
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of a pair of O-fac/O-mer 12-P-6 alkyloxaphosphates with a P–O–C–C four-membered ring†
Xin-Dong
Jiang
b,
Yuya
Toya
a,
Shiro
Matsukawa
a,
Satoshi
Kojima
a,
J. Oscar C.
Jimenez-Halla
 c,
Rong
Shang
a,
Masaaki
Nakamoto
a and
Yohsuke
Yamamoto
*a
c,
Rong
Shang
a,
Masaaki
Nakamoto
a and
Yohsuke
Yamamoto
*a
aDepartment of Chemistry, Graduate School of Science, Hiroshima University, Higashi-hiroshima, 7398526, Japan. E-mail: yyama@sci.hiroshima-u.ac.jp
bCollege of Applied Chemistry, Shenyang University of Chemical Technology, Shenyang, 110142, China
cDepartment of Chemistry, Division of Natural and Exact Sciences, University of Guanajuato, Campus Gto, Noria Alta s/n 36050, Guanajuato, Mexico
First published on 19th February 2019
Abstract
Structurally characterized hexacoordinate organophosphorus compounds remain rare due to their highly reactive nature and thermal instability. Herein we report the first synthesis of a pair of O-facial and O-meridional hexacoordinate oxaphosphates (5B and 5D) obtained from the O-apical and O-equatorial β-hydroxyalkylphosphoranes 3 and 4. This was achieved by using the bulky C2F5-groups on the ortho-substituted aryl backbone. Calculations of the relative energies of possible isomers indicate 5B and 5D are thermodynamic products. Although the mechanisms of their formation and the determining factor of stereo-selectivity are still unclear, their isolation and structure conformation contributes to a formulation of a viable strategy for diastereoselective synthesis of heteroleptic hexacoordinate organophosphates.
Introduction
Hypervalent phosphorus compounds, closely related to the phosphoryl transfer reaction in biological systems,1,2 have been attractive subjects for both experimental and theoretical chemists.3,4 In the context of synthetic organic chemistry, pentacoordinate phosphoranes have been the centre of studies related to Wittig reactions5 since the first report of the pentaphenylphosphorane (Ph5P),6 revealing their characteristic apicophilicity7–11 and facile stereomutation.12–15 This, subsequently, leads to the later development of geometrically constrained T-shaped phosphorus(III) compounds16 for small molecule activation and catalysis,17 as well as the increasing number of applications of phosphonium as Lewis acids in Frustrated Lewis Pair (FLP) chemistry.18,19In contrast to the diverse applications derived from bond cleavage and formation processes of the phosphorane chemistry, hexacoordinate phosphates have mostly used as chiral moieties for resolving enantiomeric species,20–25 and weakly coordinating anions for stabilizing highly reactive cationic species.26 The stereoselective synthesis and isolation of different diastereomers of heteroleptic systems remains a challenge, with few examples of hexacoordinate organophosphates that have been structurally confirmed to date (selected examples are shown in Chart 1a).1,27–30 This has been a persistent bottleneck for the understanding of bonding characteristics and the reactivity of hexacoordinate organophosphates, preventing them from finding wider applications in organic synthesis.31
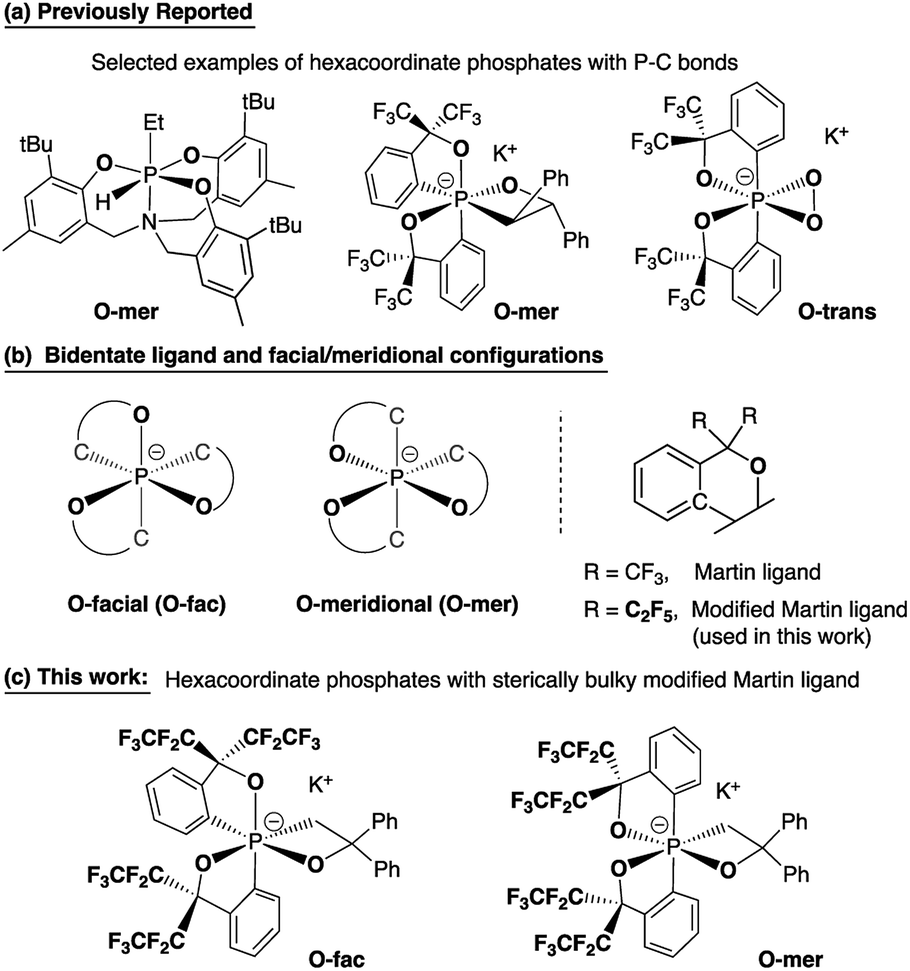 | ||
| Chart 1 Bidentate ligands, O-facial/O-meridional steric configurations, and design strategies for O-fac/O-mer hexacoordinate phosphates. | ||
The most general method of generating an octahedral hexacoordinate phosphate is by intramolecular nucleophilic addition to a pentacoordinate phosphorane center. With three bidentate ligands such as Martin's ortho-substituted aryl ligand (Chart 1b),32–37 both facial (fac-) and meridional (mer-) isomers may be formed theoretically. Typically, the meridional isomer is obtained because of the kinetic preference to the lower lying 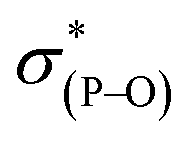 orbital over the
orbital over the 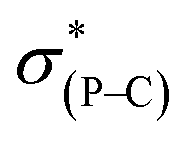 orbital.27 In spite of the fact that both fac- and mer-isomers of hexacoordinate silicon complexes and metal chelates38–40 are well documented, to the best of our knowledge, only one facial derivative of hexacoordinate phosphate has been reported, by Gates and coworkers, based on a homoleptic O/N ligand system.41 Until now, strategies for controlling the stereochemistry of heteroleptic hexacoordinate systems have not been achieved.
orbital.27 In spite of the fact that both fac- and mer-isomers of hexacoordinate silicon complexes and metal chelates38–40 are well documented, to the best of our knowledge, only one facial derivative of hexacoordinate phosphate has been reported, by Gates and coworkers, based on a homoleptic O/N ligand system.41 Until now, strategies for controlling the stereochemistry of heteroleptic hexacoordinate systems have not been achieved.
Herein we report the first stereo-pure isolation of a pair of O-fac and O-mer isomers of 12-P-6 oxaphosphates (Chart 1c) from the corresponding pentacoordinate O-equatorial/O-apical phosphoranes.32–36,42,43 This was achieved by using a modified Martin ligand37,44–46 with two bulky C2F5 groups to slow down the Berry pseudorotation (BPR) of the precursors.37,44,45 Both experimental and theoretical evidence on their structures and formation will be presented, which contributes to the formulation of a viable strategy for diastereoselective synthesis of hexacoordinate phosphates and call for further investigations into their isomerization mechanisms and reactivity.
Results and discussion
Previously, by using the pentafluoroethyl derivative of the Martin ligand to increase the steric hindrance in the BPR process, we were able to isolate both O-equatorial (1) and O-apical pentacoordinate methylphosphoranes (2).37 Compound 1 is thermodynamically less stable than 2 and readily isomerizes to 2 upon heating.37 Deprotonation of 1 and 2 followed by the addition of benzophenone lead to the formation of 3 and 4, which are precursors to our target hexacoordinate phosphates (Scheme 1). X-ray crystallographic analysis of single crystals confirmed the preservation of the O-equatorial and O-apical geometries (Fig. 1 and Table S1†).4719F{1H} and 13C{1H} and 1H NMR experiments of 3 in chloroform carried out at room temperature showed equivalent signals of the ligand, indicating an equilibrium between the pair of one-step BPR isomers of 3 in solution.48 In contrast, no dynamic behaviours were detected from the NMR spectra of 4, suggesting a single isomer in solution at room temperature (see the spectra in the ESI†). The 31P{1H} NMR spectra of 3 and 4 showed a singlet at −2.1 ppm and −15.7 ppm respectively, consistent with pentacoordinate phosphorus environments. At elevated temperatures in benzene, the O-equatorial isomer 3 slowly converts to the O-apical isomer 4 quantitatively (Scheme 1), consistent with previous observations.11,37,48 The calculated energy difference between 3 and 4 is 6.8 kcal mol−1 ((SMD:thf)ω-B97XD/def2-tzvpp//ω-B97XD/def2-svp),49 in agreement with the experimental observations.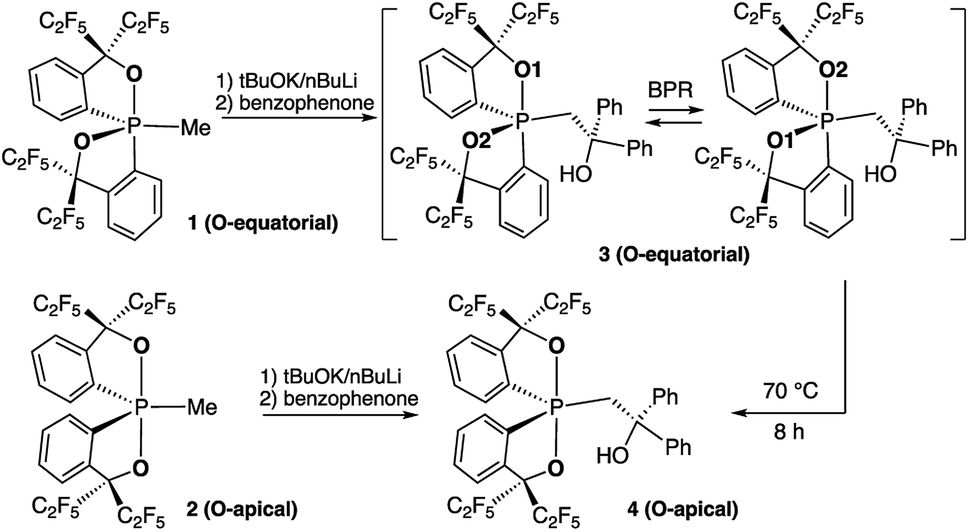 | ||
| Scheme 1 Synthesis of pentacoordinate alkyloxaphosphoranes 3 and 4with a –CH2CPh2OH group. | ||
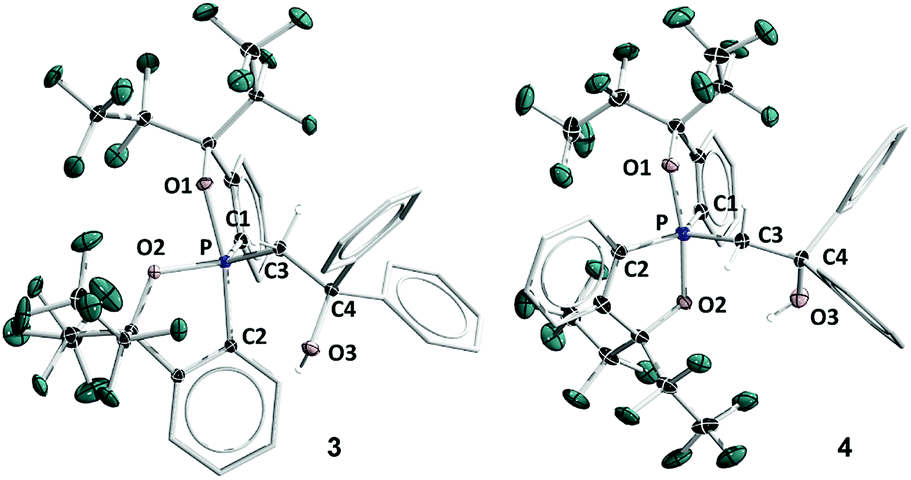 | ||
| Fig. 1 ORTEP diagrams of 3 (ref. 50) and 4 showing thermal ellipsoids at the 30% probability level. Aryl carbon and hydrogen atoms are omitted for clarity. Selected distances (Å) for 4: P1–O1, 1.753(2); P1–O2, 1.790(2); P1–C1, 1.831(3); P1–C2, 1.830(3); P1–C3, 1.838(3); C3–C4, 1.561(4); O3–C4, 1.422(4). CCDC: 1856674 for 3; 1856675 for 4. | ||
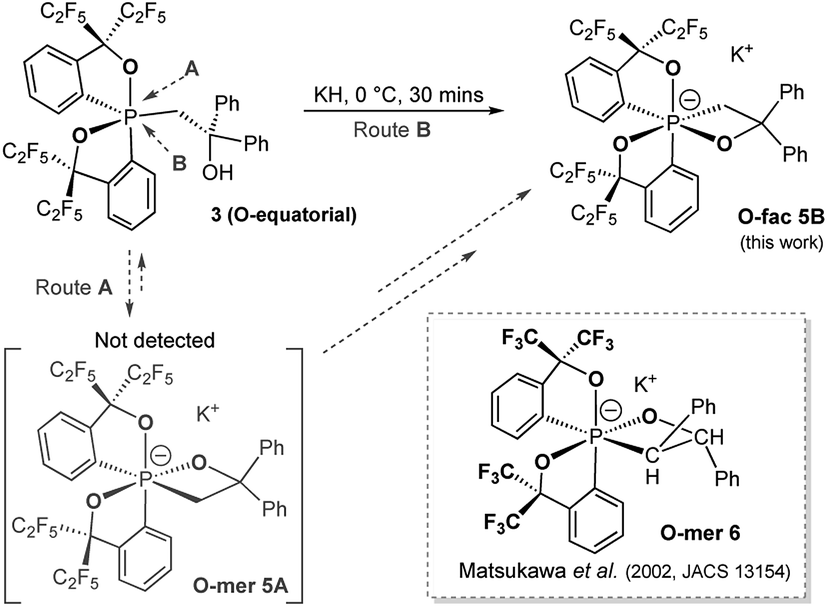
Deprotonation of pentacoordinate precursors 3 and 4 in THF by KH at 0 °C followed by intramolecular nucleophilic attack of the oxide lead to isolation of hexacoordinate phosphates. The reactions were monitored by 31P{1H} NMR spectroscopy. In the reaction of the O-equatorial phosphorane 3, the reaction completes within 30 minutes to afford a new compound 5B in 85% isolated yield. The 31P NMR spectrum of 5B in CD3CN at room temperature showed a singlet at −107.8 ppm, characteristic of a hexacoordinate phosphate (ESI†). Furthermore, in the 1H NMR spectrum, the originally equivalent hydrogen atoms (by BPR) on the methylene protons (CH2) observed in 3 (δ = 3.84 (d, 2JHP = 15.6 Hz, 2H) at 25 °C) gave rise to two sets of distinct proton signals at 4.19 ppm (dd, 2JHP = 30 Hz, 2JHH = 12 Hz, 1H) and 3.21 ppm (dd, 2JHP = 12 Hz, 2JHH = 12 Hz, 1H), consistent with a restricted P–C3 bond in a four-membered ring. Also, the 19F NMR spectrum showed four distinguishable quartets for –CF3 groups. These observations suggest a hexacoordinate structure of 5B in CD3CN at room temperature.
The structure of 5B was confirmed from single-crystal X-ray crystallographic analysis, which revealed a hexacoordinate O-facial geometry. The solid-state structure of 5B (Fig. 2 and Table S1†) shows a facial coordination of the potassium to all the oxygen atoms. This cation coordination can be prevented by addition of 18-crown-6 to the reaction mixture, which suggests that the facial coordination of potassium cation is not a crucial factor responsible for the formation of O-facial geometry.51 A solid-state structure of 5B with an independent anion and crown ether-captured cation is shown in Fig. S2.†
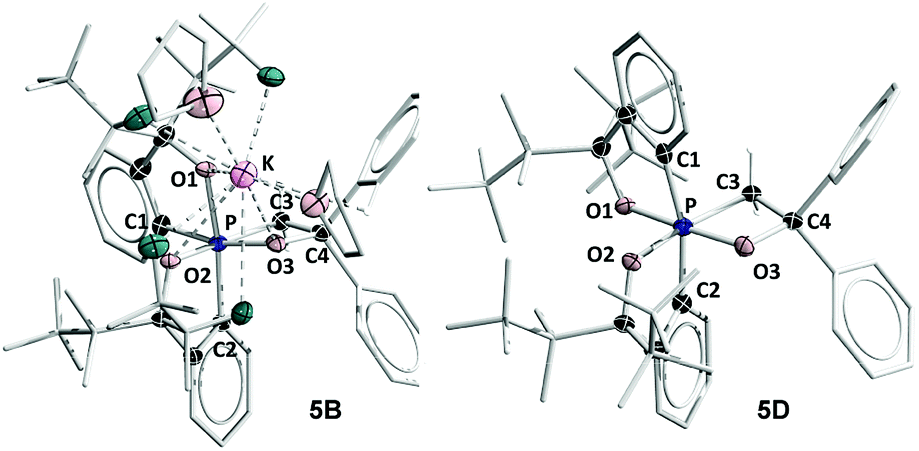 | ||
| Fig. 2 ORTEP diagram of hexacoordinate oxaphosphates O-fac5B and O-mer5D38 showing thermal ellipsoids at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) for O-fac5B: P–O1, 1.835(2); P–O2, 1.805(2); P–O3, 1.742(2); P–C1, 1.862(4); P–C2, 1.869(3); P–C3, 1.871(3); C3–C4, 1.529(5); O3–C4, 1.450(4). 5D: P–O1, 1.811(3); P–O2, 1.809(3); P–O3, 1.732(3); P–C1, 1.865(4); P–C2, 1.867(4); P–C3, 1.880 (4); C3–C4, 1.539(5); O3–C4, 1.445(5). CCDC: 1856677 for 5B; 1856676 for 5D. | ||
Compound 5B represents a rare structural example of an O-facial isomer, despite its predicted thermal stability.52,53 The formation of 5B is in contrast to the previous report by Matsukawa et al. using the smaller trifluoromethyl-substituted Martin ligand, from which the O-mer isomer 6 was isolated (Scheme 2 and Table S2†). Energetically, the lower lying 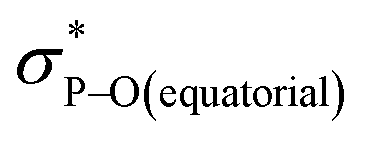 orbital (in comparison to the
orbital (in comparison to the 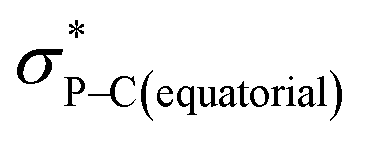 orbital) should be more susceptible to the nucleophilic attack (route A) and thus form 5A as a kinetic product. The reversible reaction and the kinetically less favourable route B would lead to the formation of the thermodynamic product 5B. However, 5A was not detected by NMR under our reaction conditions, and thus we could not eliminate the alternative route B. This direct formation of 5B from 3 through nucleophilic attack at the
orbital) should be more susceptible to the nucleophilic attack (route A) and thus form 5A as a kinetic product. The reversible reaction and the kinetically less favourable route B would lead to the formation of the thermodynamic product 5B. However, 5A was not detected by NMR under our reaction conditions, and thus we could not eliminate the alternative route B. This direct formation of 5B from 3 through nucleophilic attack at the 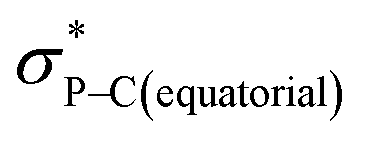 orbital (route B) may be possible due to the restricted rotation due to the large steric hindrance of the pentafluoroethyl substituents. Indeed, the difference in the chemoselectivity in deprotonation of 1 between the two ligand systems has been documented.47,54 In addition, direct conversion from 5A to 5Bvia other bond cleavage mechanisms or a one step Ray–Dutt twist also have not been probed. Isomerization of hexacoordinate phosphates by non-bond rupture, twist mechanisms has not been investigated experimentally so far. However, those of other hexacoordinate main-group compounds55,56 as well as transition metal complexes57–59 have been reported plausible based on both experimental and computational studies.60
orbital (route B) may be possible due to the restricted rotation due to the large steric hindrance of the pentafluoroethyl substituents. Indeed, the difference in the chemoselectivity in deprotonation of 1 between the two ligand systems has been documented.47,54 In addition, direct conversion from 5A to 5Bvia other bond cleavage mechanisms or a one step Ray–Dutt twist also have not been probed. Isomerization of hexacoordinate phosphates by non-bond rupture, twist mechanisms has not been investigated experimentally so far. However, those of other hexacoordinate main-group compounds55,56 as well as transition metal complexes57–59 have been reported plausible based on both experimental and computational studies.60
 | ||
| Scheme 2 Synthesis of hexacoordinate oxaphosphate O-fac5B using a –C2F5 substituted Martin ligand and the structure of Matsukawa's 12-P-6 oxaphosphate O-mer6. | ||
The reaction of the O-apical isomer 4 with KH in THF at room temperature leads to immediate generation of 5C, detected by 31P{1H} NMR spectroscopy as a singlet at −116.9 ppm. This hexacoordinate species then readily isomerizes to 5D at room temperature, which gives rise to a singlet at −114.9 ppm. An accelerated reaction at 50 °C was monitored by 31P{1H} NMR (Fig. 3(i and ii)). Furthermore, a differential NOE experiment of 5D in CD3CN showed enhancement of different proton signals in the phenyl region when the top and bottom ethylene protons (on C3) were irradiated independently (Fig. S1†), implying spatial proximity between the ethylene protons with aryl protons of the two distinct bidentate ligands, thus suggesting an O-mer structure. This has been confirmed by X-ray analysis (Fig. 2 and Table S1†). Although single crystals of 5C could not be obtained for X-ray analysis, we propose an O-meridional structure from a direct nucleophilic attack at a trans position to the equatorial carbon atoms. The corresponding analogue structure, O-mer7, was obtained and confirmed by crystallography from the trifluoromethyl substituted Martin ligand system under similar conditions. Although the structure and stereochemistry of this hexacoordinate compound 7 have been discussed in two reports previously,27,53 this is the first confirmation by crystallography (Fig. S3 and Table S3†). Its synthesis and characterization are described in detail in the ESI.†
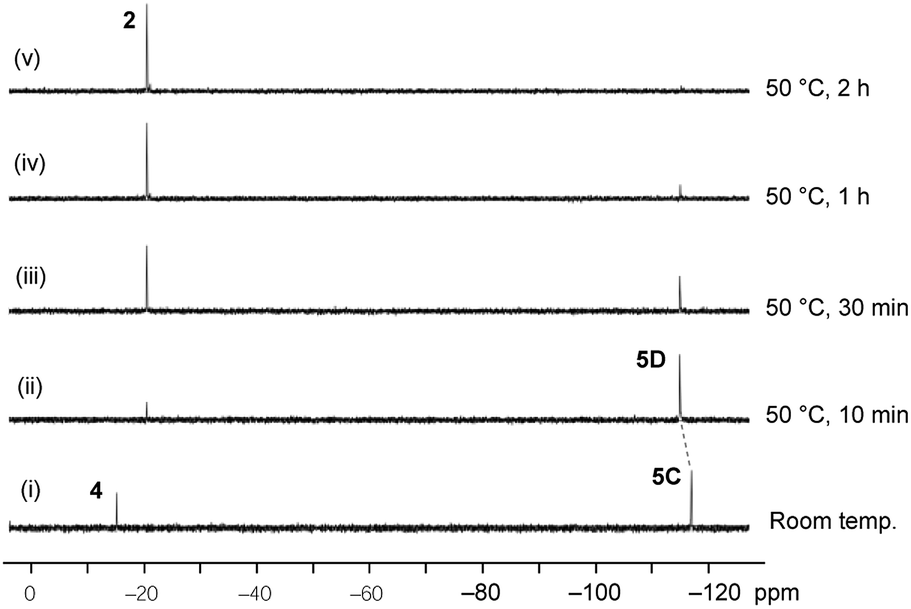 | ||
| Fig. 3 Time course of the 31P{1H} NMR for the conversion of O-mer5D and its thermal decomposition at 50 °C in THF. | ||
We can conclude that the final products of deprotonation of the pentacoordinate O-apical isomer 4 and its CF3– derivative have different O-mer geometries (5D and 7, respectively). The mechanism of the formation of 5D remains unclear. If the proposed structure of 5C is correct, the formation of 5D may be via a one-step Bailar rotation without any bond cleavage.55,56 Alternatively, through a bond-rupture pathway, the reaction equilibrium may include the unstable O-equatorial intermediate 4′. Although its concentration in the reaction mixture would be lower than that of 4 due to its higher energy (ca. 15 kcal mol−1 higher than 4),61 the kinetic barrier for intramolecular nucleophilic attack at the anti-oxygen positions is expected to be much lower, and thus trap 4′ to form the thermodynamic product 5D (Scheme 3). A similar 10-P-5 O-equatorial intermediate has also been proposed previously by Kawashima et al.53,62
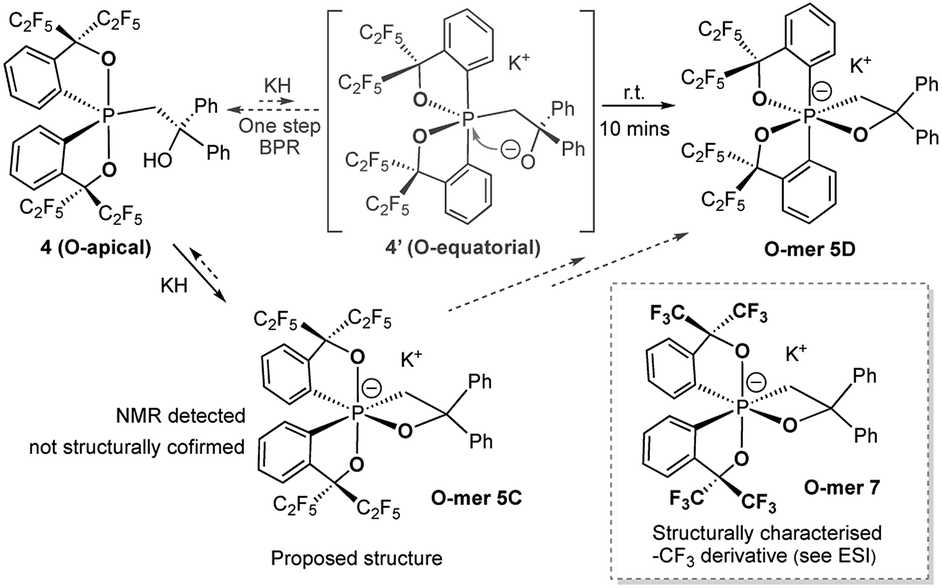 | ||
| Scheme 3 Synthesis of hexacoordinate oxaphosphate O-mer5D in the –C2F5 system and the structures of 12-P-6 oxaphosphates 5C and 7. | ||
Theoretical calculations at the (SMD:thf)ω-B97XD/def2-tzvpp//ω-B97XD/def2-svp level were carried out to estimate the relative energies of 5A–5D with and without potassium counter ions (Chart 2). The results show that 5B and 5D were lower in energy than their expected kinetic isomers 5A and 5C, consistent with the earlier conclusion that they were the thermodynamic products. Although the coordination to potassium cation changes the relative energy gap within both isomeric pairs, the stabilizing effect does not alter the relative energies of the respective pairs, which supports the conclusion that the coordination to the potassium cation is not the main driving force of isolation of the O-fac isomer 5B (Scheme 4).
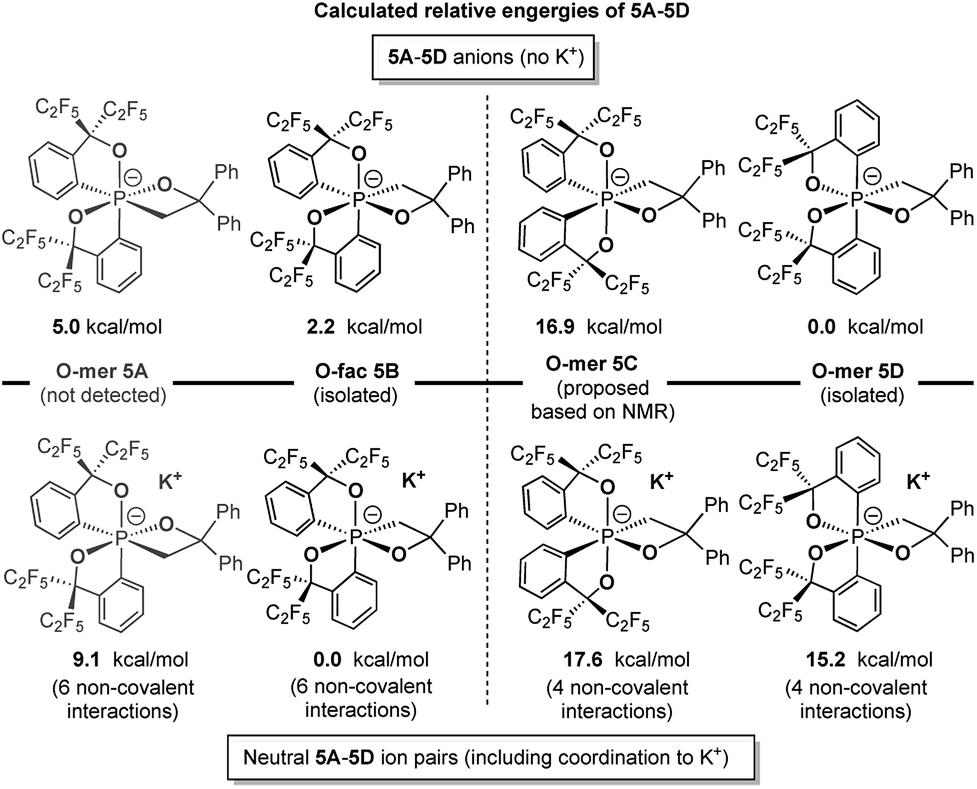 | ||
| Chart 2 Calculated relative energies of 5A–5D anions and ion pairs at the (SMD:thf)ω-B97XD/def2-tzvpp//ω-B97XD/def2-svp level.49 | ||
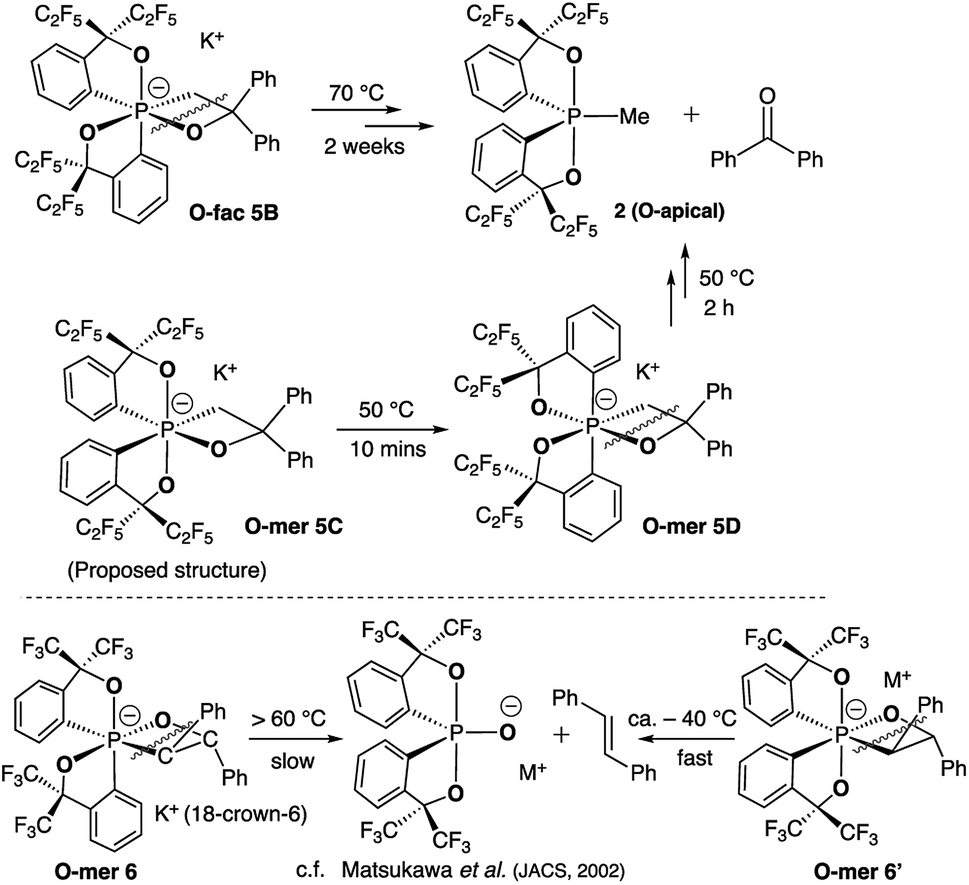 | ||
| Scheme 4 Thermal decomposition of isomers 5B and 5D. | ||
At elevated temperatures (70 °C) in solution, the O-fac isomer 5B slowly converts to 2 and benzophenone over 2 weeks. A similar conversion was also observed for 5D under milder conditions (50 °C) in solution within 2 hours (Fig. 3(ii–v)). Both thermal decomposition leads to cleavage of the P–O bond, which is different to those observed for the CF3-substituted O-mer isomers 6 and 6′.27 In the latter case, trans-stilbene and hydroxylphosphorane were formed. The difference in the reactivity is likely due to the presence of traces of water.
To verify this, we calculated the energies of fragmentation pathways of the two possible routes from O-fac5B (the lowest-in-energy isomer) to the benzophenone (Scheme 5, eqn (1)) and trans-stilbene products (Scheme 5, eqn (2)). The ketone product is much more favoured than the alkene, because of the salt effect of KOH when it is formed in the solution. Investigations to elucidate the different thermal decomposition pathways without water are underway.
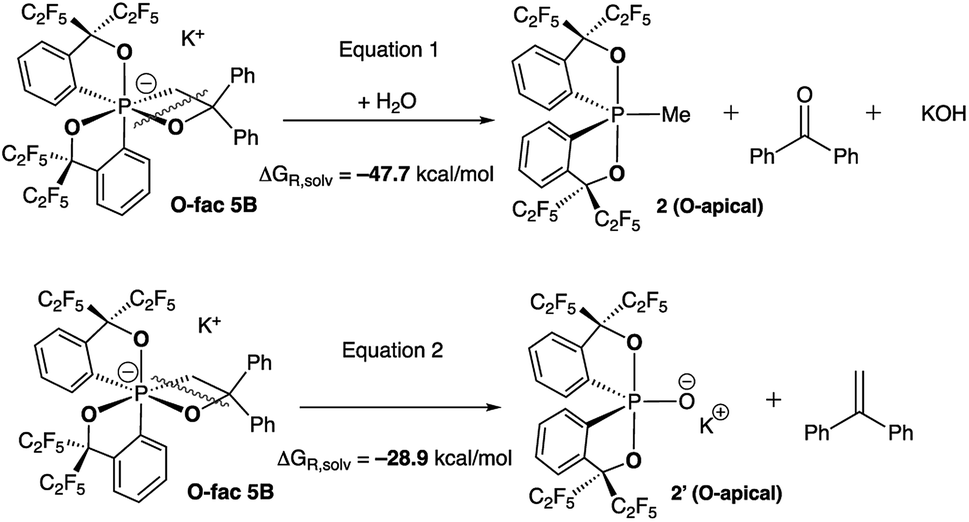 | ||
| Scheme 5 Calculated fragmentation pathways of O-fac5B in the presence and absence of water at the (SMD:thf)ω-B97XD/def2-tzvpp//ω-B97XD/def2-svp level.49 | ||
Conclusions
In conclusion, by using the pentafluoroethyl-substituted derivative of the Martin ligand, the isomeric pair of O-equatorial and O-apical pentacoordinate phosphoranes 3 and 4 was obtained by the reaction of 1 and 2 with tBuOK/nBuLi and benzophenone. Subsequent deprotonation of 3 and 4 does not yield the expected products 5A and 5C based on the kinetically favoured nucleophilic addition pathways, but leads to isolation of the first pair of O-fac and O-mer hexacoordinate oxaphosphates 5B and 5D. Calculations confirmed that 5B and 5D are thermodynamic products of their respective isomeric pairs. Their structures were confirmed by X-ray analysis. Although their formation mechanisms remain to be investigated to a full extent, their isolation confirms a strategy to synthesise a hexacoordinate oxaphosphate isomer selectively. The unexpected isolation of the O-fac isomer 5B prompts further experimental and computational studies into both bond-rupture pathways via pentacoordinate phosphoranes as well as non-dissociative twist mechanisms. In addition, both the O-fac5B and the O-mer5D afforded benzophenone and the menthylphosphorane 2 by thermal decomposition. This contrasts with the –CF3 system, which gave trans-stilbene and hydroxylphosphorane, most likely due to the presence of water.Experimental
All reactions were carried out under N2 or Ar using standard Schlenk techniques. 1H NMR (400 MHz), 13C NMR (100 MHz), 19F NMR (376 MHz), and 31P NMR (162 MHz) were recorded using a JEOL EX-400 or a JEOL AL-400 spectrometer. 1H NMR chemical shifts (δ) are given in ppm downfield from Me4Si, determined by residual chloroform (δ 7.26). 19F NMR chemical shifts (δ) are given in ppm downfield from external CFCl3. 31P NMR chemical shifts (δ) are given in ppm downfield from external 85% H3PO4. The elemental analyses were performed using a Perkin-Elmer 2400 CHN elemental analyzer. Melting points were measured using a Yanaco micro melting point apparatus. Tetrahydrofuran (THF) and diethyl ether (Et2O) were freshly distilled over CaH2. Merck silica gel 60 was used for column chromatography.O-equatorial phosphorane 3
Under Ar, n-BuLi (1.55 M n-hexane solution, 0.09 mL, 0.139 mmol) was added to a mixture of phosphorane 1 (47.1 mg, 0.0645 mmol) and t-BuOK (1.0 M THF solution, 0.13 mL, 0.13 mmol) suspended with n-hexane (5 mL) at 0 °C. The mixture was stirred for 5 min at 0 °C. Benzophenone (48.7 mg, 0.267 mmol) was added at 0 °C. The mixture was then stirred for 4 h at room temperature. The resulting solution was treated with aqueous NH4Cl at 0 °C and the crude products were extracted with Et2O (10 mL × 3). The combined organic layer was dried over MgSO4 and filtered. After evaporation of the solvent, the residue was purified by TLC (silica gel, n-hexane/CH2Cl2 = 4/1) to give 3 (18.5 mg, 0.02 mmol, 31%) as white solids. Compound 3 was recrystallized from n-hexane/CH2Cl2 to yield colourless crystals. Mp: 135.2–136.0 °C (decomp.); 1H NMR (CDCl3): δ = 7.73 (d, 3JHH = 8 Hz, 2H), 7.59 (t, 3JHH = 8 Hz, 2H), 7.37–7.43 (m, 4H), 7.22–7.26 (m, 4H), 7.13–7.19 (m, 6H), 3.84 (d, JPH = 16 Hz, 2H), 2.34 ppm (s, 1H); 19F NMR (CDCl3): δ = −78.9 (s, 12F), −114.2 (br d, JFF = 288 Hz, 2F), −115.5 (br d, JFF = 288 Hz, 4F), −115.9 ppm (br d, JFF = 288 Hz, 4F); 31P NMR (CDCl3): δ = −2.1 ppm; 13C NMR (CDCl3): δ = 148.0, 133.2, 130.0, 129.9, 128.3, 128.2, 128.0, 126.9, 125.1, 123.1, 75.3, 75.1,74.8, 53.7, 52.6 ppm. E.A.: calcd (%) for C36H21PF20O3: C 47.39, H 2.32; found: C 47.35, H 2.06.O-apical phosphorane 4
Under Ar, n-BuLi (1.63 M n-hexane solution, 0.14 mL, 0.228 mmol) was added to a mixture of phosphorane 2 (80.9 mg, 0.110 mmol) and t-BuOK (1.0 M THF solution, 0.22 mL, 0.22 mmol) suspended with n-hexane (5 mL) at 0 °C. The mixture was stirred for 5 min at 0 °C. Benzophenone (80.7 mg, 0.442 mmol) was added at 0 °C. The mixture was then stirred for 4 h at room temperature. The resulting solution was treated with aqueous NH4Cl at 0 °C and the crude products were extracted with Et2O (10 mL × 3). The combined organic layer was dried over MgSO4 and filtered. After evaporation of the solvent, the residue was purified by TLC (silica gel, n-hexane/CH2Cl2 = 4/1) to give 4 (41.8 mg, 0.0458 mmol, 41%) as white solids. Compound 4 was recrystallized from n-hexane/CH2Cl2 to yield colorless crystals. Mp: 72.0–73.0 °C (decomp.); 1H NMR (CDCl3): δ = 8.39 (m, 2H), 7.61–7.69 (m, 6H), 7.30 (dd, 3JHH = 8 Hz, 4JHH = 1.5 Hz, 2H), 7.18 (td, 3JHH = 8 Hz, 4JHH = 1.5 Hz, 2H), 7.12 (td, 3JHH = 8 Hz, 4JHH = 1.5 Hz, 2H), 6.97–7.00 (m, 2H), 6.91–6.95 (m, 2H), 4.32 (s, 1H), 3.59 (dd, JPH = 26 Hz, JHH = 15 Hz, 1H), 3.55 ppm (dd, JPH = 23 Hz, JHH = 15 Hz, 1H); 19F NMR (CDCl3): δ = −78.3 (s, 6F), −79.2 (t, J = 14 Hz, 6F), −114.2 (br d, JFF = 290 Hz, 2F), −116.3 (s, 4F), −118.1 ppm (dq, JFF = 290 Hz, JFF = 14 Hz, 2F); 31P NMR (CDCl3): δ = −15.7 ppm; 13C NMR (CDCl3): δ = 149.1, 146.1, 137.4, 135.1, 134.9, 133.3, 132.7, 131.7, 131.3, 131.1, 128.0, 76.6, 76.0, 51.8, 50.8 ppm. MS(EI(+)): m/z = 912 [M]+, 913 [M + 1]+, 914 [M + 2]+, 715 [M–CH2Ph2OH].O-fac5B
A THF solution of 3 (140 mg, 0.153 mmol) was added to a THF (5 mL) suspension of KH (100 mg, 30% oil dispersion), then the mixture was stirred for 30 min at 0 °C. The supernatant was transferred to a new Schlenk flask. After concentration in vacuo, a white solid of 5B was obtained. 5B was recrystallized from n-hexane/THF to yield colorless crystals (142.5 mg, 0.130 mmol, 85%). Mp: 123.0–124.0 °C (decomp.); 1H NMR (CDCl3): δ = 7.87 (dd, JHH = 7 Hz, JPH = 11 Hz, 1H), 7.31–7.40 (m, 6H), 6.99–7.17 (m, 6H), 6.80–6.98 (m, 5H), 6.60 (dd, J = 7 Hz, J = 14 Hz, 1H), 4.19 (dd, J = 12 Hz, J = 30 Hz, 1H), 3.21 (dd, J = 12 Hz, J = 12 Hz, 1H); 19F NMR (CDCl3): δ = −76.7 (s, 3F), −77.4 (s, 3F), −77.9 to −78.3 (m, 6F), −109.1 (d, J = 283 Hz, 2F), −109.8 (d, J = 283 Hz, 2F), −111.6 to −112.3 (m, 1F), −111.2 to −112.8 (m, 1F), −113.0 to −114.1 (m, 2F); 31P NMR (CDCl3): δ = −107.8 ppm; 13C NMR (CDCl3): δ = 154.0, 153.9, 152.4, 151.7, 151.6, 151.3, 149.5, 133.1, 129.5, 129.0, 128.5, 128.2, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.3, 127.2, 127.1, 126.6, 126.4, 126.0, 125.8, 125.5, 125.4, 125.3, 124.9, 124.5, 75.8, 65.3 ppm; E.A.: calcd (%) for C48H44PF20KO9 (5D·K·18-c-6): C 47.45, H 3.65; found: C 47.86, H 4.29.O-mer5D
A THF solution of 4 (65 mg, 0.071 mmol) and 18-crown-6 (18.7 mg, 0.071 mmol) were added to a suspension of KH (73 mg, 30% oil dispersion), then the mixture was stirred for 30 min at 0 °C. The supernatant was transferred to a new Schlenk flask. After concentration in vacuo, a white solid of 5D was obtained. 5D was recrystallized from n-hexane/THF to yield colorless crystals (10 mg, 0.0074 mmol, 10%). Mp: 131.0–133.0 °C (decomp.); 1H NMR (CDCl3): δ = 7.72 (dd, JHH = 7 Hz, JPH = 14 Hz, 1H), 7.30–7.39 (m, 4H), 7.02–7.17 (m, 4H), 6.92–7.00 (m, 4H), 6.62–6.85 (m, 5H), 3.94 (dd, J = 2 Hz, J = 14 Hz, 1H), 2.54 (ddd, J = 2 Hz, J = 10 Hz, J = 14 Hz, 1H); 19F NMR (CDCl3): δ = −76.5 (q, J = 8 Hz, 3F), −77.0 (q, J = 8 Hz, 3F), −78.0 (m, 6F), −104.2 (d, J = 286 Hz, 1F), −107.9 (d, J = 290 Hz, 1F), −110.4 (d, J = 286 Hz, 1F), −111.2 to −112.8 (m, 3F), −113.0 to −114.1 (m, 3F); 31P NMR (CDCl3): δ = −118.1 ppm; 13C NMR (CDCl3): δ = 156.1, 154.3, 153.9, 153.8, 154.1, 153.5, 152.3, 130.0, 128.6, 128.4, 128.1, 127.7, 127.6, 126.8, 126.2, 126.1, 125.6, 125.5, 125.4, 124.6, 75.0, 55.5 ppm. E.A.: calcd (%) for C56H60PF20KO11 (5B·K·18-c-6·2THF): C 49.49, H 4.45; found: C 48.98, H 4.48.Thermal conversion of 3 to 4
A solution of compound 3 (11.8 mg, 0.0129 mmol) in benzene (1.0 mL) was heated at 80 °C for 8 h. After concentration in vacuo, compound 4 was obtained (11.2 mg, 0.0123 mmol, 98%) as a white solid. The spectral data were consistent with those of the product obtained in the synthesis of 4.Reaction of 3 with KH to give benzophenone by heating
A THF (1 mL) solution of 3 (13.8 mg, 0.0151 mmol) was added to a THF (0.5 mL) suspension of KH (excess), then the mixture was stirred for 10 min at 0 °C. Then, the mixture was heated for two weeks at 70 °C. The mixture was extracted with Et2O (2 × 40 mL), and the organic layer was washed with brine (2 × 30 mL) and dried over anhydrous MgSO4. After filtering the organic layer through SiO2 and removing the solvents by evaporation, the residue was separated by reversed-phase HPLC (CH3CN) to afford 2 (RT = 41.6 min: 10.4 mg, 0.0142 mmol, 94%) as a white solid and benzophenone (RT = 19.7 min: 0.0261 mg, 0.0143 mmol, 95%) as a white solid. Benzophenone: 1H NMR (CDCl3): δ = 7.81 (d, 3JHH = 8.0 Hz, 4H), 7.59 (t, 3JHH = 8.0 Hz, 2H), 7.48 ppm (t, 3JHH = 8.0 Hz, 4H). The data of 2 are consistent with that in the reported paper.37Reaction of 4 with KH to give benzophenone by heating
A THF (0.5 mL) solution of 4 (28.2 mg, 0.0309 mmol) was added to a THF (0.3 mL) suspension of KH (excess), then the mixture was stirred for 10 min at 0 °C. The solution was transferred to an NMR tube under N2, and the mixture was heated for 2 hours at 50 °C. The NMR spectra were recorded. The mixture was extracted with Et2O (2 × 40 mL), and the organic layer was washed with brine (2 × 30 mL) and dried over anhydrous MgSO4. After filtering the organic layer through SiO2 and removing the solvents by evaporation, the residue was separated by reversed-phase HPLC (CH3CN) to afford 2 (RT = 41.2 min: 22.1 mg, 0.0302 mmol, 98%) as a white solid and benzophenone (RT = 19.2 min: 5.4 mg, 0.0296 mmol, 96%) as a white solid. Benzophenone: 1H NMR (CDCl3): δ = 7.81 (d, 3JHH = 8.0 Hz, 4H), 7.59 (t, 3JHH = 8.0 Hz, 2H), 7.48 ppm (t, 3JHH = 8.0 Hz, 4H). The data of 2 are consistent with that in the reported paper.37Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NNSFC (21542004), Young and middle-aged scientific and technological innovation talents of Shenyang Science and Technology Bureau (RC170140), the Liaoning Province Natural Science Foundation (20170540721), Basic research on the application of Industrial Development of Shenyang Science and Technology Bureau (18013027), the Distinguished Professor Project of Liaoning province, and the JSPS KAKENHI Grant (24109002) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We also thank the Chinese Scholarship Council (20183058).Notes and references
- N. V. Timosheva, A. Chandrasekaran and R. R. Holmes, Inorg. Chem., 2006, 45, 10836–10848 CrossRef CAS PubMed.
- R. R. Holmes, Acc. Chem. Res., 2004, 37, 746–753 CrossRef CAS PubMed.
- S. D. Lahiri, G. Zhang, D. Dunaway-Mariano and K. N. Allen, Science, 2003, 299, 2067–2071 CrossRef CAS PubMed.
- R. R. Holmes, Pentacoordinated Phosphorus: Structure and Spectroscopy, American Chemical Society, 1980 Search PubMed.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- G. Wittig and M. Rieber, Liebigs Ann., 1949, 562, 187–192 CrossRef CAS.
- S. Matsukawa, K. Kajiyama, S. Kojima, S.-Y. Furuta, Y. Yamamoto and K.-Y. Akiba, Angew. Chem., Int. Ed., 2002, 41, 4718–4722 CrossRef CAS PubMed.
- S. Kumaraswamy, C. Muthiah and K. C. K. Swamy, J. Am. Chem. Soc., 2000, 122, 964–965 CrossRef CAS.
- J. A. Deiters, R. R. Holmes and J. M. Holmes, J. Am. Chem. Soc., 1988, 110, 7672–7681 CrossRef CAS.
- P. Wang, Y. Zhang, R. Glaser, A. Streitwieser and P. v. R. Schleyer, J. Comput. Chem., 1993, 14, 522–529 CrossRef CAS.
- K. Kajiyama, M. Yoshimune, M. Nakamoto, S. Matsukawa, S. Kojima and K.-y. Akiba, Org. Lett., 2001, 3, 1873–1875 CrossRef CAS PubMed.
- R. S. Berry, J. Chem. Phys., 1960, 32, 933–938 CrossRef CAS.
- I. Ugi, D. Marquarding, H. Klusacek, P. Gillespie and F. Ramirez, Acc. Chem. Res., 1971, 4, 288–296 CrossRef CAS.
- P. Gillespie, P. Hoffman, H. Klusacek, D. Marquarding, S. Pfohl, F. Ramirez, E. A. Tsolis and I. Ugi, Angew. Chem., Int. Ed. Engl., 1971, 10, 687–715 CrossRef CAS.
- S. Matsukawa, H. Yamamichi, Y. Yamamoto and K. Ando, J. Am. Chem. Soc., 2009, 131, 3418–3419 CrossRef CAS PubMed.
- A. J. Arduengo, C. A. Stewart, F. Davidson, D. A. Dixon, J. Y. Becker, S. A. Culley and M. B. Mizen, J. Am. Chem. Soc., 1987, 109, 627–647 CrossRef CAS.
- A. Brand and W. Uhl, Chem.–Eur. J., 2019, 25, 1391–1404 CrossRef CAS PubMed.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
- D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 5984–5992 CrossRef CAS PubMed.
- D. Hellwinkel, Chem. Ber., 1966, 99, 3642–3659 CrossRef CAS.
- D. Hellwinkel, Chem. Ber., 1966, 99, 3628–3641 CrossRef CAS.
- D. Hellwinkel, Angew. Chem., Int. Ed. Engl., 1965, 4, 356 CrossRef.
- D. Hellwinkel, Chem. Ber., 1965, 98, 576–587 CrossRef CAS.
- J. Lacour and D. Linder, Chem. Rec., 2007, 7, 275–285 CrossRef CAS PubMed.
- J. Lacour, C. Ginglinger, C. Grivet and G. Bernardinelli, Angew. Chem., Int. Ed. Engl., 1997, 36, 608–610 CrossRef CAS.
- T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC.
- For an example of an asymmetric catalysis by a hexacoordinate chrial phosphate species see Ooi and coworkers' work: D. Uraguchi, H. Sasaki, Y. Kimura, T. Ito and T. Ooi, J. Am. Chem. Soc., 2018, 140, 2765–2768 CrossRef CAS PubMed.
- S. Matsukawa, S. Kojima, K. Kajiyama, Y. Yamamoto, K.-y. Akiba, S. Re and S. Nagase, J. Am. Chem. Soc., 2002, 124, 13154–13170 CrossRef CAS PubMed.
- M. Nakamoto and K.-y. Akiba, J. Am. Chem. Soc., 1999, 121, 6958–6959 CrossRef CAS.
- N. V. Timosheva, A. Chandrasekaran, R. O. Day and R. R. Holmes, J. Am. Chem. Soc., 2002, 124, 7035–7040 CrossRef CAS PubMed.
- K. V. P. P. Kumar, N. S. Kumar and K. C. K. Swamy, New J. Chem., 2006, 30, 717–728 RSC.
- C. Martin, Science, 1983, 221, 509–514 CrossRef PubMed.
- J. C. Martin and E. F. Perozzi, Science, 1976, 191, 154–159 CrossRef CAS PubMed.
- E. F. Perozzi and J. C. Martin, J. Am. Chem. Soc., 1979, 101, 1591–1593 CrossRef CAS.
- F. Medici, G. Gontard, E. Derat, G. Lemiere and L. Fensterbank, Organometallics, 2018, 37, 517–520 CrossRef CAS.
- H. Lenormand, J.-P. Goddard and L. Fensterbank, Org. Lett., 2013, 15, 748–751 CrossRef CAS PubMed.
- X.-D. Jiang, K.-i. Kakuda, S. Matsukawa, H. Yamamichi, S. Kojima and Y. Yamamoto, Chem.–Asian J., 2007, 2, 314–323 CrossRef CAS PubMed.
- I. Richter, M. Penka and R. Tacke, Inorg. Chem., 2002, 41, 3950–3955 CrossRef CAS PubMed.
- I. Serrano, X. Sala, E. Plantalech, M. Rodriguez, I. Romero, S. Jansat, M. Gomez, T. Parella, H. Stoeckli-Evans, X. Solans, M. Font-Bardia, B. Vidjayacoumar and A. Llobet, Inorg. Chem., 2007, 46, 5381–5389 CrossRef CAS PubMed.
- F. Julia, D. Bautista, J. M. Fernandez-Hernandez and P. Gonzalez-Herrero, Chem. Sci., 2014, 5, 1875–1880 RSC.
- C. Zhan, Z. Han, B. O. Patrick and D. P. Gates, Dalton Trans., 2018, 47, 12118–12129 RSC.
- K. C. K. Swamy and N. S. Kumar, Acc. Chem. Res., 2006, 39, 324–333 CrossRef CAS PubMed.
- J. Kobayashi, K. Goto, T. Kawashima, M. W. Schmidt and S. Nagase, J. Am. Chem. Soc., 2002, 124, 3703–3712 CrossRef CAS PubMed.
- X.-D. Jiang, S. Matsukawa, S. Kojima and Y. Yamamoto, Inorg. Chem., 2012, 51, 10996–11006 CrossRef CAS PubMed.
- X.-D. Jiang, S. Matsukawa, K.-i. Kakuda, Y. Fukuzaki, W.-L. Zhao, L.-S. Li, H.-B. Shen, S. Kojima and Y. Yamamoto, Dalton Trans., 2010, 39, 9823–9829 RSC.
- X.-D. Jiang, S. Matsukawa, H. Yamamichi and Y. Yamamoto, Inorg. Chem., 2007, 46, 5480–5482 CrossRef CAS PubMed.
- X.-D. Jiang, S. Matsukawa, H. Yamamichi and Y. Yamamoto, Heterocycles, 2007, 73, 805–824 CrossRef CAS.
- K. Kajiyama, M. Yoshimune, S. Kojima and K.-y. Akiba, Eur. J. Org. Chem., 2006, 2739–2746 CrossRef CAS.
- For computational details, please see ESI.†.
- Due to the minimal collection strategy, the data set provided a reasonable solution for structural confirmation. However, bonding parameters are not reliable for discussion..
- The addition of crown ether before, after or at the same time of the deprotonating reagent did not affect the reaction results..
- R. S. Michalak, S. R. Wilson and J. C. Martin, J. Am. Chem. Soc., 1984, 106, 7529–7539 CrossRef CAS.
- T. Kawashima, K. Watanabe and R. Okazaki, Tetrahedron Lett., 1997, 38, 551–554 CrossRef CAS.
- K.-y. Akiba, S. Matsukawa, K. Kajiyama, M. Nakamoto, S. Kojima and Y. Yamamoto, Heteroat. Chem., 2002, 13, 390–396 CrossRef CAS.
- H. S. Rzepa and M. E. Cass, Inorg. Chem., 2007, 46, 8024–8031 CrossRef CAS PubMed.
- M. Amati and F. Lelj, Theor. Chem. Acc., 2008, 120, 447–457 Search PubMed.
- J. G. Gordon and R. H. Holm, J. Am. Chem. Soc., 1970, 92, 5319–5332 CrossRef CAS.
- N. A. P. Kane-Maguire, T. W. Hanks, D. G. Jurs, R. M. Tollison, A. L. Heatherington, L. M. Ritzenthaler, L. M. McNulty and H. M. Wilson, Inorg. Chem., 1995, 34, 1121–1124 CrossRef CAS.
- R. A. Palmer, R. C. Fay and T. S. Piper, Inorg. Chem., 1964, 3, 875–881 CrossRef CAS.
- S. Alvarez, Chem. Rev., 2015, 115, 13447–13483 CrossRef CAS PubMed.
- S. Kojima, K. Kajiyama, M. Nakamoto and K.-y. Akiba, J. Am. Chem. Soc., 1996, 118, 12866–12867 CrossRef CAS.
- H. Miyake, N. Kano and T. Kawashima, Inorg. Chem., 2011, 50, 9083–9089 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and computational details, X-ray crystallographic data and NMR spectra. CCDC 1856674–1856678. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05158e |
| This journal is © The Royal Society of Chemistry 2019 |