
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating C–C coupling in thermocatalytic and electrocatalytic COx conversion based on surface science
Yawen
Jiang
,
Ran
Long
and
Yujie
Xiong
 *
*
Hefei National Laboratory for Physical Science at Microscale, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), School of Chemistry and Materials Science, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: yjxiong@ustc.edu.cn
First published on 5th July 2019
Abstract
Heterogeneous thermocatalytic and electrocatalytic conversion of COx including CO and CO2 to value-added products, which can be performed through three promising approaches – syngas conversion, CO2 hydrogenation and CO2 electroreduction, are highly important to achieving a carbon-neutral cycle associated with the continuing consumption of fossil fuels. Toward the formation of value-added C2+ products, precise regulation of C–C coupling requires rational design of catalysts in all the three approaches, which usually share similar fundamentals from the viewpoint of surface science. In this article, we outline the recent advances in catalyst design for controlling C–C coupling in syngas conversion, CO2 hydrogenation and CO2 electroreduction from the viewpoint of surface science. Specifically, the fundamental insights are provided for each conversion approach, which makes a connection between thermocatalysis and electrocatalysis in terms of catalytic site design. Finally, the challenges and opportunities are discussed in the hope of inspiring new ideas to achieve more efficient C–C coupling in thermocatalytic and electrocatalytic COx conversion.
 Yawen Jiang | Yawen Jiang was born in Anhui, China, in 1995. He received his B.S. in chemistry in 2018 from Xiamen University. Since then he has been a graduate student under the tutelage of Professor Yujie Xiong at the University of Science and Technology of China. His research interests focus on the design of electrocatalysts for carbon dioxide reduction. |
 Ran Long | Ran Long was born in Anhui, China, in 1987. She received her B.S. in chemistry in 2009 and Ph.D. in inorganic chemistry under the tutelage of Professor Yujie Xiong in 2014, both from the University of Science and Technology of China (USTC). After postdoctoral training with Professors Yujie Xiong and Li Song, she is currently an associate professor working at the USTC. Her research interests focus on controlled synthesis and catalytic applications of metal nanocrystals. |
 Yujie Xiong | Yujie Xiong received his B.S. in chemical physics in 2000 and Ph.D. in inorganic chemistry in 2004, both from the University of Science and Technology of China (USTC). After four-year postdoctoral training at the University of Washington in Seattle and the University of Illinois at Urbana-Champaign, he joined the NSF-NNIN at Washington University in St. Louis as the Principal Scientist. Starting from 2011, he has been a Professor of Chemistry at the USTC. He has published 170 papers with over 18 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations (H-index 67). His research interests include inorganic materials and devices for carbon dioxide reduction, nitrogen fixation, methane conversion, water splitting and chemical production.
000 citations (H-index 67). His research interests include inorganic materials and devices for carbon dioxide reduction, nitrogen fixation, methane conversion, water splitting and chemical production.1. Introduction
At present, fossil fuels are the main energy source in the world leading to energy and environmental issues. The reserves of fossil fuels on the earth are limited but the demand for energy for human development is never-ending. In the meantime, continuing consumption of fossil fuels has led to excess emission of carbon dioxide (CO2). Consequently, the atmospheric CO2 concentration has exceeded the safety limit of 350 ppm, and will predictably reach nearly 600 ppm by the end of this century.1,2 It has been proposed that environmental issues such as global warming and ocean acidification are very relevant to such massive CO2 emission.3,4 For this reason, reducing the dependence on fossil fuels and the atmospheric CO2 concentration are two urgent issues for the future development of mankind.Chemical conversion of COx (x = 1, 2) to value-added products has gained increasing research interest, given its potential roles in addressing both the energy and environmental issues aforementioned. To achieve such chemical conversion, three heterogeneous catalytic processes – syngas conversion, CO2 hydrogenation and CO2 electroreduction – have been extensively explored. Syngas conversion produces hydrocarbons and oxygenates which can be used as liquid fuels or building-block chemicals.5,6 The liquid fuels and chemicals produced from syngas are almost free of sulfur, aromatic compounds and other toxic impurities compared to those derived from crude oil so syngas has been considered as an ideal non-petroleum energy resource.7 CO2 hydrogenation offers a variety of products such as carbon monoxide (CO), methane (CH4), formic acid (HCOOH), methanol (CH3OH), hydrocarbons and higher alcohols.8 This process can not only contribute to reducing the atmospheric CO2 concentration, but also helps to shift energy consumption away from fossil fuels. In recent years, CO2 electroreduction driven by renewable electricity under mild conditions has gained increasing research interest.9 As the required electricity can be generated through photovoltaics or wind power, this approach can be easily combined with renewable energy. Moreover, various products such as CO, CH4, HCOOH, and CH3OH, and multi-carbon products can be obtained from electrocatalytic CO2 reduction.
Product selectivity is a highly important parameter to chemical conversion of COx. As compared with C1 products, converting COx to more valuable C2+ products (including C2 products) is more attractive. However, the formation of targeted C2+ products is largely bottlenecked by the high kinetic barrier of C–C coupling and the competition with H–H and C–H bond formation, setting a grand challenge.10 In order to overcome the challenge, the important key is to rationally design catalysts which can precisely regulate C–C coupling to produce the targeted C2+ products with high activity and selectivity. The catalyst design for regulating C–C coupling is typically achieved by controlling crystal facets, tuning catalyst sizes, adding promoters and other strategies, all of which have a great influence on the structural and electronic properties of catalysts based on surface science.
In recent years, significant breakthroughs have been made in syngas conversion, CO2 hydrogenation and CO2 electroreduction, between which a strong correction can be sorted out from the viewpoint of surface science. In this article, we will focus on the recently developed strategies for regulating C–C coupling to produce C2+ products in thermocatalysis and electrocatalysis. In the following sections, the fundamentals and typical catalyst design strategies based on surface science will be outlined for syngas conversion, CO2 hydrogenation and CO2 electroreduction. Finally, we will propose the challenges and opportunities for regulating C–C coupling by making a connection between thermocatalysis and electrocatalysis.
2. Regulating C–C coupling in syngas conversion
2.1 A brief overview of syngas conversion to C2+ products
Syngas is a mixture of CO and H2 that can be obtained from natural gas, coal and biomass.5 The research of syngas conversion has a history of more than 100 years. In 1902, Sabatier and Senderens synthesized methane through the catalytic hydrogenation of carbon monoxide.11 In 1923, Fischer and Tropsch successfully obtained long-chain hydrocarbons from the hydrogenation of CO over Fe/ZnO and Co/Cr2O3 catalysts.11 From then on, the production of long-chain hydrocarbons from syngas conversion became known as the Fischer–Tropsch (FT) synthesis. The FT technology has been put into practice in large-scale industrial production worldwide. Higher alcohols may also be obtained via syngas conversion. However, only two additional small-scale processes for higher alcohol synthesis (HAS) have been reported to reach commercialization based on syngas conversion.6In the past century, various catalysts have been explored for syngas conversion.7 Cobalt-, iron- and ruthenium-based catalysts are most suitable for the Fischer–Tropsch synthesis as the balance between CO dissociation and H2 dissociation abilities can be achieved on the surface of these three metals.7 In particular, ruthenium-based materials are the most active catalysts for syngas conversion. They offer a high selectivity for long-chain hydrocarbons and a low production of methane, but their high price hinders large-scale industrial applications. Iron catalysts show a higher selectivity to lighter hydrocarbons and have a high activity for the water–gas shift reaction (eqn (1), WGS), while cobalt catalysts are preferred to produce heavier hydrocarbons.11,12 Essentially, the active phases of the two catalyst categories are iron carbides and metallic cobalt, respectively.11,13 To obtain higher alcohols, the used catalysts can be classified into four categories: Rh-based, Mo-based, modified FT synthesis and modified methanol synthesis systems.6
| CO + H2O ⇌ CO2 + H2 | (1) |
To better design catalysts, it is imperative to understand the mechanisms of syngas conversion. However, the reaction process of syngas conversion is very complicated so the exact mechanism is still not fully understood. Fig. 1a illustrates a widely accepted carbide-based mechanism on the surface of traditional FT catalysts.6,7,14–17 During the formation of hydrocarbons, CO is dissociated to form atomic species. Subsequently, the surface carbon species are hydrogenated to yield CHx intermediates (x = 0–3). The generated CHx can undergo further hydrogenation to produce methane or C–C coupling to form alkyl intermediates with different carbon numbers. The alkyl intermediates may be converted into paraffins and olefins through H addition termination and β-CH cleavage, respectively.6 If the adsorbed CO* is further combined with the alkyl species, higher alcohols will be formed by the subsequent protonation.18 In parallel, methanol can be generated by direct hydrogenation of the adsorbed CO*. As the C–C coupling of CHx intermediates is uncontrollable on the catalyst surface, the hydrocarbon products are statistically distributed, which can be described by the Anderson–Schulz–Flory (ASF) model.7,19,20 This model can be represented by the following formula:
| Mn = (1 − α)αn−1 | (2) |
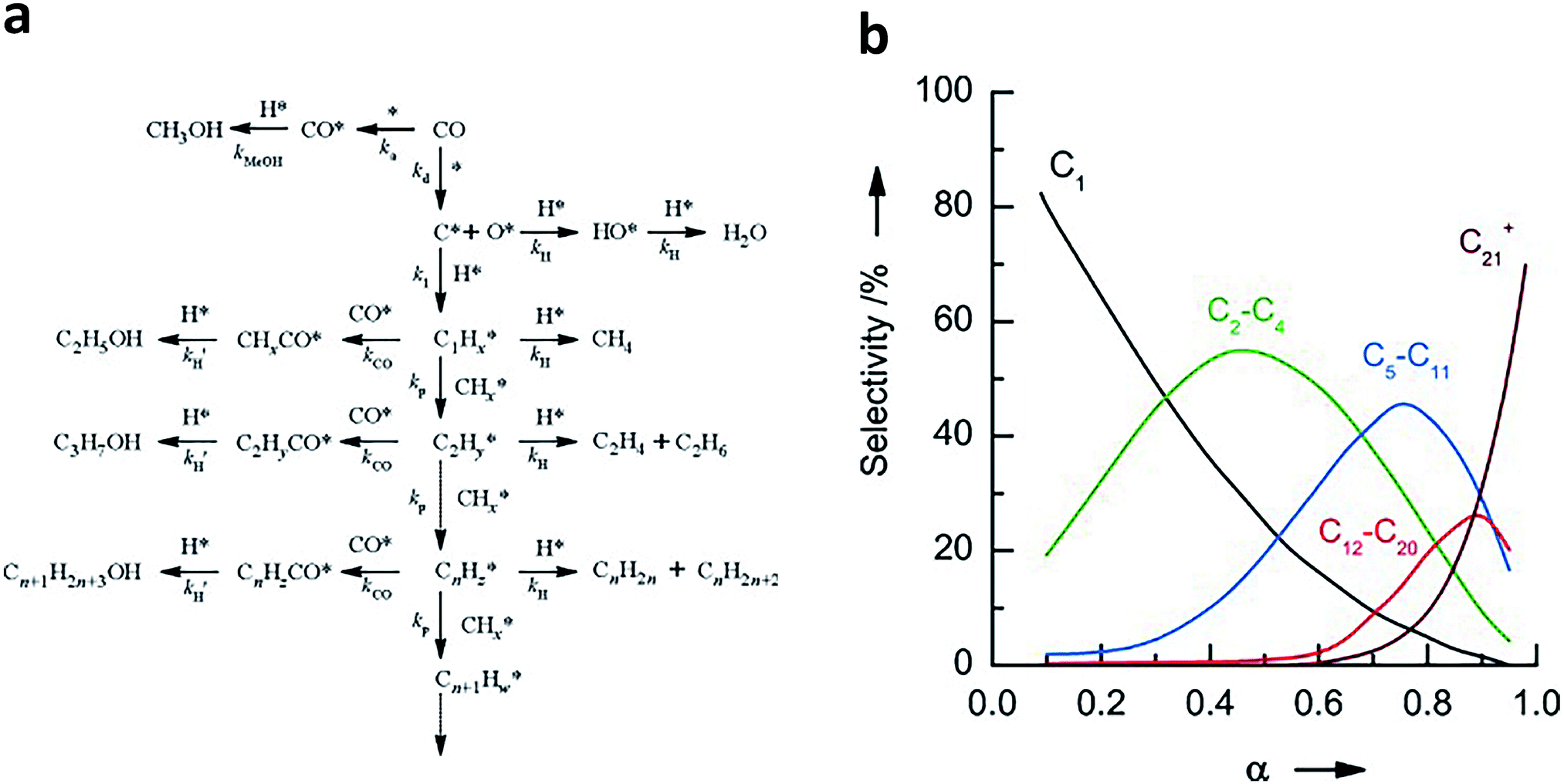 | ||
| Fig. 1 (a) The carbide-based reaction mechanism for syngas conversion to hydrocarbons and alcohols. Reprinted from ref. 17 with permission from Elsevier. (b) Typical product distribution of syngas conversion predicted by the ASF model. The product selectivity is expressed as the molar percentage of a particular range of products on a carbon basis. Reprinted from ref. 20 with permission from John Wiley & Sons. | ||
As shown above, the process of syngas conversion involves a series of steps so it is very difficult to precisely control the process by using a single type of active site (e.g., Fe or Co). According to the ASF law, the predicted maximum selectivities for C2–C4 (including both olefins and paraffins), C5–C11 (gasoline), C8–C16 (jet fuel) and C10–C20 (diesel) hydrocarbons are 58%, 48%, 41% and 40%, respectively.21 In recent years, the reaction coupling strategy based on bifunctional (or multifunctional) catalysts has been extensively studied in syngas conversion, achieving many significant breakthroughs. Notably, the C–C coupling mechanisms involved in the bifunctional catalyst system are different from carbide-based mechanisms on the surfaces of traditional FT catalysts. In general, one component in the bifunctional catalysts converts syngas to intermediates such as hydrocarbons, ketenes, methanol and dimethyl ether, and subsequently, the reaction intermediates enter another component (usually a zeolite) and undergo hydrocracking/isomerisation, hydrogenolysis, C–C coupling or dehydrogenative aromatisation to form liquid fuels, olefins or aromatics as final products. Such bifunctional catalyst systems and reaction mechanisms for syngas conversion were summarized and discussed in detail in a recent review by the Wang research group.21
2.2 Controlling crystal facets
Heterogeneous catalysis occurs on the surface of catalysts, where reactant molecules are adsorbed, activated and converted to final products through a series of elemental reaction steps. The adsorption and activation of reactant molecules and reaction intermediates are highly dependent on the exposed catalyst facets, due to the fact that crystal facets provide a knob for tailoring many parameters such as the surface atomic arrangement and surface electronic state.22As such, controlling the crystal facets of the catalyst surface allows tuning C–C coupling to form specific C2+ products during the syngas conversion process. For example, Sun and coworkers reported a Co2C nanoprism which exhibited unexpected activity for syngas conversion.23 Using the Co2C nanoprism as a catalyst, short-chain olefins can be produced with a high selectivity of up to 60% under mild Fischer–Tropsch reaction conditions, beyond the classical ASF distribution. Meanwhile, the selectivity for undesired methane was limited to about 5%. In sharp contrast, cobalt carbide had been generally considered inactive for C–C coupling as it tremendously produced methane according to earlier reports.24 Transmission electron microscopy characterization (Fig. 2a and b) and DFT calculation (Fig. 2c and d) revealed that the (101) and (020) facets preferentially exposed on Co2C nanoprisms were responsible for the high selectivity for the production of short-chain olefins and the low selectivity for methane formation. Based on DFT calculation results, CH2CH2 intermediates are well stabilized on Co2C(101), and the step of CHx species hydrogenation to methane has high energy barriers on both Co2C(101) and Co2C(020). As such, exposing appropriate facets of catalysts can stabilize some key intermediates and shift selectivity to a specific range of products in syngas conversion.
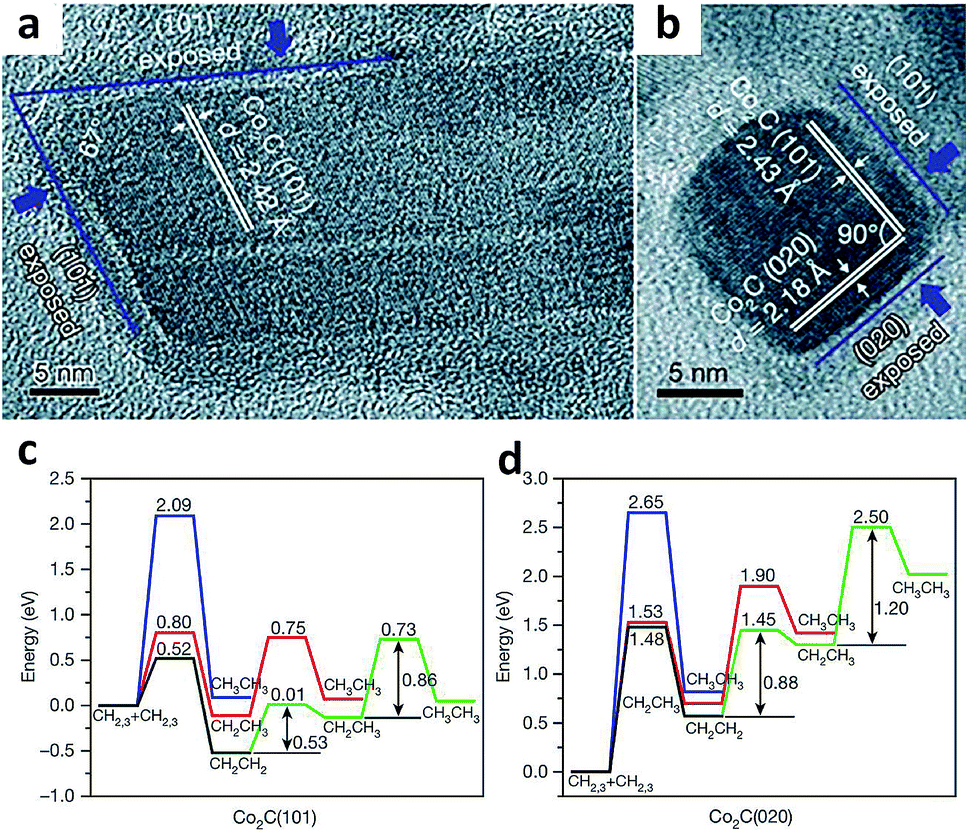 | ||
| Fig. 2 (a, b) High-resolution TEM images of Co2C nanoprisms. Energy profiles for pathways that lead to the formation of CH2CH2 and CH3CH3 on the (c) Co2C(101) surface and (d) Co2C(020) surface. Reprinted from ref. 23 with permission from Nature Publishing Group. | ||
2.3 Tuning the catalyst size
Size effects play an important role in heterogeneous catalysis. Tuning the particle size can not only change the surface-to-volume ratio of catalysts, but also alter their surface structure. For example, shrinking the size of catalysts can increase the fractions of corner and edge atoms,25 forming more highly under-coordinated atoms at the exposed surface. In the meantime, the electronic state of the catalyst surface and the adsorption energy of reactant molecules can be varied through tuning the size.22The size effects of cobalt catalysts have been extensively studied in the Fischer–Tropsch synthesis.25 The size control offers the capability of tuning C–C coupling on the catalyst surface. de Jong and coworkers systematically studied the size effect of cobalt particles on the activity and product selectivity of the Fischer–Tropsch synthesis. They chose graphitic carbon nanofibers (CNFs) as an inert support material for loading Co particles with different sizes.26 It turned out that the cobalt particles with a smaller size (<5 nm) showed a very high selectivity toward methane and a low turnover frequency (TOF). Under industrially relevant conditions (35 bar), the selectivity of C5+ products was promoted from 51 wt% to 85 wt% by increasing the cobalt particle size from 2.6 nm to 16 nm while the CH4 formation was suppressed. Later, de Jong and coworkers further used steady-state isotopic transient kinetic analysis (SSITKA), which could resolve the coverage and surface residence time for reactants and reaction intermediates, to identify the origin of size effects in the FT synthesis.27 The analysis revealed that forming Co particles with a small size (<6 nm) could reduce the surface coverage of CHx species while promoting H coverage due to the increased fraction of highly under-coordinated atoms at the exposed surface. As a result, the C–C coupling and growth of alkyl chains became more difficult while methane formation was preferred. The investigation suggested that the cobalt catalysts should be controlled in the range of 6–8 nm to obtain a high selectivity to C5+ products.
2.4 Adding promoters
The addition of promoters is another way to tune C–C coupling in syngas conversion. The added promoters can help to increase catalyst activity, selectivity or stability.28 Generally, promoters are classified into two categories according to their working mechanisms – electronic promoters and structural promoters, which can modify catalyst surface by changing surface electronic properties, blocking undesired active sites and altering the structure of the active phase. As a result, the addition of promoters can enhance the concentration of active surface intermediates,29 increase the probability of chain growth30 and facilitate the formation of active facets.31 Typical promoters used in syngas conversion include alkali metals (e.g., Na and K), transition metals (e.g., Zn, Mn, Ti, and V) and nonmetallic elements (S).Johnson et al. studied the effects of Mn promoters on the Co/SiO2 catalyst for the Fischer–Tropsch synthesis.28 The investigation revealed that, on Mn-promoted catalysts, the active sites near the interface between Co metal and MnO enhanced CO adsorption. Meanwhile, CO dissociation was facilitated by weakening the C–O bond through Lewis acid–base interaction near the interface, increasing the coverage of CHx intermediates on the surface of catalysts. As such, fewer H species were available for methanation and paraffin chain termination. Consequently, the selectivity of C5+ products was promoted while excluding methane formation. However, excessive Mn loading may block more fraction of Co active sites.
In another case, de Jong and coworkers demonstrated that the addition of S and Na promoters to an Fe/α-Al2O3 catalyst achieved a high selectivity to C2–C4 olefins (about 50%).30,32 Specifically, the Na promoter increased the probability of chain growth while the S promoter selectively blocked hydrogenation sites. Taken together, the two agents synergistically promoted the formation of C2–C4 olefins and suppressed the production of methane. Ma and coworkers fabricated Zn- and Na-modified Fe catalysts using a simple coprecipitation/washing method.33 As shown in Fig. 3, the modified catalyst achieved an improved selectivity toward alkenes up to 70% for hydrocarbons. In such a catalyst, Zn served as a structural promoter to shrink Fe crystals, exposing more surface-active sites. In the meantime, Na acted as an electronic promoter to enable electron transfer from Na to Fe species, enriching electrons in the iron carbide active phase. The alteration of the surface electronic structure restrained the hydrogenation of double bonds and promoted the desorption of products, resulting in a high selectivity to alkenes.
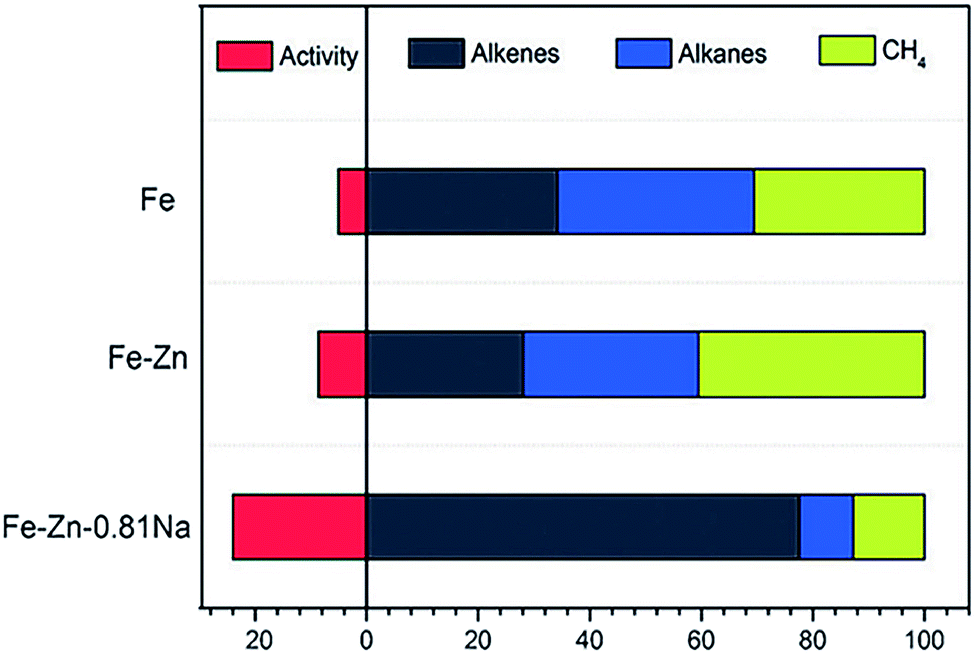 | ||
| Fig. 3 The activity and product selectivity of Fe–Zn–0.81Na, Fe–Zn, and Fe catalysts. Reprinted from ref. 33 with permission from John Wiley & Sons. | ||
2.5 Forming bimetallic catalysts
As compared with single metal catalysts, bimetallic catalysts often show enhanced catalytic performance as two metals may have synergistic effects on the surface reaction process. According to the mixing pattern of two metals, bimetallic catalysts can be classified into three main categories (Fig. 4): core–shell structures, heterostructures, and intermetallic or alloyed structures.34 The three configurations in turn enable different surface structures. The bimetallic core–shell structure exposes one metal on the surface and confines the other as an inner core, in which the catalytic properties of the outer surface may be influenced by the inner metal. The bimetallic heterostructure forms a mixed interface between two regions of different metals, which is exposed as an active site for catalysis. The intermetallic or alloyed structures are homogeneous mixtures of two metals, between which the major difference is the distribution of metal atoms. The atoms of two metals are randomly distributed on the surface of the alloyed structure, while the surface of intermetallic structures is in a long-range atomic order.34 Both atomic distribution configurations would impact catalytically active sites.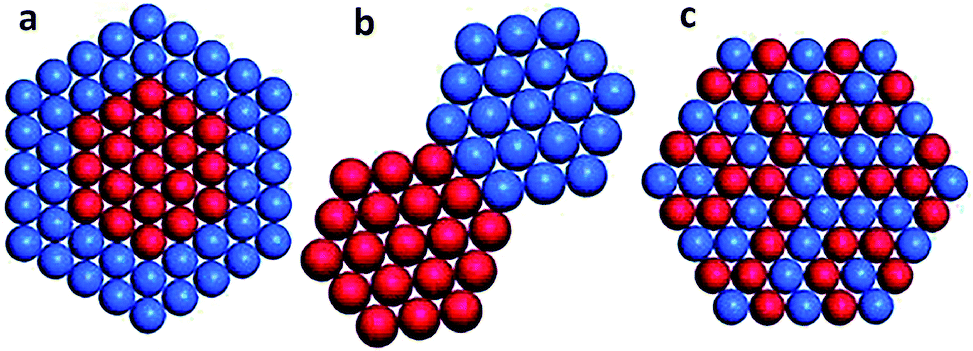 | ||
| Fig. 4 Bimetallic nanocrystals with different configurations: (a) core–shell, (b) heterostructure, and (c) intermetallic or alloyed structures. Reprinted from ref. 34 with permission from John Wiley & Sons. | ||
Developing core–shell structures with expensive Ru or Co as a shell and a cheap metal as a core is a promising strategy for reducing the cost of catalysts while maintaining high selectivity and activity. For instance, Haghtalab et al. developed core–shell structured Co@Ru/γ-Al2O3 catalysts.35 The core–shell structured catalyst showed enhanced activity and selectivity for long-chain hydrocarbons in syngas conversion as compared with the Co/γ-Al2O3 catalyst owing to the higher intrinsic activity and C5+ selectivity of Ru. Increasing the thickness of the Ru shell can further improve the selectivity of C5+ products. Moreover, the catalytic performance of the shell metal may be influenced by the inner core metal. Calderone et al. developed a core–shell Fe@Co catalyst with the mean size of the magnetite core at 7 nm and the thickness of the cobalt shell at 1 nm.36 After being supported on Al2O3 and further activation, the core–shell catalyst achieved a selectivity of about 40% for C5–C27 hydrocarbons, lower than that of bare Co catalysts; however, the selectivity for oxygenates (10%) and olefins (20%) was higher than that of traditional Co-based catalysts. This observation demonstrated that the catalytic performance of the cobalt shell was maneuvered by the inner iron; however, the mechanism behind the phenomenon still remains unclear and needs further investigation.
Differently from the case of core–shell structures where one may affect the other, the alloyed structure of bimetallic catalysts typically shows a synergistic effect on syngas conversion by providing two active sites: one site for CO dissociation and the other for C–C coupling. Abatzoglou and coworkers found that the introduction of 4 wt% iron into a cobalt catalyst dramatically enhanced the selectivity for alcohol production from 2.3% to 26.3%.37 In sharp contrast, the monometallic iron catalyst only offered a selectivity of 10.3%. The high selectivity toward alcohol formation was ascribed to the formation of a Co–Fe alloy. On the surface of the alloyed structure, Co and Fe were active sites for CO dissociation and CO insertion, respectively, so the optimization of Fe/Co ratios promoted the formation of higher alcohols.18
2.6 Designing bifunctional catalysts
In recent years, many breakthroughs have been made for converting syngas to targeted C2+ products such as olefins,38,39 heavy hydrocarbons (C5+)40 and aromatics41 using bifunctional catalysts. Usually, a bifunctional catalyst consists of a FT catalyst and zeolite. There exist four typical integration manners of the two components in the bifunctional catalyst system: dual-bed reactor model, physically mixing, core–shell structure, and loading of FT metal particles on a zeolite (Fig. 5).7 The spatial locations of the two components in the bifunctional catalyst has a great impact on the selectivity for final products.42 It should be emphasized that surface science also plays an important role in the design of bifunctional catalysts for regulating C–C coupling.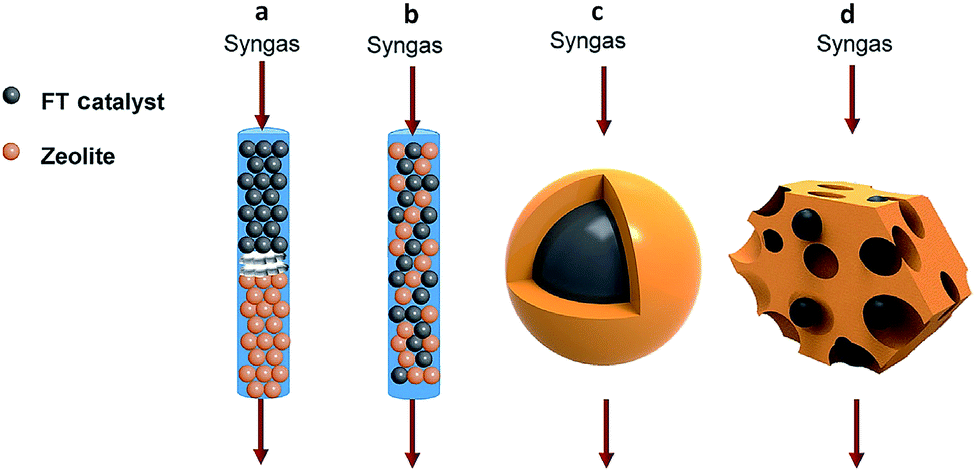 | ||
| Fig. 5 Integration manners of bifunctional catalysts consisting of a FT catalyst component and zeolite: (a) dual-bed reactor, (b) physically mixing, (c) core–shell structure, (d) loading of FT metal particles on the zeolite. Reprinted from ref. 7 with permission from Elsevier. | ||
Recently, Bao and coworkers developed a stable composite catalyst containing ZnCrOx and a mesoporous SAPO zeolite (MSAPO), which achieved 80% selectivity for C2=–C4= olefins and 94% selectivity for C2–C4 hydrocarbons at 17% CO conversion.38 The selectivity of this process (oxide–zeolite, namely OX–ZEO) was far beyond the maximum (only 58% for C2–C4 hydrocarbons) predicted by the classical ASF model38 (Fig. 6a–c). Oxygen vacancies on the surface of ZnCrOx played an important role in promoting CO activation to form CO2 and surface *C species. The surface *C was in turn hydrogenated to CH2 species, which underwent C–C coupling with CO to form a less reactive ketene (CH2CO). The CH2CO intermediate, which was detected by highly sensitive synchrotron-based vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) during the in situ investigation of syngas conversion over ZnCrOx (Fig. 6d), went into zeolite pores and was finally converted to olefins inside the pores. The C–C coupling could be manipulated by changing the strength of surface acidity on the zeolite. Specifically, the medium acidity strength of the SAPO zeolite led to a high C2=–C4= selectivity. It is worth pointing out that the confinement effects of zeolite pores also played a crucial role in tuning the selectivity for products.
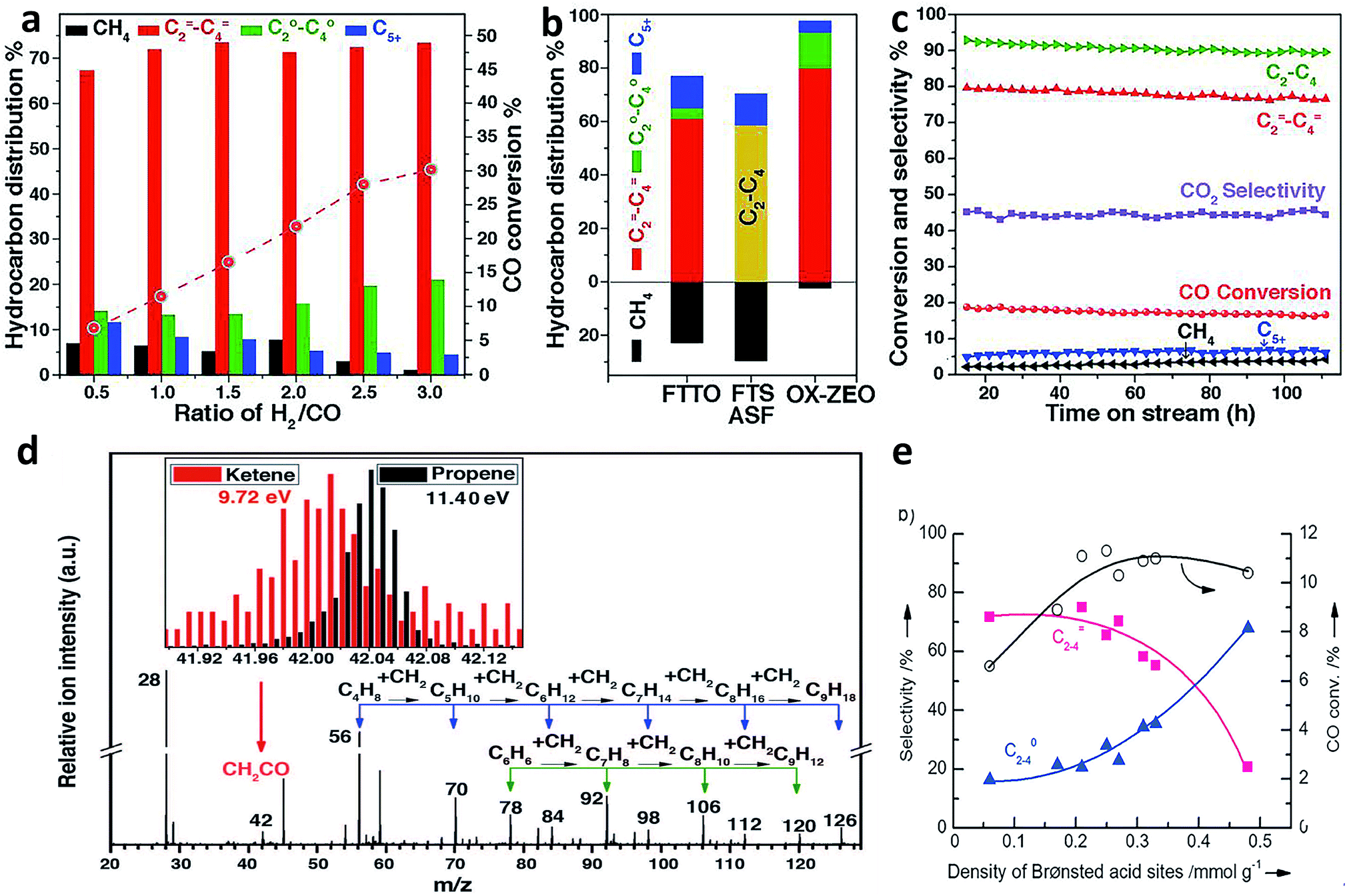 | ||
| Fig. 6 (a) Catalytic performance of the ZnCrOx/MSAPO bifunctional composite catalyst at different ratios of H2/CO. (b) Comparison of hydrocarbon product distribution among OX-ZEO, FTTO32 and FTS predicted by the ASF model at a chain growth probability of 0.46. (c) A stability test of a ZnCrOx/MSAPO composite catalyst. (d) Detection of CH2CO intermediate by highly sensitive synchrotron-based vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS). Reprinted from ref. 38 with permission from the American Association for the Advancement of Science. (e) Syngas conversion over composite catalysts with SAPO-34 of different acidities. Reprinted from ref. 39 with permission from John Wiley & Sons. | ||
The Wang research group found that the combination of a Zr–Zn binary oxide with a molecular sieve SAPO-34 could produce C2–C4 olefins with around 70% selectivity at about 10% CO conversion, also breaking the ASF distribution.39 The Zr–Zn catalyst could efficiently convert CO to methanol and dimethyl ether (DME) in a wide temperature range, which were further transformed into C2–C4 olefins by SAPO-34. In this case, the density of Brønsted acid sites on the surface of SAPO-34 obviously affected the C2–C4 olefin selectivity. The Brønsted acidity of SAPO-34 originated from the substitution of Si for P or Al in the framework of the molecular sieve. As such, the Si contents could be tailored to prepare a series of SAPO-34 samples with different densities of Brønsted acid sites. According to the measurements of methanol conversion, the density of Brønsted acid sites on the surface of SAPO-34 determined the ratio of C2–C4 olefin/paraffin. A larger density of Brønsted acid sites led to a lower selectivity for C2–C4 olefins (Fig. 6e).
Apart from the two examples above, there is another strategy for designing bifunctional catalysts to regulate C–C coupling in syngas conversion, which integrates the active sites for CO activation and C–C coupling with the Brønsted acid sites in zeolites for C–C cleavage. For example, Tsubaki and coworkers fabricated mesoporous Y-type zeolite-supported cobalt catalysts (Co/Ymeso), bifunctional catalysts which could produce various liquid fuels with high selectivities by simply tuning the properties of Ymeso.40 The Co catalyst converted syngas to a variety of hydrocarbons obeying the ASF distribution, and the Ymeso zeolite catalyzed the C–C cleavage of heavier hydrocarbons. In general, the Brønsted acidity of zeolites can lead to hydrocracking/isomerization of Fischer–Tropsch wax (C21+). Guided by this assumption, an acidic zeolite Ymeso–H was prepared by a NH4+ exchange technique; however, it turned out that the Co/Ymeso–H catalyst showed high selectivity for undesired CH4 and C2–C4 because excessive Brønsted acidity on Co/Ymeso–H led to overcracking of heavy hydrocarbons. By incorporating different cations into Ymeso, the Brønsted acidity could be tuned so as to regulate the degree of hydrocracking/isomerization of heavier hydrocarbons. Specifically, Co/Ymeso–Ce and Co/Ymeso–La possessed mild Brønsted acidities, which offered 74% selectivity for gasoline and 72% selectivity for jet fuel, respectively. In comparison, Co/Ymeso–K without Brønsted acidity produced a diesel fuel with 58% selectivity.
3. Regulating C–C coupling in CO2 hydrogenation
3.1 A brief overview of CO2 hydrogenation to C2+ products
CO2 is a very stable molecule (ΔfGθ = −396 kJ mol−1). For this reason, thermocatalytic reduction of CO2 is a process that requires high energy input. The reaction of CO2 with H2 which has higher free Gibbs energy should make CO2 conversion more thermodynamically favorable.43 Moreover, this process has relatively faster kinetics compared with electrocatalytic CO2 reduction. As such, CO2 hydrogenation to fuels and valuable chemicals is regarded as a promising way to mitigate the energy crisis and reduce the environmental problems caused by elevated atmospheric CO2 concentration.Typically, C2+ products can be generated from CO2 hydrogenation via two intermediate routes: CO intermediate route and CH3OH intermediate route (Fig. 7a).12 In the CO intermediate route, CO2 is first transformed into CO via the reverse water–gas shift (eqn (3), RWGS) reaction. As a result, the more reactive CO is subsequently hydrogenated to hydrocarbons (or oxygenates). The mechanism of CO hydrogenation to C2+ products has been discussed above. For the latter route, the CH3OH intermediate can be obtained using a catalyst for methanol synthesis, which is further converted to hydrocarbons by methanol-to-hydrocarbon (MTH) catalysts. Specifically, the MTH includes methanol-to-olefin (MTO), methanol-to-propene (MTP) and methanol-to-gasoline (MTG) processes. Various mechanisms have been proposed for the formation of C2+ products in the MTH processes, including the oxonium ylide mechanism, carbine mechanism, carbocationic mechanism, free radical mechanism and hydrocarbon pool mechanism.12 Among the mechanisms, the hydrocarbon pool mechanism in which aromatics and alkenes are important hydrocarbon pool compounds has been widely accepted. Larger hydrocarbons are formed after alkenes and aromatics are methylated with methanol (or dimethyl ether), and then crack or dealkylate to produce light alkenes and regenerate the starting compounds. The division between two classes of intermediates is usually referred to as the dual-cycle concept,44 as shown in Fig. 7b. In such a mechanism, higher alkenes may be transformed into aromatics and alkanes through cyclisation and hydride transfer reactions, while light alkenes generated from aromatics may enter into the alkene cycle.
| CO2 + H2 ⇌ CO + H2O | (3) |
 | ||
| Fig. 7 (a) Schematic illustration of CO2 hydrogenation to C2+ products via the CO intermediate route and CH3OH intermediate route. Reprinted from ref. 12 with permission from the Royal Society of Chemistry. (b) General scheme of the dual-cycle mechanism during the MTH process. Reprinted from ref. 44 with permission from Elsevier. | ||
The above two routes for CO2 hydrogenation can be achieved indirectly by using two-stage reactors;12 however, direct hydrogenation of CO2 to hydrocarbons or oxygenates is more economical and environmentally friendly. An ideal catalyst for the direct hydrogenation of CO2 to hydrocarbons or oxygenates should possess high activity for both CO/CH3OH formation and subsequent C–C coupling.45 Iron-based catalysts are often used in CO2 hydrogenation because they show outstanding catalytic properties for both RWGS and FT synthesis. In parallel, three categories of catalysts, including Cu-based catalysts, noble metal catalysts (Pd and Pt) and oxygen-deficient catalysts (In2O3 and ZrO2), are usually utilized to produce methanol, followed by the MTH process catalyzed by acidic zeolites.12 Although the catalysts based on noble metals (Rh, Pt, and Ru) and transition metals (Cu, Fe, and Co) have been widely employed to synthesize higher alcohols from CO2 hydrogenation,10,46 the reported activity and selectivity for higher alcohols are quite limited.45
3.2 Adding promoters
Like syngas conversion, the utilization of promoters is also a common strategy for tuning the selectivity for products in CO2 hydrogenation. It is known that CO2 is an acidic oxide so its adsorption occurs on the basic sites of catalysts. Adding an alkali metal such as K can increase the basicity of the catalyst surface, thereby enhancing CO2 adsorption and suppressing H2 adsorption. Choi et al. investigated the promoting effects of K based on an Fe/Al2O3 catalyst.47 According to chemisorption studies, H2 was only adsorbed on Fe while CO2 was most likely adsorbed on the K sites. The addition of K enhanced the ability of CO2 chemisorption and blocked the Fe sites for H2 adsorption. As such, the coverage of CHx species generated from CO2 was increased on the catalyst surface while reducing the H coverage, promoting the C–C bond formation. Xu and coworkers reported that the Na promoter could not only enhance the surface basicity of the Fe3O4 catalyst to promote CO2 chemisorption and inhibit the hydrogenation of double bonds, but also acted as a structure promoter to form an active iron carbide phase.48Other transition metal promoters (e.g., Mn, Cr, and Mo) can improve selectivity to long-chain alkanes. Dorner et al. suggested that the addition of Mn promoters to supported Fe-based catalysts led to an increase in the selectivity for unsaturated higher-chain hydrocarbons by repressing methane formation.49 Such a change in product distribution was caused by blocking hydrogenation active sites on the Fe surface with Mn promoters. Without sufficient H coverage, methanation and paraffin chain termination would be suppressed to facilitate the production of higher-chain hydrocarbons.
3.3 Forming bimetallic catalysts
Direct hydrogenation of CO2 to valuable C2+ products with high activity is a highly challenging task. Single metal catalysts are often incapable of offering the function to overcome the limitation of chemical equilibrium during the process of CO2 hydrogenation. Specifically, the CO generated from RWGS has low partial pressure in a CO2/H2 atmosphere due to thermodynamic constraints, limiting C–C coupling.50 Forming bimetallic catalysts can potentially circumvent the limitation of chemical equilibrium and improve the catalytic activity and selectivity for CO2 hydrogenation, benefiting from the synergistic effects on the surface reaction process mentioned above.For instance, Wang et al. synthesized an Fe–Cu bimetallic catalyst which improved the production of C2–C7 hydrocarbons and suppressed methane formation in comparison with the corresponding monometallic catalyst.51 The selectivity to C2+ hydrocarbons was enhanced by the synergistic effects of bimetallic metals. Cu functioned as an active site for the generation of CO or CO-like intermediates via RWGS, while Fe sites catalyzed C–C coupling of the CO intermediate to form hydrocarbons. The CO intermediates generated on Cu sites were subsequently consumed on Fe sites. As a result, the driving force for the RWGS could be enhanced, generating more CO intermediates on the catalyst surface. The improved surface CO coverage led to forming more CHx species on Fe sites, eventually promoting the production of long-chain hydrocarbons.
3.4 Designing bifunctional composite catalysts
In recent years, more research efforts have been devoted to the conversion of CO2 to olefins, gasoline and other valuable C2+ products using bifunctional catalysts,52–54 given that the bifunctional catalysts have achieved such a big success in syngas conversion. Typically, a bifunctional catalyst for CO2 hydrogenation consists of a catalyst for methanol synthesis and a zeolite for further conversion of methanol intermediates to final products.4,53,55 In parallel, the process that is not mediated by methanol has also been reported.52It is worth mentioning that the oxygen vacancies and surface acidity of the zeolite play vital roles in the design of bifunctional catalysts for CO2 hydrogenation. Sun and coworkers recently reported an In2O3/HZSM-5 bifunctional catalyst with excellent durability which could yield C5+ hydrocarbons with a high selectivity of 78.6% while the selectivity for methane was limited to only 1% (Fig. 8a and b).53 DFT calculations showed that CO2 was chemisorbed at the oxygen-vacancy sites on the surface of reducible In2O3 and hydrogenated to CH3OH through several intermediates (Fig. 8c). The CH3OH intermediate entered into the HZSM-5 zeolite and was further converted to hydrocarbon products at the surface acidic sites of the zeolite via a hydrocarbon-pool mechanism (Fig. 8d). Wang and coworkers developed a bifunctional catalyst composed of ZnGa2O4 and SAPO-34 which achieved 86% selectivity for C2–C4 olefins at 13% conversion of CO2.55 In that case, the oxygen vacancies on the surface of ZnGa2O4 were also responsible for CO2 activation.
 | ||
| Fig. 8 (a) Catalytic performance of CO2 hydrogenation over various bifunctional catalysts that contained Cu-based catalysts or In2O3 and HZSM-5 with different mass ratios, in reference to the stand-alone In2O3 catalyst and HZSM-5. C5+, red; C2–4, blue; CH4, grey. (b) A stability test of the In2O3/HZSM-5 composite catalyst. (c) Schematic of formation of CH3OH at the oxygen-vacancy site on the In2O3 catalyst surface. (d) Schematic of transformation of the CH3OH intermediate into a hydrocarbon at the acidic site inside the pores of the HZSM-5 catalyst via a hydrocarbon-pool mechanism. Reprinted from ref. 53 with permission of Nature Publishing Group. | ||
It should be noted that the water generated through the RWGS reaction is detrimental to the process of CO2 hydrogenation.4 Excessive water may cause the deactivation of surface acidic sites on the zeolite, which severely holds back C–C bond formation and leads to a low production of C2+ products. For this reason, it is necessary to remove the generated water timely. To this end, the hydrophobic modification of the zeolite surface helps to solve the problem caused by the produced water. Fujiwara et al. developed a composite catalyst consisting of a Cu–Zn–Al oxide and HB zeolite.56 The Cu–Zn–Al oxide was a catalyst for methanol synthesis, while the HB zeolite was used for the further transformation of methanol intermediates. However, the composite catalyst turned out to inefficiently yield C2+ hydrocarbons. After the HB zeolite was modified with 1,4-bis(hydroxydimethylsilyl)benzene, the zeolite surface was turned hydrophobic to significantly improve the yield of C2+ hydrocarbons. The hydrophobic surface of the HB zeolite suppressed the deactivation of strong acidic sites, thereby enhancing the catalytic selectivity to C2+ hydrocarbons.
4. Regulating C–C coupling in CO2 electroreduction
4.1 A brief overview of CO2 electroreduction
Compared to syngas conversion and CO2 hydrogenation, CO2 electroreduction proceeds under milder reaction conditions, which does not require hydrogen feeding, high temperature and high pressure any more. Moreover, CO2 electroreduction can be driven by the electricity generated by solar and wind energy, which contributes to the full utilization of geographical, seasonal and intermittent renewable energy sources.1,9 For this reason, CO2 electroreduction is a particularly appealing approach to CO2 conversion which has been extensively explored in recent years.Electrocatalytic CO2 reduction is typically performed in a three-electrode H-cell consisting of a working electrode, a counter electrode and a reference electrode. In order to obtain high currents, electrocatalysts can be dispersed onto a gas diffusion electrode and function in a flow cell.57,58 Metal-based catalysts are commonly used for CO2 electroreduction, whose products highly depend on catalyst compositions. Au, Ag, Zn and Pd mainly generate CO products, while Pb, In, Sn and Bi are typical catalysts for the production of formic acid or formate (in basic electrolytes).1,9 Cu is the only known metal which can catalyze CO2 electroreduction to C2+ products with reasonably high efficiencies.59 Although some carbon materials can also generate C2+ products, the current density of C2+ products is relatively low compared to that of Cu.60 Other Cu-based compounds and composites such as Cu2S,61 Cu3N62 and Cu–C3N4 (ref. 63) have also been explored for electrocatalytic CO2 reduction. Moreover, the electrochemical conversion of CO, regarded as a key intermediate to form C2+ products in the CO2 electroreduction process, can also be catalyzed by Cu.64 In both cases, the electroreduction of CO2/CO has to compete with the undesired side reaction – hydrogen evolution reaction (HER) – because most electrochemical reaction cells use inorganic salt (e.g., KHCO3, NaHCO3, and Na2SO4) aqueous solutions as electrolytes.9 Interestingly, C2+ products cannot be produced on Cu catalysts thermochemically while C–C coupling can be achieved electrochemically. In fact, Cu catalysts have been widely studied for the RWGS reaction,51 but the Cu surface only provides active sites for nondissociative activation of the CO generated from CO2. As a result, the CHx species cannot be formed for a further C–C coupling process. In an electrochemical reaction, the CO intermediates generated from CO2 also undergo nondissociative activation, but C–C coupling and the subsequent protonation can be driven by applying electrical energy on the Cu surface.
Specifically during CO2 electroreduction, the CO2˙− intermediate is firstly formed by transferring one electron to CO2. As rearranging a linear CO2 molecule to a bent radical anion needs to overcome a high energy barrier, and this step is regarded as the rate-determining step (RDS) for most transition metal-based catalysts. The highly reactive *CO2˙− intermediate then undergoes proton-coupled electron–transfer reactions to form different products. Pb, In, Sn and Bi-based catalysts primarily generate HCOO− as their surfaces show weak binding to the *CO2˙− intermediate. In comparison, Au, Ag, Zn and Pd-based catalysts bind to the *CO intermediate too weakly so CO is the major product. Among various catalysts, Cu is a unique one which has the moderate binding energy of the *CO intermediate, and as such, the following hydrogenation and C–C coupling processes can occur on its surface.
Until now, 18 products including C1 and C2+ products have been detected for CO2 electroreduction using Cu-based catalysts,65–67 among which C2H4, C2H6, CH3CH2OH and n-CH3CH2CH2OH are four main C2+ products reported in the literature. The mechanisms for CO2 electroreduction on the surface of Cu-based catalysts have been investigated by many theoretical and experimental studies.68–72 However, the formation mechanism for deep reduction products (>2e− transfer, including CH4, CH3OH, HCHO and C2+ products) has not been fully understood. According to the mainstream view, *CO is regarded as a key intermediate for the formation of deep reduction products. During the CO2 electroreduction process, CO2 is first activated and reduced to adsorbed CO (*CO), which can then be converted to CH4 and CH3OH through *COH and *CHO intermediates, respectively. As for the formation of C2+ products, several reaction pathways have been proposed,69,73,74 including (1) *CO + *CO → *COCO → C2+ products, (2) *CO → *CHO; *CHO + *CO → *COCHO → C2+ products, and (3) *CO → *COH → CHx → C2+ products. The first pathway, named the *CO dimerization pathway, is widely accepted for the formation of main C2+ products. In this mechanism, the C–C coupling through *CO dimerization that forms a negatively charged CO–CO− species is regarded as the rate-determining step.75 To improve the formation of C2+ products, the surface coverage of *CO intermediates should be enhanced. The CO–CO− intermediate is further protonated to generate the *CO–COH intermediate, which has been confirmed by Koper et al. through in situ Fourier transform infrared spectroscopy (FTIR).76 Then *CO–COH intermediates undergo a series of protonation and electron transfer steps to yield different products. According to theoretical calculation, the C2H4 pathway and C2H5OH pathway share the same intermediate, *CH2CHO.71 Nevertheless, tuning the energetics of *CH2CHO intermediate binding can shift the C2H4 pathway to the C2H5OH pathway. As for improving the formation of n-CH3CH2CH2OH, a high surface coverage of C2 intermediates should be ensured to promote the coupling of C1–C2 intermediates. The possible mechanistic pathways of CO2 electroreduction to C1 and C2+ products are summarized in Fig. 9.
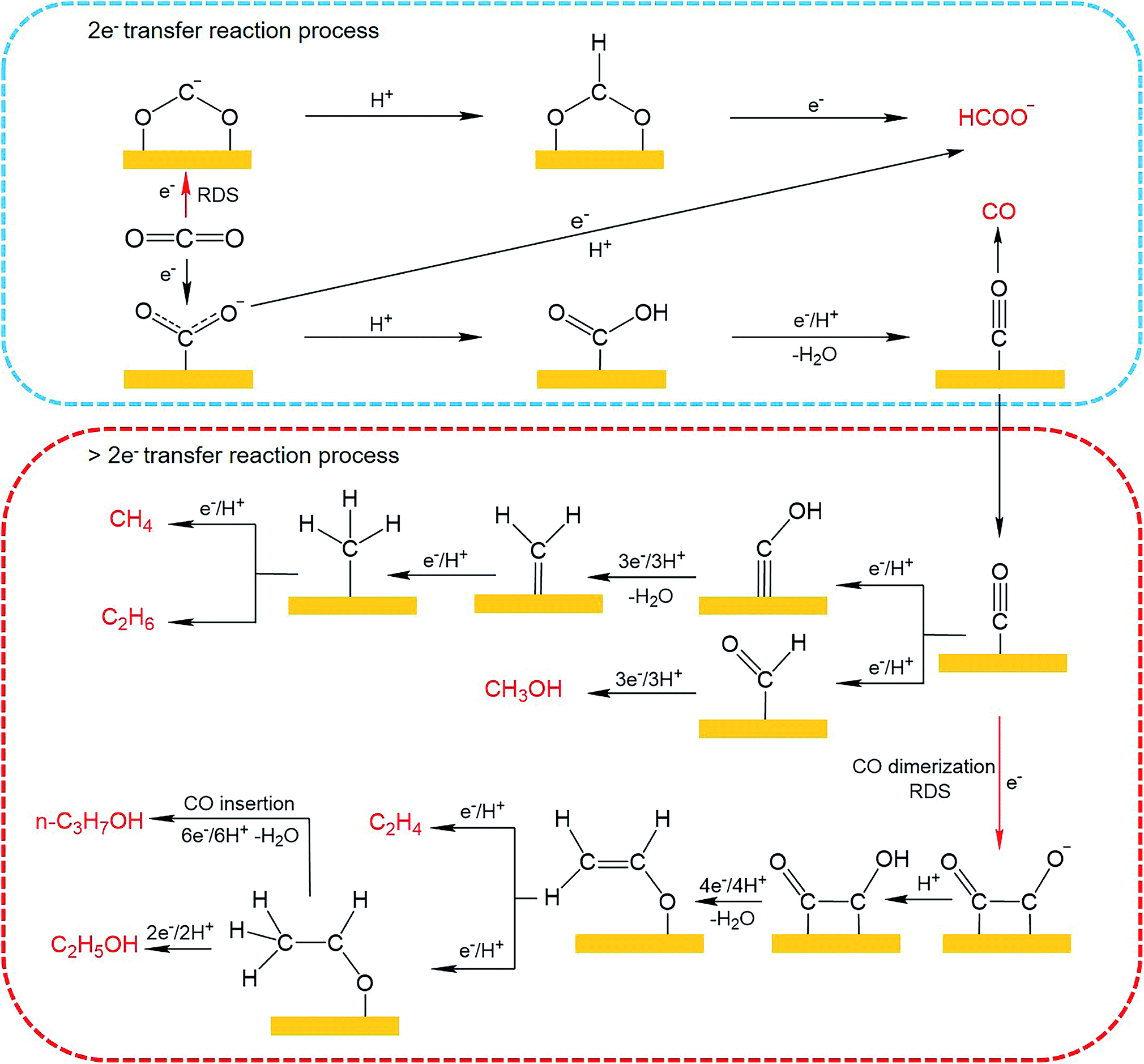 | ||
| Fig. 9 Possible reaction pathways for CO2 electroreduction on Cu-based catalysts toward various products. | ||
4.2 Controlling crystal facets
The facet effects of Cu crystals are quite evident for electrocatalytic CO2 reduction. Cu(111) facets predominantly produce CH4 while Cu(100) facets preferentially generate C2H4. After early recognized by Hori et al., this conclusion has been confirmed by many experiments and theoretical calculations. The DFT calculations showed that the atoms on Cu(100) stabilized the dimer of CO due to their unique orientation, selectively promoting C2H4 formation.77,78 In sharp contrast, Cu(111) facets favor the protonation of CO to COH,74,79,80 mainly producing CH4. Koper and coworkers used online electrochemical mass spectrometry (OLEMS) to detect gaseous products formed on Cu(100) and Cu(111) surfaces during the CO electroreduction.81 They found that ethylene was formed at −0.3 V on Cu(100), while a potential of −0.6 V was required for Cu(111), confirming that Cu(100) facts prefer to produce ethylene (Fig. 10). Cu(110) facets were found to promote the formation of hydrocarbons in electrocatalytic CO2 reduction. Yin and coworkers successfully synthesized Cu nanocrystals with a rhombic dodecahedral shape and enriched high-energy (110) facets by a chemical etching method.67 As compared with the Cu nanocubes mainly enclosed by (100) facets, the obtained Cu rhombic dodecahedra exhibited higher faradaic efficiencies toward CH4, C2H4, C2H6 and C3H8. This suggests that the high-energy (110) facets more favor the formation of hydrocarbons than (100) facets. In addition, some high-index planes such as (911) and (711) have been predicted to promote C2H4 formation and suppress CH4 formation.81,82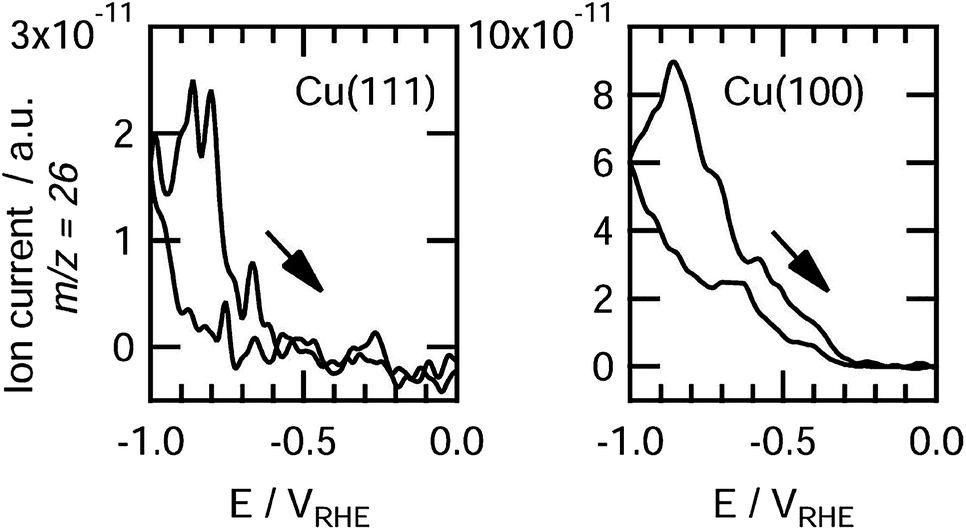 | ||
| Fig. 10 Detection of ethylene on Cu (111) (left) and Cu (100) (right) during CO electroreduction by OLEMS. Reprinted from ref. 81 with permission from American Chemical Society. | ||
4.3 Tuning catalyst sizes
The size effects of Cu spheroidal particles and Cu cubes have been studied in previous reports. Strasser and coworkers first explored the size effects of Cu nanoparticles in electrocatalytic CO2 reduction.83 They prepared a series of Cu spheroidal nanoparticles in the mean size range of 2–15 nm. While the obtained Cu spheroidal nanoparticles were tested in CO2 electroreduction, a spherical particle model was built to gain deep insights into experimental trends in the activity and selectivity for CO2 electroreduction as a function of particle size (Fig. 11a and b). When the size was below 2 nm, the number of under-coordinated atoms with a coordination number (CN) < 8 was drastically increased on the surface of Cu particles. This enabled the strong binding of intermediate reaction species such as CO and H to the catalyst surface. As a result, the faradaic efficiencies for CO and H2 production were substantially higher compared to that of Cu foil (Fig. 11c and d). When the Cu particle sizes were between 5 and 15 nm, the spherical particle model predicted that the populations of (100) (CN = 9) and (111) (CN = 8) facets were kept low and constant. This trend was consistent with the corresponding constant faradaic efficiencies for hydrocarbons. In the case of larger Cu spherical particles, the weaker binding of CO and H to their surface favored the formation of methane and ethylene.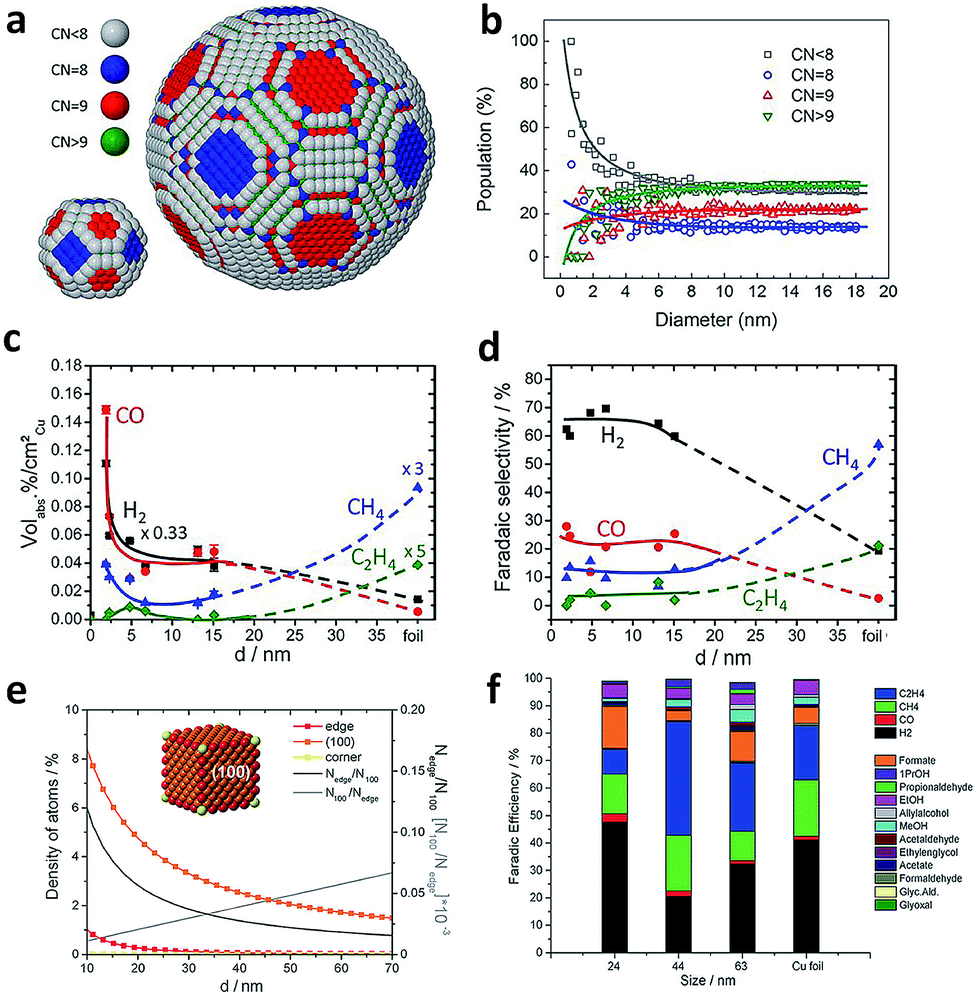 | ||
| Fig. 11 (a) Models for the surface atomic coordination of spherical Cu nanoparticles with 2.2 and 6.9 nm diameters. CN < 8, gray; CN = 8, blue; CN = 9, red; CN > 9, green. (b) Relative population ratio of surface atoms with a specific CN versus particle diameter. (c) The contents of gaseous products during CO2 electroreduction over spherical Cu nanoparticles with different diameters. (d) Faradaic selectivity for gaseous products during CO2 electroreduction on spherical Cu nanoparticles with different diameters. Reprinted from ref. 83 with permission from American Chemical Society. (e) Density of adsorption sites in Cu cubes (left axis) and trend of Nedge/N100 and N100/Nedge (right axis) is plotted as a function of the edge length. Nedge, the number of atoms at edges; N100, the number of atoms on the (100) plane. (f) Faradaic efficiencies for the products obtained using different sizes of Cu cubes and Cu foil at −1.1 V vs. RHE. Reprinted from ref. 84 with permission from John Wiley & Sons. | ||
In another example, Loiudice et al. prepared Cu cubes with edge lengths of 24 nm, 44 nm and 63 nm which exposed (100) facets predominantly.84 During electrocatalytic CO2 reduction, the cubes with the 44 nm edge length offered the best performance, with 80% selectivity toward CO2 reduction and 41% faradaic efficiency for C2H4 production (Fig. 11f). Fig. 11e shows the analysis for a simple Cu nanocube model. As the size increases, the relative number of atoms on (100) planes is promoted at the expense of corner and edge atoms. Specifically, the ratio of edge sites over (100) plane sites can be optimized to achieve the highest selectivity for CO2 reduction and C2H4 formation.
4.4 Forming bimetallic catalysts
Cu-based bimetallic catalysts have been widely explored for CO2 electroreduction.85 Typically, bimetallic catalysts offer two active sites on the surface – one for CO generation and the other for C–C coupling, which promote the production of C2+ products. For instance, Ren et al. reported that an oxide-derived Cu4Zn catalyst could produce ethanol with a remarkably high faradaic efficiency (29.1%, among the highest reported values for ethanol production) at −1.05 V vs. RHE.86 In that case, Zn acted as the site for CO production so the generated CO migrated to neighboring Cu active sites and underwent C–C coupling to produce ethanol.Similarly to syngas conversion (Fig. 4), the surface atomic mixing pattern of two metals plays a crucial role in C–C coupling to generate C2+ products in CO2 electroreduction. For instance, Ma et al. synthesized a series of Cu–Pd bimetallic catalysts with ordered, disordered and phase-separated atomic arrangements (Fig. 12a and b).87 As shown in Fig. 12c and d, the phase-separated sample exhibited the highest faradaic efficiency (up to 63%) to C2 products including ethylene and ethanol, while the ordered sample mainly generated C1 products and showed the lowest faradaic efficiency (<5%) to ethylene and ethanol. This work proposed that neighboring Cu atoms on the surface of phase-separated Cu–Pd catalysts favored the formation of C2 products while the alternating Cu–Pd sites on the surface of ordered and disordered samples promoted CH4 production.
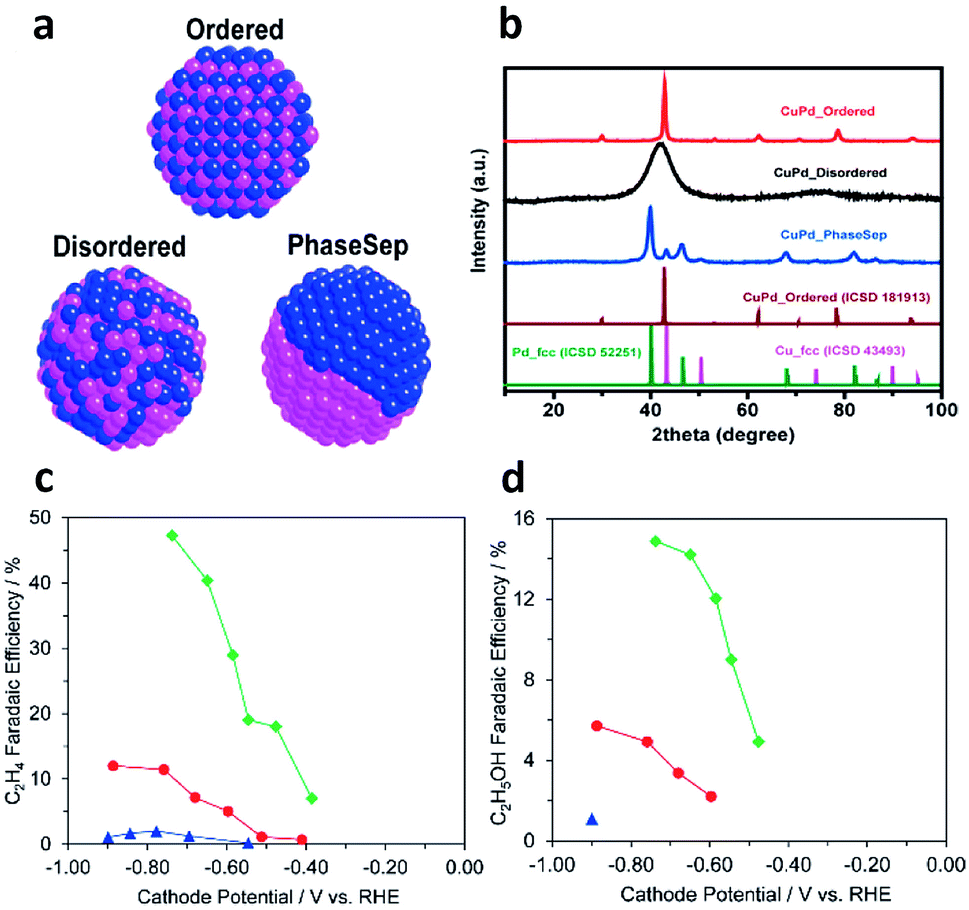 | ||
| Fig. 12 (a) Schematic illustration of CuPd nanoalloys with ordered, disordered and phase-separated structures. (b) XRD patterns of the prepared CuPd nanoalloys. Faradaic efficiencies of (c) C2H4 and (d) C2H5OH for bimetallic Cu–Pd catalysts with different mixing patterns: ordered, blue; disordered, red; phase-separated, green. Reprinted from ref. 87 with permission from American Chemical Society. | ||
It is worth pointing out that the compressive strain induced by the formation of a surface alloy may have a great influence on C–C coupling, leading to a significant change in the distribution of products. According to the work by Bell and coworkers,88 the formation of a Cu–Ag surface alloy induced compressive strain between surface Cu atoms. As a result, the compressive strain modified the electronic structure of the catalyst by shifting the valence band structure of Cu to deeper levels, reducing the binding energies of H and O relative to that of CO. Thus, the H2 production was obviously suppressed (60–75% reduction) while the faradaic efficiency of C2+ products was increased by 10–15%. Moreover, the production of multi-carbon carbonyl-containing products was enhanced at the expense of ethylene, as the modification of the electronic structure reduced the coverage of adsorbed H atoms and the oxophilicity of compressively strained Cu sites.
4.5 Confining catalysis on the internal surface
Porous catalysts such as nanopores and nanocavities offer a confined environment. The diffusion, adsorption and desorption of reactants, intermediates and products are strongly affected due to the geometrical constraints, thereby tuning the selectivity and activity for some specific reactions. Owing to the confinement effects, the reactions occurring on the internal surface of nanopores and nanocavities show unique characteristics. As such, fabricating nanoporous electrocatalysts has the potential to improve the production of C2+ products.For instance, Sargent and coworkers proposed that C2 intermediate species could potentially be concentrated inside a nanocavity structure owing to steric confinement.89 As such, the desorption of C2 intermediates was limited by the internal surface of the nanocavity so further conversion into a C3 product could be promoted. This argument was supported by simulations using the finite-element method. The simulations revealed that the suppression of C2 intermediate desorption by the cavity increased the surface coverage and residence time of the intermediates, favoring C3 formation. Guided by this simulation finding, they fabricated a series of nanocavity Cu catalysts with different hole sizes for CO electroreduction (Fig. 13a and b). Using the nanocavity copper catalyst with an appropriate hole size, the faradaic efficiency of propanol could reach 21 ± 1% at −0.56 V versus RHE, with a partial current density of 7.8 ± 0.5 mA cm−2. In another case, Yang et al. fabricated three Cu mesopore electrodes with mesopores of 30 nm width and 40 nm depth (30 nm/40 nm), 30 nm width and 70 nm depth (30 nm/70 nm), and 300 nm width and 40 nm depth (300 nm/40 nm) for CO2 electroreduction.90 Compared to the 300 nm/40 nm electrode, the 30 nm/40 nm electrode exhibited enhanced ethylene formation with faradaic efficiency from 8% to 38%. As the pore depth was further increased to 70 nm (30 nm/70 nm electrode), the major C2 product was converted to ethane, giving 46% faradaic efficiency. This change in product selectivity was ascribed to the alteration of local pH and retention time of key intermediates inside the pores caused by the confinement effect.
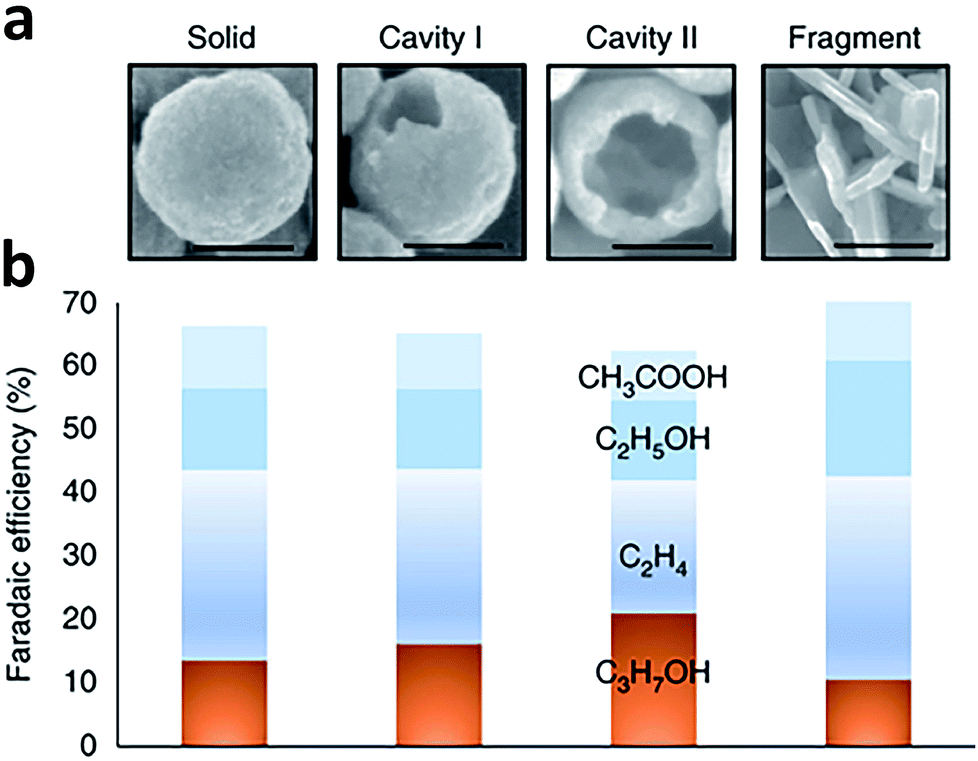 | ||
| Fig. 13 (a) SEM images of Cu-based catalysts with a morphology of solid, cavity I, cavity II and fragment. Scale bars, 100 nm. (b) Faradaic efficiency of C2 and C3 products during CO electroreduction over Cu-based catalysts with the four types of morphologies at an applied potential of −0.56 V versus RHE. Reprinted from ref. 89 with permission from Nature Publishing Group. | ||
4.6 Engineering catalyst defects
Defects, which can be classified into point defects, line defects, plane defects and bulk defects, exist widely in materials. Creating defects on the surface of catalysts can alter the electronic and surface properties of the catalyst, thereby influencing the catalytic activity and selectivity.91,92 In terms of CO2 electroreduction, engineering catalyst defects, such as heteroatom dopants, vacancies and grain boundaries, is a promising approach to promote C–C coupling toward improved selectivity for C2+ products.91As a powerful approach, doping heteroatoms can modify the surface electronic structure of Cu sites, enabling control over CO adsorption and dimerization. The Sargent research group demonstrated that doping boron on a Cu-based catalyst surface could induce and stabilize Cuδ+ sites,93 regarded as the active sites responsible for C2 product production (Fig. 14a).94,95 The ratio of Cuδ+ to Cu0 active sites could be tuned by varying the boron dopant content. As the average copper valence state was tuned to +0.35, a maximum faradaic efficiency (nearly 80%) for C2 products was achieved on boron-doped Cu catalysts at −1.1 V versus RHE (Fig. 14b and c).
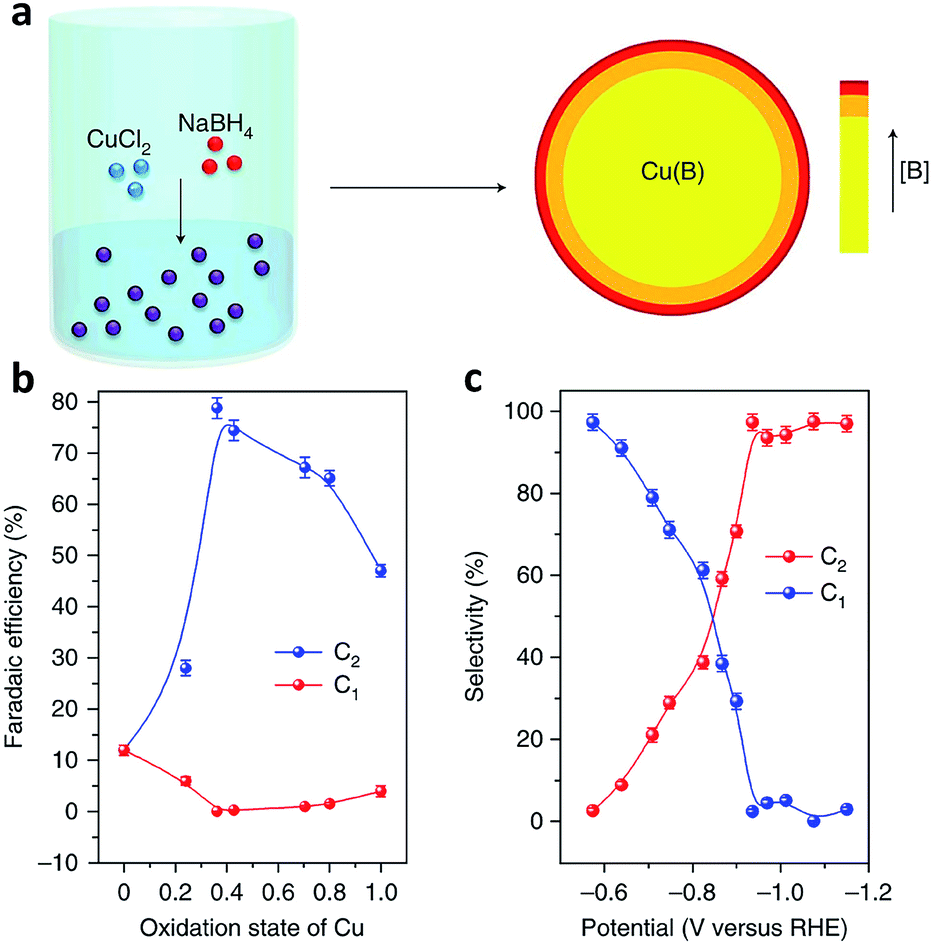 | ||
| Fig. 14 (a) Schematic illustration of the synthesis of B-doped Cu by a wet-chemical process. (b) Faradaic efficiency of C2 and C1 products versus the oxidation state of Cu. (c) Selectivity for C2 and C1 products at different potentials on B-doped Cu. Reprinted from ref. 93 with permission from Nature Publishing Group. | ||
Vacancy defect engineering is another versatile strategy for tailoring the electronic structure of neighboring atoms, thereby altering the energy barriers of the rate-limiting reaction intermediates.96 As such, creating vacancy defects on the catalyst surface has the potential to drive CO2 reduction to specific C2+ products. Sargent and coworkers reported that a Cu2S–Cu–V structure, with a Cu2S core and a shell containing Cu vacancies (Fig. 15a–g), significantly shifted the product selectivity away from ethylene toward ethanol and propanol.61 Previous mechanism studies indicated that the reaction intermediate *C2H3O could follow two different pathways to form ethylene and ethanol, respectively.71 Theoretical simulations revealed that the creation of vacancies on the Cu shell with a Cu2S core increased the energy barrier in the ethylene pathway (1.148 eV), while the ethanol formation pathway was mostly unaffected (0.427 eV). Such a design achieved a faradaic efficiency of 32% for C2+ alcohols (C2H5OH 25 ± 1% and C3H7OH 7 ± 0.5%) by steering post-C–C coupling selectivity through vacancy defect engineering.
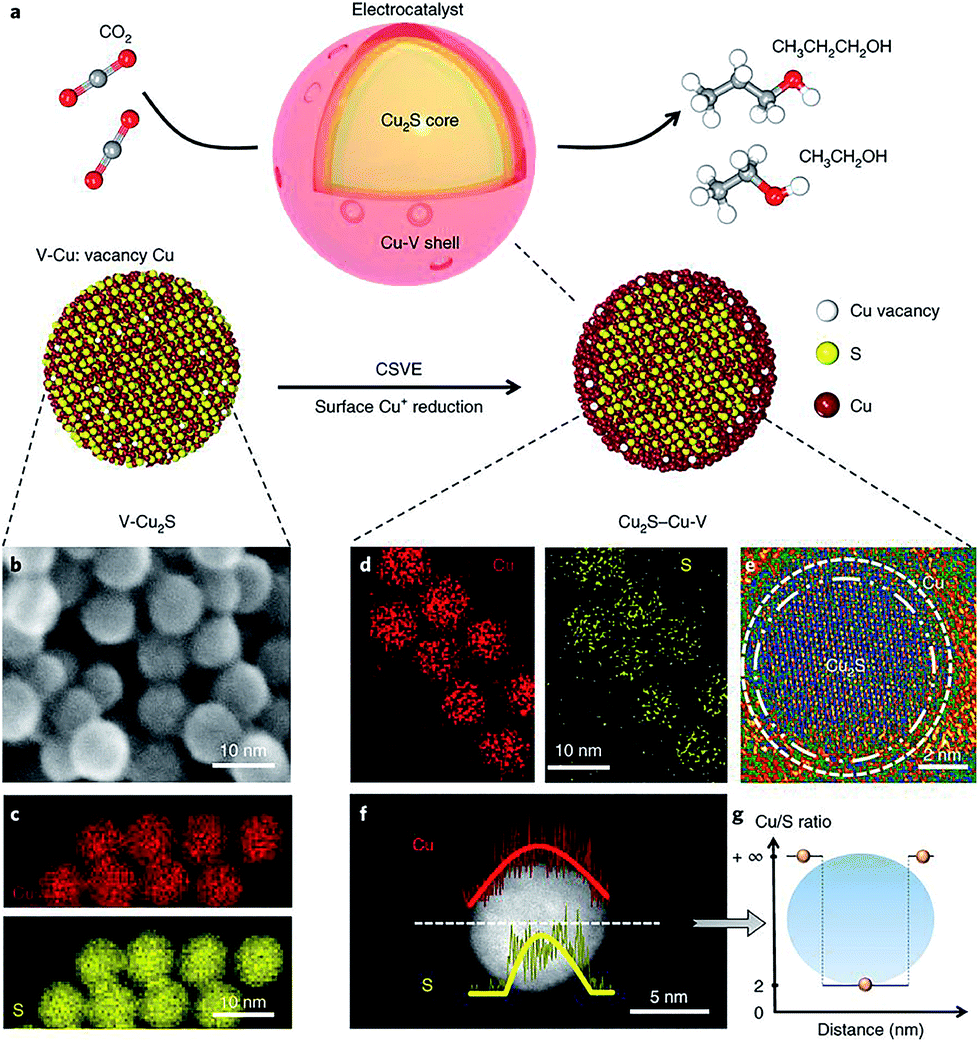 | ||
| Fig. 15 (a) Schematic illustration of Cu2S–Cu–V electrocatalyst design for CO2 electroreduction to produce multi-carbon alcohols. (b) TEM and (c) EDS mapping of original V–Cu2S nanoparticles; (d) EDS mapping, (e) high-resolution TEM, (f) EDS line scan and (g) the ratio of Cu/S concentration of the reduced Cu2S–Cu–V electrocatalyst after electrochemical reduction. Reprinted from ref. 61 with permission from Nature Publishing Group. | ||
Defect engineering has also been proven effective for tuning CO electroreduction. The Kanan research group demonstrated that up to 57% Faraday efficiency of C2+ oxygenates (ethanol, acetate and n-propanol) could be achieved for CO electroreduction on oxide-derived Cu catalysts at modest potentials.64 The excellent selectivity to C2+ oxygenates was enabled by the participation of grain boundary surfaces in the CO electroreduction process. The same research group further proved that the activity for CO electroreduction was directly correlated with the density of grain boundaries in Cu nanoparticles. Increasing the grain boundary density would promote the selectivity to ethanol and acetate linearly.97 Using isotope labelling, Ager and coworkers found that there may exist three different types of active sites on oxide-derived Cu catalysts, which accounted for the formation of C2+ products – ethylene, ethanol/acetate and 1-propanol, respectively.65 As proposed in their work, three product-specific active sites may be formed by the three different types of grain boundary termination.
5. Conclusions and outlook
COx conversion is a highly important research theme for achieving the carbon cycle. As estimated in a most recently published perspective article,98 an ideal catalytic process needs to be powered by electricity emitting less than 0.2 kg of CO2 per kW h to achieve a net reduction in CO2. To make electrocatalytic CO2 reduction appealing for practical applications, the reaction rates for CO2 conversion should be elevated by two orders of magnitude. While this estimation was mainly based on the product of methanol, it highlights the necessity of substantially improving the reaction activity. Nevertheless, very differently from many other catalytic reactions (e.g., ammonia synthesis), the major challenges for COx conversion originate from both reaction activity and selectivity. Once activated, CO2 and CO molecules may evolve into many different products. For this reason, fundamental research for COx conversion is often focused on the control over product selectivity, which relies on catalytically active sites from the perspective of surface science. The central theme for controlling product selectivity in such a reaction system is to precisely achieve C–C coupling.In this sense, rational design of catalysts based on surface science is a key strategy for precisely achieving C–C coupling in both thermocatalytic and electrocatalytic COx conversion. In this article, we have reviewed typical catalyst design strategies based on surface science for tuning C–C coupling in syngas conversion, CO2 hydrogenation and CO2 electroreduction, aiming to obtain the targeted C2+ products such as olefins, long-chain hydrocarbons and higher alcohols with high selectivity. Such surface science-based design strategies include the means of controlling crystal facets, tuning catalyst sizes, forming bimetallic catalysts and others, which can significantly alter the structural and electronic properties of thermocatalysts and electrocatalysts.
As a matter of fact, it still remains the biggest challenge to precisely regulating C–C coupling toward the targeted C2+ products in thermocatalytic and electrocatalytic COx conversion. This challenge has been especially emphasized for the approach of CO2 electroreduction. The selectivity for hydrocarbons or higher alcohols produced from CO2 electroreduction is too low and far from meeting the requirements of industrialization. Syngas conversion has witnessed a long research history and been put into practice in large-scale industrialization. Until now, the research on syngas conversion has continued its success, and many breakthroughs have been made by optimizing catalyst design based on surface science, especially using bifunctional catalysts. As such, many studies on CO2 hydrogenation have drawn lessons or been enlightened from the experience of syngas conversion, greatly promoting the development of CO2 hydrogenation. We think that the significant research progress of syngas conversion and CO2 hydrogenation will provide very instructive information for the development of CO2 electroreduction particularly from the viewpoint of C–C coupling. The design of bifunctional catalysts based on surface science has the potential to make another breakthrough in CO2 electroreduction, just like the case of Jaramillo and coworkers where a tandem gold-on-copper electrocatalyst offers gold for CO generation and copper for further CO reduction to alcohols.99
Although the three reaction systems share many similar working mechanisms, catalysts should be specifically designed for a final application as the material requirements differ from case to case owing to reaction phases and energy input. As a result, the specific working mechanisms should be fully examined for each case, which can be facilitated through collaborative research at the intersection of controlled synthesis, advanced characterization and theoretical simulation. This multidisciplinary research mode has demonstrated its success in COx conversion in the past few years. In many cases, the bottleneck for mechanistic studies comes from the limitation of operando spectroscopic techniques. The advanced spectroscopic techniques help in situ characterization of the dynamic evolution of active sites and reaction intermediates on the catalyst surface at atomic/molecular levels. Future development of characterization techniques would be more focused on spatial and temporal resolution. Certainly these challenges will be accomplished, and plentiful opportunities exist in these fields. The importance of COx conversion to society and industry will continue motivating the research toward controllable and scalable production.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported in part by the National Key R&D Program of China (2017YFA0207301), NSFC (21725102, U1832156, and 21601173), CAS Key Research Program of Frontier Sciences (QYZDB-SSW-SLH018), CAS Interdisciplinary Innovation Team, and Chinese Universities Scientific Fund (WK2310000067).References
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- L. Guo, J. Sun, Q. Ge and N. Tsubaki, J. Mater. Chem. A, 2018, 6, 23244–23262 RSC.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- H. T. Luk, C. Mondelli, D. C. Ferre, J. A. Stewart and J. Perez-Ramirez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- K. Cheng, J. Kang, D. L. King, V. Subramanian, C. Zhou, Q. Zhang and Y. Wang, in Advances in Catalysis, ed. C. Song, 2017, vol. 60, pp. 125–208 Search PubMed.
- J. Artz, T. E. Mueller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS.
- Y. Gao, S. Liu, Z. Zhao, H. Tao and Z. Sun, Acta Phys. Sin., 2018, 34, 858–872 CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, Catal. Sci. Technol., 2017, 7, 4580–4598 RSC.
- T. Riedel, M. Claeys, H. Schulz, G. Schaub, S. S. Nam, K. W. Jun, M. J. Choi, G. Kishan and K. W. Lee, Appl. Catal., A, 1999, 186, 201–213 CrossRef CAS.
- R. A. van Santen, A. J. Markvoort, I. A. W. Filot, M. M. Ghouri and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2013, 15, 17038–17063 RSC.
- R. A. van Santen, I. M. Ciobica, E. van Steen and M. M. Ghouri, in Advances in Catalysis, eds. B. C. Gates and H. Knozinger, Elsevier Academic Press Inc, San Diego, 2011, vol. 54, pp. 127–187 Search PubMed.
- X. Xiaoding, E. B. M. Doesburg and J. J. F. Scholten, Catal. Today, 1987, 2, 125–170 CrossRef.
- K. Xiao, Z. H. Bao, X. Z. Qi, X. X. Wang, L. S. Zhong, K. G. Fang, M. G. Lin and Y. H. Sun, Chin. J. Catal., 2013, 34, 116–129 CrossRef CAS.
- M. Ao, G. H. Pham, J. Sunarso, M. O. Tade and S. Liu, ACS Catal., 2018, 8, 7025–7050 CrossRef CAS.
- Q. H. Zhang, J. C. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- Q. Zhang, K. Cheng, J. Kang, W. Deng and Y. Wang, ChemSusChem, 2014, 7, 1251–1264 CrossRef CAS.
- W. Zhou, K. Cheng, J. C. Kang, C. Zhou, V. Subramanian, Q. H. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- S. Cao, F. Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84 CrossRef CAS.
- M. Claeys, M. E. Dry, E. van Steen, E. du Plessis, P. J. van Berge, A. M. Saib and D. J. Moodley, J. Catal., 2014, 318, 193–202 CrossRef CAS.
- J.-X. Liu, P. Wang, W. Xu and E. J. M. Hensen, Engineering, 2017, 3, 467–476 CrossRef.
- G. L. Bezemer, J. H. Bitter, H. Kuipers, H. Oosterbeek, J. E. Holewijn, X. D. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef CAS.
- J. P. den Breejen, P. B. Radstake, G. L. Bezemer, J. H. Bitter, V. Froseth, A. Holmen and K. P. de Jong, J. Am. Chem. Soc., 2009, 131, 7197–7203 CrossRef CAS.
- G. R. Johnson, S. Werner and A. T. Bell, ACS Catal., 2015, 5, 5888–5903 CrossRef CAS.
- N. Lohitharn and J. G. Goodwin, J. Catal., 2008, 260, 7–16 CrossRef CAS.
- H. M. T. Galvis, A. C. J. Koeken, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Catal., 2013, 303, 22–30 CrossRef.
- Z. J. Li, L. S. Zhong, F. Yu, Y. L. An, Y. Y. Dai, Y. Z. Yang, T. J. Lin, S. G. Li, H. Wang, P. Gao, Y. H. Sun and M. Y. He, ACS Catal., 2017, 7, 3622–3631 CrossRef CAS.
- H. M. T. Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y.-W. Li, Q. Zhang, X.-D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS.
- A. Haghtalab and A. Mosayebi, Int. J. Hydrogen Energy, 2014, 39, 18882–18893 CrossRef CAS.
- V. R. Calderone, N. R. Shiju, D. Curulla-Ferre, S. Chambrey, A. Khodakov, A. Rose, J. Thiessen, A. Jess and G. Rothenberg, Angew. Chem., Int. Ed., 2013, 52, 4397–4401 CrossRef CAS.
- A. Tavasoli, M. Trepanier, R. M. M. Abbaslou, A. K. Dalai and N. Abatzoglou, Fuel Process. Technol., 2009, 90, 1486–1494 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS PubMed.
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787–793 CrossRef CAS.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245 CrossRef CAS.
- C. S. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- M. Westgard Erichsen, S. Svelle and U. Olsbye, Catal. Today, 2013, 215, 216–223 CrossRef.
- J. Li, L. Wang, Y. Cao, C. Zhang, P. He and H. Li, Chin. J. Chem. Eng., 2018, 26, 2266–2279 CrossRef.
- Z. He, Q. Qian, J. Ma, Q. Meng, H. Zhou, J. Song, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 737–741 CrossRef CAS.
- P. H. Choi, K. W. Jun, S. J. Lee, M. J. Choi and K. W. Lee, Catal. Lett., 1996, 40, 115–118 CrossRef CAS.
- J. Wei, J. Sun, Z. Wen, C. Fang, Q. Ge and H. Xu, Catal. Sci. Technol., 2016, 6, 4786–4793 RSC.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Appl. Catal., A, 2010, 373, 112–121 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, J. CO2 Util., 2013, 3–4, 102–106 CrossRef CAS.
- W. Wang, X. Jiang, X. Wang and C. Song, Ind. Eng. Chem. Res., 2018, 57, 4535–4542 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, ACS Catal., 2018, 8, 571–578 CrossRef CAS.
- X. Liu, M. Wang, C. Zhou, W. Zhou, K. Cheng, J. Kang, Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2018, 54, 140–143 RSC.
- M. Fujiwara, T. Satake, K. Shiokawa and H. Sakurai, Appl. Catal., B, 2015, 179, 37–43 CrossRef CAS.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Q. Zou, R. Quintero-Bermudez, Y. J. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- S. C. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, D. Cao Thang, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2019, 2, 86–93 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Z. Wang, G. Yang, Z. Zhang, M. Jin and Y. Yin, ACS Nano, 2016, 10, 4559–4564 CrossRef CAS.
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- J. H. Montoya, C. Shi, K. Chan and J. K. Norskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- H. Xiao, T. Cheng and W. A. Goddard III, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. L. Zhou, Y. Jiao, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS.
- E. Perez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- A. A. Peterson and J. K. Norskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- K. J. P. Schouten, E. P. Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- H. Xie, T. Wang, J. Liang, Q. Li and S. Sun, Nano Today, 2018, 21, 41–54 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, D. Cao Thang, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and N. Ki Tae, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. J. J. Zheng, Joule, 2018, 2, 1–32 CrossRef.
- D. F. Yan, Y. X. Li, J. Huo, R. Chen, L. M. Dai and S. Y. Wang, Adv. Mater., 2017, 29, 20 Search PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- H. Xiao, W. A. Goddard III, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. Roldan Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef.
- I. Kasatkin, P. Kurr, B. Kniep, A. Trunschke and R. Schlogl, Angew. Chem., Int. Ed., 2007, 46, 7324–7327 CrossRef CAS.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS.
- B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |