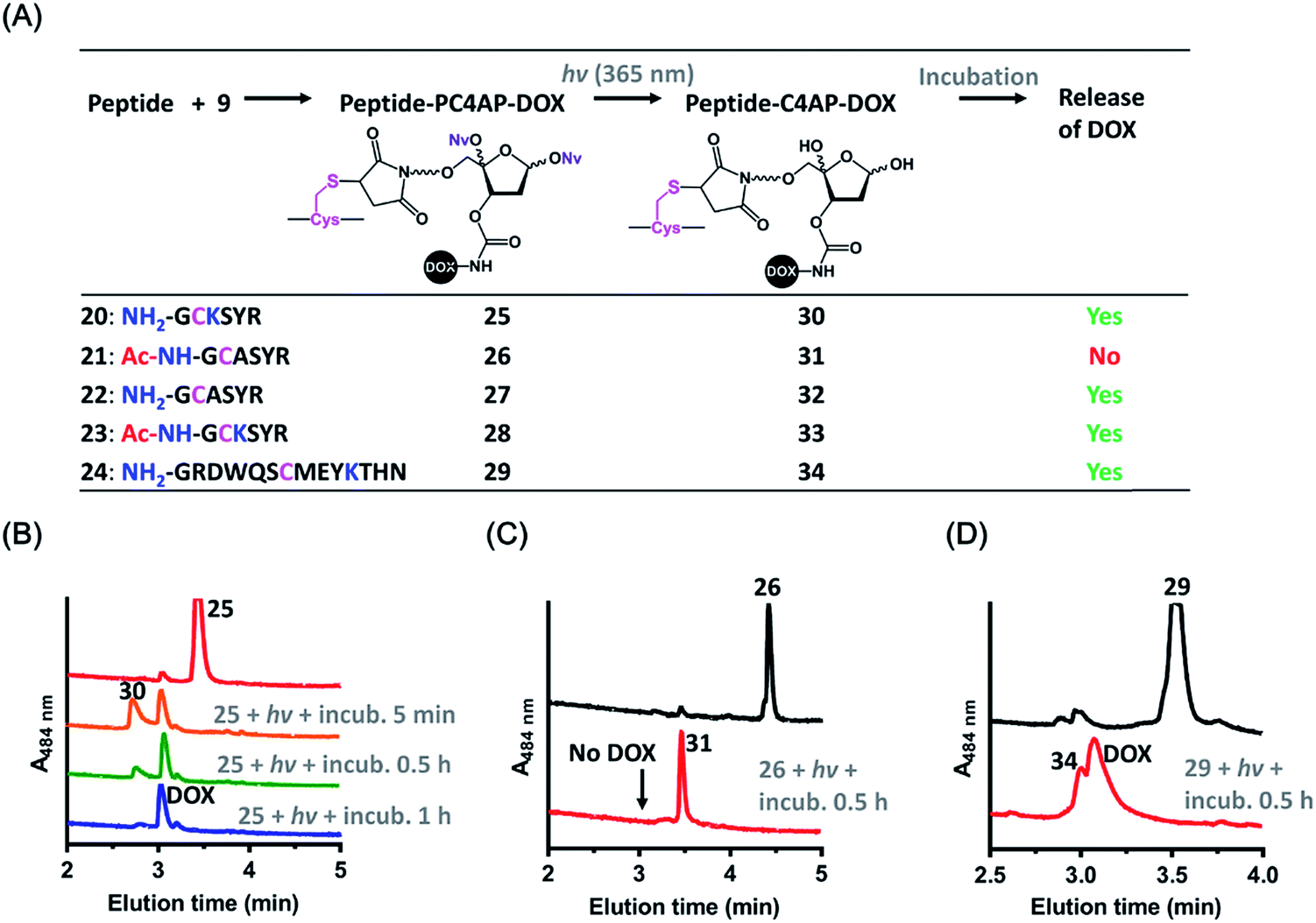 | ||
| Fig. 3 Light-triggered release of DOX from peptide-PC4AP-DOX conjugates. (A) Overview of DOX release from peptide-PC4AP-DOX conjugates 25–29 upon photoirradiation. (B) UPLC-MS analyses of light-triggered decomposition of conjugate 25. (C) UPLC-MS analyses of light-triggered decomposition of conjugate 26. (D) UPLC-MS analyses of light-triggered decomposition of conjugate 29. Reaction conditions: a solution of each conjugate (1 mM) in phosphate buffer (20 mM, pH 7.5) was photoirradiated at 365 nm for 3 min and then incubated in the dark. | ||
We also demonstrated that DOX release from peptide-PC4AP-DOX conjugates was not noticeably affected by the distance between the Cys anchor and the Lys or the N-terminal amine. For instance, even though the Lys and the N-terminal Gly of peptide-PC4AP-DOX conjugate 29 are four and six residues, respectively, away from the Cys anchor, photoirradiation of 29 followed by incubation for 0.5 h led to release of 70% of the DOX (Fig. 3D), suggesting that the drug-release efficiency of this conjugate was similar to that of 25. Given that the PC4AP linker is anchored to the side chain of Cys via a flexible 25-atom-long chain, distance-independent drug release is reasonable.
Efficient cellular delivery of DOX by means of an H3-PC4AP-DOX conjugate
Cell-penetrating peptides (CPPs), which are short peptides that can translocate across cell membranes, have been extensively used for intracellular cargo transport both in vitro and in vivo.36 We therefore conjugated 9 to a small CPP, histone H3,38 and demonstrated the utility of the resulting conjugate for controlled cellular uptake of DOX. Specifically, the H3 mutant H3-V35C, which contains 13 Lys residues but only one Cys (at position 35),37 was treated with 1.1 equiv. of 9 in HEPES buffer (pH 7.5) to afford H3-PC4AP-DOX (36) after purification by gel filtration (Fig. 4A and S6‡). A solution of 36 in phosphate buffer (20 mM, pH 7.5) was irradiated at 365 nm for 3 min and then incubated at 37 °C for 2 h. UPLC-MS analysis of the reaction mixture showed that more than 70% of the DOX was released (Fig. 4B). The remaining carrier protein exhibited a [MH]+ value of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 746, which is consistent with the proposed cyclic structure 38 ([MH]+, calcd 15746). A very small amount of photodecaged intermediate 37 ([MH]+, calcd 16351, found 16351) was observed as well (Fig. S7‡).
746, which is consistent with the proposed cyclic structure 38 ([MH]+, calcd 15746). A very small amount of photodecaged intermediate 37 ([MH]+, calcd 16351, found 16351) was observed as well (Fig. S7‡).
 | ||
| Fig. 4 Preparation and light-triggered disassembly of H3-PC4AP-DOX (36). (A) Structures of 36 and its decomposition products. (B) UPLC-MS analyses of light-triggered disassembly of 36 in phosphate buffer (20 mM, pH 7.5) at 37 °C. (C) Kinetics and photoirradiation-dependence of DOX release from 36 in phosphate buffer (20 mM, pH 7.5) at 37 °C. | ||
We also assessed the kinetics of DOX release. Specifically, a solution of 36 in phosphate buffer (20 mM, pH 7.5) was irradiated at 365 nm for 5 min and then incubated in the dark at 37 °C. Aliquots were periodically removed, quenched with NaBH3CN, and analyzed by means of 15% SDS-PAGE (Fig. S8‡). Quantification of the fluorescence intensity of each band revealed that 75% of the DOX was released after 5 min of incubation, and the amount reached 85% at 1 h (Fig. 4C). In contrast, little, if any, DOX was released when the irradiation step was omitted. Taken together, these results confirm that the PC4AP linker was stable under neutral conditions but after photodecaging, the self-immolative process occurred spontaneously, releasing the cargo from the carrier protein rapidly and efficiently.
Any intermediate 37 present in the aliquots removed from the reaction would have been quenched by the NaBH3CN to afford a stabilized product (as 16 in Fig. 2A), which would have prevented release of DOX from H3 during SDS-PAGE analysis. Thus, rapid release of DOX observed by SDS-PAGE analysis can be attributed solely to intramolecular Lys-catalyzed reactions. DOX release from H3-PC4AP-DOX (36) upon photoirradiation was even faster than release from peptide-PC4AP-DOX conjugate 25. This result led us to speculate that in addition to the Lys residue and the N-terminal amine group, other residues in the carrier protein may also have promoted linker disassembly. For instance, acidic residues may have catalyzed Schiff base formation between the linker and a primary amine, and basic residues may have catalyzed the elimination step (see Fig. 2A).
To demonstrate the utility of our strategy for cellular delivery of drugs, we treated HeLa cells with 10 μM H3-PC4AP-DOX (36) or free DOX for 3 h at 37 °C. Fluorescence confocal microscopy analysis of the treated cells showed that, as expected,38 free DOX was efficiently transported to the nucleus, where it bound to genomic DNA (Fig. 5A, left panel); in contrast, in cells treated with 36, the red fluorescence of DOX was observed predominantly in the cytoplasm, indicating that the protein-drug conjugate successfully transported DOX to the cytoplasm, where it was not released in the absence of irradiation (Fig. S9‡). After medium removal and addition of fresh DMEM (Dulbecco's Modified Eagle Medium), the 36-treated cells were irradiated at 365 nm for 5 min and then incubated at 37 °C for another 3 h; under these conditions, DOX was transported to the nucleus (Fig. 5A, middle panel). Given that DOX remained in the cytoplasm of unirradiated cells (Fig. 5A, right panel), the DOX transport to the nucleus of the irradiated cells can be attributed to photo triggered release of free DOX from the H3 carrier protein.
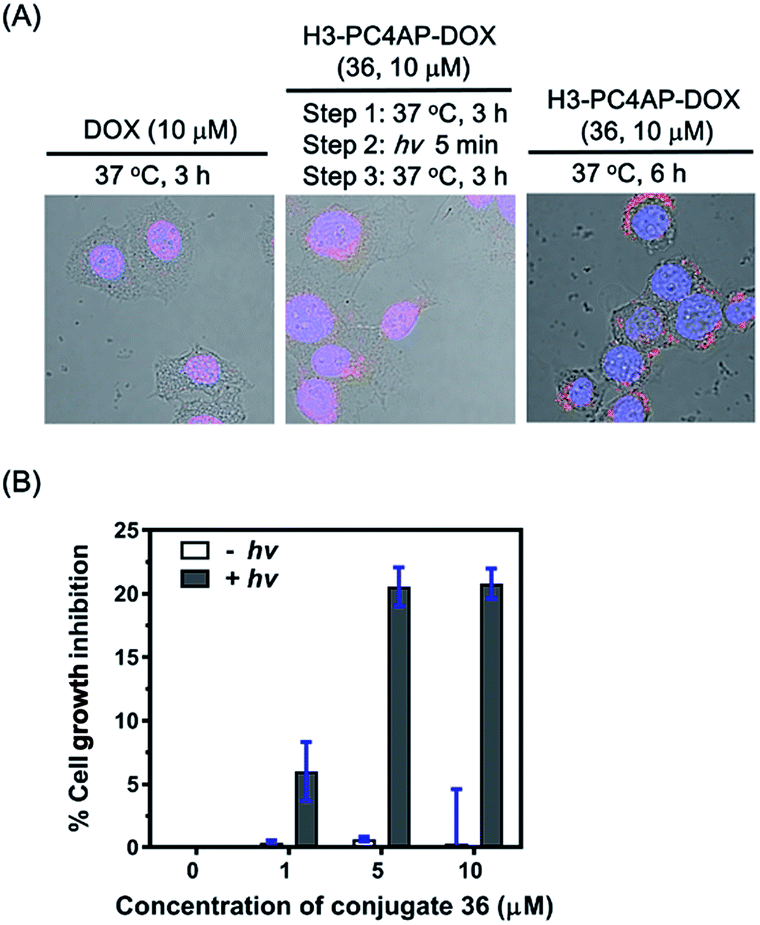 | ||
| Fig. 5 Controlled delivery of DOX into HeLa cells by H3-PC4AP-DOX (36). (A) Fluorescence confocal microscopy analyses showing the cellular location of DOX after treatment of HeLa cells with DOX and 36. The images were obtained by overlaying the DAPI channel (excitation at 408 nm and emission at 425–475 nm), the DOX channel (excitation at 561 nm and emission at 570–620 nm), and the DIC channel. (B) Light-dependence of 36 cytotoxicity to HeLa cells. | ||
Cell counting kit-8 assays showed that incubation of HeLa cells with H3-PC4AP-DOX (36) followed by photoirradiation inhibited cell growth by up to 20%. In contrast, no cytotoxicity was observed in the absence of light, even at high concentrations of the conjugate (Fig. 5B). These results once again confirm that 36 was stable inside cells but that it released active DOX upon photoirradiation.
Targeted cellular delivery of DOX by an antibody-PC4AP-DOX conjugate
The above-described results inspired us to determine whether the PC4AP linker could be used for targeted drug delivery with an antibody as the carrier.39,40 As the antibody, we chose trastuzumab, a clinical monoclonal IgG that recognizes and induces internalization of HER2, a receptor that is highly expressed in some breast cancer cell lines.41,42 To prepare trastuzumab-DOX conjugates with the PC4AP linker, we treated trastuzumab with 4 equiv. of tris(2-carboxyethyl)phosphine (TCEP) in phosphate-buffered saline for 2 h to reduce the interchain disulfide bonds, and then incubated it with 8 equiv. of Mal-PC4AP-DOX (9) at 37 °C for 1 h to give antibody-drug conjugate (ADC) 39, which was purified by gel filtration. LC-MS analysis indicated the drug/antibody ratio to be 8 (Fig. 6A and S10‡).The linker of an ADC must be stable enough to prevent off-target effects. To test the stability of the PC4AP linker in ADC 39, we incubated it in human serum at 37 °C. Aliquots were removed periodically, quenched with NaBH3CN, and analyzed by means of 12% reducing glycine-SDS-PAGE (Fig. S11‡). The heavy and light chains appeared as two separated bands in the gel, and quantification of the fluorescence intensities of the bands revealed that DOX was released very slowly from both chains (Fig. 6B and S11‡); approximately 85% of the DOX remained attached to the antibody after 72 h of incubation. Note, however, that DOX release from the antibody was not attributed to cleavage of the PC4AP linker, given that no released free DOX was observed. Instead, we detected some slower-migrating fluorescent bands (Fig. S11‡), which we attributed to transfer of the conjugated moiety (Mal-PC4AP-DOX, 9) from the antibody to other serum proteins (e.g., human serum albumin) via thiol exchange reactions.43,44 That is, the PC4AP linker itself was stable in human serum.
Photoirradiation of 39 at 365 nm for 5 min and subsequent incubation in human serum led to release of 80% of the free DOX from the antibody after incubation for 10 min (Fig. 6C and S12‡), confirming that once the photocage was removed, self-immolative disassembly of the C4AP linker in the ADC was rapid and efficient.
Zhang et al. conjugated DOX to trastuzumab via an acid-labile MMCCH linker.42 At pH 7.4, both linker degradation and drug release were rather slow (the release rate was 25% per day), but in a lysosomal environment (pH 5.4), 80% of the DOX was released from the ADC after 24 h. Our trastuzumab-PC4AP-DOX conjugate was much more stable than the trastuzumab-MMCCH-DOX conjugate under physiologically relevant conditions. In addition, after being triggered by a biocompatible stimulus, drug release was more efficient from the former than from the latter. Thus, the PC4AP linker can be expected to be useful for ADC development.
Next, we evaluated the cytotoxicity of the trastuzumab-PC4AP-DOX conjugate (39). After incubation of two fixed breast cancer cell lines, SK-BR-3 (HER2 positive) and MCF7 (HER2 negative), with ADC 39, an immunofluorescence assay showed that 39 bound specifically to the antigen HER2 on the surface of the SK-BR-3 cells (Fig. S13‡). Having confirmed the antigen-specific binding, we determined the cytotoxicity of 39 to the SK-BR-3 cells over a wide concentration range. ADC 39 showed marginal cytotoxicity in the absence of light (Fig. 6D), whereas incubation of the cells with 39 and subsequent irradiation led to decreased cell viability in a dose-dependent manner; the potency of 39 (IC50 = 203 nM) was comparable to that of free DOX (IC50 = 186 nM). Furthermore, we found that Mal-PC4AP-DOX (9) was efficiently taken up by the SK-BR-3 cells but showed no obvious cytotoxicity either in the presence or in the absence of light. Given that the free amino group of DOX is vital for its cytotoxicity,45 this result indicates that irradiation did not release active DOX from 9. That is, photodecaging was not sufficient to release DOX from the PC4AP linker in the absence of intramolecular catalysis by a primary amine inside the cells. In other words, light-triggered cellular release of DOX from the antibody-PC4AP-DOX conjugate (39) occurred by means of intramolecular catalysis, as we proposed.
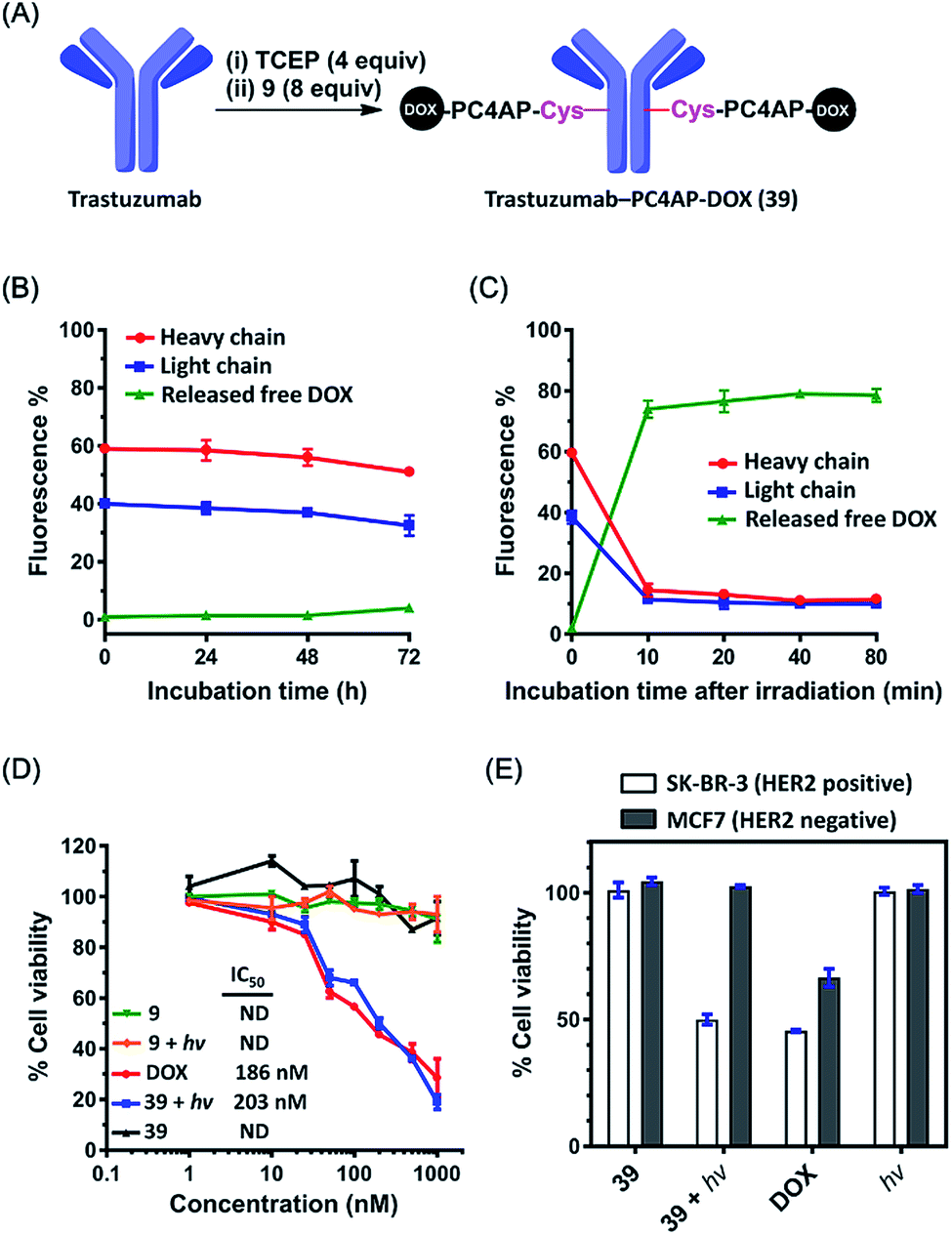 | ||
| Fig. 6 Targeted delivery of DOX to breast cancer cell lines via a trastuzumab-PC4AP-DOX conjugate (39). (A) Preparation of 39. (B) DOX fluorescence quantification revealed the kinetics of DOX release from the heavy and light chains of 39 upon incubation in human serum. (C) DOX fluorescence quantification revealed the kinetics of DOX release from the heavy and light chains of 39 upon photoirradiation and subsequent incubation in human serum. (D) Light- and concentration-dependent cytotoxicities of 9, 39, and DOX against SK-BR-3 cells (HER2 positive). (E) Comparison of the cytotoxicities of 39 (200 nM) against SK-BR-3 (HER2 positive) and MCF7 (HER2 negative) cell lines. | ||
Finally, we evaluated the cell line specificity of ADC 39 (Fig. 6E). SK-BR-3 (HER2 positive) and MCF7 (HER2 negative) cells were preincubated with 200 nM 39 for 6 h, the medium was changed to fresh DMEM, and the cells were photoirradiated and then incubated for an additional 48 h. Cell viability assays showed that the irradiated SK-BR-3 cells displayed remarkably decreased cell viability, whereas little, if any, effect on the MCF7 cells was observed, whether they were irradiated or not. This result confirms that conjugation of the drug to the antibody via the PC4AP linker enabled targeted drug delivery in a controlled manner.
Conclusions
In summary, we have described the use of a PC4AP as a light-responsive, self-immolative linker for controlled drug delivery. Amine- or hydroxyl-bearing drugs can be easily introduced at C3 of the linker by means of a carbamate or carbonate bond, and the C5–OH is functionalized for bioconjugation to a carrier peptide, a protein, or an antibody. After cellular uptake of the conjugate, photoirradiation removes the photolabile O-nitrobenzyl groups to generate a C4AP intermediate. Any nearby primary amine, including the ε-amine of Lys and the N-terminal amine, on the carrier peptide or protein spontaneously catalyzes release of the drug via an addition–elimination cascade.The PC4AP linker described herein has several merits. First, because it is stable under physiologically relevant conditions, off-target effects induced by unwanted release of the payload are minimal. Second, after photodecaging, self-catalyzed drug release is rapid and efficient. No toxic side products are generated during linkage disassembly. Third, the PC4AP linker is cleaved by photoirradiation. Because of the high spatiotemporal resolution and noninvasive nature of light-mediated cleavage, the PC4AP linker has great potential utility for manipulation of biological processes in vivo. Moreover, changing the O-nitrobenzyl group to some other protecting group would be an easy way to make the system responsive toward other stimuli or to shift the activation wavelength, indicating the versatility of this linker. Fourth, Lys is the most abundant amino acid residue in typical proteins; it is widely distributed in CPPs and on the outer surface of proteins. In addition to Lys, any other primary amine, including the N-terminal amine, can catalyze disassembly of the PC4AP linker. Therefore, this linker could be used with a broad range of carriers, including CPPs, proteins, antibodies, amine-bearing polymers such as polyethyleneimine, and amine-functionalized nanoparticles. These advantages make the PC4AP linker invaluable for controlled cargo delivery, and it can be expected to find a wide range of practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21877064 and 21572109), the National Key Research and Development Program of China (2017YFD0200501 and 2017YFA0207900) and the Fundamental Research Funds for the Central Universities, Nankai University (63191523).Notes and references
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- X. Ji, Z. Pan, B. Yu, L. K. De la Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC.
- Y. Dai, X. Chen and X. Zhang, Polym. Chem., 2019, 10, 34–44 RSC.
- X. He, J. Li, S. An and C. Jiang, Ther. Delivery, 2013, 4, 1499–1510 CrossRef CAS PubMed.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066–7070 CrossRef CAS PubMed.
- M. H. Lee, E.-J. Kim, H. Lee, H. M. Kim, M. J. Chang, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 16380–16387 CrossRef CAS PubMed.
- R. Mooney, A. A. Majid, J. Batalla, A. J. Annala and K. S. Aboody, Adv. Drug Delivery Rev., 2017, 118, 35–51 CrossRef CAS PubMed.
- J. C. Kern, M. Cancilla, D. Dooney, K. Kwasnjuk, R. Zhang, M. Beaumont, I. Figueroa, S. Hsieh, L. Liang, D. Tomazela, J. Zhang, P. E. Brandish, A. Palmieri, P. Stivers, M. Cheng, G. Feng, P. Geda, S. Shah, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, P. G. Schultz, Y. Sun and R. M. Garbaccio, J. Am. Chem. Soc., 2016, 138, 1430–1445 CrossRef CAS PubMed.
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768–2798 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., Int. Ed., 2015, 54, 13635–13638 CrossRef CAS PubMed.
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388–3432 CrossRef CAS PubMed.
- T. Ramasamy, H. B. Ruttala, B. Gupta, B. K. Poudel, H.-G. Choi, C. S. Yong and J. O. Kim, J. Controlled Release, 2017, 258, 226–253 CrossRef CAS PubMed.
- R. J. Amir, N. Pessah, M. Shamis and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 4494–4499 CrossRef CAS PubMed.
- A. Alouane, R. Labruere, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed.
- M. Gisbert-Garzaran, M. Manzano and M. Vallet-Regi, Chem. Eng. J., 2018, 340, 24–31 CrossRef CAS.
- P. Wang, Y. Song, L. Zhang, H. He and X. Zhou, Curr. Med. Chem., 2005, 12, 2893–2913 CrossRef CAS PubMed.
- R. V. Kolakowski, K. T. Haelsig, K. K. Emmerton, C. I. Leiske, J. B. Miyamoto, J. H. Cochran, R. P. Lyon, P. D. Senter and S. C. Jeffrey, Angew. Chem., Int. Ed., 2016, 55, 7948–7951 CrossRef CAS PubMed.
- B. Fan, J. F. Trant, G. Hemery, O. Sandre and E. R. Gillies, Chem. Commun., 2017, 53, 12068–12071 RSC.
- M. Aso, M. Kondo, H. Suemune and S. M. Hecht, J. Am. Chem. Soc., 1999, 121, 9023–9033 CrossRef CAS.
- M. Aso, K. Usui, M. Fukuda, Y. Kakihara, T. Goromaru and H. Suemune, Org. Lett., 2006, 8, 3183–3186 CrossRef CAS PubMed.
- C. Z. Zhou, J. T. Sczepanski and M. M. Greenberg, J. Am. Chem. Soc., 2013, 135, 5274–5277 CrossRef CAS PubMed.
- Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 6146–6151 CrossRef CAS PubMed.
- B. Yang, A. Jinnouchi, K. Usui, T. Katayama, M. Fujii, H. Suemune and M. Aso, Bioconjugate Chem., 2015, 26, 1830–1838 CrossRef CAS PubMed.
- C. Gatanaga, B. Yang, Y. Inadomi, K. Usui, C. Ota, T. Katayama, H. Suemune and M. Aso, ACS Chem. Biol., 2016, 11, 2216–2221 CrossRef CAS PubMed.
- J. A. Barltrop, P. J. Plant and P. Schofield, Chem. Commun., 1966, 822–823 RSC.
- A. Patchornik, B. Amit and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 6333–6335 CrossRef CAS.
- J. Kim, J. M. Gil and M. M. Greenberg, Angew. Chem., Int. Ed., 2003, 42, 5882–5885 CrossRef CAS PubMed.
- P. T. Wong, S. Tang, J. Cannon, D. Chen, R. Sun, J. Lee, J. Phan, K. Tao, K. Sun, B. Chen, J. R. Baker and S. K. Choi, Bioconjugate Chem., 2017, 28, 3016–3028 CrossRef CAS PubMed.
- S. Ki Choi, T. Thomas, M.-H. Li, A. Kotlyar, A. Desai and J. J. R. Baker, Chem. Commun., 2010, 46, 2632–2634 RSC.
- M. Michael Dcona, Q. Yu, J. A. Capobianco and M. C. T. Hartman, Chem. Commun., 2015, 51, 8477–8479 RSC.
- G. Yu, X. Zhao, J. Zhou, Z. Mao, X. Huang, Z. Wang, B. Hua, Y. Liu, F. Zhang, Z. He, O. Jacobson, C. Gao, W. Wang, C. Yu, X. Zhu, F. Huang and X. Chen, J. Am. Chem. Soc., 2018, 140, 8005–8019 CrossRef CAS PubMed.
- O. Minenkova, L. Vesci, R. De Santis, D. Santapaola, R. Cincinelli, L. Musso, S. Dallavalle and G. Giannini, Bioorg. Med. Chem. Lett., 2018, 28, 3312–3314 CrossRef CAS PubMed.
- Z. Paryzek, R. Joachimiak, M. Piasecka and T. Pospieszny, Tetrahedron Lett., 2012, 53, 6212–6215 CrossRef CAS.
- F. Wang, Y. Wang, X. Zhang, W. Zhang, S. Guo and F. Jin, J. Controlled Release, 2014, 174, 126–136 CrossRef CAS PubMed.
- G. Li and J. Widom, Nat. Struct. Mol. Biol., 2004, 11, 763–769 CrossRef CAS PubMed.
- G. Speelmans, R. W. H. M. Staffhorst, H. G. Steenbergen and B. de Kruijff, Biochim. Biophys. Acta, 1996, 1284, 240–246 CrossRef.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- C. D. Austin, A. M. D. Mazière, P. I. Pisacane, S. M. v. Dijk, C. Eigenbrot, M. X. Sliwkowski, J. Klumperman and R. H. Scheller, Mol. Biol. Cell, 2004, 15, 5268–5282 CrossRef CAS PubMed.
- N. Zhang, M. E. Klegerman, H. Deng, Y. Shi, E. Golunski and Z. An, J. Cancer Ther., 2013, 04, 308–322 CrossRef.
- S. Kolodych, O. Koniev, Z. Baatarkhuu, J.-Y. Bonnefoy, F. Debaene, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjugate Chem., 2015, 26, 197–200 CrossRef CAS PubMed.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the 100th anniversary of Nankai University. |
| ‡ Electronic supplementary information (ESI) available: General experimental methods, spectroscopic data and supplementary figures (PDF). See DOI: 10.1039/c9sc03016f |
| § In compound 16, the DOX moiety was reduced to DOXR by NaBH3CN. Treatment of free DOX with NaBH3CN gave the same result (Fig. 2D). UPLC-MS analysis showed that DOXR contains two more hydrogen atoms than DOX (Fig. S3‡). |
| This journal is © The Royal Society of Chemistry 2019 |