
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence5,10-Dimesityldiindeno[1,2-a:2′,1′-i]phenanthrene: a stable biradicaloid derived from Chichibabin's hydrocarbon†
Marcin A.
Majewski
 a,
Piotr J.
Chmielewski
a,
Alan
Chien
b,
Yongseok
Hong
c,
Tadeusz
Lis
a,
Maciej
Witwicki
a,
Dongho
Kim
*c,
Paul M.
Zimmerman
*b and
Marcin
Stępień
*a
a,
Piotr J.
Chmielewski
a,
Alan
Chien
b,
Yongseok
Hong
c,
Tadeusz
Lis
a,
Maciej
Witwicki
a,
Dongho
Kim
*c,
Paul M.
Zimmerman
*b and
Marcin
Stępień
*a
aWydział Chemii, Uniwersytet Wrocławski, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: marcin.stepien@chem.uni.wroc.pl; Web: http://www.mstepien.edu.pl
bDepartment of Chemistry, University of Michigan, 930 N. University Ave, Ann Arbor, MI 48109, USA. E-mail: paulzim@umich.edu
cDepartment of Chemistry, Yonsei University, 50 Yonsei-ro, Seoul 120-749, Korea. E-mail: dongho@yonsei.ac.kr
First published on 7th February 2019
Abstract
A diindenophenanthrene biradicaloid, formally derived from Chichibabin's hydrocarbon, is obtained in a short, scalable synthesis. The present system is electron-rich and devoid of conjugated substituents, and still exhibits very good stability under ambient conditions. The introduction of the diindeno[1,2-a:2′,1′-i] phenanthrene ring framework results in a singlet biradicaloid system with an easily accessible triplet state (ΔES–T = −1.30 kcal mol−1) and a small electronic bandgap (1.39 V). The stability limits of the title hydrocarbon were explored systematically in the solid state, to reveal an unusual thermally initiated hydrogen-scrambling oligomerization process.
Introduction
The use of proaromatic motifs is a commonly applied strategy to create open-shell π-conjugated organic molecules.1 In particular, quinoidal structures, which regain benzenoid aromaticity upon formal unpairing of two electrons, have been explored in a variety of molecular settings.2 A classical example of a quinoidal biradicaloid is found in Chichibabin's hydrocarbon (1, Scheme 1),3,4 which recovers two Clar sextets in its open-shell resonance form. Simple biradicaloid structures, such as 1 itself,4 tend to be unstable under ambient conditions, and extensive effort has been given to developing chemically robust open-shell systems. The diindeno[1,2-b:1′,2′-i]anthracene (DIAn) derivative 3, reported recently by Haley et al.5,6 exemplifies key strategies used to stabilize π-biradicaloids, namely extended ring fusion, steric blocking, and the use of conjugated substituents, such as ethynyl groups. 3 represents the family of stabilized diindenoarene biradicaloids,7–9 which comprises indenofluorenes10–14 and their larger congeners, e.g. ones containing naphthalene,15–18 pyrene,19 perylene,20 and bischrysene21 cores, or extended by benzenoid fusion.9,22–24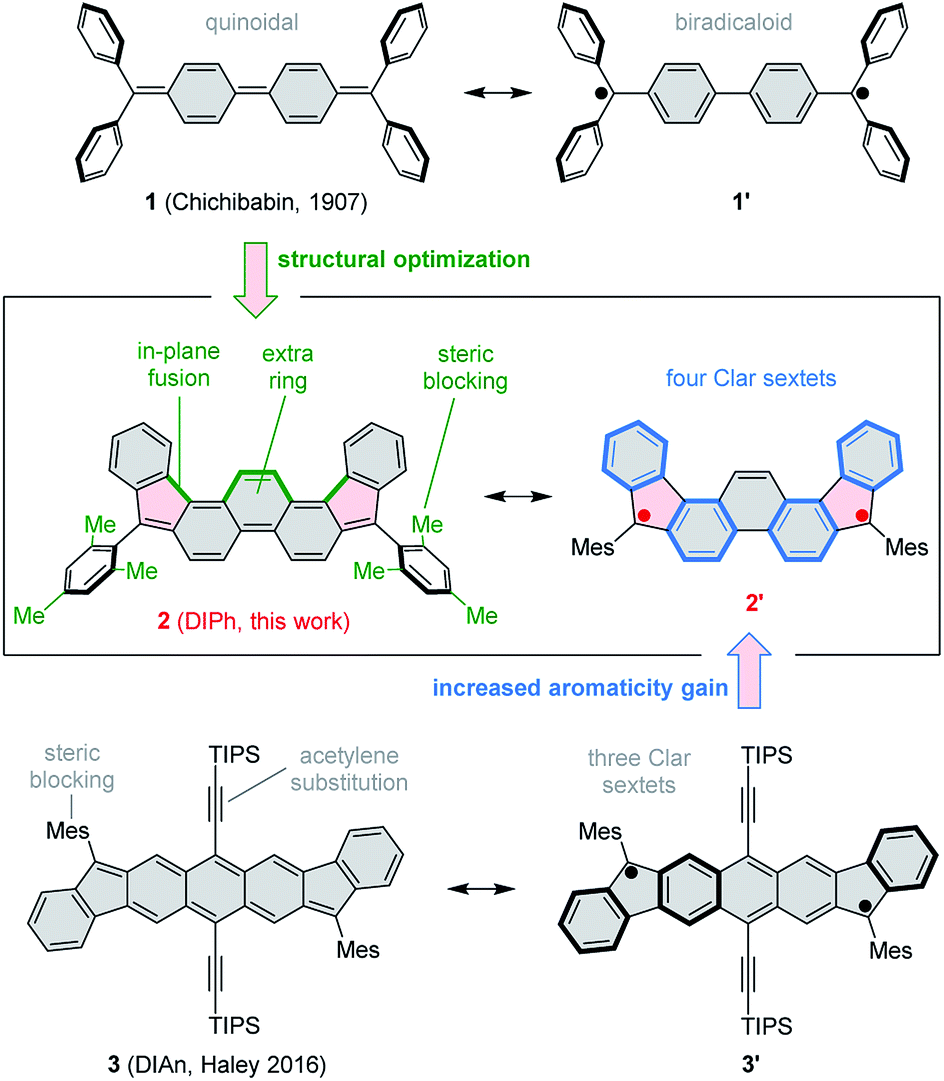 | ||
| Scheme 1 5,10-Dimesityldiindeno[1,2-a:2′,1′-i]phenanthrene (2) and its structural relationship to Chichibabin's hydrocarbon (1) and Haley's diindenoanthracene (3). Mes = mesityl, TIPS = tri(isopropyl)silyl. | ||
Here we show that by a judicious yet simple modification of the classical structure of Chichibabin's hydrocarbon 1, it is possible to create a much more stable biradicaloid system. This effect is achieved by (a) fusion of an extra benzene ring in the biphenyl section of 1, (b) pentannulation of two phenyl substituents, and (c) steric blocking of the remaining two phenyl groups in the form of bulky mesityl substituents. The resulting molecule, 5,10-dimesityldiindeno[1,2-a:2′,1′-i]phenanthrene 2 features a fused ring system (DIPh) isomeric to the DIAn framework of 3. Advantageously, 2 achieves ambient stability in the absence of bulky TIPS-acetylene substituents. This structural simplification results in a pure hydrocarbon system with a significantly lower molecular weight. The type of pentannulation introduced in 2 is also more effective than earlier modifications of 1 in which adjacent phenyl groups were fused to form terminal fluorene subunits. Those systems would either spontaneously polymerize25–27 or required multiple benzo fusion to achieve ambient stability.28
Results and discussion
Compound 2 was obtained in three steps from the previously reported 1,8-dibromophenanthrene-2,7-dicarbaldehyde 4 (ref. 29) (Scheme 2), and isolated as an air-stable dark-blue crystalline powder. The overall route from commercially available materials (six steps) is efficient and high-yielding: 2 is easily obtained in quantities exceeding 100 mg. In comparison with 3, the absence of TMS-ethynyl substituents in 2 makes the present design structurally simpler, furnishing the DIPh ring system as a heteroatom-free hydrocarbon derivative. The use of the phenanthrene ring system for elaboration of open-shell aromatics has so far been limited, with a single example provided by the recently reported [4]chrysaorene, a fully conjugated coronoid ring system with a tetraradicaloid character.30,31 Diffraction-quality crystals of 2 were grown without difficulty, and the structure of the new hydrocarbon was unambiguously confirmed using X-ray crystallographic analysis.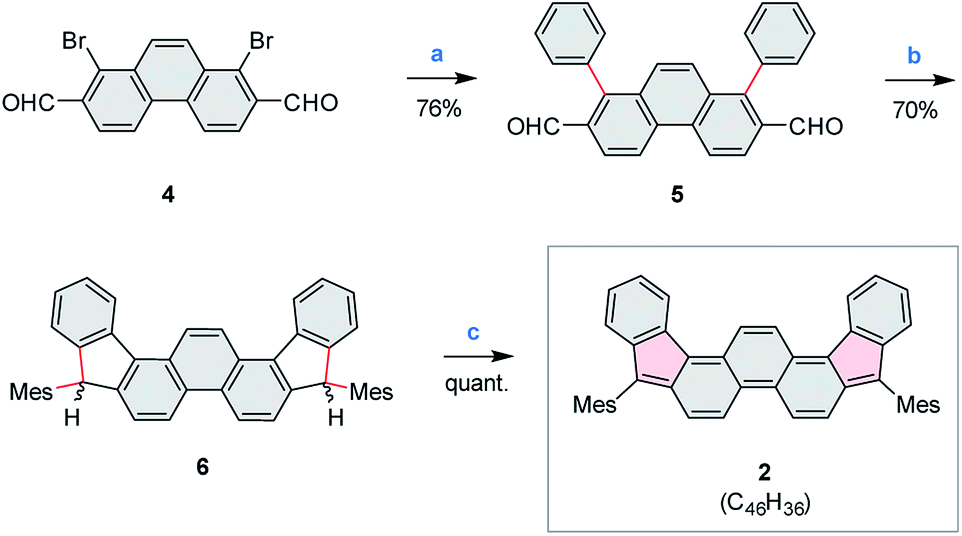 | ||
| Scheme 2 Synthesis of 5,10-dimesityldiindeno[1,2-a:2′,1′-i]phenanthrene (2). Reagents and conditions: (a) PhB(OH)2, Pd(PPh3)4, Na2CO3 (aq), dioxane, reflux, overnight. (b) (1) MesMgBr, THF, rt, overnight. (2) BF3·Et2O, DCM, rt, 10 min. (c) 2.5 equiv. DDQ, toluene, rt, overnight. | ||
In the solid state, the diindenophenanthrene core of 2 is found to deviate slightly from planarity, apparently as a result of steric repulsions in the two cove-like regions of the fused ring system (Fig. 1). DFT-optimized geometries of 2 are however planar, indicating that the experimentally observed distortion might be promoted by packing forces in the crystal. No π-stacking of DIPh cores was observed in the solid-state structure, in line with the bulky Mes substitution of the fused ring system. The “quinomethane” bonds in 2 (1.395(4) and 1.408(4) Å) are longer than expected of formally double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, a feature that correlates with the increasing biradicaloid character of diindenoarenes.7 The above bond lengths are similar to the corresponding distances determined for 3 (ref. 5) and 1 (ref. 4) (1.406 and 1.415(3) Å, respectively), in line with the presumed biradicaloid character of 2.
C bonds, a feature that correlates with the increasing biradicaloid character of diindenoarenes.7 The above bond lengths are similar to the corresponding distances determined for 3 (ref. 5) and 1 (ref. 4) (1.406 and 1.415(3) Å, respectively), in line with the presumed biradicaloid character of 2.
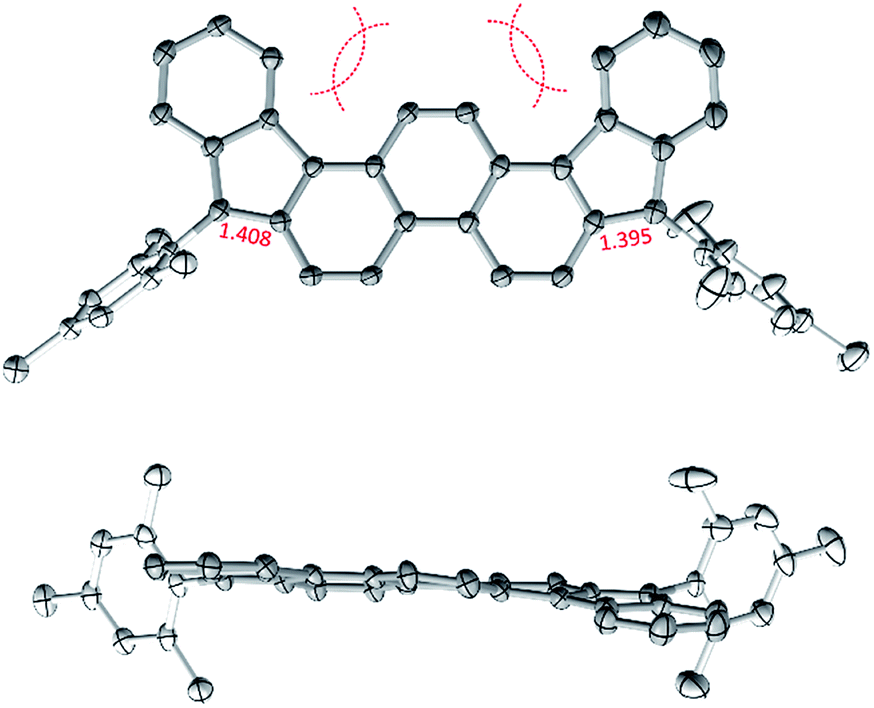 | ||
| Fig. 1 Molecular structure of 2 determined using X-ray crystallographic analysis. An n-hexane solvent molecule and hydrogen atoms are omitted for clarity. | ||
In the electronic spectrum of 2, the most intense band (λmax = 600 nm, ε = 4.1 × 104 M−1 cm−1, Fig. 2) is responsible for the deep blue color of the compound, and appears at a longer wavelength than that reported for 1 (λmax = 574 nm).4 While the main absorption in 2 is hypsochromically shifted relative to the corresponding band of 3 (690 nm), the absorption edge is observed at longer wavelengths (1050 nm vs. ca. 900 nm for 3), indicating a reduced optical bandgap of 2. According to a broken-symmetry TD-DFT calculation (ESI†), the first transition, dominated by the HOMO–LUMO excitation, is dipole-forbidden, in accord with the weak intensity of the corresponding experimental absorption.
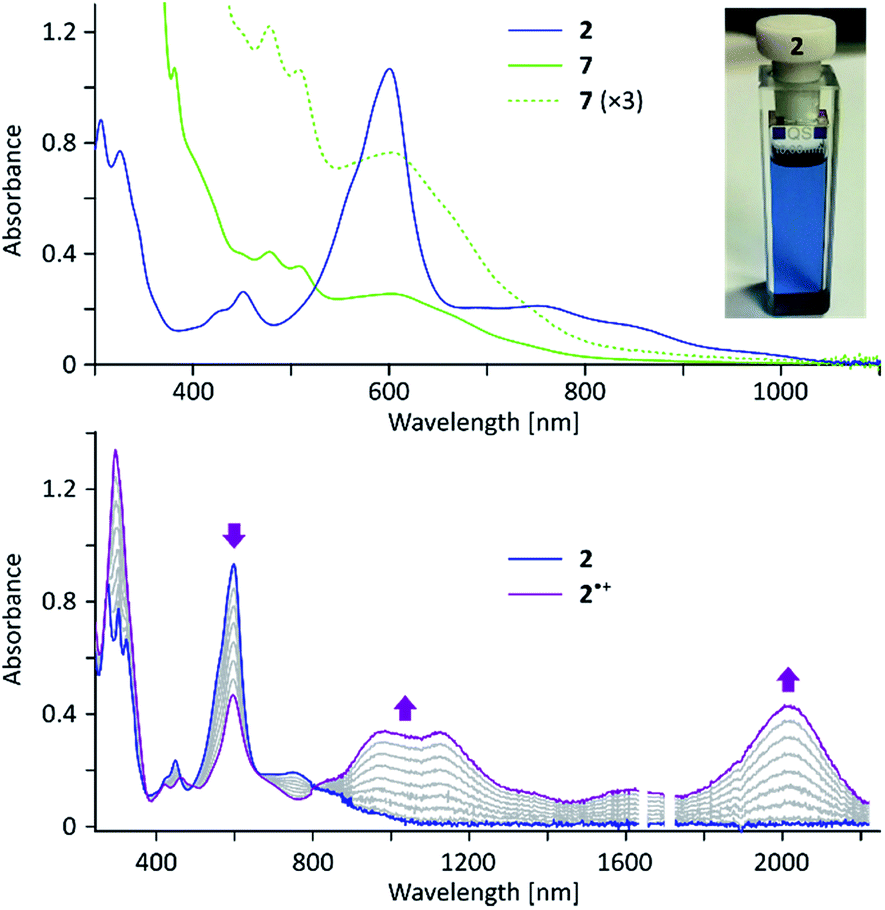 | ||
| Fig. 2 (Top) Electronic absorption spectrum of 2 (dichloromethane, ca. 2 × 10−5 M, blue trace) and 7 (dichloromethane, green traces). The photo shows the appearance of 2 in a dichloromethane solution. (Bottom) Formation of the radical cation 2˙+ (purple) upon oxidation with magic blue. Intermediate titration stages are shown in gray. | ||
In cyclic voltammetry measurements (Fig. 3), 2 undergoes three one-electron oxidations (0.26, 0.66, and 1.11 V vs. Fc+/Fc), of which only the first one is fully reversible. Good reversibility was however observed for two reductions (−1.13 and −1.42 V). All of the above redox events are shifted by up to 0.15 V to higher potentials relative to those determined for 3. The electrochemical bandgap of 1.39 V determined for 2 is smaller than the already uniquely narrow gap of 3 (1.45 V). Quinoidal molecules containing isomeric ring fusion-patterns often yield large variations of optical signatures and biradicaloid characters.12,14,32 In particular, the number of Clar sextets in the open-shell valence structure of DIPh is increased relative to DIAn (four in the open-shell formula 2′vs. three in 3′, cf.Scheme 1), and such a change has been demonstrated to enhance open-shell characters of hydrocarbon biradicaloids.33 In the present case, the bandgap change is likely induced not only by the different topologies of the ring systems in 2 and 3, but also by the difference in substitution patterns. TIPS-ethynyl groups were previously shown to reduce the optical bandgap of pentacene by 0.27 eV, an effect attributed to disproportionate lowering of the LUMO energy.34 The influence of substituents on the bandgap of 3 has not been investigated experimentally; however, if the response of 3 to ethynyl substitution is qualitatively similar to that observed in pentacene, the bandgap-reducing effect of replacing anthracene with phenanthrene in 2 would be even greater than indicated by electrochemistry data.
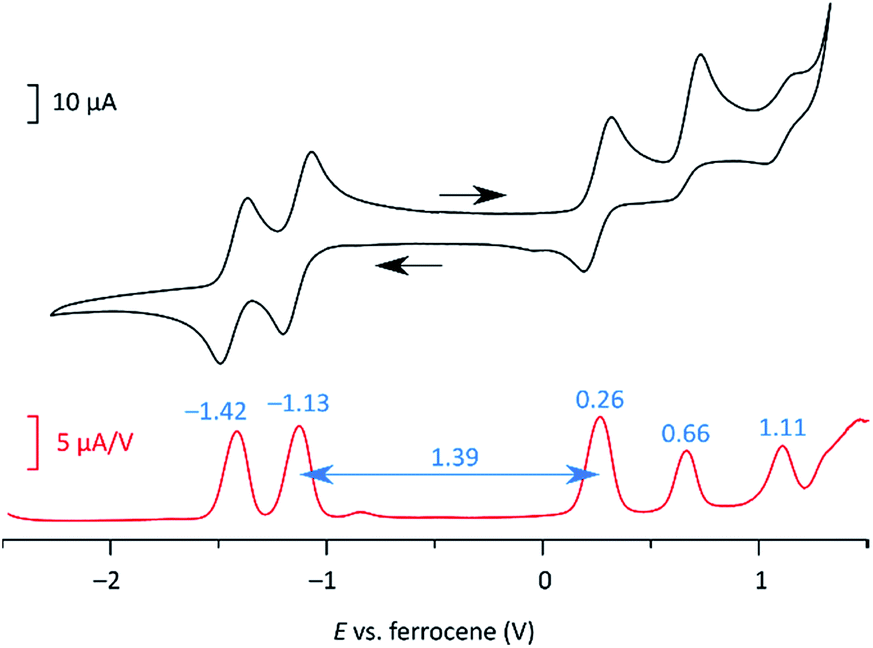 | ||
| Fig. 3 Cyclic voltammetry (top) and differential pulse voltammetry scans (bottom) obtained for 2 (dichloromethane, tetrabutylammonium hexafluorophosphate as a supporting electrolyte). | ||
At 220 K, 2 produces a sharp 1H NMR spectrum in the CDCl3 solution, enabling unambiguous assignment of all signals (Fig. 4). When the sample was heated to 360 K, the spectrum showed progressive yet reversible line broadening, accompanied by shortening of T1 relaxation times, observed already at 300 K (Fig. S10 and S11†). These spectral changes were indicative of chemical exchange with a paramagnetic species, which was presumed to be the thermally populated triplet state of 2, in analogy to the reported behavior of 3 and related systems.
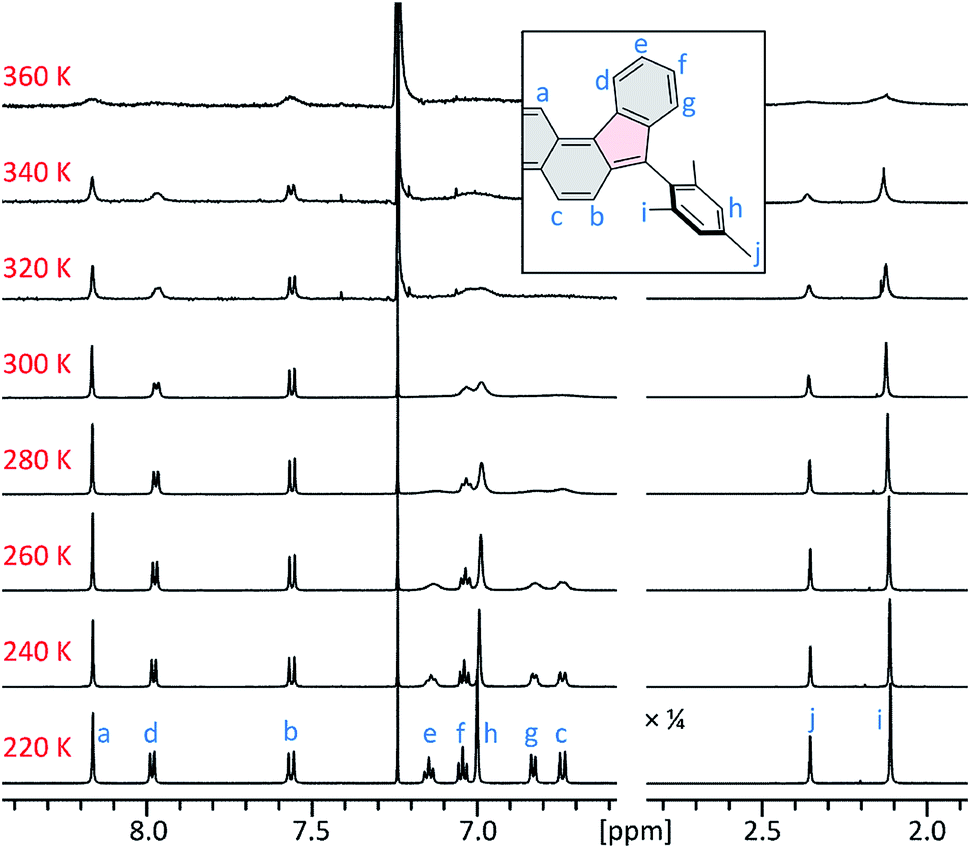 | ||
| Fig. 4 Temperature-dependent 1H NMR spectra of 2 (CDCl3, 600 MHz). The signals were assigned at 220 K using 2D spectroscopic methods (see the ESI†). | ||
Solid-state samples of 2 yielded a well-defined ESR line (g = 2.00248), however, without resolved triplet components. The lack of fine structure is apparently consistent with the relatively large distance between the two quinomethane carbons in 2 (9.84 Å vs. 10.32 Å in 3 (ref. 5) and 10.05 Å in 1 (ref. 4)). In the point-dipole approximation,35 this distance gives a value of the zero-field splitting (ZFS) parameter |D| = 29 G (0.003 cm−1). This estimate ignores electron delocalization36,37 and can be taken as the lower bound of the actual ZFS interaction. DFT calculations performed for 2 at various levels of theory produced somewhat variable, but noticeably higher estimates of D (50 to 100 G, 0.005 to 0.009 cm−1, see ESI† for computational details). These higher values reflect the effect of spin delocalization but nevertheless suggest that the ZFS in 2 may indeed be at the limit of measurability.38 Changes of the doubly integrated ESR signal intensity recorded in the 143–333 K range could be fitted with the Bleaney–Bowers equation to provide a singlet–triplet gap of ΔES–T = −1.30 kcal mol−1. The measurement of bulk magnetic susceptibility using the Evans method (for a toluene-d8 solution), showed a similar increase of the triplet contribution, yielding a ΔES–T value of −1.19 kcal mol−1, respectively. It was however observed that prolonged heating of the samples above 340 K resulted in further non-reversible increase of the magnetism, which was linked to the thermally induced reactivity of 2, discussed below.
2 can be chemically oxidized to a stable radical cation 2˙+ using [N(p-C6H4Br)3][SbCl6] (magic blue, Scheme 3, Fig. 2 and S20†). Further oxidation was observable when a stronger oxidant, (NO)[SbF6], was employed, but the resulting product was not stable. When treated with an excess of sodium metal in THF-d8, 2 cleanly produced the brownish dianion 22−,6,14 which was extremely unstable to traces of moisture and oxygen. The dianion yielded clearly defined, diamagnetic 1H and 13C NMR spectra, which were analyzed in detail with the help of 2D correlation methods (Fig. 5, S13 and S14†). The 1H resonance pattern of 22− is characterized by significant downfield relocations of the a, b, and c resonances relative to their positions in the spectrum of 2, suggesting that the aromaticity of the phenanthrene core is restored in the dianion. 13C NMR signals of quinomethane carbons in 22− were unambiguously identified at 99.0 ppm on the basis of 1H–13C HMBC correlations. This upfield shift, reproduced in a GIAO calculation (95.4 ppm), is characteristic of π-conjugated carbanions,39 and provides strong evidence for the charged nature of 22−. Interestingly, the bond length pattern obtained for 22− in a DFT calculation is very similar to that observed experimentally in the fluorenide anion,40 indicating a possible similarity of the electronic structures of these two systems. When traces of water were present during the Na-induced reduction of 2, the protonated monoanion (2-H)− was obtained instead of 22−. This new species, identified in situ on the basis of its 1D and 2D 1H NMR spectra (Fig. S12†), could be partly converted back to the neutral 2 by exposure to air.
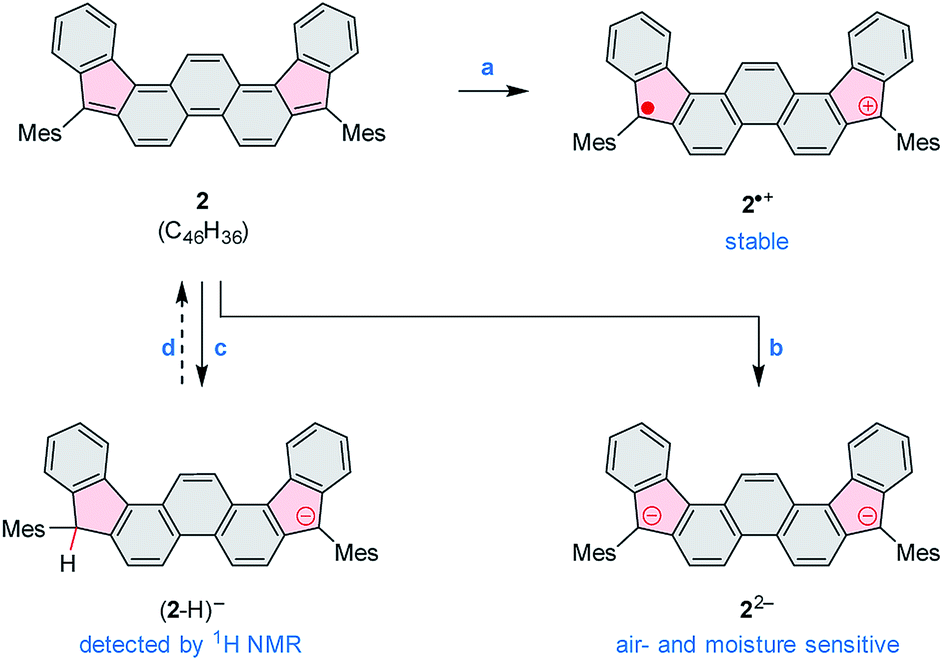 | ||
| Scheme 3 Redox reactivity of 2. Reagents and conditions: (a) [N(p-C6H4Br)3][SbCl6] (magic blue). (b) Na metal, THF-d8. (c) Na metal, THF-d8, traces of moisture. (d) Air (partial recovery). | ||
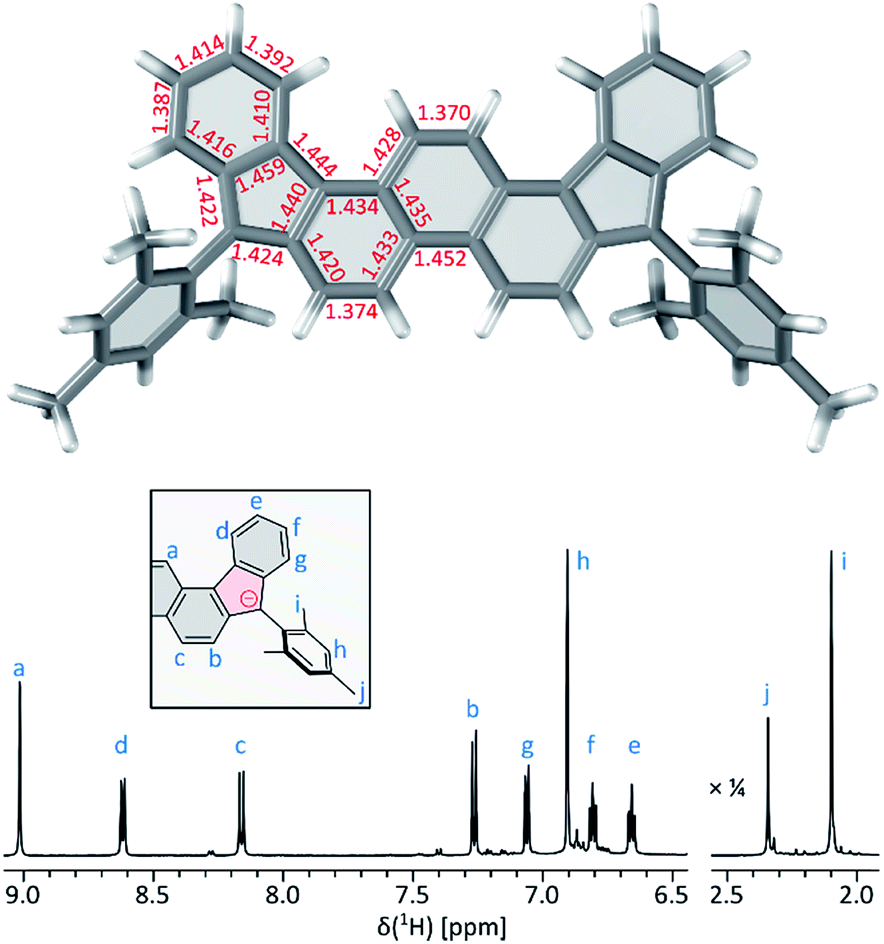 | ||
| Fig. 5 1H NMR spectrum (THF-d8, 600 MHz, 300 K, bottom) and DFT-optimized geometry of 22− (PCM(THF)/B3LYP/6-31G(d,p), top). | ||
Femtosecond transient absorption (fs-TA) measurements performed for 2 (Fig. 6A) revealed ultrafast relaxation dynamics, similar to those observed in other quinodimethane biradicaloids.7,8 Evolution-associated spectra and time traces of population ratios, obtained by global analysis (Fig. 6B and S18†), showed an initial decay with a time constant τ1 = 200 ± 20 fs corresponding to an internal conversion to the S1 state, followed by rapid deactivation to the ground state (τ2 = 7.0 ± 0.7 ps), possibly admixed with vibrational cooling. The short excited-state lifetime of 2 is comparable with those of other diindenoarenes,41 and may be similarly linked to non-radiative decay via an S1/S0 conical intersection.
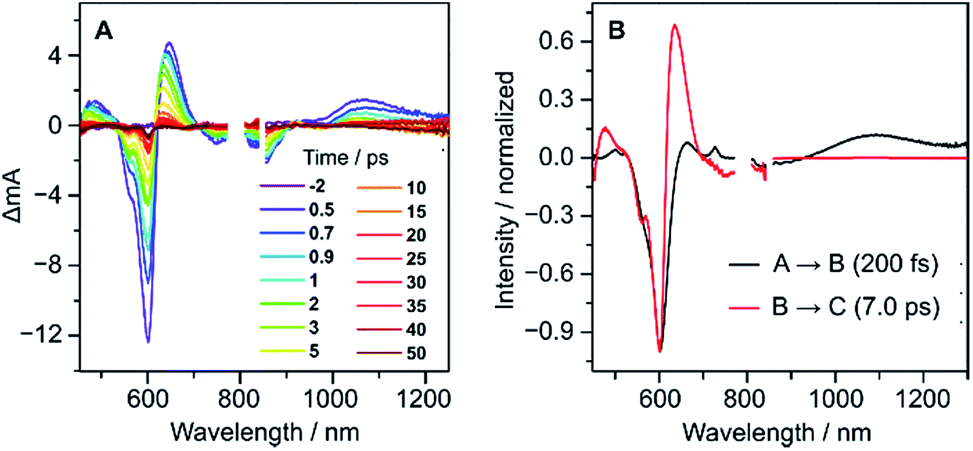 | ||
| Fig. 6 fs-TA spectra (A) and evolution-associated spectra (B) of 2 in THF with the excitation of 600 nm. | ||
The biradicaloid nature of 2 was explored computationally using the spin-flip approach.42 A RAS(2,2)-SF/cc-pVTZ calculation43,44 was performed on the substituent-free DIPh core of 2 optimized at the CAM-B3LYP/6-311++G(d,p) level of theory. A value of the ΔES–T gap of −1.43 kcal mol−1 was obtained, in remarkably good agreement with the results of ESR and Evans measurements. The strong biradicaloid character of the singlet state is indicated by the RAS-SF natural orbital occupancies, which gave a diradical index γ0 of 0.69. In both the S and T states, the odd-electron density, obtained from the RAS-SF calculation, is localized principally at the five-membered rings (Fig. 7B), in line with the open-shell configuration 2′. The validity of 2′ was further verified using 1D and 2D NICS scans performed on the DIPh core (Fig. 7A and S17†), which showed that the aromaticity of the central phenanthrene unit increases in the triplet state. In the singlet state, the terminal six-membered rings of DIPh remain aromatic, whereas the five-membered rings show a weak antiaromatic ring current. This current is apparently responsible for the upfield relocation of the o-Me resonance of 2 (signal i, Fig. 4).
 | ||
| Fig. 7 (A) NICS(1.5) maps (CAM-B3LYP/6-311++G(d,p)) and (B) RAS(4,4)-SF/cc-pVTZ odd-electron density (0.008 a.u. isovalue) calculated for the substituent-free DIPh ring system in the singlet (left) and triplet (right) states. | ||
The stability of 2 in solution depends on the choice of solvent and on storage environment (Fig. S1 and S2†). Remarkably, a 20 μM solution of 2 in dichloromethane showed no changes of the absorption spectrum after 25 days under ambient conditions. In THF, DMF, DMSO, and toluene, complete bleaching typically occurred within one day, when the sample was exposed to light. In the dark, however, THF solutions could be stored for over a week without significant decomposition, even under aerobic conditions. Solid 2 can be heated at ca. 70–110 °C for several hours without noticeable degradation. However, at these temperatures, the overall concentration of radicals in the sample, estimated using ESR, is seen to increase irreversibly, indicating a very slow, thermally activated transformation of 2. The onset of decomposition is additionally marked by the appearance of hyperfine structure in the ESR signal, which is particularly well resolved for solution samples. Line shape simulations suggest that this signal may correspond to a monoradical (or a family of structurally related species) formed by addition to one of the quinomethane carbons of 2 (ESI†). Formation of doublet radicals via addition to sp2 centers in biradicals was postulated in earlier work.38,45,46
Above 150 °C, the decomposition of 2 becomes faster, yielding an exotherm in a TGA–DTA scan (Tonset = 178 °C, Fig. S3†), with no significant mass loss. To drive the process to completion in a controlled way, solid 2 was sealed under argon and heated at 240 °C for 14 h. When so treated, 2 produced a greenish, fluorescent material (7, ca. 60% isolated yield), which was accompanied by ca. 5% of recovered 2 (displaced in the reaction tube by sublimation) and, unexpectedly, by ca. 25% of the dihydro precursor 6 (Scheme 4). While indene-based biradicaloids can be prone to hydrogenation,47 the present instance is of interest because of the absence of an external hydrogen source, and the bulky substitution at the five-membered rings. The 1H NMR spectrum of 7 is poorly defined but could be used to confirm the presence of saturated C(H)Mes moieties analogous to those in 6 (Fig. S4†). This structural feature, and the concurrent formation of 6 indicate that considerable scrambling of hydrogens occurs in the above thermally induced transformation.
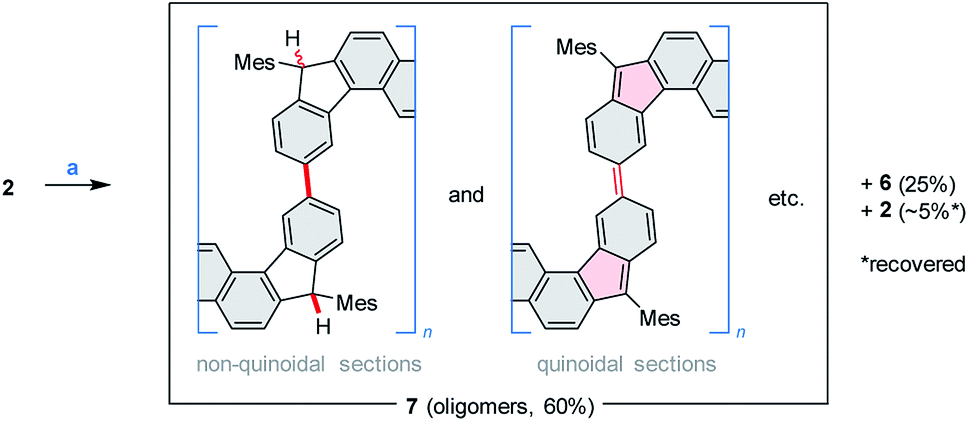 | ||
| Scheme 4 Thermally induced oligomerization of 2. Reagents and conditions: (a) argon, pressure tube, 240 °C, 14 h. | ||
The oligomeric nature of 7 was inferred from 1H DOSY maps and mass spectrometry (Fig. S6, S7 and S32†). Oligomer sizes of up to n = 15 were detectable in linear-mode MALDI analyses. High-resolution mass spectra of the lower oligomers (n ≤ 6) revealed a complex m/z pattern, which corresponded to a mixture of singly charged molecular ions with the general formula [(C46H36)n – xH + yO]+, with the major components characterized by 0 ≤ x ≤ n and y = 0 to 5. Since the oligomerization takes place in the absence of dioxygen, the oxidized species are presumed to form either during workup or in the ion source. The absorption spectrum of 7 (Fig. 2) resembles that of 2, with the absorption onset at ca. 1000 nm, except that the vis-NIR part is broadened and has a diminished intensity relative to the UV part of the spectrum. All these spectral features of 7 support an oligomer structure combining quinoidal and non-quinoidal sections (Scheme 4).
We initially hypothesized that the oligomerization may start with the formation of σ-dimers such as 8 (Scheme 5), which would undergo subsequent hydrogen scrambling to produce the oligomeric products. Analogous additions have been documented for open-shell systems for both inter-48 and intramolecular cases,49–51 with the resulting sp3-linked products undergoing facile dehydrogenation (if possible). Thermal induction of the oligomerization process was at first presumed to indicate that the triplet state of 2 (T-2)is the initial reactive species.52 Using DFT, all possible symmetrical σ-dimers of 2 were optimized (a–a, b–b, etc.), taking into account the different spin states (singlet vs. triplet) and stereoisomers (racemo vs. meso). The e–e dimer 8 was found to have much lower energy than all other structures (26.84 kcal mol−1 for the meso stereoisomer, triplet state), indicating that position e might be the preferred addition site. However, it was also found that the barriers to dimerization are very high on both the broken-symmetry singlet (bsS) and triplet hypersurfaces (ΔG‡ = 39.97 and 40.20 kcal mol−1, respectively), indicating that direct dimerization should be thermally inaccessible.
 | ||
| Scheme 5 Potential oligomerization pathways for 2. Gibbs free energies (red labels, kcal mol−1, UCAM-B3LYP/6-31G(d,p), gas phase, 298 K) are given relative to S-2 + S-2, unless indicated otherwise. S, D, and T indicate respectively the broken-symmetry-singlet, doublet and triplet wavefunctions. r and m indicate the racemic pair and the meso isomer, respectively. | ||
Looking for a more feasible pathway, we considered addition of the neutral 2 to the radical cation 2˙+, which is expected to form in small amounts in bulk samples of 2. Such an addition would produce the radical-cation intermediate 8˙+ (Scheme 5), which yields broken-symmetry geometries characteristic of a Robin–Day class-I or -II mixed-valence (MV) system.53 The calculated barrier for this process (ΔG‡ = 31.82 kcal mol−1, doublet hypersurface) is significantly lower than those obtained for dimerization of the neutral 2. While the value is still relatively high, it does not bear a quantitative significance because the accuracy of the calculation is potentially limited by the MV character of the system,53 the neglect of solvation and dispersion phenomena, and the choice of the standard state.
The calculated ΔG value for the electron transfer process 8˙+ + 2 → 2˙+ + 8 are low (0 to 2 kcal mol−1) indicating that in principle, the electron can be transferred between chains of various lengths. The oligomerization can thus propagate via a sequence of hydrogen, proton, and electron transfer steps. Importantly, while the initial adduct 8 is high in energy, the formation of its rearranged isomers, such as 9a–b, which contain hydrogenated 5-membered rings, is predicted to be significantly exergonic. A similar stabilization is predicted for 9a˙+ (ΔG of ca. −25 kcal mol−1vs. S-2 + 2˙+). The overall reactivity described above would be distinct from reported cases of biradicaloid oligo- and cyclooligomerizations, in which the addition typically involves sites with highest spin densities, sometimes relatively hindered,26,27,52,54–56 or conjugated acetylene substituents.57 DFT shows that, while the initial barrier to dimerization may be relatively high, the proposed oligomerization is a strongly exergonic process, and should be feasible for neat 2 at high temperatures. Further growth of the oligomer chain can produce a variety of species containing random sequences of closed-shell and open-shell sections. It is worth noting that in such sequences, terminal fluorenyl radicals may form (as seen in the idealized oligomer 11), which can be expected to produce a doublet ESR spectrum, qualitatively similar to that observed for 2 at the beginning of its decomposition (vide supra).
Conclusions
We have reported here a simple analogue of Chichibabin's hydrocarbon, characterized by very good ambient stability. The present design is simpler than those explored previously,28,58–61 and achieves the stabilizing effect without increasing the electronic bandgap or introduction of heteroatoms. The hydrogen-scrambling oligomerization, described herein, is relevant as a prospective method of synthesizing open-shell polymeric materials, but it also provides a guiding principle for designing stable electron-rich biradicaloids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Science Center of Poland (DEC-2012/07/E/ST5/00781 to M. S. and DEC-2015/19/N/ST5/00760 to M. A. M.) and the Foundation for Polish Science (START fellowship to M. A. M.) is gratefully acknowledged. Quantum chemical calculations were performed in the Wrocław Center for Networking and Supercomputing. P. M. Z. thanks the Alfred P. Sloan Foundation for support of this project. The work at Yonsei University was supported by Strategic Research (NRF-2016R1E1A1A01943379) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT (Information and Communication Technologies) and Future Planning. We thank Prof. Frank Würthner (Würzburg), Dr Yi-Lin Wu (Northwestern University), and anonymous referees for helpful comments.Notes and references
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- J. Casado, Top. Curr. Chem., 2017, 375, 73 CrossRef PubMed.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef CAS.
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004–6011 CrossRef CAS PubMed.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS PubMed.
- G. E. Rudebusch, G. L. Espejo, J. L. Zafra, M. Peña-Alvarez, S. N. Spisak, K. Fukuda, Z. Wei, M. Nakano, M. A. Petrukhina, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 12648–12654 CrossRef CAS PubMed.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS PubMed.
- Y. Tobe, Top. Curr. Chem., 2018, 376, 12 CrossRef PubMed.
- T. Kubo, Chem. Lett., 2015, 44, 111–122 CrossRef.
- D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127–1130 CrossRef CAS PubMed.
- A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906–6910 CrossRef CAS PubMed.
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed.
- A. G. Fix, P. E. Deal, C. L. Vonnegut, B. D. Rose, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 15, 1362–1365 CrossRef CAS PubMed.
- J. J. Dressler, Z. Zhou, J. L. Marshall, R. Kishi, S. Takamuku, Z. Wei, S. N. Spisak, M. Nakano, M. A. Petrukhina and M. M. Haley, Angew. Chem., Int. Ed., 2017, 56, 15363–15367 CrossRef CAS PubMed.
- B. D. Rose, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Org. Lett., 2012, 14, 2426–2429 CrossRef CAS PubMed.
- H. Miyoshi, S. Nobusue, A. Shimizu, I. Hisaki, M. Miyata and Y. Tobe, Chem. Sci., 2013, 5, 163–168 RSC.
- J. E. Barker, C. K. Frederickson, M. H. Jones, L. N. Zakharov and M. M. Haley, Org. Lett., 2017, 19, 5312–5315 CrossRef CAS PubMed.
- H. Miyoshi, M. Miki, S. Hirano, A. Shimizu, R. Kishi, K. Fukuda, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Nakano and Y. Tobe, J. Org. Chem., 2017, 82, 1380–1388 CrossRef CAS PubMed.
- D. Hibi, K. Kitabayashi, K. Fujita, T. Takeda and Y. Tobe, J. Org. Chem., 2016, 81, 3735–3743 CrossRef CAS PubMed.
- K. Sbargoud, M. Mamada, J. Marrot, S. Tokito, A. Yassar and M. Frigoli, Chem. Sci., 2015, 6, 3402–3409 RSC.
- J. Ma, J. Liu, M. Baumgarten, Y. Fu, Y.-Z. Tan, K. S. Schellhammer, F. Ortmann, G. Cuniberti, H. Komber, R. Berger, K. Müllen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3280–3284 CrossRef CAS.
- C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827–16838 CrossRef CAS PubMed.
- J. Liu, J. Ma, K. Zhang, P. Ravat, P. Machata, S. Avdoshenko, F. Hennersdorf, H. Komber, W. Pisula, J. J. Weigand, A. A. Popov, R. Berger, K. Müllen and X. Feng, J. Am. Chem. Soc., 2017, 139, 7513–7521 CrossRef CAS PubMed.
- J. Melidonie, J. Liu, Y. Fu, J. J. Weigand, R. Berger and X. Feng, J. Org. Chem., 2018, 83, 6633–6639 CrossRef CAS PubMed.
- W. Theilacker, H. Schulz, U. Baumgarte, H.-G. Drössler, W. Rohde, F. Thater and H. Uffmann, Angew. Chem., 1957, 69, 322–333 CrossRef.
- J. Ipaktschi, R. Hosseinzadeh and P. Schlaf, Angew. Chem., Int. Ed., 1999, 38, 1658–1660 CrossRef CAS.
- D. Beaudoin, O. Levasseur-Grenon, T. Maris and J. D. Wuest, Angew. Chem., Int. Ed., 2016, 55, 894–898 CrossRef CAS.
- Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef CAS PubMed.
- M. Shimizu, Y. Tomioka, I. Nagao, T. Kadowaki and T. Hiyama, Chem.–Asian J., 2012, 7, 1644–1651 CrossRef CAS.
- X. Lu, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Q. Jiang, C. Liu, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 13052–13056 CrossRef CAS.
- H. Gregolińska, M. Majewski, P. J. Chmielewski, J. Gregoliński, A. Chien, J. Zhou, Y.-L. Wu, Y. J. Bae, M. R. Wasielewski, P. M. Zimmerman and M. Stępień, J. Am. Chem. Soc., 2018, 140, 14474–14480 CrossRef PubMed.
- A. Konishi, Y. Okada, M. Nakano, K. Sugisaki, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2017, 139, 15284–15287 CrossRef CAS PubMed.
- Z. Sun, S. Lee, K. H. Park, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, S. Das, Y. Li, C. Chi, R.-W. Li, K.-W. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 18229–18236 CrossRef CAS.
- I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- P. Bertrand, C. More, B. Guigliarelli, A. Fournel, B. Bennett and B. Howes, J. Am. Chem. Soc., 1994, 116, 3078–3086 CrossRef CAS.
- C. Riplinger, J. P. Y. Kao, G. M. Rosen, V. Kathirvelu, G. R. Eaton, S. S. Eaton, A. Kutateladze and F. Neese, J. Am. Chem. Soc., 2009, 131, 10092–10106 CrossRef CAS PubMed.
- C. Wentrup, M. J. Regimbald-Krnel, D. Müller and P. Comba, Angew. Chem., Int. Ed., 2016, 55, 14600–14605 CrossRef CAS PubMed.
- K. Müllen, Chem. Rev., 1984, 84, 603–646 CrossRef.
- S. Filipponi, J. N. Jones, J. A. Johnson, A. H. Cowley, F. Grepioni and D. Braga, Chem. Commun., 2003, 2716–2717 RSC.
- B. D. Rose, L. E. Shoer, M. R. Wasielewski and M. M. Haley, Chem. Phys. Lett., 2014, 616–617, 137–141 CrossRef CAS.
- A. I. Krylov, Chem. Phys. Lett., 2001, 338, 375–384 CrossRef CAS.
- P. M. Zimmerman, F. Bell, M. Goldey, A. T. Bell and M. Head-Gordon, J. Chem. Phys., 2012, 137, 164110 CrossRef PubMed.
- F. Bell, P. M. Zimmerman, D. Casanova, M. Goldey and M. Head-Gordon, Phys. Chem. Chem. Phys., 2013, 15, 358–366 RSC.
- H. Sakurai, H. Tobita, M. Kira and Y. Nakadaira, Angew. Chem., Int. Ed. Engl., 1980, 19, 620 CrossRef.
- A. Kostenko, B. Tumanskii, M. Karni, S. Inoue, M. Ichinohe, A. Sekiguchi and Y. Apeloig, Angew. Chem., Int. Ed., 2015, 54, 12144–12148 CrossRef CAS PubMed.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134–1140 CrossRef CAS PubMed.
- J. L. Zafra, L. Qiu, N. Yanai, T. Mori, M. Nakano, M. P. Alvarez, J. T. L. Navarrete, C. J. Gómez-García, M. Kertesz, K. Takimiya and J. Casado, Angew. Chem., Int. Ed., 2016, 55, 14563–14568 CrossRef CAS PubMed.
- K. Uchida, S. Ito, M. Nakano, M. Abe and T. Kubo, J. Am. Chem. Soc., 2016, 138, 2399–2410 CrossRef CAS.
- P. Ravat, T. Šolomek, M. Rickhaus, D. Häussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183–1186 CrossRef CAS PubMed.
- T. Šolomek, P. Ravat, Z. Mou, M. Kertesz and M. Juríček, J. Org. Chem., 2018, 83, 4769–4774 CrossRef PubMed.
- A. Kikuchi, F. Iwahori and J. Abe, J. Am. Chem. Soc., 2004, 126, 6526–6527 CrossRef CAS PubMed.
- C. Sutton, T. Körzdörfer, V. Coropceanu and J.-L. Brédas, J. Phys. Chem. C, 2014, 118, 3925–3934 CrossRef CAS.
- T. Itoh, Prog. Polym. Sci., 2001, 26, 1019–1059 CrossRef CAS.
- W. S. Trahanovsky and S. P. Lorimor, J. Org. Chem., 2006, 71, 1784–1794 CrossRef CAS.
- J. Inoue, K. Fukui, T. Kubo, S. Nakazawa, K. Sato, D. Shiomi, Y. Morita, K. Yamamoto, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2001, 123, 12702–12703 CrossRef CAS.
- X. Fu and D. Zhao, Org. Lett., 2015, 17, 5694–5697 CrossRef CAS PubMed.
- H. Kataoka and M. Nakagawa, Bull. Chem. Soc. Jpn., 1963, 36, 799–806 CrossRef CAS.
- T. Kawase, N. Ueno and M. Oda, Tetrahedron Lett., 1992, 33, 5405–5408 CrossRef CAS.
- A. Lichtblau, H. D. Hausen, W. Schwarz and W. Kaim, Inorg. Chem., 1993, 32, 73–78 CrossRef CAS.
- C. Jiang, Y. Bang, X. Wang, X. Lu, Z. Lim, H. Wei, S. El-Hankari, J. Wu and Z. Zeng, Chem. Commun., 2018, 54, 2389–2392 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectra of new compounds, and details of computational studies. CCDC 1861241. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00170k |
| This journal is © The Royal Society of Chemistry 2019 |