
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn enzyme-free molecular catalytic device: dynamically self-assembled DNA dendrimers for in situ imaging of microRNAs in live cells†
Shuzhen
Yue
a,
Xinyue
Song
b,
Weiling
Song
c and
Sai
Bi
 *a
*a
aCollege of Chemistry and Chemical Engineering, Qingdao University, Qingdao 266071, P. R. China. E-mail: bisai11@126.com
bShandong Province Key Laboratory of Detection Technology for Tumor Makers, College of Chemistry and Chemical Engineering, Linyi University, Linyi 276000, P. R. China
cLaboratory of Optic-electric Sensing and Analytical Chemistry for Life Science, MOE, Shandong Key Laboratory of Biochemical Analysis, Key Laboratory of Analytical Chemistry for Life Science in Universities of Shandong, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China
First published on 4th December 2018
Abstract
DNA has become a promising material to construct high-order structures and molecular devices owing to its sequence programmability. Herein, a DNA machine based on branched catalytic hairpin assembly (bCHA) is introduced for dynamic self-assembly of DNA dendrimers. For this system, a Y-shaped hairpin trimer tethered with three kinds of hairpins (H1, H2 and H3) is constructed. The introduction of an initiator (I) triggers a cascade of CHA reactions among hairpin trimers, leading to the formation of DNA dendrimers. Through labeling fluorophore/quencher pairs in the hairpin trimers, this catalytic DNA machine is applied as a versatile amplification platform to analyze nucleic acids using microRNA-155 (miR-155) as a model analyte. Benefiting from the “diffusion effect”, the proposed bCHA achieves a greatly improved sensitivity in comparison with traditional CHA. This catalytic amplifier exhibits high sensitivity toward miR-155 detection with a dynamic range from 2.5 nM to 500 nM and demonstrates excellent selectivity to distinguish the single-base mismatched sequence from the perfectly complementary one, which is further applied to detect low-abundance miR-155 spiked in complex matrices with minimal interference. This method is further applied for in situ imaging of miR-155 in different live cells. The bCHA reaction can be specifically triggered by intracellular miR-155, achieving monitoring of the dynamic miRNA expression and distribution. Overall, our proposed enzyme-free dynamic DNA self-assembly strategy provides a versatile approach for the development of DNA nanotechnology in biosensing and bioimaging, and monitoring the cellular miRNA-related biological events.
Introduction
Deoxyribonucleic acids (DNAs) have been considered as promising building blocks to fabricate a variety of nanostructures and devices owing to their sequence programmability and specific recognition properties.1 The applications of DNAs as engineering materials provide a solid foundation for the development of DNA nanotechnology and materials science.2 DNA nanotechnology can be mainly divided into two categories: structural DNA nanotechnology and dynamic DNA nanotechnology, in which DNA strands are employed to program the spatial and temporal distribution of matter.3 Based on ‘bottom-up’ engineering approaches, structural DNA nanotechnology has realized the fabrication of 2D and 3D DNA assemblies with various sizes and spatial structures, such as DNA lattices,4,5 DNA origami,6 DNA tetrahedron structures,7–9 DNA nanotubes10,11 and so on. Unlike structural DNA nanotechnology, dynamic DNA nanotechnology lays emphasis on the non-equilibrium dynamics, in which the formation of DNA nanostructures results from successive dynamic assembly of DNA motifs.3 That is, the nanostructures formed by dynamic DNA nanotechnology can present mechanical operation processes. To date, a variety of dynamic DNA devices have been constructed, which are propelled by DNAzymes,12 specific DNA-modifying enzymes13,14 and toehold-mediated strand displacement (TMSD) reactions.15–17The TMSD reaction, a concept pioneered by Yurke et al., occurs when an invading strand displaces a target strand on a double-stranded complex with the help of a single-stranded sequence (termed a toehold).18 By means of the toehold sequence, the DNA invasion reaction can be accelerated and realize the kinetically controlled self-assembly of DNA.3,19 Thus, the TMSD reaction has been used as powerful tool to program enzyme-free DNA circuits and DNA nanomachines.20,21 To date, synthetic molecular machines have spurred substantial research efforts in the fields of biosensing for detecting a variety of targets such as miRNA, proteins and so on, and bioimaging for accurate miRNA imaging in living cells.34,35 Catalytic hairpin assembly (CHA) can be considered as one of the most prominent TMSD reactions. In CHA, DNA hairpins are kinetically trapped and exist metastably. Upon the introduction of an initiator, successive assembly steps are triggered among DNA hairpins to form branched junctions via cascades of strand displacement reactions, accompanied by a disassembly step in which the initiator is displaced to catalyze another CHA process.17 Since one initiator can be continuously replaced and recycled to trigger a new reaction, CHA has been applied as a powerful enzyme-free signal transducer for isothermal amplification analysis of a wide range of targets from nucleic acids, small molecules to proteins and even cancer cells.22–26 Very recently, CHA has been further applied to construct diverse DNA structures and molecular machines.27
MicroRNAs (miRNAs) are a group of small noncoding RNA molecules that play important roles in a series of cellular processes such as cell differentiation, proliferation, apoptosis and so on.28–30 More and more research has demonstrated that the cellular dysregulated expression of miRNAs is related to the genesis of many cancers.31–33 Therefore, it is necessary to sensitively detect miRNAs for early diagnosis of cancers and cellular level research. Currently, benefiting from the advantages of CHA, some CHA-based detection methods have been established for quantitative detection and analysis of miRNA expression.36–39 However, effective signal amplification of miRNAs is still greatly needed due to their low expression level in cells.
Herein, we construct an enzyme-free catalytic device based on bCHA for dynamic self-assembly of DNA dendrimers. This strategy involves the formation of a Y-shaped hairpin trimer which contains a Y-scaffold and three kinds of hairpins. The assembly into explicit DNA dendrimers is initiated and mediated via a series of CHA processes among the hairpin trimers. By using the target-triggered mechanism, we demonstrate the versatile applications of the proposed bCHA for signal amplification biosensing and in situ imaging of miR-155 in live cells with high sensitivity and selectivity. For all we know, this is the first time an intracellular CHA process is reported for in situ monitoring the dynamic expression and distribution of miRNA in live cells. Compared with other molecular devices, especially traditional CHA, the major advantage of our proposed bCHA is the significantly improved amplification efficiency which benefited from the “diffusion effect”. In bCHA, three hairpins are localized on a Y-scaffold DNA, which thus provides an addressable substrate for those released initiators, achieving greater control for cascade hairpin assembly.
Results and discussion
Principle of bCHA
The proposed bCHA strategy consists of a Y-scaffold DNA and three kinds of hairpins (HP1, HP2 and HP3). The sequences and the secondary structures of the oligonucleotides used in this work are listed in Table S1 and Fig. S1,† respectively. The assembly pathways are shown in Fig. 1a. The Y-scaffold DNA is composed of three single-stranded DNAs (ssDNAs), Y1, Y2 and Y3, in which each strand contains three segments: a long “sticky end” to interact with its complementary sequence on the corresponding hairpin and two parts to hybridize with the other two strands. On the basis of Watson-Crick base pairs, the Y-shaped hairpin trimer is formed via the hybridization of the “sticky ends” between the Y-scaffold DNA and hairpins (Fig. 1a, reaction 1). The formed hairpin trimer can metastably coexist in solution, and acts as a building block to self-assemble DNA dendrimers upon addition of the initiator to activate the dynamic self-assembly process of bCHA. During the bCHA, the initiator hybridizes and docks to the exposed toehold domain a of H1 and subsequently displaces the parts of x, b, and y from the duplex of the stem in H1 via a TMSD reaction, opening the loop of H1 (Fig. 1a, reaction 2). The opened loop makes the branch migration irreversible. Meanwhile, the newly exposed domain b* of H1 hybridizes with domain b of H2, which is the component of a proximal hairpin trimer in solution, to operate the branch migration, resulting in the opening of the loop of H2 and the formation of the hybrid of two three-arm branched hairpin structures (Fig. 1a, reaction 3). Another proximal hairpin trimer then invades the hybrid because the toehold domain c of H3 can dock to the newly exposed domain c* of the hybrid. And then a disassembly step is performed, in which H3 displaces I from the complex to catalyze another dynamic self-assembly process (Fig. 1a, reaction 4). One CHA reaction is completed as above. Theoretically, this branching chain growth would continue to form DNA dendrimers after a cascade of CHA processes among the hairpin trimers (Fig. 1a, reaction 5), which is thus termed branched catalytic hairpin assembly (bCHA).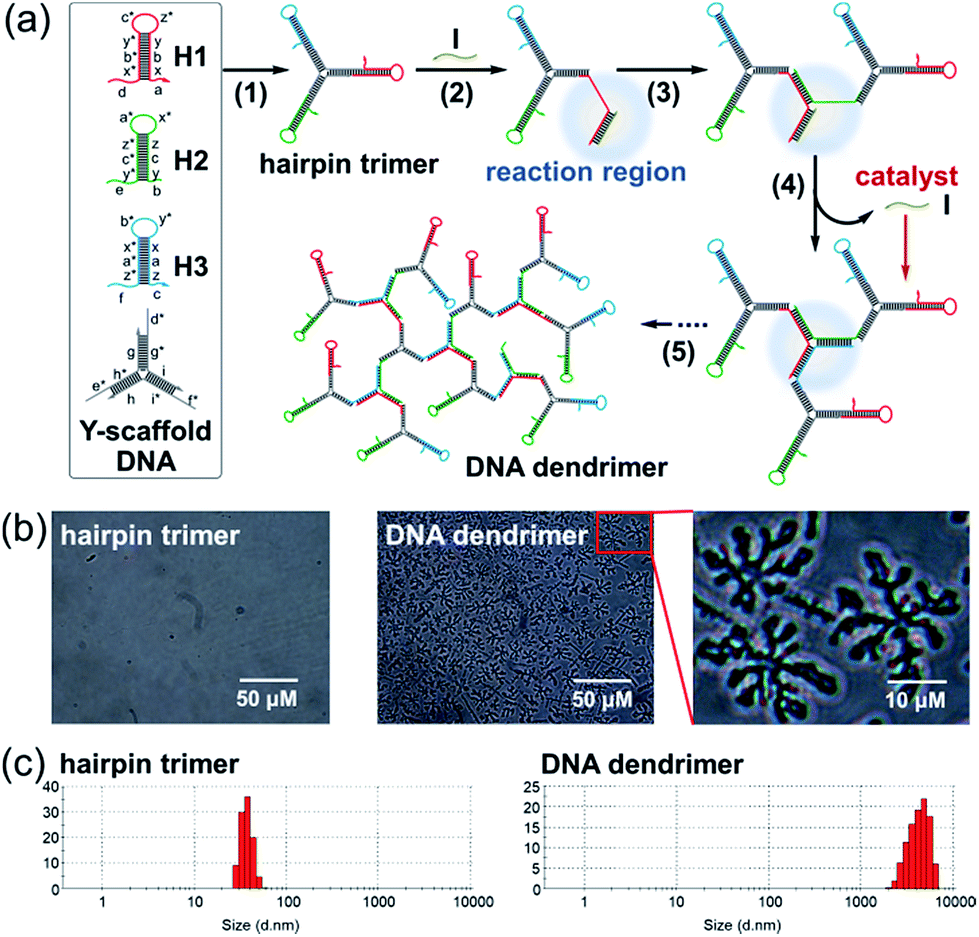 | ||
| Fig. 1 (a) Components and reaction pathways of the bCHA process triggered by an initiator (I) for dynamic self-assembly of DNA dendrimers. Letters marked with * represent the complementary domains to the corresponding unmarked ones. (b) Microscope images and (c) size distribution of hairpin trimers before and after incubation with a 0.1 μM initiator. The concentration of the hairpin trimers is 1.5 μM. Scale bar in (b): 50 μm. | ||
The morphology of the DNA dendrimers assembled via bCHA was characterized by microscope imaging (Fig. 1b). In the absence of an initiator, no assembly event of hairpin trimers occurs. In contrast, a large number of two-dimensional hyperbranched DNA structures appear upon adding the initiator into the hairpin trimers. The size distribution of the hairpin trimers before and after being incubated with the initiator was further measured by DLS (Fig. 1c). The size of hairpin trimers is determined to be 45 ± 15 nm, whereas, it significantly increases to 4.5 ± 2.5 μm upon addition of the initiator, which are consistent with the observation in microscopy images. Therefore, the above results fully demonstrate the successful dynamic self-assembly of DNA dendrimers via bCHA as anticipated. Furthermore, the zeta potential of the DNA dendrimers was measured to be −8.35 ± 3.44 mV, which can be attributed to the negatively charged phosphate backbones of the packed DNA strands constituting the DNA dendrimers (Fig. S2†).
Native polyacrylamide gel electrophoresis (PAGE)
As a proof-of-concept experiment, the assembly of Y-scaffold DNA was verified by native PAGE (Fig. 2a). The bands in lanes 1, 2 and 3 correspond to Y1, Y2 and Y3, respectively, which migrate faster than the assembled Y-scaffold DNA (lane 7). The bands representing Y1 + Y2, Y1 + Y3, and Y2 + Y3 (lanes 4, 5, and 6) are in between. These results indicate the effectivity of the assembly process, and Y-scaffold DNA has been formed as designed. Subsequently, we compared the assembly pathways of traditional CHA and the proposed bCHA by native PAGE. For the traditional CHA, three-arm DNA junctions are formed through self-assembly of H1, H2 and H3 triggered by the initiator, while in bCHA the DNA dendrimers are generated using hairpin trimers as building blocks. As shown in Fig. 2b, lanes 10–12 are H1, H2 and H3, respectively, while lane 9 corresponds to the mixture of H1, H2 and H3 without an initiator. These bands are almost at the same migration rate. In the presence of the initiator, three-arm DNA junctions are formed (lane 8), which migrate slower than hairpin species. The formation of Y-scaffold DNA in lane 13 is almost the same as that shown in Fig. 2a. When the Y-scaffold DNA is mixed with H1, H2 and H3 at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1, the hybrid of the Y-shaped hairpin trimer is formed in lane 14. With the addition of the initiator, the band corresponding to the hairpin trimer disappears and the assembly products with much lower mobility are observed in lane 15, demonstrating the formation of DNA dendrimers with high molecular weight via the bCHA.
:1:1:1, the hybrid of the Y-shaped hairpin trimer is formed in lane 14. With the addition of the initiator, the band corresponding to the hairpin trimer disappears and the assembly products with much lower mobility are observed in lane 15, demonstrating the formation of DNA dendrimers with high molecular weight via the bCHA.
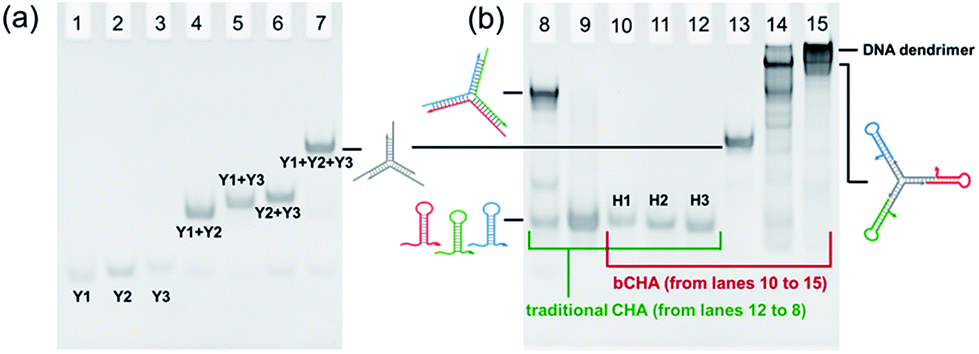 | ||
| Fig. 2 Native PAGE to demonstrate (a) the formation of Y-scaffold DNA and (b) the reaction mechanism of traditional CHA for assembly of three-arm DNA junctions (lanes 8–12) and bCHA for assembly of DNA dendrimers (lanes 10–15). Lanes 8–12: H1 + H2 + H3 + initiator to form a three-arm DNA junction, H1 + H2 + H3, H1, H2 and H3, respectively; lane 13: Y-scaffold DNA; lane 14: Y-scaffold DNA + H1 + H2 + H3 to form hairpin trimer; lane 15: hairpin trimer + initiator to form a DNA dendrimer. The initial concentration of each DNA species is 10 μM. The experiments are implemented in TE buffer (10 mM Tris–HCl, 1 mM EDTA-2Na and 12.5 mM MgCl2, pH 8.0) with a final volume of 12 μL. | ||
Amplification biosensing of microRNA
Since the initiator can be displaced to continuously trigger a new cycle of assembly, we further employed the bCHA strategy as a catalytic amplifier for sensitive and selective detection of microRNA. It has been reported that miR-155 plays an important role in various physiological and pathological processes, and can be considered as an important biomarker for diagnosis, staging, progression, and prognosis of cancers.40,41 To demonstrate the feasibility of the bCHA for amplified detection of miR-155, H1 is modified with a fluorophore (FAM) at the 3′ terminus and a quencher (Dabcyl) at the appropriate position (Fig. 3a). In the absence of miR-155, the efficient quenching effect resulted from the fact that the closely positioned Dabcyl quencher makes the fluorescent emission from the FAM minimal. In contrast, the introduction of the miR-155 initiates dynamic self-assembly among hairpin trimers, resulting in the opening of H1 to remove the quencher from the fluorophore. Thus, the significantly amplified fluorescent signal output can be monitored for highly sensitive detection of miR-155.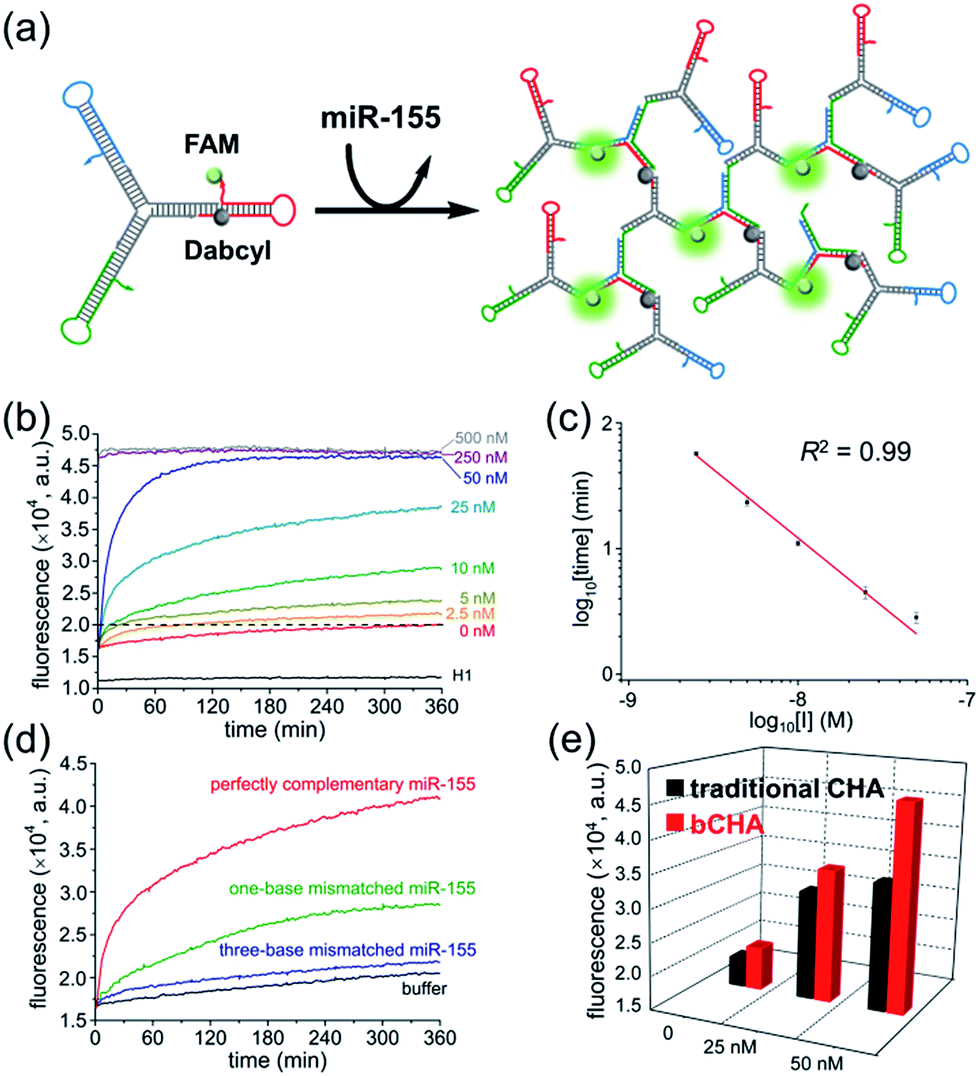 | ||
| Fig. 3 Amplification biosensing of miR-155 based on bCHA. (a) Schematic illustration of the strategy. (b) Fluorescence kinetic curves of bCHA triggered by different concentrations of miR-155. (c) The plot of the logarithm of time needed to reach a fluorescence intensity of 2.0 × 104 a.u. against the logarithm of the initial concentration of miR-155. The error bars represent the standard deviation of three independent measurements. (d) Fluorescence kinetic curves of bCHA triggered by different targets. The concentration of each analyte is 25 nM. (e) Comparison of traditional CHA and bCHA in response to different concentrations of the initiator. The fluorescence intensities are recorded at 360 min. The concentration of hairpin trimers is 1.5 μM. | ||
The real-time fluorescence triggered by different concentrations of miR-155 was measured to investigate the growth kinetics of the proposed bCHA (Fig. 3b). At the beginning of the reaction, the annealed hairpins on trimers are in a closed structure so that the fluorophore and the quencher are brought into close proximity, resulting in an “off” fluorescence signal. This phenomenon is verified by monitoring the fluorescence intensity of the control sample consisting of only H1. For the blank sample without miR-155, the fluorescence intensity should be almost the same as the control since no DNA dendrimer is generated and the fluorophore cannot separate from the quencher. However, a slightly increased fluorescence is observed, which can be attributed to the system leakage induced by few imperfectly annealed hairpin species. Thankfully, the fluorescence intensity of the blank sample remains at a low level which has a negligible influence on the reaction, demonstrating that the Y-shaped hairpin trimers can metastably coexist in the absence of an initiator. Upon the introduction of target miR-155, the self-assembly process of bCHA is activated, resulting in the formation of hyperbranched DNA structures. In this case, the fluorescence of H1 is recovered with an enlarged spatial distance between the fluorophore and the quencher, resulting in the “fluorescence-on” mode. The increased fluorescence intensity is in direct proportion to the concentration of miR-155. In particular, the emission intensity rapidly reaches a plateau when the concentrations of miR-155 are at 250 nM and 500 nM, indicating a large proportion of reactants can be depleted when the initiator is at high concentrations. The bCHA protocol has revealed high amplification efficiency for quantitative analysis of miR-155. As low as 2.5 nM miR-155 can be well distinguished from the blank. This sensitivity is satisfactory and comparable with other molecular devices.43,44 To further assess the quantitative behavior of the proposed method, we choose 20000 a.u. as the threshold, at which all samples can be taken into consideration. A log-linear trend between the initial concentration of miR-155 and the time to reach a fluorescence threshold of 20000 a.u. is shown as Fig. 3c. This relationship is presented as log10[time] = −7.63 − 1.1log10[C] with a correlation coefficient (R2) of 0.99. Furthermore, five parallel measurements are performed to study the repeatability of this strategy by adding 50 nM miR-155 to trigger the bCHA. The relative standard deviation (RSD) of 3.01% indicates the acceptable repeatability of the proposed catalytic amplification platform for biosensing of miRNAs.
Moreover, the specificity of the proposed bCHA strategy for miRNA analysis was confirmed by using one-base mismatched, three-base mismatched, and perfectly complementary miR-155 as the analytes, respectively (Fig. 3d). Under the same reaction conditions, this method could completely distinguish mismatched miR-155, even one-base mutant, showing high sequence specificity toward the target miRNA due to the precise hybridization properties of TMSD reactions. To assess the reliability of this assay in real samples, the standard addition method was implemented by detecting miR-155 with different concentrations (5 nM, 25 nM, and 50 nM) spiked in 100-fold diluted healthy human serum. As shown in Table S2,† the recoveries of miR-155 range from 95.6% to 108.0% with the RSD of 2.8–3.3%. The results indicate that the bCHA strategy has a high ability to prevent interference for miRNA analysis, which thus is available for the development of diagnostic systems in clinical applications.
Amplification efficiency of bCHA
To demonstrate the amplification efficiency of bCHA, control experiments were carried out using free DNA hairpins instead of hairpin trimers to perform the traditional CHA, in which the miR-155 triggered a cascade of assembly steps with H1, H2 and H3 to form a three-arm DNA junction (Fig. S3a†). The comparison of real-time fluorescence intensities between CHA and bCHA triggered by different concentrations of miR-155 could be observed (Fig. S3b†). In the absence of an initiator, the fluorescence intensities of both systems remain at low levels. The slightly higher fluorescence of bCHA can be attributed to the system leakage induced by the mutual interference among multiple DNA sequences. The corresponding fluorescence intensity at the reaction time of 360 min is presented in Fig. 3e and S3c.†The fluorescence intensities of bCHA can be obviously distinguished from those of CHA upon the addition of miR-155 with the same concentrations (25 nM and 50 nM). In particular for high concentration of the initiator (50 nM), the signal amplification capability of bCHA was calculated as (FbCHA(50 nM) − FbCHA(0 nM))/(FCHA(50 nM) − FCHA(0 nM)), showing that the bCHA had about a 1.68 times higher fluorescence signal than the traditional CHA. Thus, the proposed bCHA system provides significantly improved amplification efficiency for the detection of initiators (target miR-155 detection in this assay). We can attribute this phenomenon to the “diffusion effect”.42 Briefly, in a traditional CHA system the released initiators during reaction may interact with other hairpins present in solution. Nevertheless, the released initiators from bCHA are much more likely to trigger the nearby hairpin molecules attached on the Y-scaffold DNA for cascade hairpin assembly, which thus achieves greater control over directing reaction pathways.In situ imaging of miR-155 in live cells
It has been reported that miR-155 is overexpressed in many types of cancer cells, especially breast cancer.41 Thus, MCF-7 cells (human breast adenocarcinoma cell line) were selected as the model to investigate the optimal time and monitor the dynamics of the bCHA in live cells (Fig. 4a). We first test the stability of hairpin trimers, in which free H1 and hairpin trimers were treated with 100-fold diluted healthy human serum for 6 h, respectively. The result shows that the fluorescence recovery from H1 in hairpin trimers was much lower than that of free H1 (Fig. S4†), demonstrating that the structure of the hairpin trimer could protect hairpins from nuclease degradation to a certain degree. Subsequently, the transfection times required for bCHA activation by miR-155 in MCF-7 cells were investigated (Fig. 4b and S5†). On prolonging the transfection time, the fluorescence intensities increase gradually, and robust fluorescence signals produced by intracellular miR-155 can be readily observed at 4 h. 3D confocal fluorescence imaging further confirmed that the hairpin trimers have successfully entered MCF-7 cells and have been activated by miR-155 in the cytoplasm (Fig. 4c). We further designed a mutated H1 (six-base mismatched H1), named mH1, to verify the mechanism of bCHA in living cells. Almost no fluorescence was observed since the mH1 cannot hybridize with miR-155 to form DNA dendrimers, demonstrating that the observed fluorescence was indeed produced by target-induced assembly of hairpin trimers among H1, H2 and H3 (Fig. S6†).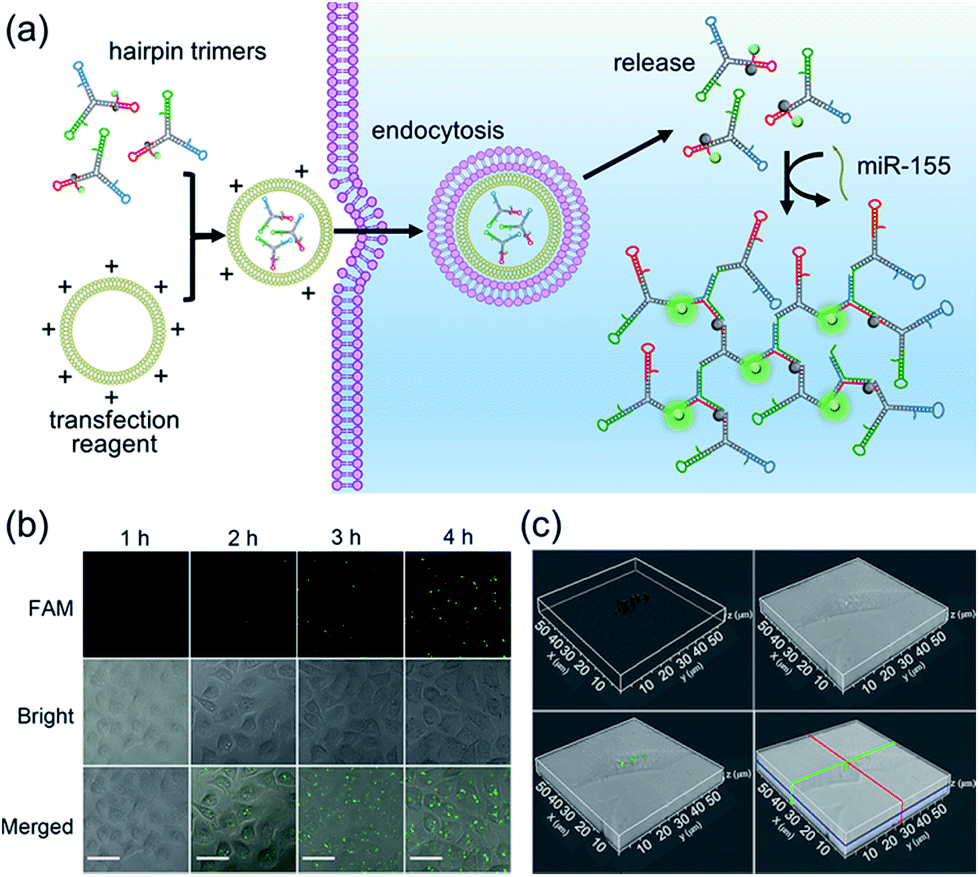 | ||
| Fig. 4 (a) In situ imaging of miR-155 in live cells. (b) Time course confocal laser scanning microscope (CLSM) images of in situ miR-155 analysis in MCF-7 cells via bCHA. MCF-7 cells are transfected with equal amounts of hairpin trimers for different times and the CLSM images are recorded at specific time points. The concentrations of hairpin trimers are 200 nM. Scale bar: 30 μm. (c) 3D fluorescence imaging of MCF-7 cells in which the bCHA is triggered by intracellular miR-155. MCF-7 cells are transfected with 200 nM hairpin trimers for 4 h at 37 °C. | ||
To demonstrate the amplification efficiency of the proposed bCHA for monitoring intracellular miR-155, MCF-7 cells were transfected with only H1, the mixture of hairpin monomers (H1, H2 and H3) and hairpin trimers to perform direct fluorescence in situ hybridization (FISH) which involved the use of a single-stranded DNA probe modified with FAM and Dabcyl, traditional CHA and bCHA, respectively. (Fig. 5a and b). When MCF-7 cells were transfected with only H1, since H1 was opened by miR-155 via TMSD reactions with a ratio of 1:1, a weak fluorescence signal was observed. However, for the transfection of hairpin monomers H1, H2 and H3 together, the traditional CHA reaction occurred by intracellular miR-155, showing a clear fluorescence signal due to the isothermal target recycling amplification of CHA. Interestingly, in comparison with traditional CHA, thanks to the “diffusion effect” discussed above, the strongest fluorescence signal could be observed when MCF-7 cells were treated with hairpin trimers, demonstrating the feasibility of bCHA for in situ imaging of intracellular miRNAs with enhanced signals. Flow cytometry analysis was further carried out (Fig. 5c and S7†). Compared with MCF-7 cells treated with either H1 or three hairpin monomers (H1, H2 and H3), the hairpin trimer-transfected MCF-7 cells showed a significantly enhanced fluorescence signal, which was well consistent with the confocal microscopy results.
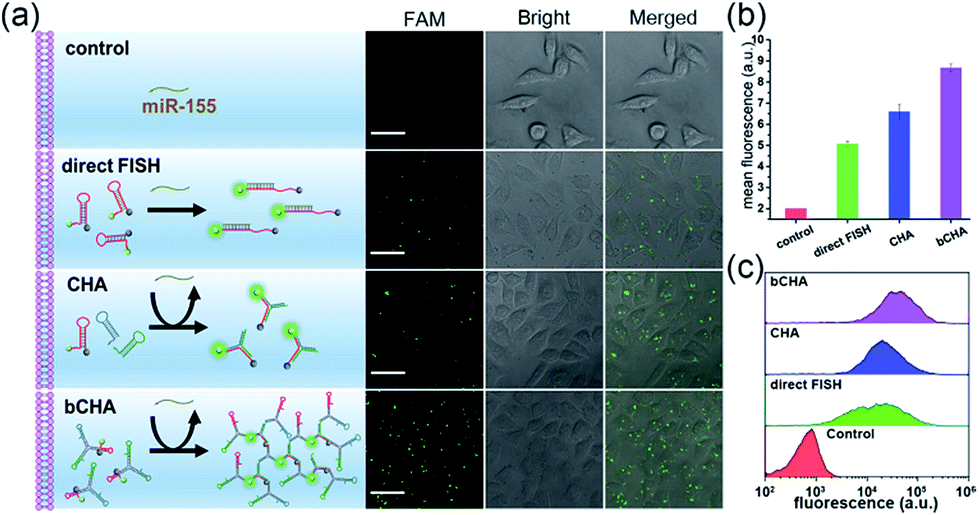 | ||
| Fig. 5 (a) In situ imaging of intracellular miR-155 using CLSM to demonstrate the amplification efficiency of the proposed bCHA. MCF-7 cells are treated with only H1, the mixture of hairpin monomers (H1, H2 and H3) and hairpin trimers for 4 h at 37 °C, respectively. The concentration of each DNA strand is 200 nM. Scale bar: 30 μm. (b) Mean fluorescence intensity as derived from (a). (c) Flow cytometry results of MCF-7 cells treated with only H1, the mixture of hairpin monomers (H1, H2 and H3) and hairpin trimers for 4 h at 37 °C. The concentration of each DNA strand is 200 nM. The error bars represent the standard deviation of three independent measurements. | ||
To further demonstrate the effectivity of bCHA, control experiments were performed in which MCF-7 cells were pretreated with miR-155 mimics and the miR-155 inhibitor, respectively, and MCF-7 cells without treatment acted as the control group (Fig. 6). As anticipated, no fluorescence readout was observed by transfecting the anti-miRNA antisense inhibitor oligonucleotide because it can bring down the content of intracellular miR-155. In contrast, MCF-7 cells treated with miR-155 mimics to imitate the high expression of miRNA-155 showed much more intense green fluorescence than the untreated cells. It was clearly demonstrated that the fluorescence readout of the bCHA system was closely related to the miR-155 concentration in living cells, and the signal increased with increasing miR-155 concentration.
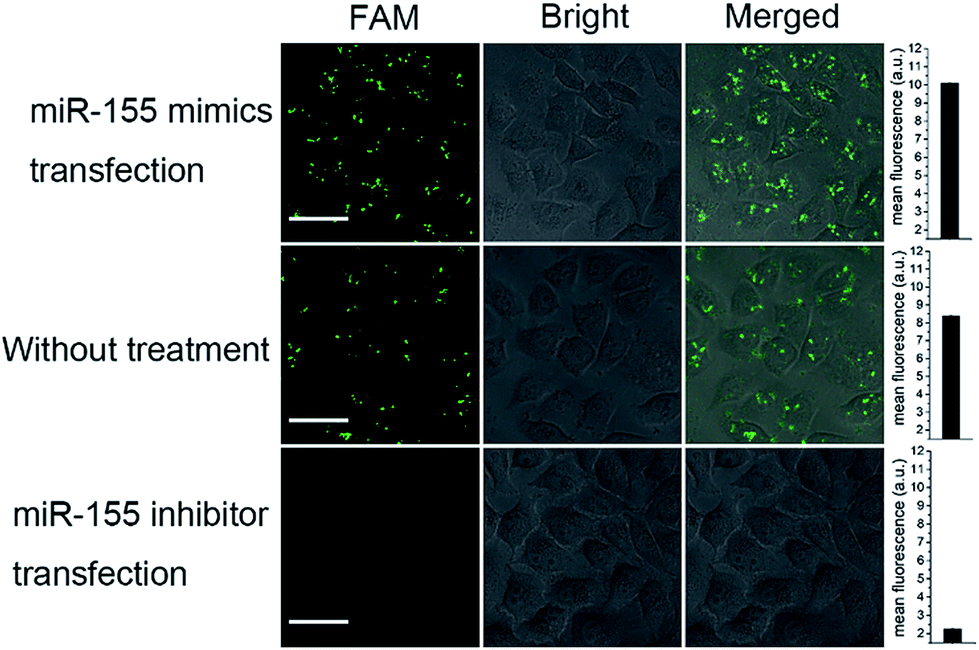 | ||
| Fig. 6 The CLSM images of MCF-7 cells pretreated with 100 nM miR-155 mimics and a 100 nM miR-155 inhibitor for 1 h at 37 °C, respectively. The MCF-7 without treatment is the control group. The concentrations of hairpin trimers are 200 nM. The error bars represent the standard deviation of three independent measurements. | ||
To investigate the capacity of the bCHA strategy to evaluate intracellular miRNA expression levels in different live cells, four types of cells, including normal human hepatocytes (L-02), human cervical cancer cells (HeLa), and breast cancer cells (MCF-7 and MDA-MB-231) were respectively transfected with hairpin trimers with identical concentrations (200 nM) at 37 °C for 4 h (Fig. 7). From Fig. 7, it can be seen that the expressions of miR-155 in different cell lines are various. Because the expression level of miR-155 in normal L-02 cells is very low,45 the fluorescence signal is emerged hardly in L-02 cells. In contrast, the fluorescence signals can be readily observed in cancer cells since miR-155 is overexpressed in cancerous processes.46 In addition, MCF-7 and MDA-MB-231 cells show stronger fluorescence intensities than HeLa cells, which is in accordance with previous reports that the expression level of miR-155 is higher in MCF-7 and MDA-MB-231 cells than in HeLa cells.47,48 Moreover, the bCHA system can be applied as a general platform for the detection of other miRNAs, such as miR-21 (Fig. S8†). It has been proven that miR-21 is an oncogene which is overexpressed in many cancers, such as breast cancer, lung cancer and so on.49,50 Compared with the fluorescence signal induced by miR-155 in MCF-7 cells under the same conditions (CLSM imaging of MCF-7 cells in Fig. 7), a stronger fluorescence signal could be observed because the expression level of miR-21 is higher than that of miR-155 in MCF -7 cells.51,52 In addition, traditional CHA and direct FISH are also performed in which bCHA exhibits a stronger fluorescence signal than CHA and direct FISH, demonstrating the proposed bCHA strategy can be used as a general platform for detecting various miRNA targets with high sensitivity and selectivity.
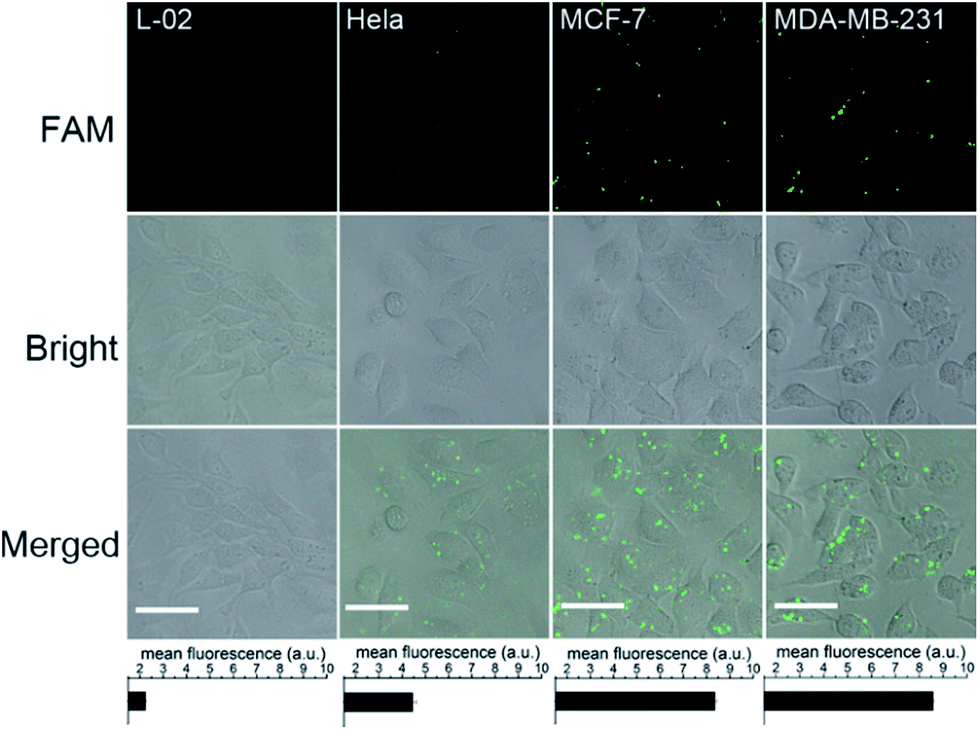 | ||
| Fig. 7 In situ imaging of miR-155 in different cell lines via bCHA. The concentrations of hairpin trimers are 200 nM. Scale bar: 20 μm. The error bars represent the standard deviation of three independent measurements. | ||
Conclusions
In summary, we have successfully demonstrated an enzyme-free DNA catalytic device of bCHA for dynamic self-assembly of DNA dendrimers. In comparison with traditional CHA, the signal amplification efficiency of bCHA is significantly improved by means of the “diffusion effect”, achieving sensitive and selective in vitro detection of miRNAs and in situ imaging of miRNAs in live cells. Given the versatility of DNA, this dynamic DNA catalytic device can be readily used for amplified detection of a wide range of analytes through combining with aptamer recognition, such as proteins, small molecules and even tumor cells. Therefore, the proposed bCHA strategy holds great potential not only in the construction of complex DNA structures but also as a versatile amplification platform in the fields of biosensing and bioimaging.Experimental section
Native polyacrylamide gel electrophoresis (PAGE)
The reaction pathways of the catalytic DNA device were confirmed using 8% acrylamide gel. Firstly, 2.7 mL of 30% acrylamide/bis-acrylamide gel solution (29:1), 1 mL of 10× TAE buffer, 90 μL of 10% ammonium persulfate (APS), 10 μL of N,N,N′,N′-tetramethylethylenediamine (TEMED) and 6.2 mL of double-distilled deionized ultrapure water were mixed to prepare the hydrogel. After polymerization for 30 min at room temperature, the gel was soaked in 1× TAE buffer (pH 8.0). Subsequently, 12 μL of each sample was mixed with 2 μL of 10× loading buffer and added to the resulting 8% native polyacrylamide gel for electrophoresis. The PAGE was run at the voltage of 170 V for 5 min and 110 V for about 45 min. Then, the gels were stained with diluted 4S Red Plus (Shanghai Sangon Biotech, China) for 40 min at room temperature. Finally, the images of the stained gels were recorded using a Tanon 2500R gel imaging system (Shanghai, China).
Morphology characterization
For microscope imaging, 2 μL of each annealed H1, H2 and H3 (10 μM for each) were added to 6 μL of the as-prepared Y-scaffold DNA, resulting in the formation of hairpin trimers after reaction at 25 °C for 1 h. The hairpin trimers as the reaction precursor were further mixed with an isometric volume of the initiator (0.1 μM), followed by incubation at 25 °C for 1.5 h. The resultant products were washed by centrifugation at 10000 rpm for 3 min with double-distilled deionized ultrapure water. The washing step was repeated three times to remove salt ions in solution. Then, 10 μL of the sample was dropped onto the glass slides. Finally, the images were taken on a microscope (Leica DM4 P, Germany) in transmission mode.
Dynamic light scattering (DLS) and zeta potential
The size distribution and zeta potential of the hairpin trimers and DNA dendrimers were measured on a Zetasizer Nano Series ZEN3700 (Malvern Instruments, UK). The DNA dendrimers were prepared according to the method mentioned above, which were diluted to 700 μL with double-distilled deionized ultrapure water for measurement.Fluorescence monitoring
2 μL of the initiator with different concentrations was rapidly mixed with 12 μL of hairpin trimers (the preparation method was the same as mentioned above) and 26 μL of TE buffer. The real-time fluorescence intensity was recorded immediately using a LineGene 4800 Real-Time detection system (Hangzhou, China) with intervals of 30 s. And the reaction was performed at 37 °C for 6 h.Cell culture
MCF-7 cells, HeLa cells, L-02 cells and MDA-MB-231 cells were all cultured in DMEM supplemented with 10% fetal calf serum, penicillin (100 mg mL−1), and streptomycin (100 mg mL−1) in a humidified and 5% CO2 incubator at 37 °C. PBS buffer (0.01 M, pH 7.4) was used to wash cells.Intracellular imaging
An amount of 1 × 105 cells was plated in a glass bottom confocal dish (diameter of 30 mm) for 16 h. Transfection was performed by adding hairpins using the LipoHigh transfection reagent (Sangon, Shanghai) in Opti-MEM® I Reduced Serum Medium (Thermo Fisher Scientific, Nanjing) at 37 °C. Hairpin trimers were transfected at a final concentration of 200 nM. After 4 h transfection, the transfection reagent was removed from the confocal dishes and the culture cells were washed three times with 1× PBS. Then, the plates were filled with 1× PBS and placed on a Leica TCS SP8 inverted confocal microscope (Leica, Germany) to obtain images.Flow cytometry
MCF-7 cells were seeded in a six-well culture plate at 1 × 105 cells per well and cultured in DMEM (10% FBS) for 16 h. After washing the cells with PBS, the medium was changed to 1 mL of Opti-MEM® I Reduced Serum Medium containing 4 μL of LipoHigh transfection reagent and hairpin trimers with the final concentration of 200 nM. After incubation for 4 h, cells were washed three times with 1× PBS and detached from the six-well culture plate by trypsin. After thoroughly washing the cells with PBS and centrifuging at 1000 rpm for 3 min, flow cytometry was performed using a CytoFLEX system (Beckman Coulter, US.) under 488 nm excitation. Fluorescence intensities represented 104 analyzed cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21722505 and 21535002), the Special Funds of the Taishan Scholar Program of Shandong Province (tsqn20161028), and the Natural Science Foundation of Shandong Province (ZR2017JL009), and the Foundation of Key Laboratory of Sensor Analysis of Tumor Marker, Ministry of Education, Qingdao University of Science and Technology (SATM201803).Notes and references
- J. Li, A. A. Green, H. Yan and C. Fan, Nat. Chem., 2017, 9, 1056 CrossRef CAS PubMed.
- E. Stulz, G. Clever, M. Shionoya and C. Mao, Chem. Soc. Rev., 2011, 40, 5633 RSC.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103 CrossRef CAS PubMed.
- I. Manuguerra, G. Grossi, R. P. Thomsen, J. Lyngsø, J. S. Pedersen, J. Kjems, E. S. Andersen and K. V. Gothelf, ACS Nano, 2017, 11, 9041 CrossRef CAS PubMed.
- M. Wang, H. Huang, Z. Zhang and S.-J. Xiao, Nanoscale, 2016, 8, 18870 RSC.
- H. Jabbari, M. Aminpour and C. Montemagno, ACS Comb. Sci., 2015, 17, 535 CrossRef CAS PubMed.
- J. P. Sadowski, C. R. Calvert, D. Y. Zhang, N. A. Pierce and P. Yin, ACS Nano, 2014, 8, 3251 CrossRef CAS PubMed.
- L. Liang, J. Li, Q. Li, Q. Huang, J. Shi, H. Yan and C. Fan, Angew. Chem., Int. Ed., 2014, 53, 7745 CrossRef CAS PubMed.
- M. Lin, J. Wang, G. Zhou, J. Wang, N. Wu, J. Lu, J. Gao, X. Chen, J. Shi, X. Zuo and C. Fan, Angew. Chem., Int. Ed., 2015, 54, 2151 CrossRef CAS PubMed.
- Q. Liu, D. Wang, M. Yuan, B. F. He, J. Li, C. Mao, G. S. Wang and H. Qian, Chem. Sci., 2018, 9, 7562 RSC.
- A. M. Mohammed, P. Šulc, J. Zenk and R. Schulman, Nat. Nanotechnol., 2017, 12, 312 CrossRef CAS PubMed.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153 RSC.
- G. Zhu, R. Hu, Z. Zhao, Z. Chen, X. Zhang and W. Tan, J. Am. Chem. Soc., 2013, 135, 16438 CrossRef CAS PubMed.
- S. Yue, T. Zhao, S. Bi and Z. Zhang, Biosens. Bioelectron., 2017, 98, 234 CrossRef CAS PubMed.
- T. Omabegho, R. Sha and N. C. Seeman, Science, 2009, 324, 67 CrossRef CAS PubMed.
- L. Qian and E. Winfree, Science, 2011, 332, 1196 CrossRef CAS PubMed.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills Jr, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605 CrossRef CAS PubMed.
- A. J. Genot, D. Y. Zhang, J. Bath and A. J. Turberfield, J. Am. Chem. Soc., 2011, 133, 2177 CrossRef CAS PubMed.
- D. Yao, H. Li, Y. Guo, X. Zhou, S. Xiao and H. Liang, Chem. Commun., 2016, 52, 7556 RSC.
- L. Wang, R. Deng and J. Li, Chem. Sci., 2015, 6, 6777 RSC.
- A. P. K. K. K. Mudiyanselage, Q. Yu, M. A. Leon-Duque, B. Zhao, R. Wu and M. You, J. Am. Chem. Soc., 2018, 140, 8739 CrossRef PubMed.
- Y. Guo, J. Wu and H. Ju, Chem. Sci., 2015, 6, 4318 RSC.
- S. Liu, C. Cheng, H. Gong and L. Wang, Chem. Commun., 2015, 51, 7364 RSC.
- H. Xie, Y. Chai, Y. Yuan and R. Yuan, Chem. Commun., 2017, 53, 8368 RSC.
- X. Li, J. Xie, B. Jiang, R. Yuan and Y. Xiang, ACS Appl. Mater. Interfaces, 2017, 9, 5733 CrossRef CAS PubMed.
- D. Li, W. Cheng, Y. Li, Y. Xu, X. Li, Y. Yin, H. Ju and S. Ding, Anal. Chem., 2016, 88, 7500 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2004, 116, 281 CrossRef CAS PubMed.
- R. H. A. Plasterk, Cell, 2006, 124, 877 CrossRef CAS PubMed.
- C. Xue, S.-X. Zhang, C.-H. Ouyang, D. Chang, B. J. Salena, Y. Li and Z.-S. Wu, Angew. Chem., Int. Ed., 2018, 57, 9739 CrossRef CAS PubMed.
- G. A. Calin and C. M. Croce, Nat. Rev. Cancer, 2006, 6, 857 CrossRef CAS PubMed.
- L. He and G. J. Hannon, Nat. Rev. Genet., 2004, 5, 522 CrossRef CAS PubMed.
- X. Meng, K. Zhang, W. Dai, Y. Cao, F. Yang, H. Dong and X. Zhang, Chem. Sci., 2018, 9, 7419 RSC.
- L. Wang, R. Deng and J. Li, Chem. Sci., 2015, 6, 6777 RSC.
- P.-Q. Ma, C.-P. Liang, H.-H. Zhang, B.-C. Yin and B.-C. Ye, Chem. Sci., 2018, 9, 3299 RSC.
- H. Wang, C. Li, X. Liu, X. Zhou and F. Wang, Chem. Sci., 2018, 9, 5842 RSC.
- S. Yu, Y. Wang, L.-P. Jiang, S. Bi and J.-J. Zhu, Anal. Chem., 2018, 90, 4544 CrossRef CAS PubMed.
- S. Bi, X. Bao, J. Ye and Y. Dong, ACS Appl. Mater. Interfaces, 2015, 7, 23310 CrossRef CAS PubMed.
- Q. Wei, J. Huang, J. Li, J. Wang, X. Yang, J. Liu and K. Wang, Chem. Sci., 2018, 9, 7802 RSC.
- D. Kong, S. Bi, Z. Wang, J. Xia and F. Zhang, Anal. Chem., 2016, 88, 10667 CrossRef CAS PubMed.
- S. Volinia, G. A. Calin, C. G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R. L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C. C. Harris and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2257 CrossRef CAS PubMed.
- H. Bui, S. Shah, R. Mokhtar, T. Song, S. Garg and J. Reif, ACS Nano, 2018, 12, 1146 CrossRef CAS PubMed.
- Q. Zhao, J. Piao, W. Peng, Y. Wang, B. Zhang, X. Gong and J. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 3324 CrossRef CAS PubMed.
- D. Li, W. Cheng, Y. Li, Y. Xu, X. Li, Y. Yin, H. Ju and S. Ding, Anal. Chem., 2016, 88, 7500 CrossRef CAS PubMed.
- P. Zhang, X. Wu, R. Yuan and Y. Chai, Anal. Chem., 2015, 87, 3202 CrossRef CAS PubMed.
- D. Kong, S. Bi, Z. Wang, J. Xia and F. Zhang, Anal. Chem., 2016, 88, 10667 CrossRef CAS PubMed.
- F. Chen, M. Bai, Y. Zhao, K. Cao, X. Cao and Y. Zhao, Anal. Chem., 2018, 90, 2271 CrossRef CAS PubMed.
- W. Zhou, D. Li, C. Xiong, R. Yuan and Y. Xiang, ACS Appl. Mater. Interfaces, 2016, 8, 13303 CrossRef CAS PubMed.
- J. Ribas, X. Ni, M. Castanares, M. M. Liu, D. Esopi, S. Yegnasubramanian, R. Rodriguez, J. T. Mendell and S. E. Lupold, Nucleic Acids Res., 2012, 40, 6821 CrossRef CAS PubMed.
- S. Campuzano, R. M. Torrente-Rodríguez, E. López-Hernández, F. Conzuelo, R. Granados, J. M. Sánchez-Puelles and J. M. Pingarrón, Angew. Chem., Int. Ed., 2014, 53, 6168 CrossRef CAS PubMed.
- Q. Liu, D. Wang, M. Yuan, B. F. He, J. Li, C. Mao, G. S. Wang and H. Qian, Chem. Sci., 2018, 9, 7562 RSC.
- Q. Guo, F. Bian, Y. Liu, X. Qu, X. Hu and Q. Sun, Chem. Commun., 2017, 53, 4954 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental section, zeta potential, real sample assay, amplification efficiency of bCHA stability test and additional cell experiments. See DOI: 10.1039/c8sc04756a |
| This journal is © The Royal Society of Chemistry 2019 |