
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalysed enantioselective intramolecular etherification of propargylic esters: synthetic approach to chiral isochromans†
Shiyao Liua,
Kazunari Nakajima b and
Yoshiaki Nishibayashi*a
b and
Yoshiaki Nishibayashi*a
aDepartment of Systems Innovation, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ynishiba@sys.t.u-tokyo.ac.jp
bFrontier Research Centre for Energy and Resources, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 17th June 2019
Abstract
Enantioselective synthesis of chiral isochromans bearing a terminal alkyne moiety has been accomplished by copper-catalysed enantioselective intramolecular propargylic substitution reactions of propargylic esters with alcoholic nucleophiles. This method represents the first successful example which directly introduced a terminal alkyne group into chiral isochromans.
Optically active isochromans, a type of oxygen-containing heterocycle, are ubiquitous structures in many natural and synthetic bioactive products, and are also well-known as pharmaceutical and agrochemical candidate compounds (Fig. 1).1,2 Although the classical methods such as diastereoselective cyclisation or optical resolution of racemates can furnish the synthesis of optically active isochromans, the limitation of the scope of substrates and the complexity of the synthetic routes delayed the progress of the synthesis of optically active isochromans.3 Recently, rapidly emerging methodologies for constructing chiral isochromans by applying asymmetric catalysts are increasingly attracting attention. Typically, two kinds of strategies are listed for providing chiral isochromans by asymmetric catalysis, where one is the enantioselective C–C bond formation (Fig. 2a) and the other is the enantioselective C–O bond formation (Fig. 2b). In 2008, Jacobsen and co-workers pioneered enantioselective thiourea-catalysed addition of silyl ketene acetals to racemic chloroisochromans.4 This group reported a brand-new strategy, where carbon nucleophiles enantioselectively attack the in situ generated oxocarbenium ion intermediates.5 Since then, according to the same strategy, some research groups6–8 have reported the enantioselective synthesis of chiral α-substituted isochromans using other carbon nucleophiles.
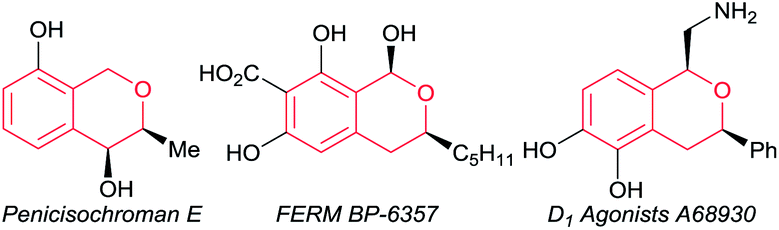 | ||
| Fig. 1 Bioactive compounds containing optically active isochroman motifs. | ||
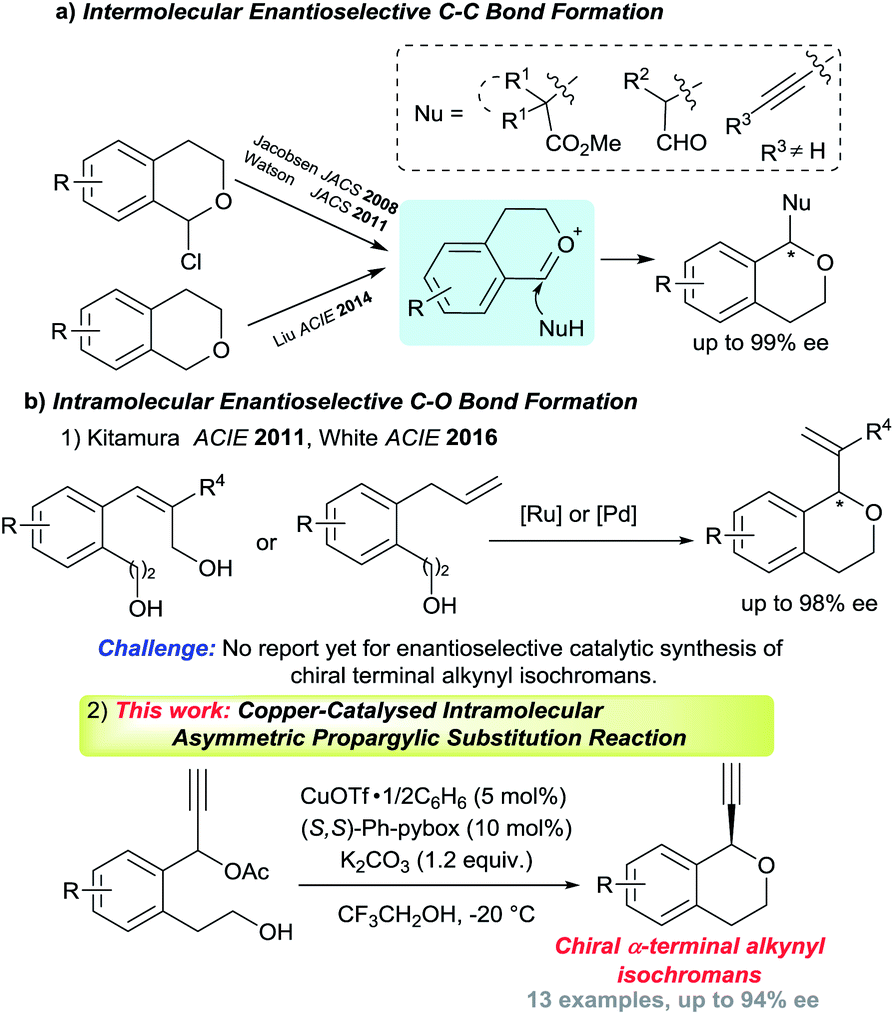 | ||
| Fig. 2 Overview of enantioselective synthesis of chiral isochromans and this work. | ||
On the other hand, reports of the synthesis of chiral isochromans via enantioselective C–O bond formation,9 where alcohols enantioselectively attack the electrophile moieties, are very scarce. To date, only two research groups found the synthesis of chiral isochromans according to the allylic substitution reactions with intramolecular alcohols. In 2011, Kitamura and co-workers reported the Ru-catalysed enantioselective intramolecular allylic etherification of allylic alcohols. In 2016, White and co-workers reported the Pd-catalysed allylic etherification of alkenes (Fig. 2b-1).10,11
Although several chiral isochroman motifs have been constructed successfully, reactions which directly introduce a terminal alkyne group into chiral isochroman motifs have not yet been achieved until now. The terminal alkyne group is one of the most diversified functional group in organic chemistry and the enantioselective introduction of the terminal alkyne group into compounds represents a considerably tough challenge in synthetic chemistry.12 Meanwhile, as a promising method to introduce a terminal alkyne group to substrates, Cu-catalysed enantioselective propargylic substitution reactions have gained considerable attention in recent years.13,14 Recently, we have achieved enantioselective intermolecular etherification of propargylic esters with alcohols,15 however, successful examples of enantioselective intramolecular etherification of propargylic esters with alcohols has not yet been reported until now. As an extensive study, we have envisaged Cu-catalysed enantioselective intramolecular etherification of propargylic esters with alcohols as a novel synthetic method for chiral isochromans bearing a terminal alkyne moiety (Fig. 2b-2). Herein, preliminary results are described.
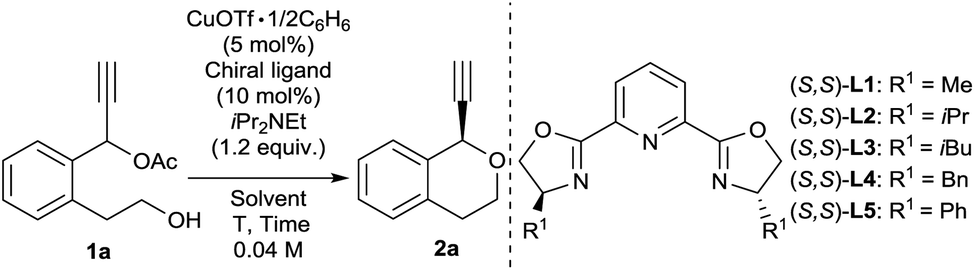
We commenced our study by using 1-(2-(2-hydroxyethyl)phenyl)prop-2-yn-1-yl acetate (1a) as a model substrate. We consider that intramolecular cyclisation of 1a may proceed to give the corresponding isochroman (2a) as a desired product. Treatment of 1a with 1.2 equivalent iPr2NEt in the presence of catalytic amounts of CuOTf·1/2C6H6 (5 mol%) and various (S,S)-pyboxs (10 mol%) as optically active ligands in MeOH at 25 °C gave 1-ethynylisochromane (2a) in good yields. At first, we investigated various (S,S)-pyboxs such as L1–L5 (Table 1, entries 1–5). In all cases, reactions proceeded smoothly. As a result, the use of L5 as an optically active ligand gave 2a with the best enantioselectivity (65% ee) (Table 1, entry 5). When other solvents such as EtOH, iPrOH, and CF3CH2OH were used in place of MeOH under the same reaction conditions (Table 1, entries 6–8), the highest enantioselectivity (88% ee) was observed in CF3CH2OH as a solvent (Table 1, entry 8). The reaction proceeded smoothly even at −20 °C to give 2a with a higher enantioselectivity (90% ee) (Table 1, entry 9). Unfortunately, the use of other copper salts such as Cu(NCCH3)4BF4, CuBr·SMe2, and Cu(OAc)2·H2O as catalysts afforded 2a with a slightly lower enantioselectivity (Table 1, entries 10–12). After fine-tuning of the effect of bases (Table 1, entries 13–15), we found that an inorganic base K2CO3 worked as the best base in the present reaction system (Table 1, entry 14).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T (°C) | Time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.1 mmol) with iPr2NEt (0.12 mmol, 1.2 equiv.) in the presence of copper salt catalyst (0.005 mmol) and chiral ligand (0.01 mmol) in solvent (2.5 mL).b Isolated yield.c Determined by chiral HPLC analysis.d Use Cu(NCCH3)4BF4 as copper salt catalyst.e Use CuBr·SMe2 as copper salt catalyst.f Use Cu(OAc)2·H2O as copper salt catalyst.g Use Et3N as base.h Use K2CO3 as base.i Use NaHCO3 as base. | ||||||
| 1 | (S,S)-L1 | MeOH | 25 | 68 | 82 | 35 |
| 2 | (S,S)-L2 | MeOH | 25 | 46 | 90 | 58 |
| 3 | (S,S)-L3 | MeOH | 25 | 47 | 76 | 59 |
| 4 | (S,S)-L4 | MeOH | 25 | 54 | 85 | 43 |
| 5 | (S,S)-L5 | MeOH | 25 | 50 | 76 | 65 |
| 6 | (S,S)-L5 | EtOH | 25 | 41 | 91 | 76 |
| 7 | (S,S)-L5 | iPrOH | 25 | 43 | 89 | 76 |
| 8 | (S,S)-L5 | CF3CH2OH | 25 | 53 | 88 | 88 |
| 9 | (S,S)-L5 | CF3CH2OH | −20 | 86 | 88 | 90 |
| 10d | (S,S)-L5 | CF3CH2OH | −20 | 112 | 87 | 85 |
| 11e | (S,S)-L5 | CF3CH2OH | −20 | 112 | 80 | 86 |
| 12f | (S,S)-L5 | CF3CH2OH | −20 | 112 | 81 | 88 |
| 13g | (S,S)-L5 | CF3CH2OH | −20 | 52 | 91 | 92 |
| 14h | (S,S)-L5 | CF3CH2OH | −20 | 51 | 89 | 93 |
| 15i | (S,S)-L5 | CF3CH2OH | −20 | 69 | Trace | — |
With optimized reaction conditions in hand, we investigated reactions of various 1 as substrates. Typical results are shown in Scheme 1. A variety of substituents on the benzene ring of 1 are well tolerated for the intramolecular etherification. In fact, reactions of propargylic acetates bearing substituents such as fluoro, trifluoromethyl, methyl, and methoxy groups at the 5-, 6-, and 7-positions of the benzene ring gave the corresponding products in good yields with a high enantioselectivity (up to 94% ee). Unfortunately, introduction of methyl group at the 8-position and chloro group at the 7-position of the benzene ring slightly decreased the enantioselectivity. The use of 1-(2-(2-hydroxyethyl)naphthalen-1-yl)prop-2-yn-1-yl acetate 1m as a substrate under the same reaction conditions gave 1-ethynyl-3,4-dihydro-1H-benzo[h]isochromene 2m in 57% yield with 83% ee. Although we have not yet obtained the exact reason why the use of compounds 1k and 1l decreased the lower enantioselectivity, we consider that this is due to the steric hindrance between the substrates and catalyst.
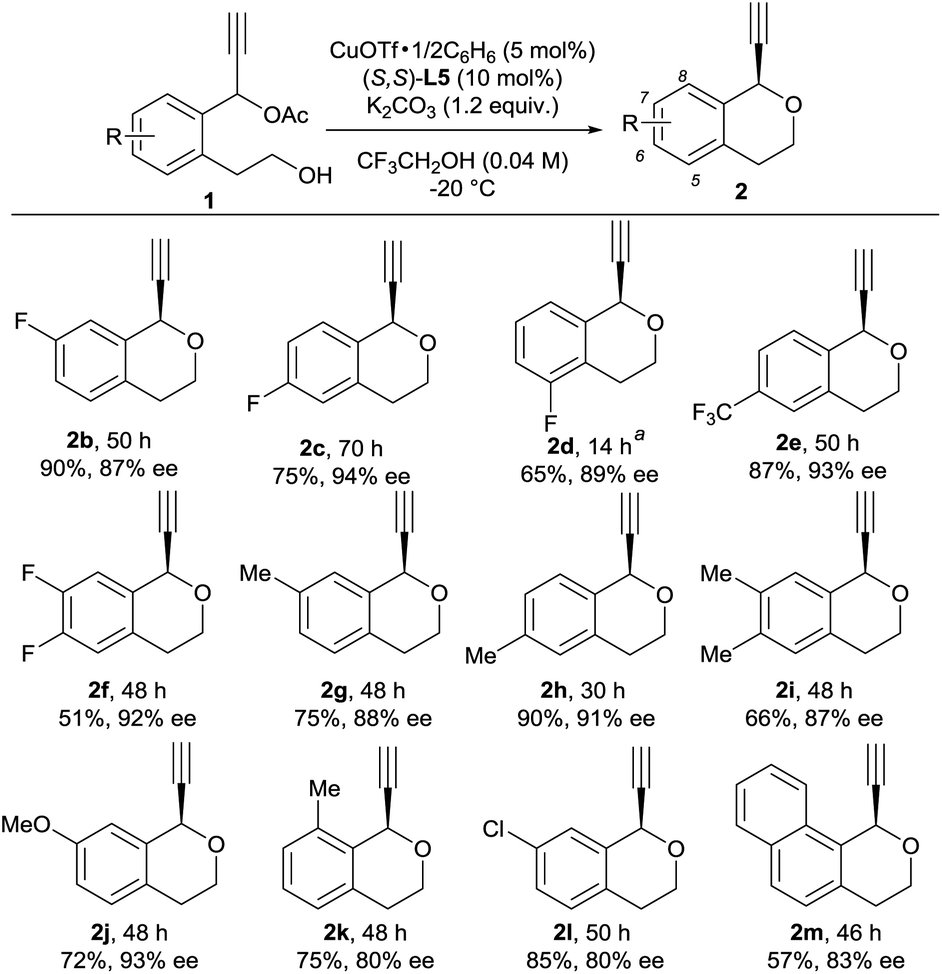 | ||
| Scheme 1 Scope of substrates. Reactions were performed on a 0.1 mmol scale with CuOTf·1/2C6H6 (5 mol%), (S,S)-L5 (10 mol%) and K2CO3 (1.2 equiv.) in CF3CH2OH (2.5 mL) at −20 °C. Yields are for the isolated products. The ee values were determined by chiral HPLC analysis. a1d (0.3 mmol), CF3CH2OH (5 mL), reaction temperature 25 °C. | ||
To further investigate the utility of the present reaction, we carried out a larger scale reaction of 1a (2 mmol). The reaction of 1a at −20 °C for 48 h gave 2a in 65% yield with 90% ee together with 1a recovered in 23% (Scheme 2a). Additionally, we investigated further transformation of 2a because the terminal alkyne moiety in 2a is a synthetically versatile as a functional group. As typical transformation of terminal alkyne, the Huisgen cycloaddition16 and the Sonogashira coupling17 of 2a were carried out (Scheme 2b). To our delight, both catalytic reactions proceeded smoothly to give the corresponding products (3 and 4, respectively) in excellent yields without the loss of the optical purity. To determine the absolute configuration of products 2, we carried out the Huisgen cycloaddition of chiral isochroman 2l with benzyl azide to give the corresponding cycloaddition product S16 successfully. After the recrystallisation, we confirmed the absolute configuration of S16 by X-ray diffraction analysis (Fig. S1†). We determined that the absolute configuration of the chiral isochroman 2l is R at the propargylic position.18
 | ||
| Scheme 2 Larger scale and diversified synthesis. Conditions: a2a (0.23 mmol), BnN3 (0.25 mmol), CuI (10 mol%), iPr2NEt (2 equiv.), THF, 45 °C, 6 h. b2a (0.24 mmol), 1-chloro-4-iodobenzene (0.29 mmol), CuI (10 mol%), Pd(PPh3)4 (5 mol%), Et3N (4 equiv.) THF, rt, 16 h. | ||
Next, to get information on the reaction mechanism, we examined a relationship between the enantiomeric purity of L5 and that of 2a (Fig. 3). As shown in Fig. 3, we obtained a positive non-linear relationship between them. This result indicates that a dicopper-allenylidene species worked as a key reactive intermediate in the present reaction system, as the similar phenomena as we observed in our previous reaction systems.15
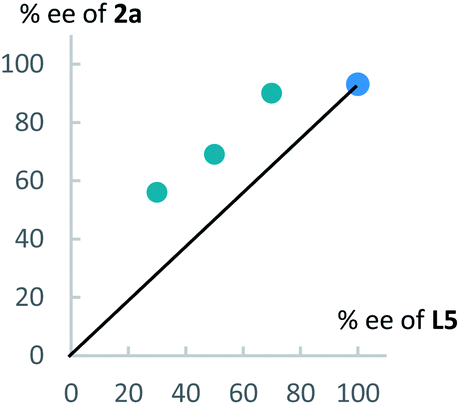 | ||
| Fig. 3 Nonlinear relationship between the enantiopurity of L5 and 2a. | ||
On the basis of the experimental results described in this paper and our previous studies, a proposed reaction pathway for the enantioselective intramolecular etherification is shown in Scheme 3a. At first, dicopper-acetylide complex (A) is formed from the dicopper complex and 1 in the presence of K2CO3 as a base. Then, dicopper-acetylide complex (B) may be formed by the elimination of acetate anion from A. Next, enantioselective intramolecular nucleophilic attack by alcohol moiety occurs to afford the corresponding dicopper-acetylide complex (C). Deprotonation of complex C occurs under the base condition to generate complex (D). Finally, complex D is converted into the starting complex A by the ligand exchange with another substrate 1 together with the formation of propargylic ether 2 as a product.
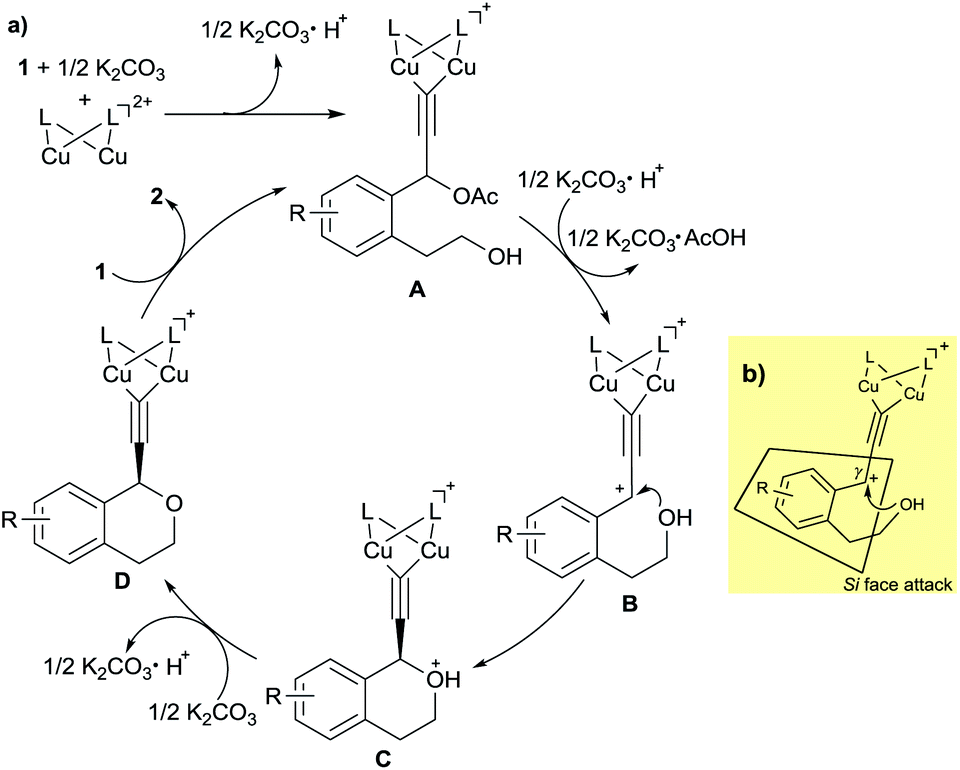 | ||
| Scheme 3 Proposed reaction pathway and transition state of dicopper-acetylide complex. | ||
To account for the enantioselective formation of propargylic ethers, we have proposed a transition state involving the dicopper-acetylide complex as shown in Scheme 3b. The absolute configuration at the propargylic position in 2 indicates that the attack of alcohol on the cationic γ-carbon in the dicopper-acetylide complex occurs from the Si face to the dicopper-acetylide ligand.
Based on results of optimisation, we believe that CF3CH2OH solvent plays a critical role to achieve the high enantioselectivity in present reaction system. Schepp and co-workers found that the use of CF3CH2OH as solvent stabilised the allylic cation.19a In their report, the rate of nucleophilic attack to allylic cations slowed down with increasing of the concentration of CF3CH2OH.19 As a result, we consider that a similar propargylic cation stabilisation might occur when CF3CH2OH is used as solvent in our present reaction system. We assume that the achievement of the high enantioselectivity in CF3CH2OH as solvent might relate to the lower reaction rate which results from the cation stabilisation. Further work is necessary to support our hypothesis.
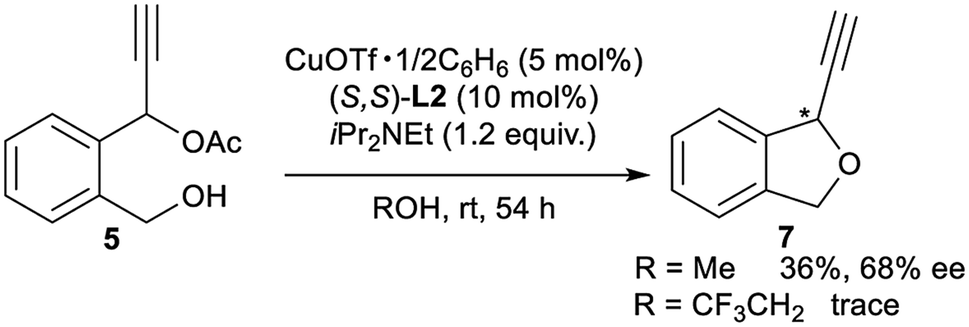
Finally, for further application of enantioselective intramolecular etherification, we attempted the use of other types of substrates bearing different lengths of carbon chains in the alcohol moiety. After the optimisation, reactions of 5 and 6 under the best reaction conditions only gave the corresponding 5-membered ring cyclic ethers as 1-ethynyl-1,3-dihydroisobenzofuran (7) and 7-membered ring cyclic ethers as 1-ethynyl-1,3,4,5-tetrahydrobenzo[c]oxepine (8) in 36% yield with 68% ee and 40% yield with 29% ee, respectively (eqn (1) and (2)). Separately, we confirmed that no reactions proceeded in CF3CH2OH as solvent. These experimental results are in sharp contrast to the enantioselective intramolecular 6-membered etherification (vide supra). Although the exact reason why the formation of 5- or 7-membered ring cyclic ethers proceeded with a low to moderate reaction rate and enantioselectivity is not so clear, we assume that it may attribute to the very delicate relationship between the lengths of carbon chains in the alcohol moiety and the protic solvent at the enantio-determining step, where 6-membered ring cyclic ethers are well-matched and 5- or 7-membered ring cyclic ethers are mismatched.
 | (1) |
 | (2) |
In summary, we have successfully established a copper-catalysed enantioselective intramolecular propargylic etherification of propargylic acetates with alcohols to give the corresponding 6-membered ring cyclic ethers in good yields with a high enantioselectivity. We believe that this is the first successful example of the enantioselective formation of chiral isochromans bearing a terminal alkyne moiety. Further exploration by using current strategy is currently now in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is supported by CREST, JST (JPMJCR1541). We thank Grants-in-Aid for Scientific Research (No. JP17H01201, JP15H05798, and JP18K19093) from JSPS and MEXT. S. L. is a recipient of the JSPS Predoctoral Fellowships for Young Scientists.Notes and references
- (a) J. Ralph, J. Peng and F. Lua, Tetrahedron Lett., 1998, 39, 4963–4964 CrossRef CAS; (b) G. He, H. Matsuura, T. Takushi, S. Kawano and T. Yoshihara, J. Nat. Prod., 2004, 67, 1084–1087 CrossRef CAS PubMed.
- (a) M. P. DeNinno, R. Schoenleber, K. E. Asin, R. MacKenzie and J. W. Kebabian, J. Med. Chem., 1990, 33, 2948–2950 CrossRef CAS PubMed; (b) K. Trisuwan, V. Rukachaisirikul, Y. Sukpondma, S. Phongpaichit, S. Preedanon and J. Sakayaroj, Tetrahedron, 2010, 66, 4484–4489 CrossRef CAS.
- E. L. Larghi and T. S. Kaufman, Synthesis, 2006, 187–220 CAS.
- S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198–7199 CrossRef CAS PubMed.
- For selected examples on asymmetric catalysis via oxocarbenium ions, see: (a) I. Coric, S. Vellalath and B. List, J. Am. Chem. Soc., 2010, 132, 8536–8537 CrossRef CAS PubMed; (b) Z. Sun, G. A. Winschel, A. Borovika and P. Nagorny, J. Am. Chem. Soc., 2012, 134, 8074–8077 CrossRef CAS PubMed; (c) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed; (d) G. C. Tsui, L. Liu and B. List, Angew. Chem., Int. Ed., 2015, 54, 7703–7706 CrossRef CAS PubMed.
- (a) P. Maity, H. D. Srinivas and M. P. Watson, J. Am. Chem. Soc., 2011, 133, 17142–17145 CrossRef CAS PubMed; (b) S. Dasgupta, T. Rivas and M. P. Watson, Angew. Chem., Int. Ed., 2015, 54, 14154–14158 CrossRef CAS PubMed.
- (a) Z. Meng, S. Sun, H. Yuan, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 543–547 CrossRef CAS PubMed; (b) M. Wan, S. Sun, Y. Li and L. Liu, Angew. Chem., Int. Ed., 2017, 56, 5116–5120 CrossRef CAS PubMed; (c) Y. Li, M. Wan, S. Sun, Z. Fu, H. Huang and L. Liu, Org. Chem. Front., 2018, 5, 1280–1283 RSC.
- S. Das, L. Liu, Y. Zheng, M. W. Alachraf, W. Thiel, C. K. De and B. List, J. Am. Chem. Soc., 2016, 138, 9429–9432 CrossRef CAS PubMed.
- For selected enantioselective C–O bond formation reactions, see: (a) G. Zhong, Angew. Chem., Int. Ed., 2003, 42, 4247–4250 CrossRef CAS PubMed; (b) C. Shu and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4794–4797 CrossRef CAS PubMed; (c) S. F. Kirsch and L. E. Overman, J. Am. Chem. Soc., 2005, 127, 2866–2867 CrossRef CAS PubMed; for reviews on enantioselective C–O bond formation, see: (d) C. F. Nising and S. Braese, Chem. Soc. Rev., 2008, 37, 1218–1228 RSC; (e) C. F. Nising and S. Braese, Chem. Soc. Rev., 2012, 41, 988–999 RSC.
- (a) K. Miyata, H. Kutsuna, S. Kawakami and M. Kitamura, Angew. Chem., Int. Ed., 2011, 50, 4649–4653 CrossRef CAS PubMed; (b) K. Miyata and M. Kitamura, Synthesis, 2012, 2138–2146 CAS.
- S. E. Ammann, W. Liu and M. C. White, Angew. Chem., Int. Ed., 2016, 55, 9571–9575 CrossRef CAS PubMed.
- (a) L. Bradsma, Preparative Acetylene Chemistry, Elsevier, Amsterdam, 2nd edn, 1988 Search PubMed; (b) P. J. Stang and F. Diederich, Modern acetylene chemistry, John Wiley & Sons, 2008 Search PubMed; (c) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2004, 4095–4105 CrossRef CAS.
- For enantioselective propargylic substitution reviews, see: (a) Y. Nishibayashi, Synthesis, 2012, 489–503 CrossRef CAS; (b) D.-Y. Zhang and X.-P. Hu, Tetrahedron Lett., 2015, 56, 283–295 CrossRef CAS; (c) k. Sakata and Y. Nishibayashi, Catal. Sci. Technol., 2018, 8, 12–25 RSC.
- For selected recent copper-catalysed enantioselective propargylic substitution reactions, see: (a) R.-Z. Li, H. Tang, K.-R. Yang, L.-Q. Wan, X. Zhang, J. Liu, Z. Fu and D.-W. Niu, Angew. Chem., Int. Ed., 2017, 56, 7213–7217 CrossRef CAS PubMed; (b) R.-Z. Li, H. Tang, L. Wan, X. Zhang, Z. Fu, J. Liu, S. Yang, D. Jia and D.-W. Niu, Chem, 2017, 3, 834–845 CrossRef CAS; (c) K. Zhang, L.-Q. Lu, S. Yao, J.-R. Chen, D.-Q. Shi and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 12847–12854 CrossRef CAS PubMed; (d) Z. Fu, N. Deng, S. N. Su, H. Li, R. Z. Li, X. Zhang, J. Liu and D.-W. Niu, Angew. Chem., Int. Ed., 2018, 57, 15217–15221 CrossRef CAS PubMed; (e) J. E. Gómez, A. Cristofol and A. W. Kleij, Angew. Chem., Int. Ed., 2019, 58, 3903–3907 CrossRef PubMed.
- K. Nakajima, M. Shibata and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 2472–2475 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- K. Sonogashira, Metal-Catalyzed Cross-Coupling Reactions, 1998, pp. 203–229 Search PubMed.
- See the ESI† in details.
- (a) G. Hallett-Tapleya, F. L. Cozensa and N. P. Schepp, J. Phys. Org. Chem., 2009, 22, 343–348 CrossRef; a similar benzylic cation stabilisation by 1,1,1,3,3,3-hexafluoroisopropanol, see: (b) J. Ammer and H. Mayr, J. Phys. Org. Chem., 2013, 26, 59–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1910939 (S16). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03880a |
| This journal is © The Royal Society of Chemistry 2019 |